 Jun Li
Jun Li Patrick X. Zhao
Patrick X. Zhao- Bioinformatics Lab, Plant Biology Division, The Samuel Roberts Noble Foundation, Ardmore, OK, USA
Identification of functional modules/sub-networks in large-scale biological networks is one of the important research challenges in current bioinformatics and systems biology. Approaches have been developed to identify functional modules in single-class biological networks; however, methods for systematically and interactively mining multiple classes of heterogeneous biological networks are lacking. In this paper, we present a novel algorithm (called mPageRank) that utilizes the Multiplex PageRank approach to mine functional modules from two classes of biological networks. We demonstrate the capabilities of our approach by successfully mining functional biological modules through integrating expression-based gene-gene association networks and protein-protein interaction networks. We first compared the performance of our method with that of other methods using simulated data. We then applied our method to identify the cell division cycle related functional module and plant signaling defense-related functional module in the model plant Arabidopsis thaliana. Our results demonstrated that the mPageRank method is effective for mining sub-networks in both expression-based gene-gene association networks and protein-protein interaction networks, and has the potential to be adapted for the discovery of functional modules/sub-networks in other heterogeneous biological networks. The mPageRank executable program, source code, the datasets and results of the presented two case studies are publicly and freely available at http://plantgrn.noble.org/MPageRank/.
Introduction
The advent of systems biology, which often integrates microarray- or RNA-Seq-based transcriptomics, proteomics, and metabolomics analyses, has made this an opportune time to determine how biological processes and complex phenotypes (also called traits) are regulated in living cells. In the post-genomics era, the development of high-throughput “omics” technologies has generated vast amounts of mRNA, protein, and metabolite profiles for many eukaryotic species, and much of this “big data” has been made publicly accessible through data repositories (Pruitt and Maglott, 2001; Parkinson et al., 2005; Barrett et al., 2007; Brandao et al., 2009). “Big data” has the potential to provide unprecedented insights into the various biological processes, including trait-regulation, leading to the discovery of vast amounts of novel biological information.
Bioinformatics and systems biology approaches, which include biological network analyses, have great potential to elucidate the fundamental mechanisms that govern dynamic cell organization and function. In the past decade, a number of experimental (Rual et al., 2005; Arabidopsis Interactome Mapping Consortium, 2011) and computational (Ma et al., 2007; Brandao et al., 2009; Stark et al., 2011; Li et al., 2013, 2014) approaches have been developed to generate and predict protein-protein interaction networks, transcriptional regulatory networks, gene-gene co-expression networks, and metabolic networks in humans, animals and plants. The current challenge, however, is how to effectively identify significant functional modules or sub-networks in these extensive global networks. Complex biological processes in livings cell are carried out through interactions between multiple functional modules at various levels (Barabasi and Oltvai, 2004; Cancer Genome Atlas Research Network, 2008), and members of the same functional module are often more densely connected than those across functional modules (Hartwell et al., 1999). Based on these observations, various clustering approaches, including hierarchical clustering, k-mean clustering, and Markov clustering, have been applied to identify function-specific modules in single-class biological networks (Eisen et al., 1998; Wu, 2008; Shih and Parthasarathy, 2012).
Due to the dynamic characteristics of living cells, networks generated from a single data source are usually limited, and can only reveal partial, static snapshots of the cell. Additionally, current technologies are often plagued by noise, leading to biases and low confidence in networks constructed by these technologies. To provide an accurate and comprehensive understanding of biological systems, the integration of different types of biological data has become an important and popular strategy. A number of approaches (Ideker et al., 2002; Chuang et al., 2007; Dittrich et al., 2008; Inoue et al., 2010) have been developed to facilitate the identification of context-dependent active functional modules/sub-networks by integrating protein-protein interaction (PPI) networks with gene expression data. On the basis of the topology structure of a PPI network, most of these methods first devise a function to score interacting edges or nodes in PPI networks while taking into account the gene expression data, and then employ optimization algorithms or graphical clustering algorithms to search the high-scoring modules/sub-networks (D'haeseleer et al., 2000; Enright et al., 2002; Langfelder and Horvath, 2008; Inoue et al., 2010). These methods have achieved some success in identifying biologically significant sub-networks, but have noticeable limitations. First, high confidence PPI networks are far from complete, especially in plants, and significant proportions of biologically significant genes/proteins may be overlooked in the PPI networks. For example, the PPI data included in the database of Arabidopsis thaliana protein interaction networks (Brandao et al., 2009) only include 201,699 interactions among 15,426 genes, while the genome of the organism encodes more than 20,000 genes. Second, cellular processes are the result of elegant coordination among gene regulation, signal transduction, and protein-protein interactions, etc. Some key proteins or genes that mediate or control these interactions among other cellular processes may be overlooked when utilizing a single network, even when the data are integrated with other types of biological data. Therefore, systematic and comprehensive mining of functional modules in heterogeneous biological networks, such as protein-protein interaction/protein-DNA interactions and gene-gene expression network, etc., is a better approach for understanding the complex mechanisms that govern the dynamic organization and function of living cells.
The Multiplex PageRank method (Halu et al., 2013), which is an extension of the widely used PageRank method (Langville and Meyer, 2006), leverages network transitivity to rank nodes in heterogeneous social networks. It simulates “bias random walking” on multiple networks to rank nodes in multiplex systems, in which the importance of a node in one network is affected by the importance that node gained in another network. The more important a node is in network A, the more important that node, and the nodes connected to it, in network B. The Multiplex PageRank method also considers the importance of the node's in-neighbors in the corresponding A/B network. The method has been shown to be highly accurate in community discovery in social networks (De Domenico et al., 2015). Recent studies (Girvan and Newman, 2002; Zhu et al., 2007; Vashisht et al., 2013) indicate significant similarities between biological networks and social networks in term of network properties, including the small-world property, power-law degree distribution, and network transitivity.
