 Zhao Li1,2†
Zhao Li1,2† Guanghui Hu1,2,3†Xiangfeng Liu1
Guanghui Hu1,2,3†Xiangfeng Liu1 Yao Zhou1Yu Li4Xu Zhang2Xiaohui Yuan2,5Qian Zhang1Deguang Yang1*Tianyu Wang4*Zhiwu Zhang1,2*
Yao Zhou1Yu Li4Xu Zhang2Xiaohui Yuan2,5Qian Zhang1Deguang Yang1*Tianyu Wang4*Zhiwu Zhang1,2*- 1Agronomy College of Northeast Agricultural University, Harbin, China
- 2Department of Crop and Soil Sciences, Washington State University, Pullman, WA, USA
- 3Institute of Maize Research, Heilongjiang Academy of Agricultural Sciences, Harbin, China
- 4Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing, China
- 5Department of Computer Science, Wuhan University of Technology, Wuhan, China
Originating in a tropical climate, maize has faced great challenges as cultivation has expanded to the majority of the world's temperate zones. In these zones, frost and cold temperatures are major factors that prevent maize from reaching its full yield potential. Among 30 elite maize inbred lines adapted to northern China, we identified two lines of extreme, but opposite, freezing tolerance levels—highly tolerant and highly sensitive. During the seedling stage of these two lines, we used RNA-seq to measure changes in maize whole genome transcriptome before and after freezing treatment. In total, 19,794 genes were expressed, of which 4550 exhibited differential expression due to either treatment (before or after freezing) or line type (tolerant or sensitive). Of the 4550 differently expressed genes, 948 exhibited differential expression due to treatment within line or lines under freezing condition. Analysis of gene ontology found that these 948 genes were significantly enriched for binding functions (DNA binding, ATP binding, and metal ion binding), protein kinase activity, and peptidase activity. Based on their enrichment, literature support, and significant levels of differential expression, 30 of these 948 genes were selected for quantitative real-time PCR (qRT-PCR) validation. The validation confirmed our RNA-Seq-based findings, with squared correlation coefficients of 80% and 50% in the tolerance and sensitive lines, respectively. This study provided valuable resources for further studies to enhance understanding of the molecular mechanisms underlying maize early freezing response and enable targeted breeding strategies for developing varieties with superior frost resistance to achieve yield potential.
Introduction
Maize (Zea mays L.) is one of the most important food, feed, and biofuel crops worldwide. Originating from tropical regions, maize is highly sensitive to low temperatures, especially during the early seedling stage (Presterl et al., 2007). Freezing (<0°C) is an extreme abiotic stress for maize (Doherty et al., 2009; Guo et al., 2013) and is the most limiting factor for early planting in temperate regions (Adamczyk and Królikowski, 1998). For example, in Northeast China, cold damage occurs, on average, every 3–4 years. Maize production losses can reach 20% in the most severe cold-weather years (Ma et al., 2003). Thus, demand is high for the development of cultivars with improved freezing tolerance for longer growth periods, so that yield potentials can be fully realized in high-latitude cultivation areas.
Significant progress has been made in understanding freezing-tolerance mechanisms in plants. Several transcription factors involved in cold acclimation have been discovered. For example, C-repeat/dehydration-responsive element Binding Factors (CBFs) have been identified as key regulators of cold response that activate the expression of cold-response genes in Arabidopsis (Jaglo-Ottosen et al., 1998; Kasuga et al., 1999). These factors are also known as dehydration-responsive element binding factor proteins (DREBs). Among this gene family, ZmDREB1A can enhance freezing tolerance in Arabidopsis. This gene was also cloned in maize (Qin et al., 2004).
ICE1, a bHLH transcription factor, was identified as an inducer of CBF expression and a key regulator of CBF genes under cold conditions. Studies have demonstrated that ICE1 can improve freezing tolerance in transgenic plants (Chinnusamy, 2003). Another example is OST1 (OPEN STOMATA 1), also known as SnRK2.6/SnRK2E. OST1 is a Ser/Thr protein kinase that controls stomatal movement under stress conditions and can be activated by abscisic acid (ABA) (Grondin et al., 2015). Recently, Ding et al. (2015) found that when OST1 is activated by cold stress, it phosphorylates ICE1. In turn, the stability and transcriptional activity of ICE1 is enhanced, leading to improved freezing tolerance in Arabidopsis.
Besides the well-known CBF pathway involved in freezing tolerance, a novel molecular pathway has also been identified. TCF1 (Tolerant to Chilling and Freezing 1), a RCC1 family protein, was shown to regulate lignin biosynthesis in response to freezing stress, acting independently of the CBF pathway (Ji et al., 2015).
In addition to studies on gene functions, responses were also investigated at protein level. First identified in polar fish, ice-binding proteins are a family of protective proteins induced by freezing temperatures that have been found in many overwintering plants, conferring anti-freezing activity (Zhang et al., 2010). Recently, transgenic plants with ice-binding proteins also showed enhanced freezing tolerance (Bredow et al., 2016). Better understanding of the freezing responses at both gene and protein levels will provide more information to help uncover freezing response mechanisms.
Although maize originated in tropical regions, genetic variation in chilling tolerance exists in maize germplasms. For example, European flint and highland tropical germplasm material are more chilling tolerant than dent material from the Corn Belt (Leipner, 2009). Their genetic basis was investigated through linkage analyses and genome-wide association studies (GWAS) to map quantitative trait locus (QTL) related to maize cold tolerance. For example, a study using genome-wide association mapping for maize cold tolerance identified 19 significant association signals (Strigens et al., 2013). Many mapped QTLs are associated with seedling vigor and basic physiological processes such as photosynthesis. However, due to the limitations of phenotypic characterization, less is known about the molecular basis for adaptation to cold environments (Fracheboud et al., 2004; Hund et al., 2004). Only recently, based on multi-year field data of different maize inbred lines' cold-sensitivity and transcriptome profiling of the lines' cold response by microarray analysis, three mechanisms responsible for chilling tolerance were identified—photosynthetic apparatus modification, cell wall properties, and developmental process (Sobkowiak et al., 2016).
The transcriptome is the overall set of transcribed regions of a genome. Previously, microarray chip hybridization experiments (gene chip) were used to explore gene expression profiles (Trzcinska-Danielewicz et al., 2009; Yang et al., 2011; An et al., 2012). Next-generation, high-throughput DNA sequencing technologies now provide powerful tools for mapping and quantifying transcriptome, creating opportunities to better understand the molecular basis of cold adaptation. For example, RNA-sequencing (RNA-seq) technology can be used to characterize the entire transcriptome and to detect areas of alternative splicing (AS) and novel transcribed regions (NTRs) (Maniatis and Tasic, 2002; Shen et al., 2012; Teoh et al., 2013).
Compared to gene chip analysis, RNA-seq analysis has many advantages such as lower background signal and higher suitability for both known transcripts and new genes. Thus, RNA-seq analysis has become an efficient tool for analyzing the transcriptomic responses of many plant species to various biotic and abiotic stresses and studying the associated molecular mechanisms (Liu et al., 2012; Zhang et al., 2012; Fu et al., 2013; Shan et al., 2013; Opitz et al., 2014; Wang et al., 2014). Recently, RNA-seq analysis was also used to identify mild freezing shock response pathways in barley (Wang et al., 2016). In maize, RNA-seq analysis has been used to study the plant's response to water deficit; differentially expressed genes and significantly enriched gene ontology (GO) terms were identified (Kakumanu et al., 2012; Opitz et al., 2014). However, little is known about the transcriptomic responses of maize to freezing stress.
Because maize seedlings are usually the most vulnerable to freezing stress, responses to cold stress at the seedling stage can be used to identify important candidate genes. In this study we focused on the seedling stage of maize and used RNA-seq analysis to accomplish the following objectives: (1) exam 30 elite maize inbred lines adapted to northern China for their responses to freezing stress at seedling stage; (2) categorize genes according differentiated expression related to freezing treatment; and (3) narrow down to the potential candidate genes for future cloning studies. Increasing our understanding of the molecular mechanisms underlying freezing response in maize can enable targeted breeding strategies to accelerate the development of freezing tolerant cultivars.
Results
Freezing Tolerance Analysis
A total of 30 maize inbred lines, commonly cultivated in Heilongjiang Province, China, were selected for freezing tolerance evaluation. We selected seedling survival rate as the measurement of freezing tolerance. Seedlings grown to the 3-leaf stage were subjected to a temperature of −1°C for 3 h, followed by recovery for 3 days under normal conditions (25°C). Afterward, seedling survival rates were recorded. ANOVA analysis identified significant differences between the inbred lines (Figure 1A, Table S1). Among the 30 lines, KR701 exhibited the highest seedling survival rate (97.3%), while Hei8834 exhibited the lowest (5.01%). Therefore, these two lines, KR701 (most tolerant) and Hei8834 (most sensitive), with significant difference (p = 5.81E-10), were selected for further analysis.
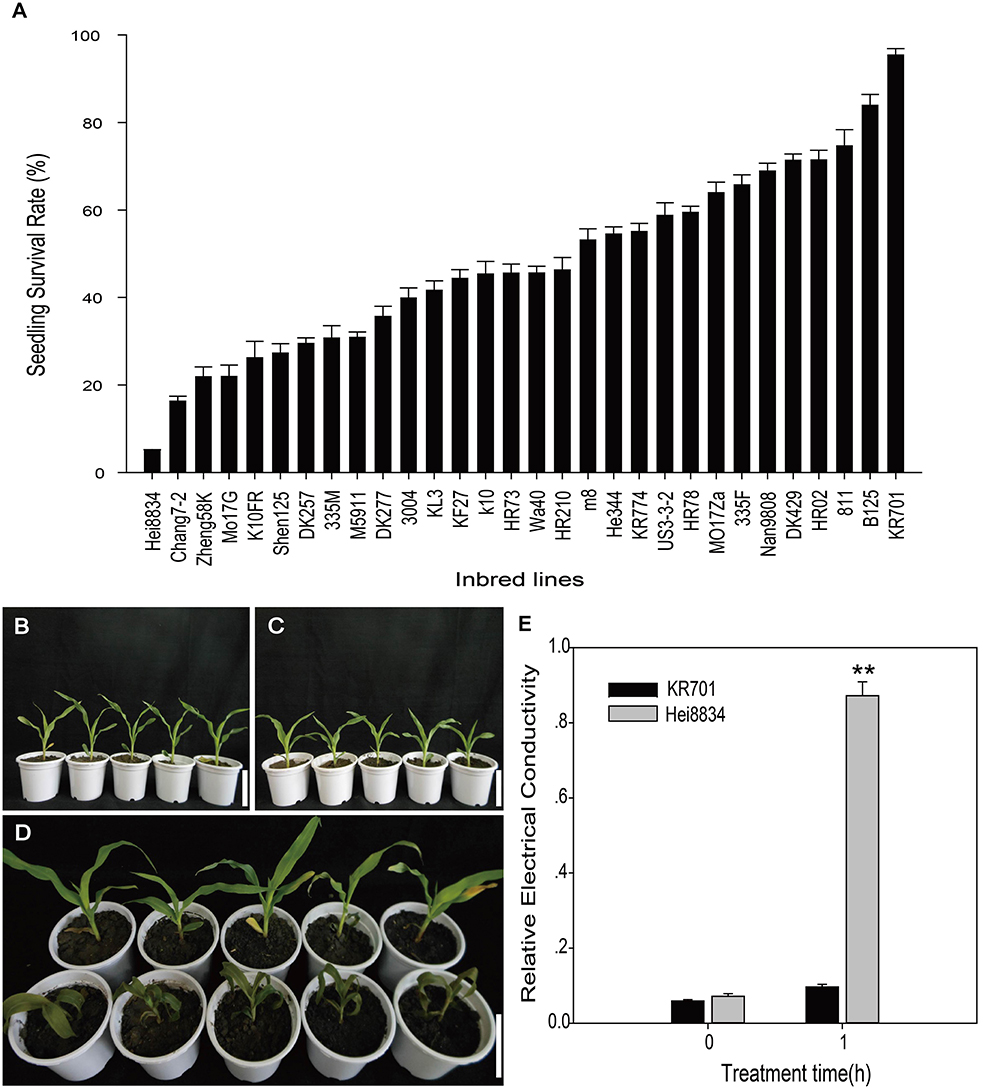
Figure 1. Seedling performances under freezing treatment. The performances were measured as survival rate and relative electrical conductivity. Survival rates were measured on the third day at 25°C after freezing treatment at −1°C for 3 h for 30 Chinese elite maize inbred lines (A). Seedlings before freezing treatment are demonstrated by the two extreme lines, freezing tolerant KR701 (B) and freezing sensitive Hei8834 (C). Visible differences were observed on the third day of recovery at 25°C after freezing treatment (D) between KR701 (upper row) and Hei8834 (lower row). Relative electrical conductivity was measured for KR701 and Hei8834 before freezing treatment and after 1 h of freezing treatment at −1°C (E). Double asterisks indicate a statistically significant difference (p < 0.01) between KR701 and Hei8834. Mean values are calculated from three biological replicates; error bars represent the standard error of the mean.
Physiological responses were recorded between these two inbred lines, including visual observations and relative electrical conductivity. The two inbred lines KR701 and Hei8834 showed little difference before freezing treatment (Figures 1B,C). After the 3-h freezing treatment (−1°C), the KR701 seedlings displayed little phenotypic change, maintaining fully expanded green leaves and intact plant architecture. In contrast, the Hei8834 seedlings showed obvious phenotypic damage, including severely wilted leaves and softened stems (Figure 1D). Relative electrical conductivity is a widely used indicator for membrane damage assay. The measurements of relative conductivity also confirmed Hei8834's severe physiological damage caused by the freezing treatment (Figure 1E).
RNA-Seq Analysis
We analyzed the transcriptomes of the freezing tolerant line (KR701, T) and freezing sensitive line (Hei8834, S) in response to freezing stress. The libraries of cDNA were prepared from these two inbred maize lines, before (Control, C) and 1 h after freezing treatment (F), and then subjected to RNA-seq profiling on the Illumina HiSeq 2000 platform. The raw data were deposited in the NCBI Sequence Read Archive (SRA) under the accession number SRX1434956. A total of about 154.4 million sequencing reads with lengths of 101 bp were generated, of which 78.84% (121.8 million reads) were mapped to the maize reference genome Zea_mays_Ensembl_AGPv3 (Table S2). Due to a technical failure, one replicate (R1) of the tolerant line after freezing stress (FT) was retrieved. All the rest three treatments (CT, CS, FS) had two replicates (R1, R2). The Q20 values of all samples, an indicator of the overall reproducibility and quality of the assay, were greater than 98% for all the replicates (Table S2). The normalized reads, referred to as fragments per kilobase of exon model per million mapped reads (FPKM), were used to estimate the total number of genes expressed in the seven samples, according to Mortazavi et al. (2008).
To analyze the similarities and differences between the seven samples, cluster analysis of all the genes using FPKM was performed. The cluster results showed clear separation between the freezing tolerant and freezing sensitive lines. And, the replicates of each treatment clustered together (Figure S1). PCA analysis of all samples also showed the same tendency as the cluster results (Figure S2). These results demonstrated that this experiment was reproducible and reliable.
Transcriptomic Responses
FPKM values ≥ 1 were used to determine genes expressed. A total of 19,794 annotated transcripts were identified in the four treatments with Cufflinks (Trapnell et al., 2010). The number of genes specifically expressed in each treatment, genes shared between each treatment, and genes shared among all combinations of treatments are illustrated in Figure 2. Of these transcripts, 73% (14,415) of the genes were represented in all treatments. Before freezing stress, 84% (16,642) and 87% (17,292) of the genes were expressed in the tolerant line (KR701, CT) and the sensitive line (Hei8834, CS), respectively. After freezing stress, 85% (16,761) and 88% (17,352) were expressed in KR701 (FT) and Hei8834 (FS), respectively (Table S3).
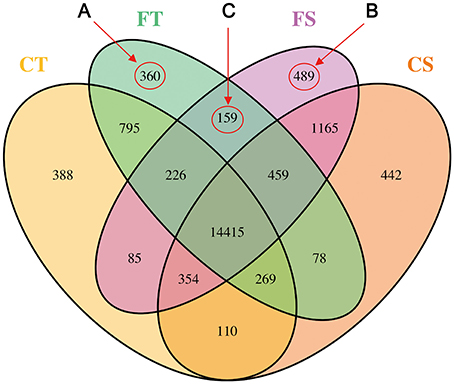
Figure 2. Profile of gene expression by inbred line and freezing treatment. The gene expression profile is illustrated as the number of transcriptomic responses by using a Venn diagram. A total of 19,794 genes were expressed. Freezing treatments are labeled Control (C) and Freezing (F). The Tolerant line (KR701) and Sensitive line (Hei8834) are labeled “T” and “S,” respectively. The biological samples of four combinations are CT, FT, CS, and FS, respectively. The area labeled “A” represents the genes specifically expressed in tolerant line KR701 after freezing treatment (FT). The area labeled “B” represents the genes specifically expressed in sensitive line Hei8834 after freezing treatment (FS). The area labeled “C” represents the freezing responsive genes shared by the tolerant and sensitive lines.
A total of 360 genes (Group A in Figure 2) were specifically expressed in tolerant line KR701 after freezing treatment (FT). GO annotation analysis (agriGO, http://bioinfo.cau.edu.cn/agriGO/analysis.php) of this group showed that GO: 0007186 (G-protein coupled receptor protein signaling pathway) and GO: 0008152 (metabolic process) were the most significantly enriched GO terms in the biological process category. Within the molecular function category, GO terms concerning nucleotide binding, ion binding, and catalytic activity were significantly enriched (Table S4).
Group B, containing 489 genes, represents the genes specifically expressed in sensitive line Hei8834 after freezing treatment (FS). In this group, GO: 0050826 (response to freezing), GO: 0045449 (regulation of transcription), and GO: 0051171 (regulation of nitrogen compound metabolic process) were the most significantly enriched GO terms in biological process. GO: 0004713 (protein tyrosine kinase activity) and GO: 0005488 (binding) were the most enriched in molecular function (Table S5).
Group C represents the expressed genes after freezing treatment that were shared by the tolerant and sensitive lines. In this group, GO: 0005488 (binding) and GO: 0004713 (protein tyrosine kinase activity) were also significantly enriched (Table S6).
Differential Expression Analysis
The software package Cuffdiff (Trapnell et al., 2012) was used to explore the differentially expressed genes (DEGs) between different treatments. Before freezing treatment, a total of 3,158 genes were differentially expressed between the tolerant and sensitive lines (see the circle labeled “CS_CT” in Figure 3). Of these DEGs, 1649 had higher expression levels in the tolerant line compared to the sensitive line (Table 1). After freezing treatment, we found 2,354 DEGs between the tolerant and sensitive lines (see the circle labeled “FS_FT” in Figure 3). Of these DEGs, 1084 had higher expression levels in the tolerant line compared to the sensitive line (Table 1). In the tolerant line, 439 genes were differentially expressed before and after freezing treatment (see the circle labeled by “CT_FT” in Figure 3); 322 of these 439 were up-regulated (Table 1). In the sensitive line, we found 852 DEGs before and after freezing treatment (see the circle labeled by “CS_FS” in Figure 3); 632 of these 852 were up-regulated (Table 1).
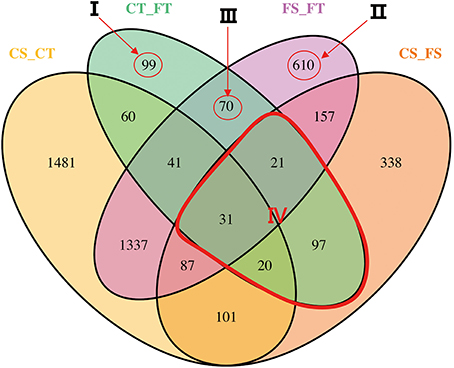
Figure 3. Profile of differentially expressed genes (DEGs). The differentiations were compared between inbred lines under each freezing treatment, or between freezing treatments in each inbred line. Freezing treatments are labeled Control (C) and Freezing (F). The tolerant line (KR701) and sensitive line (Hei8834) are labeled “T” and “S,” respectively. The four treatment-line biological samples are control-tolerant (CT), freezing-tolerant (FT), control-sensitive (CS), and freezing-sensitive (FS). Each compared combination is separated by an underscore (e.g., CT_FT). In the Venn diagram, the numbers of DEGs are illustrated across the intersection areas among the compared combinations. In total, we found 4550 DEGs from all the areas. Some of the areas are more important than others. Four critical areas, labeled I, II, III, and IV, totally contain 948 DEGs. Area I contains the tolerant treatment response (TTR) DEGs, excluding others. Area II contains the line response under freezing (LRF) DEGs, excluding others. Area III contains both tolerance treatment response and line response under freezing (TRLR) DEGs, excluding others. Area IV contains the treatment response (TR) DEGs within line.
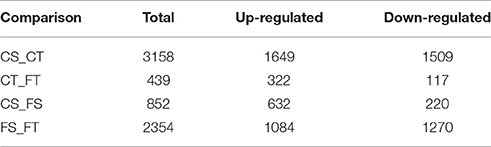
Table 1. Number of DEGs from the four experimental comparisons.
In total, we found 4550 DEGs among the four comparison groups, as indicated by the Venn diagram (Figure 3). The combinations of the four groups reflect the impact of lines or treatment. Some of the combinations are more important than others in aspect of freezing tolerance. Area I represents specific DEGs of CT_FT, that is, the specific freezing responsive DEGs of the freezing tolerant line. Of these 99 DEGs, 57 were up-regulated and 42 were down-regulated. Area II represents specific DEGs of FS_FT, that is, specific DEGs shared between the freezing sensitive and freezing tolerant lines after freezing treatment. Of these 610 DEGs, 244 were up-regulated and 366 were down-regulated. Area III represents the 70 specifically shared DEGs between CT_FT and FS_FT, that is, freezing responsive DEGs of the tolerant line that were also differentially expressed between the tolerant and sensitive lines after freezing treatment.
To investigate the biological functions of these DEGs, GO enrichment analysis was performed with agriGO (Du et al., 2010). For the 99 specific DEGs in area I (Figure 4B), GO: 0042592 (homeostatic process) and GO: 0008152 (metabolic process) were significant GO terms in biological process; GO: 0005488 (binding) and GO: 0003824 (catalytic activity) were significant GO terms in molecular function. In area II, significant GO terms in biological process included GO: 0050826 (response to freezing) and GO: 0042592 (homeostatic process), which validated our predicted biological functions for this DEG area (Figure 4A). In area III, GO terms concerning ion binding and hydrolase activity were significantly enriched (Figure 4C), suggesting DEGs with those molecular functions may contribute to the freezing response of the tolerant line and the different freezing responses between the two lines.
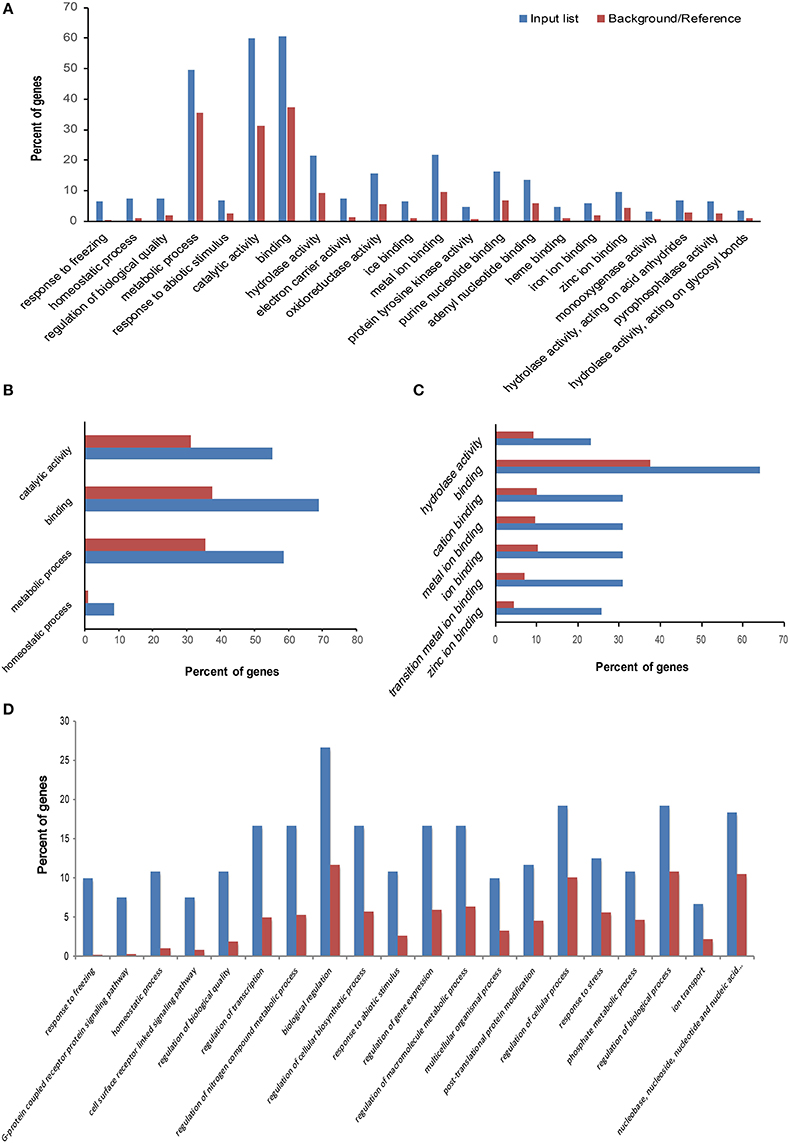
Figure 4. Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs). The GO analyses were performed on four sets of specific DEGs, labeled areas I, II, III, and IV in Figure 3; each area contained 99, 610, 70, and 169 DEGs, respectively. The observed gene frequency in each GO term is contrasted with the expected frequency under the null distribution (background/reference). The genes in areas II, I, III, and IV correspond to line response under freezing (LRF) (A), tolerance treatment response (TTR) (B), tolerance treatment response and line response under freezing (TRLR) (C), and freezing treatment response in both lines (TR) (D), respectively.
Genetic Difference between the Tolerant and Sensitive Inbred Lines at Transcriptomic Level
We anticipated that the transcriptomic differences between the tolerant and sensitive inbred lines before freezing treatment also relate the genetic difference between the two lines in responses to freezing stress. The GO enrichment analyses were performed on CT-upregulated and CS-upregulated genes of the CS_CT DEGs. The top 10 significant GO terms in biological process and molecular function were further compared (Tables 2, 3). In biological process, GO terms “metabolic process,” “response to cold,” “homeostatic process,” “response to temperature stimulus,” and “regulation of biological quality” were shared between the two lines. However, more CT-upregulated genes were enriched in these GO terms, compared to CS-upregulated genes. In CT-upregulated genes, GO terms related to metabolic process and protein phosphorylation were enriched specifically. In CS-upregulated genes, GO terms related to transcription, G-protein signaling pathway and nitrogen compound metabolic process were enriched. In molecular function, however, most of the enriched GO terms were shared. Therefore, the different number of genes in shared GO terms and the differentially enriched GO terms in biological process may provide the basis for the different freezing responses of the tolerant and sensitive inbred lines.
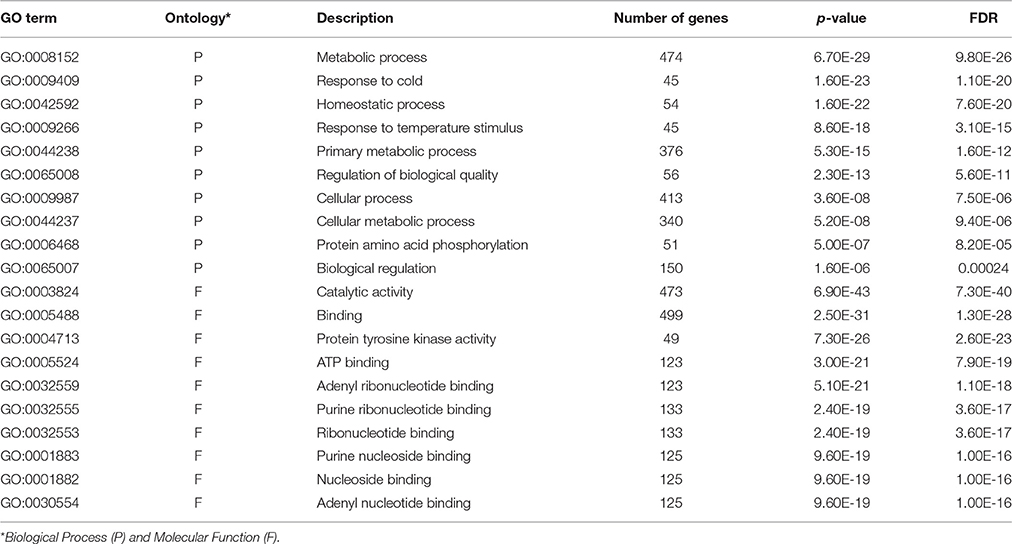
Table 2. Enriched GO terms of CT-upregulated genes.
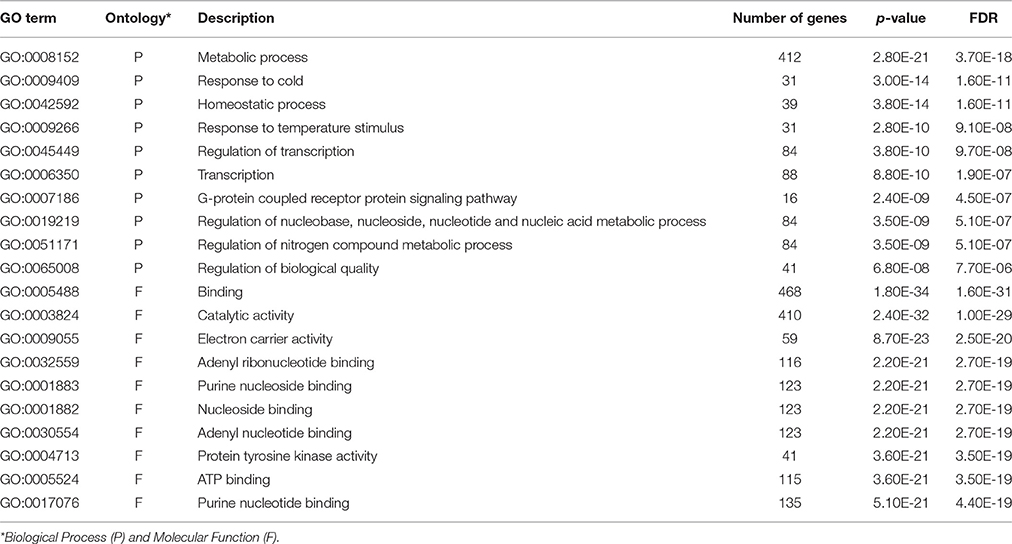
Table 3. Enriched GO terms of CS-upregulated genes.
Common Freezing Responsive Genes
As shown in Figure 3, area IV represents the 169 DEGs shared by CT_FT and CS_FS. GO enrichment analysis for these DEGs is illustrated in Figure 4D. Of these shared genes, 151 were up-regulated in both CT_FT and CS_FS (UU genes); 13 were down-regulated in both CT_FT and CS_FS (DD genes); two were up-regulated in CT_FT, but down-regulated in CS_FS (UD genes); and three were down-regulated in CT_FT, but up-regulated in CS_FS (DU genes). We designated the 151 UU and 13 DD genes as common freezing responsive genes between the tolerant and sensitive lines. Results from the gene ontology analysis of these common freezing responsive genes are provided in Table S7. For biological process, GO terms “response to freezing,” “G-protein coupled receptor protein signaling pathway,” “homeostatic process,” and “regulation of transcription” were significantly enriched. These results suggest that these processes may participate in the response of maize seedlings to freezing conditions. For molecular function, genes which function in binding (ice binding, DNA binding, metal ion binding, etc.), catalytic activity (protein tyrosine kinase activity, hydrolase activity, phosphatase activity, etc.), and receptor activity were significantly enriched. The GO term “integral to membrane part” was the only one enriched relative to cellular component.
Common Freezing Responsive Genes with Differential Response Levels
Some of the 164 common freezing responsive genes exhibited differential response levels between the tolerant and sensitive lines. We calculated the ratio of CT_FT log2 fold change to CS_FS log2 fold change. Using ratios ≥ 2 or ≤ 0.5 as selection criteria, we found a total of eight common freezing responsive genes with significant differential response levels.
With these eight genes, and the two UD genes and three DU genes, we performed a homolog search in Arabidopsis (Table S8). Previous studies report that many of the homologous Arabidopsis genes we identified are involved in abiotic stress response processes, including cold stress. For example, AT1G72360 (homolog of GRMZM2G148333) encodes a member of the ethylene response factor (ERF) subfamily B-2 of the ERF/AP2 transcription factor family and participates in low oxygen signaling (Licausi et al., 2010). GRMZM2G148333's homolog in rice, LOC_Os09g26420, has been identified as OsBIERF1, which can be induced by treatments with BTH (benzothiadiazole), salicylic acid, salt, cold, drought, and wounding. This finding suggests that OsBIERF1 may participate in different signaling pathways that mediate both biotic and abiotic responses (Cao et al., 2006). AT1G80920 (homolog of GRMZM2G086841) encodes a chloroplast-targeted DnaJ protein, AtJ8, which is negatively regulated by light and stress (Chen et al., 2011). Additionally, AT1G80920 is down-regulated in the Arabidopsis mutant sfr6, which shows more freezing sensitivity after cold acclimation compared to its wild counterpart (Boyce et al., 2003). AT3G28210 (homolog of GRMZM2G125775) is also a stress-associated protein 12 (SAP12) and is strongly induced under cold and salt stress (Ströher et al., 2009). AT5G56550 (homolog of GRMZM2G076844) encodes OXIDATIVE STRESS 3 (OXS3), a putative N-acetyltransferase or thioltransferase, which participates in heavy metal and oxidative stress (Blanvillain et al., 2009). AT2G36460 (homolog of GRMZM2G057823) encodes fructose-bisphosphate aldolase 6, which is involved in response to cadmium ion, salt stress, and pentose-phosphate shunt (Lu et al., 2012).
In addition, many gene homologs participate in plant hormone signaling. For example, AT1G18350 (homolog of GRMZM2G344388), also known as ATMKK7 or BUD1, encodes a MAP kinase kinase. BUD1 is a negative regulator of polar auxin transport and its overexpression activates both basal and systemic acquired resistance (Zhang et al., 2008). AT1G76680 (homolog of GRMZM2G000236) encodes a 12-oxophytodienoic acid reductase, which is up-regulated by senescence and jasmonic acid (He and Gan, 2001). AT4G08950 (homolog of GRMZM2G149422), also known as EXORDIUM (EXO), responds to brassinosteroid stimulus (Müssig et al., 2006).
DEGs Related to “Response to Freezing”
Of the 17 DEGs in area II (Figure 3) that were assigned GO term “response to freezing,” nine genes had higher expression levels and eight genes had lower expression levels in the tolerant line compared to the sensitive line. Detailed information about these genes, including their homologs in Arabidopsis, can be found in Table S9.
Of the nine genes with higher expression levels in the freezing tolerant line, GRMZM2G075974 and GRMZM2G115422 encode transferase. GRMZM2G075974 encodes a glutamine amidotransferase and may be involved in defense response. AT2G39980, the Arabidopsis homolog of GRMZM2G115422, encodes a HXXXD-type acyl-transferase, which may be involved in response to karrikin (Nelson et al., 2010). Two genes encode ion-binding proteins. AT4G14040, the homolog of GRMZM2G103812, encodes a selenium-binding protein and is involved in cadmium detoxification processes (Dutilleul et al., 2008). GRMZM2G459663, which encodes a Calcium-binding EF-hand family protein, may be involved in abiotic and biotic stress (Davletova et al., 2005; Ascencio-Ibanez et al., 2008). GRMZM2G079956, which encodes a BAG family protein, was up-regulated in the freezing tolerant line. Plant BAG proteins have been reported to participate in both abiotic biotic stress (Doukhanina et al., 2006). GRMZM2G348452 encodes a cytokinin dehydrogenase and was also up-regulated in the freezing tolerant line. Cytokinin dehydrogenase catalyzes the irreversible degradation of cytokinins isopentenyladenine, zeatin, and their ribosides in a single enzymatic step by oxidative side-chain cleavage. Thus, this enzyme plays an important role in regulating cytokinin functions and participates in plant aging and senescence processes (Schmülling et al., 2003; Mýtinová et al., 2011). AT2G41475 (homolog of GRMZM2G061932), also known as ATS3, is involved in stomatal closure (Van Hove et al., 2015).
Eight genes had higher expression levels in the freezing treated sensitive line compared to the tolerant line. This finding suggests a differential response strategy or simply a response caused by different degrees of freezing damage. AT1G12520 (homolog of GRMZM2G175728) encodes a copper chaperone for SOD1 and is up-regulated in response to copper and senescence (Huang et al., 2012; Kuo et al., 2013). GRMZM2G044194 encodes phytosulfokine, which is an important small peptide signal (Yang et al., 2001). AT1G56010 (homolog of GRMZM2G114850), also known as NAC1, encodes a NAC transcription factor, which is involved in auxin signaling (Xie et al., 2000). AT1G56010's homolog in Brassica napus is involved in response to multiple biotic and abiotic stresses, such as physical wounding, pathogen infection, low temperature, and drought (Hegedus et al., 2003). GRMZM2G180335 encodes a Dynamin GTPase effector, which localizes to the chloroplast, mitochondrion, and peroxisome and is involved in peroxisome and mitochondria fission (Arimura et al., 2008; Aung and Hu, 2009). GRMZM2G178038, which encodes a Zinc finger family protein, may participate in protein degradation (Boisson et al., 2003).
Quantitative Real-time RT-PCR (qRT-PCR) Confirmation
To confirm our findings based on the RNA-seq data, we conducted a validation experiment by using quantitative real-time PCR (qRT-PCR). We made the selection of genes based on these criteria: highly differentiated in response to freezing and reported to be potentially associated with cold resistance. In total, 30 genes were selected from the identified DEGs. Results of the qRT-PCR analysis confirmed our findings based on RNA-seq data (Table S11). The patterns of RNA-seq expression on 28 genes were replicated by the qRT-PCR approach. Only two genes (ZmGRMZM2G076844 and ZmGRMZM2G476685) showed different expression patterns in the sensitive line. Correlation of fold changes before and after freezing treatment between qRT-PCR and RNA-seq are shown in Figure 5.
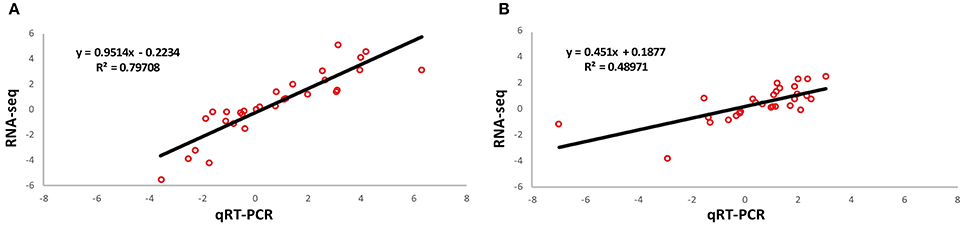
Figure 5. Validation of RNA-seq expression through qRT-PCR. Validation was performed in both freezing tolerant maize inbred line KR701 (A) and freezing sensitive maize inbred line Hei8834 (B). The plots demonstrate the expression ratio between before and after freezing treatment in Log scale with base of two.
Materials and Methods
Freezing Resistance Evaluation of Maize Inbred Lines
Thirty maize (Zea mays) inbred lines (provided by Heilongjiang Academy of Agricultural Sciences) were used for assessing seedling freezing tolerance. Seeds were surface sterilized with 75% (v/v) ethanol for 3 min, then rinsed three times with distilled water. Following, seeds were laid between two layers of damp paper at 25°C and left to germinate in the dark for about 3 days. Uniformly germinated seeds with 2–3 cm coleoptiles were selected and sown in pots filled with peat, vermiculite, and perlite (10:1:1 by vol.) Seedlings were grown in an incubator (RXZ-0450, Ningbo Jiangnan Instrument Factory) at 25/20°C (day/night), 450 lmol m−2 s−1 light density, and a 14/10 h (light/dark) photoperiod for 2 weeks until three leaves had developed. The seedlings were then subjected to the freezing stress experiment, using a treatment of −1°C for 3 h (Shou et al., 2004) followed by 25°C for 3 days. Afterward, seedling survival rates were recorded.
Physiological Analyses of Freeze-Treated Maize Seedlings
To analyze the physiological changes of maize seedlings under the freezing treatment, electrical conductivities (both before and after freezing stress) were measured (Griffith and Mclntyre, 1993). Two replicates of 0.1 g fresh leaves were sampled and cut into 2-cm pieces. The leaf samples were then rinsed in pure water to remove cellular proteins from the cut ends and placed in tubes with 10 ml distilled water. After 20 h, the initial electrical conductivity (R1) was measured at room temperature using a METTLER TOLEDO FE30 CONDUCTIVITY (METERMettler Toledo Instruments Co., LTD, Shanghai). The samples were then boiled for 30 min and measured again for the final electrical conductivity (R2). The relative electrical conductivity was calculated as R1/R2.
Total RNA Isolation, cDNA Library Construction, and Sequencing
Maize inbred lines KR701 (freezing tolerant) and Hei8834 (freezing sensitive) were grown according to the method above. For isolation of total RNA, maize seedlings at the three-leaf stage were exposed to a freezing condition (−1°C) for only 1 h (h). The third leaves of both KR701 and Hei8834 were sampled at 0 h (prior to freezing) and after 1 h freezing treatment with liquid nitrogen and then stored in −80°C for further transcriptome analysis. The total RNA of the leaf samples was extracted using Trizol reagent (Invitrogen) and purified using the RNeasy Plant kit (Invitrogen, CA, USA). The RNA quality was checked by a NanoDrop 1000 spectrophotometer and an agarose gel electrophoresis system. The cDNA library construction and sequencing were conducted by BGI Company (Shenzhen, China). In brief, poly-A-containing mRNA was isolated and then subjected to cDNA synthesis. Short mRNA fragments were purified for end reparation and single nucleotide A addition, and then connected with adapters. Next, the cDNA library was obtained by PCR amplification and sequenced on the Illumina Hiseq 2000 platform with 100 cycles of paired-end (2 × 101 bp) sequencing. Raw sequence data were obtained for further analysis. The sequence reads were submitted to GenBank GEO database under accession number SRP065916.
Processing and Mapping of Illumina Reads
The raw data (raw reads) were filtered with FASTQ_Quality_Filter tool from the FASTX-toolkit. The clean data were used for further analysis.
After preprocessing the RNA-seq data, the reads were mapped to the maize reference genome version 3 (B73 RefGen_v3) (Hubbard et al., 2002) using a spliced aligner Tophat, which was downloaded from http://ftp.maizesequence.org/current/ (Trapnell et al., 2009; Kakumanu et al., 2012).
Transcript Assembly and Differential Expression Analysis
The Sequence Alignment generated by Tophat was then processed by the software Cufflinks (Trapnell et al., 2010) to assemble the alignments in the Sequence Alignment/Map file into transcript fragments (transfrags). FPKM was used as the unit of measurement to estimate transcript abundance. Differential expression analysis of four samples was performed using the Cuffdiff program (Trapnell et al., 2013). P-value was adjusted using the q-value (Storey, 2003).
Gene Ontology (GO) Analysis of Selected DEGs
Candidate DEGs were submitted to agriGO (GO analysis toolkit and database for the agriculture community; http://bioinfo.cau.edu.cn/agriGO/index.php) for gene ontology analysis. Enriched GO terms were selected using Singular Enrichment Analysis (SEA) with the maize reference genome B73 as background (Maizesequence, version: 5b). The overrepresented terms in the three categories—biological process, cellular component, and molecular function—were filtered by statistical information, including Fisher's exact test and the Bonferroni multi-test adjustment method (Du et al., 2010; Han et al., 2015).
Quantitative Real-time RT-PCR
Quantitative real-time PCR (qRT-PCR) primers were designed for 30 genes. Gene-specific primers were designed online (https://www.genscript.com/ssl-bin/app/primer) (Table S10). Total RNA was isolated from seedling leaves. For cDNA synthesis, 1 μg of total RNA was reverse-transcribed in a total volume of 25 μL, using One-step gDNA Removal and cDNA Synthesis Super Mix (TRansScript). Synthesized cDNA samples were diluted 10 times prior to qRT-PCR. ZmActin1 was used as the internal reference gene in this study. qRT-PCR was performed with 2 μl of template cDNA, 0.5 μl of forward primer (50 pmol), 0.5 μl of reverse primer (50 pmol), and 10 μl of SYBR Green mix (TOYOBO, Japan) in a total reaction volume of 20 μl. qRT-PCR was carried out in an iQ5 (Bio-Rad) thermocycler. Each sample had three technical replicates.
Discussion
Along with the climate changes caused by greenhouse gas emissions, freezing and other abiotic stresses are posing ever-growing threats to global food security. Plant responses to these stresses are complicated processes with evolutionary wisdom. Understanding the molecular mechanisms underlying stress responses and applying this knowledge to plant breeding and agricultural practice is of great significance. Transcriptomic analysis using RNA-seq is a powerful tool to monitor global gene expression status and to discover genes responsible for molecular functions and biological processes. Therefore, more and more researchers use RNA-seq analysis to study the abiotic stress response of different species (e.g., An et al., 2012; Raney et al., 2014; Sobkowiak et al., 2014).
Originating in a tropical area, maize is inherently frost-sensitive. However, natural variations in freezing and chilling tolerance exist in different maize populations. As shown with our 30 different inbred lines, seedling survival rate after 3 h of freezing treatment can range from 5.01 to 97.3%. Therefore, investigating the molecular mechanisms that contribute to the freezing response variation in maize is both reasonable and important. Four different groups of RNA-seq data were prepared and analyzed, using two different maize inbred lines (freezing tolerant and freezing sensitive) and two treatments (before and after freezing conditions). By making multiple comparisons of these four groups, we were able to isolate functional information for both the common and differential responsive genes.
In general, plant cells sense low temperatures (chilling or freezing) at the plasma membrane (Pearce, 1999). After signal reception, downstream signaling pathways are activated, including hormone signaling, Ca2+ signaling, and ROS signaling (Miura and Furumoto, 2013).
Hormone Signaling in Freezing Stress Response
Previous studies have found that plant hormones participate in low temperature (including chilling and freezing) responses of plants. For example, a number of genes involved in the biosynthesis or signaling of plant hormones, such as abscisic acid, gibberellic acid, and auxin, are regulated by cold stress (Lee, 2005). Several auxin-responsive genes are differentially regulated under various abiotic stress conditions, suggesting that auxin may play an important role in abiotic stress signaling (Jain and Khurana, 2009).
We identified several DEGs involved in auxin signaling such as GRMZM2G344388 (Arabidopsis homolog AT1G18350, also known as ATMKK7 or BUD1, encodes a MAP kinase kinase) and GRMZM2G114850 (Arabidopsis homolog AT1G56010, also known as NAC1, encodes a NAC family transcription factor). Genes involved in the regulation of jasmonic acid, brassinosteroid, cytokinin, and ethylene were also identified, suggesting these plant hormones may also play important roles in freezing stress signaling. Additionally, we identified GRMZM2G044194, which encodes phytosulfokine, suggesting that small peptide signaling may also be an important pathway in freezing stress signaling.
Ca2+-Mediated Freezing Stress Signaling
Ca2+-mediated signal transduction is one of the most focused signaling pathways in plants and animals. Cold-shock causes a transient rise in cytosolic calcium levels, which probably results (at least in part) from cold-induced opening of plasma membrane calcium channels (Monroy and Dhindsa, 1995; Lewis et al., 1997). In mammals, Ca2+ channels can interact with heterotrimeric guanine nucleotide-binding protein (G protein) complexes in response to stress (Wang and Chong, 2010). Recently, Ma et al. (2015) found that the QTL COLD1, which may encode a regulator of G-protein signaling (RGS) with GTPase-accelerating activity, interacts with G protein to activate the Ca2+ channel for temperature sensing in rice. Consequently, the expression of stress-responsive genes such as transcription factors, metabolic enzymes, and ion transporters are up-regulated. That is, the up-regulated calcium ion binding protein can activate the downstream cascade kinases, leading to the stimulation of cold-responsive genes. More importantly, their study identified a SNP in COLD1 that may explain the cold tolerance differences between two subspecies of Asian cultivated rice.
In our study, the GO term “G-protein coupled receptor protein signaling pathway” was one of the significantly enriched GO terms in the common freezing responsive genes. For terms associated with molecular function, genes involved in binding (ice binding, DNA binding, and metal ion binding, etc.), catalytic activity (protein tyrosine kinase activity, hydrolase activity, and phosphatase activity, etc.), and receptor activity were significantly enriched. For example, we identified DEG GRMZM2G459663, which encodes a Calcium-binding EF-hand family protein. These data further support the important role of calcium signaling in the freezing stress signaling of plants.
ROS Signaling in Freezing Stress Response
Reactive Oxygen Species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals accumulate under chilling conditions (Suzuki and Mittler, 2006). To prevent the lethal damage caused by ROS, plants have evolved complex antioxidant systems. Several ROS scavenging enzymes have been identified, including catalase (CAT), superoxide dismutase (SOD), and glutathione transferase (GST).
In our RNA-seq data, we also identified genes involved in ROS scavenging. For example, GRMZM2G175728, which encodes a copper chaperone for SOD1, is up-regulated in response to copper and senescence. GRMZM2G075974, which encodes a glutamine amidotransferase, may be involved in defense response. AT5G56550 (Arabidopsis homolog of GRMZM2G076844), also known as OXIDATIVE STRESS 3 (OXS3), is involved in tolerance to heavy metals and oxidative stress.
Ultimately, in order to fully understand the mechanisms of freezing tolerance, plant hormones, Ca2+ level, and ROS level should be thoroughly investigated during the cloning process and functional analysis of these potential candidate genes.
The Transcriptomic Difference between Tolerant and Sensitive Lines before Freezing Treatment
From our RNA-seq data, we found that, for the two selected inbred lines, more DEGs existed before freezing treatment compared to after freezing treatment. This finding suggests that the different molecular basis between the tolerant and sensitive lines before freezing treatment are important for their different freezing stress responses. Detailed GO enrichment analysis of CT-upregulated genes and CS-upregulated genes also showed differences in the GO terms enriched in the biological process category. In the tolerant line, GO terms related to metabolic process and protein phosphorylation were enriched. In the sensitive line, GO terms related to transcription, G-protein signaling pathway, and nitrogen compound metabolic process were enriched. Thus, these biological processes may play major roles in the different freezing responses between these two inbred lines. As for the shared GO terms, the genes and the number of genes in each term were different, which may also explain the lines' different freezing sensitivities.
In conclusion, through a freezing-tolerance screening of maize inbred lines from the Heilongjiang Province with the highest latitude in China, we identified two inbred lines with extreme, but opposite, responses—highly tolerant and highly sensitive. Using these two lines and an RNA-seq approach, we obtained candidate genes with different expression patterns and with enrichment on specific gene ontologies. The top 30 candidate genes were repeatedly confirmed by using QRT-PCR, which resulted in high correlation with our RNA-seq-based findings. This study provided valuable resources for further studies to enhance understanding of the molecular mechanisms underlying the freezing response of maize. Ultimately, a greater understanding of these molecular mechanisms will enable targeted breeding strategies for developing superior maize varieties able to resist frost and to achieve their full yield potential.
Author Contributions
Conceived and designed the experiments: ZZ, DY, and TW. Performed the experiments: ZL, GH, XL, QZ. Analyzed the data: ZL, GH, YZ, XZ, XY, and YL. Wrote the paper: ZL, GH, and ZZ. Revised the paper: ZZ. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants from the Agriculture Department of International Cooperation Project “Cold resistance identification and utilization of the recent introduction of America maize germplasm resource” (2013); Agricultural research outstanding talents and innovation team (2015); China Postdoctoral Science Foundation (2015M571383); an Emerging Research Issues Internal Competitive Grant from the Agricultural Research Center at Washington State University, College of Agricultural, Human, and Natural Resource Sciences; and the Endowment and Research Project (No. 126593) from the Washington Grain Commission. The authors thank Linda R. Klein for writing advice and editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01477
Availability of Supporting Data
Illumina sequence data are available from NCBI under Short Read Archive accession SRP065916.
References
Adamczyk, J., and Królikowski, Z. (1998). “From open pollinated varieties to single crosses: rapid development in Polish maize breeding,” in Crop Development for Cool and Wet European Climate, eds P. Sowiński, B. Zagdańska, A. Aniol, and K. Pithan (Brussels: European Cooperation in the Field of Scientific and Technical Research, Commission of European Communities), 65–69.
An, D., Yang, J., and Zhang, P. (2012). Transcriptome profiling of low temperature-treated cassava apical shoots showed dynamic responses of tropical plant to cold stress. BMC Genomics 13:64. doi: 10.1186/1471-2164-13-64
Arimura, S., Fujimoto, M., Doniwa, Y., Kadoya, N., Nakazono, M., Sakamoto, W., et al. (2008). Arabidopsis ELONGATED MITOCHONDRIA1 is required for localization of DYNAMIN-RELATED PROTEIN3A to mitochondrial fission sites. Plant Cell 20, 1555–1566. doi: 10.1105/tpc.108.058578
Ascencio-Ibáñez, J. T., Sozzani, R., Lee, T.-J., Chu, T.-M., Wolfinger, R. D., Cella, R., et al. (2008). Global analysis of Arabidopsis gene expression uncovers a complex array of changes impacting pathogen response and cell cycle during geminivirus infection. Plant Physiol. 148, 436–454. doi: 10.1104/pp.108.121038
Aung, K., and Hu, J. (2009). The Arabidopsis peroxisome division mutant pdd2 is defective in the DYNAMIN-RELATED PROTEIN3A (DRP3A) gene. Plant Signal. Behav. 4, 542–544. doi: 10.1111/j.1365-313X.2008.03677.x
Blanvillain, R., Kim, J. H., Wu, S., Lima, A., and Ow, D. W. (2009). OXIDATIVE STRESS 3 is a chromatin-associated factor involved in tolerance to heavy metals and oxidative stress. Plant J. 57, 654–665. doi: 10.1111/j.1365-313X.2008.03717.x
Boisson, B., Giglione, C., and Meinnel, T. (2003). Unexpected protein families including cell defense components feature in the N-myristoylome of a higher eukaryote. J. Biol. Chem. 278, 43418–43429. doi: 10.1074/jbc.M307321200
Boyce, J. M., Knight, H., Deyholos, M., Openshaw, M. R., Galbraith, D. W., Warren, G., et al. (2003). The sfr6 mutant of Arabidopsis is defective in transcriptional activation via CBF/DREB1 and DREB2 and shows sensitivity to osmotic stress. Plant J. 34, 395–406. doi: 10.1046/j.1365-313X.2003.01734.x
Bredow, M., Vanderbeld, B., and Walker, V. K. (2016). Ice-binding proteins confer freezing tolerance in transgenic Arabidopsis thaliana. Plant Biotechnol. J. [Epub ahead of print]. doi: 10.1111/pbi.12592
Cao, Y., Song, F., Goodman, R. M., and Zheng, Z. (2006). Molecular characterization of four rice genes encoding ethylene-responsive transcriptional factors and their expressions in response to biotic and abiotic stress. J. Plant Physiol. 163, 1167–1178. doi: 10.1016/j.jplph.2005.11.004
Chen, K.-M., Piippo, M., Holmström, M., Nurmi, M., Pakula, E., Suorsa, M., et al. (2011). A chloroplast-targeted DnaJ protein AtJ8 is negatively regulated by light and has rapid turnover in darkness. J. Plant Physiol. 168, 1780–1783. doi: 10.1016/j.jplph.2011.04.001
Chinnusamy, V. (2003). ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 17, 1043–1054. doi: 10.1101/gad.1077503
Davletova, S., Schlauch, K., Coutu, J., and Mittler, R. (2005). The zinc-finger protein Zat12 plays a central role in reactive oxygen and abiotic stress signaling in Arabidopsis. Society 139, 847–856. doi: 10.1104/pp.105.068254.1
Ding, Y., Li, H., Zhang, X., Xie, Q., Gong, Z., and Yang, S. (2015). OST1 kinase modulates freezing tolerance by enhancing ICE1 stability in Arabidopsis. Dev. Cell 32, 278–289. doi: 10.1016/j.devcel.2014.12.023
Doherty, C. J., Van Buskirk, H. A., Myers, S. J., and Thomashow, M. F. (2009). Roles for Arabidopsis CAMTA transcription factors in cold-regulated gene expression and freezing tolerance. Plant Cell 21, 972–984. doi: 10.1105/tpc.108.063958
Doukhanina, E. V., Chen, S., van der Zalm, E., Godzik, A., Reed, J., and Dickman, M. B. (2006). Identification and functional characterization of the BAG protein family in Arabidopsis thaliana. J. Biol. Chem. 281, 18793–18801. doi: 10.1074/jbc.M511794200
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Dutilleul, C., Jourdain, A., Bourguignon, J., and Hugouvieux, V. (2008). The Arabidopsis putative selenium-binding protein family: expression study and characterization of SBP1 as a potential new player in cadmium detoxification processes. Plant Physiol. 147, 239–251. doi: 10.1104/pp.107.114033
Fracheboud, Y., Jompuk, C., Ribaut, J. M., Stamp, P., and Leipner, J. (2004). Genetic analysis of cold-tolerance of photosynthesis in maize. Plant Mol. Biol. 56, 241–253. doi: 10.1007/s11103-004-3353-6
Fu, J., Cheng, Y., Linghu, J., Yang, X., Kang, L., Zhang, Z., et al. (2013). RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4:2832. doi: 10.1038/ncomms3832
Griffith, M., and Mclntyre, H. C. H. (1993). The interrelationship of growth and frost tolerance in winter rye. Physiol. Plant. 87, 335–344. doi: 10.1111/j.1399-3054.1993.tb01739.x
Grondin, A., Rodrigues, O., Verdoucq, L., Merlot, S., Leonhardt, N., and Maurel, C. (2015). Aquaporins contribute to ABA-triggered stomatal closure through OST1-mediated phosphorylation. Plant Cell 27, 1945–1954. doi: 10.1105/tpc.15.00421
Guo, L., Yang, H., Zhang, X., and Yang, S. (2013). Lipid transfer protein 3 as a target of MYB96 mediates freezing and drought stress in Arabidopsis. J. Exp. Bot. 64, 1755–1767. doi: 10.1093/jxb/ert040
Han, Y., Lv, P., Hou, S., Li, S., Ji, G., Ma, X., et al. (2015). Combining next generation sequencing with bulked segregant analysis to fine map a stem moisture locus in sorghum (Sorghum bicolor L. Moench). PLoS ONE 10:e0127065. doi: 10.1371/journal.pone.0127065
He, Y., and Gan, S. (2001). Identical promoter elements are involved in regulation of the OPR1 gene by senescence and jasmonic acid in Arabidopsis. Plant Mol. Biol. 47, 595–605. doi: 10.1023/A:1012211011538
Hegedus, D., Yu, M., Baldwin, D., Gruber, M., Sharpe, A., Parkin, I., et al. (2003). Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol. Biol. 53, 383–397. doi: 10.1023/B:PLAN.0000006944.61384.11
Huang, C. H., Kuo, W. Y., and Jinn, T. L. (2012). Models for the mechanism for activating copper-zinc superoxide dismutase in the absence of the CCS Cu chaperone in Arabidopsis. Plant Signal. Behav. 7, 428–430. doi: 10.4161/psb.19192
Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., Clark, L., et al. (2002). The Ensembl genome database project. Nucleic Acids Res. 30, 38–41. doi: 10.1093/nar/30.1.38
Hund, A., Fracheboud, Y., Soldati, A., Frascaroli, E., Salvi, S., and Stamp, P. (2004). QTL controlling root and shoot traits of maize seedlings under cold stress. Theor. Appl. Genet. 109, 618–629. doi: 10.1007/s00122-004-1665-1
Jaglo-Ottosen, K. R., Gilmour, S. J., Zarka, D. G., Schabenberger, O., and Thomashow, M. F. (1998). Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science 280, 104–106. doi: 10.1126/science.280.5360.104
Jain, M., and Khurana, J. P. (2009). Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 276, 3148–3162. doi: 10.1111/j.1742-4658.2009.07033.x
Ji, H., Wang, Y., Cloix, C., Li, K., Jenkins, G. I., Wang, S., et al. (2015). The Arabidopsis RCC1 family protein TCF1 regulates freezing tolerance and cold acclimation through modulating lignin biosynthesis. PLoS Genet. 11:e1005471. doi: 10.1371/journal.pgen.1005471
Kakumanu, A., Ambavaram, M. M. R., Klumas, C., Krishnan, A., Batlang, U., Myers, E., et al. (2012). Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 160, 846–867. doi: 10.1104/pp.112.200444
Kasuga, M., Liu, Q., Miura, S., Yamaguchi-Shinozaki, K., and Shinozaki, K. (1999). Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat. Biotech. 17, 287–291. doi: 10.1038/7036
Kuo, W., Huang, C., and Jinn, T. (2013). Chaperonin 20 might be an iron chaperone for superoxide dismutase in activating iron superoxide dismutase (FeSOD). Plant Signal. Behav. 8:e23074. doi: 10.4161/psb.23074
Lee, B. -H., Henderson, D. A., and Zhu, J. -K. (2005). The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell Online 17, 3155–3175. doi: 10.1105/tpc.105.035568
Leipner, J. (2009). Chilling Stress in Maize: From Physiology to Genetics and Molecular Mechanisms. Doctoral and Habilitation Theses, Eidgenössische Technische Hochschule Zürich, Department of Agricultural and Food Science, Zurich.
Lewis, B. D., Karlin-Neumann, G., Davis, R. W., and Spalding, E. P. (1997). Ca2+-activated anion channels and membrane depolarizations induced by blue light and cold in Arabidopsis seedlings. Plant Physiol. 114, 1327–1334. doi: 10.1104/pp.114.4.1327
Licausi, F., Van Dongen, J. T., Giuntoli, B., Novi, G., Santaniello, A., Geigenberger, P., et al. (2010). HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. Plant J. 62, 302–315. doi: 10.1111/j.1365-313X.2010.04149.x
Liu, H., Ouyang, B., Zhang, J., Wang, T., Li, H., Zhang, Y., et al. (2012). Differential modulation of photosynthesis, signaling, and transcriptional regulation between tolerant and sensitive tomato genotypes under cold stress. PLoS ONE 7:e50785. doi: 10.1371/journal.pone.0050785
Lu, W., Tang, X., Huo, Y., Xu, R., Qi, S., Huang, J., et al. (2012). Identification and characterization of fructose 1,6-bisphosphate aldolase genes in Arabidopsis reveal a gene family with diverse responses to abiotic stresses. Gene 503, 65–74. doi: 10.1016/j.gene.2012.04.042
Ma, S., Xi, Z., and Wang, Q. (2003). Risk evaluation of cold damage to corn in Northeast China. J. Nat. Disasters 12, 137–141.
