Abstract
Small RNA (sRNA)-mediated gene silencing represents a conserved regulatory mechanism controlling a wide diversity of developmental processes through interactions of sRNAs with proteins of the ARGONAUTE (AGO) family. On the basis of a large phylogenetic analysis that includes 206 AGO genes belonging to 23 plant species, AGO genes group into four clades corresponding to the phylogenetic distribution proposed for the ten family members of Arabidopsis thaliana. A primary analysis of the corresponding protein sequences resulted in 50 sequences of amino acids (blocks) conserved across their linear length. Protein members of the AGO4/6/8/9 and AGO1/10 clades are more conserved than members of the AGO5 and AGO2/3/7 clades. In addition to blocks containing components of the PIWI, PAZ, and DUF1785 domains, members of the AGO2/3/7 and AGO4/6/8/9 clades possess other consensus block sequences that are exclusive of members within these clades, suggesting unforeseen functional specialization revealed by their primary sequence. We also show that AGO proteins of animal and plant kingdoms share linear sequences of blocks that include motifs involved in posttranslational modifications such as those regulating AGO2 in humans and the PIWI protein AUBERGINE in Drosophila. Our results open possibilities for exploring new structural and functional aspects related to the evolution of AGO proteins within the plant kingdom, and their convergence with analogous proteins in mammals and invertebrates.
Introduction
Small RNA (sRNA)-mediated gene silencing has proven to be one of several important mechanisms that regulate plant growth and development. Its action relies on the production of 20–30 nucleotides (nt) double stranded RNAs (dsRNA) that are produced by proteins of the DICER-LIKE (DCL) family after cleavage of long single-stranded RNA precursors that are copied by a family of RNA-dependent RNA polymerases (RDRs) to generate long dsRNA. sRNAs can bind to one or several ARGONAUTE (AGO) proteins and use complementary base pairing to identify and silence their targets after binding single-stranded sRNAs. The mechanisms of AGO-mediated target silencing include transcript cleavage, translational repression, methylation, and subsequent chromatin modifications (Ghildiyal and Zamore, 2009). AGO proteins of plants and animals are classified in three categories based on their association with different types of sRNAs. Whereas, some AGOs in plants and animals interact primarily with microRNAs and short-interfering RNAs (siRNAs), others exclusive of the animal kingdom associate with so-called piwi-interacting RNAs (piRNAs; also known as PIWI proteins). A third group is composed of AGO proteins only present in the worm Caenorhabditis elegans (Vaucheret, 2008). Several studies have revealed that AGO proteins contain an N-terminal domain, a PAZ and MID domains, and a PIWI domain that adopts an RNaseH conformation with endonuclease activity (Song et al., 2004); however, to this date no three dimensional conformation of a plant AGO protein has been elucidated, and therefore the evolutionary trends that have shaped the structure and function of these proteins remains to be determined.
Several phylogenetic studies have analyzed the evolutionary relationships of major gene families involved in sRNA mediated silencing in animals and plants (Cerutti and Casas-Mollano, 2006; Kapoor et al., 2008; Murphy et al., 2008; de Jong et al., 2009; Tabach et al., 2013; Swarts et al., 2014). In flowering plants, phylogenetic studies of AGO, RNA-DEPENDENT RNA POLYMERASE (RDR), and other gene components of the silencing machinery have been restricted to a few species such as Arabidopsis thaliana (Arabidopsis), Populus trichocarpa (poplar), and Oryza sativa (rice; Kapoor et al., 2008; Qian et al., 2011; Bai et al., 2012; Huang et al., 2015), resulting in a structure coalescing around four clades that follows the distribution of the ten AGO genes found in the Arabiodpsis genome. Expression and functional analysis of members of particular clades relates these phylogenetic relationships to their potential function (Nonomura et al., 2007; Kapoor et al., 2008; Havecker et al., 2010); however, the evolutionary implications of this phylogenetic architecture cannot be understood without a in-depth analysis that incorporates genes from a wide array of land plants, including algae, bryophytes, and flowering plants for which a complete genome sequence is available.
Here we present a phylogenetic approximation of 206 AGO genes contained in 23 different genomes, including algae, bryophytes, gymnosperms, and flowering plants. After showing that all genes were included in one of four major clades encompassing all the diversity of plant AGOs, we performed a sequence analysis that included 185 proteins corresponding to genes included in our phylogeny. We show that the AGO4/6/8/9 and AGO1/10 clades show the most conserved linear organization of 50 identified amino acid sequences (named blocks), whereas this linear organization is more variable in members of the AGO2/3/7 and AGO5 clades. In addition to components of the PIWI, PAZ, and DUF1785 domains, we identified several blocks of unknown function that show a predictable position within a conserved primary structure and are exclusive of proteins from a specific clade. Our analysis also revealed specific block series that are conserved between flowering plants and animal organisms, including mammals, insects, nematodes, and fungi. In particular, we identified blocks containing specific motifs involved in posttranslational modifications of AGO2 and AUBERGINE in humans and Drosophila, respectively. Our overall results expand the phylogenetic understanding of AGO proteins in plants by establishing new linear patterns associated with their evolutionary relationship.
Materials and methods
Sequence selection and construction of the primary database
Database searching was conducted in Phytozome version 5.0 (http://www.phytozome.net) for 15 of the 23 genomes included in this study, using the keywords ARGONAUTE and PIWI in the biomart tool. BLAST searches, using the Arabidopsis lyrata AGO genes as query, were conducted for the A. thaliana, O. sativa, and Zea mays genomes, under default parameters in TAIR v.9 (http://www.arabidopsis.org/), TIGR (http://blast.jcvi.org/euk-blast/index.cgi?project=osa1), and Maizesequence.org (http://maizesequence.org/index.html), respectively. Additional AGO sequences were obtained by assembling ESTs coming from Pinus taeda, Pinus radiata, Lactuca sativa, Citrus sinensis, and Amborella trichopoda from the KEGG database and added to the sequences used for the subsequent analysis. Nucleotide sequences (coding sequence) were gathered from all selected genomes (Supplementary File 1). To confirm that the selected sequences belong to the AGO family, the coding region was translated into amino acid sequence using Seaview (Gouy et al., 2010) and used to perform a domain annotation using the batch search tool in Pfam protein database (Finn et al., 2010; Supplementary File 2), discarding the sequences that did not contain the canonical domains reported for AGO proteins (PAZ and PIWI), likely due to misannotations in the original databases.
