 Jun Cao
Jun Cao- Institute of Life Science, Jiangsu University, Zhenjiang, China
Prefoldin is a hexameric molecular chaperone complex present in all eukaryotes and archaea. The evolution of this gene family in plants is unknown. Here, I identified 140 prefoldin genes in 14 plant species. These prefoldin proteins were divided into nine groups through phylogenetic analysis. Highly conserved gene organization and motif distribution exist in each prefoldin group, implying their functional conservation. I also observed the segmental duplication of maize prefoldin gene family. Moreover, a few functional divergence sites were identified within each group pairs. Functional network analyses identified 78 co-expressed genes, and most of them were involved in carrying, binding and kinase activity. Divergent expression profiles of the maize prefoldin genes were further investigated in different tissues and development periods and under auxin and some abiotic stresses. I also found a few cis-elements responding to abiotic stress and phytohormone in the upstream sequences of the maize prefoldin genes. The results provided a foundation for exploring the characterization of the prefoldin genes in plants and will offer insights for additional functional studies.
Introduction
Molecular chaperones can stabilize, interact with, or assist other proteins to acquire functionally active conformations, without being present in their final structures (Hartl, 1996). Chaperonins play a role in protein assembly, folding, trafficking and degradation, and are crucial for cellular development (Hartl et al., 2011). Group I chaperonins are found in eubacteria and endosymbiotic organelles, whereas group II ones are present in archaea and eukaryotes. They share a common structure with different functions (Yébenes et al., 2011).
Prefoldin is a group II chaperonin. Archaea prefoldin possesses two subunits (prefoldin α and β) and polymerizes to an α2β4 hexamer (Leroux et al., 1999). Eukaryotic prefoldin consists of six subunits; two α subunits (PFD3 and PFD5) and four β subunits (PFD1, PFD2, PFD4, and PFD6) (Siegert et al., 2000). The structure confers selective substrate specificity for target proteins. They form a jellyfish-like heterohexameric complex to deliver newly synthesized unfolded proteins to cytosolic chaperonins containing TCP-1 (CCT) for protein folding. In addition, prefoldin also protects unfolded proteins during CCT capturing and releasing proteins (Siegert et al., 2000; Möckli et al., 2007).
Previous studies have indicated that prefoldin plays a central role in cellular development. Through binding to tubulin and actin, yeast prefoldin facilitated productive folding and binding inside the chaperonin cavity. Deleting of single or multiple subunits of prefoldin in yeast usually results in disruption of cytoskeletal structures (Vainberg et al., 1998). Similarly, studies on prefoldin function have revealed that down regulation of prefoldin can decreased the level of endogenous α-tubulin by 95%, and that of actin by 30% (Lundin et al., 2008). Deletion of prefoldin genes results in cytoskeletal defects, slow growth and cold sensitivity in yeast (Geissler et al., 1998; Siegers et al., 1999). In Caenorhabditis elegans, reduction of functional prefoldins caused embryonic lethality, and silencing of prefoldin subunits 1, 2, 3, and 6 exhibited reduced microtubule growth (Lundin et al., 2008). Mutation of prefoldin 5 or prefoldin 1 leads to a variety of neurodegenerative effects through a reduction of microfilaments and microtubules, such as mucus clearance defects, hydrocephaly, and loss of nerve bundles, in mice (Cao et al., 2008; Lee et al., 2011). In Arabidopsis, lesions in prefoldin 6 resulted in impaired microtubule organization and dynamics, which was associated with reduced plant size, defects in cell division, and so on (Gu et al., 2008). In addition, prefoldin 3 and prefoldin 5 mutants also displayed important changes in microtubule organization and developmental patterns. Furthermore, both mutants were sensitive to salt, implying important role of cytoskeleton in plant tolerance to salt stress (Rodríguez-Milla and Salinas, 2009). DELLA proteins directly interact with the prefoldin complex in a gibberellin (GA)-dependent manner. When GA is present, the complex is localized to the cytoplasm. When GA is absent, it stays in the nucleus, and then affects microtubule orientation (Locascio et al., 2013). A recent study has shown that prefoldin can bind chromatin and plays an important role in gene transcription (Millán-Zambrano et al., 2013). Bud27, an ATP-independent prefoldin-like molecular chaperone, can also regulate the gene expression transcribed by the RNA pol II in yeast (Mirón-García et al., 2014).
Six prefoldin members [PFD1 (At2g07340). PFD2 (At3g22480), PFD3 (At5g49510), PFD4 (At1g08780), PFD5 (At5g23290), and PFD6 (At1g29990)] have been identified in the Arabidopsis genome (Hill and Hemmingsen, 2001). Phylogenetic analysis showed that these six genes were divided into different evolutionary branches, suggesting the functional divergence among them (Hill and Hemmingsen, 2001). Moreover, only several of them have been functionally identified (Gu et al., 2008; Rodríguez-Milla and Salinas, 2009). Although prefoldin can form jellyfish-shaped hexameric complexes consisting of two α-type and four β-type subunits (Siegert et al., 2000; Martín-Benito et al., 2002), smaller individual motifs and more divergent patterns are not recognized among each prefoldin proteins. The recent availability of genome sequences of some models plant species provides an opportunity to study the evolution of prefoldin gene family. In this study, I have identified the prefoldin gene family from 14 plant species, and each species comprises 6–24 genes. Considering their significant developmental and physiological role, it is of considerable interest to us to study the evolution of plant prefoldin genes. Here, I performed integrated analyses to unravel the evolutionary mechanisms of the plant prefoldin protein family especially for maize. It will provide a useful basis for further functional studies of this gene family.
Materials and Methods
Prefoldin Sequence Retrieval and Identification in 14 Plant Species
To identify potential prefoldin gene in 14 completely sequenced plant genomes, I first used six Arabidopsis prefoldin sequences previous identified (Hill and Hemmingsen, 2001) as queries to perform BLAST searches against the phytozome database1 with -1 expect (E) threshold. In addition, a keyword “prefoldin” was also used to perform searching in this study. The Pfam database (Punta et al., 2012) was used to confirm the encoded prefoldin based on the presence or not of the prefoldin subunit domain. Next, the ProtParam tool2 and the CELLO v2.5 server3 (Yu et al., 2004) were used to determine the physicochemical parameters and subcellular localization of the prefoldin proteins, respectively.
Phylogenetic Analyses of the Prefoldin Gene Family
To further explore the evolutionary relationship of plant prefoldins, multiple sequence alignments and phylogenetic analysis of the prefoldin proteins were performed with MUSCLE 3.52 (Edgar, 2004) and MEGA v5 (Tamura et al., 2011), respectively. And neighbor-joining (NJ) method was used to perform phylogenetic analyses of the prefoldin proteins with bootstrap analysis of 1,000 replicates (Cao, 2012). Furthermore, maximum likelihood and PhyML methods were also used to construct additional trees for validating the result from NJ tree.
Chromosomal Location, Genomic Duplication and Inference of Duplication Time
I used the annotation data of the prefoldin genes on MaizeSequence4 for chromosomal location. Paralogous regions of the genomes were first predicted with SyMAP v3.4 (Soderlund et al., 2011). If two genes coming from the paraloguous regions were also located at the terminal evolutionary branch in the phylogenetic tree, they were thought to be derived from common ancestral duplication. To calculate the duplication time of the prefoldin paralogs, pairwise alignment of the maize prefoldin gene pairs was performed using an embedded program ClustalW (codons) in MEGA v5 (Tamura et al., 2011). Next, the Ka and Ks values of paralogous genes were estimated using K-Estimator 6.0 program (Comeron, 1999). The Ks value was used to calculate the duplication date (T = Ks/2λ), assuming clock-like rates (λ) of 6.5 × 10-9 for maize (Gaut et al., 1996).
Exon-Intron Structure and Conserved Motifs Analysis
The organizations of prefoldin genes were analyzed in these plant lineages by comparing their coding and genomic sequence information in the Phytozome5 and NCBI databases. In addition, MEME program6 (Bailey et al., 2006) was used to identify finer motifs in the candidate plant prefoldin protein sequences. Parameter of maximum number of motifs is 15.
Functional Divergence and Gene Co-expression Analyses
In the process of protein evolution, some residues are highly conserved, while others are highly variable. To further investigate the divergence between different groups of prefoldin proteins, DIVERGE (version 2.0) (Gu, 1999, 2001) was used to analyze the type-I functional divergence. The functional divergence between two groups was measured as the coefficient of functional divergence (θ). When the coefficient equals 0; it means that the evolutionary rate of the duplicate genes at each site is entirely consistent. Vice versa, when the coefficient is greater than 0, the evolutionary rate of the duplicate genes at some critical amino-acid residues is different. The software will predict these sites responding for the functional divergence.