Here we present a novel method, named mPageRank, which adopts the Multiplex PageRank strategy to mine functional modules in heterogonous biological networks, including expression-based gene-gene association networks and protein-protein interaction networks. We demonstrated the effectiveness of our method by benchmarking against other existing methodologies, using both simulated data and experimental data. Compared with other methods and tools such as the jActiveModule method (Ideker et al., 2002), kwalks method (Faust et al., 2010), and DMSP method (Maraziotis et al., 2007), ours was the most accurate. We further demonstrated the effectiveness of our method by identifying cell division cycle related and plant signaling defense-related functional modules/sub-networks in Arabidopsis thaliana. These results suggest that the mPageRank is a promising approach for mining functional modules in heterogeneous biological networks.
Materials and Methods
Analysis Procedures
We developed a novel Multiplex PageRank-based algorithm to extract biologically significant functional modules in heterogonous biological networks, including gene expression-based gene-gene association networks and protein-protein interaction networks. Starting from seed genes, we iteratively simulated biased random walks on a gene-gene association network and a protein-protein interaction network to prioritize those genes related to the seed genes. We then extract the sub-networks based on the rank scores of all the genes in both networks. The analysis flow is illustrated in the Figure 1.
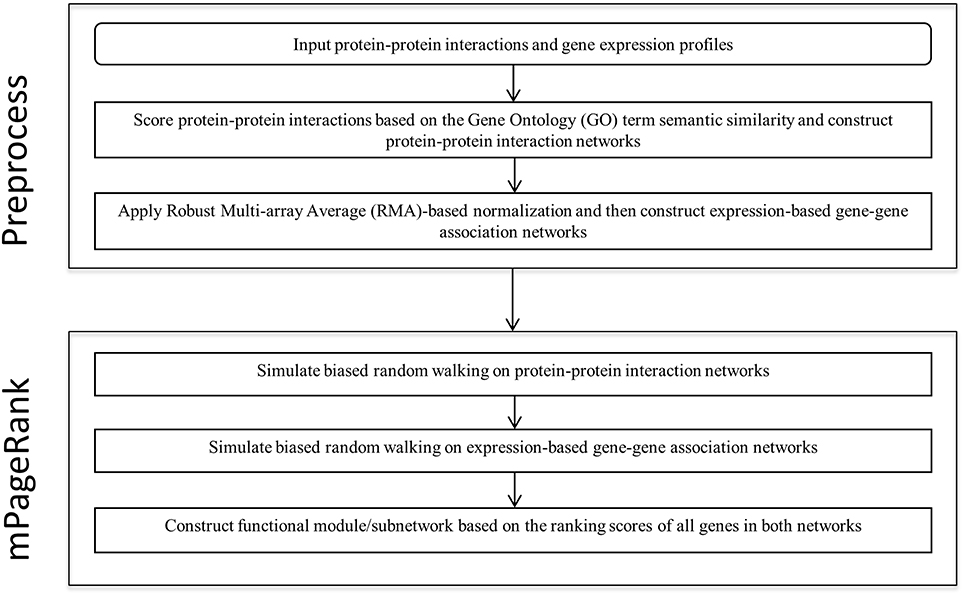
Figure 1. The key steps in the functional module/sub-network identification analysis.
Construction of the Expression-Based Gene-Gene Association Networks
To construct gene expression-based gene-gene association networks for Arabidopsis thaliana, a total of 4162 microarray hybridization-based gene expression profiles were downloaded from the ArrayExpress data repository (Parkinson et al., 2005). The dataset was firstly normalized using the Robust Multi-array Averaging (RMA) method (Irizarry et al., 2003). The gene association networks, including 22,497 nodes (genes) and 2,106,763 edges (links between genes), were then reconstructed by our DeGNServer (Li et al., 2013), which is a powerful high performance web server developed for large-scale gene association network (GAN) construction and analysis. Reconstruction was performed using the Spearman-based Context Likelihood of Relatedness (CLR) with the z-score threshold set at 4.3.
Scoring Arabidopsis thaliana Protein-Protein Interactions Based on the Gene Ontology (GO) Term Semantic Similarity
The Arabidopsis protein-protein interaction dataset was downloaded from the AtPIN database-Arabidopsis thaliana protein interaction network (Brandao et al., 2009). The Arabidopsis thaliana gene ontology (GO) annotations were downloaded from https://www.arabidopsis.org/portals/genAnnotation/functional_annotation/go.jsp. The protein-protein binary interactions were scored with Resnik implemented in GoSemSim (Yu et al., 2010), which is an R package for weighing binary protein-protein interactions by measuring the semantic similarity among GO terms of gene products (GO-term based protein annotations).
mPageRank Method
Here, we define the expression-based gene-gene association network as network A and the protein-protein interaction network as network B.
We first constructed the transition matrix, WA, based on the gene-gene correlation values for network A, and the transition matrix, WB, based on the GO term semantic similarity scores for the proteins in the network. The transition probability from gene i to gene j was calculated using the following equation (Equation 1),
where c(i, j) is the correlation value or similarity score between gene i and j; if there is no interaction between gene i and j, then c(i, j) is zero. n is the number of genes that interact with the gene i.
On network A, we performed multiple iterations of biased random walks to rank genes using the following equation (Equation 2),
where p(i) is set to 1/k for each seed gene, with k indicating the number of seed genes, and where is set to 1/m, with m indicating the total number of genes in the network. Here, known genes in a biological pathway or a functional module may be used as the seed genes. p(i) specifies preference for node i. αA denotes the probability of “returning” to one of the seed genes.
The error tolerance was defined as the Euclidean distance between the current iteration and the previous iteration: Error tolerance = Distance (, ), where n is the number of iterations.
In our experiments, an error tolerance threshold at 1e-10 was empirically tested sufficient to reach the convergence. Therefore, the error tolerance threshold is set at 1e-10 by default in the software. However, a user defined error tolerance threshold is also allowed. The output rank score for each gene i was reflected as the node importance score, denoted .
On network B, we performed the biased random walk iteration process to rank genes as applied to network A, using a similar equation (Equation 3, below). As with network A, iterations were performed until the error tolerance was less than 1e-10.
As in Equation 2, p(i) was set to 1/k for each seed gene, with k indicating the number of seed genes, and where is set to 1/m, with m indicating the total number of genes in the network. αA and αB were set to the value of 0.85 based on an empirical value (Halu et al., 2013). The output rank score for each gene i was reflected as the node importance score, denoted .