Sequence edition and phylogenetic reconstruction
Nucleotide sequences were edited to extract conserved domains, and used to construct phylogenetic trees under both Bayesian inference (BI) and maximum likelihood (ML) frameworks. Amino acid guided alignments were generated using MUSCLE in SeaView (Gouy et al., 2010). Because many inter-domain regions showed inconsistencies such as long-branch attraction and obvious misannotations resulting in misplaced taxon outgroups, a perl script (Supplementary File 3) was designed to extract the DNA sequence encoding the domains DUF1785 (recently renamed as Argonaute linker 1 domain), PAZ and PIWI from the total sequences, using the coordinates found in the Pfam batch search output. This resulted in an elimination of inter-domain regions, reducing the total length of analyzed sequences (Supplementary File 4).
Multiple sequence alignments were generated using Muscle (Edgar, 2004) by translating DNA into amino acid sequence before performing the alignment and then untranslating the sequences to maintain the coding frame at the DNA level using SeaView. Following alignment verification and editing between species, intra-domain regions showing obvious misalignments were trimmed using Gblocks (Talavera and Castresana, 2007; Supplementary File 5). For each phylogenetic reconstruction by BI and ML, the evolutionary model was chosen using Modelgenerator (Keane et al., 2006), resulting in the selection of the General Time Reversible (GTR) model, including the proportion of invariable sites and the gamma distribution into four categories (GTR+I+G). The ML analysis was conducted using PhyML version 3 (Guindon et al., 2009); nucleotide frequencies, alpha value, and proportion of invariable sites were fixed according to the values obtained from Modelgenerator, and the rest of parameters were optimized in the ML framework. Statistical support for the analysis was obtained by performing 2000 bootstrap replicates. Bayesian analyses were conducted using MrBayes 3.1 (http://mrbayes.csit.fsu.edu/; Ronquist and Huelsenbeck, 2003) as follows: two independent runs were performed for 10 million generations with eight chains (two cold and six heated), using fixed priors and sampling frequency of each 100 generations, and discarding 25% as burn-in and computing a majority-rule consensus of the trees sampled during the run. Chlamydomonas reinhardtii AGO sequences were selected as the outgroup. To support the the Bayesian inference search and ensure accurate tree sampling, the trace files generated by MrBayes were analyzed by Tracer v1.6 (Rambaut and Drummond, 2007), generating estimates, density and analysis of variance values (Supplementary Figure 1). An independent BI analysis was conducted, in which sequences from Bryophytes (Sellaginella moellendorffii and Physcomitrella patens) and Gymnosperms (P. taeda) were excluded, using the same parameters as described above (Supplementary Figures 1, 2). Both ML and BI trees were visualized and edited in FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Identification of conserved amino acid sequence blocks and comparison with AGOs from other organisms
The MEME-suite (Bailey et al., 2009) was used to search for relatively short sequences of conserved amino acids (named blocks) among plant AGOs, and subsequently compare their patterns of distribution to AGOs from other non-plant organisms. Selected full-length coding sequences of plant AGOs were translated into amino acid sequences and searched for conserved sequence blocks using MEME in two independent runs (maximum number of motifs = 50, minimum width = 8, maximum width = 50). The motifs found after the second run were used as query to search against Genbank and Swissprot to look for their distribution in both databases using MAST tool. To generate a “Block Consensus Sequence” within each clade, all proteins were carefully analyzed to identify the sequence of blocks represented in at least 90% of all members within each clade. Blocks exclusively present in at least one member of a clade but not in any member of a different clade were named “Clade-Specific Blocks.” On the basis of the block consensus sequences for all four clades and the MAST results, we created the “Viridiplantae Consensus” by selecting all blocks present in at least two out four of the clades. The Viridiplantae consensus was compared to the representation of a random selection of 10 AGO proteins belonging to mammals, insects, nematodes, and fungi, to identify equivalent sequence blocks. The MEME block in LOGOs format and MAST-suite HTML output files are available upon request.
Results
Phylogenetic relationships within the plant ARGONAUTE gene family
Previous analyses showed that the plant AGO family is monophyletic (Cerutti and Casas-Mollano, 2006; Kapoor et al., 2008; Murphy et al., 2008). The AGO phylogenetic gene tree illustrated in Figure 1 is represented by 206 sequences obtained by both Bayesian inference (BI) and maximum likelihood (ML) analyses, subdivided into four clades: AGO2/3/7, AGO4/6/8/9, AGO5, and AGO1/10, following the nomenclature of the ten AGO genes of Arabidopsis. In comparison to Kapoor et al. (2008), our phylogenetic analysis grouped AGO4/6/8/9 closer to the AGO5-AGO1/10 clade. In both BI and ML, the clade distribution was equivalent, supporting the robustness of the analysis (Supplementary Figure 1), and patterns of AGO distribution reflecting the Angiosperm phylogeny divided in the two major groups of monocots and dicots. Little changes in topology or statistical values were observed when sequences from Bryophytes (P. patens and Selaginella moellendorffii), Gymnosperms (P. taeda and P. radiata) and genomes with an incomplete set of genes (L. sativa, C. sinensis, and A. trichopoda) were excluded, suggesting that our topology is robust and that evolutionary relationships are well defined in spite of a over-representation of Angiosperm lineages (Supplementary Figure 2). Remarkably, almost all clades exhibited the presence of paralogs coming from S. moellendorffii and P. patens, suggesting that this clade organization pre-dates the emergence of vascular plants (Gymnosperms and Angiosperms).
Figure 1
The number of AGO genes per genome ranged from 2 in C. reinhardtii to 20 predicted for Z. mays (maize), reflecting the great diversity of the family size. The genome of other species such as Carica papaya and Cucumis sativus exhibited reduced copy number in several clades of the tree, which is correlated with an absence of recent genome duplications (Ming et al., 2008; Huang et al., 2009), in contrast with those of monocots and some dicots such as Arabidopsis, Glycine max, and Vitis vinifera that experienced extensive genome duplications (Van de Peer et al., 2009). Additionally, it was common to find in different groups multiple paralogs for most of the AGO genes from Arabidopsis, suggesting that there is a trend for increasing gene number in Angiosperms. In almost all clades, monocots exhibited a considerable number of paralogs for almost all AGO genes from Arabidopsis, indicating that lineage-specific gene duplications have occurred during evolution. Following the same trend, previous genomic analysis pointed to the presence of a genome triplication in V. vinifera (grapevine), and gene pairs such as AGO2-AGO3 and AGO8-AGO9 seem to be the result of lineage-specific gene duplication within the Arabidopsis genus, contrary to C. papaya in which this type of duplications are absent. Due to the lack of sequences from other closely related species, we are not able to discard the possibility of a wider, older event of gene duplication within the Brassicaceae. In contrast, given the distribution of the phylogeny with regard to sequences from Pinus, the duplication that gave rise to the AGO1 and AGO10 lineages seems to pre-date the emergence of vascular plants. Finally, there are certain lineages, such as the one represented by maize and sorghum (Sorghum bicolor), but also in poplar (P. trichocarpa), in which several AGO1 or AGO10-like paralogs are present, mostly likely coming from duplications that occurred after the divergence between monocots and dicots. In summary, our phylogenetic results indicate that clade distribution of AGO family pre-dates the emergence of Angiosperms, and that there is a lineage-specific evolution of the different clades of the family mediated by lineage- and species-specific gene duplications, suggesting that one-to-one functional analogies among AGO proteins might not be predictable on the basis of protein sequence, due to significant diversification within the AGO gene family.
