 Kumar Vasudevan
Kumar Vasudevan Casiana M. Vera Cruz2
Casiana M. Vera Cruz2 Wilhelm Gruissem
Wilhelm Gruissem Navreet K. Bhullar
Navreet K. Bhullar- 1Plant Biotechnology, Department of Biology, ETH Zurich, Switzerland
- 2International Rice Research Institute, Los Banos, Philippines
Rice blast is caused by Magnaporthe oryzae, which is the most destructive fungal pathogen affecting rice growing regions worldwide. The rice blast resistance gene Pib confers broad-spectrum resistance against Southeast Asian M. oryzae races. We investigated the allelic diversity of Pib in rice germplasm originating from 12 major rice growing countries. Twenty-five new Pib alleles were identified that have unique single nucleotide polymorphisms (SNPs), insertions and/or deletions, in addition to the polymorphic nucleotides that are shared between the different alleles. These partially or completely shared polymorphic nucleotides indicate frequent sequence exchange events between the Pib alleles. In some of the new Pib alleles, nucleotide diversity is high in the LRR domain, whereas, in others it is distributed among the NB-ARC and LRR domains. Most of the polymorphic amino acids in LRR and NB-ARC2 domains are predicted as solvent-exposed. Several of the alleles and the unique SNPs are country specific, suggesting a diversifying selection of alleles in various geographical locations in response to the locally prevalent M. oryzae population. Together, the new Pib alleles are an important genetic resource for rice blast resistance breeding programs and provide new information on rice-M. oryzae interactions at the molecular level.
Introduction
Rice is a staple food for more than half of the world population. Several biotic and abiotic stresses constantly threaten global rice production. Rice blast caused by the fungal pathogen Magnaporthe oryzae is the most destructive rice disease that can damage the whole plant (Wang et al., 2014), including the root (Sesma and Osbourn, 2004). It is one of the most widespread rice diseases found in over 85 rice growing countries (Spence et al., 2014). Recently, M. oryzae has also established itself in wheat agro-ecological systems in Argentina, which is alarming as the blast disease is becoming a significant threat to wheat production as well (Perelló et al., 2015).
Utilization of host resistance (R) genes is the most efficient environment-friendly and economically sustainable approach to control rice blast. To date about 100 major R genes and over 350 quantitative trait loci (QTLs) have been identified for rice blast resistance. Twenty-one of the R genes have been cloned and characterized (Liu et al., 2013; Zhai et al., 2014), including Pi64 that confers resistance to both leaf and neck blast (Ma et al., 2015). Most of the blast resistance genes belong to the coiled-coil nucleotide binding site leucine-rich repeats (CC-NBS-LRR) class of R genes. The LRR domain is reported to determine the resistance specificities of various plant R proteins, whereas, the NBS domain functions as a molecular-switch in regulating the active/inactive states of an R protein (McHale et al., 2006; Tameling et al., 2006; Williams et al., 2011). There are also exceptions to NBS-LRR type, such as Pid2 that encodes a receptor-like kinase and the recessive gene pi21, which encodes a proline-rich protein (Chen et al., 2006; Fukuoka et al., 2009). Some of the blast R genes such as Pikm1-TS/Pikm2-TS, Pi5-1/Pi5-2, Pikp-1/Pikp-2, Pikh-1/Pikh-2, and RGA4/RGA5 are present adjacently and both genes are essential for functional resistance (Ashikawa et al., 2008; Lee et al., 2009; Okuyama et al., 2011; Yuan et al., 2011; Césari et al., 2014; Zhai et al., 2014). Despite the large number of available rice blast resistance genes, the rapidly evolving M. oryzae can frequently overcome resistance. Because of the severity of the disease and the role as an interesting model organism, M. oryzae was the first fungal pathogen whose genome was sequenced (Dean et al., 2005). To better understand the frequent breakdown of resistance, the genomes of two additional M. oryzae field isolates were also re-sequenced. The M. oryzae genomes revealed 100s of isolate-specific genes and gene duplication events. Thousands of loci contain transposon-like elements and about 200 genes were disrupted by transposable elements in all the three sequenced M. oryzae strains (Xue et al., 2012). Nine of more than 40 characterized M. oryzae Avr genes have been cloned so far (Zhang et al., 2015). Most of these Avr genes and their allelic variants exhibit presence/absence of transposon element (TE) insertion, frequent presence/absence polymorphisms and high rate of shared non-synonymous substitutions among different M. oryzae strains (Huang et al., 2014; Zhang et al., 2015). Together, such high incidence of transposon-mediated inactivation of genes involved in host-specificity, the high rates of non-synonymous mutations as well as frequent gain and loss of avirulence (Avr) genes can explain the dynamic nature of M. oryzae to overcome host resistance (Dean et al., 2005; Yoshida et al., 2009; Xue et al., 2012).
Considering the rapid evolution of M. oryzae, it is critically important to broaden and diversify rice blast resistance sources by identifying novel resistance genes and allelic variants of the known resistance genes. Seed banks represent a valuable resource for exploring the genetic diversity present in rice cultivars, landraces and wild relatives. Advancements in molecular marker and DNA sequencing technologies have greatly accelerated the identification of allelic variants of R genes from genetically diverse accessions (Kilian and Graner, 2012). Allele mining in various crop germplasm collections has identified several functional allelic forms of Mla (powdery mildew resistance gene in barley), L (rust resistance gene in flax), Pm3 (powdery mildew resistance gene in wheat) and Pi54 (rice blast resistance gene) genes that confer race-specific or broad spectrum resistance (Ellis et al., 1999; Bhullar et al., 2009; Seeholzer et al., 2010; Thakur et al., 2015; Vasudevan et al., 2015). Sequence and functional analysis of these alleles revealed several sites that are under positive selection and domains involved in recognition specificities (Ellis et al., 1999; Bhullar et al., 2009; Seeholzer et al., 2010). In case of rice blast resistance genes, it has also become evident that small sequence variations among individual R genes and between different R gene alleles often have a major functional impact on the resistance specificities. For example, eight amino acid changes differentiate resistance specificities of Pi2 and Piz-t (Zhou et al., 2006), and a single amino acid change distinguish resistant and susceptible alleles of both Pita and Pid2 (Bryan et al., 2000; Chen et al., 2006). Many recently cloned blast R genes such as Pid3-A4, Pi54rh, Pi54of, and Pi35 are alleles/orthologs of known blast R genes (Das et al., 2012; Lv et al., 2013; Devanna et al., 2014; Fukuoka et al., 2014). These new alleles show varying patterns of resistance compared to the originally cloned R genes. Utilization of multiple alleles could increase the field durability of resistance by reducing the selection pressure on the pathogen (Fukuoka et al., 2014).
We have investigated the allelic diversity of Pib in rice accessions from 12 major rice-growing countries. Pib was the first cloned rice blast resistance gene. It was identified in the indica type Malaysian rice cultivar Engkatek and cloned from the near isogenic line Tohoku IL9 (Wang et al., 1999). Pib is an NBS-LRR type of R gene that confers broad spectrum resistance against a wide range of blast isolates present in South and Southeast Asian countries including Japan, China, Indonesia, and Korea (Wang et al., 2001; Yokoo, 2005; Roychowdhury et al., 2012). Pib is effective against M. oryzae race IE1k, which caused breakdown of the broad-spectrum blast R gene Pita, and race IB1 that is virulent against Piz, which also conferred stable blast resistance (Roychowdhury et al., 2012). We identified 25 new alleles of Pib in which most sequence variations are present in the NB-ARC and LRR domains. These new alleles represent a valuable genetic resource for rice blast resistance breeding programs and increase our understanding of the geographic dynamics of rice-M. oryzae interactions at the molecular level.
Results
Selection of Rice Genotypes for Mining Pib Allele Diversity
We screened 4246 rice accessions originating from 13 major rice growing countries and identified 3176 blast-resistant accessions of which 2494 scored ‘highly resistant’ (score 0 on standard evaluation scale of 0–9 for leaf blast; Vasudevan et al., 2014). For Pib allele mining, we selected 467 rice accessions with a phenotypic score of 0 in a uniform nursery screening and against at least two of five tested M. oryzae isolates (Figure 1). These 467 accessions were from 12 of the 13 selected countries because none of the South Korean accessions met our scoring criteria. The selected accessions were screened for the presence of Pib (Figure 1) using the gene-specific Nsb molecular marker (Cho et al., 2007). A total of 337 rice accessions with the corresponding band of 629 bp for Pib were selected as candidates for Pib allele mining (Figure 1; Supplementary Table S1). The 337 accessions represent the three major rice varietal groups, indica (295), javanica (30), and japonica (12).
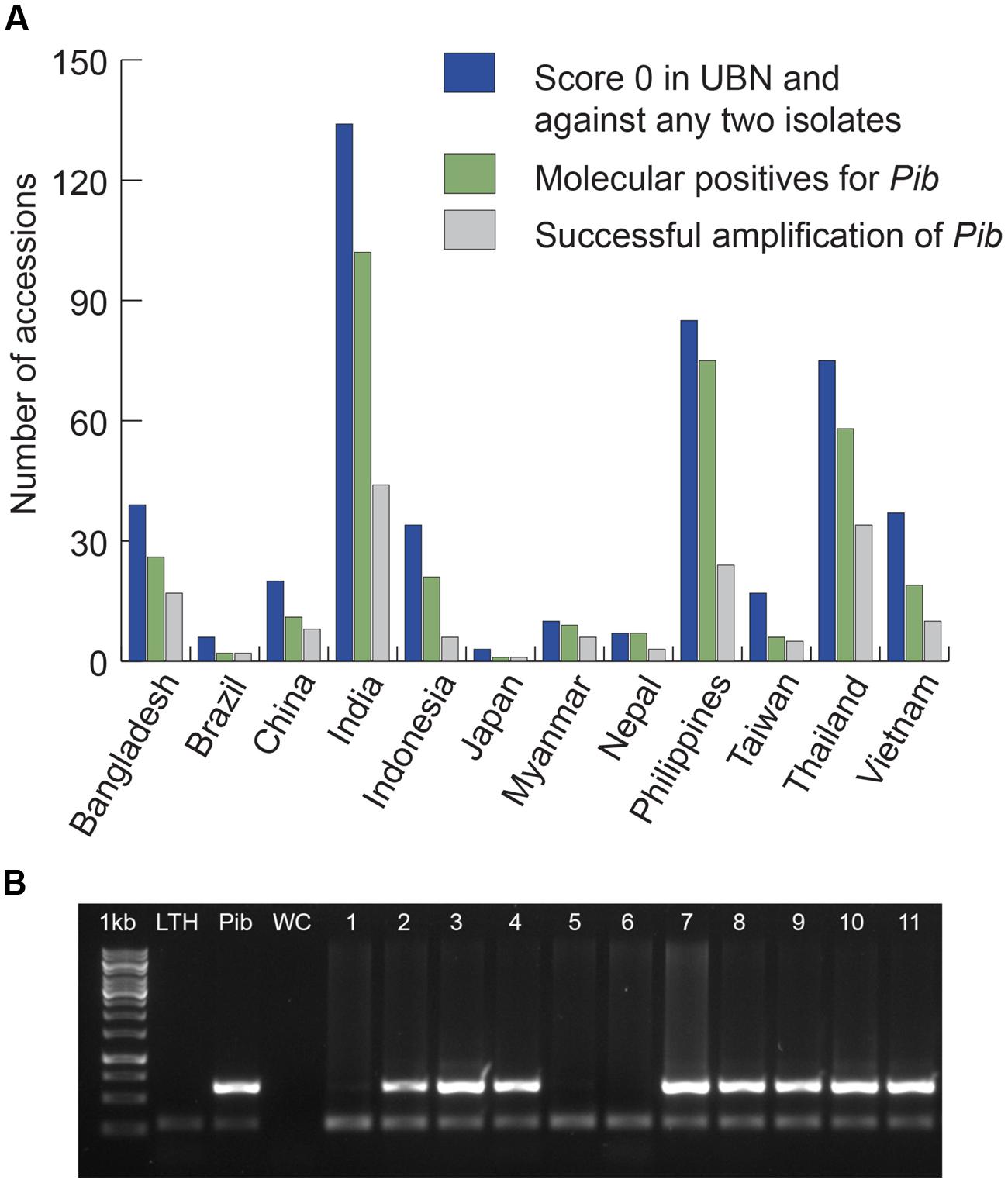
FIGURE 1. Selection of candidates for Pib allele mining. Accessions from 12 major rice growing countries that were chosen based on their phenotype (score 0) in UBN and against at least two of the five pure blast isolates are presented (A). The accessions that are molecular positives for Pib based on Nsb marker screening were chosen as candidates, and the candidates from which Pib was successfully amplified are also presented. Sample picture of molecular screening of rice accessions for selection of Pib allele mining candidates is shown (B). 1 kb, DNA marker; LTH, negative control; Pib, positive control (Pib-monogenic line); WC, water control; 1–11, test accessions. The accessions positive for Pib are 2 (IRGC-4471), 3 (IRGC-4552), 4 (IRGC-4553), 7 (IRGC-4574), 8 (IRGC-4619), 9 (IRGC-4633), 10 (IRGC-4634), and 11 (IRGC-4642). The upper band (629 bp) indicates the presence of Pib and the lower band (∼250 bp) shows the actin band, which is an internal control for the PCR.
