 Lexuan Gao
Lexuan Gao Hongxing Yang
Hongxing Yang Hongfeng Liu
Hongfeng Liu Ji Yang
Ji Yang Yonghong Hu
Yonghong Hu- 1Shanghai Key Laboratory of Plant Functional Genomics and Resources, Shanghai Chenshan Plant Science Research Center, Chinese Academy of Sciences, Shanghai, China
- 2School of Landscape Architecture, Beijing Forestry University, Beijing, China
- 3Center for Evolutionary Biology and Institute of Biodiversity Science, Fudan University, Shanghai, China
Tree peonies are a group of traditional ornamental plants, especially in East Asia, with Paeonia ostii as one of the most important ancestral species. P. ostii has flowers with varying colors, ranging from nearly white, light pink to deep pink. However, few studies have been done to unravel the molecular mechanisms underlying the flower color intensity variation in plants. Based on comparative analyses of the pigment composition and transcriptomes of P. ostii with different flower color intensities, we found that the anthocyanin concentration was significantly correlated with the flower color intensity in P. ostii. Transcriptome analysis by RNA-Sequencing revealed 7187 genes that were differentially expressed between flowers with different color intensities. Functional enrichment analysis of differentially expressed genes revealed multiple pathways possibly responsible for color intensity variation in P. ostii, including flavonoid biosynthesis, fatty acid oxidation, carbohydrate metabolism, and hormone-mediated signaling. Particularly, while anthocyanin biosynthesis genes showing positive correlations between their expression and anthocyanin concentration in flowers, two transcription factors, PoMYB2 and PoSPL1, seem to negatively regulate anthocyanin accumulation by affecting the activation capacity of the MYB-bHLH-WDR complex, exhibiting an inverse relationship between their expression and anthocyanin accumulation. Our results showed that, although anthocyanin biosynthesis had a direct effect on the pigmentation of the P. ostii flower, other metabolic and hormone-mediated signaling pathways were also contributed to the flower color intensity variation in P. ostii, suggesting complex coordinated changes in the transcriptional network. Differential expression of genes encoding anthocyanin repressors seems to be the major factor responsible for the intensity variation in anthocyanin pigmentation in P. ostii.
Introduction
Flower color is one of the most attractive sceneries in nature. It confers flowers with diverse functions (Winkel-Shirley, 2001; Steyn et al., 2002; Nagata et al., 2003), and is of paramount importance to plant evolution (Davies et al., 2012; Schiestl and Johnson, 2013; Sobel and Streisfeld, 2013). Angiosperms exhibit an astonishing polymorphism in flower colors. The phenotypic polymorphism in flower pigmentation is typically manifested in two types: (i) variation in pigment intensity determined by the concentration of pigment, and (ii) variation in floral hue, which is generally determined by the distinction of pigment types or the absence/presence of co-pigments (Wessinger and Rausher, 2012; Sobel and Streisfeld, 2013). In contrast to a wealth of knowledge on the genes related to qualitative variations between different pigment hues and between absence and presence of pigments (Zufall and Rausher, 2003; Hopkins and Rausher, 2011; Wessinger and Rausher, 2012; Sobel and Streisfeld, 2013), very few studies have been performed to characterize the genetic and molecular mechanisms determining the quantitative variation in flower color intensity (Schwinn et al., 2006; Ohno et al., 2013; Wang et al., 2014).
Paeonia ostii is a perennial shrub in the genus Paeonia and reproduces sexually (Li et al., 2011). It has been reported that P. ostii is one of the most important ancestral species of the cultivated tree peony, which is an important ornamental crop in the word and is crowned the “king of flowers” in China (Zhang et al., 2012; Zhou et al., 2014b). Flowers of P. ostii exhibit color polymorphism within populations, ranging from nearly white, light pink to deep pink. The pigmentation characteristics of P. ostii flowers make them an excellent model for studying the molecular basis of the intensity variation in pigmentation.
In most plant species, flower coloration is primarily caused by flavonoids, particularly anthocyanins (Grotewold, 2006). The anthocyanin biosynthesis pathway is one of the best characterized secondary metabolism pathway in plants, and is highly conserved in structural and regulatory components (Feller et al., 2011; Hichri et al., 2011). Genes encoding enzymes committed to flavonoid biosynthesis, such as chalcone synthase (CHS), chalcone isomerase (CHI), flavanone 3-hydroxylase (F3H), flavonoid 3′-hydroxylase (F3′H), dihydroflavonol reductase (DFR), leucoanthocyanidin dioxygenase (LDOX), and UDP flavonoid glucosyl transferase (UFGT), and genes encoding transporter proteins involved in transportation and storage of floral pigment have been well characterized in many plants (Grotewold, 2006; Chiu et al., 2010; Chen et al., 2011; Zhao et al., 2011; Tanaka and Brugliera, 2013; Li et al., 2014; Zhou et al., 2014a). In addition to the structural components of the pathway, the regulatory mechanisms of the anthocyanin production have also been characterized in several model plants, including petunia (Petunia hybrida), snapdragon (Antirrhinum majus), Arabidopsis thaliana and maize (Zea mays) (Cone et al., 1986; Goodrich et al., 1992; Quattrocchio et al., 1998, 1999; Walker et al., 1999; Spelt et al., 2002; Carey et al., 2004; Schwinn et al., 2006; Albert et al., 2011). The anthocyanin biosynthesis genes are mainly activated by an activation complex, consisting of R2R3-MYB, basic helix-loop-helix (bHLH) and WD-repeat (WDR) proteins (MBW complex), at the transcriptional level (Koes et al., 2005; Ramsay and Glover, 2005). Moreover, several repressors that limit the expression of anthocyanin biosynthesis genes have been identified. For instance, Arabidopsis R3-MYB protein MYBL2 and Petunia R2R3-MYB protein MYB27 can inhibit anthocyanin biosynthesis by forming a MBW inhibitory complex (Kranz et al., 1998; Albert et al., 2014); Mimulus R3-MYB factor ROSE INTENSITY1 (ROI1), Petunia R3-MYB factor MYBx, Arabidopsis R3-MYB factor CAPRICE (CPC) and TRIPTYCHON (TRY), Arabidopsis SBP-box protein (SPL9) and JA-ZIM domain proteins can repress the anthocyanin production by inhibiting the formation of MBW activation complex through competing for bHLH or R2R3-MYB partners (Wang et al., 2008; Wester et al., 2009; Zhu et al., 2009; Albert et al., 2011, 2014; Gou et al., 2011; Qi et al., 2011; Yuan et al., 2013); Arabidopsis LATERAL ORGAN BOUNDARY DOMAIN (LBD) transcription factors LBD37, LBD38, and LBD39, have also been identified as repressors of anthocyanin biosynthesis (Rubin et al., 2009). However, the information on the regulation of flower pigmentation in tree peonies is lacking, although previous studies have shown that the broad color series in tree peony were primarily determined by the anthocyanin content and types in the petal tissues (Wang et al., 2001; Zhang et al., 2007, 2014; Zhou et al., 2011, 2014a; Zhao et al., 2015). The knowledge from model plants provided useful references for approaching the factors determining flower color intensity in P. ostii.
Generally, the variation in anthocyanin concentration is responsible for the flower color intensity (Grotewold, 2006; Tanaka et al., 2008). The quantitative change in anthocyanin intensity is accompanied by the amount alteration of flux through the pathway (Sobel and Streisfeld, 2013). It has been proposed that either increasing the functional activity of pathway enzymes and activation regulators or removing the repressors of anthocyanin production in flowers can result in an increase in anthocyanin intensity (Sobel and Streisfeld, 2013). It is unclear, however, whether intensity variation in flower pigmentation in P. ostii is generated by alterations of expression of anthocyanin biosynthesis genes or anthocyanin repressor genes. In this study, we compared the pigment composition and transcriptomes of P. ostii flowers with different intensity of coloration. We aimed to explore the correlations between color intensity and anthocyanin concentration, and to identify transcriptional changes and candidate genes potentially responsible for the control of pigmentation intensity in P. ostii. The results would provide insights into the molecular basis underlying the intensity variation in flower pigmentation in P. ostii.
Materials and Methods
Plant Materials
P. ostii was grown in the peony planting base of Fenghuangshan, Tongling, Anhui, China (Figure 1A). At full-bloom stage, flower color was analyzed following the International Commission on Illumination (CIE) system. The L* (lightness), a* (redness and greenness), and b* (yellowness and blueness) were measured using a hand-held spectrophotometer (NF333, Nippon Denshoku Industries Co., Ltd., Tokyo, Japan). For each flower, three areas of the medial surface were measured. For each individual plant, the measurement was performed with three petals from three independent flowers. The L* is an indicator of flower color intensity, as lower lightness generally means deeper color. Four classes of color intensity were chosen for this study: L* ranges of >85, 72–75, 65–68, and 57–60 corresponded to color class I, II, III, and IV, respectively. For each color intensity class, four petal samples were collected from different plants and immediately frozen in liquid nitrogen and stored at −80°C until required for anthocyanin analysis and RNA extractions. Each sample contained three to four full-bloom flowers.
