 Aparupa Bose Mazumdar
Aparupa Bose Mazumdar Sharmila Chattopadhyay
Sharmila Chattopadhyay- Organic and Medicinal Chemistry Division, Plant Biology Laboratory, Council for Scientific and Industrial Research-Indian Institute of Chemical Biology, Kolkata, India
Phyllanthus amarus Schum. and Thonn., a widely distributed annual medicinal herb has a long history of use in the traditional system of medicine for over 2000 years. However, the lack of genomic data for P. amarus, a non-model organism hinders research at the molecular level. In the present study, high-throughput sequencing technology has been employed to enhance better understanding of this herb and provide comprehensive genomic information for future work. Here P. amarus leaf transcriptome was sequenced using the Illumina Miseq platform. We assembled 85,927 non-redundant (nr) “unitranscript” sequences with an average length of 1548 bp, from 18,060,997 raw reads. Sequence similarity analyses and annotation of these unitranscripts were performed against databases like green plants nr protein database, Gene Ontology (GO), Clusters of Orthologous Groups (COG), PlnTFDB, KEGG databases. As a result, 69,394 GO terms, 583 enzyme codes (EC), 134 KEGG maps, and 59 Transcription Factor (TF) families were generated. Functional and comparative analyses of assembled unitranscripts were also performed with the most closely related species like Populus trichocarpa and Ricinus communis using TRAPID. KEGG analysis showed that a number of assembled unitranscripts were involved in secondary metabolites, mainly phenylpropanoid, flavonoid, terpenoids, alkaloids, and lignan biosynthetic pathways that have significant medicinal attributes. Further, Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values of the identified secondary metabolite pathway genes were determined and Reverse Transcription PCR (RT-PCR) of a few of these genes were performed to validate the de novo assembled leaf transcriptome dataset. In addition 65,273 simple sequence repeats (SSRs) were also identified. To the best of our knowledge, this is the first transcriptomic dataset of P. amarus till date. Our study provides the largest genetic resource that will lead to drug development and pave the way in deciphering various secondary metabolite biosynthetic pathways in P. amarus, especially those conferring the medicinal attributes of this potent herb.
Introduction
P. amarus Schum. and Thonn., a member of family Euphorbiaceae is used in the traditional system of medicine like Ayurveda for over the centuries because of its rich medicinal values and ethnomedicinal importance. P. amarus Schum. and Thonn. is a small, erect annual herb whose stem has a green, smooth capsule, and grows up to 10–50 cm high. Over the last few decades, P. amarus has gained global recognition for its medicinal properties after several studies that were conducted to understand its therapeutic potential, yielding exciting results. However, studies about the DNA or protein sequences of this species are very limited. P. amarus, popularly known as “bhuiamlaki” is distributed worldwide. In Spain, this plant is best known by the common name “chanca piedra,” which means stone-breaker. The most significant hepatoprotective role of P. amarus has long been reported (Thyagarajan et al., 1988; Blumberg et al., 1990). The genus Phyllanthus has prospective beneficial therapeutic actions in the management of hepatitis B, nefrolitiase, and in painful disorders (Calixto et al., 1998). Recent studies have also reported the hepatoprotection property of P. amarus (Chirdchupunseree and Pramyothin, 2010; Krithika et al., 2014). In addition to this, P. amarus has also shown to exhibit antioxidant (Harish and Shivanandappa, 2006), antitumor and anticarcinogenic activities (Rajeshkumar et al., 2002). Besides, anti-allodynic and antioedematogenic properties, as well as antimicrobial potentiality (Mazumder et al., 2006) of this herb, have also been reported. Report of α-amylase inhibitory properties of P. amarus in treating diabetes has also been shown (Ali et al., 2006). The wide variety of secondary metabolites, that attribute to these medicinal properties are present mainly in the leaves and include lignans mainly phyllanthin and hypophyllanthin (Chopra et al., 1956; Rao and Bramley, 1971; Somanabandhu et al., 1993) besides nirtetralin, niranthin, diarylbutane, nyrphyllin, and a neolignan, phyllnirurin; geraniin and flavonoids like quercetin, astralgin, quercetrin, isoquercetin, and rutin (Umezawa, 2003; Kassuya et al., 2006; Leite et al., 2006). It also contains minor compounds like hydrolysable tannins like phyllanthusiin D (Foo and Wong, 1992), amariin, amarulone (Foo, 1993), amarinic acid (Foo, 1995) and alkaloids like entnorsecurinine, isobubbialine, and epibubbialine (Houghton et al., 1996).
A number of reports addressed the genetic diversity of P. amarus for application in the cultivar identification using PCR and sequencing based techniques viz. RFLP, RAPD, ISSR, SCAR, and AFLP (Jain et al., 2003; Senapati et al., 2011; Bandyopadhyay and Raychaudhuri, 2013). Despite its global medicinal importance genomic sequence resources available for P. amarus are extremely scarce. As of July 2015, only 119 ESTs, 105 genome survey sequences, and 188 nucleotide sequences are available at the National Center for Biotechnology Information (NCBI) database. Out of the 119 ESTs, 57 sequences were reported in our previous study on P. amarus leaves (Chattopadhyay and Bose Mazumdar Ghosh, 2014). In view of this extremely limited genome sequence, an in-depth study of transcriptome might facilitate the analysis of functional genes and thereby unravel the transcripts involved in several biological processes and mainly help understand the various metabolic pathways involved in the phytotherapeutic attributes of P. amarus.
The recent emergence of the next-generation sequencing (NGS) technology has made the rapid transcriptome sequencing more feasible. Previous studies have shown that development of RNA sequencing (RNA-seq) methodology has facilitated the analysis of transcriptomes of a number of models as well as non-model crop and medicinal plants (Nakasugi et al., 2013; Lehnert and Walbot, 2014; Rastogi et al., 2014). The main advantage of RNA-seq compared with the whole genome sequencing is that only transcribed regions of the genome are analyzed in the former. It is among the most popular techniques of NGS and this methodology still remains the golden standard for both coding and non-coding gene annotation. RNA-seq method offers a comprehensive and integrated view of the transcriptome revealing SNPs, novel transcribed regions, as well as the precise location of transcription boundaries (Wilhelm et al., 2010). Furthermore, RNA-seq data with NGS technologies help in assessing the process of different forms of alternative splicing from both plant and mammalian genomes as well (Rogers et al., 2012). The approach of eukaryotic transcriptome analysis is expected to get highly altered by the advanced RNA-seq technology.
In the present study, an attempt has been made to annotate and analyze the leaf transcriptome of P. amarus since the vast array of secondary metabolites are present substantially in the leaf tissues. We performed de novo transcriptome sequencing using the Illumina Miseq platform as prior genome information on P. amarus is unavailable. Here for RNA-seq analysis, we chose a Miseq platform because compared to other Illumina platforms the longer length of the sequencing reads generated from the Miseq platform considerably enhances the accuracy of the subsequent de novo assembly, besides being a rapid and cost-effective platform for transcriptome assembly and analyses. To the best of our knowledge, this is the first report of de novo sequencing and transcriptome analysis of P. amarus which will serve for the discovery of different genes involved in various metabolic pathways, especially the putative members of medicinally important secondary metabolites biosynthetic pathways and also help in the development of molecular markers like Simple Sequence Repeat (SSR) for enhancing the medicinal traits of this herb.
Materials and Methods
Ethics Statements
All necessary permits for plant sample collection for our present study were obtained. CSIR-Indian Institute of Chemical Biology, Kolkata is the authority responsible for P. amarus cultivation in its medicinal plant garden which provides permission to collect the samples for our scientific research.
Sample Preparation
Leaf samples of P. amarus Schum. and Thonn. cultivar, taxonomically identified by the Botanical Survey of India, Shibpur, Howrah, as PA202 were chosen for our study. Leaf samples from young, healthy plants were collected. RNA was extracted separately from leaf samples of the two samplings using “Roche High Pure RNA Isolation Kit,” (Product no.11828665001). The purity and concentration of each RNA sample were checked by using the Agilent 2100 bioanalyzer (Agilent Technologies, USA) before proceeding to further downstream analyses. Library preparation was performed from 1 μg of total RNA, using Illumina's “TruSeq® RNA Sample Preparation v2 Guide” (Part # 15026495 Rev. F March 2014).
Illumina Sequencing and Quality Control
Illumina MiSeq system was used for sequencing the P. amarus leaf transcriptome library using Sequencing by Synthesis (SBS) technology. The library has been sequenced following manufacturer's instructions using the MiSeq Reagent Kit v2 (Part # 15034097 Rev. B). The base-calling pipeline of Illumina's MiSeq version was MiSeq Control Software 2.2.0-RTA 1.17.28.0—CASAVA-1.8.2 which was used to generate paired-end and single-end data in FastQ format. Low-quality bases result in misassemblies by interfering in the assembly process. Hence, quality filter is the first and foremost requisite for all downstream computational analyses and results interpretation (Li et al., 2015). So additional quality control of raw data using FastQC was performed. The reads were preprocessed using Trimmomatic and SeqPrep software to obtain clean paired-end and single-end MiSeq data in a FastQ format which was also subjected to quality control using FastQC. The high quality, filtered reads were used for downstream analyses.
De novo Assembly and Clustering
Transcriptome de novo assembly was carried out on three levels using both paired and unpaired high-quality reads as inputs. Velvet (version 1.2.09) and Oases (version 1.2.09) were used for first level assembly and the clean reads were split into different “k-mers” from kmer27-kmer63. The transcripts obtained from all the “kmers” were merged and assembled at level 2 using Velvet and Oases. This level 2 assembled transcript was further assembled and clustered using CD-HIT (version 4.5.4) to remove redundant transcripts (Li and Godzik, 2006). This level 3 assembly and clustering represented the final dataset of clustered non-redundant (nr) unique sequences (“unitranscripts”) for P. amarus leaf transcriptome.
Gene, Pathway Annotation, Classification
Functional annotations were performed by sequence comparison with public databases. For sequence similarity search, the annotation of unitranscripts was performed by BLASTX (Altschul et al., 1997) at NCBI using green plants (taxid: 33090) of nr protein database. The BLASTX results were imported to Blast2GO suite (Conesa and Götz, 2008) for mapping and retrieving GO terms to the assembled sequences, and further annotated with unique enzyme codes (EC) and KEGG maps (http://www.genome.jp/kegg) (Kanehisa and Goto, 2000; Kanehisa et al., 2014). GO terms are dynamic-structured, precisely defined controlled vocabulary that can be employed to describe functions of genes and gene products. These retrieved GO terms were allocated to query sequences and the extensive groups of genes present in P. amarus leaf transcriptome were classified into three categories - biological process, molecular function, and cellular component. Then WEGO (Ye et al., 2006) tool (http://wego.genomics.org.cn) was used to functionally classify GO terms and graphically represent the unitranscript functions at the macro level. Further BLASTX against the Clusters of Orthologous Groups database (http://www.ncbi.nlm.nih.gov/COG/) was performed to predict and classify functions of the assembled unitranscripts, using Autofact (http://megasun.bch.umontreal.ca/Software/AutoFACT.htm) tool (Koski et al., 2005).
Comparison of P. amarus Assembled Data with Closely Related Species
Comparison of P. amarus assembled unitranscripts was carried out with the most closely related species that were obtained after the functional annotation of the leaf transcriptome assembled data at NCBI green plants nr protein database. Comparison with the closely related species were performed using TRAPID analysis (Van Bel et al., 2013) with similarity search E-value 10e-5. Populus trichocarpa was the most closely related species followed by Ricinus communis. A comparison between both the species were performed and the latter being a member of the same spurge family, Euphorbiaceae to which P. amarus belongs.
Identification of Transcription Factor Families and Its Domain Architecture
For identification of transcription factor (TF) families and domain mapping represented in P. amarus leaf transcriptome, the representative unitranscripts were enquired against the TF protein sequences at Plant TF database (PlnTFDB; http://plntfdb.bio.uni-potsdam.de/v3.0/downloads.php) by BLASTX with an E-value cutoff 1E-06.
Identification of Simple Sequence Repeats (SSRs)
SSRs were detected using MIcroSAtellite Identification Tool (MISA).
FPKM Value Determination of Major Secondary Metabolic Pathway Genes and Reverse Transcription PCR (RT-PCR) of Selected Secondary Metabolite Biosynthetic Pathway Genes in P. amarus Leaf Sample
For estimation of mRNA or unitranscript abundance of the major identified secondary metabolic pathway genes in the present study, FPKM values were determined. FPKM values for the unitranscripts were determined using the formula FPKM = (109 × C)/(N × L), where C = Number of reads mapped to a unitranscript; N = Total mapped reads in the experiment; L = unitranscript length in base pairs. Further, RT-PCR enables the detection and identification of target mRNA transcripts. Hence, to validate our dataset, some of the assembled P. amarus unitranscripts that share sequence similarity to various secondary metabolite biosynthetic pathway genes and related TFs as identified revealing putative information of P. amarus leaf transcriptome were selected for performing RT-PCR. All primers for RT-PCR of selected secondary metabolite pathway genes were designed from the final assembled and clustered nr unique sequences (Supplementary File 1). The housekeeping gene actin was used as a control. Actin primers were designed (Acc No.: X63603) and the primer sequences were 5′-CGCGAAAAGATGACTCAAATC and 5′- AGATCCTTTCTGATATCCACG-3′. The RT-PCR products were electrophoresed on 1.2% agarose gel containing ethidium bromide.
