 Ruicai Long1
Ruicai Long1 Mingna Li2Tiejun Zhang1Junmei Kang1Yan Sun2Lili Cong1Yanli Gao1
Mingna Li2Tiejun Zhang1Junmei Kang1Yan Sun2Lili Cong1Yanli Gao1 Fengqi Liu3
Fengqi Liu3 Qingchuan Yang1*
Qingchuan Yang1*- 1Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, Beijing, China
- 2Department of Grass and Forage Science, College of Animal Science and Technology, China Agricultural University, Beijing, China
- 3Institute of Pratacultural Science, Heilongjiang Academy of Agricultural Sciences, Haerbin, China
Salt stress is an important abiotic stress that causes decreased crop yields. Root growth and plant activities are affected by salt stress through the actions of specific genes that help roots adapt to adverse environmental conditions. For a more comprehensive understanding of proteins affected by salinity, we used two-dimensional gel electrophoresis and mass spectrometry to characterize the proteome-level changes associated with salt stress response in Medicago sativa cv. Zhongmu-1 and Medicago truncatula cv. Jemalong A17 roots. Our physiological and phenotypic observations indicated that Zhongmu-1 was more salt tolerant than Jemalong A17. We identified 93 and 30 proteins whose abundance was significantly affected by salt stress in Zhongmu-1 and Jemalong A17 roots, respectively. The tandem mass spectrometry analysis of the differentially accumulated proteins resulted in the identification of 60 and 26 proteins in Zhongmu-1 and Jemalong A17 roots, respectively. Function analyses indicated molecule binding and catalytic activity were the two primary functional categories. These proteins have known functions in various molecular processes, including defense against oxidative stress, metabolism, photosynthesis, protein synthesis and processing, and signal transduction. The transcript levels of four identified proteins were determined by quantitative reverse transcription polymerase chain reaction. Our results indicate that some of the identified proteins may play key roles in salt stress tolerance.
Introduction
Plant growth and productivity are adversely affected by various natural abiotic and biotic factors, which cause considerable crop losses worldwide. These factors prevent plants from reaching their full genetic potential and limit crop productivity (Cramer et al., 2011). Salt stress is an important abiotic factor in many parts of the world, especially on irrigated lands (Munns and Tester, 2008). Soil salinity is a major abiotic stress influencing crop production, and researchers have investigated plant salt tolerance mechanisms with the aim of improving crop plants (Duzan et al., 2004). The metabolic imbalances caused by ion toxicity, osmotic stress, and nutritional deficiency due to salinity may also lead to oxidative stress (Zhu, 2002). These negative effects trigger changes to root morphology and suppression of plant growth, and can ultimately result in plant death.
Salinity regulates the expression of many plant genes at the transcriptional and post-translational levels. The molecular mechanism of plant salt tolerance is very complex (Zhu, 2001, 2002; Munns and Tester, 2008). To investigate this mechanism, several studies have been conducted in many plant models. Published analyses have helped characterize the expression profiles of many genes and proteins involved in salt stress responses in Arabidopsis thaliana, rice, wheat, soybean, tobacco, barrel medic (Medicago truncatula), and other plant species (Merchan et al., 2007; Cheng et al., 2009; Kumari et al., 2009; Razavizadeh et al., 2009; Zhang et al., 2009; Sobhanian et al., 2010; Capriotti et al., 2014; Ghaffari et al., 2014). The root is the primary tissue involved in salinity perception and is one of the first to be injured following exposure to several types of stresses. The sensitivity of the root to stress often limits the productivity of the entire plant (Steppuhn et al., 2010). Therefore, a comprehensive understanding of root molecular responses to salt stress is necessary for researchers to be able to increase crop tolerance to salt stress.
Plants differ considerably in their tolerance to salinity, as reflected by their different growth responses. For instance, several legumes, including Medicago sativa (alfalfa) and M. truncatula, have cultivars that have adapted to saline soils. This adaptive process is associated with a number of biochemical and physiological changes. The majority of these modifications are regulated by salt through alterations in gene expression (Zhu, 2002; Munns and Tester, 2008). Proteomics-based technologies have become powerful tools in the study of protein expression (Faurobert et al., 2007). For example, the combination of two-dimensional gel electrophoresis (2-DE) and mass spectrometry has been one of the most widely used techniques to study plant proteomes. Investigations into plant proteome changes during exposure to salt stress have been conducted for many plants, such as A. thaliana (Jiang et al., 2007), tomato (Manaa et al., 2011), soybean (Sobhanian et al., 2010), rice (Zhang et al., 2009; Ghaffari et al., 2014), tobacco (Razavizadeh et al., 2009), durum wheat (Capriotti et al., 2014), and barley (Witzel et al., 2014).
In this study, we explored new potential regulatory proteins of salt stress tolerance in M. sativa and M. truncatula roots. Many M. truncatula cultivars are more salt sensitive than M. sativa. Some M. sativa cultivars are highly tolerant to salinity stress (Munns and Tester, 2008), particularly M. sativa L. cv. Zhongmu-1, which we developed. High salt concentrations surrounding plant roots can induce rapid changes to cell growth and associated metabolic activities. The accumulation of salts inside plants can be toxic (Munns and Tester, 2008). There have been many genetic studies of Medicago species, but most of them focused on the model legume, M. truncatula. The objective of our study was to identify novel proteins regulated by salt stress in M. sativa and M. truncatula roots. We also aimed to determine differences in protein expression patterns between these two leguminous plants. We prepared total protein extracts from M. sativa and M. truncatula seedling roots treated with 300 mM NaCl and compared them to those of control roots using 2-DE. We identified novel salt stress-responsive root proteins and differentially expressed proteins.