Identification of New Pib Alleles
Forward and reverse primers were designed to amplify the full-length Pib coding region of 5404 bp. The Pib gene was successfully amplified from 160 rice accessions (Supplementary Table S2) while the amplification was not successful in the remaining accessions, possibly due to sequence polymorphisms at the primer binding sites. Of these 160 accessions, 124 were indica, 24 javanica and 12 japonica accessions, and 47 of these accessions were landraces. Detailed sequence analysis using the Engkatek Pib sequence as reference allele (hereafter, referred to as Pib_Engkatek) revealed 25 new Pib alleles (Table 1). They were named after the IRGC accession from which the respective allele was first isolated in our study. Figure 2A summarizes the nucleotide polymorphisms in the 25 new Pib alleles as compared to Pib_Engkatek. The accessions from India, Thailand, and China comprise a relatively higher number of diverse Pib alleles with 10, 7, and 6 different Pib alleles identified in the accessions from these countries, respectively (Table 2). Pib_23721 and Pib_40286 are the most common Pib alleles that were identified in 35 and 30 accessions, respectively. Except for Pib_41341, Pib_26038, Pib_24475, Pib_29436, Pib_23713, Pib_23712, and Pib_4868 that were found only in one rice accession, all other alleles were identified in at least two accessions (Table 2). None of the sequenced accessions had the Pib_Engkatek allele. The new Pib alleles share between 93 and 99% DNA sequence identity with Pib_Engkatek (Table 2).

TABLE 1. The IRGC accessions carrying the newly identified Pib alleles.
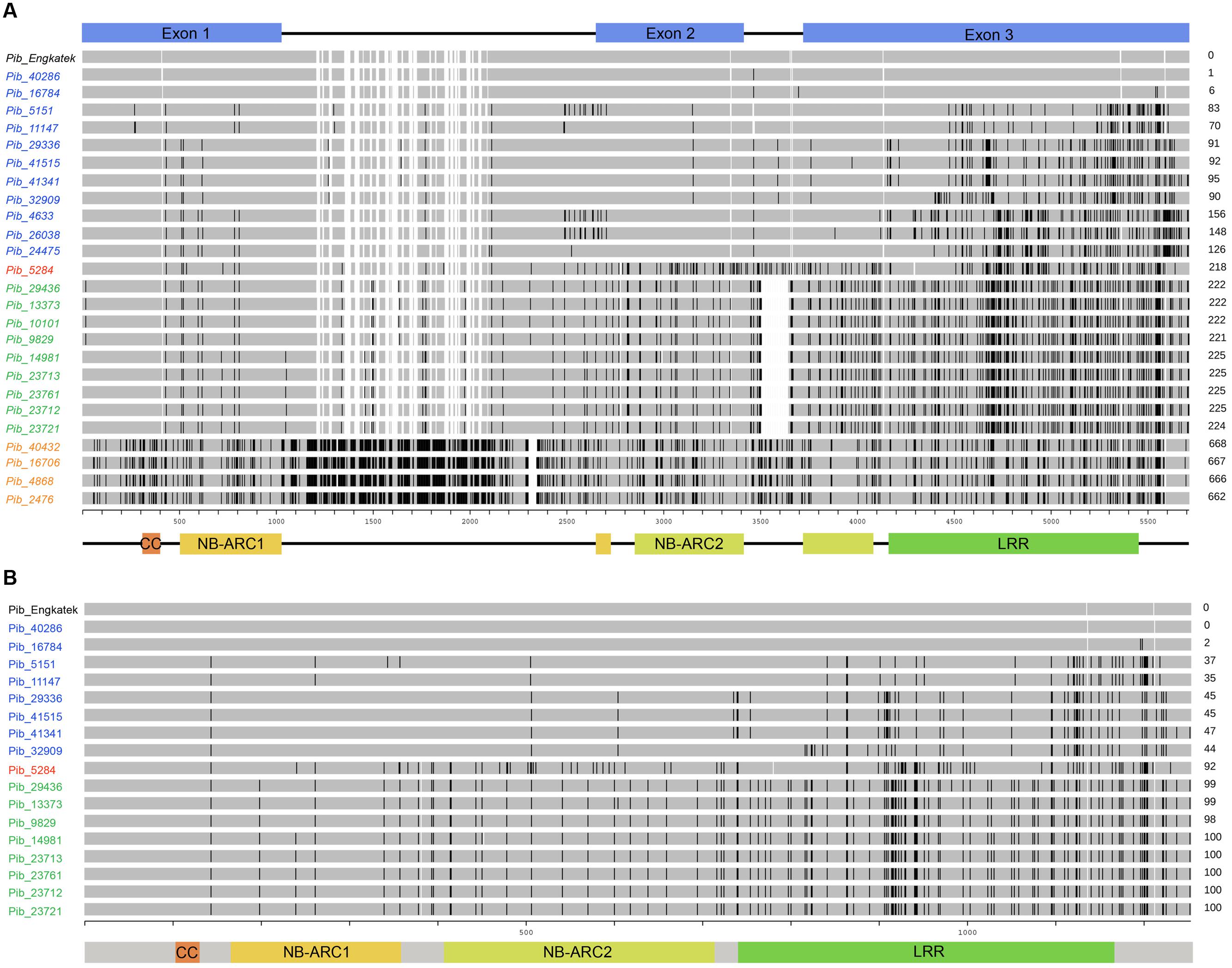
FIGURE 2. Schematic representation of sequence alignments of newly identified Pib alleles. The DNA sequence (A) and protein sequence (B) alignment of new Pib alleles is shown relative to the reference allele. The coding regions and the domains, i.e., CC, NB-ARC1, NB-ARC2, and LRR are illustrated at the bottom. The unit scale represents the nucleotide position (A) or AA position (B). The black lines on the bars as well as the numbers on the right represent polymorphisms as compared to the reference. The alleles/proteins labeled in blue, green, orange, and red indicate category I, II, III, and IV, respectively. The empty gaps in the bars for some new alleles/proteins that are not present in the reference allele bar indicate deletions. The empty gaps in the reference allele bar together with some of the new alleles/proteins appear due to presence of insertions in other compared alleles.
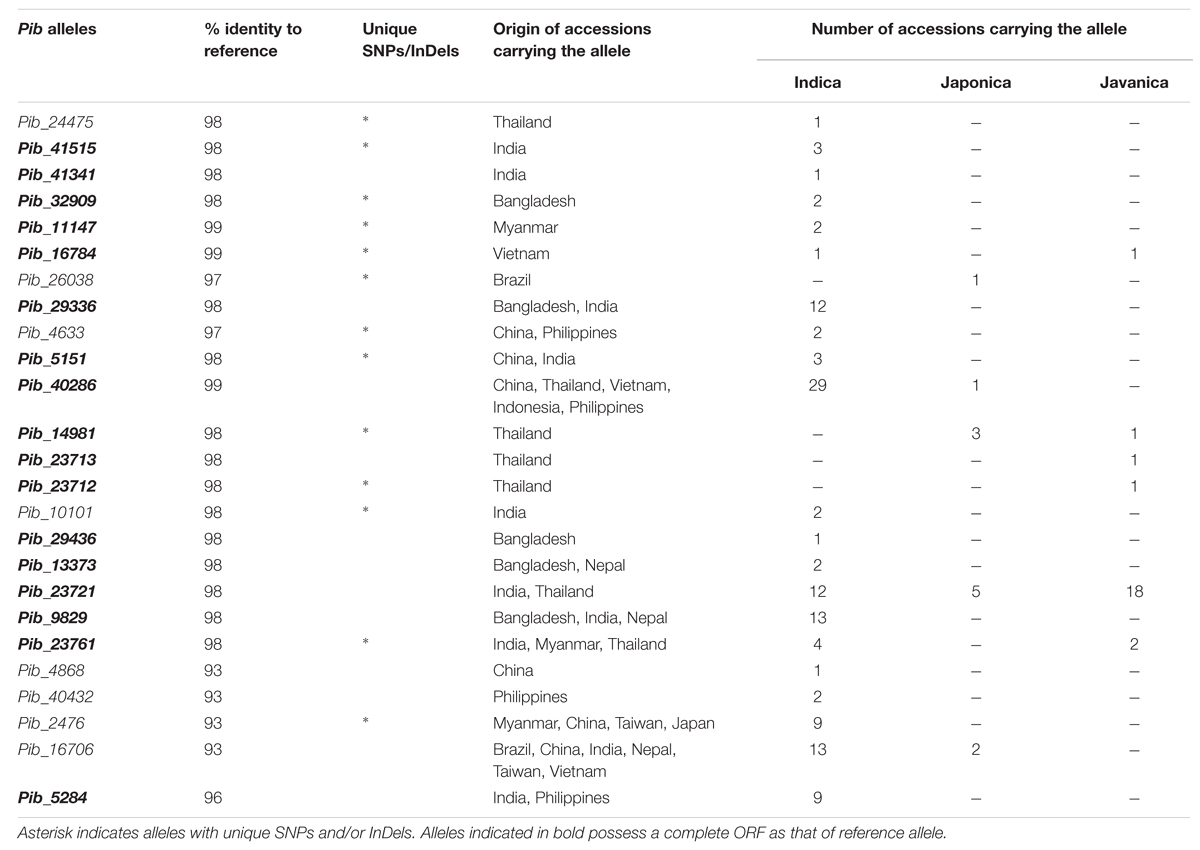
TABLE 2. Detailed information regarding the newly identified Pib alleles and the accessions carrying the respective alleles.
Sequence and Structure Diversity of New Pib Alleles
The Pib alleles were grouped into four allele categories based on the characteristics of their intron regions and nucleotide polymorphisms relative to the Pib_Engkatek reference allele (Figure 2A). Category I alleles (Pib_40286, Pib_16784, Pib_5151, Pib_11147, Pib_29336, Pib_41515, Pib_41341, Pib_32909, Pib_4633, Pib_26038, and Pib_24475) have no long sequence insertion/deletions (InDels) in their introns. Category II alleles (Pib_29436, Pib_13373, Pib_10101, Pib_9829, Pib_14981, Pib_23713, Pib_23761, Pib_23712, and Pib_23721) have a 136 nucleotide deletion in intron 2. Category III alleles (Pib_40432, Pib_16706, Pib_4868, and Pib_2476) have several insertions of varying lengths ranging from one to 28 nucleotides and a deletion of 42 nucleotides in intron 1. Category IV has only one allele, Pib_5284, without large InDels but unique nucleotide differences relative to Pib_Engkatek. Pib_5284 introns and exons partially share the sequence polymorphisms found in category I, II and III alleles (Figure 2A), suggesting that Pib_5284 is an intermediate allele of the other Pib allele categories.
Seventeen of the Pib alleles have complete open reading frames (ORFs) similar to that of Pib_Engkatek (Figure 2B). The remaining eight alleles have ORFs of variable lengths due to premature stop codon(s) resulting from point mutations and/or InDels. The ORFs of all category III alleles are disrupted by insertions that cause frame shifts or introduce alternative splicing sites. Category I alleles Pib_40286 and Pib_16784 have only minor nucleotide polymorphisms compared to Pib_Engkatek. Pib_40286 differs from Pib_Engkatek by a single nucleotide change at position 3166 in intron 2. This nucleotide change is shared among all Pib alleles except Pib_5151 and Pib_11147, which both have an eight bp deletion (Figure 2A). Except Pib_40286 and Pib_16784, all the other alleles have several nucleotide polymorphisms that are partially or completely shared within and/or between the four Pib allele categories (Figure 2A). Such partially or completely shared nucleotide polymorphisms indicate frequent sequence exchange between the alleles, possibly through gene conversion events. Thirteen alleles have one or more unique single nucleotide polymorphisms (SNPs) and/or InDels that are absent in any other Pib allele (Table 2). Pib_5284 has the most unique SNPs (Figure 2A). Nine of these 13 alleles were found in accessions of a particular country of origin (Table 2). The remaining four alleles (Pib_5151, Pib_4633, Pib_23761, and Pib_2476) were found in accessions that originate from different but geographically close countries (e.g., Pib_5151: China and India; Table 2). This suggests that such unique SNPs might be specific for resistance to prevailing M. oryzae races in certain geographical locations.
Pib Alleles Have Domain-Specific Sequence Polymorphisms Indicating Balanced and Positive Selection
The Pib N-terminal CC domain sequences of all category I, II, and IV alleles are highly conserved and have no SNPs or InDels, but category III alleles have nine SNPs (Figure 2A). Pib encodes two NB-ARC domains that are interrupted by intron 1 (1015–2354) and intron 2 (3111–3418) located 87 nucleotides 3′ of a kinase 3a motif in both NB-ARC domains. The NB-ARC domains of R proteins are involved in plant defense signaling. The NB-ARC-1 domains of category I (10 SNPs), category II (nine SNPs), and category IV (nine SNPs) alleles have fewer nucleotide changes compared to the NB-ARC-1 domains of category III alleles, which have 30 SNPs in this domain. The NB-ARC-2 domains of category II, III and IV alleles have between 35 and 52 SNPs, a three-nucleotide deletion in Pib_14981, and a three-nucleotide insertion in all category III alleles. In contrast, category I alleles have only five SNPs in this domain (Figure 2A).
The LRR domains of R genes are known to be involved in protein-protein interactions and in determining the resistance specificities. The Pib LRR domain sequences have the most nucleotide changes in the majority of the alleles. Based on the number of LRR nucleotide changes calculated as a percentage of total coding region nucleotide changes, category I alleles have the highest LRR diversity (67.1%), followed by category II (58.2%), category IV (46.9%), and category III (35.9%) alleles. Most of these nucleotide changes cause amino acid changes in the deduced protein sequence (discussed below). In case of category I and category II alleles, the highest nucleotide diversity is confined to the LRR, whereas, for category III and category IV alleles nucleotide diversity is distributed similarly between NB-ARC and LRR domain sequences (Figure 3A). Although the number of LRR polymorphic sites is very high in category I alleles, they are only partially shared among the alleles. In contrast, category II LRR polymorphisms are completely shared among the alleles (Figures 2A and 3B). Together, the polymorphism in the NB-ARC and LRR domains of Pib alleles indicate various levels of selection pressures promoting the evolution of new resistance specificities in these alleles.
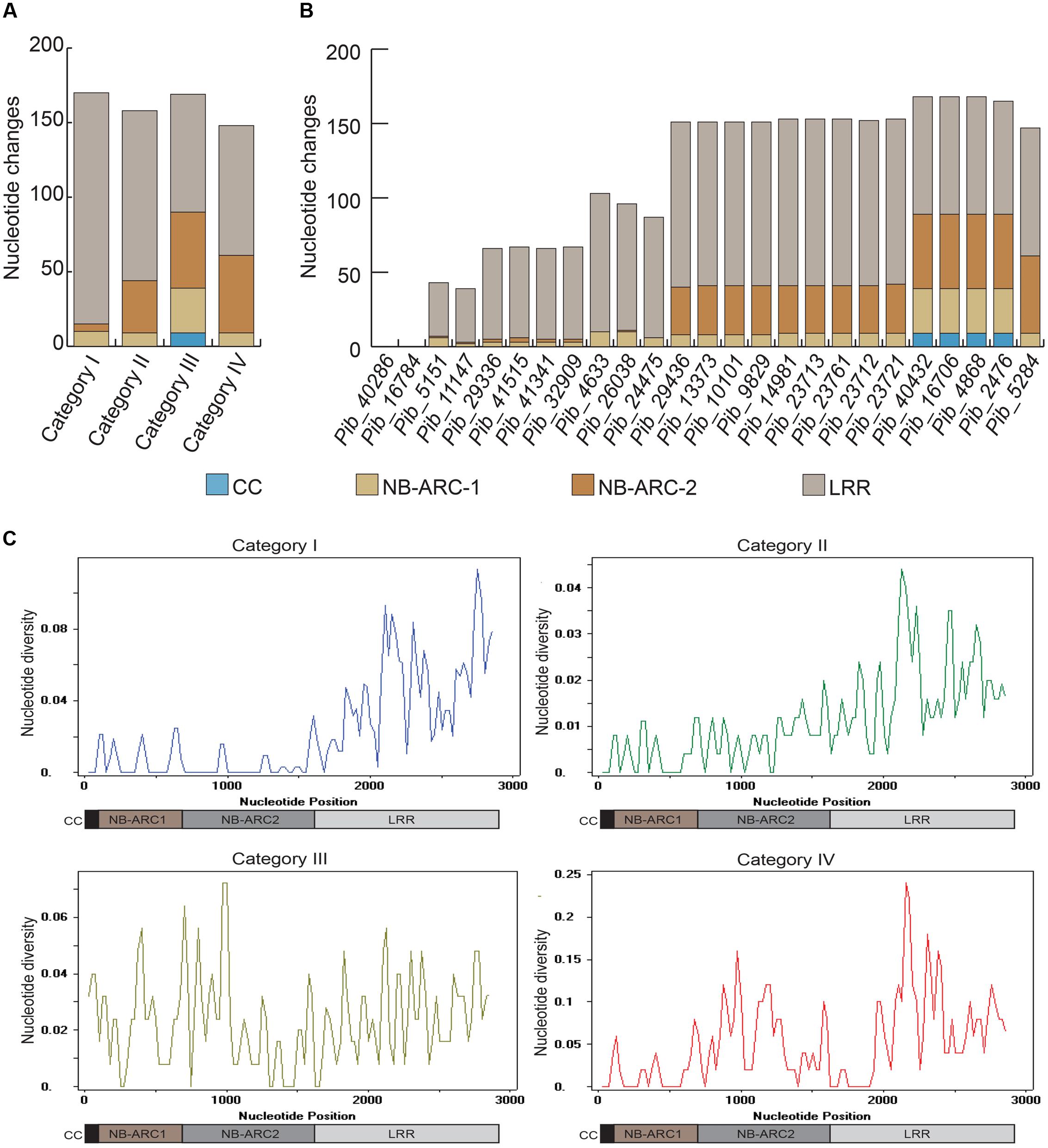
FIGURE 3. Comparison of nucleotide diversity among different domains of Pib alleles. Graphical representation of overall domain-based nucleotide changes in each of the four Pib categories are shown (A), together with domain based nucleotide changes in each of the newly identified Pib alleles (B). The sliding-window analysis of nucleotide diversity (π) in different domains of Pib alleles is shown (C). The domains are illustrated below the unit scale that represents nucleotide positions.
Sequence variations were also found in the two different Pib zinc finger motifs TTF and U1 (Gupta et al., 2012). The TTF motif sequence in exon 1 (positions 178–426) spans the CC domain while the U1 motif (positions 2976–3080) lies within the NB-ARC2 domain sequence in exon 2. TTF sequences of category I, II, and IV alleles are conserved with only two synonymous mutations in category I alleles Pib_5151 and Pib_11147 and one synonymous mutation in category IV allele Pib_5284. Category III alleles have 21 SNPs and a three-nucleotide insertion in their TTF motif. The U1 motif is conserved in category I alleles, whereas, 3, 4, and 10 SNPs were observed in category II, III, and IV alleles, respectively. In addition, a triple nucleotide insertion was found in the U1 motif of category III alleles. The Zinc finger domains determine the regulatory function of NBS-LRR proteins (Gupta et al., 2012), and our results further indicate their significant role in determining Pib function.
Domain-specific sequence analysis using DnaSP revealed selection patterns and divergence in the four allele categories. The number of mutations (η) was higher than the number of polymorphic/segregating sites (S) only in category I alleles (Table 3). Moreover, all segregating sites with more than one mutation are located within the LRR domain of category I alleles (S/LRR = 146, η/LRR = 155; Table 3). S and η values of category II and III alleles were the same, and category IV has only one allele. Sliding window analysis of nucleotide diversity (π) in each allele category showed a high rate of diversity in the LRR domain for category I, NB-ARC2 and LRR domains for category II and IV, and in all domains for category III alleles (Figure 3C). Tajima’s D value suggests that category I alleles are under balancing selection, whereas, category II and III are under positive selection (Table 3).
