 Xiuyan Feng1,2
Xiuyan Feng1,2 Jian Liu
Jian Liu Xun Gong
Xun Gong- 1Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
- 2University of Chinese Academy of Sciences, Beijing, China
The Cycas segmentifida complex consists of eight species whose distributions overlap in a narrow region in Southwest China. These eight taxa are also morphologically similar and are difficult to be distinguished. Consequently, their taxonomic status has been a matter of discussion in recent years. To study this species complex, we sequenced four plastid intergenic spacers (cpDNA), three nuclear genes and genotyped 12 microsatellites for the eight taxa from 19 different localities. DNA sequences were analyzed using Maximum Likelihood (ML) method and Bayesian Inference (BI), and microsatellites were analyzed using the Neighbor-joining (NJ) and structure inference methods. Results of cpDNA, nuclear gene GTP and microsatellites all rejected the hypotheses that this complex consisted of eight taxa or one distinct lineage (species) but two previously described species were adopted: Cycas guizhouensis K. M. Lan et R. F. Zou and Cycas segmentifida D. Y. Wang et C. Y. Deng. Cycas longlinensis H. T. Chang et Y. C. Zhong was included in C. guizhouensis and the other five taxa were included in C. segmentifida. Our species delimitation inferred from molecular data largely corresponds to morphological differentiation. However, the other two nuclear genes were unable to resolve species boundaries for this complex independently. This study offered evidences from different genomes for dealing with the species boundaries and taxonomical treatment of the C. segmentifida complex in an integrated perspective.
Introduction
Species delimitation is essential since species is regarded as the basic unit of analysis in nearly all biological disciplines, such as ecology, biogeography, conservation biology, and macroevolution (Mayr, 1982). Any incorrect species delimitation may result in more serious errors in succeeding relevant studies, which will increase the costs of species conservation or lead to an unpredictable waste of effort (Wiens, 2007). From a common accepted view, a species is considered as an evolutionarily distinct lineage which can be distinguished from other lineages due to reproductive isolation and termination of gene flow, niche differentiation and other lines of evidence (De Queiroz, 1998, 2007; Stockman and Bond, 2007; Bond and Stockman, 2008; Fujita et al., 2012; Hendrixson et al., 2013; Mckay et al., 2013). The methods of species delimitation mainly have two general categories: tree-based and non-tree-based approaches (Sites and Marshall, 2003, 2004). Delimiting species by tree-based methods are carried out by recognizing species as distinguishing clades (phylogenetic species concept), whereas delimitation species by non-tree-based methods are implemented on the basis of gene flow assessments (biological species concept; Pérez-Losada et al., 2005). However, few specific species delimitation criteria have been proposed, within which Wiens and Penkrot presented and compared three specific criteria that were based on DNA data, morphological data and character data respectively to achieve species delimitation validly, which provided a key theoretical frame for empirical studies (Wiens and Penkrot, 2002).
Cycas guizhouensis K. M. Lan et R. F. Zou (Lan and Zou, 1983) and Cycas segmentifida D. Y. Wang et C. Y. Deng (Wang and Deng, 1995) were two primarily published Cycas species from Guizhou province of China, whereas six similar taxa, Cycas longlinensis H. T. Chang et Y. C. Zhong (Zhang and Zhong, 1997), Cycas multifida H. T. Chang et Y. C. Zhong (Zhang et al., 1997), Cycas crassipes H. T. Chang, Y. Z. Zhong et Z. F. Lu (Zhang et al., 1999), Cycas xilingensis H. T. Chang et Y. C. Zhong (Zhang and Zhong, 1997), Cycas longiconifera H. T. Chang, Y. C. Zhong et Y. Y. Huang (Zhang et al., 1997), and Cycas acuminatissima H. T. Chang, Y. C. Zhong et Z. F. Lu (Zhang et al., 1997) were subsequently described, constituting the C. segmentifida complex which are all endemic to Southwest China, mainly in southwestern Guizhou, northwestern Guangxi and eastern Yunnan province. It is the complicated terrain, diversified climates and habitats that result in a high degree of diversification in the genus Cycas in this area, which even makes one continuously distributed species in different regions exhibiting different morphologies. Cycas guizhouensis is widely distributed in the valleys of the Nanpan River characterized by fusiform male cone, loose and open female cone, densely hariy sporophylls, nearly round apical lobe (the margins with numerous tapered lobes) and yellow with reddish brown mucro seeds (nearly globose; Jones, 1998). Cycas longlinensis only exists in Jinzhongshan, Longlin, Guangxi province and is characterized by narrower and longer pinnae, and fewer and broader segments in microsporophyll (Zhang and Zhong, 1997). It resembles C. guizhouensis in morphology, and the above two species have an overlapped distribution along the Nanpan River. Cycas multifida is distinguished by its numerous and glabrous lateral segments in macrosporophyll and distributed only in Bada, Xilin, Guangxi province (Zhang and Zhong, 1997). Cycas xilingensis is distinct from other taxa by its thinner and longer carpophyll and its much longer and wider terminal segment (Zhang and Zhong, 1997). Cycas crassipes is distinguished by its robust pedicels (Zhang et al., 1999) with only one population in Bianya, Longlin, Guangxi province. Cycas segmentifida is widespread, mainly distributed in southwestern Guizhou, northwestern Guangxi and eastern Yunnan province along the You River, and can be characterized by its usually pruinose annal petiole, dichotomus or sometimes forked, acuminate, aristate apically of lateral segments, and ovate-retunded terminal sterile lamina which is covered with caducous brown-tomentose (Wang and Deng, 1995). Cycas longiconifera is mainly distributed in the Baise, Guangxi province and can be distinguished by its slenderly cylindrical male cone (Zhang et al., 1997). Cycas acuminatissima is characterized by its pinnae with acuminate apex and shorter carpophylls (Zhang et al., 1997) and is distributed in northwestern Guangxi province.