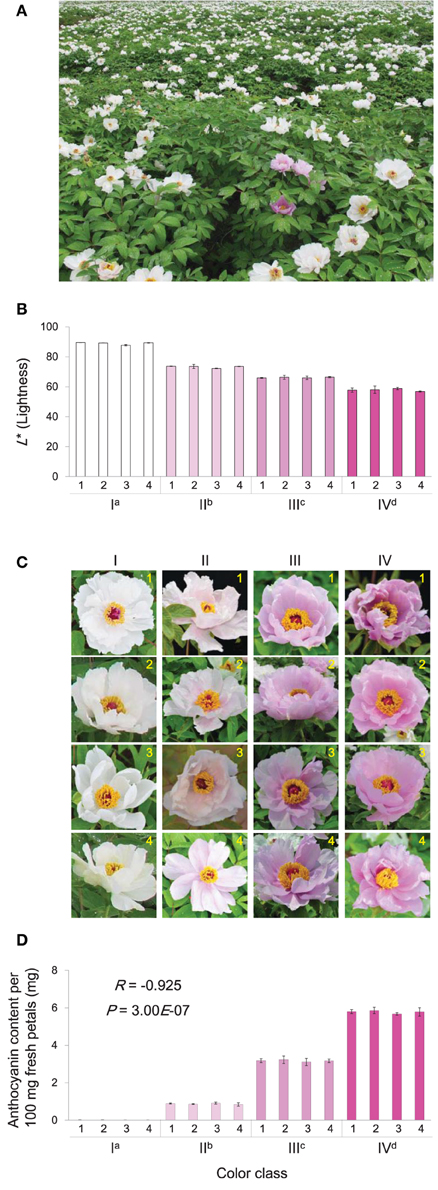
Figure 1. Characters of P. ostii flowers used in this study. (A) Typical environment of sampling location. (B) The petal L* (Lightness) of flowers in 16 plants belonged to the four color classes (I, II, III, and IV). (C) The floral phenotypes of these plants. (D) The corresponding anthocyanin concentrations of petals from these plants. In plot (B,D), each value is shown as average ± standard deviation; different superscript letters on the horizontal axis labels indicate statistically significant differences between means of different color classes, as judged by t-test (P < 0.05). In plot (D), the R- and P-values indicate the correlation between L* and anthocyanin concentration.
Pigments Extraction and High-Performance Liquid Chromatography (HPLC) Analyses
Petal samples from 16 plants belonged to four color intensity classes (four plants each) were extracted separately in acidic methanol solution (acetic acid: methanol: water = 1: 4: 5 v/v; 5 ml/100 mg tissue) for 24 h at 4°C and passed through a syringe filter (0.22 μm; Pall). Extracts were analyzed on a Nova Pak C18 column (Waters) and detected using a photodiode array detector (Waters) in the range 190–600 nm. Peonidin-3,5-di-O-glucoside (Pn3G5G) and cyanidin-3,5-di-O-glucoside (Cy3G5G) were identified using the retention time by comparing with the HPLC data obtained from the commercially available standards (Polyphenols Laboratory, Sandnes, Norway). All determinations were performed with three replicates. Peak area was recorded.
Total anthocyanins and anthoxanthins (flavone and flavonol) were determined semi-quantitatively using the peak area normalization method by employing simple linear regressions using standards Pn3G5G and Cy3G5G for anthocyanins and Rutin for anthoxanthins at 525 and 350 nm, respectively. The content of anthocyanin and anthoxanthin was calculated in mg per 100 mg of fresh petals (as a sum of quantity of Pn3G5G and Cy3G5G mg/100 mg, and as a quantity of Rutin mg/100 mg, respectively).
RNA Extraction and Transcriptome Sequencing
Total RNA was extracted from the petals using TRIzol Reagent (Life Technologies Corp., Carlsbad, CA, USA) following the manufacturer's standard protocol, then treated with RNase-free DNaseI (Ambion, USA). RNA purity was assessed using a Nanodrop 2000C spectrophotometry (Thermo scientific, USA). Another precipitation step with 0.1 volume of 3 M sodium acetate and 2.5 volumes 100% (vol/vol) ethanol, was conducted if the 260/230 absorbance ratio of the total RNA was less than 1.5. RNA quality was assessed on a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA); RNA integrity number (RIN) value was greater than 7.5.
We performed transcriptome sequencing on the bulked RNA of four plants for each flower color intensity class. cDNA library construction and Illumina sequencing were carried out at Beijing Genomics Institute (BGI)-Shenzhen, Shenzhen, China (http://www.genomics.cn/index.php) following the Illimina manufacturer's instructions (Illumina, San Diego, CA, USA). Briefly, poly-A RNA was enriched from 10 μg of total RNA using Magnetic beads with oligo (dT) and broken into short fragments with fragmentation buffer. Using these short fragments as templates, first-strand cDNA was synthesized using random hexamer primer. Then, second-strand cDNA was synthesized using buffer, dNTPs, RNase H (Invitrogen) and DNA polymerase I (Invitrogen). Following size selection and PCR amplification, the cDNA library was sequenced in a HiSeq 2000 to generate paired-end reads. After removing Illumina adapters and reads with unknown nucleotides larger than 5%, and trimming low-quality bases, the remaining high quality reads (clean reads) with an average length of 90 bp were used in this study. The raw sequence data sets were deposited in the US National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/Traces/sra) (Wheeler et al., 2008) under accession number SRP058369, including SRR2027814, SRR2027815, SRR2027817, SRR2027818, SRR2027819, SRR2027820, SRR2027821, and SRR2027822.
De novo Assembly and Functional Annotation
Transcriptome de novo assembly was performed with a short reads assembling program—Trinity (Grabherr et al., 2011). We further used a rapid clustering tool—TGICL (Pertea et al., 2003) to assemble unigenes from all four libraries to obtain a single set of non-redundant unigenes. Unigenes were annotated by BLASTX searches against the NCBI non-redundant protein (Nr) database (http://www.ncbi.nlm.nih.gov), Swiss-Prot protein database (http://www.expasy.ch/sprot) and Arabidopsis protein database at the Arabidopsis Information Resource (TAIR, http://www.arabidopsis.org) with an E-value threshold of 10−5. Only the top hit for each sequence was extracted. According to the homology annotation against Arabidopsis protein database and NCBI Nr database, gene ontology (GO, http://www.geneontology.org) annotation of unigenes was obtained using Blast2GO program (Conesa et al., 2005). Unigene sequences were also aligned to the Kyoto Encyclopedia of Genes and Genomes Pathway (KEGG; http://www.genome.jp/kegg) database (Ogata et al., 1999) by BLASTx program using an E-value cutoff of 10−5 to predict the metabolic pathway annotation.
Digital Gene Expression Profiling and Screening of Differentially Expressed Genes
To obtain digital gene expression profiles for different flower color intensity, all of the clean reads from four color classes were separately mapped to the non-redundant reference transcriptome sequences (all-unigenes). SOAPaligner/soap2 (Li et al., 2009) was used for mapping of the reads because of its high alignment accuracy and rapid alignment speed (Hatem et al., 2013; Shang et al., 2014). The number of unambiguously mapped clean reads for each gene in each sample was separately counted. The gene expression level was calculated using the Fragments Per Kilobase of exon per Million fragments mapped (FPKM) method (Mortazavi et al., 2008). Differentially expressed genes were identified based on the method described by Audic and Claverie (1997). The false discovery rate (FDR) was adopted to correct P-values in multiple hypothesis tests. A genes was judged to be differentially expressed if it had an FDR = 0.001 and the absolute value of log2 Ratio ≥ 2. Unigenes with fewer than 50 reads in all samples were excluded from analysis.
Go Enrichment Analysis
GO categories enrichment analysis was implemented using the Bioconductor topGO package (Gentleman et al., 2004; Alexa et al., 2006), with the default arguments and Fisher's exact test to evaluate statistical significance.
Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Validation and Expression Analysis
qRT-PCR analysis was performed to validate the expression pattern of selected genes identified by the digital expression analysis. The RNA separately extracted from all of the 16 petal samples was used. First strand cDNA was synthesized from 1 μg of total RNA using PrimeScript RT (Perfect Real Time) kit (TAKARA, Japan). The correctness of the gene sequences in the reference transcriptome was validated by reverse transcription PCR and TA cloning using PMD19-T vector kit (TAKARA), followed by sequencing. The qRT-PCR reactions with gene-specific primers (Supplementary Table 1) were performed using a Real-time PCR Detection Systems (RocheCycler 480, Roche, Germany) and SYBRGreen PCR Master Mix (Roche, Germany) according to the manufacturer's instructions. Three independent biological replicates were performed for each reaction. Expression levels of the selected unigenes were normalized to that of an internal reference gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which was revealed as a stably expressed gene by our Illumina sequencing data and previous studies (Wang et al., 2012). As the PCR efficiency for all the gene-specific primers ranged between 93 and 107% over 1000-fold of cDNA dilution (Supplementary Figure 1 and Supplementary Table 1), relative expression levels were calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001).
Phylogenetic Analysis
Multiple sequence alignments were produced by ClustalW using default settings, and the phylogenetic trees were created using the neighbor-joining method and bootstrap analysis (1000 replicates) in MEGA5 software (Tamura et al., 2011). The tree in Supplementary Figure 5 was constructed based on an alignment of the R2 and R3 MYB DNA-binding domains of the translated coding sequence of PoMYB2 (Unigene2047_All) and other MYB sequences. The tree in Supplementary Figure 6 was constructed based on an alignment of the SBP-domains of the translated coding sequence of PoSPL1 (Unigene24255_All) and other SPL sequences.
Results
Flower Pigmentation
The typical view of the growth location of P. ostii is shown in Figure 1A. Based on the lightness (L*) of petals, the flowers of P. ostii divided into four classes were chosen for this study. The petals of class I flowers were nearly white, with L* values greater than 85; The class II flowers have light pink petals, with L* values ranging from 72 to 75; The class III flowers have medium pink petals, with L* values ranging from 65 to 68; Deep pink petals were found in the class IV flowers, with L* values ranging from 57 to 60 (Figures 1B,C). Detection of anthocynins present in various flowers showed that the anthocyanin concentration was inversely correlated with L* (Figures 1B,D). A relatively high level of Peonidin-3,5-di-O-glucoside (Pn3G5G) and a trace amount of cyanidin-3,5-di-O-glucoside (Cy3G5G) were detected in medium and deep pink flowers. A low level of Pn3G5G was detected in the light pink flower, while Cy3G5G was almost undetectable. Both Pn3G5G and Cy3G5G were undetectable in the white flower (Supplementary Figure 2). Pn3G5G and Cy3G5G are synthesized based on peonidin and cyanidin, respectively. Cyanidin and peonidin are two similar anthocyanidins derived from the same branch of the anthocyanin biosynthesis pathway, differing in that peonidin has a methyl substitution on the B-ring.
