 Xiaopeng Li†
Xiaopeng Li† Zijian Zhou†
Zijian Zhou† Junqiang Ding†Yabin WuBo ZhouRuixia Wang
Junqiang Ding†Yabin WuBo ZhouRuixia Wang Jinliang MaShiwei WangXuecai Zhang
Jinliang MaShiwei WangXuecai Zhang Zongliang Xia
Zongliang Xia Jiafa Chen*Jianyu Wu*
Jiafa Chen*Jianyu Wu*- College of Agronomy, Synergetic Innovation Center of Henan Grain Crops and National Key Laboratory of Wheat and Maize Crop Science, Henan Agricultural University, Zhengzhou, China
Plant height (PH) and ear height (EH) are two very important agronomic traits related to the population density and lodging in maize. In order to better understand of the genetic basis of nature variation in PH and EH, two bi-parental populations and one genome-wide association study (GWAS) population were used to map quantitative trait loci (QTL) for both traits. Phenotypic data analysis revealed a wide normal distribution and high heritability for PH and EH in the three populations, which indicated that maize height is a highly polygenic trait. A total of 21 QTL for PH and EH in three common genomic regions (bin 1.05, 5.04/05, and 6.04/05) were identified by QTL mapping in the two bi-parental populations under multiple environments. Additionally, 41 single nucleotide polymorphisms (SNPs) were identified for PH and EH by GWAS, of which 29 SNPs were located in 19 unique candidate gene regions. Most of the candidate genes were related to plant growth and development. One QTL on Chromosome 1 was further verified in a near-isogenic line (NIL) population, and GWAS identified a C2H2 zinc finger family protein that maybe the candidate gene for this QTL. These results revealed that nature variation of PH and EH are strongly controlled by multiple genes with low effect and facilitated a better understanding of the underlying mechanism of height in maize.
Introduction
Maize (Zea mays L.) is one of the most cultivated crops worldwide, owing to its versatility and wide adaptability, and serves as food, animal feed, and raw material for various industrial products. Plant density is considered an important factor to increase yield (Cardwell, 1982; Zhang et al., 2014). Plant height (PH) and ear height (EH) are two important traits that directly affect plant density (Cai et al., 2012). A better understanding of the genetic architecture of PH and EH could help breeders to develop varieties with high density tolerance and, consequently, increased yield.
The mapping of quantitative trait loci (QTL) based on bi-parental populations is an effective method for detecting the genetic variation of in crops. Molecular markers have been widely used in QTL mapping for various traits in maize, including maturity (Buckler et al., 2009), disease resistance (Zwonitzer et al., 2010; Chen et al., 2012; Ding et al., 2012), abiotic stress (Agrama and Moussa, 1996), plant morphology (Xu et al., 2009; Ding et al., 2015; Chen et al., 2016), and plant color (Chen et al., 2014). More than 280 QTL have been identified for PH and EH in maize (Cai et al., 2012); Sibov et al. (2003) detected four QTL for PH and five QTL for EH that explained 24.76 and 20.91% of the phenotypic variance, respectively, using an F2:3 population with 400 families; Tang et al. (2006) detected six QTL for PH, of which four QTL were located in the same genomic region of the QTL affecting average internode length using a recombinant inbred line (RIL) population and suggested that average internode length was the main contributor to PH in maize; and Wang et al. (2006) detected 127 QTL for PH based on the maize integrated QTL map and confirmed 40 of them using meta-analysis. Additionally, Peiffer et al. (2014) showed that PH in maize has a highly polygenic genetic architecture using nested-association mapping population. Therefore, multiple models of genetic architecture, differing in polygenicity and effect sizes, can plausibly explain population variation for height in maize. All the previous studies provided valuable information to determine the genetic basis of height in maize, but only few QTL were validated in different populations. Therefore, the underlying mechanism of plant development remains unclear.
The available single nucleotide polymorphisms (SNPs) (Elshire et al., 2011; Chia et al., 2012) have been widely used in genome-wide association study (GWAS) for discovering loci controlling various traits in maize, including leaf architecture (Tian et al., 2011), disease and insect resistance (Zila et al., 2014; Samayoa et al., 2015; Li et al., 2016; Mahuku et al., 2016), and other important agronomic traits (Yang et al., 2014). Compared with traditional QTL mapping, GWAS has higher resolution and allows the identification of genes for multiple traits in a single population (Breseghello and Sorrells, 2006; Yu and Buckler, 2006; Yan et al., 2011). However, population substructure and genotyping errors can result in high false positive rates (Andersen et al., 2005; Yu et al., 2006). Therefore, the traditional QTL mapping powerfully compares pairs of alleles with low resolution, whereas association analysis provides a high-resolution evaluation of numerous alleles with uneven statistical power (Wilson et al., 2004). In this study, we combined linkage mapping and GWAS to identify QTL for PH and EH and exploit the strengths and weaknesses of both approaches for discovering causal loci across the genome.
Previous studies have cloned several genes that strongly influence PH in maize, such as brachytic2 on Chromosome (Chr.) 1, which influences polar auxin transport (Multani et al., 2003; Xing et al., 2015); ZmGA3ox2 on Chr. 3 and dwaft3 on Chr. 9 related to gibberellin synthesis (Winkler and Helentjaris, 1995; Teng et al., 2013); and dwarf8 on Chr. 1 and dwarf9 on Chr. 5 involved in gibberellin signal transduction pathways (Thornsberry et al., 2001; Lawit et al., 2010). In maize, over 40 genes have been reported for PH that are related to different biosynthesis pathways such as hormone synthesis, transport, and signaling (Wang and Li, 2008; Peiffer et al., 2014). However, the underlying mechanism of plant development remains unclear. The identification of additional loci/genes related to PH in elite inbred maize lines might help to better understand the mechanism of height development and optimize selection in breeding programs.
Here, we evaluated two bi-parental populations and one GWAS population for PH and EH in different environments and used both linkage mapping and GWAS to identify and verify QTL and candidate genes associated with the natural variation of height in maize. Our objectives were to: (1) identify QTL that significantly affect PH and EH in the two bi-parental populations; (2) discover top SNPs and their candidate genes associated with PH and EH by GWAS; (3) verify QTL in a near-isogenic line (NIL) populations; and (4) discuss the function of candidate genes.
Materials and Methods
Germplasm and Phenotype Evaluation
An F2:3 bi-parental population, consisting of 225 families were developed from a cross between Zheng58 and Chang7-2, which are the parental lines of Zhengdan958, one of the most popular maize hybrids in China with an annual acreage of 4.0 × 106 ha, accounting for 20% of total maize production (Du et al., 2006). Parental line Chang7-2 has higher PH and EH than another parental line Zheng58. The population and their parents were evaluated in Zhengzhou (34°52′N 113°37′E), Jiyuan (35°34′N 112°5′E), and Xichang (27°32′N 102°10′E) in 2007 (07Zhengzhou, 07Jiyuan, 07Xichang) and Zhengzhou and Jiyuan in 20008 (08Zhengzhou, 08Jiyuan). Another bi-parental recombinant inbred line (RIL) population consisting of 250 lines was produced from a cross between BT-1 and N6. BT-1 is an inbred line reformed by tropical Asia material with relatively high PH and EH and high resistance to various diseases, whereas N6 is a Tangsipingtou inbred line with relatively low PH and EH and low resistance to diseases. The population and their parents were evaluated in Zhengzhou in 2007 and 2008 (07Zhengzhou, 08Zhengzhou), Xuchang (34°2′N 113°81′E) in 2009 (09Xuchang), and Zhengzhou in spring and summer of 2009 (09Zhengzhou1, 09Zhengzhou2).
NIL population was developed from a cross between the low-PH line Zheng58, and the donor line Chang7-2 through four cycles of advanced backcrosses and one generation of self-crossing using marker assisted selection for the target genomic region. Two NILs, one homozygous for the QTL of the donor parent (+) and another homozygous for the QTL of the recurrent parent (−) were evaluated in Zhengzhou and Wenxian (34°95′N 113°6′E) in summer 2009.
The GWAS population, consisting of 258 maize inbred lines including the parental inbred lines of the two bi-parental populations, from the heterotic populations Tangsipingtou and Reid and some tropical lines from International Maize and Wheat Improve Center (CIMMYT), was evaluated in Zhengzhou, Wenxian and Hainan (18°21′N 109°10′E) in 2015 (15Zhengzhou, 15Wenxian, and 15Hainan, respectively).
All the populations were arranged in a randomized complete block design with three replications for each location. Sixteen plants were planted in 4 m row plots with 0.67 m row spacing. Field management was performed according to the standard agronomic practices in each location. PH was measured from the soil level to the tip of main inflorescence, and EH was measured from the soil level to the node attachment of the primary ear. The final value of each plot was the mean value of each plant inside the plot.
Phenotypic Data Analysis
Analysis of variance (ANOVA) of phenotype data was performed using the SPSS software (www.spss.com). The components of variance were estimated using a complete random effects model and broad-sense heritability was calculated as defined by Knapp et al. (1985). Best linear unbiased predictions (BLUPs) of the combined PH and EH were calculated by using a mixed linear model (lmer) in R software (R Core Team, 2015), which used replication, environments which combination location and years, and entries as a random effect. The BLUP value of each line was used for GWAS analysis. Histogram of phenotypic data was generated by using hist function in R software (R Core Team, 2015).
Linkage Mapping
The two bi-parental populations were genotyped by simple sequence repeat (SSR) markers. The linkage map of RIL population contained 207 polymorphic SSR markers had a total length of 1820.8 cM and an average distance of 11.7 cM between markers (Li et al., 2011). The F2:3 population was genotyped with 180 SSR markers, and the constructed map had a total length of 1987.7 cM and an average distance of 11.0 cM between markers (Ding et al., 2011).
Composite interval mapping (CIM) for QTL mapping was performed by Windows QTL Cartographer 2.5 (Zeng, 1994). The threshold value was set using 1000 random permutations (Churchill and Doerge, 1994). The proportion of phenotypic variation explained by a single QTL was determined by the square of the partial correlation coefficient (R2).
GWAS and Candidate Gene Annotation
A total of 955,650 SNPs were identified in the GWAS population using the genotyping-by-sequencing (GBS) method (Elshire et al., 2011; Glaubitz et al., 2014) which was conducted in Cornell University and the SNP flanking sequence and position information were available in “panzea” website (http://cbsusrv04.tc.cornell.edu/users/panzea/download.aspx?filegroupid=4). The SNPs was filtered with a missing value greater than 0.25 and a minor allele frequency (MAF) less than 0.05, resulting in 224,152 SNPs for future analysis. The principal component analysis (PCA), Kinship matrix and linkage disequilibrium (LD) between each pair of SNPs were conducted using TASSEL 5.0 software (Bradbury et al., 2007). The population structure was determined using an admixture ancestry model with correlated allele frequency in software STRUCTURE v2.3.3 (Pritchard et al., 2000). STRUCTURE was run four replicates for K (number of subpopulations) = 1–8, and two replicates for K = 9–12 with a run-length of 100,000 repetitions of Markov chain Monte Carlo following a burn-in period of 10,000 iterations. GWAS was conducted using a mixed linear model (MLM) that included BLUPs, markers, Kinship and PCA in TASSEL (Bradbury et al., 2007).
Candidate gene information were obtained from the MaizeGDB (http://www.maizegdb.org/) genome browser based on physical position of significant SNPs. Phytozome database (http://phytozome.jgi.doe.gov/pz/portal.html) were used to define relevant pathways and annotate possible functions of candidate genes.
Meta-Analysis
All QTL detected in the bi-parental populations and SNPs identified in the GWAS population were analyzed based on their physical position using R (R Core Team, 2015). The R code is provided in File S1.
Results
Phenotype Analysis
Descriptive statistics for PH and EH in the three mapping populations are presented in Table 1. There were a wide variations were observed in each population, for example, the PH of combined environments in RIL population ranged from 105 to 273 cm, whereas in the GWAS population from 106 to 293 cm. The EH in the F2:3 population ranged from 38.8 to 123 cm, and from 21.7 to 147 cm in GWAS population. The mean of PH and EH in the F2:3 families were 183.6 and 73.7 cm, respectively. The frequency of phenotypic value in all three populations for PH and EH followed an approximately normal distribution (Figure S1). The genotypic variance () and the genotype-by-environment variance () of PH and EH were significant in all three populations. Heritability for PH was 0.92 in the F2:3 population, 0.97 in the RIL population, and 0.91 in the GWAS population. Heritability for EH is similar to that for PH (Table 1). The high repeatability and heritability indicated that much of the phenotypic variance was genetically controlled in the populations and suitable for QTL mapping.
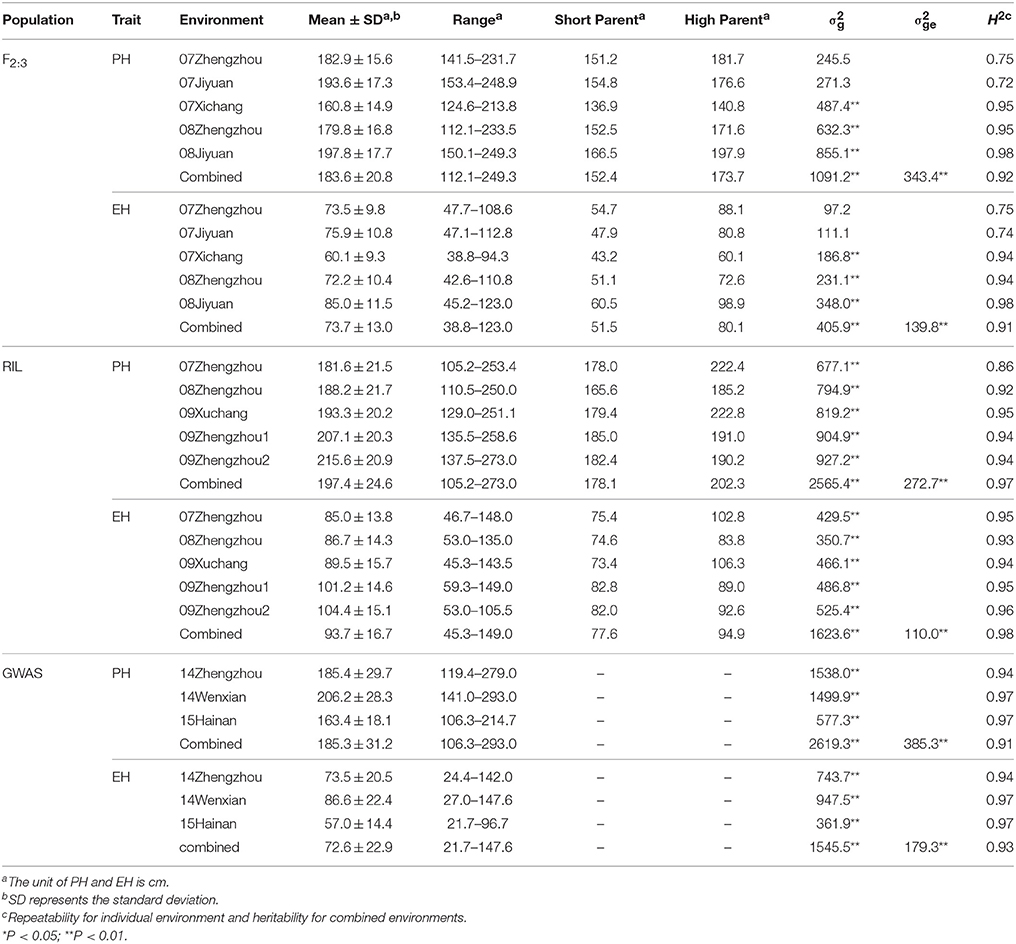
Table 1. Mean and standard deviation (SD) values, variance components and heritability of plant height (PH) and ear height (EH) in the F2:3 population, the recombinant inbred line (RIL) population, and the genome-wide association study (GWAS) population.
QTL Analysis
In the F2:3 population, nine QTL were identified for PH and EH (Table 2, Figure S2), and three of them for PH were located on Chr. 1 (bin 1.05/06), Chr. 2 (bin 2.08) and Chr. 5 (bin 5.04/05), and six QTL for EH located on Chr. 1 (bin 1.05), Chr. 2 (bin 2.09/10), Chr. 3 (bin 3.07), Chr. 5 (bin 5.05/06), Chr. 6 (bin 6.04/05), and Chr. 8 (bin 8.06/07). The increasing effect of three QTL for PH and five of the six QTL for EH were originated from the high parent Chang7-2, whereas only one QTL for EH from short parent Zheng58.
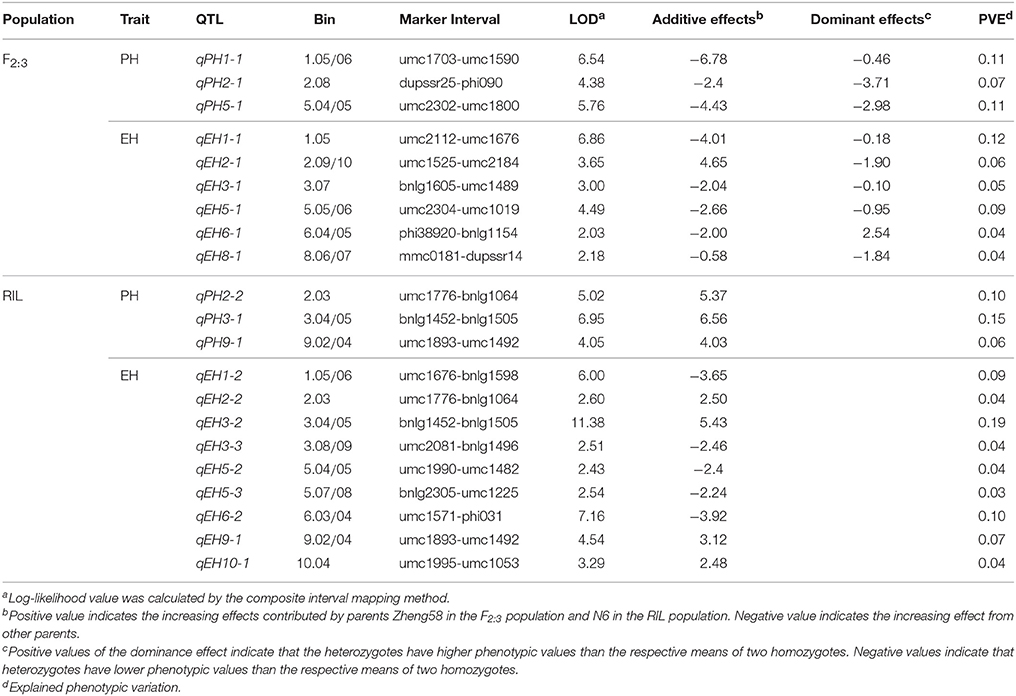
Table 2. Quantitative trait loci (QTL) for plant height (PH) and ear height (EH) identified in the F2:3 population and the recombinant inbred line (RIL) population.
In the RIL population, 12 QTL were detected for PH and EH: three QTL for PH located on Chr. 2 (bin 2.03), Chr. 3 (bin 3.04/05) and Chr. 9 (bin 9.02/9.04) and nine QTL for EH located on Chr. 1 (bin 1.05/06), Chr. 2 (bin 2.03), Chr. 3 (bin 3.04/05 and bin 3.08/09), Chr.5 (bin 5.04/05 and bin 5.07/08), Chr. 6 (bin 6.03/04), Chr. 9 (bin 9.02/04), and Chr. 10 (bin 10.04) (Table 2, Figure S3). Totally, 21 QTL were identified for PH and EH by two populations, and three common genomic regions (bin 1.05, 5.04/05, and 6.04/05) were detected by both populations.
GWAS for PH and EH
Population structure analysis showed that the LnP(D) value was stable between replications when K ≤ 3 and became unstable when at K > 3 (Figure 1A), indicating that the appropriate K-value was 3. At K = 3, 98.2% of the GWAS population was divided into Subpopulations 1, 2, and 3, containing 165, 68, and 60 lines, respectively, whereas the rest 2.8% was grouped into a mixed subpopulation (Figure 1B). Subgroup 1 was the most diverse and included tropical lines from CIMMYT; Subgroup 2 included lines from the Tangsipingtou heterotic group and Subgroup 3 included lines from the Reid heterotic group. PCA showed the lines source could separate very well by PC1 and PC2 (Figure S4A), and the first three PCs explained most of the variance (Figure S4B). PCA was in agreement with the results generated by STURCTURE, clearly showing the existence of three subgroups in the GWAS population. Hence, the first three PCs were included in the mixed model for GWAS.
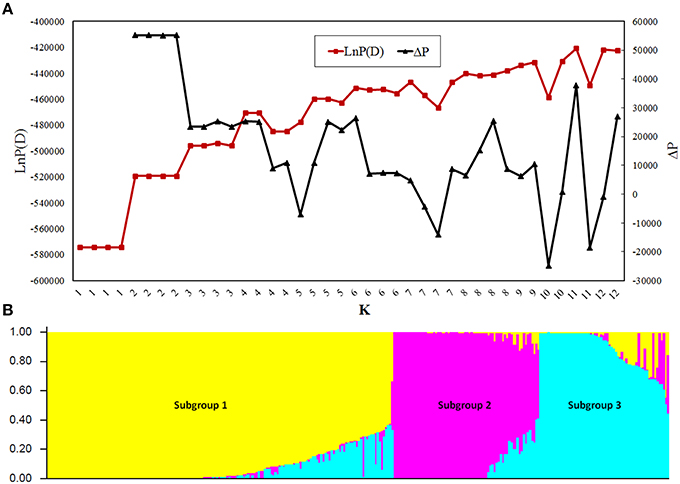
Figure 1. Population structure of the genome-wide association study (GWAS) panel. (A) Plot of LnP(D) and an ad-hoc statistic ΔP calculated for K = 1–12. LnP(D) is the output of STRUCTURE obtained by first computing the natural logarithm of the probability at each step of Markov chain Monte Carlo, ΔP is the difference of LnP(D) between adjacent K. (B) Population structure of the GWAS panel at K = 3.
Single marker-based GWAS was performed using MLM incorporating both the population structure (first three PCs) and K into the model. A total of 21 significant SNPs for PH and 20 for EH was identified with p < 1.0 × 10−4 (Figure 2), which explained 6.2–9.3% and 6.2–9.54% of the phenotypic variation for PH and EH, respectively. The most significant SNP was S6_124299082 for PH located on Chr. 6 which explained 9.3% of the phenotypic variation in the GWAS population, whereas the most significant SNP was S3_1976523 for EH located on Chr. 3 which explained 8.8% of the phenotypic variation (Table 3). The quantile-quantile (QQ) plots showed that population structure was well controlled by PCA and K (Figure S5).
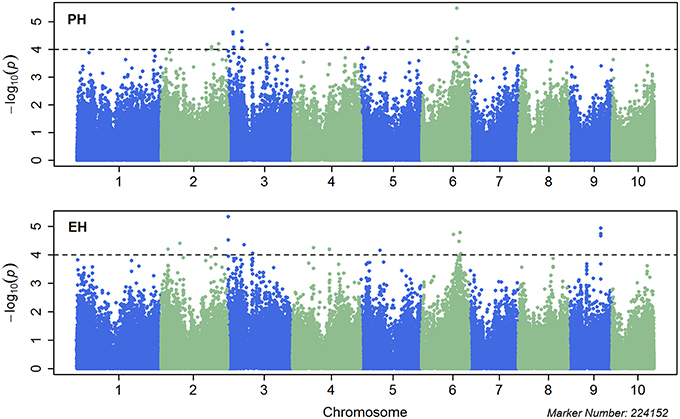
Figure 2. Manhattan plots for combined genome-wide association study (GWAS) using the mixed linear model (MLM) for plant height (upper plot) and ear height (lower plot). Chromosomes and physical positions of single nucleotide polymorphisms (SNPs) on the X-axis and −log10 p-value of each SNP derived from the association study on the Y-axis.
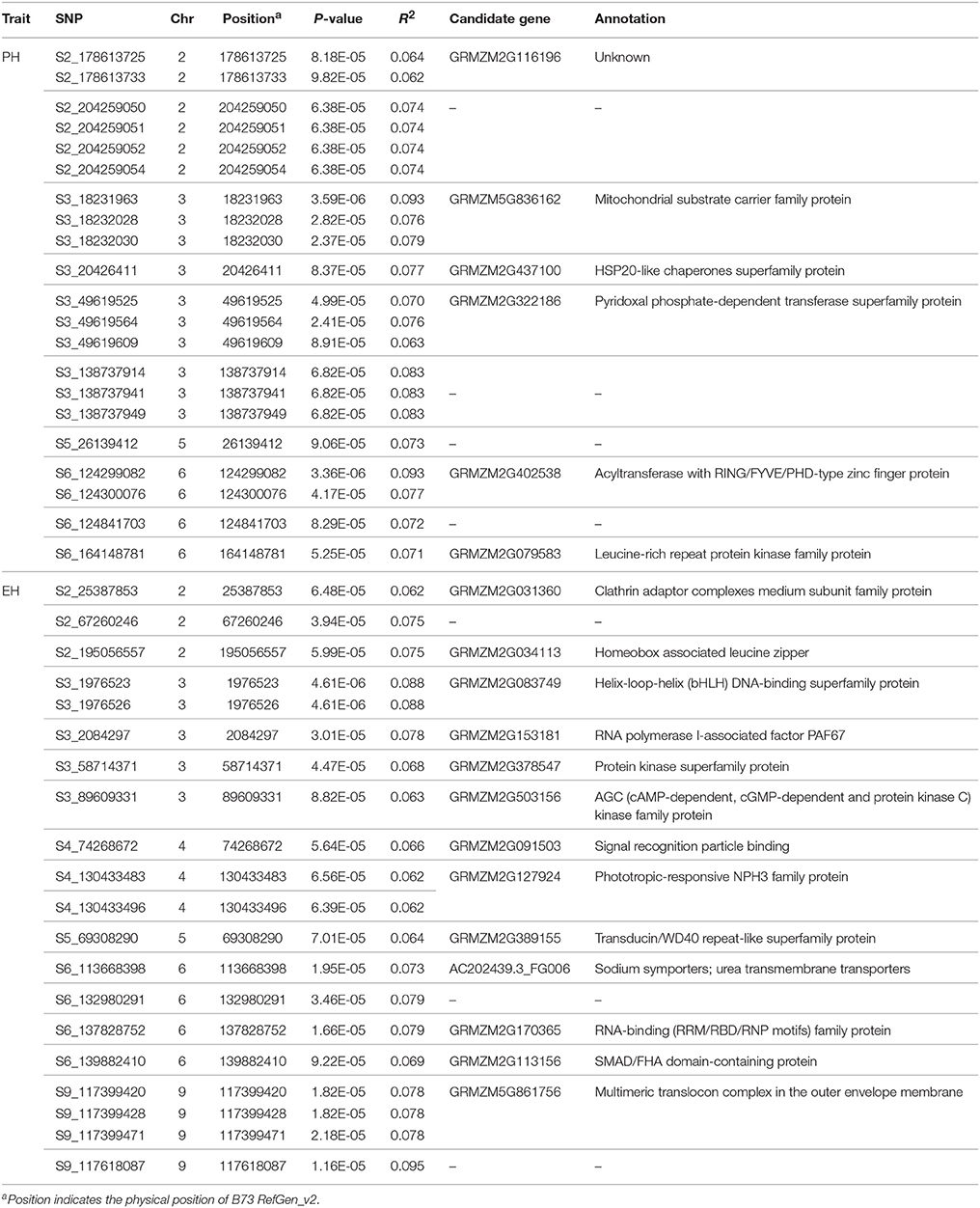
Table 3. The significant single nucleotide polymorphisms (SNPs) and their candidate genes associated with plant height (PH) and ear height (EH) identified in this study.
Based on the physical position of the significant SNPs in the B73 reference genome, 29 SNPs ware located in 19 unique candidate genes, whereas 12 SNPs in intergenic regions (Table 3). Candidate gene annotation showed that most of the candidate genes function belonged to the kinase family transport and signaling.
Meta-QTL Analysis for PH and EH
In order to identify the consistent loci for PH and EH, meta-analysis was conducted for both QTL mapping and GWAS results. Six common loci were identified on Chr. 1, Chr. 2, Chr. 3, Chr. 5, Chr. 6, and Chr. 9 (Figure 3). The QTL for PH in bin 1.05 was detected in both the two bi-parental populations, but no SNPs with p < 10 × 10−4 were identified in the GWAS population; the QTL for EH in bin 6.04 was detected in both the bi-parental populations, and one significant SNP was identified in the GWAS population; the QTL for EH and PH in bin 9.04 was detected in both the bi-parental populations, and one significant SNP was identified in the GWAS population. From meta-QTL analysis, six consistent loci were detected in more than two populations, suggesting that may have stable effects at different genetic backgrounds and environmental conditions.
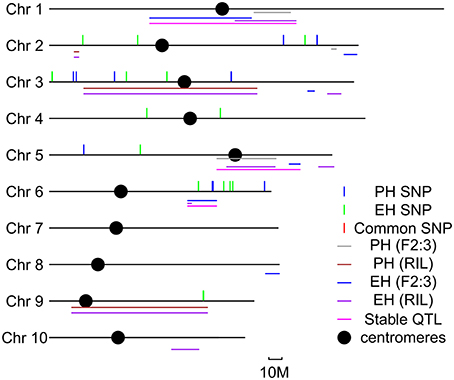
Figure 3. Meta- quantitative trait locus (QTL) analysis for QTL detected by two bi-parental populations and significant SNPs detected by genome-wide association study (GWAS) in this study. Colored lines represent different QTL or SNPs detected in this study.
QTL Verification
The qPH1-1 for PH in bin 1.05/06 share the same chromosome region of QTL qEH1-1 for EH, suggesting that they might be the same QTL, controlling both PH and EH. Furthermore, qPH1-1 had the highest LOD value in the F2:3 population. In order to fine mapping this QTL, NIL populations were developed by using marker assistance selection with flanking markers. The phenotype evaluation of NIL populations showed that positive homozygous allele (homozygous from donor parents) could increase PH by 18 cm and EH by 11 cm compared with the negative homozygous allele (Figures 4A,B), verifying QTL mapping results, and indicating that QTL in bin 1.05 could significantly increase PH and EH.
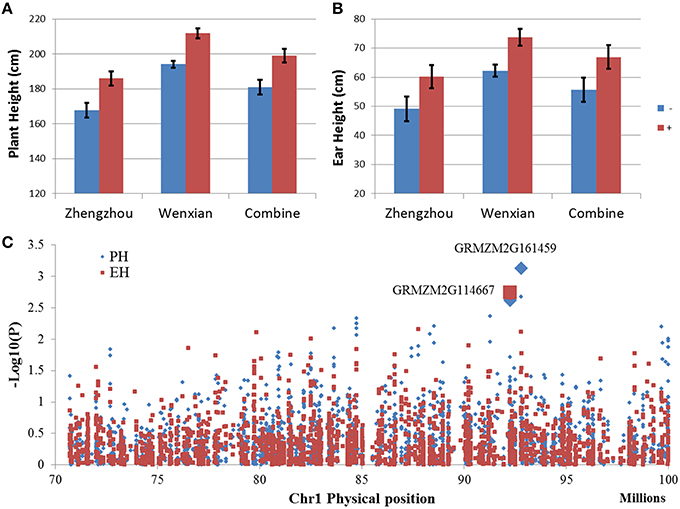
Figure 4. Quantitative trait locus (QTL) in bin 1.05 was validated using two opposite BC4F3 near isogenic line (NIL) populations and the genome-wide association study (GWAS) result in the QTL region. (A,B) Compare the two BC4F3 NIL populations for plant height (A) and ear height (B) under different environments. Blue bar indicates the target region of a homozygous allele from the recurrent parent. Red bar indicate the target region of a homozygous allele from the donor parent. (C) Manhattan plot for the GWAS result in the QTL region.
The Manhattan plot of GWAS (Figure 4C) showed that two markers with very low p-value, S1_92787084 with p = 7.4 × 10−4 for PH and p = 7.6 × 10−3 for EH and S1_92245779 with p = 2.4 × 10−3 for PH and p = 1.8 × 10−3 for EH, represented two candidate genes, GRMZM2G161459 and GRMZM2G114667, respectively. GRMZM2G161459 encodes the peptide transporter PTR2, which transports a wide spectrum of dipeptides and is involved in multiple pathways (Chiang et al., 2004; Xia et al., 2008), whereas GRMZM2G114667 encodes the C2H2 zinc finger family protein which is related to plant development and is required for the transcription of a number of genes coding proteins with stress-protective functions (Görner et al., 1998; Iuchi, 2001). Additionally, GRMZM2G114667 is highly expressed in the internode of the plant stem and may relate to the development of internodes. Compare with GRMZM2G161459, GRMZM2G114667 has higher possibility to be the candidate gene of qPH1-1, because of it is highly expressed in the internode, has a similar high effect on both PH and EH, and is highly related to plant development.
Discussion
Heredity of Height in Maize
Height is one of the most important agronomic traits and the most heritable trait in maize. However, the underlying genetic mechanisms of natural variation remain unclear. In the present study, the normal distribution and high heritability of PH and EH variation were detected in both the bi-parental and GWAS populations, suggesting that height in maize is a highly polygenic trait. The large number of markers with small effects that detected in GWAS confirmed that height in modern maize inbred lines is a polygenic trait controlled by multiple genes with low effect. These results were in agreement with previous studies (Peiffer et al., 2013, 2014).
Relationship between PH and EH
PH and EH are significantly correlated in maize breeding populations, thus some QTL for these traits are mapped in the same genomic regions due to pleiotropic effects (Tang et al., 2006). In the present study, QTL for both PH and EH were located in the same genomic region such as qPH1-1 for PH and qEH1-1 for EH (bin 1.05/06) in the F2:3 population as well as qPH3-1 for EH and qEH3-2 for EH (bin 3.04/05) in the RIL population. However, some QTL were only mapped for one of the two traits such as qEH6-2 for EH in the RIL population. These results suggested that the genetic mechanism of PH and EH may be similar, but not identical.
Genome Wide Association Study
GWAS is a powerful approach for exploring the molecular basis of phenotypic variations in plants, and the population is a foundation for GWAS. There were several GWAS populations have been reported for PH and EH research (Weng et al., 2011; Yang et al., 2011; Peiffer et al., 2014). The different population have different features, some populations used global diversity of lines which focus on global variation analysis (Yang et al., 2011; Peiffer et al., 2014), and others may only use region germplasm which focus on region variation analysis. For example, Weng et al. (2011) used a GWAS population contained Chinese elite lines identified a major QTL for height on chromosome 5, which played an important role in Chinese maize breeding programs. In the present study, the GWAS population mainly contained Chinese elite inbred lines and some tropical inbred lines from CIMMYT, it represented most of the diversity of Chinese and tropical germplasm which were the important germplasm in Chinese breeding programs. The Chinese and tropical germplasm were used for GWAS analysis, in order to reveal the genetic basis of height nature variation in these important germplasm in breeding programs. The further analysis on diversity comparison between present GWAS population and previous reported population was profitable to understand the different significant markers detected in different populations and may facilitate a better understanding of the underlying mechanism of height in maize.
GWAS has been widely used for functional gene discovery that has yielded a large number of associations between markers and various complex traits (Visscher et al., 2012; Li et al., 2016). However, some markers with high significant p-value may not be genuine whereas others with moderate or even low significant p-values can be considered genuine based on the biological trait (Panagiotou and Ioannidis, 2012). The selection of the most suitable threshold for GWAS using statistical approaches, such as the Bonferroni correction (Bland and Altman, 1995) or the false discovery rate (Benjamini and Hochberg, 1995), is important for the identification of genuine markers for target traits. A fixed cut-off value based on the number of markers and traits, such as p < 10−5 or p < 10−4, provides flexibility and is commonly used in GWAS. In the present study, no SNPs passed the Bonferroni correction threshold, probably because height is a complex traits in modern maize inbred lines controlled by multiple genes with low effect (Peiffer et al., 2014). Therefore, to identify the top SNP associated with PH and EH, we choose a fixed cut-off threshold of p < 10−4.
Comparison with QTL Identified in Previous Studies
Previous studies have shown that common QTL can be detected in different environments and populations (Beavis et al., 1991; Chen et al., 2012). In the present study, we detected nine QTL for PH and EH in the F2:3 families population and 12 QTL for PH and EH in the RIL populations. Of these, three “stable QTL” (detected in both bi-parental populations) were identified for PH and EH on Chr. 1, Chr. 5, and Chr. 6 and also reported in previous studies (Cai et al., 2012; Park et al., 2013; Sa et al., 2014; Wei et al., 2015). These results indicated that those three loci were stable QTL for PH and EH in modern maize inbred lines.
Candidate Genes for Height in Maize
The QTL for EH on Chr. 6 (bin 6.04/05) could explain 4% of the phenotypic variation in the F2:3 population and 10% of the phenotypic variation in the RIL population. However, no QTL was detected in this genomic region for PH in these two populations. The associated SNP S6_113668398 had the lowest p-value (p = 1.95 × 10−5) and explained 7.3% of the phenotypic variation in the GWAS population for EH. Candidate gene analysis showed that S6_113668398 was located in AC202439.3_FG006, which encodes a urea transmembrane transporter. The homologous genes in Arabidopsis (AtDUR3), and rice (LOC_Os10g42960.1) also encode transporters that play an important role in the uptake and use of urea (Kojima et al., 2007; Wang et al., 2008). Urea is the main source of nitrogen in modern crop production, because of its low cost and easy absorption by the plants, and provides 50% for the total weight of nitrogen fertilizer worldwide (Kojima et al., 2007; Wang et al., 2008). The efficient use of the nitrogen by the plant increases its height (Lafitte and Edmeades, 1994). Therefore, this gene could be considered an important candidate gene for maize height, and further functional verification might help to better understand the underlying genetic mechanism.
The QTL for PH and EH on Chr. 3 could explain 15 and 19% of the phenotypic variance for PH and EH, respectively, in the RIL population. An associated SNP with small p-value was detected for PH located in the annotated gene GRMZM2G322186. GRMZM2G322186 encodes a pyridoxal phosphate-dependent transferase and also has multiple catalytic functions in various organisms (Percudani and Peracchi, 2003). The protein level of GRMZM2G322186 was highly different between two parental lines BT-1 and N6 of the RIL population, and the N6 showed similar protein sequence with reference genome B73 (Figure S6). Expression analysis showed that GRMZM2G322186 has a high expression level in all the developmental stages of maize, indicating its importance in plant development (Winter et al., 2007; Sekhon et al., 2011). These results suggested that GRMZM2G322186 was the most possible candidate gene for the QTL of PH on Chr. 3.
Conclusions
In this study, two bi-parental populations and one GWAS population were used to identify and map QTL for PH and EH. A total of 21 QTL for PH and EH was detected by QTL mapping and 41 SNPs were identified by GWAS. A QTL on Chr. 1 was verified in NILs and indicated that a C2H2 zinc finger family protein might be the candidate gene for PH. One candidate gene on Chr. 6 (AC202439.3_FG006), encoding a urea transmembrane transporter, was considered as the candidate gene for EH. The candidate genes for other stable QTL were also discussed. These results confirmed that nature variation of maize height is strongly controlled by multiple genes with low effect and the QTL and candidate genes identified in this study could help to better understand the genetic basis of PH and EH in maize.
Author Contributions
JW, JC, JD, and XZ designed, led, and coordinated the overall study. XL, ZZ, YW, BZ, RW, JM, SW, and XZ perform the field experiment. ZZ, XL, and JC carried out the analysis. JC, ZZ, XL, and JW wrote the manuscript.
Funding
The work was supported by National Natural Science Foundation of China (Grant No. 31271307).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00833
Figure S1. Phenotypic distribution of plant height (PH) and ear height (EH) in the F2:3 population, the recombinant inbred line (RIL) population, and the genome-wide association study (GWAS) population. The density line is shown in black.
Figure S2. Quantitative trait loci (QTL) detected for plant height (PH) and ear height (EH) in the F2:3 population.
Figure S3. Quantitative trait loci (QTL) detected for plant height (PH) and ear height (EH) in the recombinant inbred line (RIL) population.
Figure S4. Principal components analysis (PCA) for genome-wide association study population. (A) Plot of the first two PCs separated for the heterotic group; (B) Eigenvalue of the first 15 PCs.
Figure S5. Quantile-quantile plot of genome-wide association study (GWAS) for plant height (PH) and ear height (EH).
Figure S6. Comparison of the protein sequence of the candidate gene GRMZM2G322186 between the two parental lines (BT-1 and N6) of the recombinant inbred line (RIL) population and the B73 reference genome.
File S1. R code for meta-quantitative trait locus (QTL) analysis.
References
Agrama, H. A. S., and Moussa, M. E. (1996). Mapping QTLs in breeding for drought tolerance in maize (Zea mays L.). Euphytica 91, 89–97. doi: 10.1007/BF00035278
Andersen, J. R., Schrag, T., Melchinger, A. E., Zein, I., and Lübberstedt, T. (2005). Validation of Dwarf8 polymorphisms associated with flowering time in elite European inbred lines of maize (Zea mays L.). Theor. Appl. Genet. 111, 206–217. doi: 10.1007/s00122-005-1996-6
Beavis, W. D., Grant, D., Albertsen, M., and Fincher, R. (1991). Quantitative trait loci for plant height in four maize populations and their associations with qualitative genetic loci. Theor. Appl. Genet. 83, 141–145. doi: 10.1007/BF00226242
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300.
Bland, J. M., and Altman, D. G. (1995). Multiple significance tests: the Bonferroni method. BMJ 310, 170–170.
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., and Ramdoss, Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Breseghello, F., and Sorrells, M. E. (2006). Association analysis as a strategy for improvement of quantitative traits in plants. Crop Sci. 46, 1323–1330. doi: 10.2135/cropsci2005.09-0305
Buckler, E. S., Holland, J. B., Bradbury, P. J., Acharya, C. B., Brown, P. J., Browne, C., et al. (2009). The genetic architecture of maize flowering time. Science 325, 714–718. doi: 10.1126/science.1174276
Cai, H., Chu, Q., Gu, R., Yuan, L., Liu, J., Zhang, X., et al. (2012). Identification of QTLs for plant height, ear height and grain yield in maize (Zea mays L.) in response to nitrogen and phosphorus supply. Plant Breed. 131, 502–510. doi: 10.1111/j.1439-0523.2012.01963.x
Cardwell, V. B. (1982). Fifty years of Minnesota corn production: sources of yield increase. Agron. J. 74, 984–990. doi: 10.2134/agronj1982.00021962007400060013x
Chen, J., Ding, J., Li, H., Li, Z., Sun, X., Li, J., et al. (2012). Detection and verification of quantitative trait loci for resistance to Fusarium ear rot in maize. Mol. Breed. 30, 1649–1656. doi: 10.1007/s11032-012-9748-1
Chen, J., Zhang, L., Liu, S., Li, Z., Huang, R., Li, Y., et al. (2016). The genetic basis of natural variation in kernel size and related traits using a four-way cross population in maize. PLoS ONE 11:e0153428. doi: 10.1371/journal.pone.0153428
Chen, Y., Chen, J., and Wu, J. (2014). Fine mapping of gene Rab1 for red glume collar in maize (In Chinese). Acta Agric. Boreali-Sinica 29, 7–12. doi: 10.7668/hbnxb.2014.02.002
Chia, J. M., Song, C., Bradbury, P. J., Costich, D., de Leon, N., Doebley, J., et al. (2012). Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 44, 803–807. doi: 10.1038/ng.2313
Chiang, C. S., Stacey, G., and Tsay, Y. F. (2004). Mechanisms and functional properties of two peptide transporters, AtPTR2 and fPTR2. J. Biol. Chem. 279, 30150–30157. doi: 10.1074/jbc.M405192200
Churchill, G. A., and Doerge, R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971.
Ding, J., Li, H., Wang, Y., Zhao, R., Zhang, X., Chen, J., et al. (2012). Fine mapping of Rscmv2, a major gene for resistance to sugarcane mosaic virus in maize. Mol. Breed. 30, 1593–1600. doi: 10.1007/s11032-012-9741-8
Ding, J., Ma, J., Zhang, C., Dong, H., Xi, Z., Xia, Z., et al. (2011). QTL mapping for test weight by using F2:3 population in maize. J. Genet. 90, 75–80. doi: 10.1007/s12041-011-0036-3
Ding, J., Zhang, L., Chen, J., Li, X., Li, Y., Cheng, H., et al. (2015). Genomic dissection of leaf angle in maize (Zea mays L.) using a four-way cross mapping population. PLoS ONE 10:e0141619. doi: 10.1371/journal.pone.0141619
Du, C., Cao, C., Cao, Q., Bi, M., Dong, Z., and Zhang, F. (2006). The breeding and application of maize hybrid Zhengdan 958. J. Maize Sci 14, 43–45. doi: 10.3969/j.issn.1005-0906.2006.06.011
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
Glaubitz, J. C., Casstevens, T. M., Lu, F., Harriman, J., Elshire, R. J., Sun, Q., et al. (2014). TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9:e90346. doi: 10.1371/journal.pone.0090346
Görner, W., Durchschlag, E., Martinez-Pastor, M. T., Estruch, F., Ammerer, G., Hamilton, B., et al. (1998). Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12, 586–597. doi: 10.1101/gad.12.4.586
Iuchi, S. (2001). Three classes of C2H2 zinc finger proteins. Cell. Mol. Life Sci. 58, 625–635. doi: 10.1007/PL00000885
Knapp, S. J., Stroup, W. W., and Ross, W. M. (1985). Exact confidence intervals for heritability on a progeny mean basis. Crop Sci. 25, 192–194. doi: 10.2135/cropsci1985.0011183X002500010046x
Kojima, S., Bohner, A., Gassert, B., Yuan, L., and von Wirén, N. (2007). AtDUR3 represents the major transporter for high-affinity urea transport across the plasma membrane of nitrogen-deficient Arabidopsis roots. Plant J. 52, 30–40. doi: 10.1111/j.1365-313X.2007.03223.x
Lafitte, H. R., and Edmeades, G. O. (1994). Improvement for tolerance to low soil nitrogen in tropical maize I. Selection criteria. F. Crop. Res. 39, 1–14. doi: 10.1016/0378-4290(94)90066-3
Lawit, S. J., Wych, H. M., Xu, D., Kundu, S., and Tomes, D. T. (2010). Maize DELLA proteins dwarf plant8 and dwarf plant9 as modulators of plant development. Plant Cell Physiol. 51, 1854–1868. doi: 10.1093/pcp/pcq153
Li, Z., Chen, J., Han, L., Wen, J., Chen, G., Li, H., et al. (2016). Association mapping resolving the major loci Scmv2 conferring resistance to sugarcane mosaic virus in maize. Eur. J. Plant Pathol. 145, 385–391. doi: 10.1007/s10658-015-0852-z
Li, Z. M., Ding, J. Q., Wang, R. X., Chen, J. F., Sun, X. D., Chen, W., et al. (2011). A new QTL for resistance to Fusarium ear rot in maize. J. Appl. Genet. 52, 403–406. doi: 10.1007/s13353-011-0054-0
Mahuku, G., Chen, J., Shrestha, R., Narro, L. A., Guerrero, K. V. O., Arcos, A. L., et al. (2016). Combined linkage and association mapping identifies a major QTL (qRtsc8-1), conferring tar spot complex resistance in maize. Theor. Appl. Genet. 129, 1217–1229. doi: 10.1007/s00122-016-2698-y
Multani, D. S., Briggs, S. P., Chamberlin, M. A., Blakeslee, J. J., Murphy, A. S., and Johal, G. S. (2003). Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302, 81–84. doi: 10.1126/science.1086072
Panagiotou, O. A., and Ioannidis, J. P. A. (2012). What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int. J. Epidemiol. 41, 273–286. doi: 10.1093/ije/dyr178
Park, K. J., Sa, K. J., Kim, B. W., Koh, H. J., and Lee, J. K. (2013). Genetic mapping and QTL analysis for yield and agronomic traits with an F2:3 population derived from a waxy corn × sweet corn cross. Genes Genomics 36, 179–189. doi: 10.1007/s13258-013-0157-6
Peiffer, J. A., Flint-Garcia, S. A., De Leon, N., McMullen, M. D., Kaeppler, S. M., and Buckler, E. S. (2013). The genetic architecture of maize stalk strength (I. De Smet, Ed.). PLoS ONE 8:e67066. doi: 10.1371/journal.pone.0067066
Peiffer, J. A., Romay, M. C., Gore, M. A., Flint-Garcia, S. A., Zhang, Z., Millard, M. J., et al. (2014). The genetic architecture of maize height. Genetics 196, 1337–1356. doi: 10.1534/genetics.113.159152
Percudani, R., and Peracchi, A. (2003). A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 4, 850–854. doi: 10.1038/sj.embor.embor914
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
R Core Team (2015). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Sa, K. J., Park, J. Y., Woo, S. Y., Ramekar, R. V., Jang, C. S., and Lee, J. K. (2014). Mapping of QTL traits in corn using a RIL population derived from a cross of dent corn × waxy corn. Genes Genomics 37, 1–14. doi: 10.1007/s13258-014-0223-8
Samayoa, L. F., Malvar, R. A., Olukolu, B. A., and Holland, J. B. Butrón, A. (2015). Genome-wide association study reveals a set of genes associated with resistance to the Mediterranean corn borer (Sesamia nonagrioides L.) in a maize diversity panel. BMC Plant Biol. 15:35. doi: 10.1186/s12870-014-0403-3
Sekhon, R. S., Lin, H., Childs, K. L., Hansey, C. N., Buell, C. R., de Leon, N., et al. (2011). Genome-wide atlas of transcription during maize development. Plant J. 66, 553–563. doi: 10.1111/j.1365-313X.2011.04527.x
Sibov, S. T., de Souza, C. L., Garcia, A. A. F., Silva, A. R., Garcia, A. F., Mangolin, C. A., et al. (2003). Molecular mapping in tropical maize (Zea mays L.) using microsatellite markers. 2. Quantitative trait loci (QTL) for grain yield, plant height, ear height and grain moisture. Hereditas 139, 107–115. doi: 10.1111/j.1601-5223.2003.01667.x
Tang, J. H., Teng, W. T., Yan, J. B., Ma, X. Q., Meng, Y. J., Dai, J. R., et al. (2006). Genetic dissection of plant height by molecular markers using a population of recombinant inbred lines in maize. Euphytica 155, 117–124. doi: 10.1007/s10681-006-9312-3
Teng, F., Zhai, L., Liu, R., Bai, W., Wang, L., Huo, D., et al. (2013). ZmGA3ox2, a candidate gene for a major QTL, qPH3.1, for plant height in maize. Plant J. 73, 405–416. doi: 10.1111/tpj.12038
Thornsberry, J. M., Goodman, M. M., Doebley, J., Kresovich, S., Nielsen, D., and Buckler, E. S. (2001). Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28, 286–289. doi: 10.1038/90135
Tian, F., Bradbury, P. J., Brown, P. J., Hung, H., Sun, Q., Flint-Garcia, S., et al. (2011). Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43, 159–162. doi: 10.1038/ng.746
Visscher, P. M., Brown, M. A., McCarthy, M. I., and Yang, J. (2012). Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24. doi: 10.1016/j.ajhg.2011.11.029
Wang, W. H., Köhler, B., Cao, F. Q., and Liu, L. H. (2008). Molecular and physiological aspects of urea transport in higher plants. Plant Sci. 175, 467–477. doi: 10.1016/j.plantsci.2008.05.018
Wang, Y., and Li, J. (2008). Molecular basis of plant architecture. Annu. Rev. Plant Biol. 59, 253–279. doi: 10.1146/annurev.arplant.59.032607.092902
Wang, Y., Yao, J., Zhang, Z., and Zheng, Y. (2006). The comparative analysis based on maize integrated QTL map and meta-analysis of plant height QTLs. Chinese Sci. Bull. 51, 2219–2230. doi: 10.1007/s11434-006-2119-8
Wei, X., Wang, B., Peng, Q., Wei, F., Mao, K., Zhang, X., et al. (2015). Heterotic loci for various morphological traits of maize detected using a single segment substitution lines test-cross population. Mol. Breed. 35, 1–13. doi: 10.1007/s11032-015-0287-4
Weng, J., Xie, C., Hao, Z., Wang, J., Liu, C., Li, M., et al. (2011). Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) inbred lines. PLoS ONE 6:e29229. doi: 10.1371/journal.pone.0029229
Wilson, L. M., Whitt, S. R., Ibáñez, A. M., Rocheford, T. R., Goodman, M. M., and Buckler, E. S. (2004). Dissection of maize kernel composition and starch production by candidate gene association. Plant Cell 16, 2719–2733. doi: 10.1105/tpc.104.025700
Winkler, R. G., and Helentjaris, T. (1995). The maize Dwarf3 gene encodes a cytochrome P450-mediated early step in Gibberellin biosynthesis. Plant Cell 7, 1307–1317. doi: 10.1105/tpc.7.8.1307
Winter, D., Vinegar, B., Nahal, H., Ammar, R., Wilson, G. V., and Provart, N. J. (2007). An “Electronic Fluorescent Pictograph” browser for exploring and analyzing large-scale biological data sets. PLoS ONE 2:e718. doi: 10.1371/journal.pone.0000718
Xia, Z., Turner, G. C., Hwang, C.-S., Byrd, C., and Varshavsky, A. (2008). Amino acids induce peptide uptake via accelerated degradation of CUP9, the transcriptional repressor of the PTR2 peptide transporter. J. Biol. Chem. 283, 28958–28968. doi: 10.1074/jbc.M803980200
Xing, A., Gao, Y., Ye, L., Zhang, W., Cai, L., Ching, A., et al. (2015). A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize. J. Exp. Bot. 66, 3791–3802. doi: 10.1093/jxb/erv182
Xu, D., Cai, Y., Lv, X., Dai, G., Wang, G., Wang, J., et al. (2009). QTL mapping for plant-tape traits in maize. J. Maize Sci. 17, 27–31.
Yan, J., Warburton, M., and Crouch, J. (2011). Association mapping for enhancing maize (L.) genetic improvement. Crop Sci. 51, 433–449. doi: 10.2135/cropsci2010.04.0233
Yang, N., Lu, Y., Yang, X., Huang, J., Zhou, Y., Ali, F., et al. (2014). Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet. 10:e1004573. doi: 10.1371/journal.pgen.1004573
Yang, X., Gao, S., Xu, S., Zhang, Z., Prasanna, B. M., Li, L., et al. (2011). Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breed. 28, 511–526. doi: 10.1007/s11032-010-9500-7
Yu, J., and Buckler, E. S. (2006). Genetic association mapping and genome organization of maize. Plant Biotechnol. 17, 155–160. doi: 10.1016/j.copbio.2006.02.003
Yu, J., Pressoir, G., Briggs, W. H., Vroh Bi, I., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, Q., Zhang, L., Evers, J., van der Werf, W., Zhang, W., and Duan, L. (2014). Maize yield and quality in response to plant density and application of a novel plant growth regulator. F. Crop. Res. 164, 82–89. doi: 10.1016/j.fcr.2014.06.006
Zila, C. T., Ogut, F., Romay, M. C., Gardner, C. A., Buckler, E. S., and Holland, J. B. (2014). Genome-wide association study of Fusarium ear rot disease in the U.S.A. maize inbred line collection. BMC Plant Biol. 14:372. doi: 10.1186/s12870-014-0372-6
Zwonitzer, J. C. J., Coles, N. D. N., Krakowsky, M. D., Arellano, C., Holland, J. B., McMullen, M. D., et al. (2010). Mapping resistance quantitative trait Loci for three foliar diseases in a maize recombinant inbred line population-evidence for multiple disease resistance? Phytopathology 100, 72–79. doi: 10.1094/PHYTO-100-1-0072
Keywords: plant height, ear height, association mapping, quantitative trait locus, candidate gene, maize
Citation: Li X, Zhou Z, Ding J, Wu Y, Zhou B, Wang R, Ma J, Wang S, Zhang X, Xia Z, Chen J and Wu J (2016) Combined Linkage and Association Mapping Reveals QTL and Candidate Genes for Plant and Ear Height in Maize. Front. Plant Sci. 7:833. doi: 10.3389/fpls.2016.00833
Received: 16 March 2016; Accepted: 27 May 2016;
Published: 15 June 2016.
Edited by:
Xiaowu Wang, Institute of Vegetables and Flowers, ChinaReviewed by:
Chunyi Zhang, Chinese Academy of Agricultural Sciences, ChinaCai Lin Lei, Chinese Academy of Agricultural Sciences, China
Copyright © 2016 Li, Zhou, Ding, Wu, Zhou, Wang, Ma, Wang, Zhang, Xia, Chen and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiafa Chen, Y2hlbmppYWZhMjAwNUAxNjMuY29tOw==
Jianyu Wu, d3VqaWFueXU0MEAxMjYuY29t
†These authors have contributed equally to this work.