To further analysis the relation between maize prefoldin and other genes, I also used the Co-expression Browser (COB)7 (Schaefer et al., 2014) to explore their networks. The domestication maize genotype was selected for co-expression analysis of the prefoldin genes. The network was built using expression profiles from 8-day seedlings. Expression matrices were used to generate profile correlations for the functional networks by calculating the Pearson correlation coefficient between each pair of gene expression profiles in each instance (Schaefer et al., 2014).
Microarray-Based Expression Analysis
Maize microarray data (GSE27004) (Sekhon et al., 2011) were used for the expression analysis of the prefoldin genes. Expression data were normalized and viewed in the Genesis (v 1.7.6) program (Sturn et al., 2002).
Plant Treatment, RNA Isolation, Quantitative Real-Time PCR (QRT-PCR), and Promoter Sequence Analysis
Endosperm can provide the necessary nutrition for early germination and growth when maize is grown in water. In this study, 10-day-old maize seedlings after germination were used to test the expression profiles of prefoldin genes under IAA, low temperature, drought, and salt stresses. For low temperature treatment, the maize seedlings were placed at 4°C environment for 3 h. And the seedlings were dried for 3 h between folds of tissue paper at 23 ± 1°C for drought treatment (Xia et al., 2012). The maize seedlings were put into 10 μM IAA and 150 mM NaCl solutions for 24 h for auxin and salt stress treatments, respectively. Control (CK) seedlings were normally grown at 23 ± 1°C with a photoperiod of 14 h light and 10 h dark. Three biological replicates were performed for qRT-PCR analysis. Total RNA was extracted with the TRIzol total RNA extraction kit (Sangon). RNase free DNase-I was used to remove genomic DNA. Next, I used M-MLV (TakaRa) to perform reverse transcription, followed by quantitative assays of each diluted cDNA using an ABI 7500 sequence detection system. All 13 maize prefoldin genes were selected for qRT-PCR analysis, and their primers are listed in Supplementary Table S2. Actin 1 (GRMZM2G126010) gene in maize was used as the endogenous control. And 2-ΔΔCT method (Livak and Schmittgen, 2001) was used to calculate their relative expression level. t-Test was used to perform a significant analysis.
To define the transcription start site (TSS) of each prefoldin gene in maize, I first collected their expressed sequence tag information. The TSS positions of prefoldin genes were used as references to determine their upstream promoter sequences. In this study, 1,000-bp upstream promoter sequences were acquired for further analyses. In addition, some abiotic stress- and phytohormone-responsive elements were identified in the promoter regions of the maize prefoldin genes using PLACE8 (Higo et al., 1999).
Results and Discussion
Identification of Prefoldin Genes in 14 Plant Species
To identify the prefoldin genes in plants, I first used the amino-acid sequences of Arabidopsis prefoldin (Hill and Hemmingsen, 2001) to perform BLAST searches in the phytozome database9. In addition, a keyword “prefoldin” search was also performed. All the putative prefoldin protein sequences were subjected to the Pfam analysis (Punta et al., 2012) to verify the reliability of the results based on the presence or not of the prefoldin subunit domain. As a result, I identified 140 prefoldin genes from 14 plants. The number of prefoldin genes ranged from 6 to 24 in each species (Table 1). There are 24 prefoldin genes existing in the soybean genome, while the members in other species range from about six in alfalfa, nine in Arabidopsis, sorghum, grape and Physcomitrella patens, 10 in rice, and 13 in maize and poplar. Previous study has identified 6 prefoldin genes in Arabidopsis (Hill and Hemmingsen, 2001). In this study, other three prefoldins (At1g03760, At1g26660, and At1g49245) were also found in this species. There are about 33,583, 39,454, and 42,577 genes in the Arabidopsis, maize and poplar genomes, respectively, which are 9.9, 29.2, and 39.4% larger than that of rice (30,534), respectively, implying a disproportion between the numbers of prefoldin genes and the sizes of predicted genomes. The prefoldin genes in plants encode highly hydrophilous polypeptides (from -0.003 to -0.988 in grand average hydrophobicities) with about from 16 to 612 amino acids and predicted pIs (from 4.21 to 10.2) (Supplementary Table S1). CELLO v2.5 server10 (Yu et al., 2004) was used to further predict the localization of the prefoldin proteins. The results showed that most of the candidate prefoldins were probably localized to the cytoplasm, suggesting that the prefoldin proteins participate in the cytoplasmic folding of tubulin and actin monomers (Zhao et al., 2005; Gu et al., 2008). Several plant prefoldins have also been localized in the nucleus (Supplementary Table S1), suggesting their functional relevance in DNA repair or integration, transcription and gene regulation (Millán-Zambrano and Chávez, 2014; Mirón-García et al., 2014).
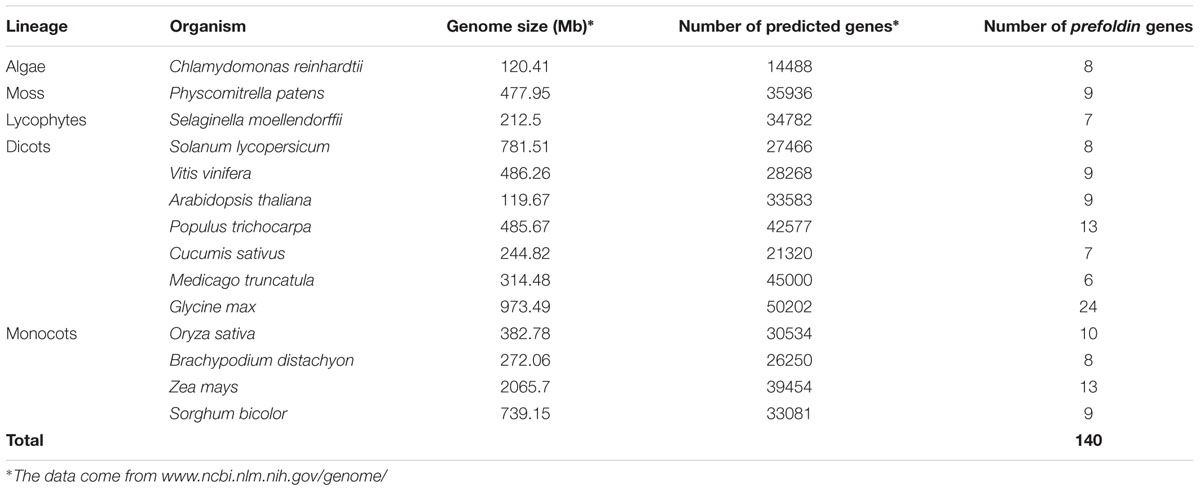
TABLE 1. Prefoldin genes identified from 14 sequenced plant genomes.
Phylogenetic Relationships, Gene Organization, and Motif Analysis
To examine the phylogenetic relationships and evolutionary history of prefoldins in plants, I first constructed an unrooted tree and classified this gene family into nine groups (Group I-IX) (Figure 1) based on the observed topological structure of tree and sequence similarity. In addition, other methods, such as, maximum likelihood, PhyML methods, were also used to reconstruct the phylogenetic trees of prefoldin family, and very similar results were got as well as the tree topology of NJ method. Here, I employed the NJ tree for further analysis. This group classification was also supported by other evidences, such as intron-exon organization, and motif composition, which will be described below. Phylogenetic relationships of some eukaryotic prefoldin subunits have been illustrated (Hill and Hemmingsen, 2001). In which, six different clusters were generated with six prefoldin subunit sequences through phylogenetic analysis. In this study, I also found that all members of Groups II and IV had high homology with eukaryotic PFD5 and PFD3, respectively, which form α subunits of the prefoldin complex. While the members of Group V, Group VII, Group VIII, and Group IX represented four β subunits of PFD2, PFG4, PFD1 and PFD6, respectively. In addition to the six prefodlin subunit sequences, I also found and used three other prefoldin genes (At1g03760, At1g26660, and At1g49245) in Arabidopsis for BLAST searching. Because less conservation exists in prefoldin sequences, these sequences from 14 plant species were divided into nine groups (Figure 1). Each group contained 6–20 members. All terrestrial plants evolved from the primitive chlorophycean species in Viridiplantae (Misumi et al., 2008). Therefore, the origin of this gene may date back to the primitive algae. My search for prefoldin genes in Chlamydomonas reinhardtii yielded about eight members. This number remained relatively constant with the complexity of the genomes in plant evolution (Figure 2). The most obvious expansion of prefoldin gene family occurred in soybean, containing about 17% (24 paralogs) of the 140 identified prefoldins, which may be due to recent genome duplication occurring about 13 MYA (Schmutz et al., 2010).