Results and Discussions
De novo Assembly and Clustering of P. amarus leaf Transcriptome
The Illumina Miseq platform generated a total of 18,060,997 raw reads for P. amarus leaf transcriptome that accounted for approximately 9 Gbases of sequence data. The raw data was also deposited in the National Center for Biotechnology Information's (NCBI) Short Read Archive (SRA) database under the accession number PRJNA248079. Raw reads were further subjected to quality control (FastQC). After quality and adaptor trimming using Trimmomatic 0.30 tool, 14,608,389 high quality paired reads, 2,291,081 (unpaired Reads R1) and 371,454 (unpaired Reads R2) were retained. All these filtered paired and unpaired reads were used in the transcriptome assembly. The summary of the filtration of total raw reads generated after RNA-seq and used further for transcriptome assembly is illustrated in Supplementary Figure 1. Filtered paired and unpaired reads were assembled using Velvet (version 1.2.09) and Oases (version 1.2.09) and the clean reads obtained were split into different k-mers from kmer27-kmer63. As a function of k-mer various output parameters were analyzed in our level 1 assembly. These parameters included—total number of transcripts, transcripts with length 100 bp and above, N50 length, longest transcript length, and average transcript length. The number of clean transcripts obtained in each kmer along with its total length, the average size and N50 value are summarized in Supplementary Table S1. For accuracy in P. amarus leaf transcriptome de novo assembly, we assembled and further merged the transcripts from kmer27-kmer63 using Velvet and Oases with long read option to obtain 360,405 transcripts in level 2 single merged assembly. To remove redundancy of the merged assembled transcripts, we used CD-HIT (version 4.5.4) to merge the level 2 assembled sequences further. Merging of the assembled transcripts resulted in 85,927 unitranscripts with maximum and minimum read lengths being 13,600 and 200 bp respectively, with an average size of the assembled unitranscripts being 1548 bp which indicated an increased coverage as well as the depth of our sequencing data. The outline of the level 2 and 3 assemblies has been summarized in Supplementary Table S2. The sequence length along with BLASTX hit and e-value distribution of P. amarus unitranscripts have been shown in Figure 1.
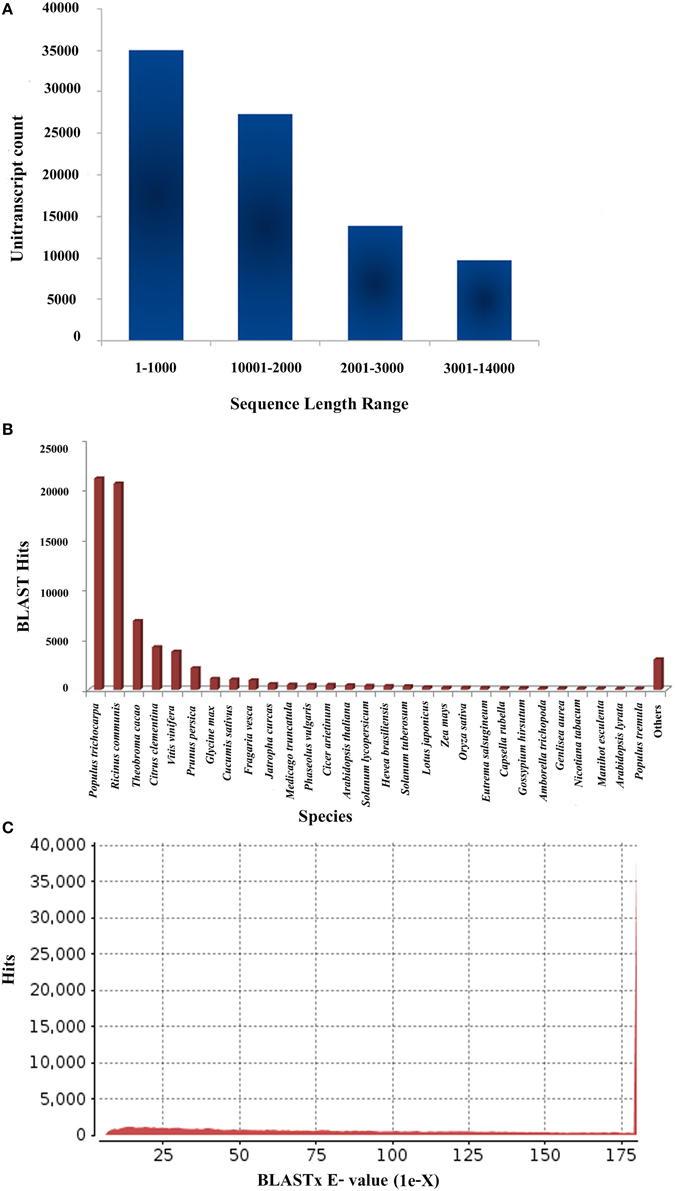
Figure 1. Sequencing, de novo assembly, annotation of P. amarus leaf transcriptome. (A) Sequence length distribution of P. amarus non-redundant unique unitranscript sequences. (B) BLASTX-Hit species distribution of P. amarus unitranscripts against nr protein database. (C) E-value distribution of BLAST hits against nr protein database.
Similarity Search and Functional Annotation of P. amarus Leaf Transcriptome
P. amarus being a non-model plant of medicinal value without any prior genome information, sequence similarity search and comparison for the assembled unitranscripts of P. amarus leaf transcriptome was carried out by BLASTX against the green plants of nr protein database at NCBI, with an E-value cut off 1E-06 (Supplementary File 2). The BLASTX hit results showed that about 60.58% of the annotated descriptions were uninformative (e.g., “unknown,” “unnamed,” “putative,” or “hypothetical” protein) as a result of inadequate P. amarus genome information. Our dataset showed that the percentage of uninformative BLASTX hit results were nearly similar to endangered medicinal plant species Chlorophytum borivilianum (Kalra et al., 2013). Also, 15,265 out of 85,927, i.e., 17.76% unitranscripts were without any hits in plant nr database. In our dataset unitranscripts showed significant similarity to Populus trichocarpa, followed by Ricinus communis, Theobroma cacao and so forth (Figure 1B). BLAST2GO suite was then used for functional annotation using the BLASTX results. Graphical representation of different levels of annotations of P. amarus unitranscripts by BLAST2GO showing mapping against different databases (UniprotKB, TAIR, etc.), annotation score distribution of assembled unitranscripts, sequence similarity distribution, distribution of annotated sequences with length, GO level distribution of annotated unitranscript sequences have been shown in Figure 2. Blast2GO is a suitable tool for plant genomics research, especially for the large-scale functional annotation and data mining of novel sequence data of non-model species. BLASTX results were used for mapping to retrieve GO terms and further annotate to retrieve the EC (EC number). To reveal molecular interaction network and metabolic pathways, KEGG pathway annotation for the assembled unitranscripts of P. amarus leaf transcriptome was performed by mapping the sequences obtained from BLAST2GO to the contents of the KEGG metabolic pathway database. Annotation summary of the assembled P. amarus unitranscripts has been specified in Supplementary Table S3.
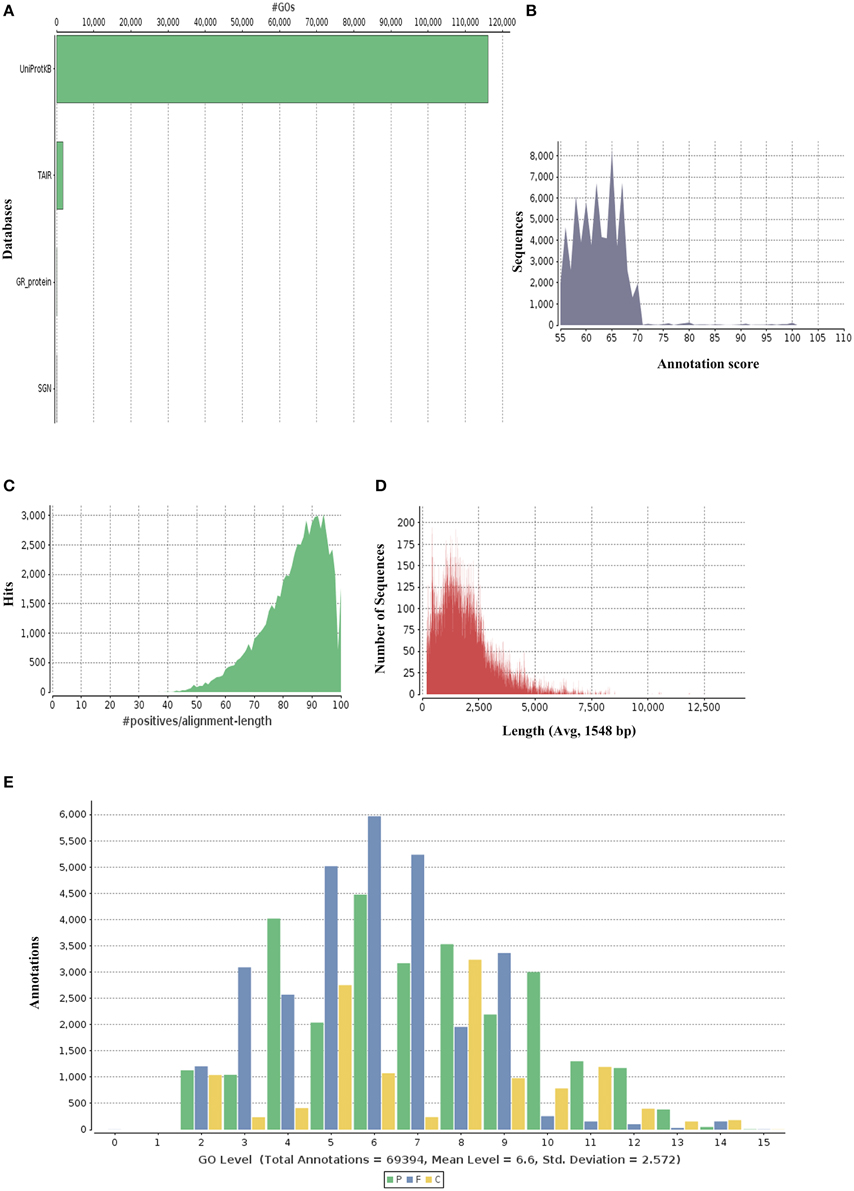
Figure 2. Graphical representations of functional annotations in P. amarus leaf transcriptome. (A) Representation of mapping databases (UniprotKB, TAIR) sources. (B) Annotation score distribution of assembled unitranscripts. (C) Sequence similarity distribution graph. (D) Distribution of annotated sequences with length. (E) GO level distribution of annotated unitranscript sequences.
Sequence Similarity and Comparison of P. amarus Data with Related Species
Functional annotation of the assembled unitranscripts of P. amarus leaf transcriptome at NCBI green plants nr protein database showed high similarity to Populus trichocarpa and Ricinus communis. So comparison of the assembled data was performed with both these species. Both the species are known to possess medicinal values like anti-inflammatory, analgesic being common to both besides other properties and also R. communis belong to the same spurge family, Euphorbiaceae like P. amarus, as already mentioned. So we aimed to compare our sequenced assembled data with both the species using TRAPID analysis, which aids in the generation of detailed gene catalogs, especially for non-model species. Out of the total 85,927 unitranscripts, 71,896 (83.7%) unitranscripts showed similarity to P. trichocarpa while that of 71,358 (83%) unitranscripts showed similarity to R. communis. The detailed comparison of P. amarus leaf transcriptome data with both the species showing the Meta annotation, Gene Family and Functional Annotation information have been shown in Supplementary Table S4.
Functional Classifications by Gene Ontology (GO)
The GO database is a significant web resource in the bioinformatics field. GO provides a set of dynamic, controlled and structured vocabularies for describing the roles of genes and their products in any organism (Ashburner et al., 2000). The three categories of the GO database are—biological process, molecular function and cellular component. P. amarus unitranscripts with nr annotations were functionally annotated with “GO terms” by BLAST2GO suite. Further WEGO software was used for the GO functional classification of the assembled P. amarus unitranscripts at the macro level.
A total of 20,582 P. amarus unitranscripts were assigned to 69,394 GO terms and one unitranscript was assigned more than one GO term. The majority of GO terms was assigned to molecular function (29,125, 41.97%), followed by biological process (27,577, 39.74%) and cellular component (12,692, 18.29%) was the least.