Materials and Methods
Plant Materials
We used M. sativa cv. Zhongmu-1 and M. truncatula cv. Jemalong A17 in this study. Zhongmu-1 is a salt-tolerant cultivar of alfalfa (M. sativa, tetraploid, 2n = 4 × = 32), which is widely cultivated in China. Jemalong A17 is a cultivar of M. truncatula (diploid, 2n = 16), which is salt-sensitive. The genome sequence of Jemalong A17 is already known. Seeds of both cultivars were surface-sterilized in 75% ethanol for 10 min followed by three washes with sterile water. Seeds were germinated on moistened Whatman filter paper placed in Petri dishes (10 cm diameter). After a week, the seedlings were transferred to hydroponic cultures containing full-strength Hoagland's solution in a growth chamber with a 16 h/8 h light/dark photoperiod at 25°C and 65% relative humidity. The Hoagland's solution was renewed every 3 days.
Physiological Analysis
One-month-old Zhongmu-1 and Jemalong A17 seedlings were treated with Hoagland's solution supplemented with 300 mM NaCl for 0, 2, 8, 24, and 48 h. They were then analyzed for relative water content (RWC), electrolyte leakage, and proline content. The RWC was used to evaluate plant water status. Leaf RWC was calculated as RWC = (FW − DW) / (WS − DW), where FW refers to fresh weight, DW refers to dry weight, and WS refers to saturated water weight. Membrane damage was assessed by measuring electrolyte leakage. For each measurement, 10 g seedlings were added to 30 ml double deionized water in 50-ml tubes. Air was removed from the tubes using a vacuum pump until all seedlings were submerged in the water. Seedlings were maintained in the water for 4 h at 25°C.
We measured the conductivity of the bathing solution with a conductivity meter (Mettler Toledo) (as L1). The tubes were then incubated at 100°C for 15 min. The conductivity of the incubated solution was measured again after cooling to room temperature (as L2). For each sample, the relative conductivity (%) was calculated as L1/L2 × 100. Samples treated with 300 mM NaCl were harvested, frozen at −80°C and ground to a fine powder in liquid nitrogen using a mortar and pestle. We measured the free proline content using a colorimetric assay as described (Bates et al., 1973). Proline concentration was determined using a calibration curve and expressed as mg proline g−1 FW.
Sample Preparation and 2-DE
To identify M. sativa and M. truncatula proteins potentially involved in regulating salt tolerance, three independent replicates of 1-month-old seedling root samples were collected from Zhongmu-1 and Jemalong A17 plants treated with 300 mM NaCl for 8 h (Long et al., 2015). Untreated roots were used as controls. Root samples were ground to a fine powder in liquid nitrogen. Total protein extracts were prepared from the ground root samples using an optimized TRIzol method (Xiong et al., 2011). The final protein pellets were washed three times in 1 ml ethanol and resuspended in 1 ml lysis buffer (8 M urea, 4% w/v CHAPS, and 2% w/v DTT). Protein samples were sonicated for 10 min (4°C) and incubated at room temperature for 2 h. The protein solutions were centrifuged (40,000 × g, 40 min, 4°C) and the supernatants were collected. The protein concentrations of the supernatants were determined using the 2-D Quant kit according to the manufacturer's protocol (GE Healthcare). We diluted protein solutions with rehydration buffer [8 M urea, 2% w/v CHAPS, 1% w/v DTT, 0.5% v/v immobilized pH gradient (IPG) buffer pH 4–7, and 0.002% w/v bromophenol blue]. We then loaded 120 mg protein (in 450 μl) onto pH 4–7 IPG strips (24 cm). Isoelectric focusing (IEF) was completed using the Ettan IPGphorII system (GE Healthcare). The IEF and second dimension sodium dodecyl sulfate polyacrylamide gel electrophoresis were performed as described (Xiong et al., 2011). The IEF running conditions were as follows: 30 V for 12 h, 150 V for 250 Vh, 200 V for 300 Vh, 500 V for 250 Vh, 1000 V for 1000 Vh, 8000 V for 3 h, and 8000 V for a total of 30,000 Vh. Gel electrophoresis was performed using 12% polyacrylamide gels and the Ettan DALTsix electrophoresis gel system (GE Healthcare). The proteins were visualized using colloidal Coomassie brilliant blue G-250.
Protein Visualization and Image Analysis
The stained gels were scanned using a UMAX Power Look 2100XL scanner (UMAX) at a resolution of 600 dots per inch. Gel images were analyzed using ImageMaster™ 2D Platinum Version 5.0 (GE Healthcare Bio-Science). We estimated the isoelectric point (pI) of the proteins based on the relative migration of the protein spots on the IPG strips. All spot volumes were normalized as a percentage of the total volume of all spots present in the gel. We used ImageMaster™ 2D Platinum Version 5.0 to perform ANOVA. Comparisons of the mean differences were completed using Duncan's multiple range test at P < 0.05. The protein spots were determined to be significantly up- or down-regulated when the abundance fold change was more than 1.5 at P < 0.05.
Protein Identification and Analysis of Function
Significantly up- or down-regulated protein spots were excised from gels and destained for 2 h at room temperature using a freshly prepared wash solution consisting of 100% acetonitrile, 50 mM NH4CHO3 (50:50 v/v). Proteins were digested using a trypsin solution according to an established method (Xiong et al., 2011). Peptide mixtures were analyzed using the 4800 Plus MALDI TOF/TOFTM Analyzer (ABI), which is a matrix-assisted laser desorption ionization time of flight (MALDI-TOF/TOF) mass spectrometer. Mass spectrometry was completed using an established method (Li et al., 2011). Proteins were identified using peak lists for searches against the NCBInr database with the Mascot search engine (http://www.matrixscience.com/). The search criteria consisted of the following: Enzyme, Trypsin; Variable modifications, Oxidation (M); Peptide tolerance, 200 ppm; MS/MS tolerance, 0.8 Da; Instrument, MALDI-TOF/TOF; and Carbamidomethyl (C) as a fixed modification for all alkylated samples. Blast2GO software was used for gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses of identified proteins (Conesa et al., 2005).