ARGONAUTE protein clades show a specific distribution of amino acid blocks
After discarding 21 sequences corresponding to AGO proteins from Bryophytes, Gymnosperms, or corresponding to incomplete sequences from a few species of flowering plants (L. sativa and Citrus sp.), a total of 185 sequences were used to generate a MEME-driven search for short but conserved amino acid sequences (named blocks), resulting in a dynamic distribution within AGO proteins that is correlated with the previously described phylogenetic architecture (Table 1 and Figure 1). The analysis yielded 50 sequence blocks with a variable length ranging between eight (block 47) and 50 (block 1) amino acids (Table 1 and Figure 2). For all clades of the phylogeny, the PIWI domain, responsible for endonuclease cleavage during AGO interaction with sRNAs, is represented by amino acid components present in blocks 1 to 5, 7, 8, 21, 25, 26, 31, 39, 40, 42, and 48; whereas the PAZ domain, responsible for anchoring sRNA during interaction with target transcripts, is represented by components present in blocks 11, 15, 18, 22, 23, 29, and 37 (Table 1). The catalytic residues DDH/DDD, previously described as included in the PIWI domain, are embedded within sequence block 5 (DGVSEGQFYQVLNYELDAIRKACA), 3 (PTIIFGMDVTHPHPGEDSSPSIAAV), and 2 (ELQTLTNNLCYTYARCTRSVSIVPPAYYAHLAA), as expected for essential components of the PIWI-dependent RNA silencing machinery (Table 1). Our analysis also identified components of the DUF1785 domain present in all AGO proteins in blocks 6, 27, and 33.
Table 1
| Blocka | Lengthb | Consensus sequencec | Domain Componentd |
|---|---|---|---|
| 1 | 50 | GNIPPGTVVDTKICHPTEFDFYLCSHAGMQGTSRPTHYHVLWDENNFTAD | PIWI |
| 2 | 33 | ELQTLTNNLCYTYARCTRSVSIVPPAYYALAA | PIWI |
| 3 | 25 | PTIIFGMVTHPHPGEDSSPSIAAV | PIWI |
| 4 | 26 | DWPEVTKYAALVCAQAHRQEMIQDLF | PIWI |
| 5 | 24 | GVSEGQFYQVLNYELDAIRKACA | PIWI |
| 6 | 27 | GFYQSFRPTQMGLSLNIDMSTTMFIEP | DUF1785 |
| 7 | 29 | KMNDQYLANVALKINAKMGGRNTVLVDAL | PIWI |
| 8 | 21 | NWQPPVTFIVVQKRHHTRLFP | PIWI |
| 9 | 27 | PQNGQWNMMNKKMVNGGTVERWACINF | – |
| 10 | 18 | YGDWKWICETDLGIVTQC | PIWI |
| 11 | 21 | RPNYLPMELCKIVEGQRYTKR | PAZ |
| 12 | 29 | YAKEFGISISEKMTQVEARVLPAPWLKYH | – |
| 13 | 15 | AYDGRKSLYTAGPLP | – |
| 14 | 27 | FKVEIKFAAKADMHHLAQFLAGRQADA | – |
| 15 | 21 | VEYFWEMYGYTIQHTHWPCLQ | PAZ |
| 16 | 15 | LPALHENVKNVMFYC | – |
| 17 | 15 | QEALQVLDIVLREHP | – |
| 18 | 17 | DWVKIKKALKNVKVEVT | PAZ |
| 19 | 50 | NVQESVARGFCHELAQMCQISGMEFNPEPVIPIYSARPDQVEKALKHVYH | – |
| 20 | 29 | PDKDLHHYDVTITPEVTSRGVNRAIMAEL | – |
| 21 | 15 | RKATGQKPQRIIFYR | PIWI |
| 22 | 16 | QITALLKMTCQRPQER | PAZ |
| 23 | 29 | NMNQKYRITGLTEQPCRELWFPMDDKNTM | PAZ |
| 24 | 15 | GTKCILKANHFFVEF | – |
| 25 | 15 | GTVDGGMIRELLISF | PIWI |
| 26 | 15 | ELQLLICILPDNNGS | PIWI |
| 27 | 15 | WRCCPVGRSFFSPDM | DUF1785 |
| 28 | 29 | FYHYSVALKYEDGRPVDGKGIGRKVIDKV | – |
| 29 | 15 | LPVIDFVAQNLNKDD | PAZ |
| 30 | 28 | SARCDVRHLVRDLIKCGMMKGIMIEPPF | – |
| 31 | 11 | FRARFYMEPEM | PIWI |
| 32 | 29 | FEENPQFRRAPPMVRVEKMFEQIQSKLPG | – |
| 33 | 11 | QDLGGGVEGWR | DUF1785 |
| 34 | 21 | FTWKEFEITLVDEDDGTGGPR | – |
| 35 | 21 | QPPPASSKSLRFPLRPGFGTV | – |
| 36 | 15 | DILQTVHHNAYHQDP | – |
| 37 | 21 | YFVNHRNIELRYSGDLPCINV | PAZ |
| 38 | 29 | AQMGQFMKFEDMSETSSSHGGHTSAGAVP | – |
| 39 | 11 | NHNDRNSTDKS | PIWI |
| 40 | 8 | RIPLVSDI | PIWI |
| 41 | 11 | YRESHLGMRLP | – |
| 42 | 21 | GNGSPNESDRKRMRRPYQSKT | PIWI |
| 43 | 15 | GGGRGGGGGGRGGGG | – |
| 44 | 21 | FINQLIQRCCQLGIFMNKNTW | – |
| 45 | 41 | SSQRIFHYNVEISPNPSKEVARMIKQKLVEENSAVLSGALP | – |
| 46 | 21 | QNKMEFTVVLEDVSSNRNNGN | – |
| 47 | 8 | KTWQDPQR | PIWI |
| 48 | 29 | SPQFEPTQVLNNVSLLESKLKKIHKAASN | – |
| 49 | 8 | DTGREKDC | – |
| 50 | 11 | TYDSELAGKDF | – |
Consensus sequence blocks of plant ARGONAUTE proteins.
Block numbers correspond to those shown in Figure 1.
Block length is given as the total number of amino acids.
The catalytic residues of the PIWI domain are shown in red and underlined.
Only blocks containing components of DUF, PAZ, and PIWI domains are indicated.
