 Xiaofeng Gu
Xiaofeng Gu Tiegang Lu
Tiegang Lu- Biotechnology Research Institute/National Key Facility for Genetic Resources and Gene Improvement, The Chinese Academy of Agricultural Sciences, Beijing, China
A Commentary on
Sequencing, de novo assembly and comparative analysis of Raphanus sativus transcriptome
by Wu, G., Zhang, L., Yin, Y., Wu, J., Yu, L., Zhou, Y., et al. (2015). Front Plant Sci. 6:198. doi: 10.3389/fpls.2015.00198
Obtaining sequence information for a plant species of interest greatly facilitates functional genomic research. However, many plant species have not been sequenced due to the complexities of their genomes and the high cost of sequencing. Therefore, the transcriptomes of several plants were sequenced using next-generation sequencing technology. RNA-seq technology is an important high-throughput tool for both gene mapping and the transcriptome analysis of non-model organisms lacking genome information (Schuster, 2008; Wang et al., 2009; Klie et al., 2012; Drogue et al., 2014; Druege et al., 2014). Additionally, RNA-seq data can help connect the genome to gene function (Adams, 2008) and have tremendously expanded the volume of transcriptome information available for non-model plants. The uniformly processed and matched nature of the transcriptome data also facilitates their integration with upstream factor-binding and chromatin-modification signals (Gerstein et al., 2014).
The transcriptome sequencing analyses of single tissues from Raphanus sativus were recently reported because it is an important Brassicaceae plant with both economic value and medicinal properties (Wang et al., 2013; Zhang et al., 2013). These studies have advanced functional genomic investigations of R. sativus. However, routine analysis and annotation methods have impeded our understanding of the functional genomics and medicinal potential of R. sativus. The traditional analysis of transcriptome data focuses on evaluating the expression changes of single genes. The accumulating volume of transcriptome data requires the development of new methods and tools for data integration. The network theory and similar methods have been widely used to analyze high-throughput data in the field of systems biology (Ashburner, 2000; Lee et al., 2010; Kleessen et al., 2013). Cytoscape is an open source software used for visualizing molecular interaction networks and biological pathways. The software can also integrate these networks with annotation and gene expression data (Shannon et al., 2003; Ma et al., 2014).
Using R. sativus as a model, Wu et al. (2015) combined the leaf sequencing data with the root sequencing data and obtained the better transcriptome assembly of R. sativus. For example, the authors obtained 68,086 unigenes with an average length of 576 bp by Trinity program, which represents a rich genome resource of R. sativus. Moreover, a total of 31,875 potential simple sequence repeats (SSRs) were identified in this article. SSRs are repeating DNA sequences of 2–6 bp and widely used in molecular marker identification, high-throughput genome mapping, and the analysis of species diversity. Thus, the identification of multiple SSRs in R. sativus will be very helpful for the functional genomic research of R. sativus in the future. The authors (Wu et al., 2015) used Cytoscape software to annotate these differentially expressed genes (Figure 1). The annotations effectively explained the GO classifications for the differentially expressed genes found in leaf and root tissues and are very important for functional genomics research of R. sativus. In addition, Wu et al. examined the TFs (transcription factors) in R. sativus and constructed a TF-based regulation network using Cytoscape software to elucidate the regulatory network controlling the development of R. sativus. This analysis may provide critical insights into the regulatory role of TFs in the development of R. sativus.
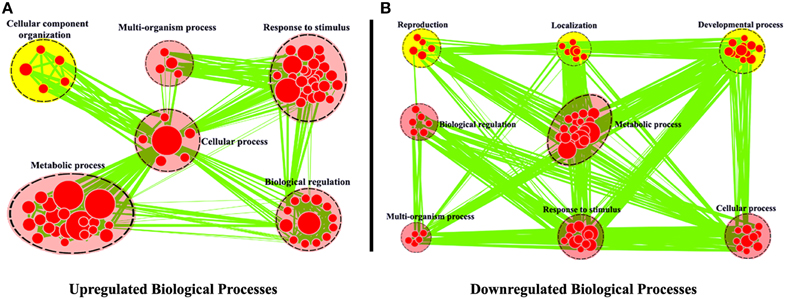
Figure 1. Biological process analysis of differentially expressed genes between leaf and root tissues. GO modules enriched with up-regulated DEGs (A) and down-regulated DEGs (B) were visualized by the EnrichmentMap in Cytoscape. The red and yellow circles indicate the common and different biological processes between up-regulated and down-regulated DEGs, respectively (From the article Wu et al., 2015).