Transcriptome De novo Assembly and Functional Annotation
In order to identify genes associated with flower color variation in P. ostii, we separately sequenced the petal transcriptomes of flowers of different color classes. De novo assembly using approximately 129, 138, 130, and 139 million paired-end clean reads generated 59,532, 58,169, 60,929 and 60,896 unigenes in class I, II, III, and IV, respectively (Table 1). A total of 66,501 non-redundant all-unigenes were obtained after clustering, with average length and N50 (50% of the assembled bases are incorporated into contigs of length N or larger) of 973 and 1475 bp respectively (Table 1). Among 66,501 all-unigenes, 41,409 (62.27%) were larger than 500 bp in length (Supplementary Table 2). The annotation results of all-unigenes were shown in Supplementary Table 3.
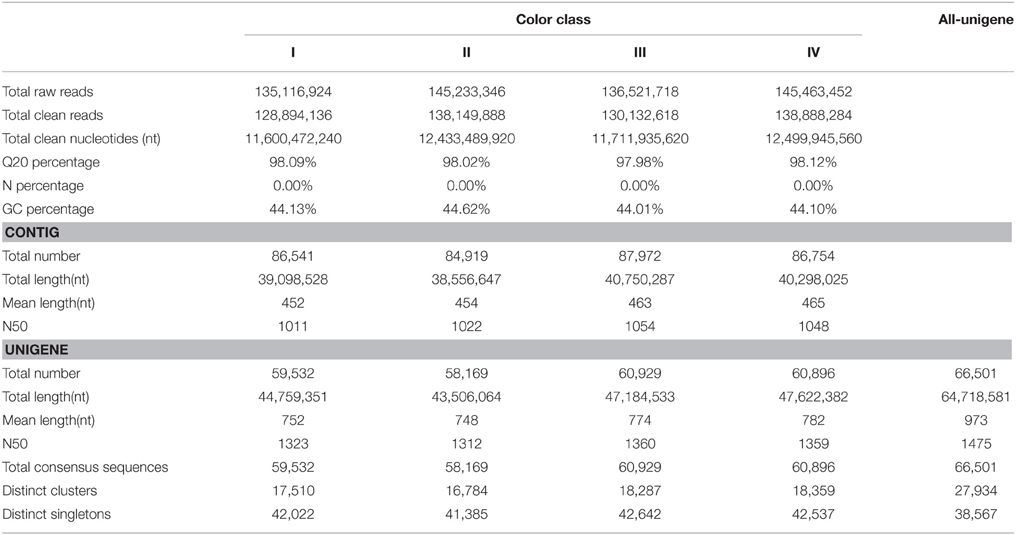
Table 1. Summary of transcriptome sequencing and assembly results in P. ostii.
Based on BLAST, 33,289 (55.92%), 33,130 (56.95%), 34,067 (55.91%), and 34,188 (56.14%) unigenes in class I, II, III, and IV respectively, showed significant similarity to known proteins (Table 2). 29,621, 29,433, 30,213, and 30,293 unigenes could be assigned to at least one Gene Ontology (GO) term in class I, II, III, and IV, respectively. Base on the GO term annotation, the unigenes were categorized into 48 functional groups (Figure 2). The distribution patterns of unigenes from the four classes were similar under different GO categories (Figure 2). Unigenes assigned to categories of “cellular process,” “catalytic activity,” and “metabolic process” were dominant. More than 470 unigenes were assigned to “flavonoid metabolic process” and “pigmentation” categories in each class of flowers. KEGG pathway analyses showed that 18,130, 18,083, 18,637, and 18,770 unigenes in class I, II, III, and IV respectively, could be assigned to 128 KEGG pathways. The most represented pathways were “metabolism” (containing around 21% unigenes in each class), “biosynthesis of secondary metabolites” (around 11% unigenes in each class) and “plant hormone signal transduction” (around 5% unigenes in each class) (Supplementary Table 4).
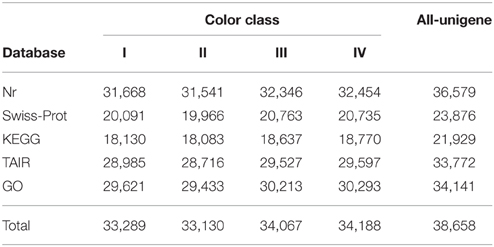
Table 2. Number of unigenes of P. ostii annotated with various databases.
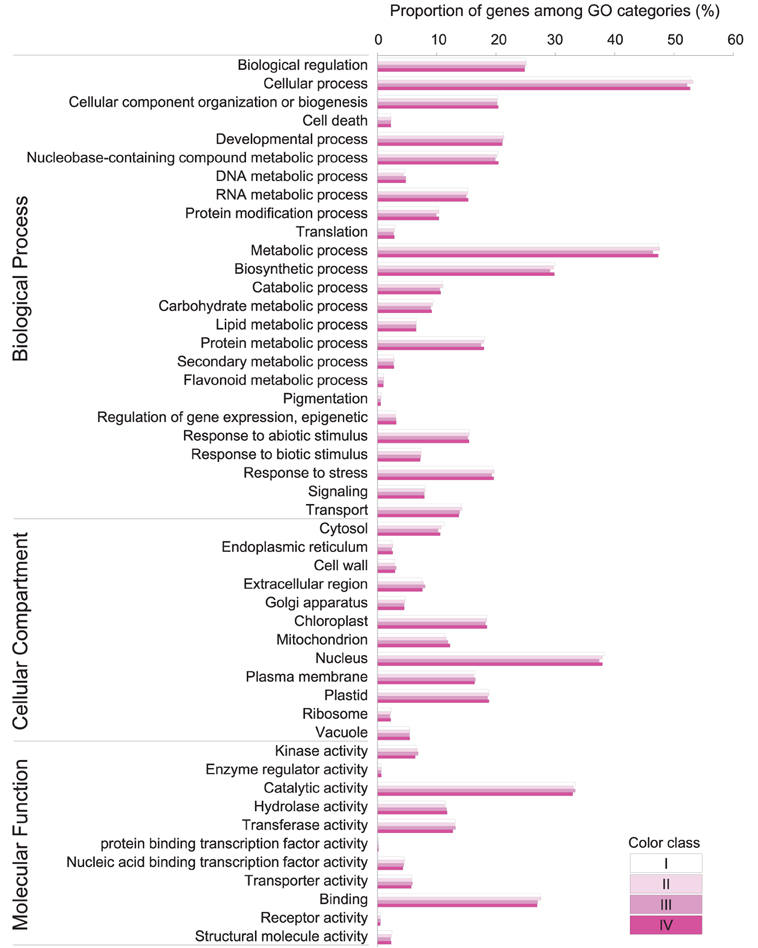
Figure 2. Gene classification based on GO for unigenes in P. ostii of different flower color classes. The results are summarized in three main categories: biological process, cellular compartment, and molecular function. The frequency of GO terms was analyzed using GO Slim Assignment. Bars indicate the ratio of each cluster.
Differentially Expressed Unigenes Between Flowers with Varied Color Intensities
Gene expression profiles of petals were compared between flowers with different color intensities. 55.50, 57.28, 55.27, and 55.47% reads of class I, II, III and IV libraries were aligned uniquely to the reference transcriptome obtained via combined assembly. A total of 7187 unigenes were differentially expressed between flowers with different color intensities (Supplementary Table 5). Most of the differentially expressed unigenes were found between pink and white flowers. The highest number of differentially expressed unigene was observed between flowers of class I and IV (Figure 3). One hundred and twenty-eight unigenes were found to be commonly up-regulated in various pink flowers, while 424 unigenes being commonly down-regulated. To validate our expression data obtained by RNA sequencing, the differential expression patterns of 19 unigenes were verified by qRT-PCR (Supplementary Figure 3). A high correlation was observed between expression levels obtained by RNA-Seq and qRT-PCR (R = 0.787, P = 1.90E-13) (Supplementary Figure 3).
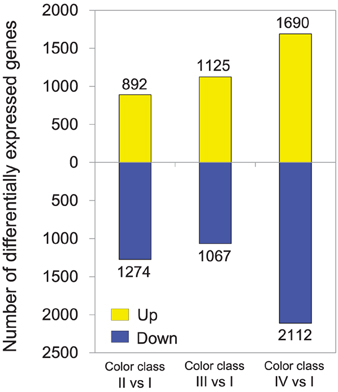
Figure 3. Changes in gene expression profile between P. ostii petals of different color classes. The number of up- and down-regulated genes between class II and class I, class III and class I, and class IV and class I are summarized. For example, “892” means the expression of 892 genes was significantly higher in color class II than in color class I; and “1274” means the expression of 1274 genes was significantly lower in color class II than in color class I.
GO enrichment was carried out on differential expressed genes (Figure 4). The GO categories enriched in the up-regulated gene set include “Fatty acid beta-oxidation,” “extracellular region,” “carboxylesterase activity,” and “fatty-acyl-CoA reductase activity.” Several categories, such as “MAPKKK cascade,” “intracellular signal transduction,” “ethylene mediated signaling pathway,” “ATP-dependent peptidase activity,” “squalene monooxygenase activity,” and “calcium ion binding,” were significantly enriched in the down-regulated gene set.