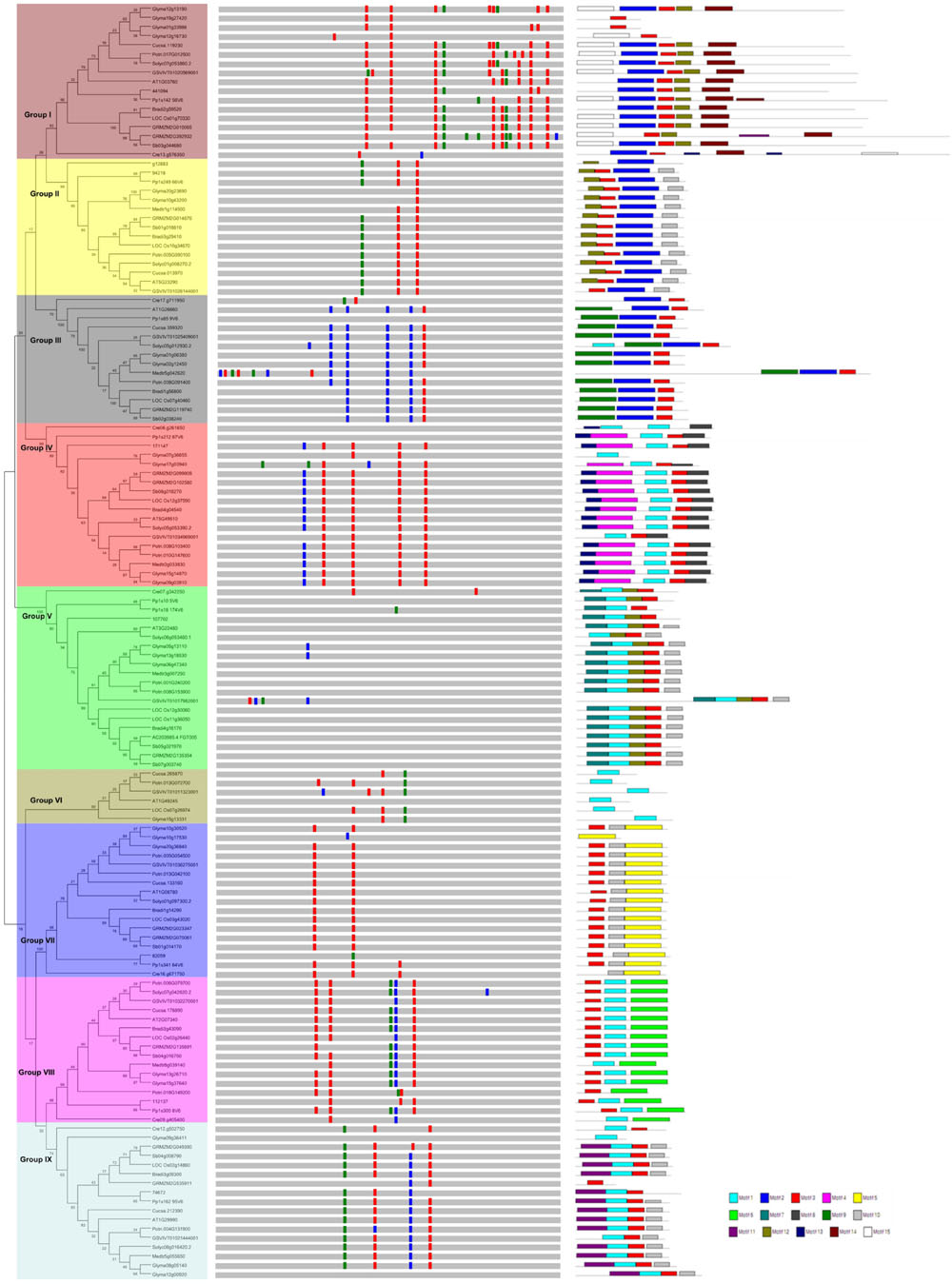
FIGURE 1. Phylogenetic relationship, gene structure and motif composition of the prefoldin genes in plants. The phylogenetic tree is constructed and classified into nine major groups (I–IX). The insertion positions of 0, 1, and 2 phase introns are marked with red, green, and blue lines, respectively. Different motifs of the prefoldin proteins are displayed by different colored boxes.
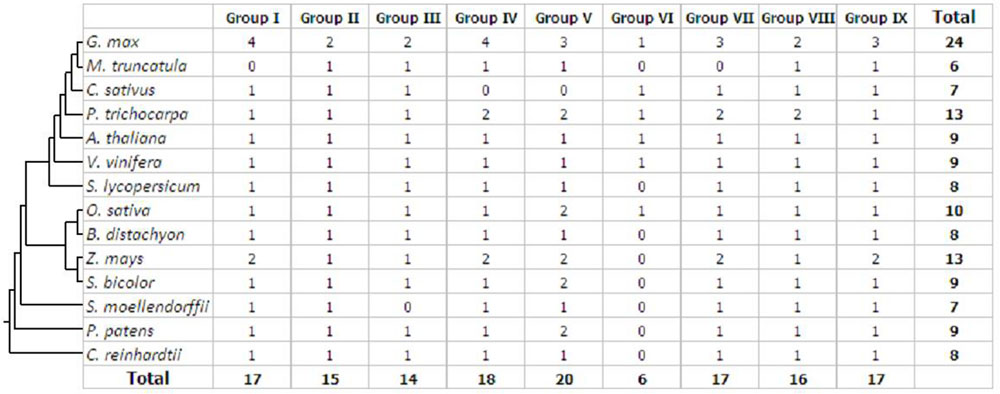
FIGURE 2. Distribution of the prefoldin genes in different plant species and groups.
In general, intron gain or loss generates structural complexity, which is a key evolutionary mechanism of most gene families (Cao, 2012; Chen and Cao, 2014; Cao et al., 2015). To further insight into the evolutionary relationships of the plant prefoldin genes, I investigated the exon-intron structure of each member and compared with each other. The position and distribution of introns of each prefoldin gene are shown in Figure 1. Each prefoldin gene contains 0–13 introns (Figure 1). Fifteen out of 20 members in Group V possessed a minimum of zero intron each, whereas GRMZM2G392932 possessed a maximum of thirteen introns. Similar exon-intron structure occurred in most prefoldin genes of same group, implying conserved evolutionary relationship. For instance, most prefoldin gene in Group VII has two introns with same insertion position and phase distribution, whereas most members within Group IV contain five introns. The structural diversity in different groups suggests multiple origins of gene ancestry.
Next, to obtain more insights into the diversity of motif compositions in the 140 prefoldin proteins, I used the MEME motif search tool (Bailey et al., 2006) to search for the conserved motifs in these proteins, and found 15 conserved motifs (Figure 1). As described above, nine groups were classified among these prefoldin genes based on the phylogenetic analyses. Noticeably, I also found common motif compositions in most members of each group, implying functional similarities between them (Figure 1). Several groups (such as Groups I, IV, and V) possess five motifs, while Group VI only has one motif. Except for members in Group VI, motif 3 was shared by most other prefoldin proteins. I also found some additional distinct motifs in specific groups, such as, motif 2 restricted in Group I, II, and III; motifs 14 and 15 in Group I; motif 9 in Group III; and motif 13, 7, 6, and 11 in Group IV, V, VIII, and IX, respectively. Previous study has indicated that low conservation exists between prefoldin sequences (Hill and Hemmingsen, 2001). My motif composition analyses of the prefoldin proteins also confirm it. The diversity of the prefoldin sequences may increase the complexity of the binding substrate, thus greatly expand the functional scope of the prefoldin complex. Further investigation may be required to determine if any of these distinct motifs across diverse groups also have a unique functional role.