Regarding the cellular component ontology, “cell” (GO: 0005623), “cell part” (GO: 0044464), and “organelle” (GO: 0043226) were the most representative category. Under molecular function ontology, results showed a high percentage of genes from “binding” (GO: 0005488) and “catalytic activity” (GO: 0003824). Some percentage of genes were also involved in “antioxidant activity” (GO: 0016209) in the molecular function ontology as well. Moreover, biological process ontology contained mainly genes involved in “metabolic process” (GO: 0008152), “cellular process” (GO: 0009987). Figure 3 shows the categorization of P. amarus unitranscripts into three main ontologies and 47 sub-groups.
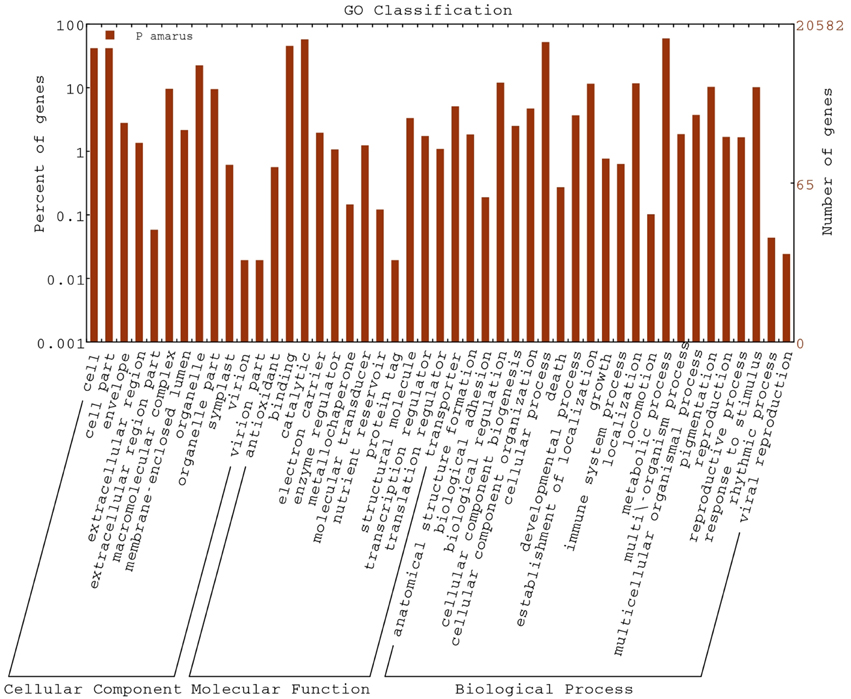
Figure 3. GO functional classifications using WEGO.
Functional Classifications by COG
The Cluster of Orthologous Groups (COG) database classifies orthologous gene products. The unitranscripts obtained in our study were aligned to the COG database to predict and classify their possible functions (Supplementary File 3). COG classification of the assembled unitranscripts showed that 28,121 (32.7%) unitranscripts were clustered into 24 functional categories (Figure 4). Among the different COG classes, the highest number of unitranscripts were clustered into the “general function prediction only” category (5003, 17.791%) followed by “posttranslational modification, protein turnover, chaperones” (4110, 14.615%), “translation, ribosomal structure and biogenesis” (2671, 9.498%), “amino acid transport and metabolism” (1889, 6.717%), “transcription” (1872, 6.657%), “energy production and conversion” (1687, 6.00%), “carbohydrate transport and metabolism” (1569, 5.579%), “lipid transport and metabolism” (1234, 4.388%), “signal transduction mechanisms” (1136, 4.04%). Only a few unitranscripts were assigned to “cytoskeleton,” “chromatin structure and dynamics,” “cell motility,” and “nuclear structure” (261, 199, 35, and 2, respectively). Also, 950 and 431 unitranscripts were clustered into “inorganic ion transport and metabolism,” and “nucleotide transport and metabolism” respectively. Interestingly our dataset also showed that 792 unitranscripts that constituted 2.8% of total unitranscripts annotated with COG database represented the “secondary metabolites biosynthesis, transport and catabolism” category which indicates the large number of secondary metabolites present in P. amarus. This finding is very similar to the previous results of KOG classification studied in another rhizomatous perennial plant Curcuma longa with significant therapeutic potentials (Annadurai et al., 2013).
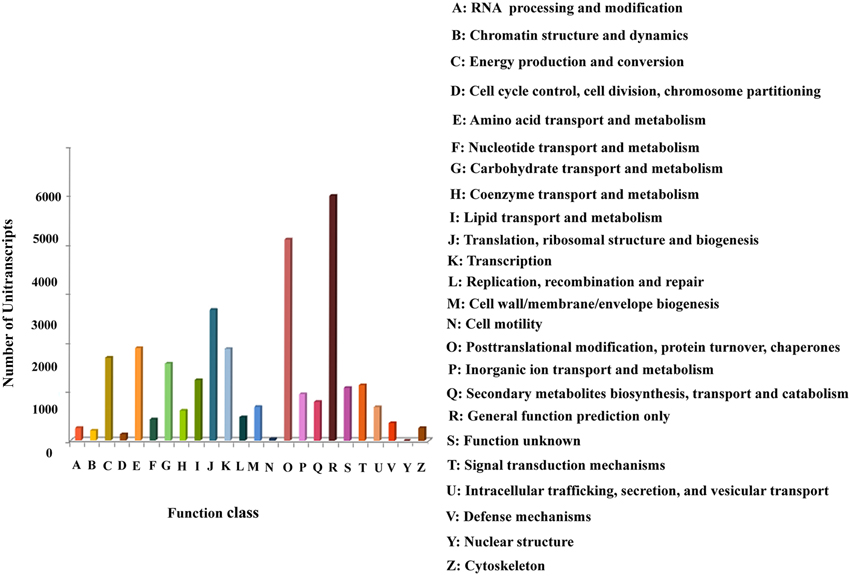
Figure 4. Clusters of Orthologous Groups (COG) classification of P. amarus unitranscripts.
Pathway Analysis by KEGG
Biological pathway studies play a key role in gaining insight into the advanced studies of genomics. KEGG is a highly integrated database providing information of the biological systems and their relationships at the molecular, cellular and organism levels, particularly via the KEGG pathway maps (Kanehisa et al., 2008). KEGG pathway annotations and EC were generated (Supplementary File 4) from the assembled unitranscripts of P. amarus leaf transcriptome that were mapped with GO terms. In total, 4,697 P. amarus unitranscripts were assigned to 134 KEGG maps and 583 EC and these EC were then used to retrieve and color the KEGG pathway maps to represent the putatively identified genes involved in several metabolic pathways. Interestingly, in our dataset it was seen that more than one unique sequence of P. amarus leaf transcriptome was annotated as the same enzyme. Enzymes encoded by annotated unitranscripts were grouped into the 5 major pathways in the KEGG pathway database (Figure 5A)—“metabolism” (9323 unitranscripts), “genetic information processing” (78), “environmental information processing” (191), “organismal systems” (303), “human diseases” (2). Apparently “metabolism” being one of the most significant and the most highly represented category in our study led to the in-depth analysis of this and has been represented in Figure 5B. In our dataset, it was seen that the maximum number of unitranscripts fell under the “carbohydrate metabolism” (2424 unitranscripts) followed by “amino acid metabolism” (1740 unitranscripts), “lipid metabolism” (1143 unitranscripts), “nucleotide metabolism” (637 unitranscripts). Nucleotide metabolism plays a vital role in plants for metabolism and development like other organisms. Besides, although lipid metabolism closely relates to oil plants mostly, and since P. amarus is a plant of therapeutic importance, lipid metabolism is also associated with plants in general (Mazliak, 1973). A number of P. amarus unique sequences (1332 unitranscripts) were involved in secondary metabolism as well (Supplementary Table S5). Out of all secondary metabolite pathways, “flavonoid biosynthesis” pathway was shown to be encoded by the highest number of P. amarus assembled unitranscripts (134 unitranscripts) followed by “phenylpropanoid biosynthesis” (125 unitranscripts). The entire functional KEGG pathway categorization of P. amarus leaf transcriptome unitranscripts have been shown in Supplementary Table S6.
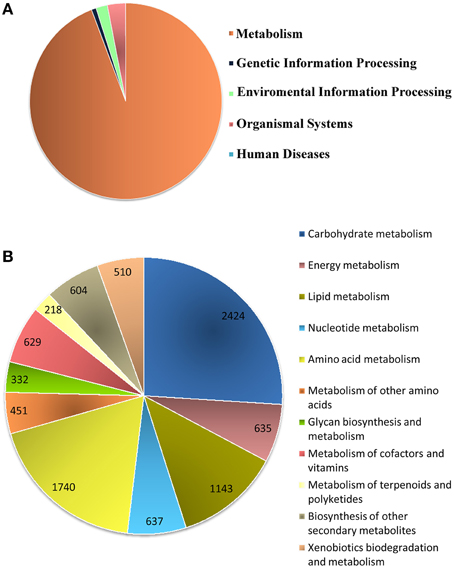
Figure 5. Annotation of P. amarus unitranscripts by KEGG database. (A) Distribution of P. amarus unitranscripts into KEGG biological categories. (B) Classification of P. amarus leaf transcriptome into KEGG “Metabolism” category.
Analysis of Secondary Metabolic Pathway Genes
Lignan Biosynthetic Genes
Phenylpropanoids which comprise a large group of plant-based natural compounds is derived from phenylalanine (Michal, 1999). Phenylpropanoid biosynthesis pathway starts with the formation of cinnamic acid from phenylalanine, which leads the formation of cinnamoyl-CoA, p-coumaroyl-CoA, feruloyl-CoA, and sinapoyl-CoA. These CoA-activated compounds are starting metabolites for the synthesis of lignans, flavonoids, flavones, and flavonols, etc. In the present study KEGG analyses of P. amarus leaf transcriptome sequences revealed the presence of 11 genes involved in the biosynthesis of different compounds of phenylpropanoid pathway (Figure 6).
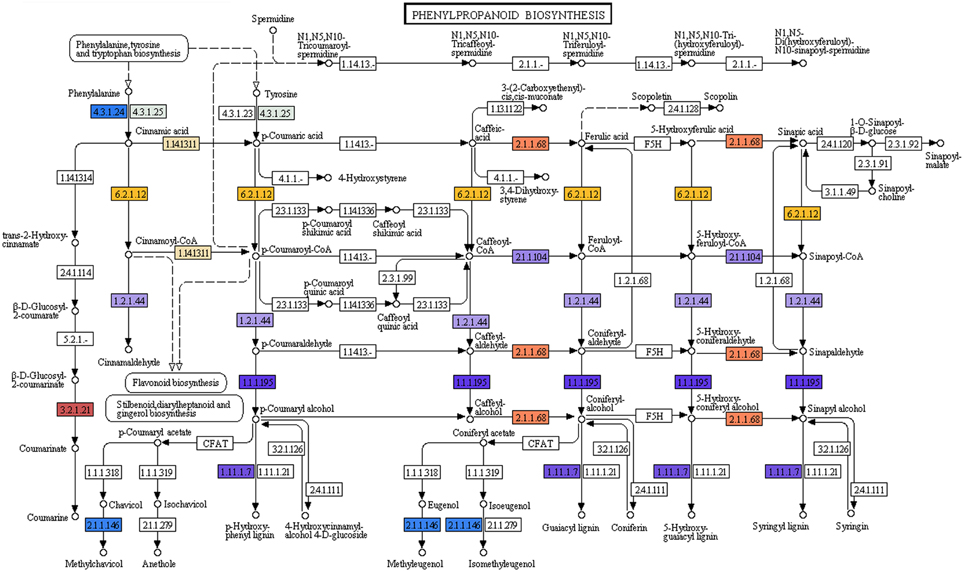
Figure 6. Phenylpropanoid biosynthesis pathway study by KEGG analysis showing the different identified enzymes (one color for each Enzyme Code or EC).
The major lignans reported in P. amarus—phyllanthin and hypophyllanthin are known to possess significant hepatoprotective and antioxidant properties. But the exact biosynthetic pathway leading to the formation of phyllanthin and hypophyllanthin is still under investigation. The structural similarity between the skeleton of secoisolariciresinol and phyllanthin is suggestive of the derivation of phyllanthin/hypophyllanthin from secoisolariciresinol. The presence of pinoresinol/lariciresinol reductase gene was indicated due to the presence of (+) secoisolariciresinol in species of Phyllanthus (Umezawa et al., 1997). In our leaf transcriptomic data, one unique sequence (unitranscript 77577) was assigned the pinoresinol reductase activity gene ontology term (GO: 0010283) after mapping the assembled P. amarus unitranscripts into BLAST2GO suite. Another six unique sequences (unitranscripts 63241; 63242; 63243; 63244; 63245; and 63246) encoding phenylcoumaran benzylic ether reductase (PCBER) - like protein were annotated the lignan biosynthetic process GO term (GO: 0009807). Besides, PCBER has also shown to have high sequence similarity with PLR, i.e., pinoresinol/lariciresinol reductase (Gang et al., 1999; Vander Mijnsbrugge et al., 2000), showing the active involvement of P. amarus leaves in lignans biosynthesis and thus complementing its phytotherapeutic properties.