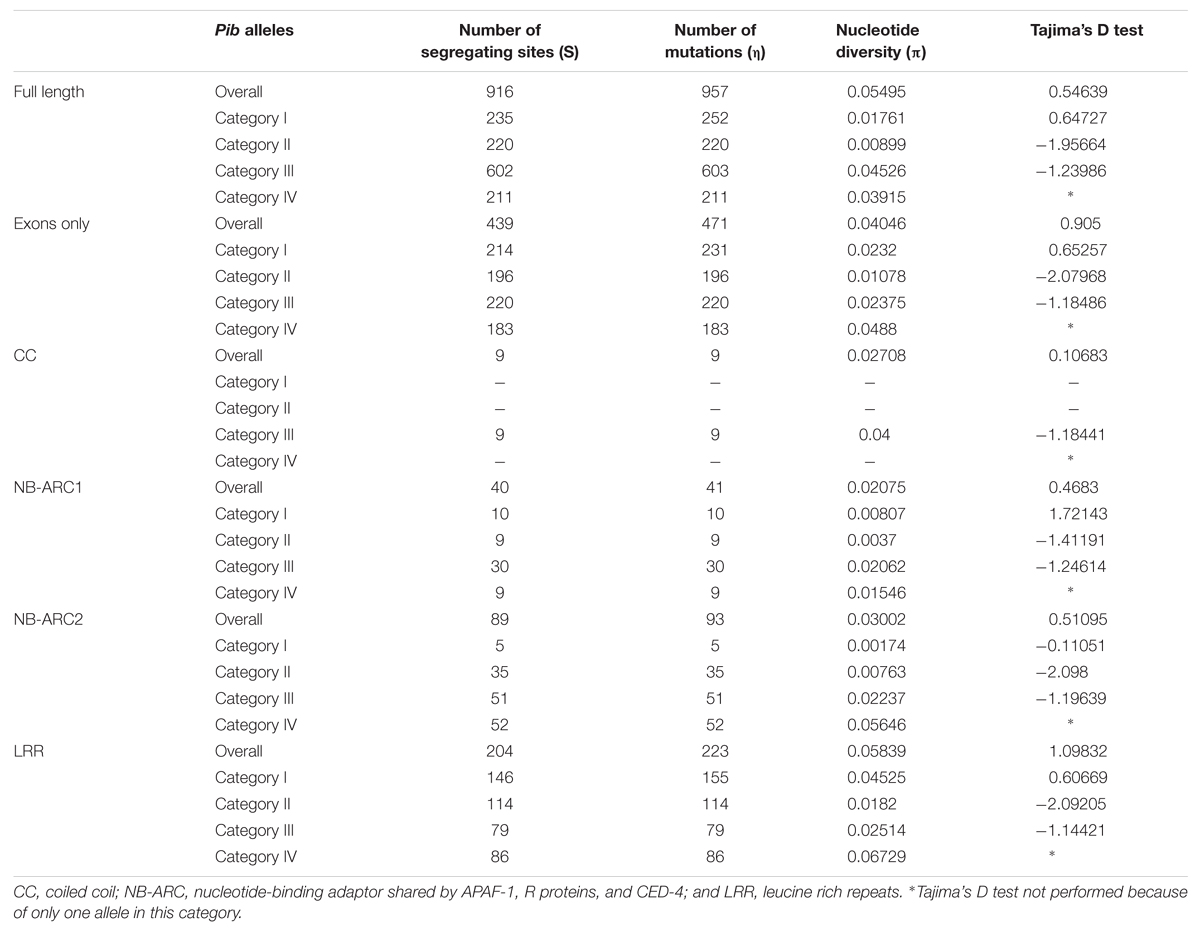
TABLE 3. Nucleotide diversity of the 25 new Pib alleles.
Pib Proteins Have Conserved Structural Domains and Post-translational Modification Sites
Protein predictions and analysis were performed only for the 17 Pib alleles with complete ORFs similar to the Pib_Engkatek reference allele (Table 2). Some of the alleles differ only by intron mutations and therefore have identical protein sequences (Pib_40286/Pib_Engkatek, Pib_29336/Pib_41515, and Pib_23713/Pib_23721; Figure 2B). In some alleles, the 3-nucleotide InDels (described above) result in addition or deletion of an amino acid (AA) without disrupting the ORF. The category I proteins Pib_29336, Pib_41515, Pib_41341, and Pib_32909 have an additional isoleucine and arginine at AA positions 1136 and 1211, respectively. Category II Pib proteins have deletions of lysine (in all category II proteins) and aspartate (only in Pib_14981) at AA positions 382 and 453, respectively. Similarly, the category IV Pib_5284 protein has deletions of lysine and histidine at AA positions 382 and 780, respectively (Figure 2B).
No AA changes were found in the CC domain of the Pib proteins. The CC domain of several R proteins contains a functional EDVID motif (Rairdan et al., 2008; Wang et al., 2015). The Pib has two possible EDVID motif variants that are EDSLQ (80–84) and EDVSQ (120–124), the former located before the CC domain and the later within the CC domain. Both the EDVID motif variants are conserved among all the analyzed Pib proteins. The NB-ARC1 domain has five polymorphic AA residues in category I, II and IV Pib proteins. In the NB-ARC2 domain, category I Pib proteins have three AA changes, but category II and IV proteins have 20 and 31 AA changes, respectively (Figure 2B). Despite these AA changes, the Kinase 1a (P-loop), Kinase 2 and Kinase 3a motifs within the NB-ARC1 and NB-ARC2 domains are highly conserved in most Pib proteins. The GLPL (619–622) motif and the MHD motifs are also conserved among the Pib proteins (Figure 4). There are two MHD motifs in Pib (750–756) separated by a single amino acid. The MHD1 (VHD in case of Pib) is highly conserved in all Pib proteins (Figure 4A), while the MHD2 is identified as MRD in most of the category I (except Pib_29336, Pib_41515, and Pib_41341) and category IV Pib proteins, and as IRD in all of the category II Pib proteins. The higher number of SNPs in the LRR domain result in 56, 65, and 46 AA changes in category I, II and IV Pib proteins, respectively (Figure 2B), which account for 65, 58, and 44% of the total AA changes in these proteins, respectively. Among the category I proteins, the highest number of AA changes were found in Pib_29336, Pib_41515, Pib_41341, and Pib_32909. The LRR of Pib_5151, Pib_11147, and Pib_32909 has several unique AA changes that are not shared with other category I alleles. However, in category II, most AA changes are shared between the Pib proteins (Figure 2B). According to NetSurfP (Petersen et al., 2009) most of the polymorphic AA changes in the NB-ARC2 (79.5%) and LRR (71.4%) domains are solvent exposed residues (Figure 4), suggesting that these AA changes might be critical for interaction with M. oryzae effector proteins and/or other cellular proteins. However it is to be noted that, the solvent exposed residue predictions are not supported by detailed structural analysis.
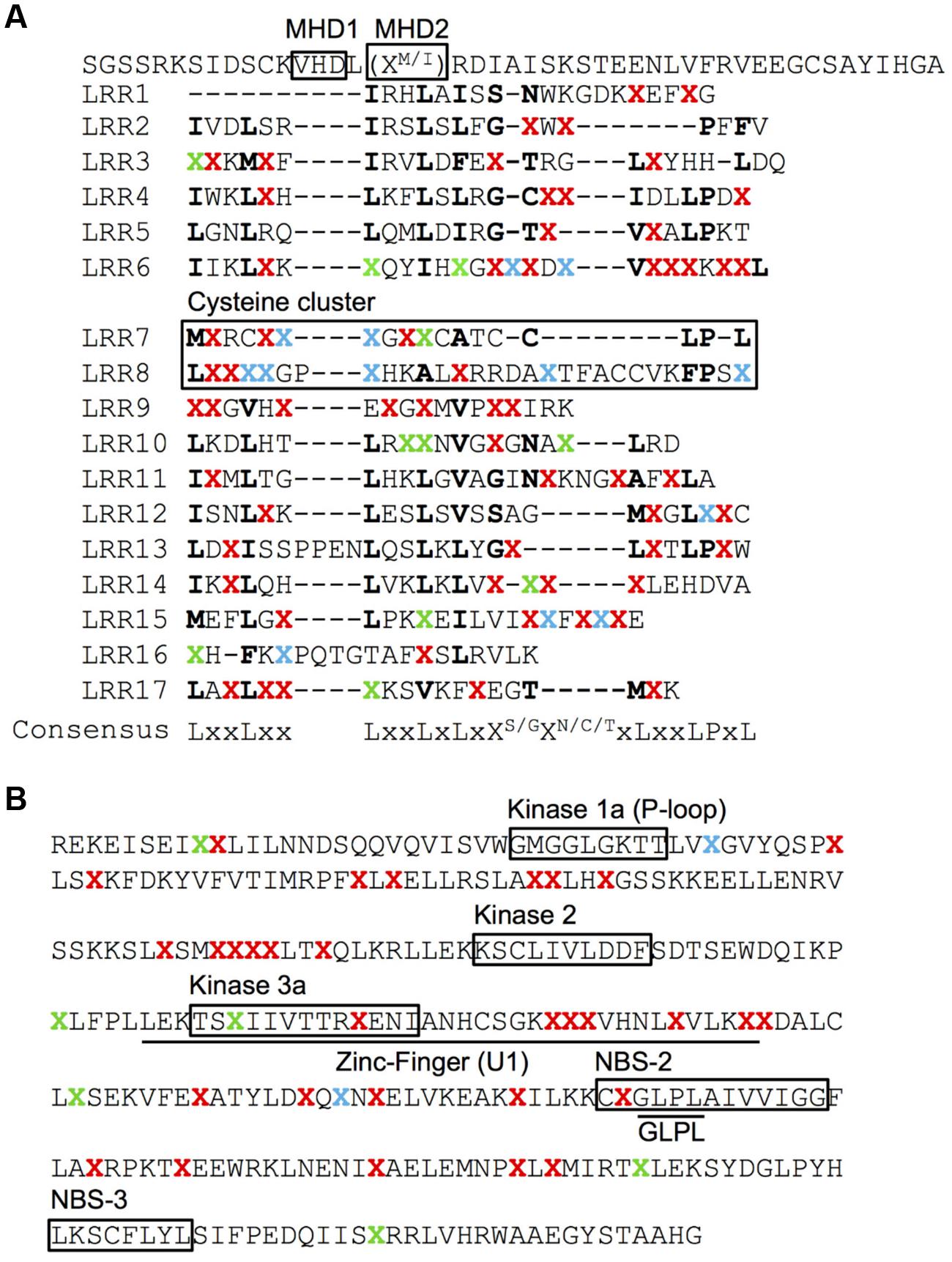
FIGURE 4. Surface accessibility prediction of the Pib proteins. Surface accessibility of AA of LRR (A) and NB-ARC2 (B) domains of Pib proteins was predicted using NetSurfP server. The LRR repeats (A) are indicated as initially described for Pib (Wang et al., 1999). ‘X’ represents polymorphic AA identified at least in one of the analyzed Pib proteins. The conserved AA are represented by their respective single letter code. The polymorphic AA predicted as solvent exposed are highlighted in red and the ones in green are predicted as buried. The polymorphic AA that are predicted as either exposed in some proteins or buried in rest of the analyzed proteins are highlighted in blue. The cysteine cluster of LRR and the functional motifs are indicated on the figure (A,B).
According to ‘PredictProtein,’ on average 25 protein-binding sites are predicted in all Pib proteins, with minor changes in their positions among the different proteins (Supplementary Figure S1). Apart from the disordered regions found in the N- and C-terminal regions of most Pib proteins, two conserved disordered regions are detected between the Kinase 1a and Kinase 2 domains of both the NB-ARC-1 and NB-ARC-2, respectively (Supplementary Figure S1). In addition, all category I Pib proteins have a conserved disordered region between their NB-ARC-1 and NB-ARC-2 domains that is absent in category II and IV Pib proteins as the result of an AA replacement (Ala to Val) at position 378 and loss of a lysine at position 381.
Analysis of Pib protein sequences using ScanProsite revealed several conserved patterns of post-translational modification sites (PTM), including potential tyrosine kinase (4), casein kinase II (26), cAMP- and cGMP-dependent protein kinase (2) and protein kinase C phosphorylation sites (26), myristoylation sites (11), glycosylation sites (4), and amidation site (1) in Pib_Engkatek (Table 4). The tyrosine kinase phosphorylation sites are the only PTM sites that are highly conserved among all the Pib proteins, indicating that they are functionally relevant. The amidation site was found only in Pib_40286, Pib_16784, Pib_5151, Pib_11147, and Pib_5284. The other PTM sites were found in all Pib proteins, however, their numbers vary between different proteins when compared to Pib_Engkatek (Table 4). These predicted PTM sites and their differences among the Pib proteins might influence the functional structure of the protein.