Figure 4. Overrepresented GO terms amongst differentially expressed genes between P. ostii petals of different color classes. Only GO terms are shown where P < 0.01 in at least one of the six gene sets (up- or down-regulated in any of these three comparisons: class II vs. class I, class III vs. class I, or class IV vs. class I).
Genes involved in anthocyanin biosynthesis and transport were identified from the P. ostii transcriptome (Supplementary Figure 4 and Supplementary Table 6), and their expression levels in different classes of flowers were measured by RNA-Seq and qRT-PCR. The results showed that genes encoding chalcone isomerase (CHI; PoCHI3), flavanone 3-hydroxylase (F3H; PoF3H13, PoF3H18, and PoF3H20), flavonoid 3′-hydroxylase (F3′H; PoF3′H11, and PoF3′H12), dihydroflavonol reductase (DFR; PoDFR6), and leucoanthocyanidin dioxygenase (LDOX; PoLDOX1) were expressed at a higher level in various pink flowers than in white ones (Figure 5). Notably, the expression levels of PoDFR6 and PoLDOX1 were well correlated with flower color intensity and anthocyanin concentration (Figure 5). These two genes were involved in the later steps of anthocyanin biosynthesis. The expression of genes encoding chalcone synthase (CHS; PoCHS1, and PoCHS2) and UDP flavonoid glucosyl transferase (UFGT; PoUFGT1, PoUFGT2, PoUFGT3, PoUFGT4, and PoUFGT5) showed no significant difference between pink and white flowers (Figure 5, Supplementary Table 6). The anthocyanins are stored in the central vacuole in plants (Kitamura, 2006). The activity of vacuolar flavonoid transporters, such as multidrug and toxin extrusion (MATE) and ATP-binding cassette (ABC) transporters, can affect anthocyanins accumulation in vacuole, which in turn affects the intensity of pigmentation in plant tissues (Goodman et al., 2004; Gomez et al., 2009; Zhao et al., 2011). The increasing tendency for anthocyanin accumulation in pink flowers of P. ostii was also accompanied by increased expression of genes encoding MATE transporters (PoMATE1, PoMATE2, and PoMATE3) and ABC transporters (PoMRP1 and PoMRP2) (Figure 6), suggesting their potential roles in promoting anthocyanins accumulation in P. ostii.
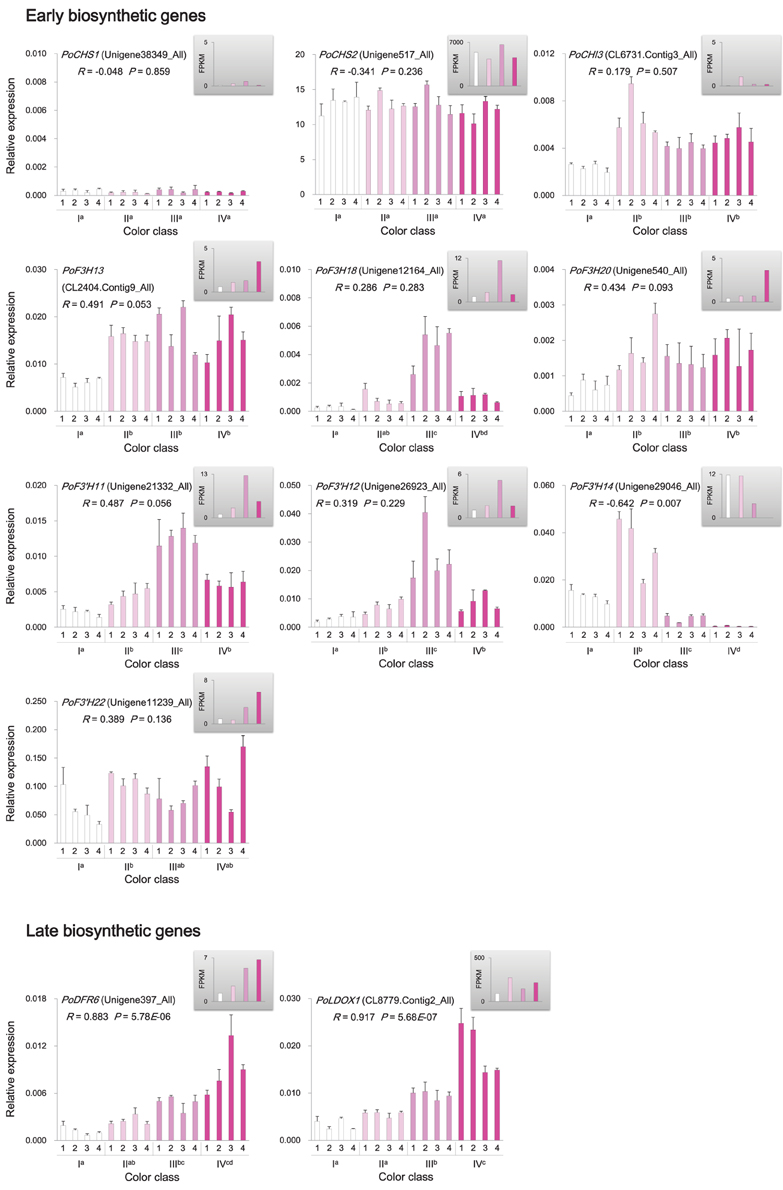
Figure 5. Expression of anthocyanin structural genes in P. ostii of different flower color classes. The expression was investigated by qRT-PCR (shown in the bigger histograms) and RNA-seq (shown in the smaller histograms in the gray shadow areas) of petal samples from the four color classes (I, II, III, and IV). The analyzed plants were shown in Figure 1. For qRT-PCR, each value was normalized relative to PoGAPDH expression and is shown as average ± standard deviation from three biological replicate sampling. Different superscript letters on the horizontal axis labels indicate statistically significant differences between means of different color classes, as judged by t-test (P < 0.05). The R- and P-values given in the plot indicate the correlation between anthocyanin concentration and gene expression. CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; DFR, dihydroflavonol 4-reductase; LDOX, leucoanthocyanidin dioxygenase.
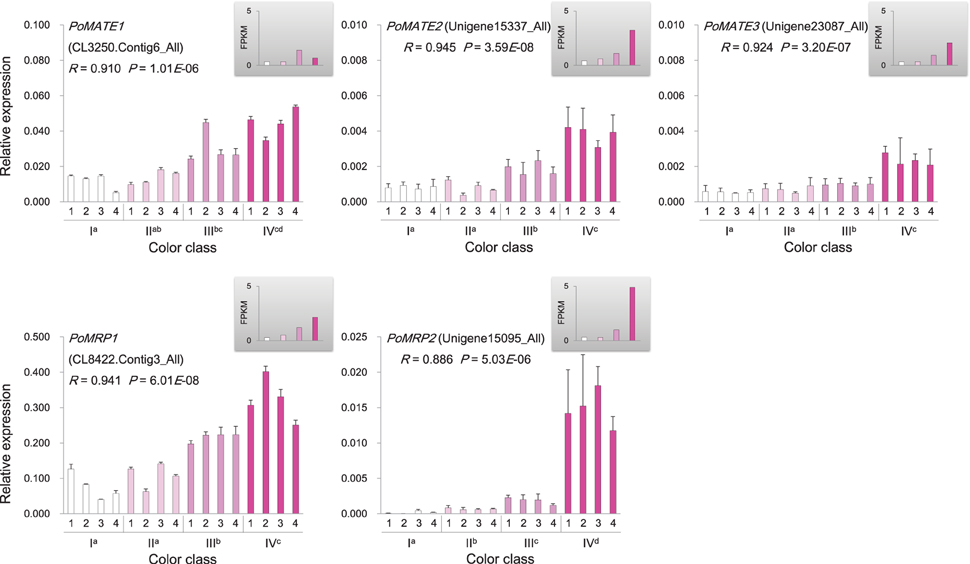
Figure 6. Expression of anthocyanin transporter genes in P. ostii of different flower color classes. The expression was investigated by qRT-PCR (shown in the bigger histograms) and RNA-seq (shown in the smaller histograms in the gray shadow areas) of petal samples from the four color classes (I, II, III, and IV). The analyzed plants were shown in Figure 1. For qRT-PCR, each value was normalized relative to PoGAPDH expression and is shown as average ± standard deviation from three biological replicate sampling. Different superscript letters on the horizontal axis labels indicate statistically significant differences between means of different color classes, as judged by t-test (P < 0.05). The R- and P-values given in the plot indicate the correlation between anthocyanin concentration and gene expression. MATE, multidrug and toxin extrusion transporter; MRP, ATP binding cassette transporter.
The MBW ternary protein complex composed of R2R3-MYB and basic helix-loop-helix (bHLH) transcription factors as well as WD-repeat (WDR) proteins has been documented as a primary regulator in anthocyanin biosynthesis (Heim et al., 2003; Baudry et al., 2004; Zimmermann et al., 2004; Koes et al., 2005; Ramsay and Glover, 2005; Lepiniec et al., 2006; Petroni and Tonelli, 2011). However, genes encoding the active MBW complex components were not significantly differentially expressed among flowers with different color intensities. Instead, a gene, PoMYB2, was identified to exhibit an inverse relationship between its expression and anthocyanin accumulation (Figure 7A). Phylogenetic analysis showed that PoMYB2 was homologous to Petunia PhMYB27 and strawberry FaMYB1 (Supplementary Figure 5), which have been suggested to act as transcriptional repressors of anthocyanin accumulation (Aharoni et al., 2001; Albert et al., 2011). A well-conserved bHLH interaction motif [D/E]Lx2[R/K]x3Lx6Lx3R (Zimmermann et al., 2004) was found in the R3 repeat domain in the predicted protein sequence of PoMYB2 (Figure 7B). A putative repression motif LNLDL conforming to the LxLxL type of ERF-associated amphiphilic repression (EAR) motif was present in the C terminal region of the predicted PoMYB2 protein sequence (Kagale and Rozwadowski, 2011) (Figure 7B). In addition to PoMYB2, PoSPL1, a gene homologous to the miR156-targeted Arabidopsis SPL protein AtSPL13 (Guo et al., 2008; Wu et al., 2009), also exhibited a negative relationship between its expression and anthocyanin concentration (Figure 8A, Supplementary Figure 6). A conserved miR156 target site in the coding sequence of PoSPL1 was also identified (Figure 8B, Supplementary Data Sheet 1).