The taxonomic status of these similar taxa within the C. segmentifida complex has been a matter of debate in recent years. The Flora of China (Chen and Stevenson, 1999) treated Cycas acuminatissima, C. longlinensis, C. multifida, and C. xilingensis in the synonymy of Cycas segmentifida, while treated Cycas guizhouensis in the synonymy of Cycas szechuanensis, and subsequently Wang (2000) proposed to classify C. longlinensis into C. guizhouensis, the remaining five taxa into C. segmentifida and considered the complex containing only two valid species (i.e., C. guizhouensis and C. segmentifida). Huang (2001) accepted these eight taxa based on their morphological character comparisons, while Whiteloek (2002) placed C. longiconifera, C. multifida, and C. xilingensis into C. segmentifida, put C. acuminatissima into C. sexseminifera and C. longlinensis into C. guizhouensis. The World List of Cycads Group placed Cycas acuminatissima, C. longlinensis, C. multifida, and C. xilingensis in the synonymy of Cycas segmentifida and accepted C. guizhouensis as a distinct taxon (Calonje et al., 2015; http://cycadlist.org/index.php). A recent study which combined morphology and ISSR methods suggested that the C. segmentifida complex only contained two species with C. longlinensis incorporated into C. guizhouensis while the remaining five species into C. segmentifida (Ma, 2005). At present, the classification of these taxa is not fully settled. According to field investigations, we found that C. longlinensis had no substantial difference with C. guizhouensis in morphology. The remaining five taxa were sympatric with C. segmentifida. Cycas guizhouensis and C. segmentifida belonged to two different basins (Nanpan vs. You River) and could be easily distinguished by their leaves and megasporophylls. In this study, species delimitation was carried out on all the above eight taxa by using DNA sequences and microsatellite markers with a tree-based haplotype aggregation methods and genetic structure inference, aiming to deal with their boundaries from a genetic and phylogenetic perspective. With the above methods, we propose the following possible hypotheses for the present taxa:
(1) Eight independent clades would be identified according to the evidence from either DNA haplotypes or genetic structure inferred by SSR data.
(2) None of the eight taxa could be distinguishable while only one single lineage was formed because of complete lineage sorting or strong gene flow and natural hybridization in this area.
(3) At least two distinct groups which corresponded to morphology and geography can be identified.
Materials and Methods
Cycas segmentifida Complex Sampling
All known species in the C. segmentifida complex were investigated and sampled in this study. A total of 311 individuals from eight taxa of this complex were collected from 19 different populations in southwestern Guizhou, northwestern Guangxi and eastern Yunnan province of Southwest China. Young and healthy leaves were dried in silica gel immediately after collection. Within the 311 samples, 5 individuals from each location, except for population BDN where only two individuals were found, were randomly selected for plastid and nuclear DNA sequencing while all the 311 individuals were used for the microsatellite study. Sampling information on each C. segmentifida complex species is summarized in Table 1 and Figure 1.
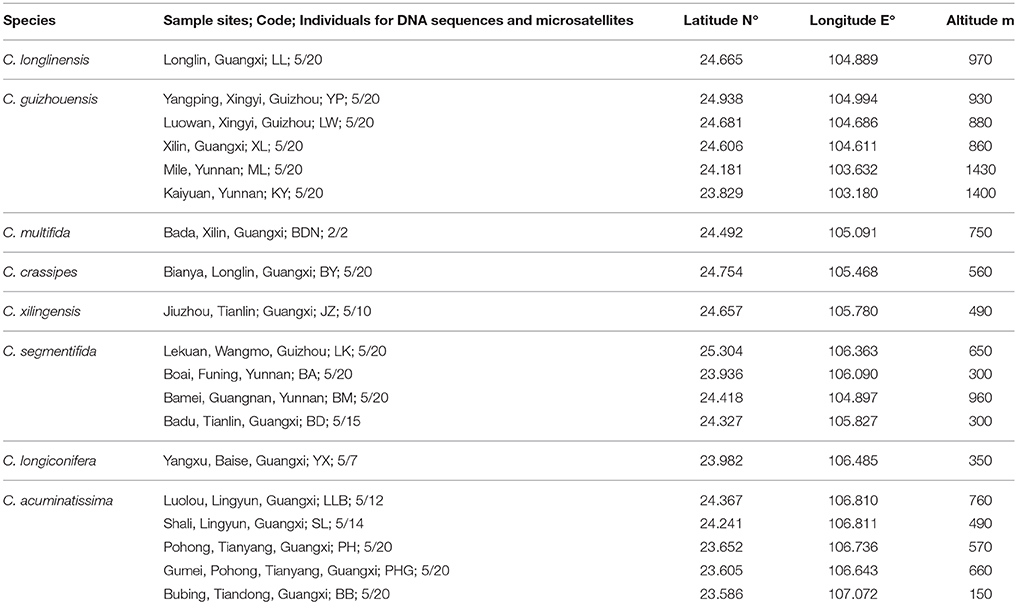
Table 1. Details of sample locations, sample sizes (n) surveyed for DNA sequences and microsatellites of eight taxa of the C. segmentifida complex.
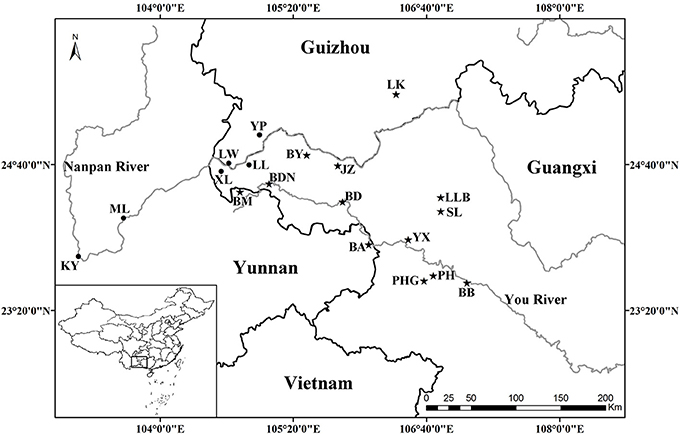
Figure 1. Geographical distribution of 19 populations of the C. segmentifida complex. Maps were drawn using the software ArcGIS version 10.2 (http://desktop.arcgis.com).
DNA Extraction, PCR Amplification, and Genotyping
We extracted genomic DNA from dried leaves using the modified CTAB method (Doyle, 1991). DNA was dissolved in TE buffer and stored at –20°C. After preliminary screening from universal plastid and nuclear primers, four cpDNA intergenic spacers, psbA-trnH (Chiang and Peng, 1998), psbM-trnD (Shaw et al., 2005), trnS-trnG (Shaw et al., 2005), and trnL-trnT (Taberlet et al., 1991) and three nuclear genes, GTP, guanosine triphosphate (GTP) gene (Salas-Leiva et al., 2014); PHYP, phytochrome P gene and PPRC, hypothetical protein gene (unpublished) were chose for complete analysis (Table 2).

Table 2. Sequences of plastid and nuclear gene primers used in this study.