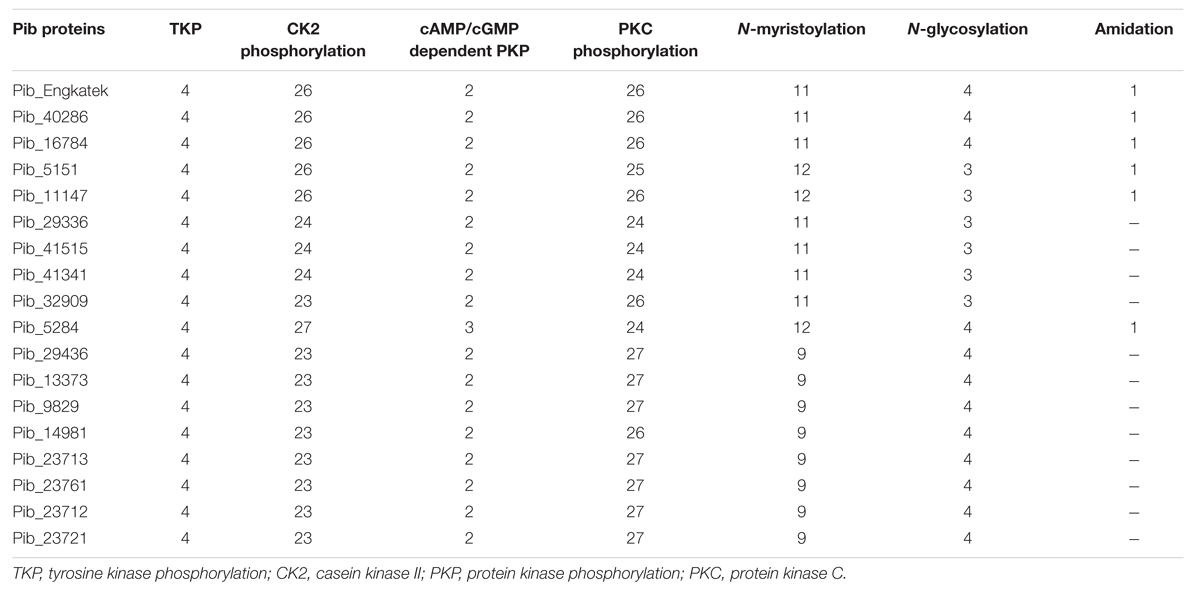
TABLE 4. Post-translational modification sites in Pib proteins predicted using ScanProsite server.
The cysteine cluster in the center of the Pib LRR (AA positions 921–967) containing eight cysteine residues (Wang et al., 1999) is highly conserved in the category I Pib proteins. Six of the eight cysteines are also conserved in the category II Pib proteins, and the two additional cysteines at positions 925 and 956 may compensate the loss of the cysteines at AA positions 929 and 940 (Figure 5). The cysteine cluster of the category IV Pib_5284 has also lost two conserved cysteines similar to category II Pib proteins but compensated these with the addition of two cysteines at AA positions 925 and 1228. This strong conservation of seven or eight cysteines in the center of the LRR suggests that the cysteine cluster in the Pib proteins has a structural and/or functional role.
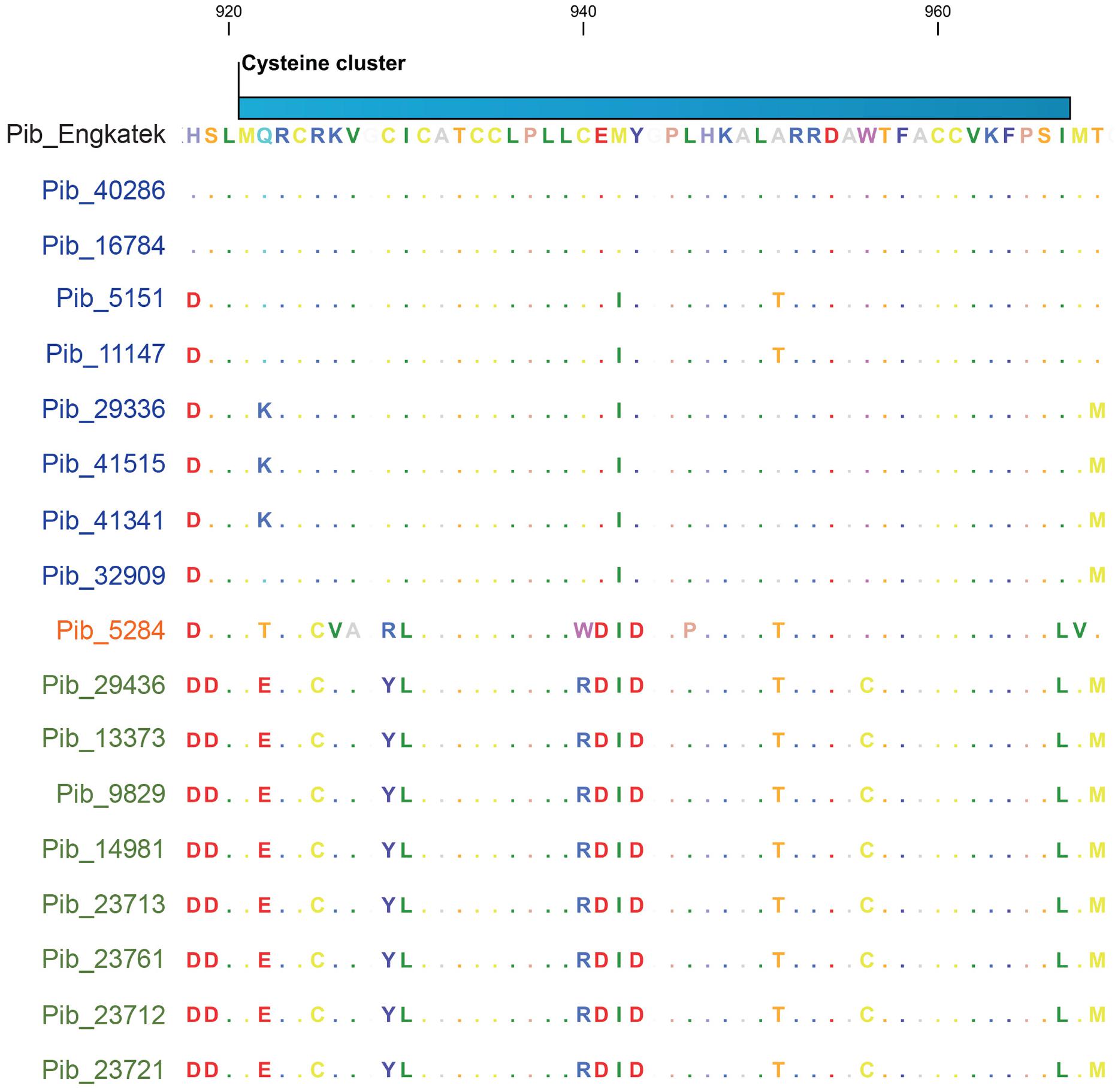
FIGURE 5. Selection window of cysteine-cluster region from the Pib protein sequence alignment. Proteins labeled in blue, orange, and green represents Pib category I, IV, and II, respectively. Scale on top indicates the amino acid position.
Distribution and Genetic Relatedness of Pib Alleles from Diverse Geographical Origins
Sixteen of the 25 Pib alleles were observed only in the indica accessions, whereas, only one and two alleles were found exclusively in the japonica and javanica accessions (Table 2). To some extent this may reflect the sampling number of accessions in the rice subspecies, but could also indicate a higher dynamics in the co-evolution of M. oryzae with the indica subspecies. Among the 25 Pib alleles, 13 alleles were identified in at least one landrace. Different landraces that have the identical Pib allele mostly originated from the same country, except for those having Pib_40286, Pib_13373, Pib_9829, and Pib_16706 alleles. The landraces having these four alleles originated from different countries, but most of them are in geographic proximity. For example, Pib_13373 is present in landraces from Bangladesh and Nepal, and Pib_9829 in landraces from India and Bangladesh (Supplementary Table S2). Except Indonesia and Japan, all other countries have accessions with more than one new Pib allele. Fourteen Pib alleles are country specific, with Thailand (4), India (3), and Bangladesh (2) having more than one such origin-specific alleles. None of the accessions from Indonesia, Japan, Nepal and Taiwan had such origin-specific allele (Table 2). The geographical origin of the new Pib alleles (Figure 6A) showed that category I alleles are widely distributed in Southeastern Asia. Of the category II alleles, three are present in Northeast India, Nepal and Bangladesh, while five are predominantly found in Thailand. The category III alleles were distributed in the Eastern Asia while the category IV allele is present only in accessions from India and the Philippines (Figure 6A).
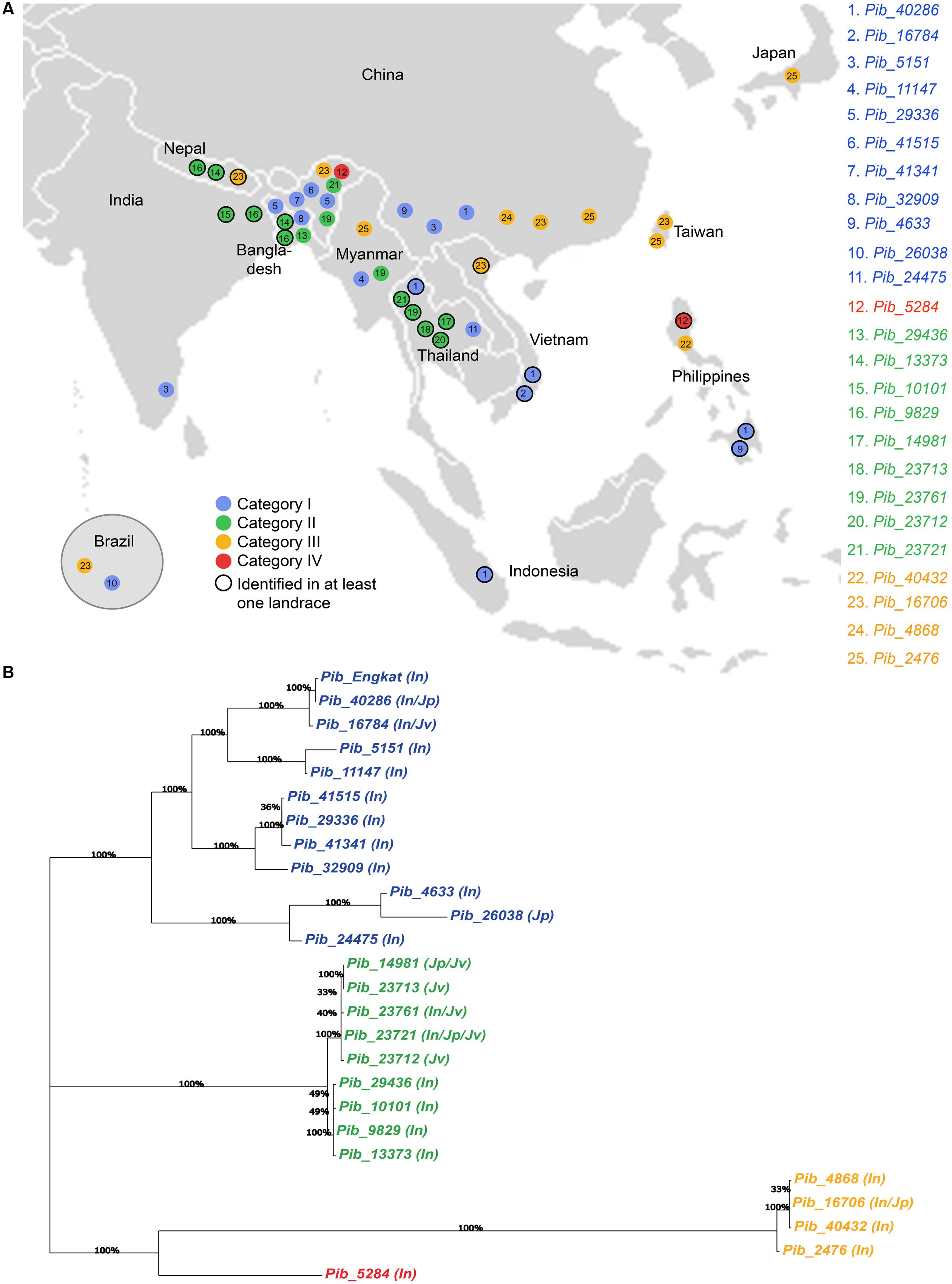
FIGURE 6. Geographical distribution of newly identified Pib alleles and their genetic relatedness. Map view of geographical distribution of newly identified Pib alleles (A). The accessions for which the information on local collection site is available in the International Rice Germplasm Collection Information System, were placed on the map accordingly. Phylogenetic analysis was performed with DNA sequences of Pib alleles together with Pib_Engkatek to analyze their genetic relatedness (B). The varietal group for accessions carrying each allele is mentioned in brackets, i.e., In, indica; Jp, japonica and Jv, javanica. Bootstrap values (100 replications) are mentioned at the branch nodes. The alleles labeled in blue, green, orange, and red represent category I, II, III, and IV, respectively.