Figure 2
With the exception of AGO proteins from the moss P. patens and the lycophyte S. moellendorffii that show a unique distribution and sequence of blocks, all four clades show a generally conserved trend of linear organization (Figure 2). In addition to the PIWI, PAZ, and DUF1785 domains, we found two new series of conserved amino acid blocks that were named domain A (comprising a variables series of blocks 13, 14, 17, 20, 24, 28, 34, and 35) and domain B (comprising a variable series of blocks 9, 12, 19, 30, 32, 36, 44, and 49). These domains are described in Table 2. The AGO1/10 clade shows the most conserved sequence of blocks of all four clades, with a linear series of four domains that includes A-1 (block sequence: 24-13-17), DUF1785-1 (block sequence: 27-6), PAZ-1 (block sequence: 18-23-15-11), B-1 (block sequence 12-9-44), and the PIWI-1 domain (block sequence: 26-10-7-3-4-21-58-1-2) that incorporated block 31 as specificity of proteins from this clade (Figure 2 and Table 2). Although, no clade-specific blocks were identified in members of the AGO1/10 clade, Figure 2 shows a highly variable number of copies of block 43 (GGGRGGGGGGRGGGG) at the N-terminal region of numerous protein members.
Table 2
| Cladea | Consensus domain sequenceb | Specific blocksc |
|---|---|---|
| AGO2/3/7 (44) | 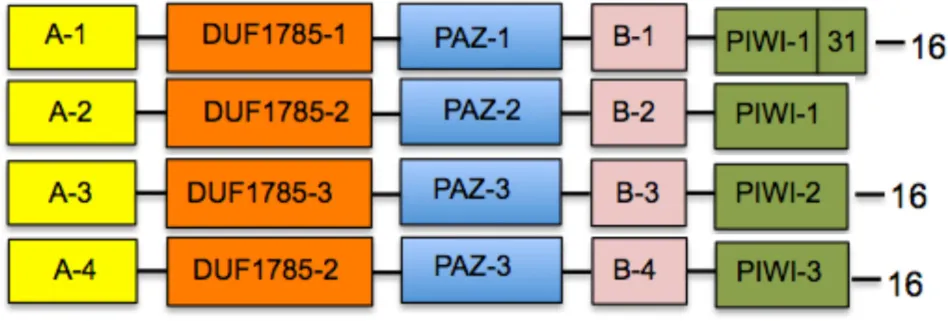 | 45, 48 |
| AGO4/6/8/9 (55) | 28, 30, 32, 37, 42, 46, 50 | |
| AGO5 (28) | none | |
| AGO1/10 (58) | none | |
| A-1: 24-13-17 | DUF1785-1: 27-6 | PIWI-1: 26-10-7-3-4-21-5-8-1-2 |
| A-2: 24-28*-13-14-17 | DUF1785-2: 27-33-6 | PIWI-2: 26-10-7-40-3-4-21-5-8-1-2-31 |
| A-3: 35-24-20-13-14-17 | DUF1785-3: 33-6 | PIWI-3: 26-10-7-40-3-4-47-21-5-39-1-2-3 |
| A-4: 35-24-20-41-13-34-14-17 | ||
| B-1: 12-9-44 | PAZ-1: 18-23-15-11 | |
| B-2: 12-9-30*-32* | PAZ-2: 29-18-15-12-22 | |
| B-3: 12-9-19 | PAZ-3: 29-18-23-15-11-22 | |
| B-4: 36-12-49-9-19 |
Consensus domain sequence distribution in four clades of plant ARGONAUTE proteins.
Clades follow the Arabidopsis thaliana nomenclature; numbers between brackets indicate the number of proteins included in each clade.
Asterisks indicate blocks present in Angiosperms but not in proteins from Physcomitrella and Selaginella sp.
Blocks exclusively found in at least one member of the corresponding clade, but not in other clades.
The AGO4/6/8/9 and AGO5 clades also show a highly conserved series of blocks that are present in at least 90% of the proteins analyzed within each clade (Figure 2 and Table 2). Whereas, the AGO4/6/8/9 clade is characterized by containing the A-2 (24-28-13-14-17), DUF1785-2 (27-33-6), PAZ-2 (29-18-15-12-22), B-2 (12-9-30-32), and PIWI-1 domains, the AGO5 clade has the A-3 (35-24-20-13-14-17), DUF1785-3 (33-6), PAZ-3 (29-18-23-15-11-22), B-3 (12-9-19), and PIWI-2 domains (26-10-7-40-3-4-21-5-8-2-31). Contrary to the AGO4/6/8/9 clade, members of the AGO1/10 clade have a strong tendency to include blocks 40 (RIPLVSDI), 47 (KTWQDPQR), 39 (NHNDRNSTDKS), and 31 (FRARFYMEPEM), all components of the PIWI domain that are also partially included in the consensus of the AGO5 (blocks 40 and 31) and AGO2/3/7 (block 31) clades (Table 2). In addition, in the AGO4/6/8/9 clade we found four exceptional proteins (one from Ricinus communis, one from Cocumis sativus, one from P. trichocarpa, and one from Manihot esculenta) containing a copy of block 20 (PDKDLHHYDVTITPEVTSRGVNRA IMAEL; of unknown function) or 21 (RKATGQKPQRIIFYR; a component of the PIWI domain) at the N-terminal region (Figure 2). Additional off-types of this clade at the N-terminal domain are Bradi2g10370 from Brachypodium dystachion that contains block 35 (QPPPASSKSLRFPLRPGFGTV; of unknown function) in the N-terminal region, and LOC Os01gA16850 from rice that is exceptionally long and contains an overall unique sequence of block distribution, suggesting a case of sequence misannotation. By contrast to the AGO4/6/8/9 and AGO2/3/7 clades, the AGO5 clade does not contain specific blocks absent from members of all other clades.
Finally, the AGO2/3/7 clade shows a conserved sequence of blocks present in at least 90% of the proteins analyzed within the clade that includes domains A-4 (35-24-20-41-13-34-14-17), DUF1785-2 (as the AGO4/6/8/9 clade), PAZ-3 (as the AGO5 clade), B-4 (36-12-49-9-19), and PIWI-3 (26-10-7-40-3-4-47-21-5-8-39-1-2-3) as shown in Table 2. Blocks 45 (SSQRIFHYNVEISPNPSKEVARMIKQKLVEENSAVLSGALP) and 48 (SPQFEPTQVLNNVSLLESKLKKIHKAASN) that contain consensus sequences of unknown function are exclusively found in this clade, clearly showing the divergence that some primary sequence components have among members of the AGO family of proteins in plants.
Although being present in < 90% of proteins analyzed, a few additional blocks are abundantly present in members of specific clades, often in multiple copies (Figure 2 and Table 1). In the case of the AGO2/3/7 clade, these include blocks 20 (PDKDLHHYDVTITPEVTSRGVNRAIMAEL), 22 (QIT ALLKMTCQRPQER), 43 (GRGGGRGRGGR), and 45 (SSQRIFHYNVEISPN PSKEVARMIKQKLVEENSAVLSGALP). Whereas, block 20 is usually present at the N-terminal region of many proteins, block 22 represents a component of the PAZ domain that is usually located in close association of block 11 (which is also a component of PAZ; Table 1). Block 45 is a large element of unknown function found at the N-terminal region, and in close association with block 13. Of particular interest is block 43, abundantly present in up to eight copies at the N-terminal region of a multitude of proteins belonging to three clades (AGO2/3/7, AGO5, and AGO1/10), but present in members of C. reinhardtii, P. patens, and S. moellendorffii, which suggests an ancestral origin for the block (Table 1). In the case of the AGO4/6/8/9 clade, block 38 is often present in at the C-terminal region of many proteins, and in close association with components of the PIWI domain (blocks 1 and 2). In the case of the AGO5 clade, blocks 35 (QPPPASSKSLRFPLRPGFGTV) and 49 (DTGREKDC) are often present in a single copy at the N-terminal or mid region, respectively. Finally, in the case of the AGO1/10 clade, block 43 is also abundantly present in multiple copies at the N-terminal region, and additionally, only in some proteins belonging to highly distinct families of flowering plants (Brassicaceae, Cucurbitaceae, Fabaceae, and Phrymaceae), also present as a single copy located at the C-terminal region, and in close association with components of the PIWI domain.
Consensus block sequences are conserved between plant and non-plant organisms
After determining the distribution of linear blocks within the plant clades, we looked for the possible presence of similar or identical primary sequence blocks in other AGO proteins belonging to non-plant organisms. Using the motif alignment and search tool (MAST) of the MEME suite, we searched protein databases such as NCBI non-redundant protein database and SWISSPROT, comparing the sequences in terms of presence or absence of specific blocks, and the organization of their linear arrangement. We performed six independent MAST runs using as input all sequence blocks, changing the parameters from standard to stringent values by modifying the sequence composition thresholds, as well as the E- and P-values. We found that specific blocks defined domains that were conserved between plants, animals, and fungi (Table 3), supporting the robustness of our analysis and extending the functional conservation of domains across kingdoms. For example, the mammalian consensus block sequence contains domains A-3, DUF1785-1, and B-3 that are also contained in the plant consensus sequence (Table 3); it also contains the PAZ-3 domain in conjunction to block 4 that correspond to a component of the PIWI domain (Table 3), and the PIWI-1 domain supplemented with block 31 (Table 3). The insect consensus block sequence also contains the DUF1785-1 and PAZ-3 domains, but is characterized by different variants of the A, B, and PIWI domains. By contrast, nematodes and fungi are characterized by consensus sequences that largely diverge from the plant consensus sequence, although in general terms, only the insertion of a few individual blocks distinguished the flowering plant consensus from these highly divergent non-plant groups of organisms. Interestingly, the consensus of the PIWI subfamily of AGO proteins was more divergent than the rest of the consensus (Tables 2, 3), suggesting that PIWI subgroup has considerably diverged as compared to the rest of the AGO proteins.
Table 3
| Organism group | Consensus domain sequencea | |
|---|---|---|
| Viridiplantae | 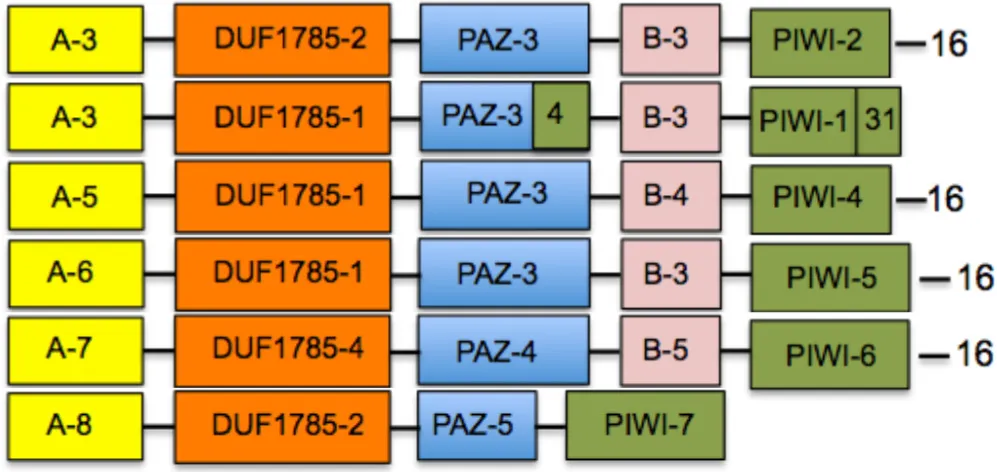 | |
| Mammals | ||
| Insects | ||
| Nematodes | ||
| Fungi | ||
| PIWI subfamily | ||
| A-5: 20-13-14-17 | DUF1785-4: 27-33 | PIWI-4: 26-10-7-3-4-21-5-8-1-2-31 |
| A-6: 24-20-13-14-17 | PIWI-5: 10-7-3-4-21-5-8-1-2-31 | |
| A-7: 43-35-24-13-14-23-17 | PAZ-4: 18-11-22 | PIWI-6: 7-3-4-21-5-8-1-2-31 |
| A-8: 43-24-20 | PAZ-5: 15-12 | PIWI-7: 10-4-21-5-8-2-1 |
| B-5: 12-9 | ||
Consensus domain sequences among ARGONAUTE proteins from different group of organisms.
The nomenclature of domains is equivalent to Table 2.
Specific functional motifs are conserved between animal and plant ago proteins
We also analyzed plant AGOs in search for motifs previously reported for animal AGO proteins, and found two types of sequences that could be related to similar or equivalent functions across kingdoms. Qi et al. (2008) reported that human AGO2 is post-translationally modified to confer stability and allow protein-protein interactions with members of the RNA interference silencing complex (RISC). This posttranslational modification depends on the proline residue of a X-P-G motif that is recognized and hydroxylated by a collagen prolyl-4-hydroxylase. Mutations that suppress this modification result in AGO2 instability and defective RISC activity. We found that the proline residue of the X-P-G motif is present in all AGO proteins containing block 8 (Figure 3A). In plants, the proline and glycine residues were highly conserved in all phylogenetic groups, suggesting that it might be a site for posttranslational modifications. Recent evidence supports the presence and function of prolyl-hydroxylases in Arabidopsis, supporting the possibility that this type of modifications could occur in plant AGO proteins (Tiainen et al., 2005; Vlad et al., 2007; Asif et al., 2009; Velasquez et al., 2015).
Figure 3
Our analysis also revealed that block 43 (GRGGGRGRGGR) is abundantly present in a variable number of consecutive copies at the N-terminal region of members of the AGO2/3/7, AGO5, and AGO1/10 clades, but completely absent from the AGO4/6/8/9 clade (Figure 2). This conservation appears to be related to mechanisms such as those regulating posttranslational modification in AUBERGINE (AUB), a PIWI protein of Drosophila melanogaster that is modified by a symmetrical arginine methylation in an amino acid motif rich in glycine and arginine residues. This modification allows AUB to interact with proteins of the TUDOR (TUD) family (Kirino et al., 2009, 2010; Siomi et al., 2010). Based on our MAST analysis, we found a close association between sequence block 43 (GRGGGRGRGGR), and the proposed region to be modified in AUB and MIWI (Siomi et al., 2010; Figure 3B), raising the possibility that the sequence corresponding to block 43 could function as an evolutionary conserved protein-protein interaction and localization signal in plant AGOs. These results suggest that non-canonical functional motifs involved AGO protein function might be shared across kingdoms, opening possibilities for new experimental assays.
Discussion
Our phylogenetic analysis based on 23 plant genomes defined that the AGO gene family is divided into four major clades that recapitulate the relationships among Arabidopsis AGO genes. Although previous studies already classified AGO into these clades, there are specific differences in the resulting clade distribution and statistical support of our phylogeny. In contrast to previous reports (Cerutti and Casas-Mollano, 2006; Kapoor et al., 2008; Murphy et al., 2008), our phylogeny placed AGO2/3/7 as a distinct sister clade to the common branch that includes all other three clades, although with moderate to weak statistical support (up to 60% and 0.9 in bootstrap and posterior probability). In our analysis, the ML and BI statistical frameworks allowed the establishment of phylogenetic relationships that use nucleotide sequences as markers of phylogenetic reconstructions. Although, we cannot rule out the influence of several parameters such as out-group selection, taxon sampling, or molecular edition in the outcome of the topology or the statistical support of the phylogenetic tree, a recent analysis based on a larger number of plant and animal genomes that included 19 of the 23 plant genomes included here resulted in a closely related but less detailed topology (Singh et al., 2015), providing independent support to our analysis.
While our phylogeny is mostly composed of genes from flowering plants, sequences from P. patens and S. moellendorffii also clustered in at least one of the four major AGO clades, suggesting that the divergence of these clades could precede the divergence of Gymnosperms and Angiosperms. Although, there is little functional information for the AGO genes of C. reinhardtii (Casas-Mollano et al., 2008), P. patens, or S. moellendorffii, we hypothesize that in these ancient plant organisms the RNAi machinery is dependent on AGO proteins that are capable of ubiquitously fulfill functions that in Angiosperms are executed separately by different AGOs from the four major clades (Bartel, 2004; Bonnet et al., 2006; Casas-Mollano et al., 2008; Cho et al., 2008). According to the hypothesis of the origins of sRNAs in eukaryotic evolution (Shabalina and Koonin, 2008) we propose that even in C. reinhardtii, AGO genes are functional and participate in basic processes such as transposon silencing and possibly gene regulation through a pathway reminiscent of the miRNA-dependent pathway of flowering plants (Molnar et al., 2007).
The evolutionary trend observed in AGO proteins could be associated with the diversification and function of key developmental processes in flowering plants (Cibrian-Jaramillo and Martienssen, 2009). The AGO4/6/8/9 clade, which in our analysis shows a high level of block conservation and possibly functional specialization, is directly involved in epigenetic silencing of heterochromatin, including transposons, retrotransposons, and other repetitive elements (Matzke et al., 2015). Specialization of this specific clade could have involved the acquisition of restricted temporal or spatial patterns of protein expression (Havecker et al., 2010; Olmedo-Monfil et al., 2010), recent genomic duplications (Takeda et al., 2008), or sRNA interaction and regulation through de novo DNA methylation. Although several RNA-DEPENDENT RNA POLYMERASE (RDR) proteins have also been implicated in these mechanisms, a possible phylogenetic relationship between AGOs and RDRs has not yet been investigated, even if a genome phylogeny that used measures of congruence suggested that AGO1 and RDR6 are of critical importance in the evolution of seed plants (Cibrian-Jaramillo et al., 2010). The presence of some consensus sequences corresponding to blocks that are elements of the PIWI and PAZ domains suggest that the clade might include canonical components that are not functionally represented in any of the three other clades. In addition to these blocks, several other newly defined domains such as A-2 and B-2 of unknown tertiary structure or function are exclusive of proteins either included in the AGO2/3/7 or AGO4/6/8/9 clades, suggesting that some of corresponding sequences might represent a clade-specific degree of specialization with possible structural or biochemical functions. Their identification could serve as the basis for subsequent experimental analysis.
The AGO1/10 members also have a tightly conserved primary sequence, probably related to their almost exclusive association with microRNAs, being AGO1 the best characterized AGO protein to this date (Kidner and Martienssen, 2005; Vaucheret, 2008). The AGO2/3/7 clade has been implicated in tasiRNA-dependent posttranscriptional regulation, particularly during leaf development (Montgomery et al., 2008; Chitwood et al., 2009); the function of members of this clade also include SHOOTLESS4 (SHL4)/SHOOT ORGANIZATION2 (SHO2) in rice (LOC_Os03g33650 in the phylogeny; Nagasaki et al., 2007), and RAGGED SEEDLING2 in maize (GRMZM2G365589 in the phylogeny; Douglas et al., 2010). In the case of the AGO5 clade, evidence in Arabidopsis and rice indicates broad sRNA binding capacity by corresponding proteins and specific functions during gametogenesis and meiosis. In rice, MEIOSIS ARRESTED AT LEPTOTENE (MEL1) is expressed during meiosis and is important for functional megaspore development as well as both male and female gametogenesis (Nonomura et al., 2007).
The structure of the two C. reinhardtii AGO sequences containing the majority of blocks found in all four major clades supports the hypothesis of a monophyletic and multifunctional ancestral origin for the family. The conservation of specific blocks and motifs across the animal and plant kingdom also suggests that specific elements of unknown function could have prevailed after the divergence and specialization of AGO proteins in plants. In some cases such as the A-3, B-3, and PAZ-3 domains conserved between plants and mammals, the corresponding block elements include sequences of unknown function associated with elements included in canonical domains, opening possibilities for experimentally testing whether this primary sequence conservation could be a consequence of functional elements that were conserved after the split between plants and animals.
Functional motifs present in animal AGO proteins were identified in one or more of the conserved sequence blocks generated by our analysis, within regions subject for posttranslational modifications such as hydroxylation (Qi et al., 2008) or arginine methylation (Kirino et al., 2009, 2010). In the former case, we found a block that is highly conserved in all AGOs from different kingdoms, which points to the possibility of a functional conservation across kingdoms. In the latter, the conserved motif was described as a site for protein-protein interaction and localization signals related to the D. melanogaster AUB protein, suggesting a function beyond a specific AGO clade or specific developmental process due to divergent functional evolution. Our overall results provide a first analysis of AGO linear sequences, providing new regions that might conserve ancestral elements related to unforeseen functions requiring experimental assessment before being further defined or elucidated.
Statements
Author contributions
DR and JV conceived and designed the research; AC and IR provided computational support; DR and JV analyzed the data, interpreted the results, and wrote the paper.
Acknowledgments
We thank Miguel Vallebueno-Estrada, Gloria León-Martínez, and Nidia Sánchez-León for helpful comments and technical help. DR and IR were recipients of a graduate scholarship from the Consejo Nacional de Ciencia y Tecnología (CONACyT). This research was funded by CONACyT and the Howard Hughes Medical Institute International Scholar program.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer MB and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01347
Sequences and script files can be downloaded from: http://datos.langebio.cinvestav.mx/~vielle/Rodriguez-Leal_etal_2016_suppData/
- ML
Maximum likelihood
- BI
Bayesian inference
- AGO
ARGONAUTE.
Abbreviations
References
1
AsifM. H.TrivediP. K.MisraP.NathP. (2009). Prolyl-4-hydroxylase (AtP4H1) mediates and mimics low oxygen response in Arabidopsis thaliana. Funct. Integr. Genomics9, 525–535. 10.1007/s10142-009-0118-y
2
BaiM.YangG. S.ChenW. T.MaoZ. C.KangH. X.ChenG. H.et al. (2012). Genome-wide identification of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analyses in response to viral infection and abiotic stresses in Solanum lycopersicum. Gene501, 52–62. 10.1016/j.gene.2012.02.009
3
BaileyT. L.BodénM.BuskeF. A.FrithM.GrantC. E.ClementiL.et al. (2009). MEME Suite: tools for motif discovery and searching. Nucleic Acids Res.37, W202–W208. 10.1093/nar/gkp335
4
BartelD. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell116, 281–297. 10.1016/S0092-8674(04)00045-5
5
BonnetE.Van de PeerY.RouzéP. (2006). The small RNA world of plants. New Phytol.171, 451–468. 10.1111/j.1469-8137.2006.01806.x
6
Casas-MollanoJ. A.RohrJ.KimE. J.BalassaE.van DijkK.CeruttiH. (2008). Diversification of the core RNA interference machinery in Chlamydomonas reinhardtii and the role of DCL1 in transposon silencing. Genetics179, 69–81. 10.1534/genetics.107.086546
7
CeruttiH.Casas-MollanoJ. A. (2006). On the origin and functions of RNA-mediated silencing: from protists to man. Curr. Genet.50, 81–99. 10.1007/s00294-006-0078-x
8
ChitwoodD. H.NogueiraF. T.HowellM. D.MontgomeryT. A.CarringtonJ. C.TimmermansM. C. (2009). Pattern formation via small RNA mobility. Genes Dev.23, 549–554. 10.1101/gad.1770009
9
ChoS. H.Addo-QuayeC.CoruhC.ArifM. A.MaZ.FrankW.et al. (2008). Physcomitrella patens DCL3 is required for 22-24 nt siRNA accumulation, suppression of retrotransposon-derived transcripts, and normal development. PLoS Genet.4:e1000314. 10.1371/journal.pgen.1000314
10
Cibrián-JaramilloA.De la Torre-BárcenaJ. E.LeeE. K.KatariM. S.LittleD. P.StevensonD. W.et al. (2010). Using phylogenomic patterns of gene onthology to identify proteins of importance in plant evolution. Genome Biol. Evol.2, 225–239. 10.1093/gbe/evq012
11
Cibrián-JaramilloA.MartienssenR. A. (2009). Darwin's “Abominable Mystery”: the role of RNA interference in the evolution of flowering plants. Cold Spring Harb. Symp. Quant. Biol.74, 267–273. 10.1101/sqb.2009.74.051
12
de JongD.EitelM.JakobW.OsigusH. J.HadrysH.DesalleR.et al. (2009). Multiple dicer genes in the early-diverging metazoa. Mol. Biol. Evol.26, 1333–1340. 10.1093/molbev/msp042
13
DouglasR. N.WileyD.SarkarA.SpringerN.TimmermansM. C.ScanlonM. J. (2010). Ragged seedling2 Encodes an ARGONAUTE7-like protein required for mediolateral expansion, but not dorsiventrality, of maize leaves. Plant Cell22, 1441–1451. 10.1105/tpc.109.071613
14
EdgarR. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics5:113. 10.1186/1471-2105-5-113
15
FinnR. D.MistryJ.TateJ.CoggillP.HegerA.PollingtonJ. E.et al. (2010). The Pfam protein families database. Nucleic Acids Res.38, D211–D222. 10.1093/nar/gkp985
16
GhildiyalM.ZamoreP. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet.10, 94–108. 10.1038/nrg2504
17
GouyM.GuindonS.GascuelO. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol.27, 221–224. 10.1093/molbev/msp259
18
GuindonS.DelsucF.DufayardJ. F.GascuelO. (2009). Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol.537, 113–137. 10.1007/978-1-59745-251-9_6
19
HaveckerE. R.WallbridgeL. M.HardcastleT. J.BushM. S.KellyK. A.DunnR. M.et al. (2010). The Arabidopsis RNA-directed DNA methylation argonautes functionally diverge based on their expression and interaction with target loci. Plant Cell22, 321–334. 10.1105/tpc.109.072199
20
HuangS.LiR.ZhangZ.LiL.GuX.FanW.et al. (2009). The genome of the cucumber, Cucumis sativus L. Nat. Genet.41, 1275–1281. 10.1038/ng.475
21
HuangY.KendallT.ForsytheE. S.Dorantes-AcostaA.LiS.Caballero-PérezJ.et al. (2015). Ancient origin and recent innovations of RNA polymerase IV and V. Mol. Biol. Evol.32, 1788–1799. 10.1093/molbev/msv060
22
KapoorM.AroraR.LamaT.NijhawanA.KhuranaJ. P.TyagiA. K.et al. (2008). Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics9:451. 10.1186/1471-2164-9-451
23
KeaneT. M.CreeveyC. J.PentonyM. M.NaughtonT. J.McLnerneyJ. O. (2006). Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol.6:29. 10.1186/1471-2148-6-29
24
KidnerC. A.MartienssenR. A. (2005). The role of ARGONAUTE1 (AGO1) in meristem formation and identity. Dev. Biol.280, 504–517. 10.1016/j.ydbio.2005.01.031
25
KirinoY.KimN.de Planell-SaguerM.KhandrosE.ChioreanS.KleinP. S.et al. (2009). Arginine methylation of Piwi proteins catalysed by dPRMT5 is required for Ago3 and Aub stability. Nat. Cell Biol.11, 652–658. 10.1038/ncb1872
26
KirinoY.VourekasA.SayedN.de Lima AlvesF.ThomsonT.LaskoP.et al. (2010). Arginine methylation of Aubergine mediates Tudor binding and germ plasm localization. RNA16, 70–78. 10.1261/rna.1869710
27
MatzkeM. A.KannoT.MatzkeA. J. (2015). RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 66, 243–267. 10.1146/annurev-arplant-043014-114633
28
MingR.HouS.FengY.YuQ.Dionne-LaporteA.SawJ. H.et al. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature452, 991–996. 10.1038/nature06856
29
MolnárA.SchwachF.StudholmeD. J.ThuenemannE. C.BaulcombeD. C. (2007). miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature447, 1126–1129. 10.1038/nature05903
30
MontgomeryT. A.HowellM. D.CuperusJ. T.LiD.HansenJ. E.AlexanderA. L.et al. (2008). Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell133, 128–141. 10.1016/j.cell.2008.02.033
31
MurphyD.DancisB.BrownJ. R. (2008). The evolution of core proteins involved in microRNA biogenesis. BMC Evol. Biol.8:92. 10.1186/1471-2148-8-92
32
NagasakiH.ItohJ.HayashiK.HibaraK.Satoh-NagasawaN.NosakaM.et al. (2007). The small interfering RNA production pathway is required for shoot meristem initiation in rice. Proc. Natl. Acad. Sci. U.S.A.104, 14867–14871. 10.1073/pnas.0704339104
33
NonomuraK.MorohoshiA.NakanoM.EiguchiM.MiyaoA.HirochikaH.et al. (2007). A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell19, 2583–2594. 10.1105/tpc.107.053199
34
Olmedo-MonfilV.Durán-FigueroaN.Arteaga-VázquezM.Demesa-ArévaloE.AutranD.GrimanelliD.et al. (2010). Control of female gamete formation by a small RNA pathway in Arabidopsis. Nature464, 628–632. 10.1038/nature08828
35
QiH. H.OngusahaP. P.MyllyharjuJ.ChengD.PakkanenO.ShiY.et al. (2008). Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature455, 421–424. 10.1038/nature07186
36
QianY.ChengY.ChengX.JiangH.ZhuS.ChengB. (2011). Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep.30, 1347–1363. 10.1007/s00299-011-1046-6
37
RambautA.DrummondA. J. (2007). Tracer v1.6. Available online at: http://beast.bio.ed.ac.uk/Tracer
38
RonquistF.HuelsenbeckJ. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics19, 1572–1574. 10.1093/bioinformatics/btg180
39
ShabalinaS. A.KooninE. V. (2008). Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol.23, 578–587. 10.1016/j.tree.2008.06.005
40
SinghR. K.GaseK.BaldwinI. T.PandeyS. P. (2015). Molecular evolution and diversification of the Argonaute family of proteins in plants. BMC Plant Biol.15:23. 10.1186/s12870-014-0364-6
41
SiomiM. C.MannenT.SiomiH. (2010). How does the royal family of Tudor rule the PIWI-interacting RNA pathway?Genes Dev.24, 636–646. 10.1101/gad.1899210
42
SongJ. J.SmithS. K.HannonG. J.Joshua-TorL. (2004). Crystal structure of Argonaute and its implications for RISC slicer activity. Science305, 1434–1437. 10.1126/science.1102514
43
SwartsD. C.MakarovaK.WangY.NakanishiK.KettingR. F.KooninE. V.et al. (2014). The evolutionary journey of Argonaute proteins. Nat. Struct. Mol. Biol.21, 743–753. 10.1038/nsmb.2879
44
TabachY.BilliA. C.HayesG. D.NewmanM. A.ZukO.GabelH.et al. (2013). Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature493, 694–698. 10.1038/nature11779
45
TakedaA.IwasakiS.WatanabeT.UtsumiM.WatanabeY. (2008). The mechanism selecting the guide strand from small RA duplexes is different among argonaute proteins. Plant Cell Physiol.49, 493–500. 10.1093/pcp/pcn043
46
TalaveraG.CastresanaJ. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol.56, 564–577. 10.1080/10635150701472164
47
TiainenP.MyllyharjuJ.KoivunenP. (2005). Characterization of a second Arabidopsis thaliana prolyl 4-hydroxylase with distinct substrate specificity. J. Biol. Chem.280, 1142–1148. 10.1074/jbc.M411109200
48
Van de PeerY.FawcettJ. A.ProostS.SterckL.VandepoeleK. (2009). The flowering world: a tale of duplications. Trends Plant Sci.14, 680–688. 10.1016/j.tplants.2009.09.001
49
VaucheretH. (2008). Plant ARGONAUTES. Trends Plant Sci.13, 350–358. 10.1016/j.tplants.2008.04.007
50
VelasquezS. M.RicardiM. M.PoulsenC. P.OikawaA.DilokpimolA.HalimA.et al. (2015). Complex regulation of prolyl-4-hydroxylases impacts root hair expansion. Mol. Plant8, 734–746. 10.1016/j.molp.2014.11.017
51
VladF.SpanoT.VladD.DaherF. B.OuelhadjA.FragkostefanakisS.et al. (2007). Involvement of Arabidopsis prolyl 4 hydroxylases in hypoxia, anoxia and mechanical wounding. Plant Signal. Behav.2, 368–369. 10.1111/j.1399-3054.2007.00915.x
Summary
Keywords
ARGONAUTE evolution, phylogenetics, MEME, maximum likelihood, Bayesian inference
Citation
Rodríguez-Leal D, Castillo-Cobián A, Rodríguez-Arévalo I and Vielle-Calzada J-P (2016) A Primary Sequence Analysis of the ARGONAUTE Protein Family in Plants. Front. Plant Sci. 7:1347. doi: 10.3389/fpls.2016.01347
Received
02 April 2016
Accepted
22 August 2016
Published
31 August 2016
Volume
7 - 2016
Edited by
Ramin Yadegari, University of Arizona, USA
Reviewed by
Jianbin Wang, University of Colorado Denver School of Medicine, USA; Mark Beilstein, University of Arizona, USA
Updates
Copyright
© 2016 Rodríguez-Leal, Castillo-Cobián, Rodríguez-Arévalo and Vielle-Calzada.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Philippe Vielle-Calzada vielle@ira.cinvestav.mx
This article was submitted to Plant Genetics and Genomics, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.