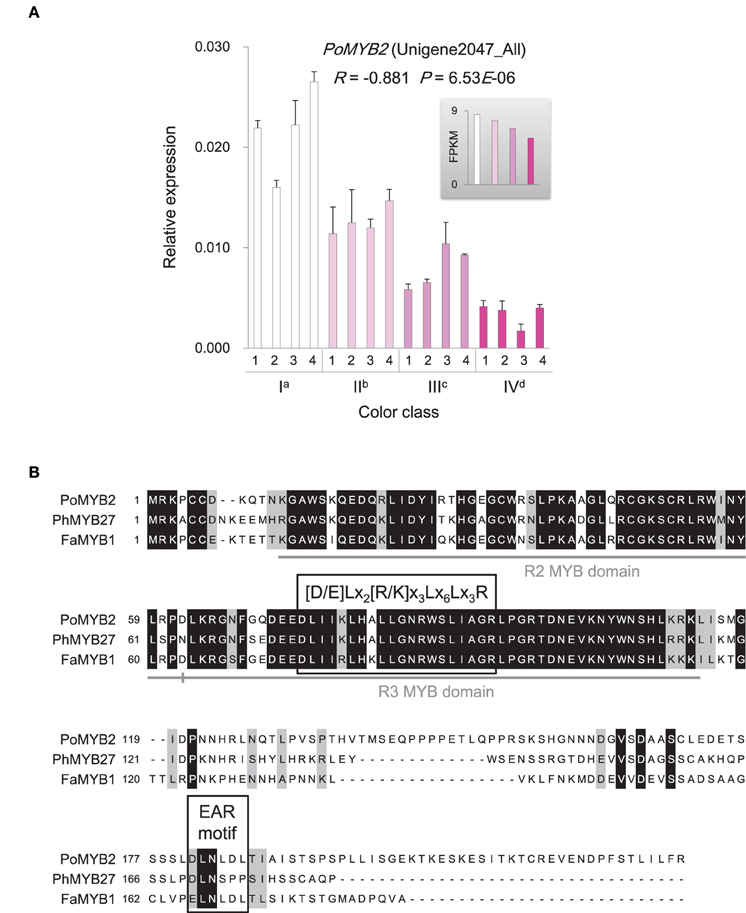
Figure 7. Expression and sequence similarity analysis of PoMYB2. (A) Expression of PoMYB2 gene in P. ostii of different flower color classes. The expression was investigated by qRT-PCR (shown in the bigger histogram) and RNA-seq (shown in the smaller histogram in the gray shadow area) of petal samples from the four color classes (I, II, III, and IV). The analyzed plants were shown in Figure 1. For qRT-PCR, each value was normalized relative to PoGAPDH expression and is shown as average ± standard deviation from three biological replicate sampling. Different superscript letters on the horizontal axis labels indicate statistically significant differences between means of different color classes, as judged by t-test (P < 0.05). The R- and P-values given in the plot indicate the correlation between anthocyanin concentration and gene expression. (B) Amino acid sequence alignments of PoMYB2, PhMYB27, and FaMYB1. The bHLH interaction motif ([D/E]Lx2[R/K]x3Lx6Lx3R) and EAR repressor motif are boxed. Identical amino acids are indicated in black, similar amino acids in gray, gaps introduced in the alignment are indicated with dashes. The full-length coding sequence of PoMYB2 is displayed in Supplementary Data Sheet 1.
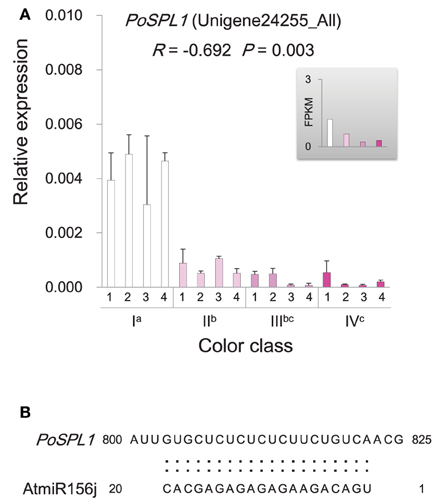
Figure 8. Expression and sequence characteristics of PoSPL1. (A) Expression of PoSPL1 gene in P. ostii of different flower color classes. The expression was investigated by qRT-PCR (shown in the bigger histogram) and RNA-seq (shown in the smaller histogram in the gray shadow area) of petal samples from the four color classes (I, II, III, and IV). The analyzed plants were shown in Figure 1. For qRT-PCR, each value was normalized relative to PoGAPDH expression and is shown as average ± standard deviation from three biological replicate sampling. Different superscript letters on the horizontal axis labels indicate statistically significant differences between means of different color classes, as judged by t-test (P < 0.05). The R- and P-values given in the plot indicate the correlation between anthocyanin concentration and gene expression. (B) The conserved miR156 target site in the coding sequence of PoSPL1. Alignment of the AtmiR156 sequence with PoSPL1. The full-length coding sequence of PoSPL1 is displayed in Supplementary Data Sheet 1.
Discussion
Anthocyanin concentration was closely correlated with the intensity of petal color in P. ostii. Anthocyanins were synthesized via the flavonoid pathway. The corresponding genes have been well characterized and been divided into early biosynthetic genes and late biosynthetic genes in dicotyledonous plants (Martin and Gerats, 1993; Mol et al., 1998; Petroni and Tonelli, 2011). The early biosynthetic genes, including CHS, CHI, F3H, and F3′H, located upstream of the anthocyanin biosynthetic pathway and led to the production of flavonols and other flavonoid compounds, while the late biosynthetic genes, including DFR and LDOX, were downstream genes in the anthocyanin biosynthetic pathway and were specifically for anthocyanin biosynthesis (Lepiniec et al., 2006; Petroni and Tonelli, 2011). In P. ostii, the late anthocyanin biosynthetic genes were differentially expressed among flowers with different color intensities, displaying a strong correlation between their expression levels and the concentration of anthocyanin in petals, whereas none of the early biosynthetic genes exhibited expression patterns significantly positively correlated with the concentration of anthocyanin (Figure 5). The increased expression of DFR (PoDFR6) in deeper color flowers probably led more dihydroflavonols into the direction of anthocyanin branch (Figure 5, Supplementary Figure 4). In contrast, the higher expression of genes encoding flavonol synthase (PoFLS1: CL8054.Contig1_All and PoFLS4: Unigene22443_All) and flavonol 3-O-glucosyltransferase (CL4664.Contig3_All) in the nearly white flowers probably promoted the conversion of dihydroflavonols to flavonols (Supplementary Table 6 and Supplementary Figure 4), resulting in the relatively higher accumulation of anthoxanthins in the nearly white flower than in deeper color flowers (Supplementary Figure 2). Previous studies have shown that inactivation of FLS promoted the accumulation of anthocyanin, while inactivation of DFR promoted the accumulation of flavonols (Pelletier et al., 1999; Owens et al., 2008; Stracke et al., 2009). Substrate competition seems to exist between FLS and DFR, controlling the metabolic flux through the branches of the flavonoid biosynthetic pathway underlying flower color intensity variation in P. ostii. A significant positive correlation was also observed between the expression levels of anthocyanin transporter genes and the anthocyanin levels in petals of different flowers (Figure 6). On the contrary, the putative anthocyanin–biosynthesis repressor genes were expressed at a high level in the nearly white petals and gradually reduced in deep color flowers, exhibiting an expression pattern inverse to anthocyanin biosynthetic and transporter genes (Figures 7, 8). The coordinated expression of genes involved in anthocyanin biosynthesis, anthocyanin transport and suppression of anthocyanin synthesis suggested a sophisticated regulatory system underlying anthocyanin pigmentation in P. ostii.