Chromosomal Position and Duplication of Prefoldin Genes in Maize
Gene duplication, including segmental duplication, tandem duplication and retroposition, plays an important role in the evolution of organism (Chen Y et al., 2014; Cao and Li, 2015). To investigate the duplication mechanisms of prefoldin genes, I first examined their physical locations on chromosomes and found that the prefoldin genes were scattered in the maize genome (Figure 3). Further, all prefoldin genes were located in the duplicated segments of chromosomes in maize. Three of 6 pairs (GRMZM2G023347/GRMZM2G070061, GRMZM2G099909/GRMZM2G102580, and GRMZM2G010065/GRMZM2G392932) were retained in the maize duplication event (Figure 3). Next, I also used Ks to estimate the evolutionary dates of duplicated prefoldin genes (Table 2). The results indicated that duplication events of maize 3/6 pairs occurred within the past 17.69–21.73 million years, consistent with the time of subsequent genome duplication events in maize (Gaut et al., 1996). In addition, the earliest segmental duplication event was also observed around 81.46 MYA in the prefoldins of maize (GRMZM2G014676/GRMZM2G119740) (Kellogg, 2001). Therefore, expansion of prefoldin genes in maize might have occurred due to the large-scale segmental duplication events in evolution. Segmental duplication contributes to the prefoldin family gene expansion in maize.
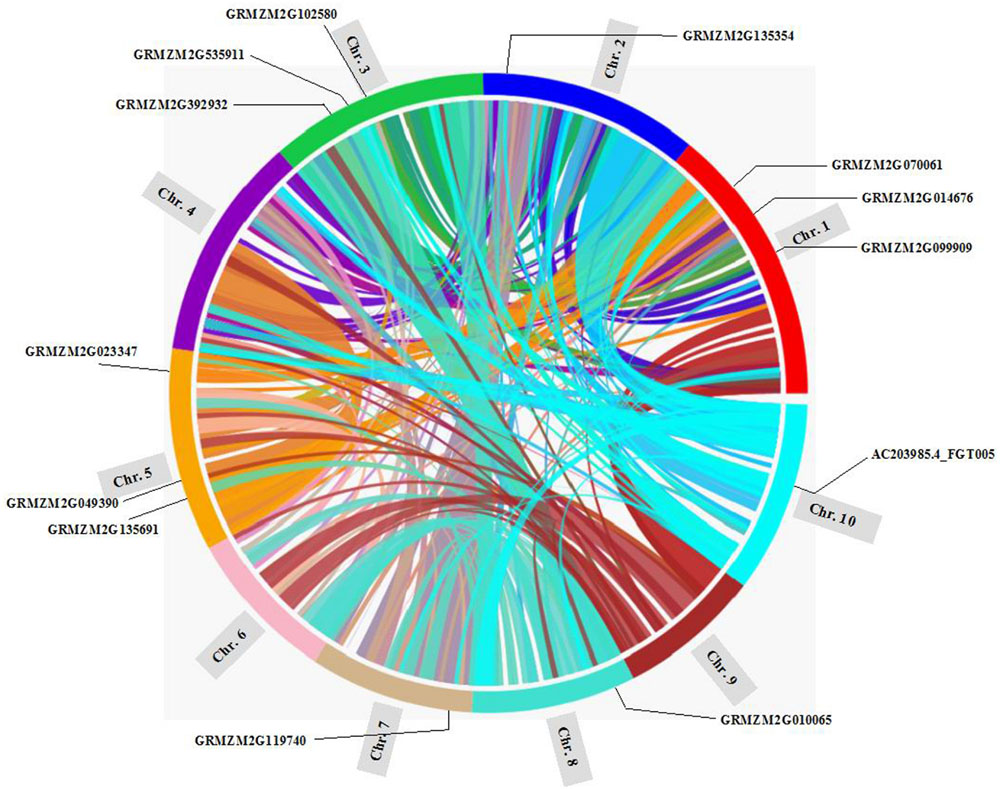
FIGURE 3. Location of the prefoldin genes and segmental duplication regions on maize chromosomes. The SyMAP database (Soderlund et al., 2011) was used to determine the segmental duplication regions.
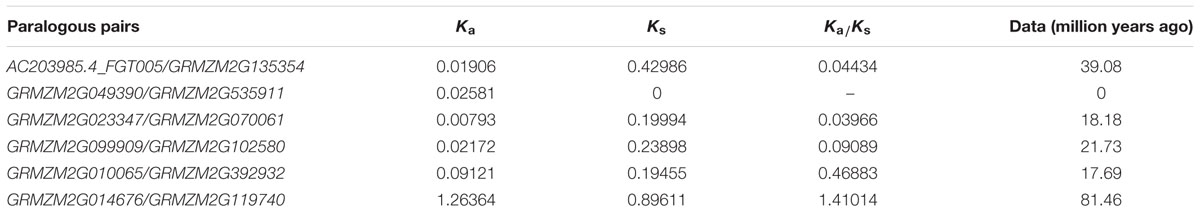
TABLE 2. Inference of duplication time of prefoldin paralogous pairs in maize.
Functional Divergence Analyses of the Prefoldin Proteins
To further study whether amino-acid substitutions cause adaptive functional diversification, the program DIVERGE (Gu, 1999, 2001) was used to estimate type-I functional divergence between different prefoldin groups. Thirty-six pairs of paralogous members were compared and estimated the evolutionary rate of each amino-acid sites. The results indicated that the coefficient of functional divergence (θ) values between 29 group pairs was less than 1 (Table 3), indicating significant site-specific alteration in selective constraints of most members of the prefoldin group pairs. Further, I also predicted a few critical residues associated with the functional divergence. For example, about 15 critical sites were predicted in Group I/II, I/VII, I/VIII, II/VII, V/VI, V/VII, VI/VIII, VII/VIII, and VII/IX pairs, while no site was predicted in the Group II/IX, IV/IX, and V/IX pairs. An example of detailed distribution of the functional divergence sites of Group VI/VIII pairs was shown in Figure 4. Further analyses indicated that one divergence site is located in the first residue. Other fourteen predicted sites are located in the N-terminal of α-helice 2 (α2). As we know, prefoldin can form a heterohexameric jellyfish like structure with coiled-coil tentacles (Siegert et al., 2000). These tentacles are involved in substrate binding. In this study, I found that some amino-acid sites on α-helice 2 (tentacle) present different sequence composition, implying an increase in substrate specificity for target proteins. Higher theta values (θ) exit in Group I/VI, I/VII, I/VIII, V/VI, V/VII, VII/VIII, and VII/IX pairs (Table 3), suggesting a higher evolutionary rate between them. Thus, due to the different evolutionary rates, the prefoldin genes diverge functionally from each other. The amino-acid mutations promoted the functional evolution and divergence of prefoldin genes, as an adaptation of the species to the changing environment (Cao et al., 2011).