Transcript Analysis Using Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
We used 1-month-old Zhongmu-1 and Jemalong A17 seedlings for transcript analyses. Total RNA was isolated from roots treated with NaCl (0, 2, 8, and 24 h) using TRIzol (Invitrogen, USA) according to the manufacturer's instructions. The RNA was then reverse transcribed and the synthesized cDNA was used as the template for qRT-PCR. The real-time fluorescent quantitative PCR was completed using the ABI 7500 system (Applied Biosystems). The β-actin gene served as a housekeeping gene to normalize target gene quantities. The real-time PCR primers used for the amplification of β-actin and the genes of ten identified proteins are listed in Supplementary Table 1. The PCR program consisted of a maximum of 40 cycles of 95°C for 15 s and 60°C for 30 s, followed by melting curve analysis. Transcript abundance for each gene was normalized to that of β-actin. The relative expression levels were calculated as follows: ratio = 2−ΔΔCt = 2−[Ct, t−Ct, r], where Ct refers to cycle threshold, Ct,t refers to Ct of the target gene and Ct,r refers to Ct of the β-actin control gene.
Statistical Analysis
All experiments were repeated with three independent biological replicates. All data obtained were subjected to a one-way ANOVA. The mean differences were compared using Duncan's multiple range t-test. Comparisons with P < 0.05 were considered significantly different. The values provided in the figures and tables are the means ± standard errors.
Results
Physiological Parameters Related to Salt Tolerance
The responses of Zhongmu-1 and Jemalong A17 to salt stress were compared in terms of leaf RWC, electrolyte leakage, and proline content. We observed that the leaf RWC of Zhongmu-1 and Jemalong A17 decreased by about 10% and over 20%, respectively, after exposure to salt stress for 48 h (Figure 1A). Relative electrolyte conductivity and proline content increased dramatically in salt stressed plants (Figure 1B). Following salt treatment, the relative electrolyte conductivity of Zhongmu-1 was lower than that of Jemalong A17. Conversely, proline accumulation in Zhongmu-1 was higher than that of Jemalong A17 (Figure 1C). Acording to the phenotypic observation the wilting degree of Jemalong A17 seedlings was much more obvious than that of Zhongmu-1 seedlings after treating with 300 mM NaCl for 8 h (Figures 1D–G). The physiological and phenotypic observations confirmed that M. sativa cv. Zhongmu-1 is more salt-tolerant than M. truncatula cv. Jemalong A17.
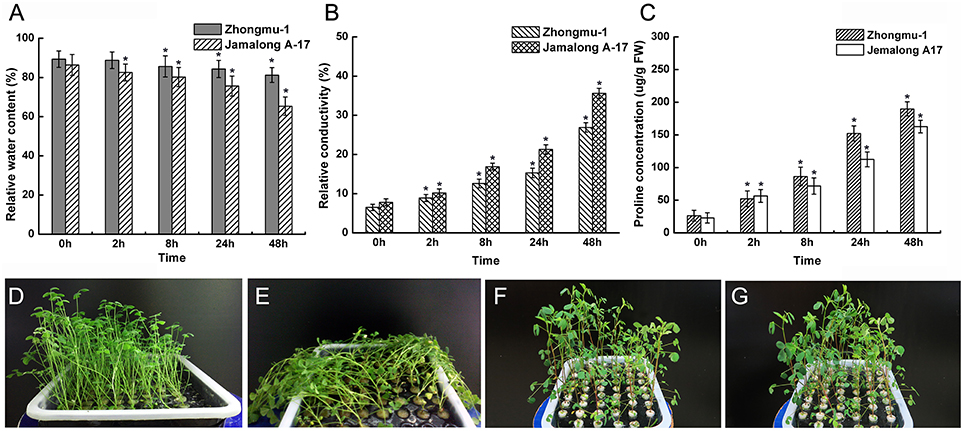
Figure 1. Physiological analysis and phenotypic observation of M. sativa cv. Zhongmu-1 and M. truncatula cv. Jemalong A17 under salt stress. One-month-old Zhongmu-1 and Jemalong A17 seedlings treated with 300 mM NaCl for 0, 2, 8, 24, and 48 h were analyzed for relative water content (A), electrolyte leakage (B), and proline content (C). (D,E) phenotypic observation of 1-month-old Jemalong A17 seedlings treated with 300 mM NaCl for 0 and 8 h. (F,G) phenotypic observation 1-month-old Zhongmu-1 seedlings treated with 300 mM NaCl for 0 and 8 h. * Indicates significant difference at p < 0.05 (Student's t-test).
Protein Responses to Salt Stress
The root is the first plant organ to be affected by salt stress. Representative 2-DE gel images of the Zhongmu-1 and Jemalong A17 root proteomes following salt treatment are presented in Figure 2. There was a broad distribution of the proteins in terms of pI (4.0–7.0) and mass (10–70 kDa). Of the approximately 800 detected Zhongmu-1 protein spots, 93 exhibited significant changes to spot abundance (P < 0.05) (Figure 2A, Supplementary Figure 1). Fifty-three of these proteins were up-regulated by salt stress, with the remaining 40 being down-regulated (Supplementary Table 2, Figure 3). Of the approximately 900 detected Jemalong A17 protein spots, 30 protein spots exhibited significant changes to spot abundance (P < 0.05) (Figure 2B, Supplementary Figure 1). Twenty-two of these proteins were up-regulated by salt stress, with the remaining eight being down-regulated (Supplementary Table 2, Figure 3).
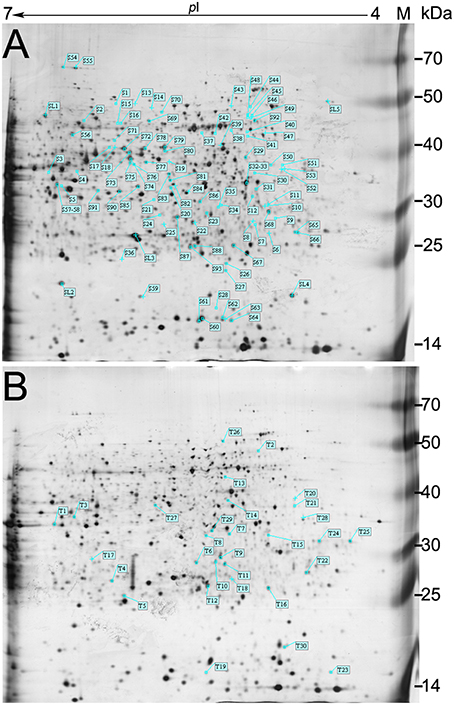
Figure 2. Representative 2-DE gel images of M. sativa cv. Zhongmu-1 and M. truncatula cv. Jemalong A17 root proteins. There were 93 and 30 protein spots in Zhongmu-1 (A) and Jemalong A17 (B), respectively, showing at least a 1.5-fold change following 300 mM NaCl treatment (P < 0.05). M, protein marker.
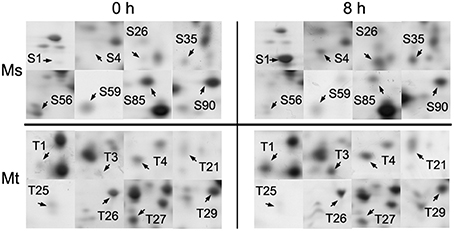
Figure 3. Enlarged 2-DE gel regions of 16 differentially accumulated root proteins in M. sativa cv. Zhongmu-1 (Ms) and M. truncatula cv. Jemalong A17 (Mt). The abundance of S1, S4, S26, S35, T1, T3, T4, and T21 increased after 8 h salt stress. The abundance of S56, S59, S85, S90, T25, T26, T27, and T29 decreased after 8 h salt stress.
Protein Identification
After mass spectrometric analysis, a total of 60 and 26 protein spots were identified in Zhongmu-1 and Jemalong A17, respectively (Table 1, S and T correspond to protein spots of Zhongmu-1 and Jemalong A17). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaino et al., 2014) partner repository with the dataset identifier PXD003761. These proteins were classified into five groups according to their molecular function as follows: Anti-oxidation, photosynthesis, metabolism, signal transduction, and protein synthesis and processing. Only the following proteins were identified in Zhongmu-1 and Jemalong A17: chaperonin CPN60-like protein (S44 and T2), fructose-bisphosphate aldolase (S3 and T1), and heat shock protein (S28 and T26). The abundance of chaperonin CPN60-like protein and fructose-bisphosphate aldolase increased in Zhongmu-1 and Jemalong A17 following salt treatment. However, the response of the heat shock protein corresponding to S28 and T26 differed between Zhongmu-1 and Jemalong A17. We observed an increase in S28 abundance and a decrease in T26 following salt treatment. Some proteins were identified in more than one spot on the same gel (Table 1). For example, S3 and S4 were identified as fructose-bisphosphate aldolase, S17 and S53 were identified as phosphopyruvate hydratase, S54 and S55 were identified as aconitate hydratase, and S61, S62, S63, and S64 were identified as a translation initiation factor (eIF-5A). Further examination of electrophoresis patterns indicated that the inferred mass and pI values of these spots differed, perhaps because of post-translational modifications or degradations.
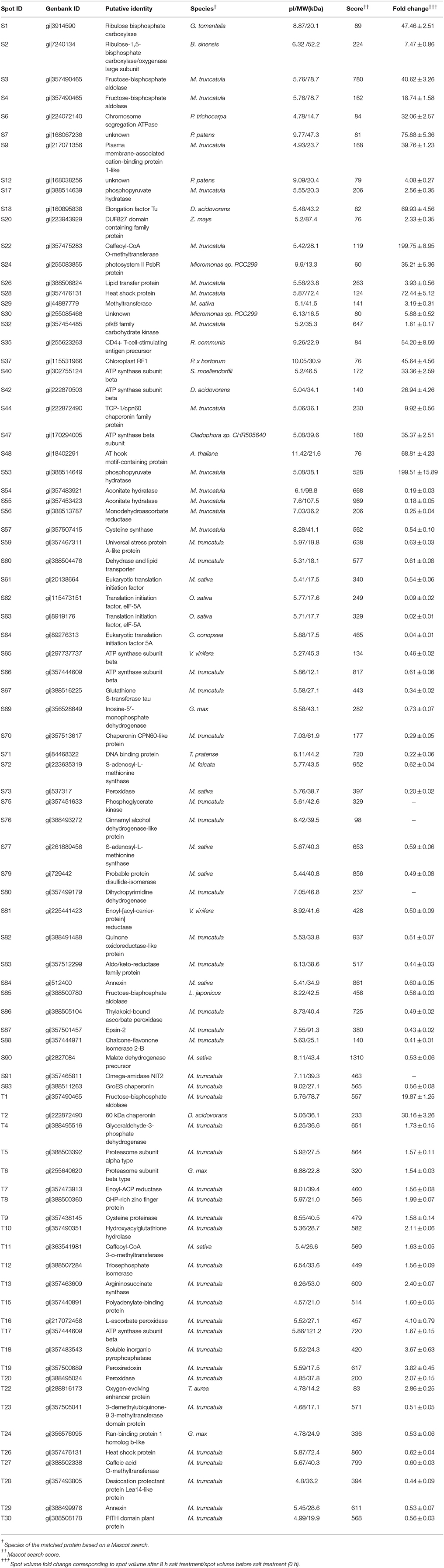
Table 1. Identities of salt-responsive proteins in M. sativa cv. Zhongmu-1 and M. truncatula cv. Jemalong A17 based on mass spectrometry and a Mascot search.