Ma, Y., Dai, X., Xu, Y., Luo, W., Zheng, X., Zeng, D., et al. (2015). COLD1 confers chilling tolerance in rice. Cell 160, 1209–1221. doi: 10.1016/j.cell.2015.01.046
Maniatis, T., and Tasic, B. (2002). Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 418, 236–243. doi: 10.1038/418236a
Miura, K., and Furumoto, T. (2013). Cold signaling and cold response in plants. Int. J. Mol. Sci. 14, 5312–5337. doi: 10.3390/ijms14035312
Monroy, A. F., and Dhindsa, R. S. (1995). Low-temperature signal transduction: induction of cold acclimation-specific genes of alfalfa by calcium at 25 degrees C. Plant Cell 7, 321–331. doi: 10.1105/tpc.7.3.321
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth. 5, 621–628. doi: 10.1038/nmeth.1226
Müssig, C., Lisso, J., Coll-Garcia, D., and Altmann, T. (2006). Molecular analysis of brassinosteroid action. Plant Biol. 8, 291–296. doi: 10.1055/s-2005-873043
Mýtinová, Z., Motyka, V., Haisel, D., Lubovská, Z., Trávníčková, A., Dobrev, P., et al. (2011). Antioxidant enzymatic protection during tobacco leaf ageing is affected by cytokinin depletion. Plant Growth Regul. 65, 23–34. doi: 10.1007/s10725-011-9571-4
Nelson, D. C., Flematti, G. R., Riseborough, J.-A., Ghisalberti, E. L., Dixon, K. W., and Smith, S. M. (2010). Karrikins enhance light responses during germination and seedling development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 107, 7095–7100. doi: 10.1073/pnas.0911635107
Opitz, N., Paschold, A., Marcon, C., Malik, W., Lanz, C., Piepho, H.-P., et al. (2014). Transcriptomic complexity in young maize primary roots in response to low water potentials. BMC Genomics 15:741. doi: 10.1186/1471-2164-15-741
Pearce, R. (1999). Molecular analysis of acclimation to cold. Plant Growth Regul. 29, 47–76. doi: 10.1023/A:1006291330661
Presterl, T., Ouzunova, M., Schmidt, W., Möller, E. M., Röber, F. K., Knaak, C., et al. (2007). Quantitative trait loci for early plant vigour of maize grown in chilly environments. Theor. Appl. Genet. 114, 1059–1070. doi: 10.1007/s00122-006-0499-4
Qin, F., Sakuma, Y., Li, J., Liu, Q., Li, Y.-Q., Shinozaki, K., et al. (2004). Cloning and functional analysis of a novel DREB1/CBF transcription factor involved in cold-responsive gene expression in Zea mays L. Plant Cell Physiol. 45, 1042–1052. doi: 10.1093/pcp/pch118
Raney, J. A., Reynolds, D. J., Elzinga, D. B., Page, J., Udall, J. A., Jellen, E. N., et al. (2014). Transcriptome analysis of drought induced stress in Chenopodium quinoa. Am. J. Plant Sci. 5, 338–357. doi: 10.4236/ajps.2014.53047
Schmülling, T., Werner, T., Riefler, M., Krupková, E., and Bartrina y Manns, I. (2003). Structure and function of cytokinin oxidase/dehydrogenase genes of maize, rice, Arabidopsis and other species. J. Plant Res. 116, 241–252. doi: 10.1007/s10265-003-0096-4
Shan, X., Li, Y., Jiang, Y., Jiang, Z., Hao, W., and Yuan, Y. (2013). Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Report 31, 1485–1491. doi: 10.1007/s11105-013-0622-z
Shen, Y., Jiang, Z., Yao, X., Zhang, Z., Lin, H., Zhao, M., et al. (2012). Genome expression profile analysis of the immature maize embryo during dedifferentiation. PLoS ONE 7:e32237. doi: 10.1371/journal.pone.0032237
Shou, H., Bordallo, P., Fan, J.-B., Yeakley, J. M., Bibikova, M., Sheen, J., et al. (2004). Expression of an active tobacco mitogen-activated protein kinase kinase kinase enhances freezing tolerance in transgenic maize. Proc. Natl. Acad. Sci. U.S.A. 101, 3298–3303. doi: 10.1073/pnas.0308095100
Sobkowiak, A., Jończyk, M., Adamczyk, J., Szczepanik, J., Solecka, D., Kuciara, I., et al. (2016). Molecular foundations of chilling-tolerance of modern maize. BMC Genomics 17:125. doi: 10.1186/s12864-016-2453-4
Sobkowiak, A., Jończyk, M., Jarochowska, E., Biecek, P., Trzcinska-Danielewicz, J., Leipner, J., et al. (2014). Genome-wide transcriptomic analysis of response to low temperature reveals candidate genes determining divergent cold-sensitivity of maize inbred lines. Plant Mol. Biol. 85, 317–331. doi: 10.1007/s11103-014-0187-8
Storey, J. D. (2003). The positive false discovery rate: a bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035. doi: 10.2307/3448445
Strigens, A., Freitag, N. M., Gilbert, X., Grieder, C., Riedelsheimer, C., Schrag, T. A., et al. (2013). Association mapping for chilling tolerance in elite flint and dent maize inbred lines evaluated in growth chamber and field experiments. Plant Cell Environ. 36, 1871–1887. doi: 10.1111/pce.12096
Ströher, E., Wang, X.-J., Roloff, N., Klein, P., Husemann, A., and Dietz, K.-J. (2009). Redox-Dependent Regulation of the Stress-Induced Zinc-Finger Protein SAP12 in Arabidopsis thaliana. Mol. Plant 2, 357–367. doi: 10.1093/mp/ssn084
Suzuki, N., and Mittler, R. (2006). Reactive oxygen species and temperature stresses: a delicate balance between signalling and destruction. Physiol. Plant. 126, 41–51. doi: 10.1111/j.1399-3054.2005.00582.x
Teoh, K. T., Requesens, D. V., Devaiah, S. P., Johnson, D., Huang, X., Howard, J. A., et al. (2013). Transcriptome analysis of embryo maturation in maize. BMC Plant Biol. 13:19. doi: 10.1186/1471-2229-13-19
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., and Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53. doi: 10.1038/nbt.2450
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Trzcinska-Danielewicz, J., Bilska, A., Fronk, J., Zielenkiewicz, P., Jarochowska, E., Roszczyk, M., et al. (2009). Global analysis of gene expression in maize leaves treated with low temperature. I. Moderate chilling (14°C). Plant Sci. 177, 648–658. doi: 10.1016/j.plantsci.2009.09.001
Van Hove, J., De Jaeger, G., De Winne, N., Guisez, Y., and Van Damme, E. J. M. (2015). The Arabidopsis lectin EULS3 is involved in stomatal closure. Plant Sci. 238, 312–322. doi: 10.1016/j.plantsci.2015.07.005
Wang, F., Li, D., Wang, Z., Dong, A., Liu, L., Wang, B., et al. (2014). Transcriptomic analysis of the rice white tip nematode, Aphelenchoides besseyi (Nematoda: Aphelenchoididae). PLoS ONE 9:e91591. doi: 10.1371/journal.pone.0091591
Wang, L., and Chong, K. (2010). “Signaling and communication in plants,” in Integrated G Proteins Signaling in Plants, eds S. Yalovsky, F. Baluska, and A. Jones (Springer), 1–25.
Wang, X., Wu, D., Yang, Q., Zeng, J., Jin, G., Chen, Z.-H., et al. (2016). Identification of mild freezing shock response pathways in barley based on transcriptome profiling. Front. Plant Sci. 7:106. doi: 10.3389/fpls.2016.00106
Xie, Q., Frugis, G., Colgan, D., and Chua, N. (2000). Arabidopsis NAC1 transduces auxin signal downstream of TIR1 to promote lateral root development. Genes Dev. 14, 3024–3036. doi: 10.1101/gad.852200
Yang, G., Zou, H., Wu, Y., Liu, H., and Yuan, Y. (2011). Identification and characterisation of candidate genes involved in chilling responses in maize (Zea mays L.). Plant Cell Tissue Organ Cult. 106, 127–141. doi: 10.1007/s11240-010-9900-8
Yang, H., Matsubayashi, Y., Nakamura, K., and Sakagami, Y. (2001). Diversity of Arabidopsis genes encoding precursors for phytosulfokine, a peptide growth factor 1. Plant Physiol. 127, 842–851. doi: 10.1104/pp.010452
Zhang, C., Fei, S. Z., Arora, R., and Hannapel, D. J. (2010). Ice recrystallization inhibition proteins of perennial ryegrass enhance freezing tolerance. Planta 232, 155–164. doi: 10.1007/s00425-010-1163-4
Zhang, T., Zhao, X., Wang, W., Pan, Y., Huang, L., Liu, X., et al. (2012). Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS ONE 7:e43274. doi: 10.1371/journal.pone.0043274
Keywords: freezing stress, maize, RNA-seq, DEGs, molecular mechanism
Citation: Li Z, Hu G, Liu X, Zhou Y, Li Y, Zhang X, Yuan X, Zhang Q, Yang D, Wang T and Zhang Z (2016) Transcriptome Sequencing Identified Genes and Gene Ontologies Associated with Early Freezing Tolerance in Maize. Front. Plant Sci. 7:1477. doi: 10.3389/fpls.2016.01477
Received: 22 April 2016; Accepted: 16 September 2016;
Published: 07 October 2016.
Edited by:
Alma Balestrazzi, University of Pavia, ItalyReviewed by:
Biswapriya Biswavas Misra, University of Florida, USARosa Rao, University of Naples Federico II, Italy
Copyright © 2016 Li, Hu, Liu, Zhou, Li, Zhang, Yuan, Zhang, Yang, Wang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deguang Yang, ZGVndWFuZ3lhbmdAc2luYS5jb20=
Tianyu Wang, d2FuZ3RpYW55dUBjYWFzLmNu
Zhiwu Zhang, emhpd3UuemhhbmdAd3N1LmVkdQ==
†These authors have contributed equally to this work.