Construction of Functional Module and P-value Estimation
Based on a user-specified output number of top-ranking nodes, n, our algorithm first grouped the top ranked nodes (in both networks) associated with the seed genes by alternative random walks into a functional module. Then, the interactions with nodes included in the functional module were added to the module as edges to denote the interactions in the gene expression-based association networks or protein-protein interaction networks. Finally, to evaluate the significance of the functional module, one million of potential modules with same number of nodes were randomly sampled, and a one-sample Z-test was then applied to calculate the Z-score using the following equation (Equation 4):
The Z-score of the identified functional module was converted to a p-value for downstream analyses.
Gene Set Enrichment Analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed using the online tool agriGo (Du et al., 2010) (http://bioinfo.cau.edu.cn/agriGO/). The significance of the GO term enrichment was determined using a Fisher's exact test with the entire Arabidopsis thaliana genome as the background reference. A Yekutieli correction was used to control for false positives.
Results and Discussion
Performance Benchmark Analysis Using Simulation Data
We benchmarked the performance of our Multiplex PageRank-based method by comparing its performance against that of jActiveModule (Ideker et al., 2002), kwalks (Faust et al., 2010), and the method originally described by Ioannis et al. (Cancer Genome Atlas Research Network, 2008). The jActiveModule is a tool for module extraction from protein-protein interaction network through combining with expression profiles. In jActiveModule, the similarity for each interaction is measured with the expression value. The other two methods were developed based on the similar idea, but with different graph search strategies. These methods may have their advantages to identify those modules with high consistence between the expression profiles and PPI interactions network. However, such type of methods does not utilize the topological relationships inferred from expression profiles. In contrast, our mPageRank method effectively utilizes the topological relationship information via converting the expression profiles into gene-gene association networks, then walking on the two graphs (PPI networks and gene-gene association networks) to identify functional modules. Therefore, besides those interactions are highly consistent between two networks could be included, those interactions that are highly confident in a single network could also be included in the extracted subnetworks or functional modules. To generate simulation data for performance benchmark analysis, a yeast cell cycle pathway related module that included 113 genes and 369 experimentally validated interactions were downloaded from KEGG pathway database (Kanehisa et al., 2014) and used as the ground truth module for our benchmark performance analysis. We also extracted yeast cell cycle specific protein-protein interaction networks from the BioGRID database (Stark et al., 2011) Combining the two datasets, we created a network with 575 total genes and 803 total interactions, 685 of which were identified from protein-protein interactions. Due to the lack of experimentally validated expression datasets that could be applied to accurately extract all known interactions from this network, we utilized the widely used software, SynTren Van Den Bulcke et al., 2006, to generate a series of simulated expression datasets for performance benchmark analyses. We define as true positive (TP) a non-seed node that is present in both the reference pathway and in the inferred module, while a false positive (FP) was defined as a non-seed node found in the inferred module but not in the reference pathway. We defined a false negative (FN) as a non-seed node present in the reference pathway but not in the inferred module, while a true negative (TN) was defined as a node absent from both the inferred module and reference pathway. Using the true/false positive and true/false negative rates, the prediction accuracy of each method was evaluated by plotting Precision-Recall (PR) curves, where the Precision was calculated as TP/(TP + FP) and the Recall was calculated as TP/(TP + FN). A series of functional modules were chosen with the variable sizes of the top-ranking nodes. The Precision and Recall value were calculated based on the confidence values of these series of functional modules. The average F-scores were calculated to estimate the accuracy (Supplemental Table 1). As illustrated in Figure 2, our method achieved the best precision under different recall rates, followed by the jActiveModule (Ideker et al., 2002) method, the Ioannis et al. method, and the kwalks method (Faust et al., 2010).
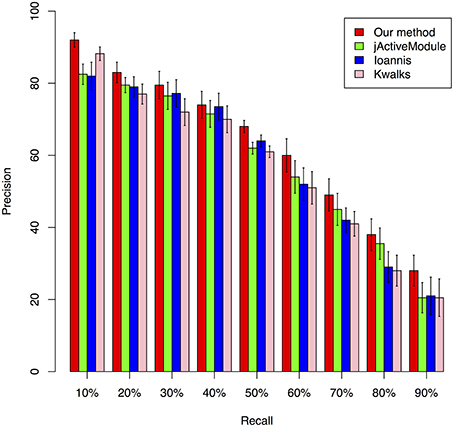
Figure 2. Comparison of the precision of four different methods under different recall rates, ranging from 10 to 90%.
Identification of a Cell Cycle Core Functional Module
To further demonstrate the performance of our proposed method, we applied our method to a group of Arabidopsis thaliana specific datasets including gene expression-based association networks and protein-protein interaction networks to identify a cell cycle core functional module. The protein-protein interaction network was the same one as described in the previous section. An Arabidopsis thaliana gene expression-based association network comprised of 15,285 genes and 2,587,457 interactions was downloaded from http://plantgrn.noble.org/GPLEXUS/Result.jsp?sessionid=Arabidopsis.
Applying our method, we successfully baited a cell division cycle related core functional module that consisted of 70 genes and 403 interactions using the KRP2 and CDC2 genes as seed genes. The functions of the proteins encoded by these 70 genes, most of which are cell division cycle related (see the list in Supplemental Table 2). Among these 70 genes, 21 genes were identified as CDK genes or core cell cycle genes. Three E2F transcription factors were also included in the module. Other genes such as WEE1, TON1, PAS2, AUR2, and SIM, which have been validated to encode proteins with cell division cycle related functions (Bach et al., 2008; Gutierrez, 2009; Cook et al., 2013), were also included in the module.
A Gene Ontology Set Enrichment Analysis on the module (Supplemental Table 3) further demonstrated that the module we produced was composed of genes and proteins involved in the cell division cycle. Among the 70 genes in our module, 46 genes were annotated with cell cycle related gene ontology categories (under GO term: GO: 0007049) and an additional 35 genes were annotated with cell cycle regulation-related gene ontology categories (under GO term: GO:0051726). A comparison of the top 12 significantly enriched gene ontology categories in our module compared to the whole-genome GO categories is shown in Figure 3.