Amplification protocols were as follows: for cpDNA, each 30μL reaction contained 15 ng DNA, 3.0μL 10 × PCR buffer, 1.5μL MgCl2 (25mM), 1.5μL dNTPs (10mM), 1.5μL DMSO, 0.45μL of each primer, 0.45μL Taq DNA polymerase (5 U/μL) (Takara, Shiga, Japan) and 19.5μL double-distilled water; for nuclear genes, the PCR reactions contained 30 ng DNA, 3.0μL 10 × PCR buffer, 2.25μL MgCl2 (25mM), 2.25μL dNTPs (10mM), 2.25 DMSO, 0.6μL of each primer, 0.5μL Taq DNA polymerase (5 U/μL) (Takara, Shiga, Japan) and 15.53μL double-distilled water. PCR amplifications were performed in a thermocycler under the following conditions: an initial 5 min denaturation at 80°C, followed by 29 cycles of 1 min at 95°C, 1 min annealing at 50°C, and a 1.5 min extension at 65°C, and a final extension for 5 min at 65°C for cpDNA intergenic spacers. For nuclear genes, we used an initial 4 min denaturation at 94°C, which was followed by 34 cycles of 45 s at 94°C, 1 min annealing at 54, 55 and 50°C for GTP, PHYP and PPRC, and a 1.5 min extension at 72°C, and a final extension for 9 min at 72°C. All PCR products of different DNA fragments were sequenced directly in both directions by the dideoxy chain-termination method, using an ABI 3730XL automated sequencer (made in Applied Biosystems) at Shanghai Meiji Biological Medicine and Technology Co Ltd. The sequencing primers were the same as those used in the amplification reactions. All the sequences were deposited in GenBank with the accession numbers KU240442-KU240491, KT824924-KT824925, KT824931-KT824932, KT824936, KT824941, KT824946-KT824948, KT824951, KT824954-KT824955, KT824960-KT824962.
Microsatellite markers were selected from recently developed nuclear microsatellites in Cycas (Cibrián-Jaramillo et al., 2008; Wang et al., 2008; Yang et al., 2008; Li et al., 2009; Zhang et al., 2009, 2010; Ju et al., 2011). PCR amplification was carried out as in the same way as in C. simplicipinna (Feng et al., 2014). After preliminary screening microsatellite loci for the C. segmentifida complex, the selected microsatellite loci were stained with fluorescent dye at the 5′ end, their PCR products were separated and visualized using an ABI 3730XL automated sequencer at Shanghai Meiji Biological Medicine and Technology Co Ltd, and their profiles were read with the GeneMapper version 4.0, software (Applied Biosystems, Foster City, CA). An individual was declared null (nonamplifying) at a locus and was treated as missing data after two or more amplification failures. Finally, we chose polymorphic microsatellite loci for the C. segmentifida complex, if the microsatellite locus had two or more alleles (Table 3).
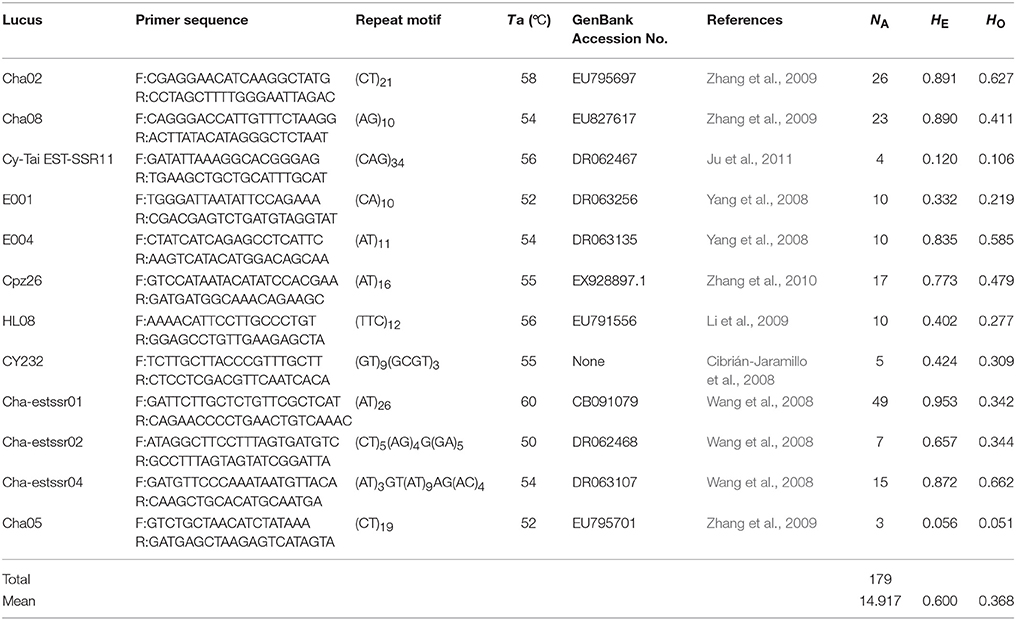
Table 3. Information of 12 microsatellite primers for species delimitation of the C. segmentifida complex.
Sequence Processing and Phylogenetic Reconstruction
Sequences were edited and assembled using SeqMan II (Swindell and Plasterer, 1997). Multiple alignments of DNA sequences were performed manually and subsequently adjusted in Bioedit, version 7.0.4.1 (Hall, 1999). We combined the four cpDNA regions and performed a congruence test in PAUP* 4.0b10 (Swofford, 2003). The testing result showed a significant rate of homogeneity (P = 1, >0.5), suggesting a high degree of homogeneity between the four cpDNA regions. The combined cpDNA sequences were therefore used in the next analyses. Three nuclear genes were analyzed separately. Nuclear genes often had heterozygous sites in some individuals, which were identified by overlapping peaks in chromatograms. We resolved the nuclear sequences by applying the algorithms of PHASE (Stephens et al., 2001; Stephens and Donnelly, 2003) in the software package DnaSP, version 5.0 (Librado and Rozas, 2009). The phased nuclear sequences were used in the following analyses. Maps were drawn using the software ArcGIS version 10.2 (http://desktop.arcgis.com).
Haplotypes were inferred from aligned DNA sequences by DnaSP, version 5.0. Phylogenetic relationships among haplotypes were inferred using Maximum Likelihood (ML) and Bayesian Inference (BI) methods with C. dolichophylla as the outgroup. The maximum likelihood analysis using Tamura-Nei model and Nearest-Neighbor-Interchange (NNI) ML heuristic method with 1000 bootstrap replications was performed on MEGA, version 5 (Tamura et al., 2011). Bayesian Inference was performed in MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003). Each Markov chain was started from a random tree and run for 106 cycles with every 100th cycle sampled from the chain. Each analysis was repeated three times for the checking stationarity.