Phylogenetic analysis of the sequence polymorphisms of the 25 Pib alleles and Pib_Engkatek revealed that category I, II III, and IV alleles form distinct clusters and that category III alleles are phylogenetically distant (Figure 6B). Most of category I and III alleles are found in indica sub-species, whereas, in category II the alleles of one sub-cluster are found exclusively in indica sub-species while the alleles in the other sub-cluster are found in javanica and japonica sub-species (Figure 6B). When comparing the LRR domain only, category II alleles are phylogenetically distinct from all other alleles. The LRR of category III alleles is closely related to category I alleles than to category II, while the category IV allele Pib-5284 clusters among the category I alleles (Supplementary Figure S2). In case of NB-ARC-1 domain only, the category III alleles cluster together with category I alleles, while the category IV allele clusters within the category II cluster forming a separate branch. While, in case of the NB-ARC-2 domains only, the overall tree-structure and clustering remains similar to that for the entire coding sequence analysis, except for a few changes in the sub-clustering of category I alleles (Supplementary Figure S2). These results suggest frequent sequence shuffling and gene conversion events at the Pib locus that create natural allele chimeras by inter-allele recombination, possibly leading to the expansion of a functional allele series and/or gene diversification.
Discussion
The extensive use of high-yielding varieties has greatly reduced the genetic diversity in breeding germplasm of major food crops, which limits future crop improvements (Tanksley and McCouch, 1997; Warschefsky et al., 2014). The analysis of nucleotide variation in Oryza sativa and its wild relatives found that only 20 and 10% of the diversity in wild rice species were retained in the cultivated indica and japonica rice sub-species, respectively (Zhu et al., 2007). Large-scale cultivation of genetically uniform varieties makes them more vulnerable to disease epidemics because of the high selection pressure on the pathogen populations. Because of changing and evolving pathogen populations, crop plant diseases are continuous problems that are influenced by various factors including selection pressure, environmental conditions and the changing climate. Consequently, crop protection requires continuous improvements to keep pace with pathogen evolution and disease severity. This implies the need for new resistance genes and alleles beyond those available in the breeding gene pool, such as genes and alleles from landraces and wild relatives. Much of the genetic diversity of crop plants is being maintained in the seed banks that have large collections of germplasm of diverse origins, but most accessions that are not yet fully annotated and their genotypic diversity is not well-understood (McCouch et al., 2012). We explored the allele diversity of Pib in 467 rice accessions originated from 12 major rice-growing countries and identified 25 new Pib alleles that represent one of the largest allelic series known among blast R genes.
Among the 25 Pib alleles, 14 alleles are country specific, suggesting the existence of distinct M. oryzae populations in different geographical regions. Accordingly, currently unknown selective pressures are causing the co-evolution of Pib alleles with sequence polymorphisms specific to a geographical region. In addition to SNPs that are partly or fully shared within or between the four Pib allele categories, several unique SNPs/InDels were identified in 13 of the Pib alleles. The shared SNPs likely resulted from gene conversion events and the unique polymorphisms could be the result of selection pressure specific to the M. oryzae populations in a particular geographical location. Studies on various M. oryzae populations collected from different parts of China and allelic variation among AvrPib alleles indicated that Pib shaped the genetic architecture of M. oryzae populations (Zhang et al., 2015). It is also reported that some rare AvrPib types were restricted to M. oryzae populations in Southern China as compared to those in Northeastern China, particularly indicating host selection pressure owing to Pib donor cultivars in these climatically distinct regions of China (Zhang et al., 2015). However, the effects of the unique SNPs/InDels observed in the Pib alleles need to be experimentally verified through functional validation and comparison of resistance specificities of alleles with shared SNPs and that of alleles with additional unique polymorphism(s). These different Pib alleles could be subjected to interaction studies against recently reported AvrPib and its allelic variants (Zhang et al., 2015), as such analysis would help in further strengthening our understanding of the molecular interactions between Pib and AvrPib as well as their co-evolution.
Nucleotide diversity, positive selection sites and selection pressure on R gene alleles can be assigned either to a single or more domains. For example, specificity differences between alleles of the flax rust resistance gene L can be determined by both the LRR and TIR regions, whereas, for the barley powdery mildew resistance gene Mla, most of the positive selection sites are found within the LRR of functional alleles (Ellis et al., 1999; Seeholzer et al., 2010). Similarly, for Pib we identified alleles in which diversity and selection pressure appears to be restricted to either LRR (category I) or NB-ARC2 and LRR (category II and IV) and in which all domains are under selection pressure (category III). Together, our results suggest that diversified selection events have occurred at the Pib locus. Moreover, category I alleles have higher diversity (unique SNPs), variable segregation sites between alleles, η > S and a positive Tajima’s D value. This indicates that category I alleles are under either balancing selection or, more likely, divergence selection, eventually resulting in further divergence, which is supported by the phylogenetic sub-clustering of alleles (Figure 6B).
The LRR domain determines the resistance specificity of several R proteins and physically interacts with cognate effector proteins. Based on the functional analysis of R proteins such as Arabidopsis RPP1 (Krasileva et al., 2010), wheat Pm3 (Bhullar et al., 2009, 2010), flax L (Ellis et al., 1999; Ravensdale et al., 2012) and barley Mla (Seeholzer et al., 2010), the LRR domain is necessary and in some cases sufficient to maintain the same recognition specificity as that of the full-length R protein (Krasileva et al., 2010). It was shown that replacing six amino acids in the LRR domain of flax P2 can confer P1 specificity (Dodds et al., 2001). The recognition of AvrL567 by L6 is LRR controlled, although, the TIR domain can affect the specificity of pathogen recognition (McHale et al., 2006; Ellis et al., 2007). We found a high rate of nucleotide polymorphisms in the LRR region of Pib alleles. If only the exon regions of the 25 alleles are considered, then 47% of the total nucleotide polymorphisms are within the LRR domain, resulting in 55% of the total amino acid changes. This suggests that the Pib LRR domain has a significant role in establishing recognition specificities for M. oryzae effector proteins. In addition, the cysteine cluster in the center of the Pib LRR (Wang et al., 1999) is strongly maintained despite the loss and gain of two cysteine residues as the result of AA changes in the category II and IV proteins. Although the function of the cysteine cluster is still unknown, it likely has a structural and/or functional role in the Pib protein.
The CC domain is also proposed to have an important role in R protein functioning (Bai et al., 2012; Hao et al., 2013; Zhai et al., 2014), and the EDVID motif is reported to be important for intramolecular interaction and function of CC domain (Rairdan et al., 2008; Wang et al., 2015). The CC domain and the two EDVID motif variants in/around CC domain are conserved among the Pib proteins, however, it is not known which of the two EDVID motif variants is critical for its function. The NB-ARC domain functions as molecular switch that stabilizes the functional structure of R proteins (McHale et al., 2006; Tameling et al., 2006; Williams et al., 2011). The disordered regions within and between the two Pib NB-ARC domains could also facilitate intramolecular interactions that stabilize the proteins. In Pid3, functional AA polymorphisms between resistant and susceptible orthologs are located in their Kinase 1a motif of NBS domains (Xu et al., 2014). In Pib, all Kinase motifs in category I proteins are highly conserved. Similarly, despite several AA changes in the NB-ARC domains of category II and IV Pib proteins, the sequences in the Kinase 1a, Kinase 2, and Kinase 3a motifs within these two domains are highly conserved, including sequences critical for ATP hydrolysis (McHale et al., 2006; Tameling et al., 2006; Chattopadhyaya and Pal, 2008). It is therefore unlikely that the minor AA changes we found in some of the category II and IV Kinase motifs would result in Pib loss-of-function proteins. The GLPL and MHD motifs of the NB-ARC domains have also been shown to affect the function of R proteins (van Ooijen et al., 2008; Williams et al., 2011; Wang et al., 2015). The GLPL motif located in the NB-ARC2 domain of Pib is highly conserved among all Pib proteins. Among the MHD motifs, the MHD1 variant (VHD) is highly conserved but the MHD2 is identified in two different forms, MRD or MID. The histidine and aspartate are the most conserved residues of MHD1 in most of the plant R proteins (van Ooijen et al., 2008) and aspartate is the most conserved residue in case of MHD2 (Wang et al., 2015). The observation of conserved residues of MHD1 and MHD2 motifs of Pib is consistent with these reports.
Most of the positive selection sites in Mla proteins are solvent-exposed AA clustered in the LRR domain (Seeholzer et al., 2010). Similarly, among the category I, II, and IV Pib proteins, 79.5 and 71.4% of the polymorphic AA in NB-ARC2 and LRR domains, respectively, are predicted to be solvent-exposed. This suggests that surface-exposed polymorphic AA in Pib proteins might have functional roles such as interactions with one or more effector proteins or metabolites that establish resistance specificities. However, these solvent-exposed residue predictions are only sequence-based and not supported by in depth structure analysis. To validate such functional roles requires complementation assays and/or screening against blast isolates from various geographical locations. Once validated, the distinct polymorphisms among the Pib alleles can be combined to generate novel resistance specificities. For example, the resistance specificities of wheat Pm3d and Pm3e alleles were successfully fused into a single chimeric allele by intragenic allele pyramiding (Brunner et al., 2010). Similarly, a recombinant flax rust resistance allele L2-L10 has a novel recognition specificity that is distinct from the L2 and L10 parental alleles (Ellis et al., 1999). The detailed understanding of Pib alleles and polymorphisms that contribute to differences in resistance specificity to M. oryzae isolates will enable us to combine specificities through inter-allelic pyramiding or using CRISPR/Cas-mediated editing of polymorphic sites.