It has been revealed that the combinatorial action of R2R3-MYB and bHLH transcription factors, along with WDR proteins, played an important role in regulating the transcription of anthocyanin genes (Heim et al., 2003; Baudry et al., 2004; Zimmermann et al., 2004; Koes et al., 2005; Ramsay and Glover, 2005; Lepiniec et al., 2006; Petroni and Tonelli, 2011; Thévenin et al., 2012; Xu et al., 2014). The action of the MBW complex is primarily specified by the activity of R2R3-MYB factors in the complex. Diverged members of the R2R3-MYB gene family had distinct roles in determining the action of the complex either to promote or inhibit the transcription of anthocyanin biosynthesis genes (Matsui et al., 2008). In P. ostii, genes homologous to the subunits of the active MBW complex were not differently expressed in flowers with different color intensity. Instead, a new R2R3-MYB gene encoding PoMYB2 highly similar to PhMYB27 (Figure 7B, Supplementary Figure 5) which can reduce anthocyanin production either by repressing the expression of key factors required for MBW complex thus preventing the MBW complex formation or by incorporating to MBW activation complexes to convert them into repressive complexes (Albert et al., 2014), was highly expressed in the nearly white flower and gradually reduced expression in flowers with increasing color intensity (Figure 7A). In line with the inverse expression pattern of PoMYB2 with that of the late anthocyanin biosynthetic genes, the PoMYB2 protein contains an amino acid motif [D/E]Lx2[R/K]x3Lx6Lx3R, which is required for interaction with bHLH partners in MBW complex (Zimmermann et al., 2004), and a C terminal EAR repressor motif (LNLDL), which mediate transcriptional repression in plants (Kagale and Rozwadowski, 2011) (Figure 7B). A similar pattern of expression was also observed for the gene PoSPL1, which was homologous to AtSPL13 (Figure 8A, Supplementary Figure 6). Although the functional evidence on AtSPL13 is lacking to date, previous studies have shown that AtSPL13 is one of the miR156-targeted SPL genes, which also include AtSPL3, AtSPL9, and AtSPL10 (Guo et al., 2008; Wu et al., 2009). Previous functional analyses revealed that the miR156-targeted SPLs shared some common regulatory functions (Yu et al., 2010). For example, both AtSPL9 and AtSPL13 are involved in the regulation of trichome production (Yu et al., 2010; Preston and Hileman, 2013). A well-conserved miR156 target site was also present in the coding sequence of PoSPL1 (Figure 8B). It has been confirmed that AtSPL9 can repress anthocyanin production through its “squelching activity” causing the depletion of transcription factors essential for activation (Gou et al., 2011). That is, AtSPL9 can serve as a competitive inhibitor that acts by binding R2R3-MYB proteins required for the formation of MBW complex for regulation of anthocyanin production (Gou et al., 2011). The expression pattern of PoSPL1 exhibited a negative correlation with the anthocyanin concentration (Figure 8A), implying a possible role of PoSPL1 in repressing anthocyanin synthesis in P. ostii. All these findings suggested the existence of a complex regulatory network controlling anthocyanin synthesis in P. ostii. Negative regulators identified in P. ostii seem to be the major factor responsible for the intensity variation of P. ostii in anthocyanin pigmentation. It is likely that decreased expression of PoMYB2 and PoSPL1 released the repression of anthocyanin structural genes, that in turn increased the flower color intensity in P. ostii.
The decreased expression of PoMYB2 and PoSPL1 in deeper color flowers was unlikely caused by loss-of-function mutations because we did not find critical differences in the coding sequences of PoMYB2 and PoSPL1 from different intensity variants. Additionally, these two genes belong to different gene families and function through distinct mechanisms. The probability is small for PoMYB2 and PoSPL1 to mutate simultaneously to result in a coordinated decrease in expression in P. ostii. PoMYB2 and PoSPL1 might be coordinately regulated by other elements. Gene expression profiling showed that many genes involved in MAPKKK cascade, respiratory burst, phytohormone biosynthesis, and various organic substance metabolic processes were differentially expressed among different intensity variants (Figure 4). The expression levels of two genes (Unigene12751_All, Unigene10302_All) encoding sucrose synthases were positively correlated with the flower color intensity (Supplementary Table 5). Sucrose can promote the anthocyanin production by suppressing the expression of the transcription factor MYBL2 which negatively regulates anthocyanin biosynthesis, while concurrently inducing the expression of the positive regulators, including PAP1, TT8, and GL3 (Solfanelli et al., 2006; Jeong et al., 2010; Das et al., 2012). Previous studies in Arabidopsis showed that the accumulation of anthocyanin in leaves is positively correlated with the increase in endogenous sucrose content (Jeong et al., 2010). Genes involved in ethylene mediated signaling pathway were down-regulated in deeper color flowers (Figure 4), that is of particular interest because it has been revealed that ethylene plays a negative regulatory role in anthocyanin biosynthesis and functions by regulating the activity of positive and negative regulators at the transcriptional level (Jeong et al., 2010; Das et al., 2011, 2012; Qi et al., 2011). Ethylene also has a suppression effect on the sucrose-induced anthocyanin pigmentation (Jeong et al., 2010). Arabidopsis mutants defective in ethylene signaling or wild-type plants treated with ethylene biosynthesis and ethylene-binding inhibitors displayed a promotion on anthocyanin accumulation (Jeong et al., 2010). The alteration of the internal environment resulted from changes in sucrose and ethylene synthesis and signaling probably had an effect on transcriptional variation of anthocyanin repressor genes in different color variants. Fatty acid beta-oxidation was also found among the enriched GO categories in the up-regulated gene sets in deeper color classes (Figure 4). The up-regulation patterns of genes encoding acyl-CoA oxidase (PoACX, Unigene12172_All) and peroxisomal 3-ketoacyl-CoA thiolase (PoPKT, Unigene29334_All, Unigene33907_All) were coordinated with the increase in flower color intensity (Supplementary Table 5). This result probably has implications for the re-allocation of acyl-CoA between fatty acid and anthocyanin biosynthesis. As the final product of fatty acid beta-oxidation, acyl-CoA can be used as a substrate in anthocyanin biosynthesis. The induction of enzymes involved in fatty acid beta-oxidation may result in split-flowing of acyl-CoA and eventually enhance the production of anthocyanin in deeper color petals.
Tree peony is an important ornamental crop in the word, especially in East Asia. Classical breeding techniques have enabled the creation of a wide range of flower colors for tree peony cultivars. It has been look forward, however, to produce more varieties with desirable and novel flower colors by genetic engineering. P. ostii is one of the most important ancestral species of the cultivars of tree peony. The results of this study provided deep insights into the molecular basis underlying the variation in flower color intensity in tree peony, providing a foundation for developing novel flower colors through manipulating the anthocyanin biosynthesis pathway.
Conclusions
Flowers of P. ostii exhibited variations in color intensity, which was significantly correlated with the anthocyanin concentration. Genome-wide transcription analysis showed that, although anthocyanin biosynthesis had a direct effect on the pigmentation of P. ostii flowers, other metabolic and hormone-mediated signaling pathways were also contributed to the color intensity variation in P. ostii. Differential expression of genes encoding anthocyanin repressors, which negatively regulate the expression of anthocyanin biosynthesis genes by affecting the activation capacity of the MYB-bHLH-WDR complex, seems to be the major factor responsible for the intensity variation in anthocyanin pigmentation in P. ostii. This study provided insights into the potential key components related to the regulation of flower color intensity in P. ostii. As P. ostii is one of the most important ancestral species of the cultivars of tree peony, these results might provide a foundation for developing novel flower colors in ornamental cultivars through manipulating the anthocyanin biosynthesis pathway.
Author Contributions
LG designed the research, performed the experiments and the data analysis, and drafted the manuscript. HY contributed analysis tools and participated in the data analysis and manuscript preparation. HL helped in the HPLC experiments and data analysis; JY participated in the design of the study, helped in data analysis and manuscript preparation. YH conceived the idea, and participated in the design of the study and in interpreting results, and manuscript preparation. All authors carefully read and approved the final manuscript.
Funding
This project was supported by the Chenshan Key Scientific Research Projects (F122431, G142421), and Key Technologies R&D Program of Shanghai (14DZ2260400).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2015.01205
Abbreviations
Pn3G5G, peonidin-3,5-di-O-glucoside; Cy3G5G, cyanidin-3,5-di-O-glucoside; PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; 4CL, 4-coumaroyl:CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; FLS, flavonol synthase; DFR, dihydroflavonol 4-reductase; LDOX, leucoanthocyanidin dioxygenase; UFGT, UDP flavonoid glucosyl transferase; MATE transporter, multidrug and toxin extrusion transporter; MRP transporter, ATP binding cassette transporter; bHLH, basic helix-loop-helix; WDR, WD-repeat protein; SPL, SBP-box transcription factor; FDR, False discovery rate; FPKM, Fragments per kilobase of exon per million fragments mapped; GO, Gene Ontology; RIN, RNA integrity number.
References
Aharoni, A., De Vos, C. H. R., Wein, M., Sun, Z., Greco, R., Kroon, A., et al. (2001). The strawberry FaMYB1 transcription factor suppresses anthocyanin and flavonol accumulation in transgenic tobacco. Plant J. 28, 319–332. doi: 10.1046/j.1365-313X.2001.01154.x
Albert, N. W., Davies, K. M., Lewis, D. H., Zhang, H., Montefiori, M., Brendolise, C., et al. (2014). A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell 26, 962–980. doi: 10.1105/tpc.113.122069
Albert, N. W., Lewis, D. H., Zhang, H., Schwinn, K. E., Jameson, P. E., and Davies, K. M. (2011). Members of an R2R3-MYB transcription factor family in Petunia are developmentally and environmentally regulated to control complex floral and vegetative pigmentation patterning. Plant J. 65, 771–784. doi: 10.1111/j.1365-313X.2010.04465.x
Alexa, A., Rahnenfüehrer, J., and Lengauer, T. (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. doi: 10.1093/bioinformatics/btl140
Audic, S., and Claverie, J. M. (1997). The significance of digital gene expression profiles. Genome Res. 7, 986–995.