This study is not only an important genomic resource for R. sativus because it provides both transcriptome sequencing and a TF-based interaction network but also will also facilitate future network-based functional genomic analyses and will provide insights into the systematic analysis of high-throughput sequencing data.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, J. (2008). Transcriptome: connecting the genome to gene function. Nat. Educ. 1:195. Available online at: http://www.nature.com/scitable/topicpage/transcriptome-connecting-the-genome-to-gene-function-605
Ashburner, M., Ball, C., Blake, J., Botstein, D., Butler, H., Cherry, J., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Drogue, B., Sanguin, H., Chamam, A., Mozar, M., Llauro, C., Panaud, O., et al. (2014). Plant root transcriptome profiling reveals a strain-dependent response during Azospirillum-rice cooperation. Front. Plant Sci. 5:607. doi: 10.3389/fpls.2014.00607
Druege, U., Franken, P., Lischewski, S., Ahkami, A. H., Zerche, S., Hause, B., et al. (2014). Transcriptomic analysis reveals ethylene as stimulator and auxin as regulator of adventitious root formation in petunia cuttings. Front. Plant Sci. 5:494. doi: 10.3389/fpls.2014.00494
Gerstein, M., Rozowsky, J., Yan, K., Wang, D., Cheng, C., Brown, J., et al. (2014). Comparative analysis of the transcriptome across distant species. Nature 512, 445–448. doi: 10.1038/nature13424
Kleessen, S., Klie, S., Nikoloski, Z. (2013). Data integration through proximity-based networks provides biological principles of organization across scales. Plant Cell 25, 1917–1927. doi: 10.1105/tpc.113.111039
Klie, S., Caldana, C., and Nikoloski, Z. (2012). Compromise of multiple time-resolved transcriptomics experiments identifies tightly regulated functions. Front. Plant Sci. 3:249. doi: 10.3389/fpls.2012.00249
Lee, I., Ambaru, B., Thakkar, P., Marcotte, E. M., and Rhee, S. Y. (2010). Rational association of genes with traits using a genome-scale gene network for Arabidopsis thaliana. Nat. Biotechnol. 28, 149–156. doi: 10.1038/nbt.1603
Ma, C., Xin, M. M., Feldmann, K. A., and Wang, X. F. (2014). Machine learning-based differential network analysis: a study of stress-responsive transcriptomes in Arabidopsis. Plant Cell 26, 520–537. doi: 10.1105/tpc.113.121913
Schuster, S. C. (2008). Next-generation sequencing transforms today's biology. Nat. Methods 5, 16–18. doi: 10.1038/nmeth1156
Shannon, P., Markiel, A., Ozier, O., Baliga, N., Wang, J., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Wang, Y., Xu, L., Chen, Y., Shen, H., Gong, Y., Limera, C., et al. (2013). Transcriptome profiling of radish (Raphanus sativus L.) root and identification of genes involved in response to lead (Pb) stress with next generation sequencing. PLoS ONE 8:e66539. doi: 10.1371/journal.pone.0066539
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet 10, 57–63. doi: 10.1038/nrg2484
Wu, G., Zhang, L., Yin, Y., Wu, J., Yu, L., Zhou, Y., et al. (2015). Sequencing, de novo assembly and comparative analysis of Raphanus sativus transcriptome. Front Plant Sci. 6:198. doi: 10.3389/fpls.2015.00198
Keywords: Raphanus sativus, transcription factors, RNA sequencing (RNA-Seq), trancriptome, simple sequence repeats (SSRs)
Citation: Gu X and Lu T (2016) Commentary: Comparative Transcriptome Analysis of Raphanus sativus Tissues. Front. Plant Sci. 6:1191. doi: 10.3389/fpls.2015.01191
Received: 05 October 2015; Accepted: 11 December 2015;
Published: 05 January 2016.
Edited by:
Elena R. Alvarez-Buylla, Universidad Nacional Autónoma de Mexico, MexicoReviewed by:
Stefan De Folter, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoCopyright © 2016 Gu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiegang Lu, bHV0aWVnYW5nQGNhYXMuY24=