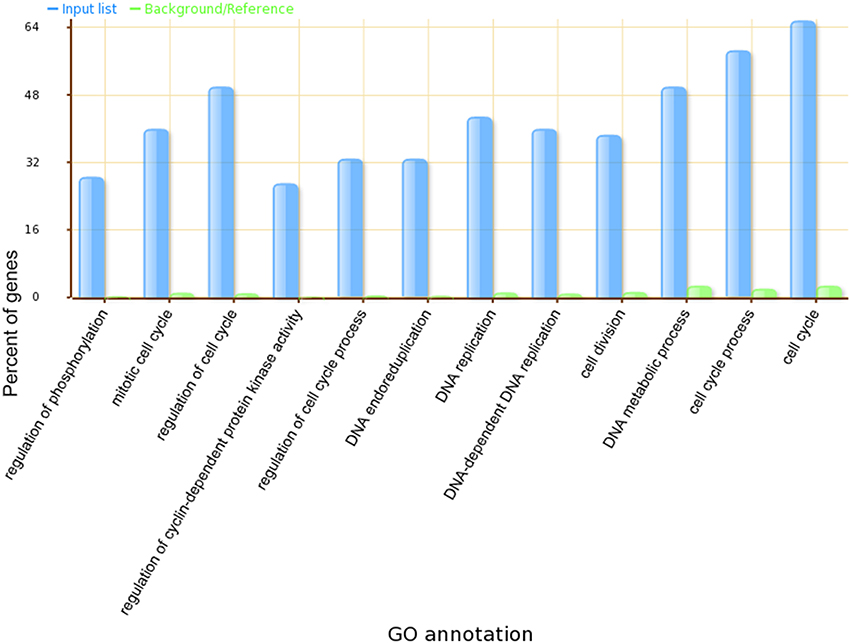
Figure 3. The top 12 significantly enriched GO categories in our identified Arabidopsis thaliana cell cycle core functional module.
We then further examined the sub-network around the core genes in the module (Figures 4, 5). Among the 403 total interactions in the module, 276 were identified from the gene expression-based network and 101 from the protein-protein interactions network. Only 26 interactions were present in both networks (Figure 4A). This is not unexpected, as protein-protein interaction networks only reveal physical interactions, in contrast to expression-based gene-gene association networks, which not only reflect direct physical interaction, but also reveal potential cell regulatory relationships.
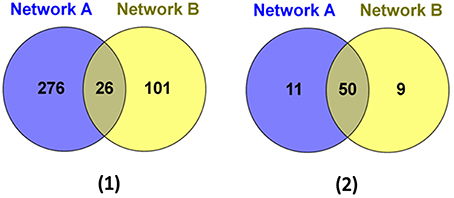
Figure 4. The genes and interactions that were identified from the Network A: expression-based gene-gene association network and the Network B: protein-protein interaction network. (1) The Venn Diagram shows the numbers of unique and shared genes in network A and B. (2) The Venn Diagram shows the numbers of unique and shared interactions in network A and B.
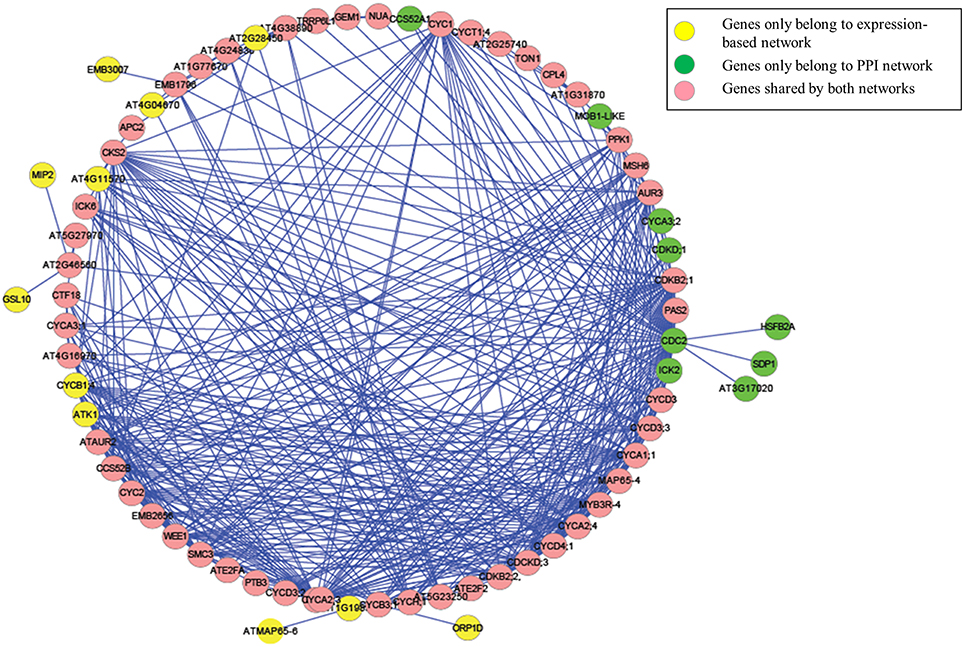
Figure 5. The genes and interactions included in our identified Arabidopsis thaliana cell cycle core functional module.
Although, the number of shared protein-protein interactions was minimal, most genes were present in both networks. As illustrated in Figure 4B, 61 genes were identified from the expression-based network and 59 were identified from the protein-protein network, while 50 genes were present in both networks. Importantly, we demonstrate that those genes existing in only one network would be missed using other methods (Ideker et al., 2002; Cancer Genome Atlas Research Network, 2008; Faust et al., 2010), which identify modules from a single network. These results suggest that mining the heterogonous datasets captured by multiple complementary technologies could provide greater insights in biology, leading to better reflection of the whole cellular activities.