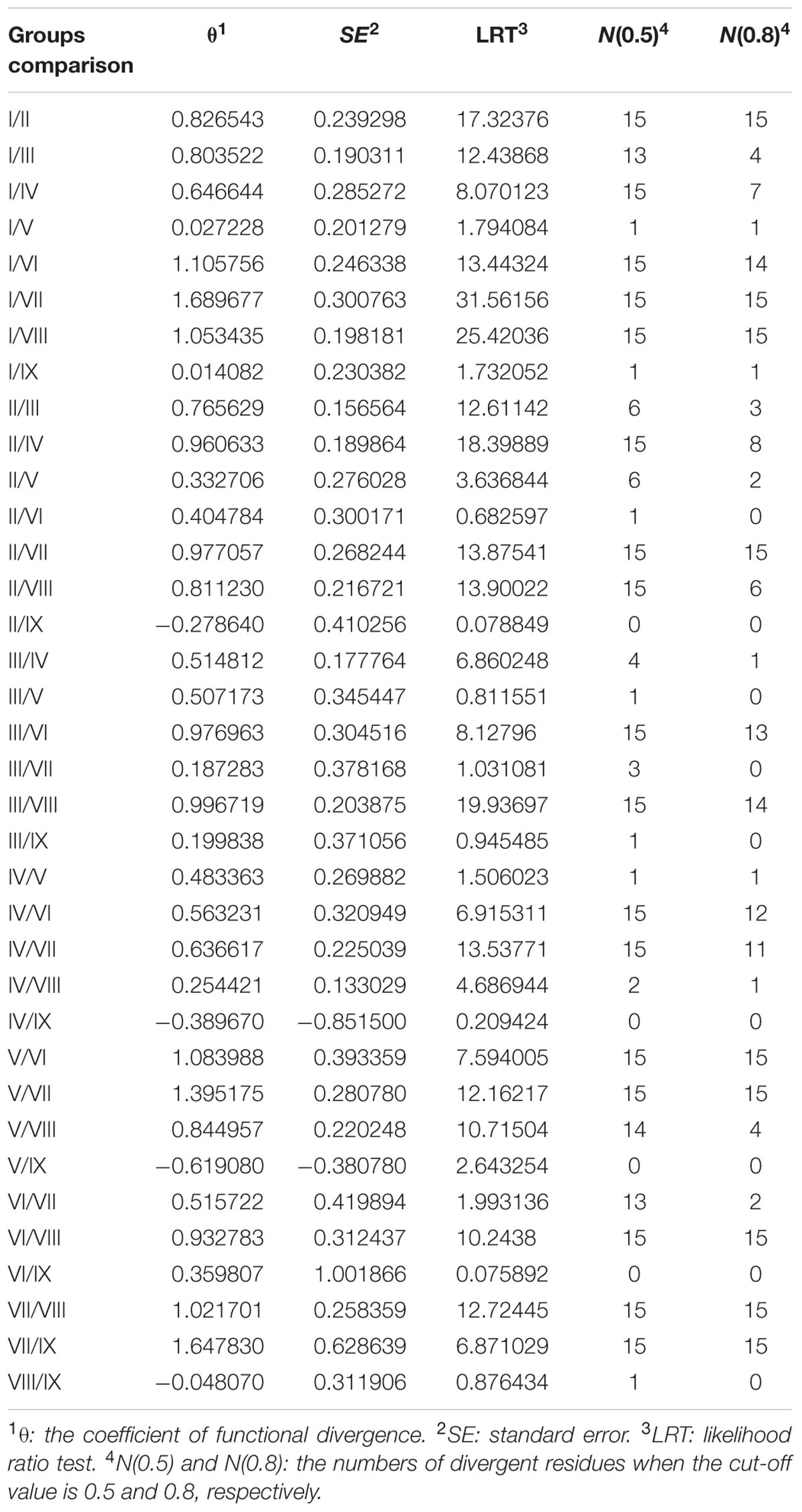
TABLE 3. Functional divergence estimated in prefoldin paralogs.
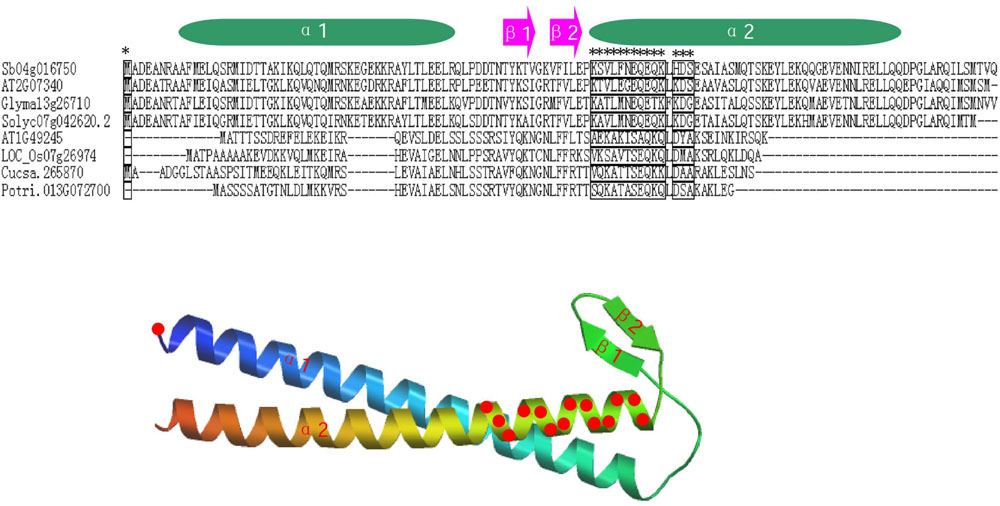
FIGURE 4. Distribution of functional divergence sites of Group VI/VIII pairs. Alignment results of some prefoldin proteins and the predicted tertiary structure are shown. Fifteen potential functional divergence sites are marked with a star in alignment results and marked with red circles on the predicted tertiary structure of the prefoldin protein.
Functional Network Analysis of the Maize Prefoldins
Genes involved in the same biological process are often coordinately expressed, and thus their co-expression information will be as a factor to understand the biological process (Eisen et al., 1998). To further explore which genes are possibly interacted by maize prefoldin, I performed a coexpression analyses with the COB11 (Schaefer et al., 2014). I found that 7 of 13 maize prefoldins were co-expressed in this network, resulting in 347 interactions exhibited by 78 genes (Figure 5). Among the 347 interactors identified, 23 and 17 genes were coexpressed with GRMZM2G014676 and GRMZM2G135691, respectively. GRMZM1G014676 is an orthologous gene of Arabidopsis PFD5 (At5g23290), while GRMZM2G135691 is an orthologous gene of Arabidopsis PFD1 (At2g07340) (Figure 1). Moreover, PFD5 plays an essential role in Arabidopsis tolerance to salt stress (Rodríguez-Milla and Salinas, 2009). They may also have similar biology functions. The network analysis of maize prefoldins has a reference value for the functional research of prefoldin genes. Some of these interactors include carrier proteins, such as equilibrative nucleoside transporter (ENT), mitochondrial phosphate carrier (MPC), acyl carrier protein (ACP), and so on. ENT (GRMZM2G002391) -mediated nucleosides transport across plasma membrane influenced Arabidopsis growth and pollen germination (Bernard et al., 2011). MPC (GRMZM2G118208) is a mitochondrial solution carrier protein, which delivers phosphate across the inner mitochondrial membrane and function in plant development and resistance (Haferkamp and Schmitz-Esser, 2012). As a small acidic protein with conserved Asp-Ser-Leu (DSL) motif, ACP (GRMZM2G149580) functions in root nodule symbiosis in soybean (Wang et al., 2014). I also found that another interactor, an auxin binding protein (ABP1), is required for binding to auxin at low concentration and involved in the embryogenesis, auxin signaling and cell division (Chen X et al., 2014; Xu et al., 2014). In addition, an Arabidopsis cyclin-dependent kinase (CDK) G2 regulated the salinity stress response and was associated with the control of flowering time (Ma et al., 2015). Coexpression analyses reflect the correlation of the expression profiles of different genes, and are suggestive in tracing the genes in the same biological process or pathway. My results indicated that some proteins possessed carrying, binding, and kinase function were usually coexpressed with maize prefoldins (Figure 5). The prefoldin genes may be involved in some related molecular processes by interacting with these interactors. Thus, whether these interactors could function with prefoldin need further experimental verification. The approaches and results open a new way to explore the molecular mechanism of prefoldin dynamics. Also, this interaction can also help to study the key regulatory steps in these processes or pathways. It will be advantageous to screen candidate genes for further functional researches.
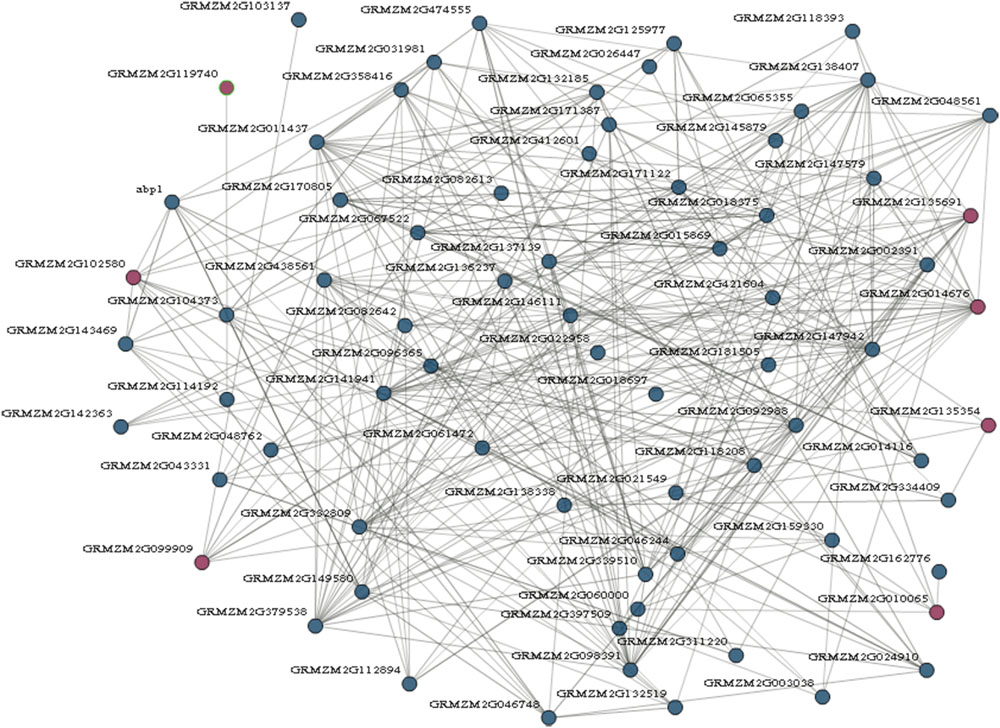
FIGURE 5. Prefoldin genes interaction network. Seven prefoldin genes are mapped to the co-expression database and reveal a total of 78 unique genes that showed 347 interactions in maize.