Functional Analysis of Identified Proteins
The identified protein sequences were blasted by BLASTP in the NCBI database. These identified proteins in Jemalong A17 were classified into 11 functional groups based on GO prediction (Figure 4A), including binding, catalytic activity, nucleotide binding, hydrolase activity, small molecule binding, protein binding, transferase activity, RNA binding, lipid binding, transporter activity, nucleic acid binding and not determined. These identified proteins in Zhongmu-1 were classified into 15 functional groups based on GO prediction (Figure 4B), including binding, catalytic activity, nucleotide binding, small molecule binding, transferase activity, hydrolase activity, protein binding, RNA binding, nucleic acid binding, translation factor activity, transporter activity, kinase activity, transferase activity, DNA binding, enzyme regulator activity and not determined. Molecule binding group and catalytic activity group were the two mainly functional groups.
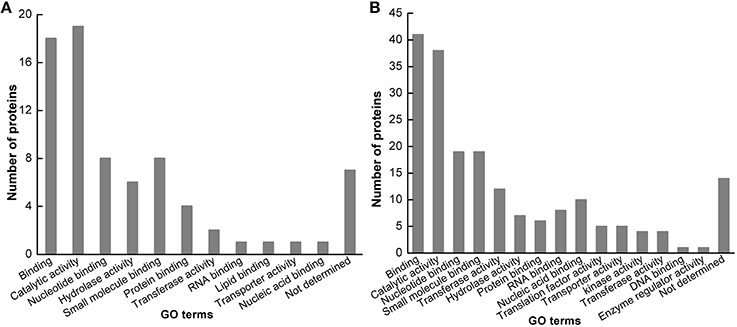
Figure 4. Functional categorization of identified proteins. The identified proteins in M. truncatula cv. Jemalong A17 (A) and M. sativa cv. Zhongmu-1 (B) were grouped into 12 and 16 functional categories, respectively.
Transcript Analysis of Selected Proteins
Ten differentially accumulated root proteins identified from Zhongmu-1 and Jemalong A17 were chosed to perform transcript expression analyses. The expression levels of fructose-bisphosphate aldolase (S3/T1), heat shock protein (S28/T26), TCP-1/cpn60 chaperonin family protein (S44/T2), and cinnamyl alcohol dehydrogenase-like protein (S76) based on qRT-PCR analyses are provided in Figure 4. The expression analyses results of all the 10 genes are provided in Supplementary Table 3. Compared with the expression level at 0 h, transcript abundance of fructose-bisphosphate aldolase, heat shock protein, and TCP-1/cpn60 chaperonin family protein increased considerably after salt treatment in Zhongmu-1. The transcript abundance of cinnamyl alcohol dehydrogenase-like protein decreased significantly (P < 0.05) after salt treatment in Zhongmu-1, whereas that of Jemalong A-17 did not significantly change, though there was a decreasing trend (Figure 5). After salt treatment, the heat shock protein transcript abundance increased more than 6-fold in Zhongmu-1, whereas that of Jemalong A-17 showed no significant changes, but did exhibit an increasing trend (Figure 5). These results along with those from the 2-DE analyses suggest that the transcript and protein level changes of most analyzed proteins were similar.
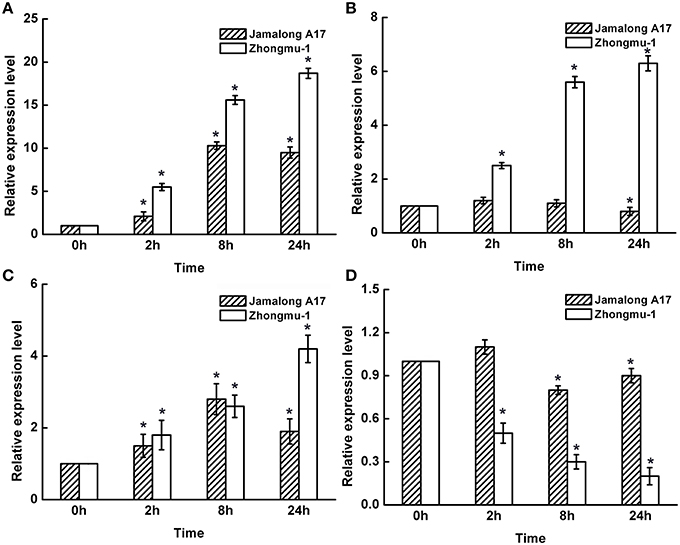
Figure 5. Transcript expression levels of fructose-bisphosphate aldolase (A), heat shock protein (B), TCP-1/cpn60 chaperonin family protein (C), and cinnamyl alcohol dehydrogenase-like protein (D) in M. sativa cv. Zhongmu-1 and M. truncatula cv. Jemalong A17 roots treated with NaCl. * Indicates significant difference at p < 0.05 (Student's t-test).
Discussion
The leaf RWC is one of the factors used to determine the extent of wilting after certain abiotic stresses. We observed that after salt treatment, the RWC of Zhongmu-1 was significantly higher than that of Jemalong A17, indicating Jemalong A17 was more wilted. Proline is an organic solute that helps regulate cellular osmolarity during plant responses to osmotic stress. Proline accumulation has been used as a drought-tolerance selection criterion related to membrane integrity in different plant species (Misra and Gupta, 2005). After salt treatment, Zhongmu-1 accumulated more proline than Jemalong A17. An increased rate of electrolyte leakage has been used as an indicator of cell membrane physical damage during exposure to abiotic stresses (Thiaw and Hall, 2004). Electrolyte leakage in Zhongmu-1 was lower than that of Jemalong A17, indicating that the cell membranes of Jemalong A17 were more damaged than those of Zhongmu-1. The physiological characteristics of Zhongmu-1 and Jemalong A17 were consistent with their salt tolerance levels. Based on their phenotypes, Zhongmu-1 was much more tolerant than Jemalong A17. The number of Zhongmu-1 protein spots exhibiting significant changes in abundance in response to salt stress was 3-fold higher than that of Jemalong A17. This result may be related to the fact that Zhongmu-1 is considerably more salt tolerant than Jemalong A17.
Some of the identified salt stress-regulated proteins were also reported in other plant species. For example, heat shock proteins, fructose-bisphosphate aldolase, peroxidase, DNA/RNA binding protein, and caffeoyl-CoA O-methyltransferase were detected in tomato roots after exposure to salt stress (Jiang et al., 2007; Manaa et al., 2011; Witzel et al., 2014). To evaluate the correlation between mRNA and the corresponding protein levels, the expression of 10 proteins with significant salt-induced changes in protein spot abundance was quantified by qRT-PCR. The qRT-PCR and 2-DE results suggest that the mRNA and protein level changes exhibit similar trends. These results also support the concept that post-transcriptional regulation plays an important role in stress-responsive gene expression, and indicates the importance of a combined transcriptomic and proteomic analyses (Mooney et al., 2006; Jiang et al., 2007).
We observed only a few salt stress-regulated proteins that were common between Zhongmu-1 and Jemalong A17 and there were differences in the expression patterns of these proteins. For intance, heat shock protein (70 kDa) was up-regulated in Zhongmu-1 (S28), but down-regulated in Jemalong A17 (T26). Heat shock proteins play important roles in a variety of cellular processes. They maintain proteins in their functional state and are also involved in protein translocations to subcellular compartments (Goswami et al., 2010). Heat shock proteins were previously reported to be up-regulated in tomato following exposure to cold stress (Page et al., 2010) and salt stress (Manaa et al., 2011).
Some proteins were identified in more than one spot in the same gel, such as fructose-bisphosphate aldolase (S3, S4, and S85) and phosphopyruvate hydratase (S17 and S53). Glycosylation, phosphorylation, and other post-translational modifications, which can alter the molecular weight and/or charge of proteins, may be responsible for these results. It is also possible that proteins were identified from multiple spots because of translation of alternatively spliced mRNAs (Yoshimura et al., 1999; Ndimba et al., 2005; Jiang et al., 2007). Proteomic studies have also shown that some proteins may be degraded during exposure to abiotic stress. For example, 19 different ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) large subunit fragments were detected in salt-treated rice roots (Yan et al., 2006). Similar phenomena have been reported in tomato root proteomes affected by salt stress (Manaa et al., 2011). The multiple fragments may also be the result of protein degradation by reactive oxygen species (ROS) during stress responses (Kingston-Smith and Foyer, 2000). Excess ROS can seriously disrupt normal plant metabolism through oxidative damage to lipids, proteins, and nucleic acids (Apel and Hirt, 2004; Askari et al., 2006; Bhushan et al., 2007). Anti-oxidative enzyme peroxiredoxin (T19) was detected in our study, which has been observed in responses to various abiotic stresses, including cold (Sarhadi et al., 2010), salinity (Ghaffari et al., 2014), and drought (Ali and Komatsu, 2006).
The proteins identified in this study are involved in various molecular processes. According to our results, some proteins associated with photosynthesis and metabolism were differentially expressed in Zhongmu-1 and Jemalong A17 following salt treatment. Ribulose-1,5-bisphosphate carboxylase/oxygenase (S1 and S2) is the most prevalent plant enzyme. It forms approximately 30-50% of the total soluble protein content in chloroplasts. The small subunit of Rubisco may be degraded because of oxidative stress (Sobhanian et al., 2010). Subsequently, the production of the large subunit may be inhibited. In our study, the abundance of the small (S1) and large (S2) subunits of Rubisco increased significantly in Zhongmu-1 8 h after salt treatment. This increase in abundance indicates that M. sativa can survive and photosynthesize even during moderate levels of salt stress. The increased activity of Rubisco subunits in tobacco and rice under salt stress has also been demonstrated (Kim et al., 2005; Razavizadeh et al., 2009). Therefore, it is possible that the accumulation of Rubisco in M. sativa reflects the increase in photorespiration during exposure to salt stress. Our results showed that the abundances of some proteins associated with energy production or transport, such as cytosolic malate dehydrogenase (S90) and glyceraldehyde-3-phosphate dehydrogenase (T2), were affected by salt in Zhongmu-1 and Jemalong A-17. Cytosolic malate dehydrogenase was reported to be responsive to salinity stress in A. thaliana roots (Jiang et al., 2007). However, the function of some identified proteins (such as S7, S12, and S30) are still unknown. Further research is needed to determine the functions of these proteins.
Conclusion
Our physiological and phenotypic observations confirmed that M. sativa cv. Zhongmu-1 is considerably more salt tolerant than M. truncatula cv. Jemalong A17. We used 2-DE to explore the changes in the root proteomes of these leguminous plants as a result of exposure to salt stress. Differentially accumulated proteins identified in Zhongmu-1 and Jemalong A17 were determined to be involved in various molecular processes, most of which belonged to molecule binding and catalytic activity. Some of the identified proteins were validated or predicted to play critical roles in salt stress regulation. The identification of salt-responsive proteins provides new insights into salt stress responses and the basis for further studies to improve the salt tolerance of alfalfa and other plants.
Author Contributions
RL and QY Conceived and designed the experiments, RL, ML, and TZ performed the experiments, JK, YS, LC, YG, FL contributed to data analysis, RL, ML, and QY wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, PK, and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This work was supported by the China Forage and Grass Research System (CARS-35-04), National Key Basic Research Program of China (2015CB943500), and Basic Scientific Research Fund of IAS-CAAS (2014ywf-zd-2).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00424
Supplementary Figure 1. The 2-DE images of 3 biological replicates for control and the treated samples.
Supplementary Table 1. Primers used for qRT-PCR to analyze transcript abundance of four proteins identified during 2-DE and mass spectrometry.
Supplementary Table 2. Protein spot volume fold changes of all proteins identified in Zhongmu-1 and Jemalong A17.
Supplementary Table 3. Transcript expression analyses results of 10 differentially accumulated root proteins in Zhongmu-1 and Jemalong A17.
References
Ali, G. M., and Komatsu, S. (2006). Proteomic analysis of rice leaf sheath during drought stress. J. Proteome Res. 5, 396–403. doi: 10.1021/pr050291g
Apel, K., and Hirt, H. (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. doi: 10.1146/annurev.arplant.55.031903.141701
Askari, H., Edqvist, J., Hajheidari, M., Kafi, M., and Salekdeh, G. H. (2006). Effects of salinity levels on proteome of Suaeda aegyptiaca leaves. Proteomics 6, 2542–2554. doi: 10.1002/pmic.200500328
Bates, L., Waldren, R., and Teare, I. (1973). Rapid determination of free proline for water-stress studies. Plant Soil 39, 205–207. doi: 10.1007/BF00018060
Bhushan, D., Pandey, A., Choudhary, M. K., Datta, A., Chakraborty, S., and Chakraborty, N. (2007). Comparative proteomics analysis of differentially expressed proteins in chickpea extracellular matrix during dehydration stress. Mol. Cell. Proteomics 6, 1868–1884. doi: 10.1074/mcp.M700015-MCP200
Capriotti, A. L., Borrelli, G. M., Colapicchioni, V., Papa, R., Piovesana, S., Samperi, R., et al. (2014). Proteomic study of a tolerant genotype of durum wheat under salt-stress conditions. Anal. Bioanal. Chem. 406, 1423–1435. doi: 10.1007/s00216-013-7549-y
Cheng, Y. W., Qi, Y. C., Zhu, Q., Chen, X., Wang, N., Zhao, X., et al. (2009). New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 9, 3100–3114. doi: 10.1002/pmic.200800340
Conesa, A., Goötz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Cramer, G. R., Urano, K., Delrot, S., Pezzotti, M., and Shinozaki, K. (2011). Effects of abiotic stress on plants: a systems biology perspective. BMC Plant Biol. 11:163. doi: 10.1186/1471-2229-11-163
Duzan, H. M., Zhou, X., Souleimanov, A., and Smith, D. L. (2004). Perception of Bradyrhizobium japonicum Nod factor by soybean [Glycine max (L.) Merr.] root hairs under abiotic stress conditions. J. Exp. Bot. 55, 2641–2646. doi: 10.1093/jxb/erh265
Faurobert, M., Mihr, C., Bertin, N., Pawlowski, T., Negroni, L., Sommerer, N., et al. (2007). Major proteome variations associated with cherry tomato pericarp development and ripening. Plant Physiol. 143, 1327–1346. doi: 10.1104/pp.106.092817
Ghaffari, A., Gharechahi, J., Nakhoda, B., and Salekdeh, G. H. (2014). Physiology and proteome responses of two contrasting rice mutants and their wild type parent under salt stress conditions at the vegetative stage. J. Plant Physiol. 171, 31–44. doi: 10.1016/j.jplph.2013.07.014
Goswami, A., Banerjee, R., and Raha, S. (2010). Mechanisms of plant adaptation/memory in rice seedlings under arsenic and heat stress: expression of heat-shock protein gene HSP70. Aob Plants 2010:plq023. doi: 10.1093/aobpla/plq023
Jiang, Y., Yang, B., Harris, N. S., and Deyholos, M. K. (2007). Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 58, 3591–3607. doi: 10.1093/jxb/erm207
Kim, D. W., Rakwal, R., Agrawal, G. K., Jung, Y. H., Shibato, J., Jwa, N. S., et al. (2005). A hydroponic rice seedling culture model system for investigating proteome of salt stress in rice leaf. Electrophoresis 26, 4521–4539. doi: 10.1002/elps.200500334
Kingston-Smith, A. H., and Foyer, C. H. (2000). Bundle sheath proteins are more sensitive to oxidative damage than those of the mesophyll in maize leaves exposed to paraquat or low temperatures. J. Exp. Bot. 51, 123–130. doi: 10.1093/jexbot/51.342.123
Kumari, S., Panjabi, V., Kushwaha, H., Sopory, S., Singla-Pareek, S., and Pareek, A. (2009). Transcriptome map for seedling stage specific salinity stress response indicates a specific set of genes as candidate for saline tolerance in Oryza sativa L. Funct. Integr. Genomics. 9, 109–123. doi: 10.1007/s10142-008-0088-5
Li, J. Y., Liu, F. J., Liu, X., Liu, J., Zhu, P., Wan, F. C., et al. (2011). Mapping of the human testicular proteome and its relationship with that of the epididymis and spermatozoa. Mol. Cell. Proteomics 10:M110.004630. doi: 10.1074/mcp.m110.004630
Long, R. C., Li, M. N., Kang, J. M., Zhang, T. J., Sun, Y., and Yang, Q. C. (2015). Small RNA deep sequencing identifies novel and salt-stress-regulated microRNAs from roots of Medicago sativa and Medicago truncatula. Physiol. Plant. 154, 13–27. doi: 10.1111/ppl.12266
Manaa, A., Ben Ahmed, H., Valot, B., Bouchet, J. P., Aschi-Smiti, S., Causse, M., et al. (2011). Salt and genotype impact on plant physiology and root proteome variations in tomato. J. Exp. Bot. 62, 2797–2813. doi: 10.1093/jxb/erq460
Merchan, F., de Lorenzo, L., Rizzo, S. G., Niebel, A., Manyani, H., Frugier, F., et al. (2007). Identification of regulatory pathways involved in the reacquisition of root growth after salt stress in Medicago truncatula. Plant J. 51, 1–17. doi: 10.1111/j.1365-313X.2007.03117.x
Misra, N., and Gupta, A. K. (2005). Effect of salt stress on proline metabolism in two high yielding genotypes of green gram. Plant Sci. 169, 331–339. doi: 10.1016/j.plantsci.2005.02.013
Mooney, B. P., Miernyk, J. A., Greenlief, C. M., and Thelen, J. J. (2006). Using quantitative proteomics of Arabidopsis roots and leaves to predict metabolic activity. Physiol. Plant. 128, 237–250. doi: 10.1111/j.1399-3054.2006.00746.x
Munns, R., and Tester, M. (2008). Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681. doi: 10.1146/annurev.arplant.59.032607.092911
Ndimba, B. K., Chivasa, S., Simon, W. J., and Slabas, A. R. (2005). Identification of Arabidopsis salt and osmotic stress responsive proteins using two-dimensional difference gel electrophoresis and mass spectrometry. Proteomics 5, 4185–4196. doi: 10.1002/pmic.200401282
Page, D., Gouble, B., Valot, B., Bouchet, J. P., Callot, C., Kretzschmar, A., et al. (2010). Protective proteins are differentially expressed in tomato genotypes differing for their tolerance to low-temperature storage. Planta 232, 483–500. doi: 10.1007/s00425-010-1184-z
Razavizadeh, R., Ehsanpour, A. A., Ahsan, N., and Komatsu, S. (2009). Proteome analysis of tobacco leaves under salt stress. Peptides 30, 1651–1659. doi: 10.1016/j.peptides.2009.06.023
Sarhadi, E., Mahfoozi, S., Hosseini, S. A., and Salekdeh, G. H. (2010). Cold acclimation proteome analysis reveals close link between the up-regulation of low-temperature associated proteins and vernalization fulfillment. J. Proteome Res. 9, 5658–5667. doi: 10.1021/pr100475r
Sobhanian, H., Razavizadeh, R., Nanjo, Y., Ehsanpour, A. A., Jazii, F. R., Motamed, N., et al. (2010). Proteome analysis of soybean leaves, hypocotyls and roots under salt stress. Proteome Sci. 8:19. doi: 10.1186/1477-5956-8-19
Steppuhn, H., Falk, K. C., and Zhou, R. (2010). Emergence, height, grain yield and oil content of camelina and canola grown in saline media. Can. J. Soil Sci. 90, 151–164. doi: 10.4141/CJSS09046
Thiaw, S., and Hall, A. E. (2004). Comparison of selection for either leaf-electrolyte-leakage or pod set in enhancing heat tolerance and grain yield of cowpea. Field Crops Res. 86, 239–253. doi: 10.1016/j.fcr.2003.08.011
Vizca´ino, J. A., Deutsch, E. W., Wang, R., Csordas, A., Reisinger, F., Rios, D., et al. (2014). ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226. doi: 10.1038/nbt.2839
Witzel, K., Matros, A., Strickert, M., Kaspar, S., Peukert, M., Muhling, K. H., et al. (2014). Salinity stress in roots of contrasting barley genotypes reveals time-distinct and genotype-specific patterns for defined proteins. Mol. Plant 7, 336–355. doi: 10.1093/mp/sst063
Xiong, J. B., Yang, Q. C., Kang, J. M., Sun, Y., Zhang, T. J., Margaret, G., et al. (2011). Simultaneous isolation of DNA, RNA, and protein from Medicago truncatula L. Electrophoresis 32, 321–330. doi: 10.1002/elps.201000425
Yan, S. P., Zhang, Q. Y., Tang, Z. C., Su, W. A., and Sun, W. N. (2006). Comparative proteomic analysis provides new insights into chilling stress responses in rice. Mol. Cell. Proteomics 5, 484–496. doi: 10.1074/mcp.M500251-MCP200
Yoshimura, K., Yabuta, Y., Tamoi, M., Ishikawa, T., and Shigeoka, S. (1999). Alternatively spliced mRNA variants of chloroplast ascorbate peroxidase isoenzymes in spinach leaves. Biochem. J. 338, 41–48. doi: 10.1042/bj3380041
Zhang, L., Tian, L. H., Zhao, J. F., Song, Y., Zhang, C. J., and Guo, Y. (2009). Identification of an apoplastic protein involved in the initial phase of salt stress response in rice root by two-dimensional electrophoresis. Plant Physiol. 149, 916–928. doi: 10.1104/pp.108.131144
Zhu, J. K. (2001). Plant salt tolerance. Trends Plant Sci. 6, 66–71. doi: 10.1016/S1360-1385(00)01838-0
Keywords: Medicago, salt stress, root, protein, 2-DE, gene expression, function
Citation: Long R, Li M, Zhang T, Kang J, Sun Y, Cong L, Gao Y, Liu F and Yang Q (2016) Comparative Proteomic Analysis Reveals Differential Root Proteins in Medicago sativa and Medicago truncatula in Response to Salt Stress. Front. Plant Sci. 7:424. doi: 10.3389/fpls.2016.00424
Received: 28 October 2015; Accepted: 18 March 2016;
Published: 31 March 2016.
Edited by:
Qingsong Lin, National University of Singapore, SingaporeReviewed by:
Pannaga Krishnamurthy, National University of Singapore, SingaporeChien-Chen Lai, National Chung Hsing University, Taiwan
Copyright © 2016 Long, Li, Zhang, Kang, Sun, Cong, Gao, Liu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingchuan Yang, cWNoeWFuZzY2QDE2My5jb20=; eG1zX2dyYXNzMTY4QDE2My5jb20=