We further examined the genes that were unique to each network. Among them, 11 (CYCB1;4, EMB3007, GSL10, ATK1, AT4G04670, ORP1D, AT1G19835, ATMAP65-6, AT4G11570, AT2G28450, and MIP2) could only be identified from the gene expression-based network, while 9 (CYCA3;2, CCS52A1, AT3G17020, SDP1, CDC2, CDKD;1, ICK2, HSFB2A, and MOB1-LIKE) could only be identified from protein-protein interaction network. Analyzing the functional descriptions of these genes indicates that all of these genes have been experimentally validated as cell cycle related genes. For example, CYCB1;4 has been validated as the core cell cycle gene (Vandepoele et al., 2002), while ATMAP65-6 encodes a protein that induces microtubules to form a mesh-like network (Mao et al., 2005). GSL10, a member of the glucan synthase-like (GSL) family, which is involved in synthesis of the cell-wall component callose at specialized locations, governs the entry of micropores into mitosis, and impaired GSL10 function leads to a perturbation in micropore division symmetry (Toller et al., 2008). Additionally, ATK1 plays a crucial role in spindle morphogenesis during meiosis in male Arabidopsis plants (Chen et al., 2002).
In the extracted module, among those identified interactions that could be experimentally validated, many of them only exist in one network. For example, the interaction between the cyclin-dependent protein kinase regulators CYCA1;1 (AT1G44110) and CYC1 (AT4G37490) was described in (De Almeida Engler et al., 2012), and the interaction between CCS52B and CDKB2;2 (AT1G20930) was reported in Van Leene et al. (2010). Nevertheless, these interactions were only present in the gene expression-based association network but not in the protein-protein interaction network. On the other hand, the experimentally validated interactions between CDC2 and ATE2FA (Boruc et al., 2010), between CDC2 and TON1 (Van Leene et al., 2007), and between ICK6 and CKS2 (Van Leene et al., 2010) were only present in the protein-protein interaction network. Therefore, by performing biased random walks on both networks, we successfully identified an integrated cell division cycle functional module.
Identification of an Immune-Related Functional Module Involved in Plant Defense Signaling
To further demonstrate the performance of our proposed method, we next applied our method to identify genes and sub-networks involved in the plant defense signaling. Plants have evolved highly sophisticated immune systems, which are critically important for plant survival in the face of life-threatening pathogens, pests, and harsh environmental challenges. Understanding the plant defense signaling pathway will help biologists to better understand the biotic and abiotic stress response mechanisms in plants. Using biased random walks with four plant seed genes (FLS2, MPK4, WRKY40, and WRKY33), we constructed a highly confident (p = 0.008) functional core module related to the MAMP signaling pathway (MAMP), the primary means by which plants detect and respond to pathogens.
Functional annotation of the genes present in this pathway revealed a number of biological features related to MAMP signaling pathway (see Supplemental Table 4 for a list of the functions of all genes in the module), providing strong evidence that the module is highly related to the plant defense signal transduction pathway. The major gene products included mitogen-activated protein (MAP) kinases, WRKY transcription factors, and vesicle-trafficking proteins, all of which have been reportedly involved in the plant defense signaling functions. Indeed, several MAP kinases involved in the plant defense response have been experimentally validated (Ligterink et al., 1997; Nuhse et al., 2000; Lee et al., 2001), and the WRKY transcription factors are involved in several immune responses in plants, including microbe-associated molecular pattern-triggered (MAMP-triggered) immunity, pathogen-associated molecular pattern-triggered (PAMP-triggered) immunity, effector-triggered immunity (ETI), and systemic acquired resistance (Eulgem et al., 2000; Xu et al., 2006; Encinas-Villarejo et al., 2009; Pandey and Somssich, 2009). We also identified BIK1, a positive regulator of plant immunity, and in our module, BIK1 functioned as a negative regulator of plant hormone brassinosteroid (BR)-mediated growth through association with the BR receptor BRI1. This dual association may contribute to the inverse functions of BIK1 previously reported in plant immunity and development (Lin et al., 2013).
GSEA (Supplemental Table 5) further validated that core genes in our module are highly related to plant defense signaling, as the GO terms defense response to bacteria, signaling transduction, cellular response to salicylic acid stimulus, and regulation of immune response were overrepresented. Almost 80 percent of genes in the module have the GO term referring to response to stimulus. A comparison of the top 12 significantly enriched gene ontology categories in our module compared to the whole-genome GO categories is shown in Figure 6.
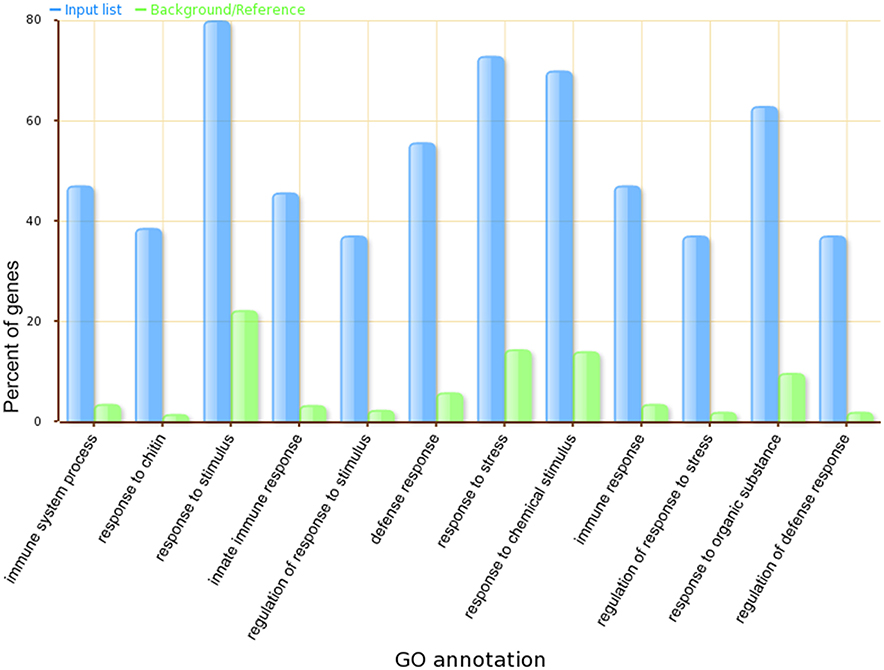
Figure 6. The top 12 significantly enriched GO categories in our Arabidopsis thaliana defense response related functional module.