The Wiens and Penkrot's protocol (Wiens and Penkrot, 2002) was used to test species boundaries between haplotypes of the C. segmentifida complex. Their approach named a sampling as focal species (the species of interest in the study; here the C. segmentifida complex) and nonfocal species (one or more closely related species; C. dolichophylla). A haplotype phylogeny tree may show the focal species to be either exclusive or not. The method needs at least two individuals per locality to make the between-population gene flow inferences. It requires a phylogeny of haplotypes (or individuals) of known locality and taxonomic designation. Specific explanations for the method refer to Wiens and Penkrot's article.
Neighbor-Joining Analysis and Structure Inference
Microsatellite data editing and formatting were performed in GenAlEx, version 6.3 (Peakall and Smouse, 2006). Genetic diversity test for microsatellite loci was performed in GenAlEx, version 6.3 by calculating common genetic diversity indices. Input files for other software analysis were exported by GenAlEx, version 6.3. The phylogenetic relationships of sampled species' populations were estimated using Nei's 1983 genetic distance with Neighbor-joining method (NJ) performed in Powermarker, version 3.25 (Lui and Muse, 2005). Confidence in the resulting relationships was assessed using the bootstrap procedure with 1000 bootstrap replicates. The consensus tree was obtained by the procedure consense in the software package Phylip, version 3.68 (Felsenstein, 2002). Genetic structure inference on the microsatellite data was conducted by STRUCTURE, version 2.2 (Pritchard et al., 2000). The combination of an admixture and a correlated-allele frequencies model was used for the analysis. The simulation was run with values of K from 1 to 20 and repeated 20 times for each set. Each run included a burn-in of 1 × 105 iterations and 1 × 105 subsequent MCMC steps. The best-fit number of grouping was evaluated using ΔK and log-likelihood value by STRUCTURE HARVESTER, version 0.6.8 (Earl, 2012).
Results
Haplotypes Distribution in the Cycas segmentifida Complex
The four combined plastid DNA regions surveyed across the 92 individuals (19 populations, Table 1) of the C. segmentifida complex were aligned, with a total length of 3198 bp. 11 haplotypes (comH1-comH11) were inferred from the cpDNA matrix in total. Of those, plastid haplotype comH1 was shared by C. longlinensis and C. guizhouensis, comH6 was shared by C. multifida, C. crassipes, C. xilingensis, and C. segmentifida and the remaining plastid haplotypes were specific to single species with plastid haplotypes comH2-5 specific to C. guizhouensis, comH7 unique for C. longiconifera and comH8-11 only fixed in C. acuminatissima. The aligned nuclear gene GTP had a length of 574 bp identifying five haplotypes (comG1-comG5), of which haplotype comG1 was shared by C. longlinensis and C. guizhouensis while comG3 was shared by the remaining six taxa. 12 haplotypes (comP1-comP12) were derived from the nuclear gene PHYP, which shared a consensus length of 930 bp. Of those, the most abundant haplotype comP1 occupied seven taxa except C. multifida, while the more frequent haplotype comP2 was predominant in C. longlinensis and C. guizhouensis and comP5 in the remaining six taxa. The nuclear gene PPRC fixed 11 haplotypes (comR1-comR11) with a unified length of 718 bp. Haplotype comR1 and comR3 were the most widely distributed haplotypes, separately occupied C. longlinensis and C. guizhouensis and the remaining six taxa. The information of plastid haplotypes and nuclear haplotypes and their distribution components in the C. segmentifida complex are shown in Table 4.
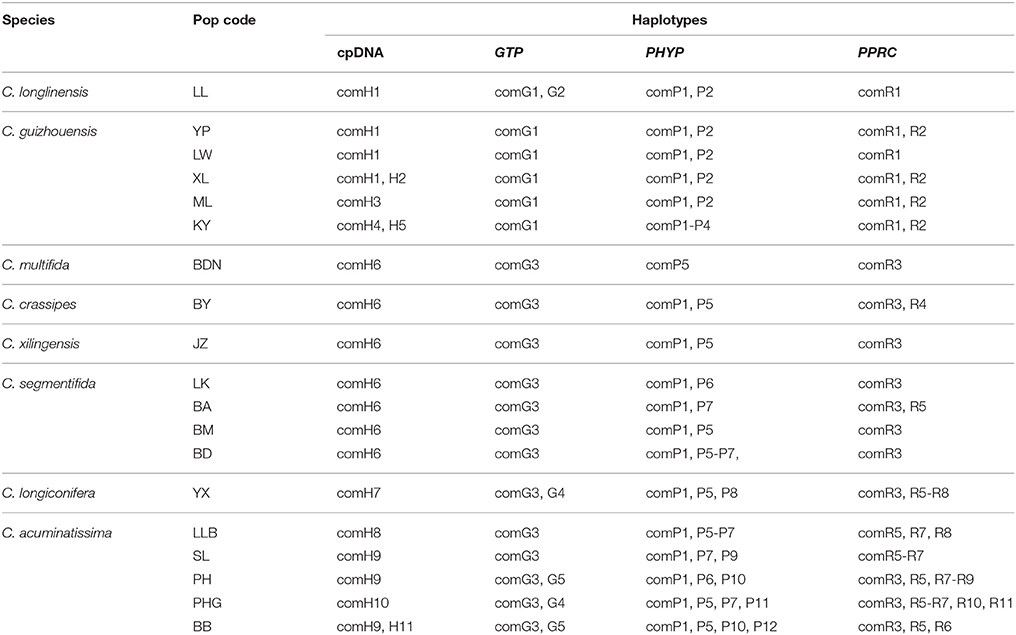
Table 4. Composition of haplotypes in populations of the C. segmentifida complex derived from combined plastid DNA and nuclear genes.
Phylogeny of Haplotypes
Our phylogenetic analyses showed no major conflicts among the ML and BI topologies with the ML tree showing more detailed internal evolutionary relationships within haplotypes (Figures 2, 3). All the plastid haplotypes inferred from the C. segmentifida complex were monophyletic and clustered together into two deep clades, one composed of plastid haplotypes from C. guizhouensis, C. longlinensis and the other clade consisted of C. multifida, C. crassipes, C. xilingensis, C. segmentifida, C. longiconifera, and C. acuminatissima. Thus, the result rejected the hypothesis that the C. segmentifida complex consisted of eight distinct taxa or one single lineage. These two assemblages detected by plastid DNA were also supported by high bootstrap (BS = 96, 94, Figure 2A) and posterior probability (PP = 1, Figure 2B) values. Interspecific relationships were unresolved in all the nuclear gene analyses except GTP, which showed a consistent cladogram with plastid DNA, suggesting the hypothesis that two lineages maybe occurred in this complex (Figures 3A,B).