Materials and Methods
Plant Material and Molecular Screening for Pib
The rice germplasm material was obtained from the International Rice GenBank (IRG) of the International Rice Research Institute (IRRI), Philippines. A set of 467 rice accessions with a phenotypic score of 0 in a uniform nursery screening and against at least two of five tested M. oryzae isolates (Vasudevan et al., 2014) was chosen for Pib allele mining. The isolates were selected based on their virulence pattern on near isogenic lines for major rice blast resistance genes (Vasudevan et al., 2014) and it is not known if these isolates have a functional AvrPib. Molecular screening for identifying the accessions carrying the Pib gene was carried out using the gene specific marker Nsb (Cho et al., 2007; Forward: 5′-ATCAACTCTGCCACAAAATCC; Reverse: 5′-CCCATATCACCACTTGTTCCCC). Genomic DNA extracted from a Pib monogenic line was used as a positive control and that from susceptible rice cultivar LTH was used as the negative control, in addition to a water control. Additionally, the primers for amplification of the rice actin gene were used in PCR reaction mixtures as an internal PCR control (Forward: 5′-TTATGGTTGGGATGGGACA; Reverse: 5′-AGCACGGCTTGAATAGCG).
Isolation and Cloning of Pib Alleles
The forward primer 5′-CCACAAAATCCATTCAAAAATAGA ACAGAGCA and reverse primer 5′-GGAGCACGGCAAAGT AACTCCAAAGGAG were used to amplify the full-length coding region of Pib. PCR was performed with an initial denaturation at 95°C for 5 min; followed by additional denaturation at 98°C for 20 s, annealing at 63°C for 20 s, extension at 72°C for 3 min (these three steps repeated for 35 cycles); followed by final extension at 72°C for 6 min, using KAPA HiFi HotStart DNA polymerase (high fidelity proof-reading enzyme). The samples yielding low amplified product were repeated with three or more replicates and the amplified products were pooled to process further for cloning. The amplified products were cloned using pJET1.2 blunt end cloning vector.
Sequencing and Sequence Analysis
In addition to the standard forward and reverse primers from the pJET1.2 cloning vector, seven additional primers were used for the complete sequence coverage of Pib alleles (Supplementary Table S3). In addition, a second set of nine primers was designed to cover the sequence gap that might arise from the first round of sequencing, as well as for the re-confirmation of SNPs and/or InDels within the overlap regions. The primers for the isolation of Pib alleles and internal primers for sequencing were designed using CLC-genomics workbench.
DNA sequencing was performed using Applied Biosystems Capillary Sequencer 3730. Sequence assembly, consensus development, and alignments were done using CLC Genomics Workbench (version-7.5). The Pib sequence from cultivar Engkatek (Accession No. AB013448.1) was used as the reference for sequence comparisons. Multiple sequence alignment was performed to identify the SNPs. Alleles with unique SNPs or InDels were re-amplified (from genomic DNA of respective rice accessions), re-cloned and re-sequenced for SNP/InDel confirmation. For alleles identified in more than four accessions (thereby representing multiple independent amplification and cloning events), no repetitions were made. Sequence polymorphism analyses such as, number of segregation sites and mutations, sliding window analysis of nucleotide diversity and Tajima’s D test were performed using ‘DnaSP, version 5′ (Librado and Rozas, 2009).
Phylogenetic Analysis
Phylogenetic analysis was performed using full-length nucleotide sequences of the newly identified Pib alleles together with reference Pib_Engkatek. Phylogenetic analysis was also performed for the NB-ARC-1, NB-ARC-2, and LRR domains separately (Supplementary Figure S2). The sequence alignments were performed using MAFFT and the tree was constructed using RAxML on an online server1, with default parameters and the bootstrap analysis was performed with 100 replicates.
Protein Prediction and Analysis
The intron and exon regions were assigned as reported for the reference Pib sequence from cv. Engkatek (Wang et al., 1999). The protein sequences of Pib alleles were obtained using the EXPASY translation tool. The kinase motifs were assigned as reported for Pib (Wang et al., 1999). The CC, NB-ARC and LRR domains were predicted using SMART server (Letunic et al., 2014). The patterns and PTM sites were predicted using ScanProsite server (de Castro et al., 2006). The secondary structure, solvent accessibility, disorders and flexibility regions were predicted and analyzed using PredictProtein server (Yachdav et al., 2014). NetSurfP server was used for sequence-based prediction of the surface accessibility of amino acid residues of Pib proteins (Petersen et al., 2009).
Accession Numbers
Sequence data from this article can be found in GenBank data libraries with accession numbers, Pib_40286 – KR527222, Pib_16784 – KR527223, Pib_5151 – KR527224, Pib_11147 – KR527225, Pib_29336 – KR527226, Pib_41515 – KR527227, Pib_41341 – KR527228, Pib_32909 – KR527229, Pib_4633 – KR527230, Pib_26038 – KR527231, Pib_24475 – KR527232, Pib_5284 – KR527233, Pib_29436 – KR527234, Pib_13373 – KR527235, Pib_10101 – KR527236, Pib_9829 – KR527237, Pib_14981 – KR527238, Pib_23713 – KR527239, Pib_23761 – KR527240, Pib_23712 – KR527241, Pib_23721 – KR527242, Pib_40432 – KR527243, Pib_16706 – KR527244, Pib_4868 – KR527245, Pib_2476 – KR527246.
Author Contributions
NB: conceived and designed the experiment, KV: carried out the experiments, CVC: supported the phenotypic screening at IRRI, KV and NB: analyzed the data, KV, WG, and NB: discussed the data, KV and NB: wrote the manuscript, NB and WG: edited the final manuscript. All authors have read the manuscript and agree with its content.
Funding
The research was supported by funds from ETH Zurich to WG and from ETH Global (previously North-South Centre) to NB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the IRG of the International Rice Research Institute (IRRI), Philippines, for providing seed material. We thank Dr. Thomas Wicker for generating the schematic representation of alleles presented in Figure 2. DNA sequencing was performed at Genetic Diversity Centre (GDC) of ETH Zurich.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00915
Footnotes
References
Ashikawa, I., Hayashi, N., Yamane, H., Kanamori, H., Wu, J., Matsumoto, T., et al. (2008). Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180, 2267–2276. doi: 10.1534/genetics.108.095034
Bai, S., Liu, J., Chang, C., Zhang, L., Maekawa, T., Wang, Q., et al. (2012). Structure-function analysis of barley NLR immune receptor MLA10 reveals its cell compartment specific activity in cell death and disease resistance. PLoS Pathog. 8:e1002752. doi: 10.1371/journal.ppat.1002752
Bhullar, N. K., Mackay, M., and Keller, B. (2010). Genetic diversity of the Pm3 powdery mildew resistance alleles in wheat gene bank accessions as assessed by molecular markers. Diversity 2, 768–786. doi: 10.3390/d2050768
Bhullar, N. K., Street, K., Mackay, M., Yahiaoui, N., and Keller, B. (2009). Unlocking wheat genetic resources for the molecular identification of previously undescribed functional alleles at the Pm3 resistance locus. Proc. Natl. Acad. Sci. U.S.A. 106, 9519–9524. doi: 10.1073/pnas.0904152106
Brunner, S., Hurni, S., Streckeisen, P., Mayr, G., Albrecht, M., Yahiaoui, N., et al. (2010). Intragenic allele pyramiding combines different specificities of wheat Pm3 resistance alleles. Plant J. 64, 433–445. doi: 10.1111/j.1365-313X.2010.04342.x
Bryan, G. T., Wu, K. S., Farrall, L., Jia, Y., Hershey, H. P., McAdams, S. A., et al. (2000). A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell 12, 2033–2046. doi: 10.2307/3871103
Césari, S., Kanzaki, H., Fujiwara, T., Bernoux, M., Chalvon, V., Kawano, Y., et al. (2014). The NB-LRR proteins RGA4 and RGA5 interact functionally and physically to confer disease resistance. EMBO J. 33, 1–19. doi: 10.15252/embj.201487923
Chattopadhyaya, R., and Pal, A. (2008). Three-dimensional models of NB-ARC domains of disease resistance proteins in tomato, Arabidopsis, and flax. J. Biomol. Struct. Dyn. 25, 357–371. doi: 10.1080/07391102.2008.10507184
Chen, X., Shang, J., Chen, D., Lei, C., Zou, Y., Zhai, W., et al. (2006). A B-lectin receptor kinase gene conferring rice blast resistance. Plant J. 46, 794–804. doi: 10.1111/j.1365-313X.2006.02739.x
Cho, Y., Kwon, S., Choi, I., and Lee, S. (2007). Identification of major blast resistance genes in Korean rice varieties (Oryza sativa L.) using molecular markers. J. Crop Sci. 10, 265–276.