We further analyzed the sub-network around the core genes in the module and found several interactions that have been experimentally validated. Again, it demonstrates the effectiveness of our method. For example, it has been demonstrated that in vivo, FLS2 and BIK1 form a complex in a specific ligand-dependent manner, and that this interaction plays a functional role PRR-dependent signaling, which initiated innate immunity (Chinchilla et al., 2007). Qiu et al. (2008) demonstrated that WRKY33 can bind in a complex with MAP kinase 4 (MPK4) and MKS1, playing a role in the immune response to Pseudomonas syringae. While the interaction between WRKY33 and MPK4 could be revealed in the PPI network, the interaction between WRKY33 and MKS1 was identified in the expression-based network only. Using our method to integrate the two networks, both of these interactions were successfully identified. However, only the interaction between WRKY33 and MPK4 could be included by other three methods because they only considered those interactions in the PPI network. As with the cell division cycle functional module, some interactions were only identified in the expression-based gene-gene association network or the protein-protein interaction network, while some were identified in both networks. Figure 7 illustrates the core interactions of the functional core module, with experimentally validated interactions highlighted in red. Again, only part of those interactions that existed in a single network could be identified by other three methods due to their inability to combine the topological relationship information in both networks. Taken them together, we demonstrated that our mPageRank algorithm was effective in combining graph topological relationship information in two heterogonous biological networks for functional module discovery.
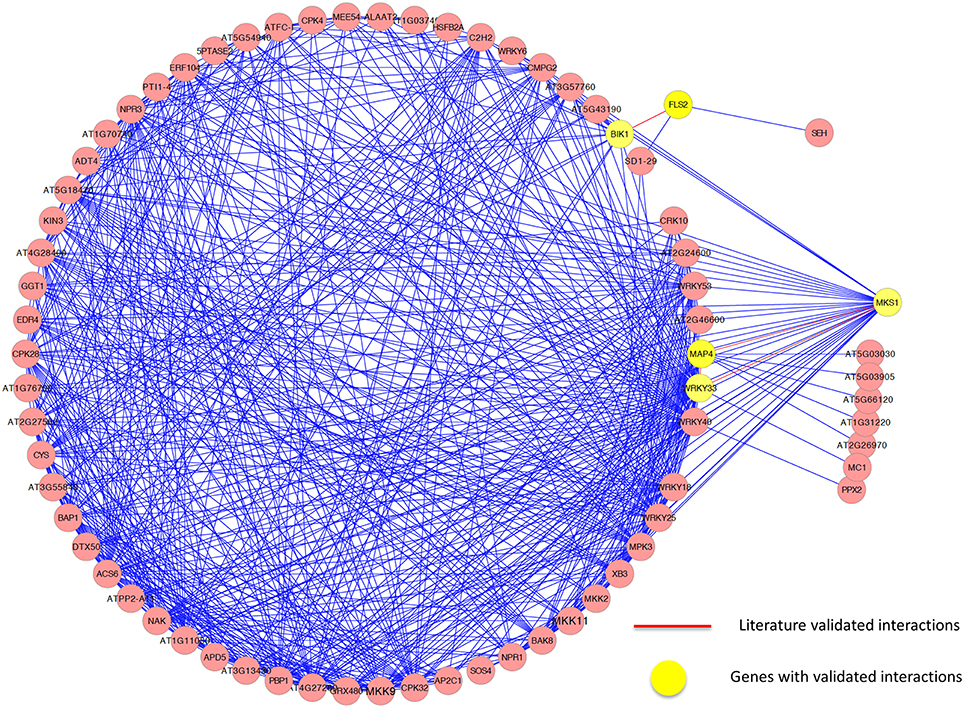
Figure 7. The genes and interactions included in the identified functional core module related to defense response in Arabidopsis thaliana.
Conclusions
We present here a novel mPageRank approach for mining functional modules in heterogeneous biological networks. Beginning with several cell division cycle related or immune-related seed genes from the model plant, Arabidopsis thaliana, our approach successfully ranked and retrieved genes involved in cell division cycle related functions and plant defense signaling related functions. These genes formed the basis for core functional modules created from global gene co-expression association networks and protein-protein interaction networks. Our benchmarking analyses using simulated data and case study analyses additionally demonstrated that our proposed mPageRank method is effective and is a promising approach for mining functional modules in heterogonous biological networks.
Availability
The program namely mPageRank, datasets and results of the presented two case studies are publicly and freely available at http://plantgrn.noble.org/MPageRank/.
Author Contributions
JL implemented the mPageRank algorithm, carried out case study analyses and wrote the manuscript. PZ conceived and directed the study, performed bioinformatics analyses and wrote the manuscript.
Funding
This work was supported by the National Science Foundation [grants DBI: 0960897 and DBI: 1458597 to PZ] and the Samuel Roberts Noble Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Xinbin Dai and Ms. Zhaohong Zhuang for their assistances in Arabidopsis thaliana gene expression data collection, compilation and curation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00903
References
Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. doi: 10.1126/science.1203877
Bach, L., Michaelson, L. V., Haslam, R., Bellec, Y., Gissot, L., Marion, J., et al. (2008). The very-long-chain hydroxy fatty acyl-CoA dehydratase PASTICCINO2 is essential and limiting for plant development. Proc Natl Acad Sci U.S.A. 105, 14727–14731. doi: 10.1073/pnas.0805089105
Barabasi, A. L., and Oltvai, Z. N. (2004). Network biology: understanding the cell's functional organization. Nat. Rev. Genet 5, 101–113. doi: 10.1038/nrg1272
Barrett, T., Troup, D. B., Wilhite, S. E., Ledoux, P., Rudnev, D., Evangelista, C., et al. (2007). NCBI GEO: mining tens of millions of expression profiles–database and tools update. Nucleic Acids Res. 35, D760–D765. doi: 10.1093/nar/gkl887
Boruc, J., Van Den Daele, H., Hollunder, J., Rombauts, S., Mylle, E., Hilson, P., et al. (2010). Functional modules in the Arabidopsis core cell cycle binary protein-protein interaction network. Plant Cell 22, 1264–1280. doi: 10.1105/tpc.109.073635
Brandao, M. M., Dantas, L. L., and Silva-Filho, M. C. (2009). AtPIN: Arabidopsis thaliana protein interaction network. BMC Bioinformat. 10:454. doi: 10.1186/1471-2105-10-454
Cancer Genome Atlas Research Network (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068. doi: 10.1038/nature07385
Chen, C., Marcus, A., Li, W., Hu, Y., Calzada, J. P., Grossniklaus, U., et al. (2002). The Arabidopsis ATK1 gene is required for spindle morphogenesis in male meiosis. Development 129, 2401–2409.