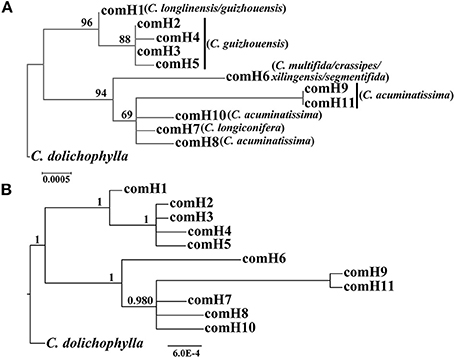
Figure 2. Phylogenetic trees of plastid haplotypes based on (A) Maximum Likelihood (ML) and (B) Bayesian Inference (BI) methods. The number (if ≥ 50 or 0.5) on each branch indicates the (A) bootstrap value (BS) and (B) posterior probability (PP).
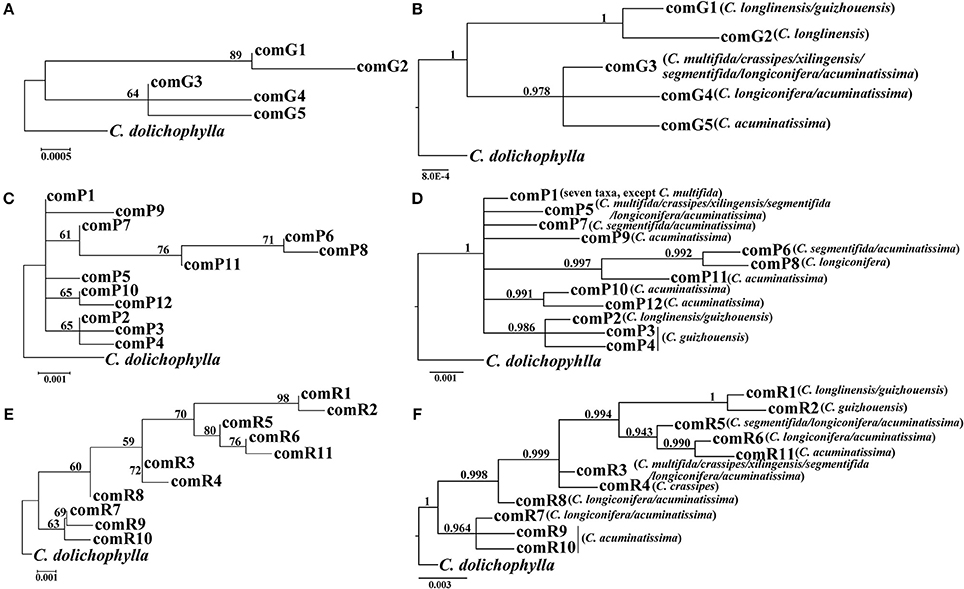
Figure 3. Phylogenetic trees of nuclear haplotypes based on (A,C,E) Maximum Likelihood (ML) and (B,D,F) Bayesian Inference (BI) methods (GTP: (A,B); PHYP: (C,D); PPRC: (E,F). The number (if ≥ 50 or 0.5) on each branch indicates the (A,C,E) bootstrap value (BS) and (B,D,F) posterior probability (PP).
NJ Tree and Structure Inference
After calculating three common genetic diversity indices, 12 microsatellite loci were selected for delimitating species boundaries within the C. segmentifida complex. The 12 microsatellite loci identified 179 alleles in the C. segmentifida complex in total with a mean value of 14.917. The number of alleles (NA) ranged from 3 to 49, expected heterozygosity (HE) ranged from 0.056 to 0.953, and observed heterozygosity (HO) ranged from 0.051 to 0.662. The levels of genetic diversity estimated from 12 loci were different. The highest level of genetic diversity was detected in the locus Cha-estssr01, while the lowest level of genetic diversity was detected in the locus Cha05 (Table 3). The 12 polymorphic microsatellites were used to investigate the phylogenetic relationships of 19 populations from the eight taxa in C. segmentifida complex.
For the result of NJ tree, all populations of the C. segmentifida complex were grouped into three main clades with two clades supported by a high BS value (99/92) (Figure 4A). Cycas longlinensis together with five populations of C. guizhouensis were clustered into a clade, five populations of C. acuminatissima together with one population C. segmentifida as well as C. longiconifera were grouped into a clade and the remaining six populations from four taxa were grouped into another clade with relative low BS value. Although three clades were clustered in the NJ tree, only two lineages were revealed. The hypothesis of a two-lineage derived pattern could also be supported by the Structure analysis based on the ΔK method, the optimal K value was K = 2, showing that the 19 populations from the C. segmentifida complex were separated into two distinct clusters (Figure 4B). One cluster contained five populations of C. guizhouensis and the only one population of C. longlinensis and another harbored 13 populations of the remaining six taxa. Furthermore, the two clusters had less or no gene flow or introgression with each other through the results displayed by Structure analysis, implying that this complex contained only two evolutionary significant units with little contact.
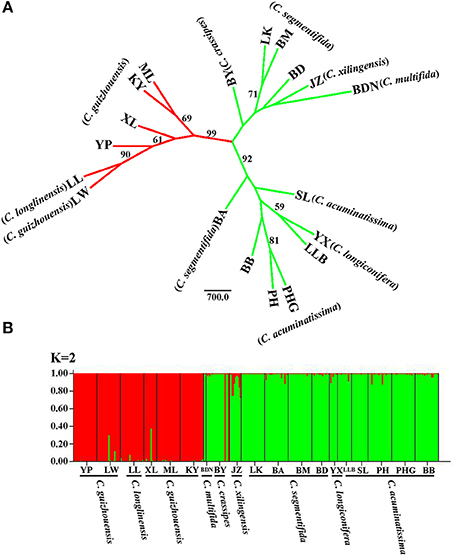
Figure 4. (A) The Neighbour-joining consensus tree (NJ tree) based on Nei's 1983 genetic distance and (B) Bayesian inference (K = 2) of microsatellite phenotype from 19 populations of 311 individuals of the C. segmentifida complex. Numbers (if ≥ 50) on branches indicated bootstrap values from 1000 replicates.