Study of Flavonoid Biosynthesis and Related Pathway Genes in the P. amarus Leaf Transcriptome
Flavonoids, a class of plant secondary metabolites, are polyphenolic compounds that are categorized into flavanone, flavones, flavonols, isoflavones, catechins, chalcones and their derivatives. Due to the diverse beneficial effects of flavonoids, we chose to study the flavonoid biosynthesis and related pathway genes that were detected in the present study (Figure 7). In this dataset, starting from initial enzymes of flavonoids biosynthesis (via the phenylpropanoid pathway) like phenylalanine ammonia lyase (EC: 4.3.1.24, EC: 4.3.1.25), cinnamate 4- monooxygenase (EC: 1.14.13.11), 4-coumarate CoA ligase (EC: 6.2.1.12), and chalcone synthase (EC: 2.3.1.74) were identified. Besides, chalcone isomerase (EC: 5.5.1.6) that catalyzes chalcone isomerisation into naringenin was also found in the present study. Further, the enzymes required for naringenin conversion to produce eriodictyol and dihydrotricetin by flavonoid 3′- monooxygenase (EC: 1.14.13.21) and flavonoid 3′, 5′-hydroxylase (EC: 1.14.13.88) respectively were also identified. In addition to these, the enzyme 6′-deoxychalcone synthase (EC: 2.3.1.170) required to convert p-Coumaroyl CoA to isoliquiritigenin to produce the flavonoid butein was also found. The enzyme chalcone isomerase (EC: 5.5.1.6) also helps butein further produce another flavonoid butin. Moreover, P. amarus leaf transcriptome dataset also contained enzymes like flavonol synthase (EC: 1.14.11.23) and leucoanthocyanidin dioxygenase (EC: 1.14.11.19). A similar set of genes of the flavonoid biosynthesis has been reported in the leaf tissues of endangered medicinal herb Chlorophytum (Kalra et al., 2013). These putative findings support our present dataset in showing how the flavonoids biosynthesis pathway genes complement the therapeutic significance of P. amarus. The KEGG pathway analysis showed that the genes reported in the present study were required for the synthesis of several other flavonoids like pinostrobin, butein, naringenin, galangin, butin, garbanzol, dihydrofisetin (futin), eriodictyol, homoeriodictyol as well as flavones and flavonols like kaempferol, astragalin, quercetin, myricetin, and luteolin. These flavonoids (including flavones and flavonols) are known to exhibit hepatoprotective, antioxidant, anti-inflammatory, antimutagenic, antiviral, etc. properties that support these reported phytotherapeutic potentials of P. amarus as well. Some of the various reported bioactive effects of these flavonoids on human health are summarized below.
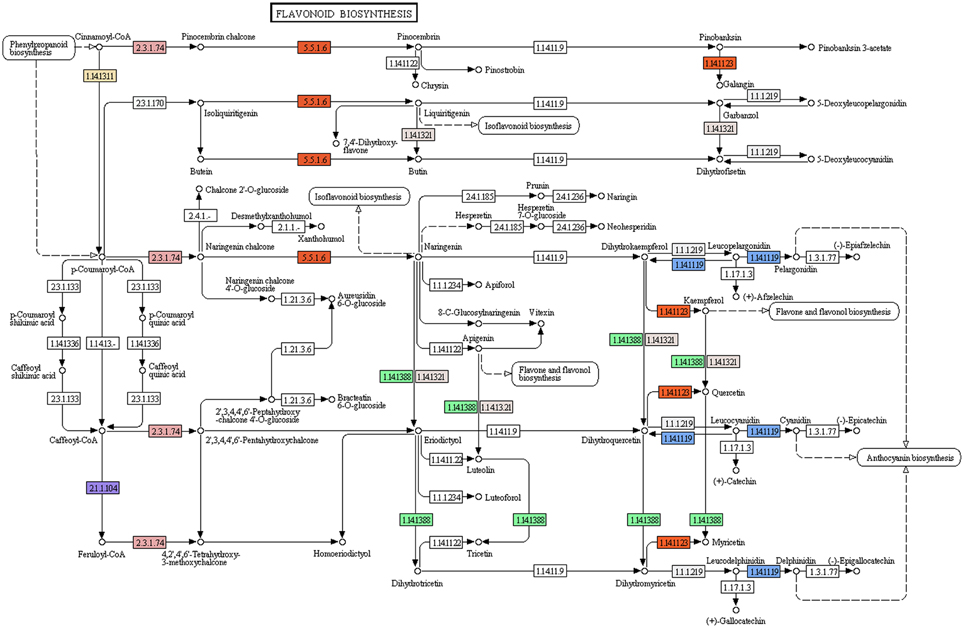
Figure 7. Flavonoid biosynthesis pathway study in P. amarus leaf transcriptome representing each of the identified colored EC.
For instance, pinostrobin has been shown to possess chemopreventive and antioxidant properties (Fahey and Stephenson, 2002), antiviral effect (Wu et al., 2011). The flavonoid naringenin has been shown to possess hepatoprotective and antioxidant effects (Hermenean et al., 2014), anti-inflammatory as well as anticancer activities showing its preventive measures in oral carcinogenesis, hepatocarcinogenesis, and colorectal cancer (Arul and Subramanian, 2013; Sulfikkarali et al., 2013; Li et al., 2014). Recent reports show naringenin to possess neuroprotective effect against Parkinson's disease-related pathology (Lou et al., 2014), iron-induced neurotoxicity and protection of ocular ischemic diseases (Kara et al., 2014). Garbanzol has been shown as an antimutagenic flavonoid (Park et al., 2004). The flavanonol dihydrofisetin (also known as fustin), a type of flavonoid showed protective effects on neuronal cell death (Park et al., 2007). Galangin, another bioflavonoid suggested as a potential candidate for further development of new drugs against Alzheimer's disease (Guo et al., 2010) also has anticancer (Zhang et al., 2013) and hepatoprotective properties (Wang et al., 2013). Butin inhibit aromatase in the human body (Park et al., 2014). Butein suppresses breast and lung cancer (Cho et al., 2014; Seo and Jeong, 2014). Another important flavonoid reported in P. amarus is quercetin, widely accepted as a potent antioxidant also shows anticarcinogenic and hepatoprotective (Ji et al., 2014) activities. Both quercetin and myricetin possess antimicrobial properties (Rashed et al., 2014). Further the well-recognized flavonoid of P. amarus, viz. kaempferol has antioxidant, hepatoprotective and anticancer effects (Huang et al., 2014; Shakya et al., 2014; Dang et al., 2015). The flavone luteolin too has anticancer potential (Lim do et al., 2007). Recent studies have also reported that luteolin along with quercetin reduces high blood cholesterol levels in vivo (Nekohashi et al., 2014). Astragalin is another flavone known to possess antioxidant effects as well (Choi et al., 2013). The pathway genes of all these flavonoids were reported in our putative leaf transcriptome dataset of P. amarus.
Terpenoids and Alkaloid Biosynthesis Pathway Genes
A number of terpenoids and alkaloid metabolism related genes were also revealed in P. amarus leaf transcriptome dataset. Like lignans, flavonoids, and other polyphenolic compounds, terpenoids have also shown to possess therapeutic effects in many clinical studies. Terpenoids are derived from geranyl pyrophosphate (GPP). GPP is synthesized via the cross talks between the cytosolic mevalonate (MVA) pathway and plastidial 2-C-methyl-D-erythritol-4-phosphate (MEP) or DOXP pathway products viz. isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP). MVA pathway starts with the formation of acetoacetyl-CoA while MEP/DOXP pathway starts with D-glyceraldehyde 3-phosphate (Eisenreich et al., 1998). Both MVA and MEP pathways are part of the terpenoid backbone biosynthesis (Figure 8). In the present study, multiple transcripts encoding some of the known enzymes involved in the MVA pathway, MEP/DOXP pathway, and the terpenoids biosynthesis pathway were identified. Genes involved in MVA pathway that were found in our leaf transcriptome included acetyl-CoA acetyltransferase (EC: 2.3.1.9), HMG-CoA synthase (EC: 2.3.3.10), HMG-CoA reductase (EC: 1.1.1.34), phosphomevalonate kinase (EC: 2.7.4.2), mevalonate diphosphate decarboxylase (EC: 4.1.1.33). MEP/DOXP pathway genes included 1-deoxy-D-xylulose-5-phosphate synthase (EC: 2.2.1.7), 1-deoxy-D-xylulose-5-phosphate reductoisomerase (EC: 1.1.1.267), 4-hydroxy-3-methylbut-2-enyl diphosphate synthase (EC: 1.17.7.1), 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (EC: 1.17.1.2), isopentenyl-diphosphate delta isomerase (EC: 5.3.3.2). Interestingly, in the terpenoid backbone biosynthesis pathway farnesyltransferase (EC: 2.5.1.58) gene was also found to be present in putative P. amarus leaf transcriptome dataset. The enzyme farnesyltransferase has been one of the most attractive and fascinating targets in cancer research over the past few decades in the development of anticancer drugs (Sousa et al., 2008). Squalene synthase (EC: 2.5.1.21) and squalene monooxygenase (EC: 1.14.13.132)—the two enzymes involved in the synthesis of terpenoids precursors viz. squalene and squalene-2, 3-epoxide (Supplementary Figure 2) were also identified in the present dataset.
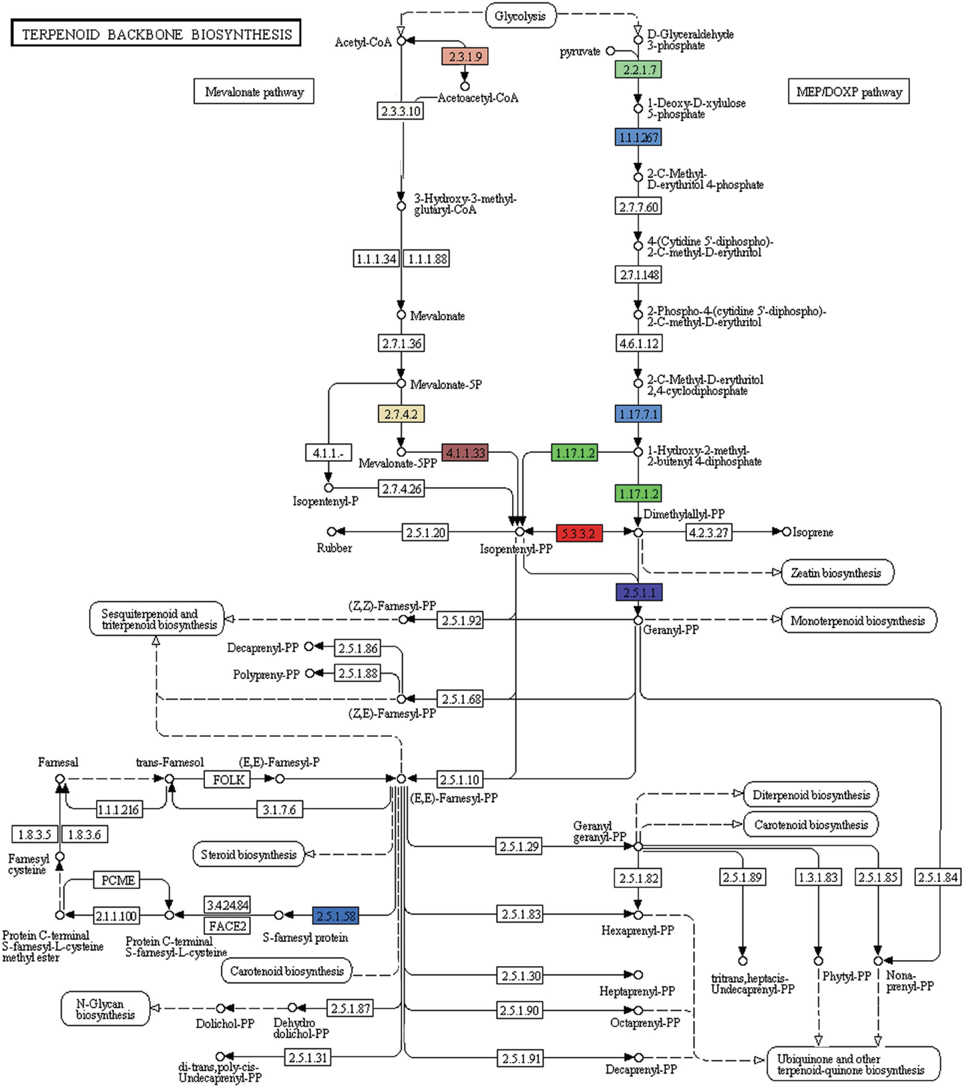
Figure 8. KEGG analysis showing genes involved in MVA, MEP pathways forming the terpenoid backbone biosynthesis (Each EC with one color).