Expression Patterns and Promoter Sequence Analysis of the Maize Prefoldin Genes
The gene expression patterns may provide important clues to their functions (Cao and Shi, 2012). Here, I first tested spatial and temporal specific expression patterns of maize prefoldin genes in 20 tissues using microarray data. I detected 10 probes of the maize prefoldin transcripts, and other three transcripts (GRMZM2G049390, GRMZM2G535911, and GRMZM2G014676) with no detectable probes were not further analyzed in this study. The results showed that the expression abundance of the prefoldin genes detected presented great variability in maize (Figure 6). Some prefoldins (AC203985.4_FGT005, GRMZM2G135354, and GRMZM2G392932) expressed exclusively in developing embryo and endosperm may improve seed yield and quality. Notably, due to the great expression in maize silk, GRMZM2G135691 may play a key role in silk development.
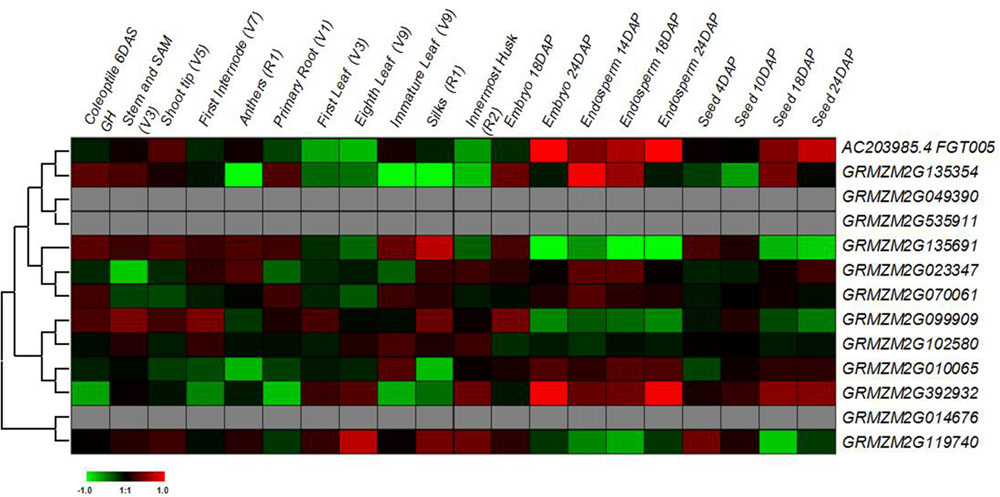
FIGURE 6. Expression profiles of the maize prefoldin genes in 20 different developmental stages.
Next, I also investigated the responses of maize prefoldin genes under drought, low temperature and salt stresses using 10-day-old seedlings. Quantitative RT-PCR was used to analyze the expression profiles of 13 prefoldin genes in maize under various stresses (Figure 7). The expression of GRMZM2G135354 and GRMZM2G049390 was significantly upregulated during drought, whereas other prefoldin genes were downregulated. The expression of over 84 percent of maize prefoldins was significantly repressed under salt, whereas GRMZM2G135354 and GRMZM2G049390 were strongly induced, suggesting that the prefoldin genes might present different responses to salinity or drought stress. Similar results also occurred with low-temperature. Under low temperature stress, most prefoldin genes were significantly downregulated (Figure 7). For instance, the expression level of GRMZM2G135691 decreased about 50 times. However, the expression of some prefoldin genes (GRMZM2G010065, and GRMZM2G049390) was induced. Previous study has shown that osmotic stress caused by drought and salinity can change microtubule orientation, and then regulates primary root elongation in Arabidopsis (Liu et al., 2014). Moreover, low temperature also leads to the depolymerization of microtubule (Bartolo and Carter, 1991). My expression profiles of maize prefoldin genes under these stresses indicated that most prefoldin members were significantly repressed. Therefore, it can be inferred that under the drought, salinity, or low temperature stress, the decreased expression of prefoldin genes may inhibit or change the development of microtubules. Next, the auxin response of these maize prefoldin genes was also investigated. Among the 13 prefoldin genes detected, three members (GRMZM2G392932, GRMZM2G014676, and GRMZM2G049390) were upregulated during IAA treatment, suggesting important roles in regulating IAA response. Nevertheless, the expression of GRMZM2G135354 and GRMZM2G535911 was unchanged after IAA treatment, whereas others were steadily decreased during the treatment (Figure 7).
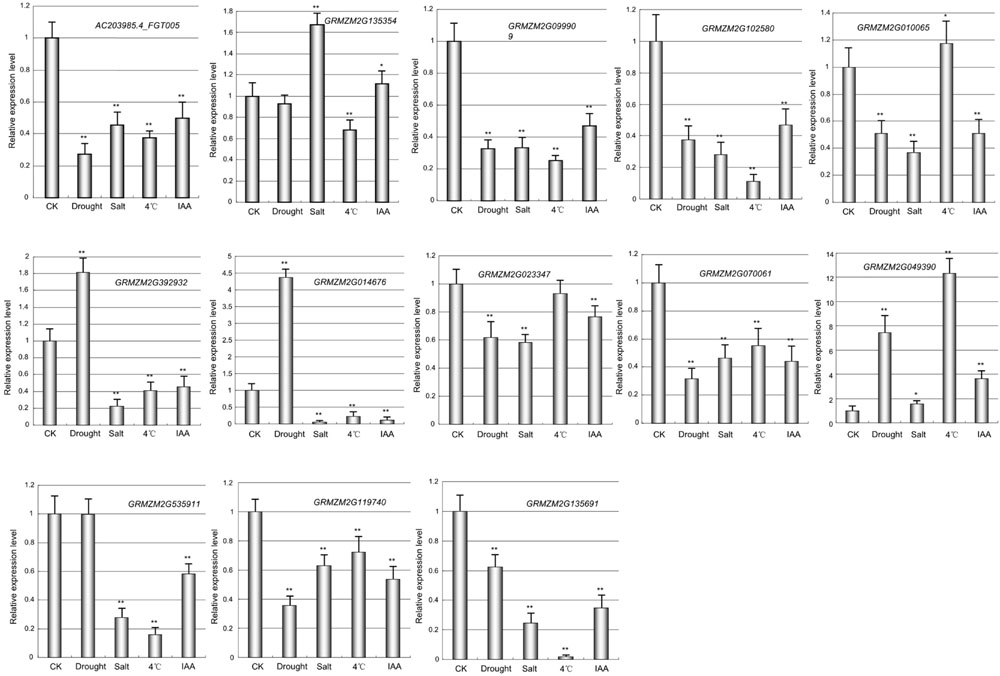
FIGURE 7. Expression profiles of the maize prefoldin genes under abiotic stresses and IAA. Experiments were conducted in triplicate. Significance was tested relative to each CK using t-test. Significance of ∗P < 0.05 and ∗∗P < 0.01. Error bar: standard deviation.
To further explore the potential regulatory mechanism of maize prefoldin genes in phytohormone and abiotic stress stimuli, I used the PLACE web server12 (Higo et al., 1999) to identify their putative cis-elements. Here, 1,000-bp promoter regions of the maize prefoldin genes were analyzed. A few phytohormones and abiotic response regulatory elements were found. They included auxin response factor (ARF)-binding site (S000270); tissue-specific expression and auxin-inducible (S000273); auxin-inducible (S000370); IAA/SA-inducible (S000024); SA-inducible (S000390); drought-inducible (S000414 and S000174); cold/drought-inducible (S000153); and salt-inducible (S000453) response regulatory elements. I also found that all maize prefoldin genes contained multiple regulatory elements in their promoter regions (Figure 8), suggesting that these phytohormones and abiotic stresses regulated the expression of maize prefoldin genes. Comparing the distribution of nine cis-elements in their promoter sequences, I found variation between all sister pairs of maize prefoldin genes, implying divergent expression profiles in the duplicated genes. It may be the reason of subfunctionalization or neofunctionalization (Prince and Pickett, 2002).