Discussion
Species Delimitation Based on Tree-Based Methods by DNA Sequences
Tree-based species delimitation methods are on the basis of the concordance between some properties such as monophyly and geography with phylogenetic tree topologies. The tree-based methods by checking DNA sequences variations were formerly recognized and applied based on various strategies in delimitating boundaries of different species (Pérez-Losada et al., 2005; Perez-Losada et al., 2009; Rosell et al., 2010; Harrington and Near, 2012; Liu et al., 2015; Su et al., 2015; Tsai et al., 2015). According to the Wiens and Penkrot's criterion (Wiens and Penkrot, 2002), if haplotypes from the same locality fail to cluster together, it may suggest gene flow between localities (i.e., focal species = single species). However, the plastid haplotype and GTP haplotype trees showed that haplotypes of the focal species, C. segmentifida complex, were exclusive and there was no gene flow between their two basal lineages in this study. On one hand, for nuclear haplotypes' distributions in the C. segmentifida complex, C. longlinensis mainly shared the same nuclear haplotypes with C. guizhouensis and the two taxa owned unique nuclear haplotypes, indicating that C. longlinensis had closer relationship with C. guizhouensis than any other species. The remaining six taxa shared same nuclear haplotypes which were specific for them, indicating the remaining six taxa were more closely related. For plastid haplotypes' distributions in the C. segmentifida complex, C. longlinensis owned the same plastid haplotype comH1 with C. guizhouensis without its unique plastid haplotypes, indicating a possible identical common ancestor between C. longlinensis and C. guizhouensis. In addition, C. multifida, C. xilingensis, C. crassipes, and C. segmentifida only occupied and shared one single unique plastid haplotype comH6, suggesting the four taxa came from the same lineage, while C. longiconifera and C. acuminatissima derived their unique plastid haplotypes not shared with other taxa. On the other hand, C. longlinensis also shared closer geographical distance with C. guizhouensis in Nanpan River basin than other members of the complex which mainly distributed along the You River basin. Therefore, based on the phylogeny of plastid haplotypes and GTP nuclear haplotypes (Brower, 1999), two sets of topologically contiguous populations can be divided into two separate species. Our conclusion supports Wang's (Wang, 2000) previous morphological study and Ma's (Ma, 2005) research based on statistical analyses of morphological variations and UPGMA dendrogram revealed by ISSR markers, but conflicts with other treatments mainly based on morphological traits (Chen and Stevenson, 1999; Huang, 2001; Whiteloek, 2002).
The other two nuclear genes cannot deal with species boundaries of this complex as their haplotype phylogenetic trees showed no clear phylogenetic clades corresponding to one or more haplotypes from continuous populations. The congruence between taxonomy and plastid genes along with one of the nuclear genes GTP but not with two others may be caused by the specific history of the recently diversified Cycas which are still sorting their lineage processes by isolation or genetic drift. As nuclear genes own a higher effective population size (Ne) that is four times higher than a given plastid gene, they can strongly influence the rate at which the haplotypes of a species become exclusive (Neigel and Avise, 1986; Moore, 1995). Therefore, on equal conditions, it takes four times longer for a nuclear gene to fix a species exclusive or distinct than its plastid genes (Moore, 1995). In addition, the disparate histories revealed by plastid and nuclear genes could be explained by the fact that the whole plastid genome is inherited all together, whereas recombination between chromosome and within chromosome usually drives to have different histories in the nuclear genes.
Species Delimitation Based on NJ-Tree and Structure Inference by Microsatellites
It is believed that STRUCTURE analysis could cluster individuals regardless of their population-of-origin based on rough consistence to Hardy-Weinberg genetic expectations. The strategy employed by STRUCTURE is straightforward and matches reasonably well the properties of meta-population lineages (Shaffer and Thomson, 2007). Meanwhile, microsatellites can also have great probability of success resolving species boundaries largely because microsatellite markers have several ten times faster evolutionary rates than DNA sequences (Wolfe et al., 1987; Graur and Li, 2000; O'Connell and Ritland, 2004; Kuchma et al., 2011). In this study, both the NJ tree and STRUCTURE analysis derived from microsatellite data revealed the same clustering for the C. segmentifida complex, two major lineages, one contained all populations of C. longlinensis and C. guizhouensis while the other included all populations of the remaining six taxa, which were in accordance with the above conclusion uncovered by DNA sequences. It is noteworthy that the NJ tree revealed two subclades in the “C. segmentifida” (BND-BB) clade. Both the two subclades occupied populations of C. segmentifida, suggesting slight or recent differentiation among populations within the BND-BB clade, while the differentiation was not enough for the two subclades to sort to morphological distinct species.
Combining evidences from nuclear and plastid DNA sequences, microsatellites analysis, as well as the geographical distribution, we propose the C. segmentifida complex to be divided into two exclusive species: Cycas guizhouensis K. M. Lan et R. F. Zou and Cycas segmentifida D. Y. Wang et C. Y. Deng. Our species delimitation within this complex can offer guidelines for further morphological taxonomic revision and conservation studies.
Taxonomic Treatment
Cycas guizhouensis K. M. Lan et R. F. Zou in Acta Phytotax. Sin. 21(2): 209-210, 1983.
Synonym: Cycas longlinensis H. T. Chang et Y. C. Zhong in Acta Sci. Nat. Univ. Sunyatseni, 36(3): 68, 1997.
Cycas segmentifida D. Y. Wang et C. Y. Deng in Encephalartos, 43: 11-14, 1995.
Synonyms: Cycas multifida H. T. Chang et Y. C. Zhong in Acta Sci. Nat. Univ. Sunyatseni, 36(3): 70, 1997; Cycas xilingensis H. T. Chang et Y. C. Zhong in Acta Sci. Nat. Univ. Sunyatseni, 36(3): 69, 1997; Cycas longiconifera H. T. Chang, Y. C. Zhong et Y. Y. Huang in Acta Sci. Nat. Univ. Sunyatseni, 37(4): 6, 1998; Cycas acuminatissima H. T. Chang, Y. C. Zhong et Z. F. Lu in Acta Sci. Nat. Univ. Sunyatseni, 37(4): 6, 1998; Cycas crassipes H. T. Chang, Y. C. Zhong et Z. F. Lu in Acta Sci. Nat. Univ. Sunyatseni, 38(3): 121-122, 1999.
Author Contributions
XF carried out the molecular genetic studies, participated in the data analysis and wrote the manuscript. JL participated in collection of study materials and DNA extraction. XG designed the research and collected study materials. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ying Zheng and Yiqing Gong for their assistance with field sampling. This research was supported by the United Fund of the NSFC and the Yunnan Natural Science Foundation (U1136602 to XG).