Das, A., Soubam, D., Singh, P. K., Thakur, S., Singh, N. K., and Sharma, T. R. (2012). A novel blast resistance gene, Pi54rh cloned from wild species of rice, Oryza rhizomatis confers broad spectrum resistance to Magnaporthe oryzae. Funct. Integr. Genomics 12, 215–228. doi: 10.1007/s10142-012-0284-1
de Castro, E., Sigrist, C. J. A., Gattiker, A., Bulliard, V., Langendijk-Genevaux, P. S., Gasteiger, E., et al. (2006). ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, 362–365. doi: 10.1093/nar/gkl124
Dean, R. A., Talbot, N. J., Ebbole, D. J., Farman, M. L., Mitchell, T. K., Orbach, M. J., et al. (2005). The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434, 980–986. doi: 10.1038/nature03449
Devanna, N. B., Vijayan, J., and Sharma, T. R. (2014). The blast resistance gene Pi54of cloned from Oryza officinalis interacts with Avr-Pi54 through its novel non-LRR domains. PLoS ONE 9:e104840. doi: 10.1371/journal.pone.0104840
Dodds, P. N., Lawrence, G. J., and Ellis, J. G. (2001). Six amino acid changes confined to the leucine-rich repeat beta-strand/beta-turn motif determine the difference between the P and P2 rust resistance specificities in flax. Plant Cell 13, 163–178. doi: 10.1105/tpc.13.1.163
Ellis, J. G., Dodds, P. N., and Lawrence, G. J. (2007). Flax rust resistance gene specificity is based on direct resistance–avirulence protein interactions. Annu. Rev. Phytopathol. 45, 289–306. doi: 10.1146/annurev.phyto.45.062806.094331
Ellis, J. G., Lawrence, G. J., Luck, J. E., and Dodds, P. N. (1999). Identification of regions in alleles of the flax rust resistance gene L that determine differences in gene-for-gene specificity. Plant Cell 11, 495–506. doi: 10.2307/3870876
Fukuoka, S., Saka, N., Koga, H., Ono, K., Shimizu, T., Ebana, K., et al. (2009). Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325, 998–1001. doi: 10.1126/science.1175550
Fukuoka, S., Yamamoto, S.-I., Mizobuchi, R., Yamanouchi, U., Ono, K., Kitazawa, N., et al. (2014). Multiple functional polymorphisms in a single disease resistance gene in rice enhance durable resistance to blast. Sci. Rep. 4, 1–7. doi: 10.1038/srep04550
Gupta, S. K., Rai, A. K., Kanwar, S. S., and Sharma, T. R. (2012). Comparative analysis of zinc finger proteins involved in plant disease resistance. PLoS ONE 7:e42578. doi: 10.1371/journal.pone.0042578
Hao, W., Collier, M. S., Moffett, P., and Chai, J. (2013). Structural basis for the interaction between the potato virus X resistance protein (Rx) and its cofactor Ran GTPase-activating protein 2 (RanGAP2). J. Biol. Chem. 288, 35868–35876. doi: 10.1074/jbc.M113.517417
Huang, J., Si, W., Deng, Q., Li, P., and Yang, S. (2014). Rapid evolution of avirulence genes in rice blast fungus Magnaporthe oryzae. BMC Genet. 15:45. doi: 10.1186/1471-2156-15-45
Kilian, B., and Graner, A. (2012). NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genomics 11, 38–50. doi: 10.1093/bfgp/elr046
Krasileva, K. V., Dahlbeck, D., and Staskawicz, B. J. (2010). Activation of an Arabidopsis resistance protein is specified by the in planta association of its leucine-rich repeat domain with the cognate oomycete effector. Plant Cell 22, 2444–2458. doi: 10.1105/tpc.110.075358
Lee, S. K., Song, M. Y., Seo, Y. S., Kim, H. K., Ko, S., Cao, P. J., et al. (2009). Rice Pi5-mediated resistance to Magnaporthe oryzae requires the presence of two coiled-coil-nucleotide-binding-leucine-rich repeat genes. Genetics 181, 1627–1638. doi: 10.1534/genetics.108.099226
Letunic, I., Doerks, T., and Bork, P. (2014). SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260. doi: 10.1093/nar/gku949
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, W., Liu, J., Ning, Y., Ding, B., Wang, X., Wang, Z., et al. (2013). Recent progress in understanding PAMP- and effector-triggered immunity against the rice blast fungus Magnaporthe oryzae. Mol. Plant 6, 605–620. doi: 10.1093/mp/sst015
Lv, Q., Xu, X., Shang, J., Jiang, G., Pang, Z., Zhou, Z., et al. (2013). Functional analysis of Pid3-A4, an ortholog of rice blast resistance gene Pid3 revealed by allele mining in common wild rice. Phytopathology 103, 594–599. doi: 10.1094/PHYTO-10-12-0260-R
Ma, J., Lei, C., Xu, X., Hao, K., Wang, J., Cheng, Z., et al. (2015). Pi64, Encoding a novel CC-NBS-LRR protein, confers resistance to leaf and neck blast in rice. MPMI 28, 558–568. doi: 10.1094/MPMI-11-14-0367-R
McCouch, S. R., McNally, K. L., Wang, W., and Hamilton, R. S. (2012). Genomics of gene banks: a case study in rice. Am. J. Bot. 99, 407–423. doi: 10.3732/ajb.1100385
McHale, L., Tan, X., Koehl, P., and Michelmore, R. W. (2006). Plant NBS-LRR proteins: adaptable guards. Genome Biol. 7:212. doi: 10.1186/gb-2006-7-4-212
Okuyama, Y., Kanzaki, H., Abe, A., Yoshida, K., Tamiru, M., Saitoh, H., et al. (2011). A multifaceted genomics approach allows the isolation of the rice Pia-blast resistance gene consisting of two adjacent NBS-LRR protein genes. Plant J. 66, 467–479. doi: 10.1111/j.1365-313X.2011.04502.x
Perelló, A., Martinez, I., and Molina, M. (2015). First report of virulence and effects of Magnaporthe oryzae isolates causing wheat blast in argentina. Plant Dis. 99, 1177. doi: 10.1094/PDIS-11-14-1182-PDN
Petersen, B., Petersen, T. N., Andersen, P., Nielsen, M., and Lundegaard, C. (2009). A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol. 9:51. doi: 10.1186/1472-6807-9-51
Rairdan, J. G., Collier, M. S., Sacco, A. M., Baldwin, T. T., Boettrich, T., and Moffett, P. (2008). The coiled-coil and nucleotide binding domains of the potato Rx disease resistance protein function in pathogen recognition and signaling. Plant Cell 20, 739–751. doi: 10.1105/tpc.107.056036
Ravensdale, M., Bernoux, M., Ve, T., Kobe, B., Thrall, P. H., Ellis, J. G., et al. (2012). Intramolecular interaction influences binding of the Flax L5 and L6 resistance proteins to their AvrL567 ligands. PLoS Pathog. 8:e1003004. doi: 10.1371/journal.ppat.1003004
Roychowdhury, M., Jia, Y., Jia, M. H., Fjellstrom, R., and Cartwright, R. D. (2012). Identification of the rice blast resistance gene Pib in the national small grains collection. Phytopathology 102, 700–706. doi: 10.1094/PHYTO-02-12-0026-R
Seeholzer, S., Tsuchimatsu, T., Jordan, T., Bieri, S., Pajonk, S., Yang, W., et al. (2010). Diversity at the Mla powdery mildew resistance locus from cultivated barley reveals sites of positive selection. Mol. Plant. Microbe. Interact. 23, 497–509. doi: 10.1094/MPMI-23-4-0497
Sesma, A., and Osbourn, A. E. (2004). The rice leaf blast pathogen undergoes developmental processes typical of root-infecting fungi. Nature 431, 582–586. doi: 10.1038/nature02880
Spence, C., Alff, E., Johnson, C., Ramos, C., Donofrio, N., Sundaresan, V., et al. (2014). Natural rice rhizospheric microbes suppress rice blast infections. BMC Plant Biol. 14:130. doi: 10.1186/1471-2229-14-130
Tameling, W. I. L., Vossen, J. H., Albrecht, M., Lengauer, T., Berden, J. A., Haring, M. A., et al. (2006). Mutations in the NB-ARC domain of I-2 that impair ATP hydrolysis cause autoactivation. Plant Physiol. 140, 1233–1245. doi: 10.1104/pp.105.073510
Tanksley, S. D., and McCouch, S. R. (1997). Seed banks and molecular maps: unlocking genetic potential from the wild. Science 277, 1063–1066. doi: 10.1126/science.277.5329.1063
Thakur, S., Singh, P. K., Das, A., Rathour, R., Variar, M., Prashanthi, S. K., et al. (2015). Extensive sequence variation in rice blast resistance gene Pi54 makes it broad spectrum in nature. Front. Plant Sci. 6:345. doi: 10.3389/fpls.2015.00345
van Ooijen, G., Mayr, G., Kasiem, M. A. M., Albrecht, M., Cornelissen, J. C. B., and Takken, L. W. F. (2008). Structure–function analysis of the NB-ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383–1397. doi: 10.1093/jxb/ern045
Vasudevan, K., Gruissem, W., and Bhullar, K. N. (2015). Identification of novel alleles of the rice blast resistance gene Pi54. Sci. Rep. 5:15678. doi: 10.1038/srep15678
Vasudevan, K., Vera Cruz, C. M., Gruissem, W., and Bhullar, N. K. (2014). Large scale germplasm screening for identification of novel rice blast resistance sources. Front. Plant Sci. 5:505. doi: 10.3389/fpls.2014.00505
Wang, D., Guo, C., Huang, J., Yang, S., Tian, D., and Zhang, X. (2014). Allele-mining of rice blast resistance genes at AC134922 locus. Biochem. Biophys. Res. Commun. 446, 1085–1090. doi: 10.1016/j.bbrc.2014.03.056
Wang, G.-F., Ji, J., Ei-Kasmi, F., Dangl, L. J., Johal, G., and Balint-Kurti, J. P. (2015). Molecular and functional analyses of a maize autoactive NB-LRR protein identify precise structural requirements for activity. PLoS Pathog. 11:e1004674. doi: 10.1371/journal.ppat.1004674
Wang, Z., Yamanouchi, U., Katayose, Y., Sasaki, T., and Yano, M. (2001). Expression of the Pib rice-blast resistance gene family is up-regulated by environmental conditions favoring infection and by chemical signals that trigger secondary plant defenses. Plant Mol. Biol. 47, 653–661. doi: 10.1023/A:1012457113700
Wang, Z.-X., Yano, M., Yamanouchi, U., Iwamoto, M., Monna, L., Hayasaka, H., et al. (1999). The Pib gene for rice blast resistance belongs to the nucleotide binding and leucine-rich repeat class of plant disease resistance genes. Plant J. 19, 55–64. doi: 10.1046/j.1365-313X.1999.00498.x
Warschefsky, E., Penmetsa, R. V., Cook, D. R., and von Wettberg, E. J. B. (2014). Back to the wilds: tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 101, 1791–1800. doi: 10.3732/ajb.1400116
Williams, J. S., Sornaraj, P., deCourcy-Ireland, E., Menz, R. I., Kobe, B., Ellis, G. J., et al. (2011). An autoactive mutant of the M flax rust resistance protein has a preference for binding ATP, whereas wild-type M protein binds ADP. MPMI 24, 897–906. doi: 10.1094/MPMI-03-11-0052
Xu, X., Lv, Q., Shang, J., Pang, Z., Zhou, Z., Wang, J., et al. (2014). Excavation of Pid3 orthologs with differential resistance spectra to Magnaporthe oryzae in rice resource. PLoS ONE 9:e93275. doi: 10.1371/journal.pone.0093275
Xue, M., Yang, J., Li, Z., Hu, S., Yao, N., Dean, R. A., et al. (2012). Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet. 8:e1002869. doi: 10.1371/journal.pgen.1002869
Yachdav, G., Kloppmann, E., Kajan, L., Hecht, M., Goldberg, T., Hamp, T., et al. (2014). PredictProtein-an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 42, W337–W343. doi: 10.1093/nar/gku366
Yokoo, M. (2005). Introduction of Piz-t and Pib genes for blast disease resistance from indica varieties and their utilization for genetic researches in rice. Jpn Agric. Res. Q. 39, 239–245. doi: 10.6090/jarq.39.239
Yoshida, K. K., Saitoh, H., Fujisawa, S., Kanzaki, H., Matsumura, H., Yoshida, K. K., et al. (2009). Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen Magnaporthe oryzae. Plant Cell 21, 1573–1591. doi: 10.1105/tpc.109.066324
Yuan, B., Zhai, C., Wang, W., Zeng, X., Xu, X., Hu, H., et al. (2011). The Pik-p resistance to Magnaporthe oryzae in rice is mediated by a pair of closely linked CC-NBS-LRR genes. Theor. Appl. Genet. 122, 1017–1028. doi: 10.1007/s00122-010-1506-3
Zhai, C., Zhang, Y., Yao, N., Lin, F., Liu, Z., Dong, Z., et al. (2014). Function and interaction of the coupled genes responsible for Pik-h encoded rice blast resistance. PLoS ONE 9:e98067. doi: 10.1371/journal.pone.0098067
Zhang, S., Wang, L., Wu, W., He, L., Yang, X., and Pan, Q. (2015). Functional and evolution of Magnaporthe oryzae avirulence gene AvrPib responding to the rice blast resistance gene Pib. Sci. Rep. 5:11642. doi: 10.1038/srep11642
Zhou, B., Qu, S., Liu, G., Dolan, M., Sakai, H., Lu, G., et al. (2006). The eight amino-acid differences within three leucine-rich repeats between Pi2 and Piz-t resistance proteins determine the resistance specificity to Magnaporthe grisea. Mol. Plant. Microbe. Interact. 19, 1216–1228. doi: 10.1094/MPMI-19-1216
Keywords: rice blast resistance, M. oryzae, Pib, allele mining, genetic diversity, SNP, NB-ARC, LRR
Citation: Vasudevan K, Vera Cruz CM, Gruissem W and Bhullar NK (2016) Geographically Distinct and Domain-Specific Sequence Variations in the Alleles of Rice Blast Resistance Gene Pib. Front. Plant Sci. 7:915. doi: 10.3389/fpls.2016.00915
Received: 30 November 2015; Accepted: 09 June 2016;
Published: 23 June 2016.
Edited by:
Benjamin Schwessinger, Australian National University, AustraliaReviewed by:
Liliana M. Cano, University of Florida, USASimon John Williams, University of Queensland, Australia
Copyright © 2016 Vasudevan, Vera Cruz, Gruissem and Bhullar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Navreet K. Bhullar, Ymh1bGxhcm5AZXRoei5jaA==