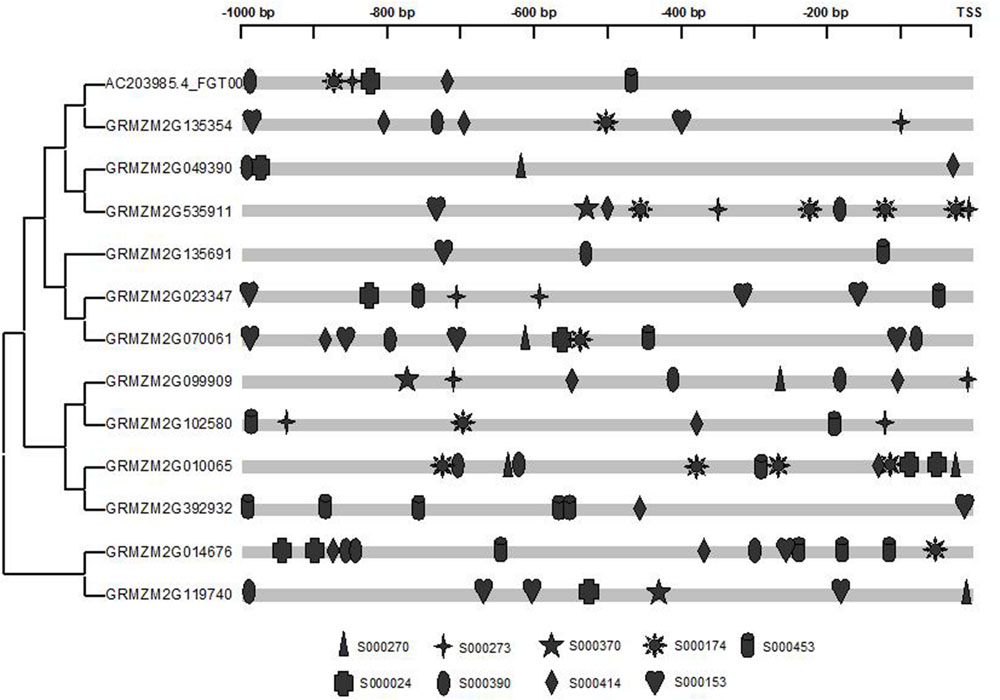
FIGURE 8. Promoter analyses of the maize prefoldin genes. Cis-regulated elements responsive to abiotic stresses and phytohormones are marked differently.
Summary
I performed a comparative analysis of the plant prefoldin gene family in this study, and found that the prefoldin family could be divided into nine groups by phylogenetic analyses. Gene organization and motif compositions of the prefoldin members were highly conserved in each group, implying their functional conservation. Segmental duplication contributed to maize prefoldin family gene expansion. A few critical residues associated with the functional divergence were predicted. Functional network analyses identified some co-expressed genes, which usually have binding, carrying and kinase activity. Furthermore, the differential expression profiles of the maize prefoldin genes suggested functional divergence during development and IAA and some abiotic stress treatments. Some cis-elements responsive to abiotic stress and phytohormone were also found in the upstream sequence of the maize prefoldin genes. These data will provide useful insights for further functional investigation of this gene family in the future.
Author Contributions
JC designed and carried out the experiments and wrote the manuscript. The author read and approved the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AB and the handling Editor declared a shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This project is supported by grants from the National Science Foundation of China (No. 31100923), the National Science Foundation of Jiangsu Province (BK2011467), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and Jiangsu University “Youth Backbone Teacher Training Project” (2012–2016).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00317
Footnotes
- ^http://www.phytozome.net
- ^http://web.expasy.org/protparam
- ^http://cello.life.nctu.edu.tw
- ^http://www.maizesequence.org
- ^http://www.phytozome.net
- ^http://meme.sdsc.edu
- ^http://csbio.cs.umn.edu/cob
- ^http://www.dna.affrc.go.jp/PLACE/signalscan.html
- ^http://www.phytozome.net
- ^http://cello.life.nctu.edu.tw
- ^http://csbio.cs.umn.edu/cob
- ^http://www.dna.affrc.go.jp/PLACE/
References
Bailey, T. L., Williams, N., Misleh, C., and Li, W. W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373. doi: 10.1093/nar/gkl198
Bartolo, M. E., and Carter, J. V. (1991). Microtubules in mesophyll cells of nonacclimated and cold-acclimated spinach: visualization and responses to freezing, low temperature, and dehydration. Plant Physiol. 97, 175–181. doi: 10.1104/pp.97.1.175
Bernard, C., Traub, M., Kunz, H. H., Hach, S., Trentmann, O., and Möhlmann, T. (2011). Equilibrative nucleoside transporter 1 (ENT1) is critical for pollen germination and vegetative growth in Arabidopsis. J. Exp. Bot. 62, 4627–4637. doi: 10.1093/jxb/err183
Cao, J. (2012). The pectin lyases in Arabidopsis thaliana: evolution, selection and expression profiles. PLoS ONE 7:e46944. doi: 10.1371/journal.pone.0046944
Cao, J., Huang, J., Yang, Y., and Hu, X. (2011). Analyses of the oligopeptide transporter gene family in poplar and grape. BMC Genomics 12:465. doi: 10.1186/1471-2164-12-465
Cao, J., and Li, X. (2015). Identification and phylogenetic analysis of late embryogenesis abundant proteins family in tomato (Solanum lycopersicum). Planta 241, 757–772. doi: 10.1007/s00425-014-2215-y
Cao, J., Li, X., Lv, Y., and Ding, L. (2015). Comparative analysis of the phytocyanin gene family in 10 plant species: a focus on Zea mays. Front. Plant Sci. 6:515. doi: 10.3389/fpls.2015.00515
Cao, J., and Shi, F. (2012). Evolution of the RALF gene family in plants: gene duplication and selection patterns. Evol. Bioinform. Online 8, 271–292. doi: 10.4137/EBO.S9652
Cao, S., Carlesso, G., Osipovich, A. B., Llanes, J., Lin, Q., Hoek, K. L., et al. (2008). Subunit 1 of the prefoldin chaperone complex is required for lymphocyte development and function. J. Immunol. 181, 476–484. doi: 10.4049/jimmunol.181.1.476
Chen, X., Grandont, L., Li, H., Hauschild, R., Paque, S., Abuzeineh, A., et al. (2014). Inhibition of cell expansion by rapid ABP1-mediated auxin effect on microtubules. Nature 516, 90–93. doi: 10.1038/nature13889
Chen, Y., and Cao, J. (2014). Comparative genomic analysis of the Sm gene family in rice and maize. Gene 539, 238–249. doi: 10.1016/j.gene.2014.02.006
Chen, Y., Hao, X., and Cao, J. (2014). Small auxin upregulated RNA (SAUR) gene family in maize: identification, evolution, and its phylogenetic comparison with Arabidopsis, rice, and sorghum. J. Integr. Plant Biol. 56, 133–150. doi: 10.1111/jipb.12127
Comeron, J. M. (1999). K-Estimator: calculation of the number of nucleotide substitutions per site and the confidence intervals. Bioinformatics 15, 763–764. doi: 10.1093/bioinformatics/15.9.763
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Natl. Acad. Sci. 95, 14863–14869.