References
Bond, J. E., and Stockman, A. K. (2008). An integrative method for delimiting cohesion species: finding the population-species interface in a group of Californian trapdoor spiders with extreme genetic divergence and geographic structuring. Syst. Biol. 57, 628–646. doi: 10.1080/10635150802302443
Brower, A. V. (1999). Delimitation of phylogenetic species with DNA sequences: a critique of Davis and Nixon's population aggregation analysis. Syst. Biol. 48, 199–213. doi: 10.1080/106351599260535
Calonje, M., Stevenson, D. W., and Stanberg, L. (2015). The World List of Cycads, online edition [Internet]. 2013-2015. [cited 2015 Dec 12]. Available online at: http://www.cycadlist.org
Chen, J. R., and Stevenson, D. W. (1999). “Cycadaceae,” in Flora of China, Vol. 4, eds C. Y. Wu and P. Raven (Beijing: Science Press/Saint Louis, MO: Missouri Botanical Garden Press), 1–7.
Chiang, T., and Peng, C. I. (1998). “Phylogeography of the endemic plants in Taiwan,” in Proceeding of the Symposium on Conservation of Endemic Species, ed S. D Yeng (Taipei: Research Institute of Taiwan Endemic Species), 148–155.
Cibrián-Jaramillo, A., Marler, T. E., Desalle, R., and Brenner, E. D. (2008). Development of EST-microsatellites from the cycad Cycas rumphii, and their use in the recently endangered Cycas micronesica. Conserv. Genet. 9, 1051–1054. doi: 10.1007/s10592-007-9447-3
De Queiroz, K. (1998). “The general lineage concept of species, species criteria, and the process of speciation: a conceptual unification and terminological recommendations,” in Endless Forms: Species and Speciation, eds D. J. Howard and S. H. Berlocher (New York, NY: Oxford University Press), 57–75.
De Queiroz, K. (2007). Species concepts and species delimitation. Syst. Biol. 56, 879–886. doi: 10.1080/10635150701701083
Doyle, J. (1991). “DNA protocols for plants-CTAB total DNA isolation,” in Molecular Techniques in Taxonomy, eds G. M. Hewitt and A. Johnston (Berlin: Springer-Verlag), 283–293.
Earl, D. A. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Felsenstein, J. (2002). PHYLIP (Phylogeny Inference Package) version 3.6. Distributed by the author. Seattle, WA: Department of Genome Sciences, University of Washington.
Feng, X. Y., Wang, Y. H., and Gong, X. (2014). Genetic diversity, genetic structure and demographic history of Cycas simplicipinna (Cycadaceae) assessed by DNA sequences and SSR markers. BMC Plant Biol. 14:187. doi: 10.1186/1471-2229-14-187
Fujita, M. K., Leache, A. D., Burbrink, F. T., Mcguire, J. A., and Moritz, C. (2012). Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 27, 480–488. doi: 10.1016/j.tree.2012.04.012
Graur, D., and Li, W. H. (2000). Fundamentals of Molecular Evolution. Sunderland, MA: Sinauer Associates.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Harrington, R. C., and Near, T. J. (2012). Phylogenetic and coalescent strategies of species delimitation in snubnose darters (Percidae: Etheostoma). Syst. Biol. 61, 63–79. doi: 10.1093/sysbio/syr077
Hendrixson, B. E., Derussy, B. M., Hamilton, C. A., and Bond, J. E. (2013). An exploration of species boundaries in turret-building tarantulas of the Mojave Desert (Araneae, Mygalomorphae, Theraphosidae, Aphonopelma). Mol. Phylogenet. Evol. 66, 327–340. doi: 10.1016/j.ympev.2012.10.004
Huang, Y. Y. (2001). The System Classification and Evolution of Cycadaceae in China. Beijing: Meteorological Press.
Ju, L. P., Kuo, C. C., Chao, Y. S., Cheng, Y. P., Gong, X., and Chiang, Y. C. (2011). Microsatellite primers in the native perennial cycad Cycas taitungensis (Cycadaceae). Am. J. Bot. 98, e84–e86. doi: 10.3732/ajb.1000504
Kuchma, O., Vornam, B., and Finkeldey, R. (2011). Mutation rates in Scots pine (Pinus sylvestris L.) from the Chernobyl exclusion zone evaluated with amplified fragment-length polymorphisms (AFLPs) and microsatellite markers. Mutat. Res. Genet. Toxicol. Environ. 725, 29–35. doi: 10.1016/j.mrgentox.2011.07.003
Lan, K. M., and Zou, R. F. (1983). A new species of Cycas Linn. from Guizhou province. Acta Phytotaxonomic Sinica 21, 209–210.
Li, L., Wang, Z. F., Jian, S. G., Zhu, P., Zhang, M., Ye, W. H., et al. (2009). Isolation and characterization of microsatellite loci in endangered gCycas changjiangensis (Cycadaceae). Conserv. Genet. 10, 793–795. doi: 10.1007/s10592-008-9664-4
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, J., Zhou, W., and Gong, X. (2015). Species delimitation, genetic diversity and population historical dynamics of Cycas diannanensis (Cycadaceae) occurring sympatrically in the Red River region of China. Front. Plant Sci. 6:696. doi: 10.3389/fpls.2015.00696
Lui, K., and Muse, S. V. (2005). PowerMarker: integrated analysis environment for genetic data. Bioinformatics 20, 2128–2129. doi: 10.1093/bioinformatics/bti282
Ma, Y. (2005). Studies on Classification and Genetic Diversity of Cycas Segmentifida Complex in Baise of Guangxi, China. Guangxi: Guangxi University.
Mayr, E. (1982). The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Cambridge, MA: Harvard University Press.
Mckay, B. D., Mays, H. L., Wu, Y., Li, H., Yao, C. T., Nishiumi, I., et al. (2013). An empirical comparison of character-based and coalescent-based approaches to species delimitation in a young avian complex. Mol. Ecol. 22, 4943–4957. doi: 10.1111/mec.12446
Moore, W. S. (1995). Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evol 49, 718–726. doi: 10.2307/2410325
Neigel, J. E., and Avise, J. C. (1986). “Phylogenetic relationships of mitochondrial DNA under various demographic models of speciation,” Evolutionary Processes and Theory, eds E. Nevo and S. Karlin (New York, NY: Academic Press), 515–534.