Alkaloids are a large class of naturally occurring organic nitrogen-containing chemical compounds found primarily in plants (Robinson, 1974). Several plant-based alkaloids like morphine, piperine, caffeine, quinine, strychnine, brucine, vinblastine, vincristine colchicine, etc. and their uses have long been reported (Kutchan, 1995). In P. amarus leaf transcriptome dataset, in addition to flavonoids (including flavones and flavonols) and terpenoids, few genes were involved in indole alkaloid (Figure 9), isoquinoline (Supplementary Figure 3) as well as tropane, piperidine and pyridine alkaloid (Supplementary Figure 4) biosynthesis pathways. Polyneuridine-aldehyde esterase (EC: 3.1.1.78), strictosidine synthase (EC: 4.3.3.2), deacetoxyvindoline 4-hydroxylase (EC: 1.14.11.20) were the genes involved in the indole alkaloid biosynthetic pathway. Unique sequences encoding for enzymes like polyphenol oxidase (EC: 1.10.3.1), primary amine oxidase (EC: 1.4.3.21), N-methylcoclaurine 3′-monooxygenase (EC: 1.14.13.71) and reticuline oxidase (EC: 1.21.3.3) in the isoquinoline alkaloid biosynthesis pathway were also revealed. Tropinone reductase I (EC: 1.1.1.206) which synthesize tropine, a derivative of tropane was also present in our dataset.
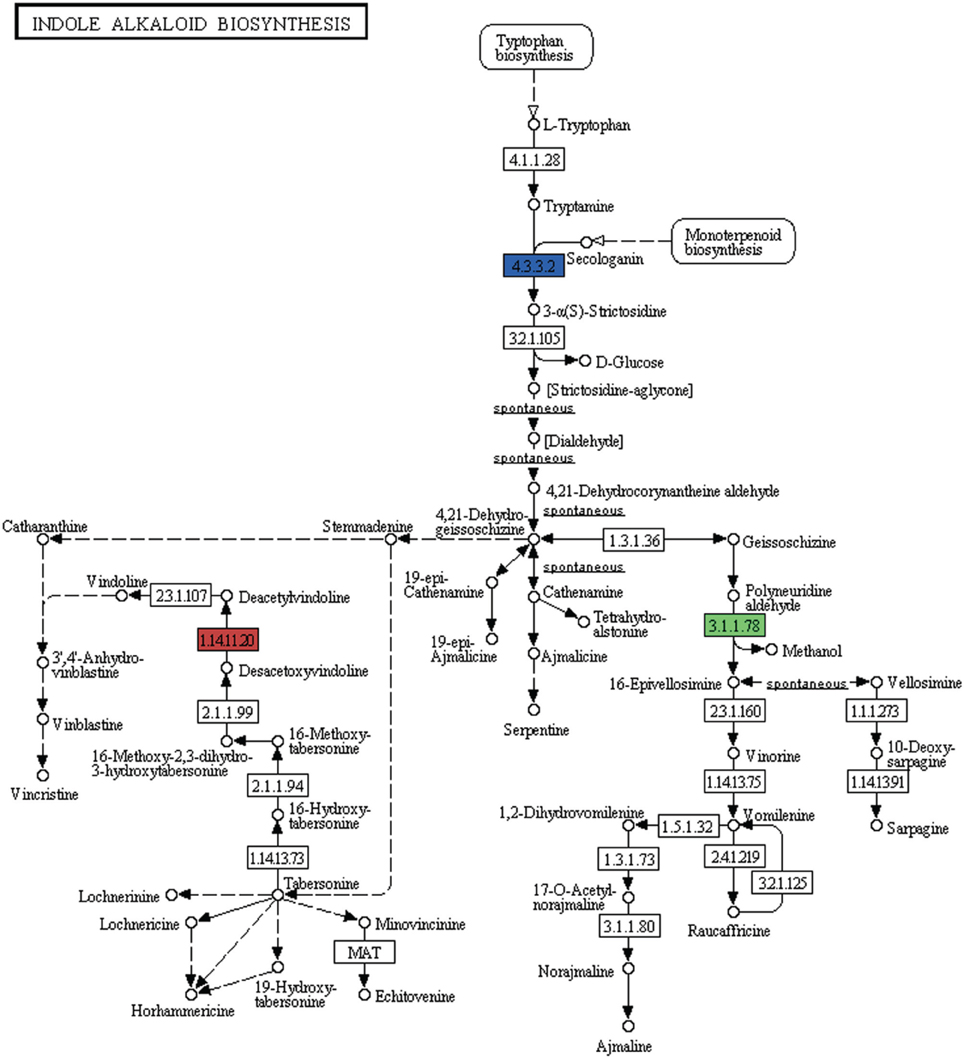
Figure 9. Indole alkaloid biosynthetic pathway genes found in P. amarus leaf transcriptome are depicted by the different colored ECs (one color for each EC).
P. amarus leaf transcriptome dataset also revealed some antimicrobial-related pathways like—“penicillin and cephalosporin biosynthesis,” “tetracycline biosynthesis,” “polyketide sugar unit biosynthesis,” “stilbenoid, diarylheptanoid and gingerol biosynthesis,” “biosynthesis of ansamycins,” “butirosin and neomycin biosynthesis,” “streptomycin biosynthesis,” “novobiocin biosynthesis.”
P. amarus possesses anticancer properties as we have already discussed, owing to the diverse pharmacological properties of the different polyphenolic compounds. Interestingly, in our present study, we reported the presence of some of the unique sequences coding for enzymes having similarity with the genes involved in taxol biosynthesis. KEGG analysis showed the presence of enzymes O-acetyltransferase (EC: 2.3.1.162), 13-alpha-hydroxylase (EC: 1.14.13.77), III-10-O-acetyltransferase (EC: 2.3.1.167) that showed similarity with the genes involved in taxol biosynthesis in diterpenoid pathway (Supplementary Figure 5). Few sequences coding enzymes like dehydrogenase (EC: 1.1.1.207 and EC: 1.1.1.208) having similarities with genes involved in the menthol biosynthesis pathway were also reported in this study in the monoterpenoid biosynthesis pathway of the KEGG database. Previous study shows that menthol has a potent anticancer property (Lu et al., 2007). A summary of a few of the putatively identified major genes involved in phenylpropanoid and flavonoids as well as terpenoids and alkaloid biosynthesis pathways has been represented in Tables 1, 2, respectively.
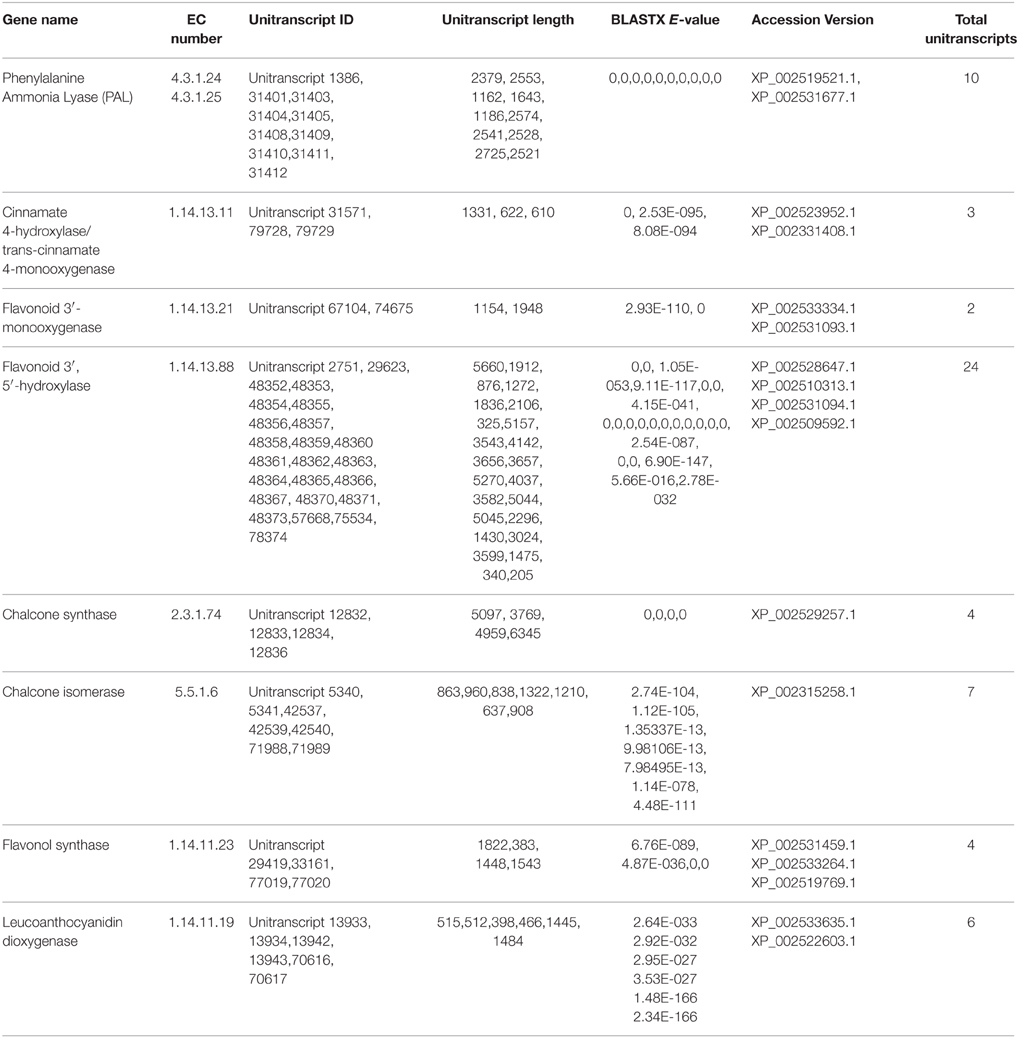
Table 1. Summary of few major genes involved in Phenylpropanoid and Flavonoid biosynthesis pathways identified putatively from P. amarus leaf transcriptome.
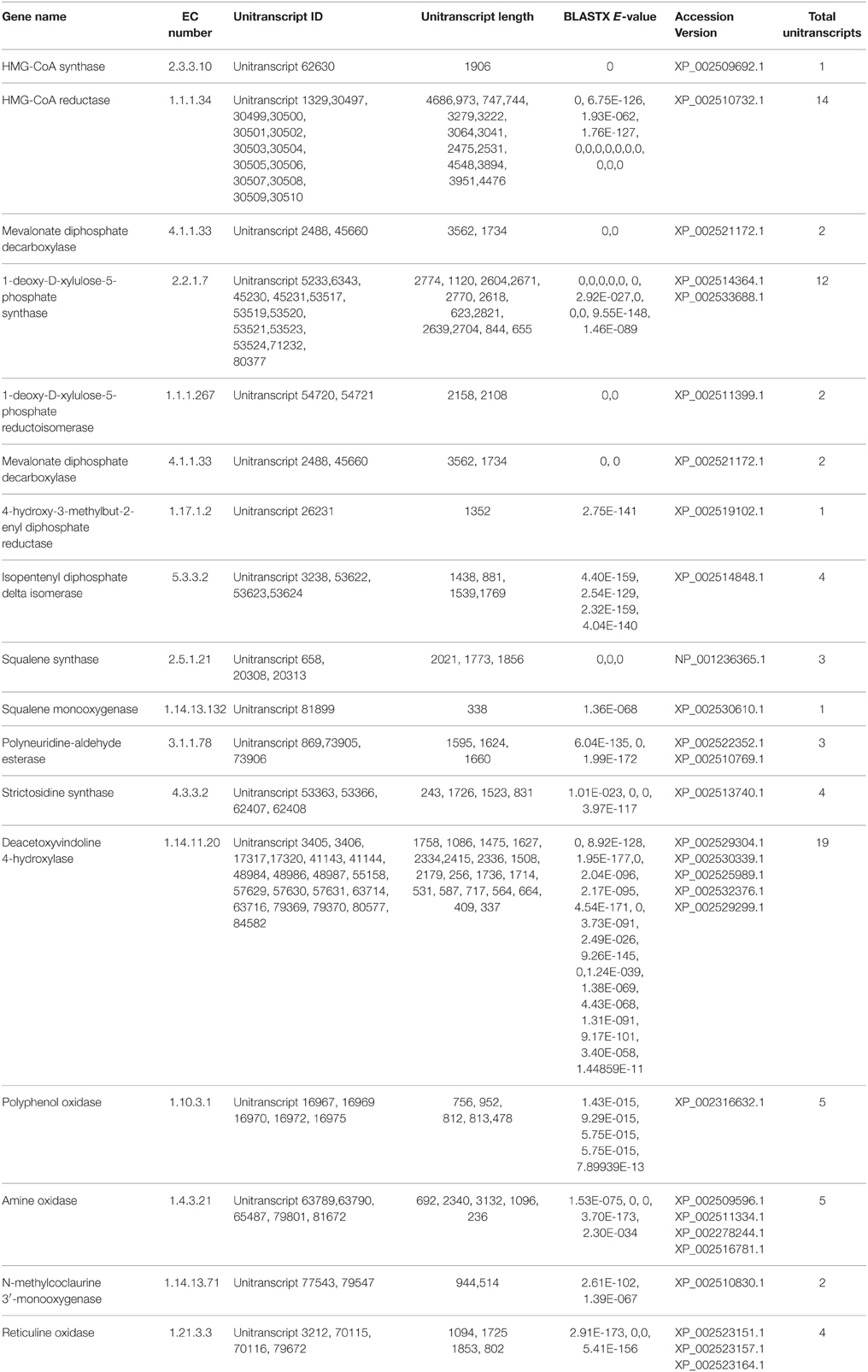
Table 2. Summary of few major genes involved in Terpenoid and Alkaloid biosynthesis pathways identified putatively from P. amarus leaf transcriptome.