Baudry, A., Heim, M. A., Dubreucq, B., Caboche, M., Weisshaar, B., and Lepiniec, L. (2004). TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 39, 366–380. doi: 10.1111/j.1365-313X.2004.02138.x
Carey, C. C., Strahle, J. T., Selinger, D. A., and Chandler, V. L. (2004). Mutations in the pale aleurone color1 regulatory gene of the Zea mays anthocyanin pathway have distinct phenotypes relative to the functionally similar TRANSPARENT TESTA GLABRA1 gene in Arabidopsis thaliana. Plant Cell 16, 450–464. doi: 10.1105/tpc.018796
Chen, W. H., Hsu, C. Y., Cheng, H. Y., Chang, H., Chen, H. H., and Ger, M. J. (2011). Downregulation of putative UDP-glucose: flavonoid 3-O-glucosyltransferase gene alters flower coloring in Phalaenopsis. Plant Cell Rep. 30, 1007–1017. doi: 10.1007/s00299-011-1006-1
Chiu, L. W., Zhou, X., Burke, S., Wu, X. L., Prior, R. L., and Li, L. (2010). The purple cauliflower arises from activation of a MYB transcription factor. Plant Physiol. 154, 1470–1480. doi: 10.1104/pp.110.164160
Cone, K. C., Burr, F. A., and Burr, B. (1986). Molecular analysis of the maize anthocyanin regulatory locus C1. Proc. Natl. Acad. Sci. U.S.A. 83, 9631–9635. doi: 10.1073/pnas.83.24.9631
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Das, P. K., Geul, B., Choi, S.-B., Yoo, S.-D., and Park, Y.-I. (2011). Photosynthesis-dependent anthocyanin pigmentation in Arabidopsis. Plant Signal. Behav. 6, 23–25. doi: 10.4161/psb.6.1.14082
Das, P. K., Shin, D. H., Choi, S.-B., and Park, Y.-I. (2012). Sugar-hormone cross-talk in anthocyanin biosynthesis. Mol. Cells 34, 501–507. doi: 10.1007/s10059-012-0151-x
Davies, K. M., Albert, N. W., and Schwinn, K. E. (2012). From landing lights to mimicry: the molecular regulation of flower colouration and mechanisms for pigmentation patterning. Funct. Plant Biol. 39, 619–638. doi: 10.1071/FP12195
Feller, A., Machemer, K., Braun, E. L., and Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Gentleman, R. C., Carey, V. J., Bates, D. M., Bolstad, B., Dettling, M., Dudoit, S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. doi: 10.1186/gb-2004-5-10-r80
Gomez, C., Terrier, N., Torregrosa, L., Vialet, S., Fournier-Level, A., Verriès, C., et al. (2009). Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters. Plant Physiol. 150, 402–415. doi: 10.1104/pp.109.135624
Goodman, C. D., Casati, P., and Walbot, V. (2004). A multidrug resistance-associated protein involved in anthocyanin transport in Zea mays. Plant Cell 16, 1812–1826. doi: 10.1105/tpc.022574
Goodrich, J., Carpenter, R., and Coen, E. S. (1992). A common gene regulates pigmentation pattern in diverse plant species. Cell 68, 955–964. doi: 10.1016/0092-8674(92)90038-E
Gou, J. Y., Felippes, F. F., Liu, C. J., Weigel, D., and Wang, J. W. (2011). Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23, 1512–1522. doi: 10.1105/tpc.111.084525
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grotewold, E. (2006). The genetics and biochemistry of floral pigments. Annu. Rev. Plant Biol. 57, 761–780. doi: 10.1146/annurev.arplant.57.032905.105248
Guo, A.-Y., Zhu, Q.-H., Gu, X., Ge, S., Yang, J., and Luo, J. (2008). Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family. Gene 418, 1–8. doi: 10.1016/j.gene.2008.03.016
Hatem, A., Bozdag, D., Toland, A. E., and Çatalyüerek, Ü. V. (2013). Benchmarking short sequence mapping tools. BMC Bioinformatics 14:184. doi: 10.1186/1471-2105-14-184
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., and Bailey, P. C. (2003). The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20, 735–747. doi: 10.1093/molbev/msg088
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., and Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hopkins, R., and Rausher, M. D. (2011). Identification of two genes causing reinforcement in the Texas wildflower Phlox drummondii. Nature 469, 411–414. doi: 10.1038/nature09641
Jeong, S.-W., Das, P. K., Jeoung, S. C., Song, J.-Y., Lee, H. K., Kim, Y.-K., et al. (2010). Ethylene suppression of sugar-induced anthocyanin pigmentation in Arabidopsis. Plant Physiol. 154, 1514–1531. doi: 10.1104/pp.110.161869
Kagale, S., and Rozwadowski, K. (2011). EAR motif-mediated transcriptional repression in plants - an underlying mechanism for epigenetic regulation of gene expression. Epigenetics 6, 141–146. doi: 10.4161/epi.6.2.13627
Kitamura, S. (2006). “Transport of flavonoids: from cytosolic synthesis to vacuolar accumulation,” in The Science of Flavonoids, ed E. Grotewald (New York, NY: Springer), 123–146.
Koes, R., Verweij, W., and Quattrocchio, F. (2005). Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 10, 236–242. doi: 10.1016/j.tplants.2005.03.002
Kranz, H. D., Denekamp, M., Greco, R., Jin, H., Leyva, A., Meissner, R. C., et al. (1998). Towards functional characterisation of the members of the R2R3-MYB gene family from Arabidopsis thaliana. Plant J. 16, 263–276. doi: 10.1046/j.1365-313x.1998.00278.x
Lepiniec, L., Debeaujon, I., Routaboul, J.-M., Baudry, A., Pourcel, L., Nesi, N., et al. (2006). Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 57, 405–430. doi: 10.1146/annurev.arplant.57.032905.105252
Li, J. J., Zhang, X. F., and Zhao, X. Q. (2011). Tree Peony of China. Beijing: Encyclopedia of China Publishing House.
Li, Q., Wang, J., Sun, H. Y., and Shang, X. (2014). Flower color patterning in pansy (Viola x wittrockiana Gams.) is caused by the differential expression of three genes from the anthocyanin pathway in acyanic and cyanic flower areas. Plant Physiol. Biochem. 84, 134–141. doi: 10.1016/j.plaphy.2014.09.012
Li, R., Yu, C., Li, Y., Lam, T.-W., Yiu, S.-M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Martin, C., and Gerats, T. (1993). Control of pigment biosynthesis genes during petal development. Plant Cell 5, 1253–1264. doi: 10.1105/tpc.5.10.1253
Matsui, K., Umemura, Y., and Ohme-Takagi, M. (2008). AtMYBL2, a protein with a single MYB domain, acts as a negative regulator of anthocyanin biosynthesis in Arabidopsis. Plant J. 55, 954–967. doi: 10.1111/j.1365-313X.2008.03565.x
Mol, J., Grotewold, E., and Koes, R. (1998). How genes paint flowers and seeds. Trends Plant Sci. 3, 212–217. doi: 10.1016/S1360-1385(98)01242-4
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nagata, T., Todoriki, S., Masumizu, T., Suda, I., Furuta, S., Du, Z., et al. (2003). Levels of active oxygen species are controlled by ascorbic acid and anthocyanin in Arabidopsis. J. Agric. Food Chem. 51, 2992–2999. doi: 10.1021/jf026179+
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/27.1.29
Ohno, S., Deguchi, A., Hosokawa, M., Tatsuzawa, F., and Doi, M. (2013). A basic helix-loop-helix transcription factor DvIVS determines flower color intensity in cyanic dahlia cultivars. Planta 238, 331–343. doi: 10.1007/s00425-013-1897-x
Owens, D. K., Alerding, A. B., Crosby, K. C., Bandara, A. B., Westwood, J. H., and Winkel, B. S. J. (2008). Functional analysis of a predicted flavonol synthase gene family in Arabidopsis. Plant Physiol. 147, 1046–1061. doi: 10.1104/pp.108.117457
Pelletier, M. K., Burbulis, I. E., and Winkel-Shirley, B. (1999). Disruption of specific flavonoid genes enhances the accumulation of flavonoid enzymes and end-products in Arabidopsis seedlings. Plant Mol. Biol. 40, 45–54. doi: 10.1023/A:1026414301100
Pertea, G., Huang, X., Liang, F., Antonescu, V., Sultana, R., Karamycheva, S., et al. (2003). TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652. doi: 10.1093/bioinformatics/btg034
Petroni, K., and Tonelli, C. (2011). Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 181, 219–229. doi: 10.1016/j.plantsci.2011.05.009
Preston, J. C., and Hileman, L. C. (2013). Functional evolution in the plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) gene family. Front. Plant Sci. 4:80. doi: 10.3389/fpls.2013.00080
Qi, T. C., Song, S., Ren, Q., Wu, D., Huang, H., Chen, Y., et al. (2011). The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 23, 1795–1814. doi: 10.1105/tpc.111.083261
Quattrocchio, F., Wing, J., van der Woude, K., Souer, E., de Vetten, N., Mol, J., et al. (1999). Molecular analysis of the anthocyanin2 gene of petunia and its role in the evolution of flower color. Plant Cell 11, 1433–1444. doi: 10.1105/tpc.11.8.1433
Quattrocchio, F., Wing, J. F., van der Woude, K., Mol, J. N. M., and Koes, R. (1998). Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 13, 475–488. doi: 10.1046/j.1365-313X.1998.00046.x
Ramsay, N. A., and Glover, B. J. (2005). MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci. 10, 63–70. doi: 10.1016/j.tplants.2004.12.011