O'Connell, L., and Ritland, K. (2004). Somatic mutations at microsatellite loci in western redcedar (Thuja plicata: Cupressaceae). J. Hered. 95, 172–176. doi: 10.1093/jhered/esh024
Peakall, R., and Smouse, P. E. (2006). GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295. doi: 10.1111/j.1471-8286.2005.01155.x
Pérez-Losada, M., Eiroa, J., Mato, S., and Domínguez, J. (2005). Phylogenetic species delimitation of the earthworms Eisenia fetida (Savigny, 1826) and Eisenia andrei Bouché, 1972 (Oligochaeta, Lumbricidae) based on mitochondrial and nuclear DNA sequences. Pedobiologia 49, 317–324. doi: 10.1016/j.pedobi.2005.02.004
Perez-Losada, M., Ricoy, M., Marshall, J. C., and Dominguez, J. (2009). Phylogenetic assessment of the earthworm Aporrectodea caliginosa species complex (Oligochaeta: Lumbricidae) based on mitochondrial and nuclear DNA sequences. Mol. Phylogenet. Evol. 52, 293–302. doi: 10.1016/j.ympev.2009.04.003
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rosell, J. A., Olson, M. E., Weeks, A., De-Nova, J. A., Lemos, R. M., Camacho, J. P., et al. (2010). Diversification in species complexes: tests of species origin and delimitation in the Bursera simaruba clade of tropical trees (Burseraceae). Mol. Phylogenet. Evol. 57, 798–811. doi: 10.1016/j.ympev.2010.08.004
Salas-Leiva, D. E., Meerow, A. W., Francisco-Ortega, J., Calonje, M., Griffith, M. P., Stevenson, D. W., et al. (2014). Conserved genetic regions across angiosperms as tools to develop single-copy nuclear markers in gymnosperms: an example using cycads. Mol. Ecol. Resour. 14, 831–845. doi: 10.1111/1755-0998.12228
Shaffer, H. B., and Thomson, R. C. (2007). Delimiting species in recent radiations. Syst. Biol. 56, 896–906. doi: 10.1080/10635150701772563
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding plastid DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Sites, J. W., and Marshall, J. C. (2003). Delimiting species: a Renaissance issue in systematic biology. Trends Ecol. Evol. 18, 462–470. doi: 10.1016/S0169-5347(03)00184-8
Sites, J. W., and Marshall, J. C. (2004). Operational criteria for delimiting species. Annu. Rev. Ecol. Evol. Syst. 35, 199–227. doi: 10.1146/annurev.ecolsys.35.112202.130128
Stephens, M., and Donnelly, P. (2003). A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 73, 1162–1169. doi: 10.1086/379378
Stephens, M., Smith, N. J., and Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68, 978–989. doi: 10.1086/319501
Stockman, A. K., and Bond, J. E. (2007). Delimiting cohesion species: extreme population structuring and the role of ecological interchangeability. Mol. Ecol. 16, 3374–3392. doi: 10.1111/j.1365-294X.2007.03389.x
Su, X., Wu, G. L., Li, L. L., and Liu, J. Q. (2015). Species delimitation in plants using the Qinghai-Tibet Plateau endemic Orinus (Poaceae: Tridentinae) as an example. Ann. Bot. 116, 35–48. doi: 10.1093/aob/mcv062
Swindell, S. R., and Plasterer, T. N. (1997). SEQMAN[M]//Sequence Data Analysis Guidebook. NewYork, NY: Springer.
Swofford, D. L. (2003). Phylogenetic Analysis Using Parsimony (* and Other Methods). Version 4. Sunderland, MA: Sinauer Associates.
Taberlet, P., Gielly, L., Pautou, G., and Bouvet, J. (1991). Universal primers for amplification of three non-coding regions of plastid DNA. Plant Mol. Biol. 17, 1105–1109. doi: 10.1007/BF00037152
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tsai, C. C., Chou, C. H., Wang, H. V., Ko, Y. Z., Chiang, T. Y., and Chiang, Y. C. (2015). Biogeography of the Phalaenopsis amabilis species complex inferred from nuclear and plastid DNAs. BMC Plant Biol. 15:202. doi: 10.1186/s12870-015-0560-z
Wang, D. Y. (2000). Morphological Structure, System Classification and Evolutionary Research of Cycadaceae. Nanjing: Nanjing Forestry University.
Wang, D. Y., and Deng, C. Y. (1995). Cycas segmentifida, a new species from China. Encephalartos, J. Cycad Soc. South. Africa. 43, 11–14.
Wang, Z. F., Ye, W. H., Cao, H. L., Li, Z. C., and Peng, S. L. (2008). Identification and characterization of EST-SSRs and cpSSRs in endangered Cycas hainanensis. Conserv. Genet. 9, 1079–1081. doi: 10.1007/s10592-007-9461-5
Wiens, J. J. (2007). Species delimitation: new approaches for discovering diversity. Syst. Biol. 56, 875–878. doi: 10.1080/10635150701748506
Wiens, J. J., and Penkrot, T. A. (2002). Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Syst. Biol. 51, 69–91. doi: 10.1080/106351502753475880
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, plastid, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Yang, Y., Li, Y., Li, L. F., Ge, X. J., and Gong, X. (2008). Isolation and characterization of microsatellite markers for Cycas debaoensis YC Zhong et CJ Chen (Cycadaceae). Mol. Ecol. Resour. 8, 913–915. doi: 10.1111/j.1755-0998.2008.02114.x
Zhang, F. M., Su, T., Yang, Y., Zhai, Y. H., Ji, Y. H., and Chen, S. T. (2010). Development of seven novel EST-SSR markers from Cycas panzhihuaensis (Cycadaceae). Am. J. Bot. 97, e159–e161. doi: 10.3732/ajb.1000377
Zhang, H. D., and Zhong, Y. C. (1997). New Cycas from Guangxi. Acta Sci. Nat. Univ. Sunyatseni 36, 67–71.
Zhang, H. D., Zhong, Y. C., Huang, Y. Y., Zheng, H. X., and Lu, Z. F. (1997). Additions to the cycadaceous flora of China. Acta Sci. Nat. Univ. Sunyatseni 37, 6–8.
Zhang, H. D., Zhong, Y. C., and Lu, Z. F. (1999). A new species of Cycas from Guangxi. Acta Sci. Nat. Univ. Sunyatseni 38, 121–122.
Keywords: Cycas segmentifida complex, delimitation, phylogeny, species, synonym
Citation: Feng XY, Liu J and Gong X (2016) Species Delimitation of the Cycas segmentifida Complex (Cycadaceae) Resolved by Phylogenetic and Distance Analyses of Molecular Data. Front. Plant Sci. 7:134. doi: 10.3389/fpls.2016.00134
Received: 02 November 2015; Accepted: 25 January 2016;
Published: 15 February 2016.
Edited by:
Federico Luebert, Rheinische Friedrich-Wilhelms-Universität Bonn, GermanyReviewed by:
Dennis William Stevenson, New York Botanical Garden, USAMarcial Escudero, University of Seville, Spain
Alan W. Meerow, United States Department of Agriculture-Agricultural Research Service, USA
Copyright © 2016 Feng, Liu and Gong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun Gong, Z29uZ3h1bkBtYWlsLmtpYi5hYy5jbg==