Discovery of Transcripts Encoding Transcription Factors and Their Domain Architecture
TFs play key roles in controlling gene expression. In plants, TFs have been employed to manipulate various types of metabolic, developmental and stress response pathways. Further TFs are also known to regulate secondary metabolism in plants at the gene and protein levels as well (Vom Endt et al., 2002). TFs that are known to regulate plant secondary metabolism include R2R3-MYB, basic helix-loop-helix (bHLH) proteins like CrMYC2, AP2/ERF family proteins, WRKY, NAC, DOF, bZIP, HD-ZIP, and TFIIIA zinc finger TFs (Bhattacharyya et al., 2013). A total of 16,344 P. amarus unitranscripts could be annotated at the Plant TF database (PlnTFDB; http://plntfdb.bio.uni-potsdam.de/v3.0/downloads.php) (Pérez-Rodríguez et al., 2009) and categorized into 59 TF categories (Figure 10). The unitranscripts with their detailed TF protein identities and their corresponding domain annotations have been shown in Supplementary File 5. Among the annotated unitranscripts, notable unitranscripts identified and related to secondary metabolism were AP2-EREBP, NAC, bHLH, MYB, or MYB related, bZIP, mTERF, WRKY, zf-HD, C2C2-CO-like, and C2C2- Dof. Similar results were also obtained in our previous study of de novo transcriptome assembly of endangered medicinal herb Podophyllum hexandrum Royle whose extract podophyllotoxin is used for production of anticancer drugs (Bhattacharyya et al., 2013). Our present study has shown that 303 and 737 unitranscripts have encoded for MYB and MYB related TFs respectively. MYB TFs that regulate the phenylpropanoid biosynthetic pathway and also identified in several plant species, mostly include the R2R3-MYB TFs (Hichri et al., 2011; Bhattacharyya et al., 2013). Besides, R2R3-MYB TF has also been shown as a flavonol-specific regulator of phenylpropanoid biosynthesis in Arabidopsis thaliana (Mehrtens et al., 2005). Previous reports also show that in the grapevine phenylpropanoid pathway (Deluc et al., 2006) and its major branch viz. flavonoid biosynthetic pathway in Prunus persica and Epimedium sagittatum are both regulated by R2R3 MYB TFs (Huang et al., 2013; Ravaglia et al., 2013). Besides this, 750 unitranscripts coding for bHLH TFs have been identified in the present study. bHLH TFs like MYB also regulate the flavonoid biosynthetic pathway in plants. (Xu et al., 2015). Besides, we identified 1 Dof TF family in our present study. Dof TFs besides regulating diverse biological processes like carbon and nitrogen assimilation, dormancy, seed maturation and germination, phytochrome signaling, salicylic acid response, guard cell-specific gene expression, photoperiodic flowering, etc. have also been reported to influence phenylpropanoid metabolism in an environmental and tissue-specific manner (Skirycz et al., 2007; Gupta et al., 2015). Another TF family viz. WRKY has also been encoded by 541 unique sequences. WRKY proteins amongst its diverse functions are also involved in the biosynthesis of secondary metabolites (Eulgem et al., 2000). Taken together, TFs identified here can be evaluated further, especially those related to a wide array of secondary metabolites biosynthesis in this potential medicinal herb.
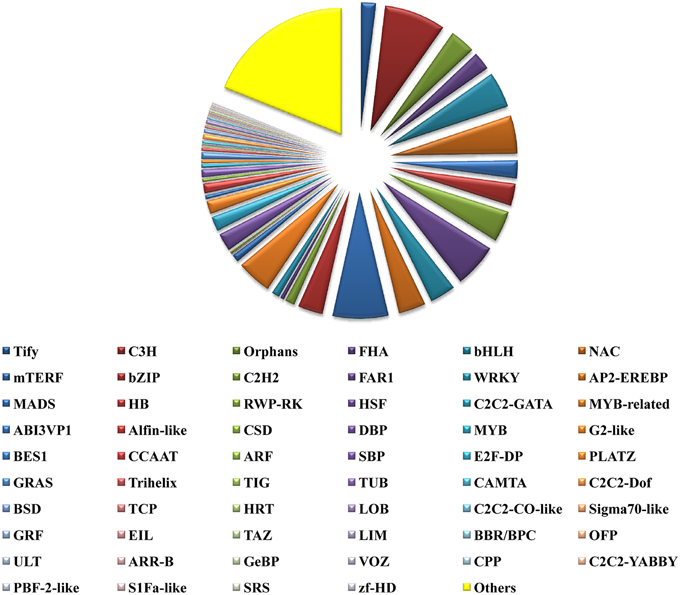
Figure 10. Transcription factors identified from leaf transcriptome of P. amarus de novo assembled unitranscripts.
In silico SSR Mining and Discovery
Microsatellites, or SSRs, are tandemly repeated short DNA sequence motifs ranging from 1 to 6 base pairs extensively distributed in eukaryotes, including the plants, animals and microorganisms, as well as in some prokaryotes (Morgante et al., 2002). SSRs have become one of the most widely used informative molecular markers because it is easy to detect and further used in several applications, including genetic diversity, evolution, genome mapping, marker assisted selection, and breeding studies. Out of 85,927 sequences that were examined by MISA tool, a total of 65,273 SSRs was identified out of which 29,652 were present in compound formation. Of the examined sequences 28,304 contained SSRs with 42% harboring more than one SSR. Statistical analysis of SSRs identified in our study has been presented in Table 3.

Table 3. Statistics of SSRs identified from P. amarus leaf transcriptome.
Frequency and Distribution of Different SSR Repeat Motifs
A summary of SSRs, including distribution of different repeat type classes, frequency of identified SSR motifs, and frequency of classified repeat types (considering sequence complementary) are shown in Supplementary File 6. Among each of the SSR classes the different possible repeat motifs were not evenly distributed. Among the identified SSRs mono-nucleotide repeat motif was the most abundant (44526, 68.215%) followed by di-nucleotide (14659, 22.458%), tri- nucleotide (5585, 8.556%), tetra-nucleotide (251, 0.385%), penta-nucleotide (236, 0.362%) and hexa-nucleotide (16, 0.025%) (Figure 11A). Of the mono-nucleotide repeats, adenine was the most abundant (65.06%) followed by thymine (34.564%), guanine (0.317%), and cytosine (0.061%) (Figure 11B1). The highest frequency observed for each of the identified mononucleotide SSR motif was 10 [(A)10, (T)10, (G)10 and (C)10]. Of the two possible types of mono-nucleotide repeats (considering sequence complementary), the most abundant was (A/T), as in most plants accounting for 99.623% as compared to the (C/G) type (Figure 11B2). The frequency for the different number of repeats of the (A/T) and (C/G) repeat types as shown was maximum for (A/T)10 and (C/G)10 accounting for 20.5 and 26.19% respectively (Figure 11B2). The dinucleotide repeat (GA)12 was the most abundant (33.079%) (Figure 11B3) with the repeat type (AG/CT)12 having the maximum frequency (Figure 11B4). With respect to tri- nucleotide repeats (GAA)8 was the most abundant (13.823%) with the repeat type (AAG/CTT)5 having the highest frequency (36.115%) (Supplementary File 6). The most frequent tetra- nucleotide repeat motif was (AGAA)6 (9.562%). (AAAG/CTTT)5 was the most abundant tetra-nucleotide repeat type occurring 28.287%. (Supplementary File 6). Among the penta- and hexa-nucleotide repeat motifs the most abundant were (TCTCT)5 (12.288%) and (AAGCCA)6 (68.75%) respectively (Supplementary File 6). (AAGAG/CTCTT)5 (20.763%) and (AAAGCC/CTTTGG)5 (68.75%) were the repeat types with highest frequency among the penta- and hexa-nucleotide repeat motifs respectively (Supplementary File 6). The details of SSR types obtained for the P. amarus unitranscripts have been shown in Supplementary File 7.
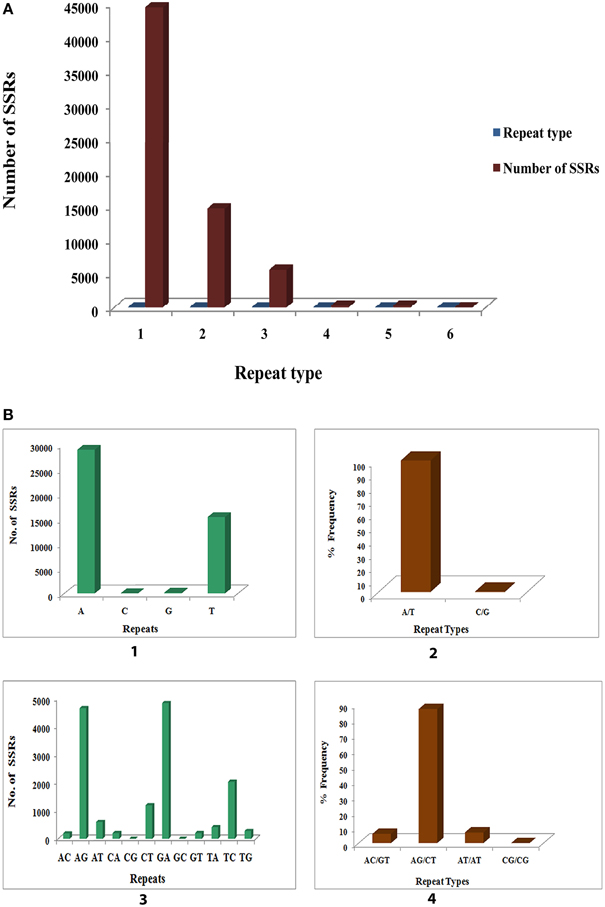
Figure 11. Identification of molecular markers (SSRs) in leaf transcriptome of P. amarus. (A) Distribution of SSR's into mono, di, tri, tetra, penta, and hexa repeat types. (B) Distribution of mono and di-nucleotide SSR motifs and percent frequency of their repeat types.
The large number of SSRs thus identified for the first time from the leaf transcriptome of this medicinal herb will facilitate gene mapping, secondary metabolite improvement, and enable genetic diversity analysis in future on P. amarus genomics study.
RT-PCR for Validation and FPKM Value Determination of the Major Secondary Metabolite Pathway Genes from P. amarus Leaf
The de novo assembled and annotated unitranscripts of P. amarus leaf transcriptome were further validated. We selected some of the unitranscripts annotated against the multiple databases that revealed putative information of P. amarus leaf transcriptome. The FPKM values of the major secondary metabolite pathway genes that were identified from the P. amarus leaf transcriptome as mentioned in Tables 1, 2 were also calculated to further correlate the putative unitranscript abundance with their expression in the leaf tissues of this herb (Supplementary File 8). Some of the significantly expressed unitranscripts with FPKM values on the higher side, identified as the major secondary metabolic pathway genes in P. amarus leaf transcriptome included—phenylalanine ammonia lyase (PAL), cinnamate 4-hydroxylase/trans-cinnamate 4-monooxygenase, flavonoid 3′,5′-hydroxylase, phenylcoumaran benzylic ether reductase -like protein, HMG-CoA synthase, mevalonate diphosphate decarboxylase, isopentenyl diphosphate delta isomerase, strictosidine synthase, deacetoxyvindoline 4-hydroxylase, 1-deoxy-D-xylulose-5-phosphate reductoisomerase, polyphenol oxidase, amine oxidase, squalene synthase, chalcone isomerase (Supplementary File 8). Based on the putative findings and correlating with the FPKM values, a few of the secondary metabolic genes were selected for performing Reverse Transcription PCR (RT-PCR) to validate the reliability and accuracy of our de novo assembled P. amarus leaf transcriptome data.
We identified 7 of the selected genes while performing RT-PCR with the leaf sample, each from the lignans biosynthetic pathway genes like phenylcoumaran benzylic ether reductase -like protein (Unitranscript 63241; GO:0009807: lignan biosynthetic process); phenylcoumaran benzylic ether reductase -like protein (Unitranscript 77577; GO:0010283: pinoresinol reductase activity); and each from phenylpropanoid, flavonoid, and alkaloid biosynthetic pathways like phenylalanine ammonia lyase, putative (Unitranscript 31404; GO:0045548: phenylalanine ammonia lyase activity); chalcone isomerase (Unitranscript 5340; GO:0009813: flavonoid biosynthetic process, GO:0045430: chalcone isomerase activity); strictosidine synthase, putative (Unitranscript 53363; GO:0016844: strictosidine synthase activity) respectively. Also, two TFs (related to the secondary metabolites biosynthesis) like R2R3-MYB TF (Unitranscript 1528) and bHLH TF (Unitranscript 14718) were selected for RT-PCR validation.
Experimentally confirmed data of gene expression provide a preferable understanding of the function and regulation of the genes. Hence, we performed RT-PCR of the above mentioned unitranscripts (Figure 12) identified in our putative leaf transcriptome dataset to confirm the gene expression of these unitranscripts along with actin gene used as a control in the leaf tissue samples of P. amarus. Our results suggested that the three-level de novo assembly and annotation data of our present study can be used in genomics study and practical experiments on P. amarus in future.
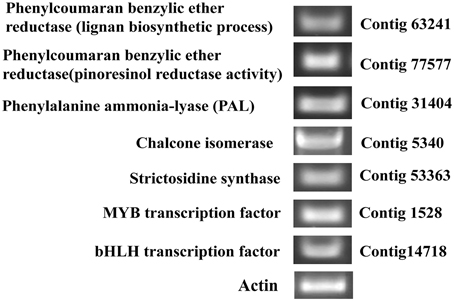
Figure 12. RT-PCR image of selected P. amarus unitranscripts expressed in leaf samples of P. amarus.
Conclusion
P. amarus transcriptome research lags that of other plants of medicinal importance. To facilitate molecular research in P. amarus we characterized the leaf transcriptome since the vast repertoire of secondary metabolites are mainly present in the leaf tissues of this herb. Our data on leaf transcriptome analysis using the Illumina MiSeq platform have instigated to the identification of a large number of transcripts, TFs, molecular/SSR markers involved in diverse processes, functions, metabolic pathways together with the transcripts involved in a number of secondary metabolic pathways, specially attributing to the phytomedicinal significance of this herb. To be more precise, the putative transcriptome information explored in our data revealing the various lignans, flavonoids, terpenoids, alkaloids and other secondary metabolites biosynthesis pathway genes adds a copious amount of information to P. amarus database and also paves the way for functional and comparative genomic studies of this highly promising medicinal plant in future. Further RT-PCR results showing expression of the few selected unitranscripts, involved in various classes of secondary metabolites synthesis, also confirmed the reliability and accuracy of our P. amarus leaf transcriptome assembly. This is the first report on a detailed study of P. amarus leaf transcriptome that was done to provide an important resource for future studies on “bhuiamalaki” thereby greatly facilitating research on non-model plants in plant biology.
Author Contributions
AB and SC designed the experiment. AB carried out the experimental work, analyzed the data and drafted the manuscript. SC supervised the analysis of NGS data and prepared the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to the Director, CSIR-Indian Institute of Chemical Biology, Kolkata for providing the necessary facilities. The authors would like to thank CSIR, New Delhi for financial support. ABM acknowledges the Department of Science and Technology (DST), New Delhi for INSPIRE Fellowship. The authors also acknowledge Nucleome Informatics Pvt. Ltd. (Hyderabad, India) for NGS data generation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.01199
Supplementary Figure 1. Summary of filtration report of total raw reads generated and used for transcriptome assembly.
Supplementary Figure 2. Terpenoid biosynthesis pathway.
Supplementary Figure 3. Isoquinoline alkaloid biosynthesis pathway.
Supplementary Figure 4. Tropane, piperidine, and pyridine alkaloid biosynthetic pathway genes identified.
Supplementary Figure 5. Putatively identified genes of diterpenoid biosynthesis pathway.
References
Ali, H., Houghton, P. J., and Soumyanath, A. (2006). α-Amylase inhibitory activity of some Malaysian plants used to treat diabetes; with particular reference to Phyllanthus amarus. J. Ethnopharmacol. 107, 449–455. doi: 10.1016/j.jep.2006.04.004
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Annadurai, R. S., Neethiraj, R., Jayakumar, V., Damodaran, A. C., Rao, S. N., Katta, M. A. V. S. K., et al. (2013). De novo transcriptome assembly (NGS) of Curcuma longa L. rhizome reveals novel transcripts related to anticancer and antimalarial terpenoids. PLoS ONE 8:e56217. doi: 10.1371/journal.pone.0056217
Arul, D., and Subramanian, P. (2013). Inhibitory effect of naringenin (citrusflavonone) on N-nitrosodiethylamine induced hepatocarcinogenesis in rats. Biochem. Biophys. Res. Commun. 434, 203–209. doi: 10.1016/j.bbrc.2013.03.039
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Bandyopadhyay, S., and Raychaudhuri, S. S. (2013). Development and comparison of RAPD, SCAR and AFLP markers for distinguishing some medicinally important species of the genus Phyllanthus. Plant Biosyst. 147, 12–20. doi: 10.1080/11263504.2011.635714
Bhattacharyya, D., Sinha, R., Hazra, S., Datta, R., and Chattopadhyay, S. (2013). De novo transcriptome analysis using 454 pyrosequencing of the Himalayan Mayapple, Podophyllum hexandrum. BMC Genomics 14:748. doi: 10.1186/1471-2164-14-748
Blumberg, B. S., Millman, I., Venkates, P. S., and Thyagarajan, S. P. (1990). Hepatitis B virus and primary hepatocellular carcinoma: treatment of HBV carriers with Phyllanthus amarus. Vaccine 8, S86–S92. doi: 10.1016/0264-410X(90)90225-B
Calixto, J. B., Santos, A. R., Cechinel Filho, V., and Yunes, R. A. (1998). A review of the plants of the Phyllanthus: their chemistry, pharmacology, and therapeutic potential. Med. Res. Rev. 18, 225–258.
Chattopadhyay, S., and Bose Mazumdar Ghosh, A. (2014). Establishment of cDNA library and EST analysis from leaves of Phyllanthus amarus. Int. J. Biochem. Res. Rev. 4, 1–15. doi: 10.9734/IJBCRR/2014/5262
Chirdchupunseree, H., and Pramyothin, P. (2010). Protective activity of phyllanthin in ethanol-treated primary culture of rat hepatocytes. J. Ethnopharmacol. 128, 172–176. doi: 10.1016/j.jep.2010.01.003
Cho, S. G., Woo, S. M., and Ko, S. G. (2014). Butein suppresses breast cancer growth by reducing a production of intracellular reactive oxygen species. J. Exp. Clin. Cancer Res. 33:51. doi: 10.1186/1756-9966-33-51
Choi, J., Kang, H. J., Kim, S. Z., Kwon, T. O., Jeong, S. I., and Jang, S. I. (2013). Antioxidant effect of astragalin isolated from the leaves of Morus alba L. against free radical-induced oxidative hemolysis of human red blood cells. Arch. Pharm. Res. 36, 912–917. doi: 10.1007/s12272-013-0090-x
Chopra, R. N., Nayar, S. L., and Chopra, I. C. (1956). Glossary of Indian Medicinal Plants. New Delhi: Council of Scientific and Industrial Research.
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Dang, Q., Song, W., Xu, D., Ma, Y., Li, F., Zeng, J., et al. (2015). Kaempferol suppresses bladder cancer tumor growth by inhibiting cell proliferation and inducing apoptosis. Mol. Carcinog. 54, 831–840. doi: 10.1002/mc.22154
Deluc, L., Barrieu, F., Marchive, C., Lauvergeat, V., Decendit, A., Richard, T., et al. (2006). Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 140, 499–511. doi: 10.1104/pp.105.067231
Eisenreich, W., Schwarz, M., Cartayrade, A., Arigoni, D., Zenk, M. H., and Bacher, A. (1998). The deoxyxylulose phosphate pathway of terpenoid biosynthesis in plants and microorganisms. Chem. Biol. 5, R221–R233. doi: 10.1016/s1074-5521(98)90002-3
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/S1360-1385(00)01600-9
Fahey, J. W., and Stephenson, K. K. (2002). Pinostrobin from honey and Thai ginger (Boesenbergia pandurata): a potent flavonoid inducer of mammalian phase 2 chemoprotective and antioxidant enzymes. J. Agric. Food Chem. 50, 7472–7476. doi: 10.1021/jf025692k
Foo, L. Y. (1993). Amarulone, a novel cyclic hydrolysable tannin from Phyllanthus amarus. Nat. Prod. Lett. 3, 45–52. doi: 10.1080/10575639308043836
Foo, L. Y. (1995). Amariinic acid and related ellagitannins from Phyllanthus amarus. Phytochemistry 39, 217–224. doi: 10.1016/0031-9422(94)00836-I
Foo, L. Y., and Wong, H. (1992). Phyllanthusiin D an unusual hydrolysable tannin from Phyllanthus amarus. Phytochemistry 31, 711–713. doi: 10.1016/0031-9422(92)90071-W
Gang, D. R., Kasahara, H., Xia, Z. Q., Vander Mijnsbrugge, K., Bauw, G., Boerjan, W., et al. (1999). Evolution of plant defense mechanisms. Relationships of phenylcoumaran benzylic ether reductases to pinoresinol-lariciresinol and isoflavone reductases. J. Biol. Chem. 274, 7516–7527. doi: 10.1074/jbc.274.11.7516
Guo, A. J., Xie, H. Q., Choi, R. C., Zheng, K. Y., Bi, C. W., Xu, S. L., et al. (2010). Galangin, a flavonol derived from Rhizoma Alpiniae Officinarum, inhibits acetylcholinesterase activity in vitro. Chem. Biol. Interact. 187, 246–248. doi: 10.1016/j.cbi.2010.05.002
Gupta, S., Malviya, N., Kushwaha, H., Nasim, J., Bisht, N. C., Singh, V. K., et al. (2015). Insights into structural and functional diversity of Dof (DNA binding with one finger) transcription factor. Planta 241, 549–562. doi: 10.1007/s00425-014-2239-3
Harish, R., and Shivanandappa, T. (2006). Antioxidant activity and hepatoprotective potential of Phyllanthus niruri. Food Chem. 95, 180–185. doi: 10.1016/j.foodchem.2004.11.049
Hermenean, A., Ardelean, A., Stan, M., Hadaruga, N., Mihali, C. V., Costache, M., et al. (2014). Antioxidant and hepatoprotective effects of naringenin and its β-cyclodextrin formulation in mice intoxicated with carbon tetrachloride: a comparative study. J. Med. Food. 17, 670–677. doi: 10.1089/jmf.2013.0007
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., and Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Houghton, P. J., Woldemariam, T. Z., O'shea, S., and Thyagarajan, S. P. (1996). Two securinega-type alkaloids from Phyllanthus amarus. Phytochemistry 43, 715–717. doi: 10.1016/0031-9422(96)00345-7
Huang, W., Sun, W., Lv, H., Luo, M., Zeng, S., Pattanaik, S., et al. (2013). A R2R3-MYB transcription factor from Epimedium sagittatum regulates the flavonoid biosynthetic pathway. PLoS ONE 8:e70778. doi: 10.1371/journal.pone.0070778
Huang, Y. B., Lin, M. W., Chao, Y., Huang, C. T., Tsai, Y. H., and Wu, P. C. (2014). Anti-oxidant activity and attenuation of bladder hyperactivity by the flavonoid compound kaempferol. Int. J. Urol. 21, 94–98. doi: 10.1111/iju.12179
Jain, N., Shasany, A. K., Sundaresan, V., Rajkumar, S., Darokar, M. P., Bagchi, G. D., et al. (2003). Molecular diversity in Phyllanthus amarus assessed through RAPD analysis. Curr. Sci. 85, 1454–1458.
Ji, L., Ma, Y., Wang, Z., Cai, Z., Pang, C., and Wang, Z. (2014). Quercetin prevents pyrrolizidine alkaloid clivorine-induced liver injury in mice by elevating body defense capacity. PLoS ONE 9:e98970. doi: 10.1371/journal.pone.0098970
Kalra, S., Puniya, B. L., Kulshreshtha, D., Kumar, S., Kaur, J., Ramachandran, S., et al. (2013). De Novo Transcriptome Sequencing Reveals Important Molecular Networks and Metabolic Pathways of the Plant, Chlorophytum borivilianum. PLoS ONE 8:e83336. doi: 10.1371/journal.pone.0083336
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kara, S., Gencer, B., Karaca, T., Tufan, H. A., Arikan, S., Ersan, I., et al. (2014). Protective effect of hesperetin and naringenin against apoptosis in ischemia/reperfusion-induced retinal injury in rats. Sci. World J. 2014:797824. doi: 10.1155/2014/797824
Kassuya, C. A. L., Silvestre, A., Menezes-de-Lima, O. Jr., Marotta, D. M., Rehder, V. L., and Calixto, J. B. (2006). Antiinflammatory and antiallodynic actions of the lignan niranthin isolated from Phyllanthus amarus: evidence for interaction with platelet activating factor receptor. Eur. J. Pharmacol. 546, 182–188. doi: 10.1016/j.ejphar.2006.07.025
Koski, L. B., Gray, M. W., Lang, B. F., and Burger, G. (2005). AutoFACT: an automatic functional annotation and classification tool. BMC Bioinformatics 6:151. doi: 10.1186/1471-2105-6-151
Krithika, R., Jyothilakshmi, V., and Verma, R. J. (2014). Phyllanthin inhibits CCL4-mediated oxidative stress and hepatic fibrosis by down-regulating TNF-α/ NF-κB and pro-fibrotic factor TGF-β1 mediated inflammatory signaling. Toxicol. Ind. Health. [Epub ahead of print]. doi: 10.1177/0748233714532996
Kutchan, T. M. (1995). Alkaloid biosynthesis-the basis for metabolic engineering of medicinal plants. Plant Cell. 7, 1059–1070. doi: 10.2307/3870057
Lehnert, E. M., and Walbot, V. (2014). Sequencing and de novo assembly of a Dahlia hybrid cultivar transcriptome. Front. Plant Sci. 5:340. doi: 10.3389/fpls.2014.00340
Leite, D. F., Kassuya, C. A. L., Mazzuco, T. L., Silvestre, A., de-Melo, L. V., Rehder, V. L., et al. (2006). The cytotoxic effect and the multidrug resistance reversing action of lignans from Phyllanthus amarus. Planta Med. 72, 1353–1358. doi: 10.1055/s-2006-951708
Li, H., Zhu, F., Chen, H., Cheng, K. W., Zykova, T., Oi, N., et al. (2014). 6-C-(E-phenylethenyl)-naringenin suppresses colorectal cancer growth by inhibiting cyclooxygenase-1. Cancer Res. 74, 243–252. doi: 10.1158/0008-5472.CAN-13-2245
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, X., Nair, A., Wang, S., and Wang, L. (2015). “Quality control of RNA-seq experiments,” in RNA Bioinformatics, Methods in Molecular Biology, Vol. 1269, ed E. Picardi (New York, NY: Springer), 137–146. doi: 10.1007/978-1-4939-2291-8_8
Lim do, Y., Jeong, Y., Tyner, A. L., and Park, J. H. Y. (2007). Induction of cell cycle arrest and apoptosis in HT-29 human colon cancer cells by the dietary compound luteolin. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G66–G75. doi: 10.1152/ajpgi.00248.2006
Lou, H., Jing, X., Wei, X., Shi, H., Ren, D., and Zhang, X. (2014). Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology 79, 380–388. doi: 10.1016/j.neuropharm.2013.11.026
Lu, H. F., Liu, J. Y., Hsueh, S. C., Yang, Y. Y., Yang, J. S., Tan, T. W., et al. (2007). (-)-Menthol inhibits WEHI-3 leukemia cells in vitro and in vivo. In Vivo 21, 285–290.
Mazliak, P. (1973). Lipid metabolism in plants. Ann. Rev. Plant Physiol. 24, 287–310. doi: 10.1146/annurev.pp.24.060173.001443
Mazumder, A., Mahato, A., and Mazumder, R. (2006). Antimicrobial potentiality of Phyllanthus amarus against drug resistant pathogens. Nat. Prod. Res. 20, 323–326. doi: 10.1080/14786410600650404
Mehrtens, F., Kranz, H., Bednarek, P., and Weisshaar, B. (2005). The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 138, 1083–1096. doi: 10.1104/pp.104.058032
Michal, G. (1999). Biochemical Pathways: An Atlas of Biochemistry And Molecular Biology. Heidelberg: Spektrum Akademischer.
Morgante, M., Hanafey, M., and Powell, W. (2002). Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat. Genet. 30, 194–200. doi: 10.1038/ng822
Nakasugi, K., Crowhurst, R. N., Bally, J., Wood, C. C., Hellens, R. P., and Waterhouse, P. M. (2013). De novo transcriptome sequence assembly and analysis of RNA silencing genes of Nicotiana benthamiana. PLoS ONE 8:e59534. doi: 10.1371/journal.pone.0059534
Nekohashi, M., Ogawa, M., Ogihara, T., Nakazawa, K., Kato, H., Misaka, T., et al. (2014). Luteolin and quercetin affect the cholesterol absorption mediated by epithelial cholesterol transporter Niemann–Pick C1-Like 1 in caco-2 cells and rats. PLoS ONE 9:e97901. doi: 10.1371/journal.pone.0097901
Park, B. C., Lee, Y. S., Park, H. J., Kwak, M. K., Yoo, B. K., Kim, J. Y., et al. (2007). Protective effects of fustin, a flavonoid from Rhus verniciflua Stokes, on 6-hydroxydopamine-induced neuronal cell death. Exp. Mol. Med. 39, 316–326. doi: 10.1038/emm.2007.35
Park, K. Y., Jung, G. O., Lee, K. T., Choi, J., Choi, M. Y., Kim, G. T., et al. (2004). Antimutagenic activity of flavonoids from the heartwood of Rhus verniciflua. J. Ethnopharmacol. 90, 73–79. doi: 10.1016/j.jep.2003.09.043
Park, M. H., Kim, I. S., Kim, S. A., Na, C. S., Hong, C. Y., Dong, M. S., et al. (2014). Inhibitory effect of Rhus verniciflua Stokes extract on human aromatase activity; butin is its major bioactive component. Bioorg. Med. Chem. Lett. 24, 1730–1733. doi: 10.1016/j.bmcl.2014.02.039
Pérez-Rodríguez, P., Riaño-Pachón, D. M., Corrêa, L. G. G., Rensing, S. A., Kersten, B., and Mueller-Roeber, B. (2009). PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res. 38, D822–D827. doi: 10.1093/nar/gkp805
Rajeshkumar, N. V., Joy, K. L., Kuttan, G., Ramsewak, R. S., Nair, M. G., and Kuttan, R. (2002). Antitumour and anticarcinogenic activity of Phyllanthus amarus extract. J. Ethnopharmacol. 81, 17–22. doi: 10.1016/S0378-8741(01)00419-6
Rashed, K., Ciric, A., Glamočlija, J., and Soković, M. (2014). Antibacterial and antifungal activities of methanol extract and phenolic compounds from Diospyros virginiana L. Ind. Crop. Prod. 59, 210–215. doi: 10.1016/j.indcrop.2014.05.021
Rastogi, S., Meena, S., Bhattacharya, A., Ghosh, S., Shukla, R. K., Sangwan, N. S., et al. (2014). De novo sequencing and comparative analysis of holy and sweet basil transcriptomes. BMC Genomics 15:588. doi: 10.1186/1471-2164-15-588
Ravaglia, D., Espley, R. V., Henry-Kirk, R. A., Andreotti, C., Ziosi, V., Hellens, R. P., et al. (2013). Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol. 13:68. doi: 10.1186/1471-2229-13-68
Robinson, T. (1974). Metabolism and function of alkaloids in plants. Science 184, 430–435. doi: 10.1126/science.184.4135.430
Rogers, M. F., Thomas, J., Reddy, A. S., and Ben-Hur, A. (2012). SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 13:R4. doi: 10.1186/gb-2012-13-1-r4
Senapati, S. K., Aparajita, S., and Rout, G. R. (2011). Identification of species-diagnostic inter simple sequence repeat markers for ten Phyllanthus species. Z. Naturforsch C 66, 167–172. doi: 10.5560/ZNC.2011.66c0167
Seo, Y. H., and Jeong, J. H. (2014). Synthesis of butein analogues and their anti-proliferative activity against gefitinib-resistant non-small cell lung cancer (NSCLC) through Hsp90 inhibition. Bull. Korean Chem. Soc. 35, 1294–1298. doi: 10.5012/bkcs.2014.35.5.1294
Shakya, G., Manjini, S., Hoda, M., and Rajagopalan, R. (2014). Hepatoprotective role of kaempferol during alcohol- and ΔPUFA-induced oxidative stress. J. Basic Clin. Physiol. Pharmacol. 25, 73–79. doi: 10.1515/jbcpp-2013-0051
Skirycz, A., Jozefczuk, S., Stobiecki, M., Muth, D., Zanor, M. I., Witt, I., et al. (2007). Transcription factor AtDOF4;2 affects phenylpropanoid metabolism in Arabidopsis thaliana. New Phytol. 175, 425–438. doi: 10.1111/j.1469-8137.2007.02129.x
Somanabandhu, A., Nitayangkura, S., Mahidol, C., Ruchirawat, S., Likhitwitayawuid, K., Shieh, H. L., et al. (1993). 1H and 13C- NMR assignments of phyllanthin and hypophyllanthin: lignans that enhance cytotoxic responses with cultured multidrug resistant cells. J. Nat. Prod. 56, 233–239. doi: 10.1021/np50092a008
Sousa, S. F., Fernandes, P. A., and Ramos, M. J. (2008). Enzyme flexibility and the catalytic mechanism of farnesyltransferase: targeting the relation. J. Phys. Chem. B 112, 8681–8691. doi: 10.1021/jp711214j
Sulfikkarali, N., Krishnakumar, N., Manoharan, S., and Nirmal, R. M. (2013). Chemopreventive efficacyof naringenin-loaded nanoparticles in 7,12-dimethylbenz(a)anthracene induced experimental oral carcinogenesis. Pathol. Oncol. Res. 19, 287–296. doi: 10.1007/s12253-012-9581-1
Thyagarajan, S. P., Subramanian, S., Thirunalasundari, T., Venkateswaran, P. S., and Blumberg, B. S. (1988). Effect of Phyllanthus amarus on chronic carriers of hepatitis B virus. Lancet 332, 764–766. doi: 10.1016/S0140-6736(88)92416-6
Umezawa, T. (2003). Diversity in lignan biosynthesis. Phytochem. Rev. 2, 371–390. doi: 10.1023/B:PHYT.0000045487.02836.32
Umezawa, T., Okunishi, T., and Shimada, M. (1997). Mechanisms of lignan biosynthesis. Annu. Rep. Interdiscipl. Res. Inst. Environ. Sci. 16, 65–71.
Van Bel, M., Proost, S., Van Neste, C., Deforce, D., Van de Peer, Y., and Vandepoele, K. (2013). TRAPID: an efficient online tool for the functional and comparative analysis of de novo RNA-Seq transcriptomes. Genome Biol. 14:R134. doi: 10.1186/gb-2013-14-12-r134
Vander Mijnsbrugge, K., Beeckman, H., De Rycke, R., Van Montagu, M., Engler, G., and Boerjan, W. (2000). Phenylcoumaran benzylic ether reductase, a prominent poplar xylem protein, is strongly associated with phenylpropanoid biosynthesis in lignifying cells. Planta 211, 502–509. doi: 10.1007/s004250000326
Vom Endt, D., Kijne, J. W., and Memelink, J. (2002). Transcription factors controlling plant secondary metabolism: what regulates the regulators? Phytochemistry 61, 107–114. doi: 10.1016/s0031-9422(02)00185-1
Wang, X., Gong, G., Yang, W., Li, Y., Jiang, M., and Li, L. (2013). Antifibrotic activity of galangin, a novel function evaluated in animal liver fibrosis model. Environ. Toxicol. Pharmacol. 36, 288–295. doi: 10.1016/j.etap.2013.04.004
Wilhelm, B. T., Marguerat, S., Goodhead, T. I., and Bähler, J. (2010). Defining transcribed regions using RNA-seq. Nat. Protoc. 5, 255–266. doi: 10.1038/nprot.2009.229
Wu, N., Kong, Y., Zu, Y., Fu, Y., Liu, Z., Meng, R., et al. (2011). Activity investigation of pinostrobin towards herpes simplex virus-1 as determined by atomic force microscopy. Phytomedicine 18, 110–118. doi: 10.1016/j.phymed.2010.07.001
Xu, W., Dubos, C., and Lepiniec, L. (2015). Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 20, 176–185. doi: 10.1016/j.tplants.2014.12.001
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Keywords: Phyllanthus amarus, next-generation sequencing (NGS), Illumina Miseq, leaf transcriptome, de novo assembly, functional annotation, secondary metabolism
Citation: Bose Mazumdar A and Chattopadhyay S (2016) Sequencing, De novo Assembly, Functional Annotation and Analysis of Phyllanthus amarus Leaf Transcriptome Using the Illumina Platform. Front. Plant Sci. 6:1199. doi: 10.3389/fpls.2015.01199
Received: 13 August 2015; Accepted: 14 December 2015;
Published: 28 January 2016.
Edited by:
Chang-Jun Liu, Brookhaven National Laboratory, USACopyright © 2016 Bose Mazumdar and Chattopadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sharmila Chattopadhyay, c2hhcm1pbGFAaWljYi5yZXMuaW4=