Chinchilla, D., Zipfel, C., Robatzek, S., Kemmerling, B., Nurnberger, T., Jones, J. D., et al. (2007). A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448, 497–500. doi: 10.1038/nature05999
Chuang, H. Y., Lee, E., Liu, Y. T., Lee, D., and Ideker, T. (2007). Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 3, 140. doi: 10.1038/msb4100180
Cook, G. S., Gronlund, A. L., Siciliano, I., Spadafora, N., Amini, M., Herbert, R. J., et al. (2013). Plant WEE1 kinase is cell cycle regulated and removed at mitosis via the 26S proteasome machinery. J. Exp. Bot. 64, 2093–2106. doi: 10.1093/jxb/ert066
De Almeida Engler, J., Kyndt, T., Vieira, P., Van Cappelle, E., Boudolf, V., Sanchez, V., et al. (2012). CCS52 and DEL1 genes are key components of the endocycle in nematode-induced feeding sites. Plant J. 72, 185–198. doi: 10.1111/j.1365-313X.2012.05054.x
De Domenico, M., Sole-Ribalta, A., Omodei, E., Gomez, S., and Arenas, A. (2015). Ranking in interconnected multilayer networks reveals versatile nodes. Nat. Commun. 6, 6868. doi: 10.1038/ncomms7868
D'haeseleer, P., Liang, S., and Somogyi, R. (2000). Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics 16, 707–726. doi: 10.1093/bioinformatics/16.8.707
Dittrich, M. T., Klau, G. W., Rosenwald, A., Dandekar, T., and Muller, T. (2008). Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics 24, i223–i231. doi: 10.1093/bioinformatics/btn161
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U.S.A. 95, 14863–14868. doi: 10.1073/pnas.95.25.14863
Encinas-Villarejo, S., Maldonado, A. M., Amil-Ruiz, F., De Los Santos, B., Romero, F., Pliego-Alfaro, F., et al. (2009). Evidence for a positive regulatory role of strawberry (Fragaria x ananassa) Fa WRKY1 and Arabidopsis At WRKY75 proteins in resistance. J. Exp. Bot. 60, 3043–3065. doi: 10.1093/jxb/erp152
Enright, A. J., Van Dongen, S., and Ouzounis, C. A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584. doi: 10.1093/nar/30.7.1575
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/S1360-1385(00)01600-9
Faust, K., Dupont, P., Callut, J., and Van Helden, J. (2010). Pathway discovery in metabolic networks by subgraph extraction. Bioinformatics 26, 1211–1218. doi: 10.1093/bioinformatics/btq105
Girvan, M., and Newman, M. E. (2002). Community structure in social and biological networks. Proc. Natl. Acad. Sci. U.S.A. 99, 7821–7826. doi: 10.1073/pnas.122653799
Gutierrez, C. (2009). The Arabidopsis cell division cycle. Arabidopsis Book 7:e0120. doi: 10.1199/tab.0120
Halu, A., Mondragon, R. J., Panzarasa, P., and Bianconi, G. (2013). Multiplex PageRank. PLoS ONE 8:e78293. doi: 10.1371/journal.pone.0078293
Hartwell, L. H., Hopfield, J. J., Leibler, S., and Murray, A. W. (1999). From molecular to modular cell biology. Nature 402, C47–C52. doi: 10.1038/35011540
Ideker, T., Ozier, O., Schwikowski, B., and Siegel, A. F. (2002). Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics 18 (Suppl. 1), S233–S240. doi: 10.1093/bioinformatics/18.suppl_1.s233
Inoue, K., Li, W., and Kurata, H. (2010). Diffusion model based spectral clustering for protein-protein interaction networks. PLoS ONE 5:e12623. doi: 10.1371/journal.pone.0012623
Irizarry, R. A., Bolstad, B. M., Collin, F., Cope, L. M., Hobbs, B., and Speed, T. P. (2003). Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31:e15. doi: 10.1093/nar/gng015
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformat. 9:559. doi: 10.1186/1471-2105-9-559
Langville, A. N., and Meyer, C. D. (2006). Google's PageRank and Beyond: The Science of Search Engine Rankings. Princeton, N.J.: Princeton University Press.