Rubin, G., Tohge, T., Matsuda, F., Saito, K., and Scheible, W. R. (2009). Members of the LBD family of transcription factors repress anthocyanin synthesis and affect additional nitrogen responses in Arabidopsis. Plant Cell 21, 3567–3584. doi: 10.1105/tpc.109.067041
Schiestl, F. P., and Johnson, S. D. (2013). Pollinator-mediated evolution of floral signals. Trends Ecol. Evol. 28, 307–315. doi: 10.1016/j.tree.2013.01.019
Schwinn, K., Venail, J., Shang, Y., Mackay, S., Alm, V., Butelli, E., et al. (2006). A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 18, 831–851. doi: 10.1105/tpc.105.039255
Shang, J., Zhu, F., Vongsangnak, W., Tang, Y., Zhang, W., and Shen, B. (2014). Evaluation and comparison of multiple aligners for next-generation sequencing data analysis. BioMed. Res. Intern. 2014, 309650–309650. doi: 10.1155/2014/309650
Sobel, J. M., and Streisfeld, M. A. (2013). Flower color as a model system for studies of plant evo-devo. Front. Plant Sci. 4:321. doi: 10.3389/fpls.2013.00321
Solfanelli, C., Poggi, A., Loreti, E., Alpi, A., and Perata, P. (2006). Sucrose-specific induction of the anthocyanin biosynthetic pathway in Arabidopsis. Plant Physiol. 140, 637–646. doi: 10.1104/pp.105.072579
Spelt, C., Quattrocchio, F., Mol, J., and Koes, R. (2002). ANTHOCYANIN1 of petunia controls pigment synthesis, vacuolar pH, and seed coat development by genetically distinct mechanisms. Plant Cell 14, 2121–2135. doi: 10.1105/tpc.003772
Steyn, W. J., Wand, S. J. E., Holcroft, D. M., and Jacobs, G. (2002). Anthocyanins in vegetative tissues: a proposed unified function in photoprotection. New Phytol. 155, 349–361. doi: 10.1046/j.1469-8137.2002.00482.x
Stracke, R., De Vos, R. C. H., Bartelniewoehner, L., Ishihara, H., Sagasser, M., Martens, S., et al. (2009). Metabolomic and genetic analyses of flavonol synthesis in Arabidopsis thaliana support the in vivo involvement of leucoanthocyanidin dioxygenase. Planta 229, 427–445. doi: 10.1007/s00425-008-0841-y
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tanaka, Y., and Brugliera, F. (2013). Flower colour and cytochromes P450. Philos. Trans. R. Soc. B Biol. Sci. 368, 20432–20432. doi: 10.1098/rstb.2012.0432
Tanaka, Y., Sasaki, N., and Ohmiya, A. (2008). Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 54, 733–749. doi: 10.1111/j.1365-313X.2008.03447.x
Thévenin, J., Dubos, C., Xu, W., Le Gourrierec, J., Kelemen, Z., Charlot, F., et al. (2012). A new system for fast and quantitative analysis of heterologous gene expression in plants. New Phytol. 193, 504–512. doi: 10.1111/j.1469-8137.2011.03936.x
Walker, A. R., Davison, P. A., Bolognesi-Winfield, A. C., James, C. M., Srinivasan, N., Blundell, T. L., et al. (1999). The TRANSPARENT TESTA GLABRA1 locus, which regulates trichome differentiation and anthocyanin biosynthesis in Arabidopsis, encodes a WD40 repeat protein. Plant Cell 11, 1337–1349. doi: 10.1105/tpc.11.7.1337
Wang, L., Albert, N. W., Zhang, H., Arathoon, S., Boase, M. R., Ngo, H., et al. (2014). Temporal and spatial regulation of anthocyanin biosynthesis provide diverse flower colour intensities and patterning in Cymbidium orchid. Planta 240, 983–1002. doi: 10.1007/s00425-014-2152-9
Wang, L. S., Hashimoto, F., Shiraishi, A., Aoki, N., Li, J. J., Shimizu, K., et al. (2001). Phenetics in tree peony species from China by flower pigment cluster analysis. J. Plant Res. 114, 213–221. doi: 10.1007/PL00013985
Wang, S., Hubbard, L., Chang, Y., Guo, J., Schiefelbein, J., and Chen, J. G. (2008). Comprehensive analysis of single-repeat R3 MYB proteins in epidermal cell patterning and their transcriptional regulation in Arabidopsis. BMC Plant Biol. 8:81. doi: 10.1186/1471-2229-8-81
Wang, Y. J., Dong, L., Zhang, C., and Wang, X. Q. (2012). Reference gene selection for real-time quantitative PCR normalization in tree peony (Paeonia suffruticosa Andr.). J. Agric. Biotechnol. 20, 521–528. doi: 10.3969/j.issn.16747968.2012.05.008
Wessinger, C. A., and Rausher, M. D. (2012). Lessons from flower colour evolution on targets of selection. J. Exp. Bot. 63, 5741–5749. doi: 10.1093/jxb/ers267
Wester, K., Digiuni, S., Geier, F., Timmer, J., Fleck, C., and Hulskamp, M. (2009). Functional diversity of R3 single-repeat genes in trichome development. Development 136, 1487–1496. doi: 10.1242/dev.021733
Wheeler, D. L., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2008). Database resources of the national center for biotechnology information. Nucleic Acids Res. 36, D13–D21. doi: 10.1093/nar/gkm1000
Winkel-Shirley, B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. doi: 10.1104/pp.126.2.485
Wu, G., Park, M. Y., Conway, S. R., Wang, J.-W., Weigel, D., and Poethig, R. S. (2009). The sequential Action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138, 750–759. doi: 10.1016/j.cell.2009.06.031
Xu, W., Grain, D., Bobet, S., Le Gourrierec, J., Thévenin, J., Kelemen, Z., et al. (2014). Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 202, 132–144. doi: 10.1111/nph.12620
Yu, N., Cai, W.-J., Wang, S., Shan, C.-M., Wang, L.-J., and Chen, X.-Y. (2010). Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell 22, 2322–2335. doi: 10.1105/tpc.109.072579
Yuan, Y. W., Sagawa, J. M., Young, R. C., Christensen, B. J., and Bradshaw, H. D. (2013). Genetic dissection of a major anthocyanin QTL contributing to pollinator-mediated reproductive isolation between sister species of Mimulus. Genetics 194, 255–263. doi: 10.1534/genetics.112.146852
Zhang, C., Wang, W. N., Wang, Y. J., Gao, S. L., Du, D. N., Fu, J. X., et al. (2014). Anthocyanin biosynthesis and accumulation in developing flowers of tree peony (Paeonia suffruticosa) ‘Luoyang Hong’. Postharvest Biol. Technol. 97, 11–22. doi: 10.1016/j.postharvbio.2014.05.019
Zhang, J. J., Shu, Q. Y., Liu, Z. A., Ren, H. X., Wang, L. S., and De Keyser, E. (2012). Two EST-derived marker systems for cultivar identification in tree peony. Plant Cell Rep. 31, 299–310. doi: 10.1007/s00299-011-1164-1
Zhang, J. J., Wang, L. S., Shu, Q. Y., Liu, Z. A., Li, C. H., Zhang, J., et al. (2007). Comparison of anthocyanins in non-blotches and blotches of the petals of Xibei tree peony. Sci. Horticul. 114, 104–111. doi: 10.1016/j.scienta.2007.05.009
Zhao, D., Tang, W., Hao, Z., and Tao, J. (2015). Identification of flavonoids and expression of flavonoid biosynthetic genes in two coloured tree peony flowers. Biochem. Biophys. Res. Commun. 459, 450–456. doi: 10.1016/j.bbrc.2015.02.126
Zhao, J., Huhman, D., Shadle, G., He, X. Z., Sumner, L. W., Tang, Y. H., et al. (2011). MATE2 mediates vacuolar sequestration of flavonoid glycosides and glycoside malonates in Medicago truncatula. Plant Cell 23, 1536–1555. doi: 10.1105/tpc.110.080804
Zhou, L., Wang, Y., and Peng, Z. H. (2011). Molecular characterization and expression analysis of chalcone synthase gene during flower development in tree peony (Paeonia suffruticosa). Afr. J. Biotechnol. 10, 1275–1284. doi: 10.5897/AJB10.599
Zhou, L., Wang, Y., Ren, L., Shi, Q., Zheng, B., Miao, K., et al. (2014a). Overexpression of PsCHI1, a homologue of the chalcone isomerase gene from tree peony (Paeonia suffruticosa), reduces the intensity of flower pigmentation in transgenic tobacco. Plant Cell Tissue Organ Cult. 116, 285–295. doi: 10.1007/s11240-013-0403-2
Zhou, S. L., Zou, X. H., Zhou, Z. Q., Liu, J., Xu, C., Yu, J., et al. (2014b). Multiple species of wild tree peonies gave rise to the 'king of flowers', Paeonia suffruticosa Andrews. Proc. R. Soc. B Biol. Sci. 281, 41687–41687. doi: 10.1098/rspb.2014.1687
Zhu, H. F., Fitzsimmons, K., Khandelwal, A., and Kranz, R. G. (2009). CPC, a single-repeat R3 MYB, is a negative regulator of anthocyanin biosynthesis in Arabidopsis. Mol. Plant 2, 790–802. doi: 10.1093/mp/ssp030
Zimmermann, I. M., Heim, M. A., Weisshaar, B., and Uhrig, J. F. (2004). Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J. 40, 22–34. doi: 10.1111/j.1365-313X.2004.02183.x
Keywords: Paeonia ostii, flower color intensity, anthocyanin pigmentation, transcriptome changes, anthocyanin repressor
Citation: Gao L, Yang H, Liu H, Yang J and Hu Y (2016) Extensive Transcriptome Changes Underlying the Flower Color Intensity Variation in Paeonia ostii. Front. Plant Sci. 6:1205. doi: 10.3389/fpls.2015.01205
Received: 22 October 2015; Accepted: 14 December 2015;
Published: 06 January 2016.
Edited by:
Tiegang Lu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Tao Sun, Stanford University, USANazgol Emrani, Christian Albrechts University of Kiel, Germany
Copyright © 2016 Gao, Yang, Liu, Yang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Yang, aml5YW5nQGZ1ZGFuLmVkdS5jbg==;
Yonghong Hu, aHV5b25naG9uZ0Bjc25iZ3NoLmNu