Gaut, B. S., Morton, B. R., McCaig, B. C., and Clegg, M. T. (1996). Substitution rate comparisons between grasses and palms: synonymous rate differences at the nuclear gene Adh parallel rate differences at the plastid gene rbcL. Proc. Natl. Acad. Sci. U.S.A. 93, 10274–10279. doi: 10.1073/pnas.93.19.10274
Geissler, S., Siegers, K., and Schiebel, E. (1998). A novel protein complex promoting formation of functional alpha- and gamma-tubulin. EMBO J. 17, 952–966. doi: 10.1093/emboj/17.4.952
Gu, X. (1999). Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 16, 1664–1674. doi: 10.1093/oxfordjournals.molbev.a026080
Gu, X. (2001). Maximum-likelihood approach for gene family evolution under functional divergence. Mol. Biol. Evol. 18, 453–464. doi: 10.1093/oxfordjournals.molbev.a003824
Gu, Y., Deng, Z., Paredez, A. R., DeBolt, S., and Wang, Z. Y. (2008). Prefoldin 6 is required for normal microtubule dynamics and organization in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 105, 18064–18069. doi: 10.1073/pnas.0808652105
Haferkamp, I., and Schmitz-Esser, S. (2012). The plant mitochondrial carrier family: functional and evolutionary aspects. Front. Plant Sci. 3:2. doi: 10.3389/fpls.2012.00002
Hartl, F. U. (1996). Molecular chaperones in cellular protein folding. Nature 381, 571–579. doi: 10.1038/381571a0
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Hill, J. E., and Hemmingsen, S. M. (2001). Arabidopsis thaliana type I and type II chaperonins. Cell Stress Chaperones 6, 190–200. doi: 10.1379/1466-1268(2001)006<0190:ATTIAI>2.0.CO;2
Kellogg, E. A. (2001). Evolutionary history of the grasses. Plant Physiol. 125, 1198–1205. doi: 10.1104/pp.125.3.1198
Lee, Y., Smith, R. S., Jordan, W., King, B. L., Won, J., Valpuesta, J. M., et al. (2011). Prefoldin 5 is required for normal sensory and neuronal development in a murine model. J. Biol. Chem. 286, 726–736. doi: 10.1074/jbc.M110.177352
Leroux, M. R., Fändrich, M., Klunker, D., Siegers, K., Lupas, A. N., Brown, J. R., et al. (1999). MtGimC, a novel archaeal chaperone related to the eukaryotic chaperonin cofactor GimC/prefoldin. EMBO J. 18, 6730–6743. doi: 10.1093/emboj/18.23.6730
Liu, J., Wang, B., Zhang, Y., Wang, Y., Kong, J., Zhu, L., et al. (2014). Microtubule dynamics is required for root elongation growth under osmotic stress in Arabidopsis. Plant Growth Regul. 74, 187–192. doi: 10.1007/s10725-014-9910-3
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Locascio, A., Blázquez, M. A., and Alabadí, D. (2013). Dynamic regulation of cortical microtubule organization through prefoldin-DELLA interaction. Curr. Biol. 23, 804–809. doi: 10.1016/j.cub.2013.03.053
Lundin, V. F., Srayko, M., Hyman, A. A., and Leroux, M. R. (2008). Efficient chaperone-mediated tubulin biogenesis is essential for cell division and cell migration in C. elegans. Dev. Biol. 313, 320–334. doi: 10.1016/j.ydbio.2007.10.022
Ma, X., Qiao, Z., Chen, D., Yang, W., Zhou, R., Zhang, W., et al. (2015). CYCLIN-DEPENDENT KINASE G2 regulates salinity stress response and salt mediated flowering in Arabidopsis thaliana. Plant Mol. Biol. 88, 287–299. doi: 10.1007/s11103-015-0324-z
Martín-Benito, J., Boskovic, J., Gómez-Puertas, P., Carrascosa, J. L., Simons, C. T., Lewis, S. A., et al. (2002). Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT. EMBO J. 21, 6377–6386. doi: 10.1093/emboj/cdf640
Millán-Zambrano, G., and Chávez, S. (2014). Nuclear functions of prefoldin. Open Biol. 4, 140085. doi: 10.1098/rsob.140085
Millán-Zambrano, G., Rodríguez-Gil, A., Peñate, X., de Miguel-Jiménez, L., Morillo-Huesca, M., Krogan, N., et al. (2013). The prefoldin complex regulates chromatin dynamics during transcription elongation. PLoS Genet. 9:e1003776. doi: 10.1371/journal.pgen.1003776
Mirón-García, M. C., Garrido-Godino, A. I., Martínez-Fernández, V., Fernández-Pevida, A., Cuevas-Bermúdez, A., Martín-Expósito, M., et al. (2014). The yeast prefoldin-like URI-orthologue Bud27 associates with the RSC nucleosome remodeler and modulates transcription. Nucleic Acids Res. 42, 9666–9676. doi: 10.1093/nar/gku685
Misumi, O., Yoshida, Y., Nishida, K., Fujiwara, T., Sakajiri, T., Hirooka, S., et al. (2008). Genome analysis and its significance in four unicellular algae, Cyanidioschyzon merolae, Ostreococcus tauri, Chlamydomonas reinhardtii, and Thalassiosira pseudonana. J. Plant Res. 121, 3–17. doi: 10.1007/s10265-007-0133-9
Möckli, N., Deplazes, A., Hassa, P. O., Zhang, Z., Peter, M., Hottiger, M. O., et al. (2007). Yeast split-ubiquitin-based cytosolic screening system to detect interactions between transcriptionally active proteins. Biotechniques 42, 725–730. doi: 10.2144/000112455
Prince, V. E., and Pickett, F. B. (2002). Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 3, 827–837. doi: 10.1038/nrg928
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Rodríguez-Milla, M. A., and Salinas, J. (2009). Prefoldins 3 and 5 play an essential role in Arabidopsis tolerance to salt stress. Mol. Plant 2, 526–534. doi: 10.1093/mp/ssp016
Schaefer, R. J., Briskine, R., Springer, N. M., and Myers, C. L. (2014). Discovering functional modules across diverse maize transcriptomes using COB, the Co-expression Browser. PLoS ONE 9:e99193. doi: 10.1371/journal.pone.0099193
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Sekhon, R. S., Lin, H., Childs, K. L., Hansey, C. N., Buell, C. R., de Leon, N., et al. (2011). Genome-wide atlas of transcription during maize development. Plant J. 66, 553–563. doi: 10.1111/j.1365-313X.2011.04527.x
Siegers, K., Waldmann, T., Leroux, M. R., Grein, K., Shevchenko, A., Schiebel, E., et al. (1999). Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J. 18, 75–84. doi: 10.1093/emboj/18.1.75
Siegert, R., Leroux, M. R., Scheufler, C., Hartl, F. U., and Moarefi, I. (2000). Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103, 621–632. doi: 10.1016/S0092-8674(00)00165-3
Soderlund, C., Bomhoff, M., and Nelson, W. M. (2011). SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 39:e68. doi: 10.1093/nar/gkr123
Sturn, A., Quackenbush, J., and Trajanoski, Z. (2002). Genesis: cluster analysis of microarray data. Bioinformatics 18, 207–208. doi: 10.1093/bioinformatics/18.1.207
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Vainberg, I. E., Lewis, S. A., Rommelaere, H., Ampe, C., Vandekerckhove, J., Klein, H. L., et al. (1998). Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873. doi: 10.1016/S0092-8674(00)81446-4
Wang, J., Tóth, K., Tanaka, K., Nguyen, C. T., Yan, Z., Brechenmacher, L., et al. (2014). A soybean acyl carrier protein, GmACP, is important for root nodule symbiosis. Mol. Plant Microbe Interact. 27, 415–423. doi: 10.1094/MPMI-09-13-0269-R
Xia, K., Wang, R., Ou, X., Fang, Z., Tian, C., Duan, J., et al. (2012). OsTIR1 and OsAFB2 downregulation via OsmiR393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS ONE 7:e30039. doi: 10.1371/journal.pone.0030039
Xu, T., Dai, N., Chen, J., Nagawa, S., Cao, M., Li, H., et al. (2014). Cell surface ABP1-TMK auxin-sensing complex activates ROP GTPase signaling. Science 343, 1025–1028. doi: 10.1126/science.1245125
Yébenes, H., Mesa, P., Muñoz, I. G., Montoya, G., and Valpuesta, J. M. (2011). Chaperonins: two rings for folding. Trends Biochem. Sci. 36, 424–432. doi: 10.1016/j.tibs.2011.05.003
Yu, C. S., Lin, C. J., and Hwang, J. K. (2004). Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 13, 1402–1406. doi: 10.1110/ps.03479604
Keywords: prefoldin, phylogeny, evolution, functional divergence, expression
Citation: Cao J (2016) Analysis of the Prefoldin Gene Family in 14 Plant Species. Front. Plant Sci. 7:317. doi: 10.3389/fpls.2016.00317
Received: 27 November 2015; Accepted: 29 February 2016;
Published: 15 March 2016.
Edited by:
Daniel Pinero, Universidad Nacional Autónoma de México, MexicoReviewed by:
Alma Armenta-Medina, Centro de Investigación y de Estudios Avanzados, MexicoAlicia Gamboa De Buen, Universidad Nacional Autónoma de México, Mexico
Copyright © 2016 Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Cao, Y2ppbmZvckAxNjMuY29t