Lee, J., Klessig, D. F., and Nurnberger, T. (2001). A harpin binding site in tobacco plasma membranes mediates activation of the pathogenesis-related gene HIN1 independent of extracellular calcium but dependent on mitogen-activated protein kinase activity. Plant Cell 13, 1079–1093. doi: 10.1105/tpc.13.5.1079
Li, J., Wei, H., Liu, T., and Zhao, P. X. (2014). GPLEXUS: enabling genome-scale gene association network reconstruction and analysis for very large-scale expression data. Nucleic Acids Res. 42, e32. doi: 10.1093/nar/gkt983
Li, J., Wei, H., and Zhao, P. X. (2013). DeGNServer: deciphering genome-scale gene networks through high performance reverse engineering analysis. Biomed. Res. Int. 2013:856325. doi: 10.1155/2013/856325
Ligterink, W., Kroj, T., Zur Nieden, U., Hirt, H., and Scheel, D. (1997). Receptor-mediated activation of a MAP kinase in pathogen defense of plants. Science 276, 2054–2057. doi: 10.1126/science.276.5321.2054
Lin, W., Lu, D., Gao, X., Jiang, S., Ma, X., Wang, Z., et al. (2013). Inverse modulation of plant immune and brassinosteroid signaling pathways by the receptor-like cytoplasmic kinase BIK1. Proc. Natl. Acad. Sci. U.S.A. 110, 12114–12119. doi: 10.1073/pnas.1302154110
Ma, S., Gong, Q., and Bohnert, H. J. (2007). An Arabidopsis gene network based on the graphical Gaussian model. Genome Res. 17, 1614–1625. doi: 10.1101/gr.6911207
Mao, T., Jin, L., Li, H., Liu, B., and Yuan, M. (2005). Two microtubule-associated proteins of the Arabidopsis MAP65 family function differently on microtubules. Plant Physiol. 138, 654–662. doi: 10.1104/pp.104.052456
Maraziotis, I. A., Dimitrakopoulou, K., and Bezerianos, A. (2007). Growing functional modules from a seed protein via integration of protein interaction and gene expression data. BMC Bioinformat. 8:408. doi: 10.1186/1471-2105-8-408
Nuhse, T. S., Peck, S. C., Hirt, H., and Boller, T. (2000). Microbial elicitors induce activation and dual phosphorylation of the Arabidopsis thaliana MAPK 6. J. Biol. Chem. 275, 7521–7526. doi: 10.1074/jbc.275.11.7521
Pandey, S. P., and Somssich, I. E. (2009). The role of WRKY transcription factors in plant immunity. Plant Physiol. 150, 1648–1655. doi: 10.1104/pp.109.138990
Parkinson, H., Sarkans, U., Shojatalab, M., Abeygunawardena, N., Contrino, S., Coulson, R., et al. (2005). ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 33, D553–555. doi: 10.1093/nar/gki056
Pruitt, K. D., and Maglott, D. R. (2001). RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res. 29, 137–140. doi: 10.1093/nar/29.1.137
Qiu, J. L., Fiil, B. K., Petersen, K., Nielsen, H. B., Botanga, C. J., Thorgrimsen, S., et al. (2008). Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 27, 2214–2221. doi: 10.1038/emboj.2008.147
Rual, J. F., Venkatesan, K., Hao, T., Hirozane-Kishikawa, T., Dricot, A., Li, N., et al. (2005). Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178. doi: 10.1038/nature04209
Shih, Y. K., and Parthasarathy, S. (2012). Identifying functional modules in interaction networks through overlapping Markov clustering. Bioinformatics 28, i473–i479. doi: 10.1093/bioinformatics/bts370
Stark, C., Breitkreutz, B. J., Chatr-Aryamontri, A., Boucher, L., Oughtred, R., Livstone, M. S., et al. (2011). The BioGRID interaction database: 2011 update. Nucleic Acids Res. 39, D698–D704. doi: 10.1093/nar/gkq1116
Toller, A., Brownfield, L., Neu, C., Twell, D., and Schulze-Lefert, P. (2008). Dual function of Arabidopsis glucan synthase-like genes GSL8 and GSL10 in male gametophyte development and plant growth. Plant J. 54, 911–923. doi: 10.1111/j.1365-313X.2008.03462.x
Van Den Bulcke, T., Van Leemput, K., Naudts, B., Van Remortel, P., Ma, H., Verschoren, A., et al. (2006). SynTReN: a generator of synthetic gene expression data for design and analysis of structure learning algorithms. BMC Bioinformatics 7:43. doi: 10.1186/1471-2105-7-43
Vandepoele, K., Raes, J., De Veylder, L., Rouze, P., Rombauts, S., and Inze, D. (2002). Genome-wide analysis of core cell cycle genes in Arabidopsis. Plant Cell 14, 903–916. doi: 10.1105/tpc.010445
Van Leene, J., Hollunder, J., Eeckhout, D., Persiau, G., Van De Slijke, E., Stals, H., et al. (2010). Targeted interactomics reveals a complex core cell cycle machinery in Arabidopsis thaliana. Mol. Syst. Biol. 6, 397. doi: 10.1038/msb.2010.53
Van Leene, J., Stals, H., Eeckhout, D., Persiau, G., Van De Slijke, E., Van Isterdael, G., et al. (2007). A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Mol. Cell Proteomics 6, 1226–1238. doi: 10.1074/mcp.M700078-MCP200
Vashisht, R., Bhardwaj, A., Osdd, C., and Brahmachari, S. K. (2013). Social networks to biological networks: systems biology of Mycobacterium tuberculosis. Mol. Biosyst. 9, 1584–1593. doi: 10.1039/c3mb25546h
Wu, F. X. (2008). Genetic weighted k-means algorithm for clustering large-scale gene expression data. BMC Bioinformat. 9 (Suppl. 6):S12. doi: 10.1186/1471-2105-9-S6-S12
Xu, X., Chen, C., Fan, B., and Chen, Z. (2006). Physical and functional interactions between pathogen-induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 18, 1310–1326. doi: 10.1105/tpc.105.037523
Yu, G., Li, F., Qin, Y., Bo, X., Wu, Y., and Wang, S. (2010). GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978. doi: 10.1093/bioinformatics/btq064
Keywords: heterogeneous biological network, sub-network, functional module, multiplex PageRank, mPageRank, gene expression association network, protein-protein interaction network, Arabidopsis thaliana
Citation: Li J and Zhao PX (2016) Mining Functional Modules in Heterogeneous Biological Networks Using Multiplex PageRank Approach. Front. Plant Sci. 7:903. doi: 10.3389/fpls.2016.00903
Received: 20 January 2016; Accepted: 08 June 2016;
Published: 22 June 2016.
Edited by:
Yasset Perez-Riverol, European Bioinformatics Institute (EMBL-EBI), UKReviewed by:
Thomas Triplet, Ciena Corporation & École Polytechnique Montréal, CanadaJason Edward Shoemaker, Japan Science and Technology Agency, Japan
Mingze Bai, Chongqing University of Posts and Telecommunications, China
Copyright © 2016 Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Xuechun Zhao, cHpoYW9Abm9ibGUub3Jn
†Present Address: Jun Li, Department of Genomics Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA