 Dong-Wen Lv1,2†
Dong-Wen Lv1,2† Shoumin Zhen1†
Shoumin Zhen1† Geng-Rui Zhu1Yan-Wei Bian1Guan-Xing Chen1Cai-Xia Han1Zi-Tong Yu3
Geng-Rui Zhu1Yan-Wei Bian1Guan-Xing Chen1Cai-Xia Han1Zi-Tong Yu3 Yue-Ming Yan1*
Yue-Ming Yan1*- 1College of Life Science, Capital Normal University, Beijing, China
- 2Department of Oral and Craniofacial Molecular Biology, VCU Philips Institute for Oral Health Research, School of Dentistry, Virginia Commonwealth University, Richmond, VA, USA
- 3State Agriculture Biotechnology Centre, Murdoch University, Perth, WA, Australia
Oxidative stress in plants can be triggered by many environmental stress factors, such as drought and salinity. Brachypodium distachyon is a model organism for the study of biofuel plants and crops, such as wheat. Although recent studies have found many oxidative stress response-related proteins, the mechanism of microRNA (miRNA)-mediated oxidative stress response is still unclear. Using next generation high-throughput sequencing technology, the small RNAs were sequenced from the model plant B. distachyon 21 (Bd21) under H2O2 stress and normal growth conditions. In total, 144 known B. distachyon miRNAs and 221 potential new miRNAs were identified. Further analysis of potential new miRNAs suggested that 36 could be clustered into known miRNA families, while the remaining 185 were identified as B. distachyon-specific new miRNAs. Differential analysis of miRNAs from the normal and H2O2 stress libraries identified 31 known and 30 new H2O2 stress responsive miRNAs. The expression patterns of seven representative miRNAs were verified by reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis, which produced results consistent with those of the deep sequencing method. Moreover, we also performed RT-qPCR analysis to verify the expression levels of 13 target genes and the cleavage site of 5 target genes by known or novel miRNAs were validated experimentally by 5′ RACE. Additionally, a miRNA-mediated gene regulatory network for H2O2 stress response was constructed. Our study identifies a set of H2O2-responsive miRNAs and their target genes and reveals the mechanism of oxidative stress response and defense at the post-transcriptional regulatory level.
Background
Many environmental stress factors, including high light, UV irradiation, heat, salinity, drought, and cold, can cause plant cells to produce reactive oxygen species (ROS), leading to acceleration of lipid peroxidation and leaf senescence (Mittler et al., 2004; Upadhyaya et al., 2007). Hydrogen peroxide (H2O2) is a kind of ROS mainly produced by the following parts of the cell: the mitochondrion, peroxisome, chloroplast, apoplast, and plasma membrane (Apel and Hirt, 2004). Superoxide radicals (), another kind of ROS, can also be rapidly dismutated to H2O2 spontaneously or catalyzed by superoxide dismutase (SOD). In contrast to other ROS, such as and hydroxyl radicals (OH−), H2O2 can easily pass through membranes (Foyer et al., 1997; Uchida et al., 2002; de Azevedo Neto et al., 2005; Wahid et al., 2007) and is relatively stable, so it is suitable for its roles as an important component of cell signaling cascades (Mittler, 2002; Neill S. et al., 2002; Neill S. J. et al., 2002; Vranová et al., 2002) and an indispensable second messenger in biotic and abiotic stress responses (Pastori and Foyer, 2002). H2O2 stress can affect fluctuation of the Ca2+ concentration in plants, thereby inducing the production of appropriate amounts of antioxidants (Rentel and Knight, 2004). Global transcript profiling under H2O2 stress in tobacco revealed that redox homeostasis associated proteins are upregulated while proteins related to normal growth and development are downregulated (Vandenabeele et al., 2003). H2O2 can also trigger acclimation and cross-tolerance phenomena (Neill S. et al., 2002; Pastori and Foyer, 2002). Overall, at low concentrations H2O2 may play a role as a signaling molecule, whereas at high concentrations H2O2 will cause programmed cell death (Quan et al., 2008).
MicroRNAs (miRNAs) are small, non-coding RNAs that have been demonstrated to be involved in many responses to and defenses against various biotic and abiotic stresses in plants (Bartel, 2004). With the development of high-throughput sequencing technology and bioinformatics, many plant miRNAs have been identified. In animals, at least 60% of protein-coding genes can be regulated by miRNAs (Friedman et al., 2009), but known target genes of miRNAs in plants are far fewer (~1% of the protein-coding genes) (Addo-Quaye et al., 2008; Li et al., 2010). Even so, the regulatory role of miRNAs in plants cannot be underestimated, because most known target genes are transcription factors (TFs) (Jones-Rhoades and Bartel, 2004; Jones-Rhoades et al., 2006). Under environmental stresses, plants up- or down-regulate certain miRNAs or synthesize new miRNAs to respond to or defend against stresses (Khraiwesh et al., 2012). Abiotic stresses, such as high light, UV, heat, heavy metals, drought, and salinity, can elevate ROS levels (Mittler et al., 2004). To date, only a few studies have focused on oxidative stress-triggered miRNA expression changes. miR398, whose target genes are Cu-Zn superoxide dismutases, is a well-studied miRNA related to the response to oxidative stress triggered by high light (Sunkar et al., 2006). Iyer et al. (2012) identified 22 ozone-induced oxidative stress miRNA families using a plant miRNA array in Arabidopsis; most of them were also reported as UV-B responsive miRNAs (Zhou et al., 2007). Jia et al. (2009) identified 24 UV-B responsive miRNAs (13 upregulated and 11 downregulated) in Populus tremula through a miRNA filter array. Some of these upregulated miRNAs (miR156, miR160, miR165/166, miR167, miR398, and miR168) were also reported in UV-B-stressed Arabidopsis (Zhou et al., 2007). Similarly, six miRNAs were identified as UV-B-responsive miRNAs in wheat (Wang et al., 2013), in which miR159, miR167a, and miR171 are upregulated and miR156, miR164, miR395 are downregulated. As a ROS, H2O2 can also act as a secondary messenger during stress response and defense signal transduction. However, only a few H2O2-responsive miRNAs (miR169, miR397, miR1425, miR408-5p, miR827, miR528, and miR319a.2) have been identified in plants (Li et al., 2011). Thus, the miRNAs related to oxidative stress caused by abiotic stressors are far from being completely elucidated in plants.
Brachypodium distachyon, as a model plant for crops such as wheat and barley, has been sequenced (Vogel et al., 2010). Several studies under abiotic stress have been performed at different levels, including the transcriptome (Priest et al., 2014), proteome, and phosphoproteome (Lv et al., 2014b). In addition, recent miRNA studies using high-throughput sequencing have identified many stress responsive miRNAs under several kinds of abiotic stress, such as cold stress (Zhang et al., 2009), dehydration stress (Budak and Akpinar, 2011), and drought stress (Bertolini et al., 2013). In particular, Jeong et al. (2013) sequenced 17 small RNA libraries that represented different tissues and stressors and identified many previously unreported and B. distachyon-specific miRNAs. However, the identified miRNAs are far from sufficient for B. distachyon, especially for oxidative stress regulated miRNAs.
Thus, in this study, we identified miRNAs and their potential target genes related to H2O2 stress using high-throughput sequencing, reverse transcription quantitative polymerase chain reaction (RT-qPCR) and 5′ RACE, combined with bioinformatics methods. The differentially expressed miRNAs observed between B. distachyon seedlings grown under control and H2O2-treated conditions, as well as the miRNA-directed regulatory network, provide new insights that will inform the genetic improvement of stress tolerance in plants.
Materials and Methods
Plant Materials
Seedlings of B. distachyon 21 (Bd21) were grown in a growth chamber at 25/20°C (16 h day/8 h night) and 70% relative humidity, as reported previously (Lv et al., 2014a). For H2O2 treatment, seedlings at the three leaves stage were treated with 20 mM H2O2 for 6 h in plastic containers and collected at 2, 4, or 6 h based on our previous study (Bian et al., 2015). Untreated seedlings were used as a control. All of the H2O2treated and untreated samples had three biological replicates and at least 100 seedlings were used in each replicate. All samples were snap-frozen in liquid nitrogen and then stored at −80°C until RNA extraction.
Total RNA Isolation
Total RNA was extracted from the frozen seedlings with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Prior to nucleic precipitation, two extra chloroform washes were performed. A 1% agarose gel stained with ethidium bromide was run to determine the preliminary integrity of the RNA. All RNA samples were quantified and examined using an ND 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) for contamination with either protein (A260/A280 ratios) or reagent (A260/A230 ratios). The RNA integrity number (RIN) was >8, as determined with a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Construction of Small RNA (sRNA) Libraries and Deep Sequencing
For sRNA library construction and deep sequencing, RNA samples were prepared as follows: equal quantities (10 mg) of total RNA isolated from Bd21 seedlings treated with 20 mM H2O2 for 2, 4, and 6 h were mixed together to construct the TS library, and 30 mg of total RNA prepared from the control sample (without H2O2 treatment) were used to construct the CS library. Then, total RNA was separated by 15% TBE-urea denaturing polyacrylamide gel electrophoresis (PAGE), and RNA molecules in the range of 18–30 nt were enriched and ligated with proprietary adapters to the 5′ and 3′ termini by T4 RNA ligase. The samples were used as templates for cDNA synthesis by Super-Script II Reverse Transcriptase (Invitrogen) and the resulting cDNA was amplified to produce sequencing libraries. The final quality of the cDNA library was ensured by examining its size, purity, and concentration with a 2100 Bioanalyzer. The sequencing was performed by the Beijing Genomics Institute (BGI, Shenzhen, China). The two libraries were run on the Illumina HiSeq™ 2000 platform side by side.
Bioinformatic Analysis of Sequencing Data
After trimming the 30-bp adaptor sequence, sequences shorter than 18 nt were excluded from further analysis. First, rRNA, scRNA, snoRNA, snRNA, and tRNA in clean reads were identified by a blastall search against the Rfam (version 10.1) database. Next, sequences were perfectly mapped onto the Bd21 genome v1.0 (http://www.phytozome.net/) using the program SOAP2 (Li et al., 2009). Known miRNAs were identified according to B. distachyon defined mature miRNAs and stem-loop miRNA precursors from miRBase (version 20; http://www.mirbase.org) (Kozomara and Griffiths-Jones, 2011). Potential novel miRNAs were identified using the MIREAP (Li et al., 2012) software (http://sourceforge.net/projects/mireap/) based on Meyers et al. (2008), and unique sequences that had more than 10 hits to the genome or matches to known non-coding RNAs were removed. The secondary structures of novel miRNA precursors were predicted by RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi; Zuker, 2003) with default parameters.
Differential Expression Analysis
The relative miRNA expression levels of the two libraries were compared and the differentially expressed miRNAs were screened based on a previously established method (Audic and Claverie, 1997). The frequency of miRNAs in the two libraries was normalized to one million by the total number of miRNAs in each library (transcripts per million (TPM) normalized expression = initial miRNA count*1,000,000/total count of clean reads). Following normalization, if the miRNA gene expression in both libraries was zero, then it was revised to 0.01; if the miRNA gene expression in both libraries was less than 1, owing to its too low expression, it was excluded from further differential expression analysis. Fold change = log2(the normalized H2O2 treatment reads/the normalized control reads). The P-value was calculated as described previously (Wu et al., 2014).
Target Gene Prediction of miRNAs and Functional Analysis
Target genes were predicted using the MIREAP program developed by the BGI, combined with psRNATarget online software (http://www.plantgrn.org/psRNATarget/) (Dai and Zhao, 2011), and obeying the rules described in Allen et al. (2005) and Schwab et al. (2005). The criteria for using MIREAP and psRNATarget followed a previous study (Bertolini et al., 2013). Only the shared predictions of the two softwares were considered as the final target genes. The biological processes, molecular functions, and cellular components of the target genes were examined using the agriGO online tool (Du et al., 2010) to perform Gene Ontology (GO) annotation and GO enrichment analysis. The statistical test method was set as Fisher and the multi-test adjustment method was set as Bonferroni. The threshold of significance was defined as p < 0.01 and the false discovery rate (FDR) as < 0.01. The dataset containing protein sequences of B. distachyon genome was set as the background dataset.
H2O2-Responsive miRNA-Mediated Network Analysis
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database of physical and functional interactions (Szklarczyk et al., 2011) was used to analyse the protein-protein interactions (PPI) of the proteins encoded by target genes of differentially expressed miRNAs. The miRNA-regulated PPI network was displayed by the Cytoscape (version 3.1.1) software (Shannon et al., 2003).
Expression Validation of miRNAs and Their Targets
We verified the patterns of expression of seven conserved B. distachyon miRNAs (miR159a-3p, miR159b-3p.1, miR160a/b/c/d-5p, miR169b, miR169d, miR397a, and miR528-5p). A miRcute miRNA First-strand cDNA Synthesis Kit (TIANGEN) was used for the RT reactions. The thermocycling program was adjusted to run for 60 min at 37°C, 5 s at 85°C, and then 4°C forever. For each miRNA, three biological replicates were used. The miR168 and 5.8S genes served as the endogenous controls (Bertolini et al., 2013; Jeong et al., 2013). All primers are listed in Table S1. RT-qPCR was conducted on a CFX96 Real-Time PCR Detection System (Bio-Rad). Each reaction included 2 μL of product from the diluted RT reactions, 1.0 μL of each primer (forward and reverse), 12.5 μL of SYBR® Premix Ex Taq™ (Perfect Real Time; TaKaRa), and 8.5 μL of nuclease-free water. The reactions were incubated in a 96-well plate at 95°C for 30 s, followed by 40 cycles of 95°C for 5 s, 60°C for 30 s, and 72°C for 10 s. All reactions were run in triplicate for each sample. All data were analyzed using the CFX Manager software (Bio-Rad). We also selected 13 target genes to validate their expression profiles in the CS and TS libraries via RT-qPCR following the method described by Lv et al. (2014b) according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. The Actin and SamDC genes served as the endogenous controls (Lv et al., 2014b). All primers are listed in Table S1. Statistical analysis was performed using the SPSS 17.0 software. Statistical differences amongst the two libraries were assessed using the independent two-sample t-test. P < 0.05 were considered statistically significant.
Modified RNA Ligase-Mediated (RLM) 5′ Race for the Mapping of mRNA Cleavage Sites
To identify cleavage sites in the target mRNAs, a modified RLM-5′-RACE was performed using a FirstChoice RLM-RACE Kit (Ambion, Austin, TX, USA). All the steps followed the manufacturer's instructions, except that the calf intestinal phosphatase treatment was omitted to maintain the cleaved transcripts. Nested PCR amplifications were performed using the general sense primers and gene specific nested antisense primers that were listed in Table S1. The amplification products were gel purified, cloned, and sequenced, and 10 independent clones were sequenced.
Results
In this work, the Illumina Solexa sequencing platform was used to investigate the genome-wide identification and expression profiles of miRNAs in B. distachyon under H2O2 stress. Two sRNA libraries were constructed using total RNAs isolated from control seedlings (CS) and H2O2-treated seedlings (TS). sRNA sequencing yielded a total of 18,220,106 and 19,373,978 high-quality raw sequence reads from the CS and TS libraries, respectively. The raw reads of the two libraries were uploaded to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA; accession numbers: SRX1542485 and SRX1542460). After removing low quality reads, adapters, poly-A sequences, and short RNA reads smaller than 18 nucleotides (nt), 17,811,109 (97.76%) and 17,708,762 (91.40%) clean reads representing 3,830,474 and 3,548,088 unique sRNAs were obtained from the CS and TS libraries, respectively (Table S2). Among the unique sequences, 1,963,866 (51.27%) and 1,689,996 (47.63%) generated from the CS and TS libraries, respectively, were mapped to the B. distachyon genome using SOAP2. To reveal the sequence distribution of the sRNAs, all clean reads were queried against the B. distachyon genome database at Phytozome (http://www.phytozome.net/), Rfam (http://rfam.sanger.ac.uk/), and miRBase v20.0 (http://www.mirbase.org/), and classified into seven annotation categories: non-coding RNAs (tRNA, rRNA, snRNA, and snoRNA), miRNA, exon-sense, exon-antisense, intron-sense, intron-antisense, and unknown sRNAs (Table S2). The length distribution of the total sRNA reads revealed that the majority of reads from each library were 20–25 nt in length, of which, 24-nt reads were the most abundant, followed by 21-nt reads (Figure 1A). Compared to the CS library, the TS library contained more 21-nt sRNAs and fewer 24-nt sRNAs. Of the unique sRNAs, 24-nt sRNAs accounted for 47.91 and 43.78% in the CS and TS libraries, respectively, while 21-nt sRNAs accounted for 5.10 and 5.33% (Figure 1B).
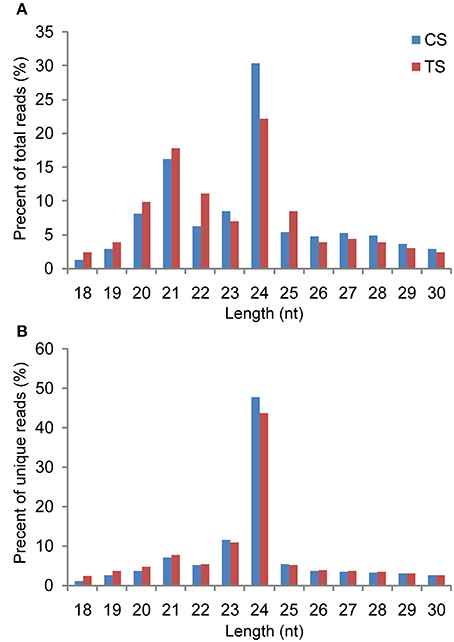
Figure 1. Length distribution of sRNAs detected in the control library and stress library. (A) Redundant sRNAs; (B) unique sRNAs.
Identification of Known and Novel miRNAs in B. distachyon
To identify known miRNAs from the CS and TS libraries, sRNA sequences generated from each library were independently aligned with currently known and experimentally validated mature B. distachyon miRNAs deposited in miRBase v20.0. Finally, a total of 144 known miRNAs were identified from the CS (133) and TS (138) libraries (Table S3); among them, 127 (88.19%) were detected in both libraries. For the total reads of each known miRNA in our study, 54.86% (79 miRNAs) had more than 100 reads, 29.17% (42) had more than 1000 reads, 12.5% (18) had more than 10,000 reads, and only 3.47% (5) had more than 100,000 reads. Among them, bdi-miR168-5p possessed the highest expression level in each library (556,771 reads in the CS library and 571,620 reads in the TS library), followed by bdi-miR156c, bdi-miR156b-5p, bdi-miR156d-5p, and bdi-miR528-5p.
After identifying the known miRNAs, the remaining sequences from the two libraries, which were classified as “unannotated” (excluding known miRNAs and other non-coding RNAs classified by Rfam), were used to discover novel and potential B. distachyon-specific miRNA candidates. To accomplish this, these small RNA sequences were aligned with the B. distachyon genome to identify genomic regions potentially harboring pre-miRNA sequences, whose hairpin-like structures are widely used to distinguish miRNAs from other small non-coding RNAs. The minimum free energy (MFE) of the secondary structures was also considered as a criterion for the prediction of potential pre-miRNAs. After aligning these unannotated sequences to the genome, we obtained a total of 221 novel miRNA candidates from the CS (156) and TS (132) libraries (Table S4). Among the 221 novel miRNA candidates, 36 had homologs in other plant species or their pre-miRNA sequences belonged to known B. distachyon pre-miRNAs (Table S4), while the remaining 185 novel miRNA candidates were B. distachyon-specific (Table S4). In agreement with previously reports, the cytosine and uracil nucleotides were dominant in the first position of the 5′ end for the majority of these newly determined putative novel miRNAs (Figure S1A). In detail, first positions included 11,629 cytosine nucleotides (67.52%) and 4279 uracil nucleotides (24.85%) in the CS library and 16,115 cytosine nucleotides (78.05%) and 3455 uracil nucleotides (16.73%) in the TS library. The first nucleotide bias analysis showed that cytosine was the most frequently used first nucleotide for novel 21-nt miRNAs and uracil was most frequent for novel 20-, 22-, and 23-nt miRNAs (Figure S1B). Our sequence analyses of the two libraries showed that the putative pre-miRNAs of each library greatly varied in length from 69 to 361 nt in the CS library and from 65 to 372 nt in the TS library. These pre-miRNA sequences were applied to predict the characteristic stem-loop secondary structure of pre-miRNA and their locations were also determined in the genomic loci (Table S4). We also calculated the minimum folding free energies of putative miRNA precursors for each library, which ranged from −18.1 to −180.64 kcal/mol with an average of −62.07 kcal/mol for the CS library and from −19.9 to −192.4 kcal/mol with an average of −60.46 kcal/mol for the TS library (Table S4). In contrast with the known miRNAs, most of the predicted novel miRNAs were expressed at very low levels. Only 9.50% (21) of the 221 novel miRNAs had more than 100 reads, and only two (novel_mir_59 and novel_mir_54) had more than 1000 reads (Table S4).
Target Gene Prediction and GO Annotation Analysis
A total of 352 putative known miRNA target transcripts (corresponding to 284 target genes) and 554 putative novel miRNA target transcripts (corresponding to 460 target genes) were obtained (Table S5). The number of targets for each known miRNA and novel miRNA varied, ranging from 1 to 54 and 1 to 61, respectively, and the percent of novel miRNAs with more than 10 predicted target transcripts was 33.08%, while that number was 7.34% for known miRNAs. For comprehensive annotation, all putative target genes were analyzed by GO terms with the aid of the Blast2GO program with default parameters. Genes with a known function were categorized by biological process, molecular function, and cellular component according to the ontological definitions of the GO terms (Figure S2). For biological process, genes were mainly in the single-organism process (10.12%), response to stimulus (9.97%), localization (6.49%), multicellular organismal process (6.17%), and developmental process (6.01%) categories. For molecular function categories, nucleic acid binding transcription factor activity (4.11%), transporter activity (3.01%), and structural molecule activity (1.42%) were highlighted. For cellular component, they were mainly localized in the organelle (26.90%), membrane-bounded organelle (25.00%), and membrane (13.61%).
Screening of H2O2-Responsive miRNAs
In this study, 31 known and 30 novel miRNAs were observed with a more than two-fold change in response to H2O2 treatment in B. distachyon seedlings (Figure 2, Tables 1, 2). As reported in a previous study (Li et al., 2011), a series of known H2O2-responsive miRNAs, including miR159, miR160, miR169, miR397, and miR528, were also identified in our study. Further analysis revealed that 10 of the 31 known miRNAs were downregulated in the TS library compared to the CS library, whereas 21 were upregulated (Figure 2A). The 10 downregulated known miRNAs were composed of three miRNA families, including five members of miR169, four members of miR160, and miR7770. Among them, miR7770 displayed a dramatic (log2 fold change = −7.92) decrease. For the 21 upregulated known miRNAs, miR395 was the major family containing 14 members. bdi-miR159b-3p.1 showed the highest upregulation (log2 fold change = 16.21).
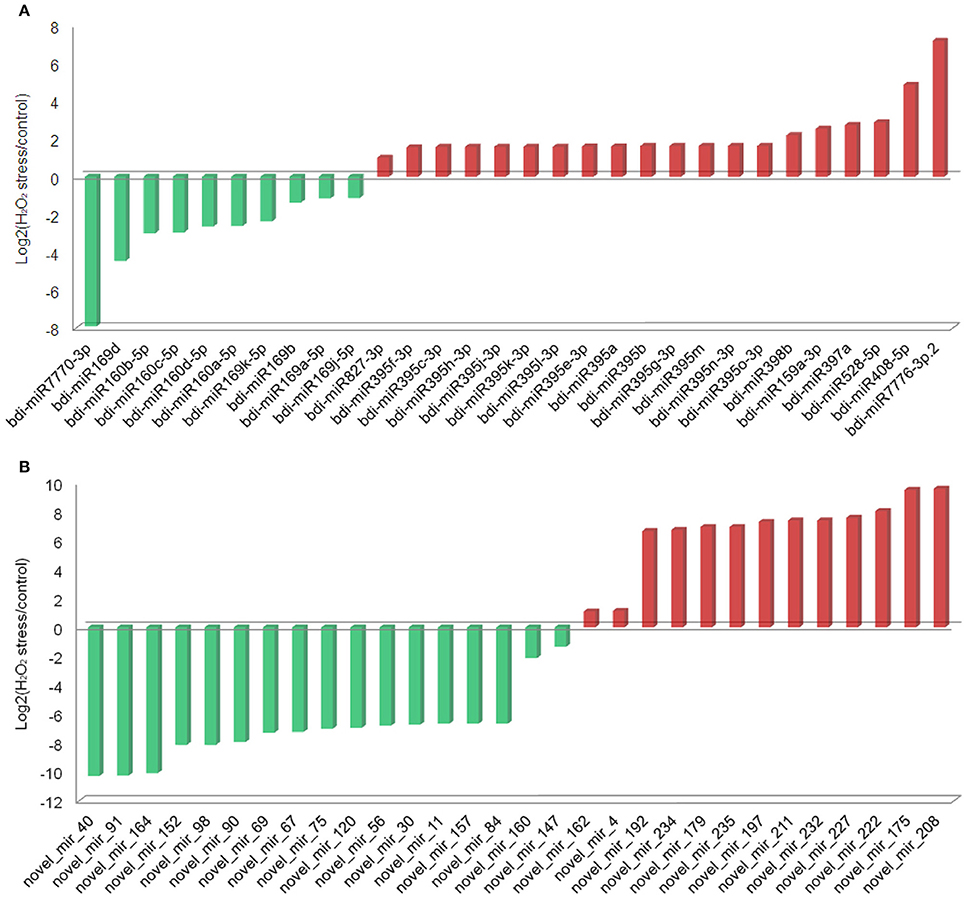
Figure 2. Known and novel miRNAs differentially expressed between the CS library and TS library. (A) Known miRNAs; (B) novel miRNAs.
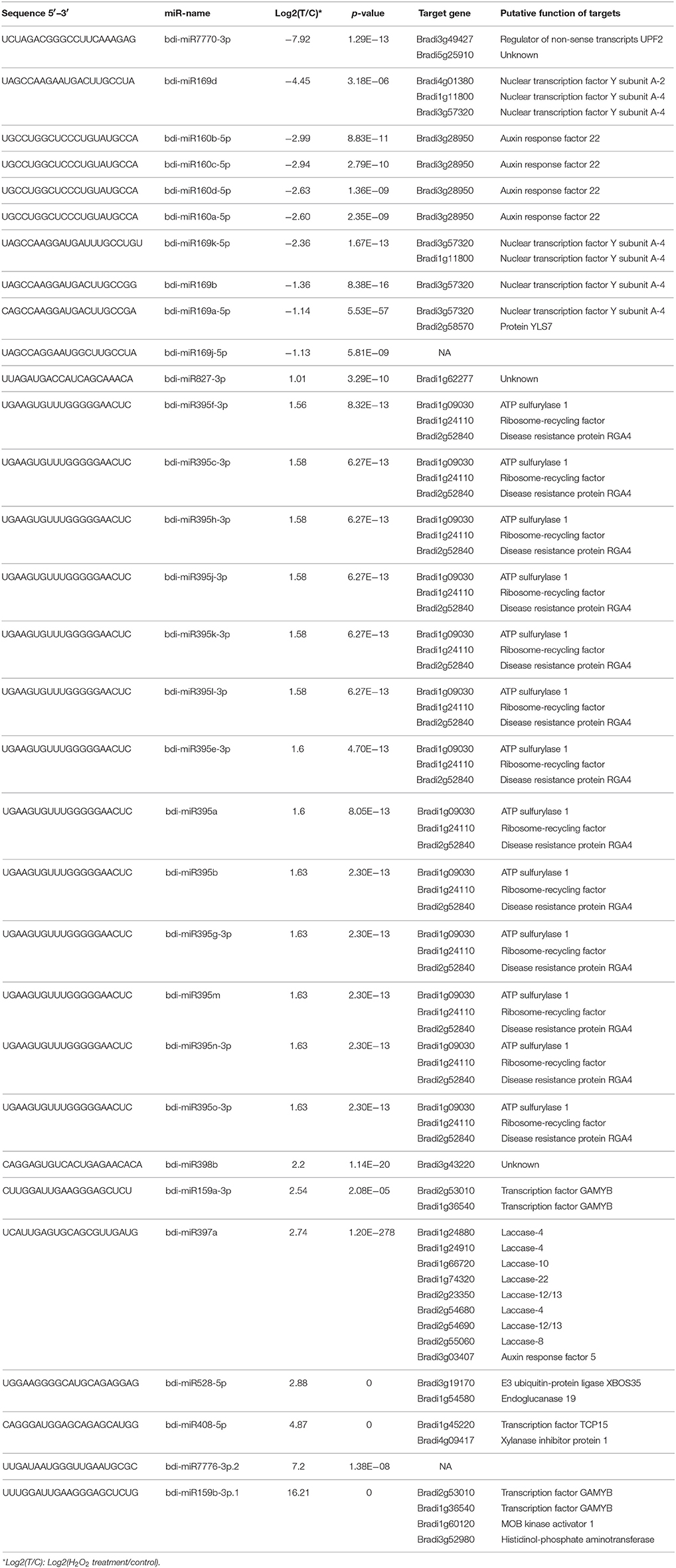
Table 1. Differential expressed known miRNA between CS and TS libraries.
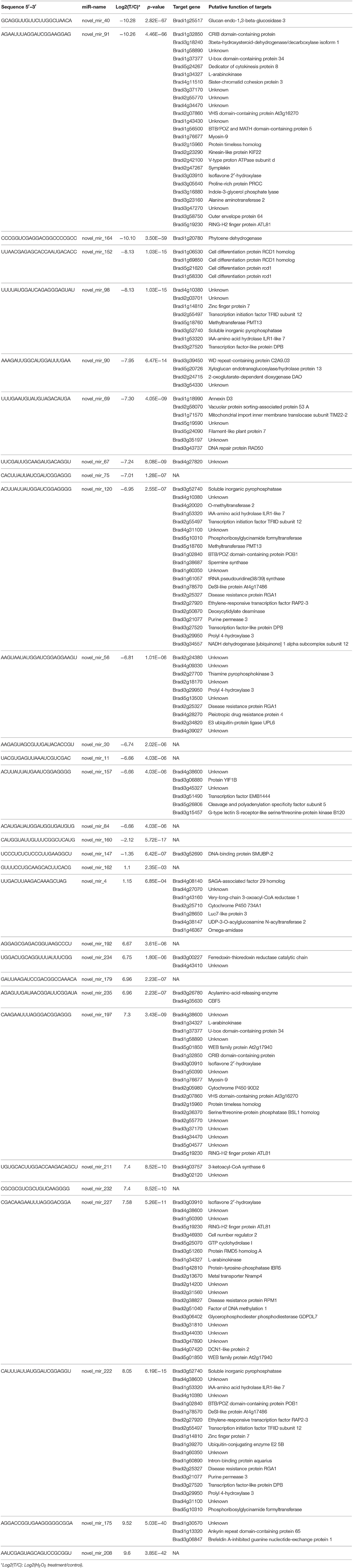
Table 2. Differential expressed novel miRNA between CS and TS libraries.
Among the 30 differential novel miRNAs, 17 downregulated and 13 upregulated miRNAs were found in the TS library compared to the CS library (Figure 2B). All but three novel miRNAs (novel_mir_4, novel_mir_148, and novel_mir_161) showed dramatic changes (log2 fold change >4 or < −4), in contrast to known differential miRNAs, of which only four exhibited dramatic changes.
We performed GO enrichment analysis for the target genes of these H2O2-responsive miRNAs and found that the proteins encoded by these target genes were mainly involved in the categories of multicellular organismal development (GO:0007275, FDR:3.1E−11), secondary metabolic process (GO:0019748, FDR:1.0E−10), reproduction (GO:0000003, FDR:2.8E−6), catabolic process (GO:0009056, FDR:8.6E−6), nucleobase, nucleoside, nucleotide and nucleic acid metabolic process (GO:0006139, FDR:4.3E−5), cellular component organization (GO:0016043, FDR:6.6E−4), and response to stress (GO:0006950, FDR:1.2E−3) (Figure 3). These proteins mainly exhibited binding function (GO:0005515, FDR:3.4E−6) and were localized in the extracellular region (GO:0005576, FDR:5.3E−15), mitochondrion (GO:0005739, FDR:5.1E−7), plasma membrane (GO:0005886, FDR:1.1E−6), and nucleus (GO:0005634, FDR:3.0E−3) (Figure 3).
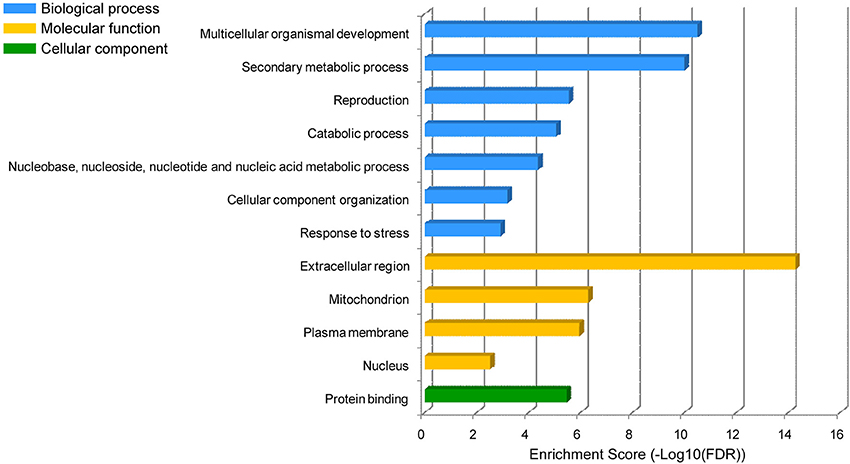
Figure 3. GO enrichment of the H2O2-responsive miRNA target genes. The dataset containing protein sequences of B. distachyon genome was set as the background dataset. The vertical axis is the enriched GO category, and the horizontal axis is GO enrichment.
A miRNA-Mediated Regulatory Network for H2O2 Stress Response
Based on the miRNA target prediction and protein-protein interaction (PPI) analysis, the H2O2-responsive miRNAs and the proteins encoded by their target genes were used to construct the miRNA-mediated regulatory network for H2O2 stress response (Figure 4). Generally, the more proteins that a protein interacts with, the more important are that protein's functions in the network. Two MYB TFs (encoded by genes Bradi2g53010 and Bradi1g36540) were centered in the network and each could interact with 24 proteins. Thus, the two MYB TFs may play critical roles during H2O2-triggered oxidative stress response. Some target genes were regulated by two or more miRNAs that belonged to the same miRNA family or different families, and the proteins encoded by these genes always interacted with more proteins. For example, Bradi2g52840 (target gene of the bdi-miR395 family) encodes disease resistance protein RGA4, which interacts with seven proteins encoded by the target genes of H2O2 stress-responsive miRNAs, including the two MYB TFs mentioned above (Figure 4). Gene Bradi2g55497 is regulated by three novel miRNAs (novel_mir_98, novel_mir_120, and novel_mir_222) and encodes transcription initiation factor TFIID subunit 12, which can interact with four proteins, including three members of the NFYA TF family (encoded by genes Bradi4g01380, Bradi1g11800, and Bradi3g57320) and a transcriptional regulator SAGA-associated factor 29 homolog (encoded by gene Bradi4g08140).
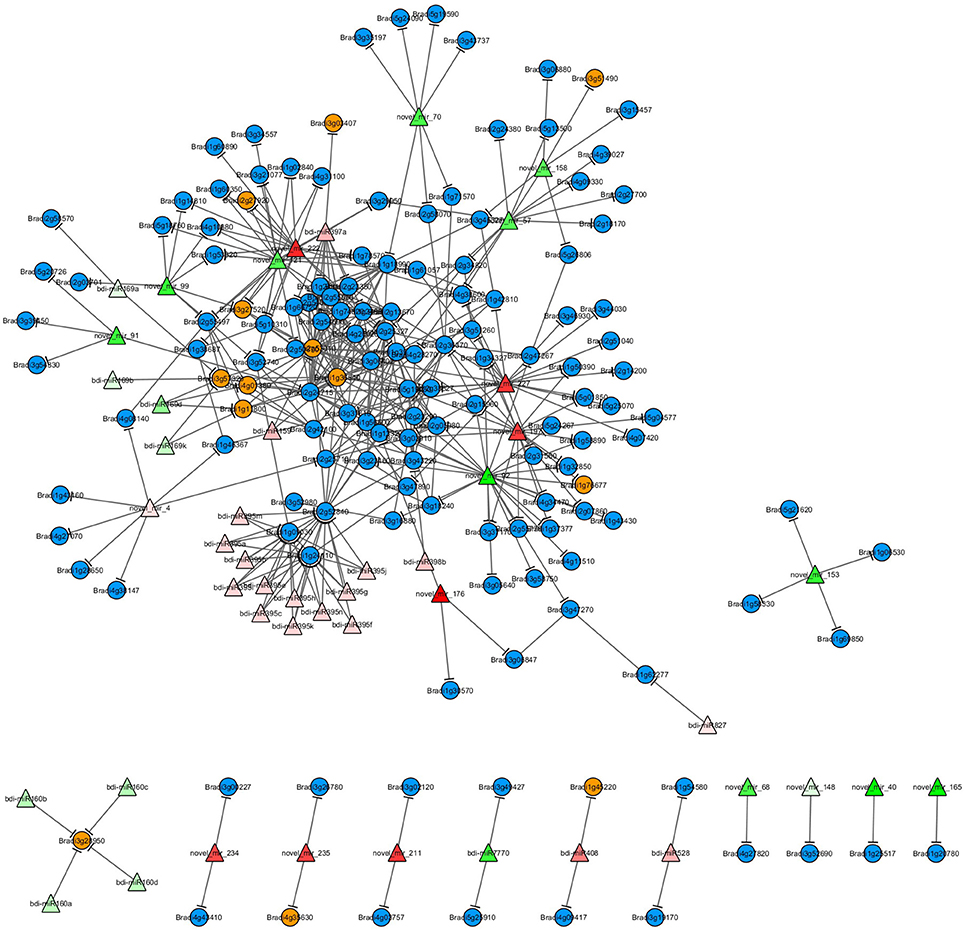
Figure 4. A miRNA-mediated H2O2-responsive regulatory network. Triangles represent the differentially expressed miRNAs and the color gradient shows the fold change (red: upregulation; green: downregulation). Blue circles represent the target genes of differentially expressed miRNAs; yellow circles represent target genes that encode transcription factors.
RT-qPCR Validation of B. distachyon miRNAs and Target Genes
We applied RT-qPCR analysis for further experimental verification of the presence of several conserved miRNAs and compared the expression patterns of these miRNAs with deep sequencing results. Analysis of seven H2O2-responsive miRNAs (miR159a-3p, miR159b-3p.1, miR160a/b/c/d-5p, miR169b, miR169d, miR397a, and miR528-5p) by RT-qPCR (Figure 5) showed that all of the relative expression profiles exhibited the same trends as their deep sequencing results, although there were some extent differences between the results obtained from deep sequencing and the RT-qPCR experiment. In detail, miR528-5p, miR397a, miR159a-3p, and miR159b-3p.1 were up-regulated under H2O2 stress, while the expression of miR160a/b/c/d-5p, miR169d, and miR169b decreased during H2O2 treatment.
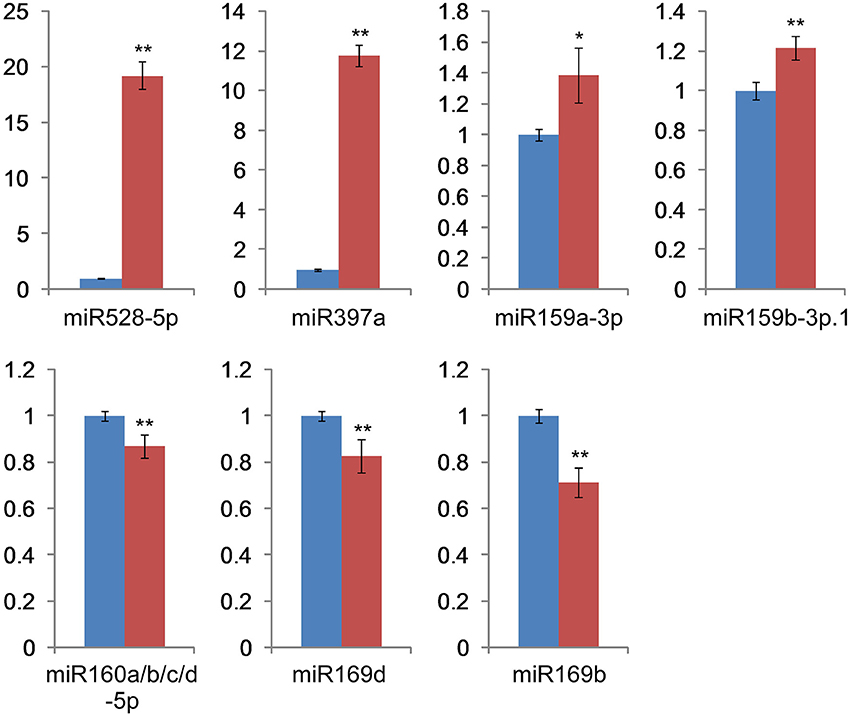
Figure 5. RT-qPCR validation of the miRNAs. Blue bar: relative gene expression level in the control library. Red bar: relative gene expression level in the H2O2 stress library. The data are derived from three biological repeats and represent mean ± standard deviation (n = 3). “*” and “**” indicate significant difference at P < 0.05 and 0.01 level, respectively.
RT-qPCR was also used for detection and quantification of the predicted targets of 13 H2O2-responsive miRNAs (Figure 6). Our results revealed that for most of the miRNAs, there was a negative correlation between the miRNA level and the levels of their target genes (Figure 6), with the exception of Bradi1g36540. Bradi1g36540 and Bradi2g53010 were both regulated by the upregulated bdi-miR159a-3p and bdi-miR159b-3p.1. Bradi2g53010 was downregulated, but Bradi1g36540 showed no significant change under H2O2 stress. Bradi2g55497 was mediated by three novel miRNAs (Figure 4). Two of the miRNAs (novel_mir_98 and novel_mir_120) were downregulated, while another (novel_mir_222) was upregulated, but the expression level of Bradi2g55497 was unexpectedly downregulated. This phenomenon was also observed in the profiles of Bradi4g10380 (the target gene of novel_mir_98, novel_mir_120, and novel_mir_222) and Bradi1g14810 (the target gene of novel_mir_98 and novel_mir_222).
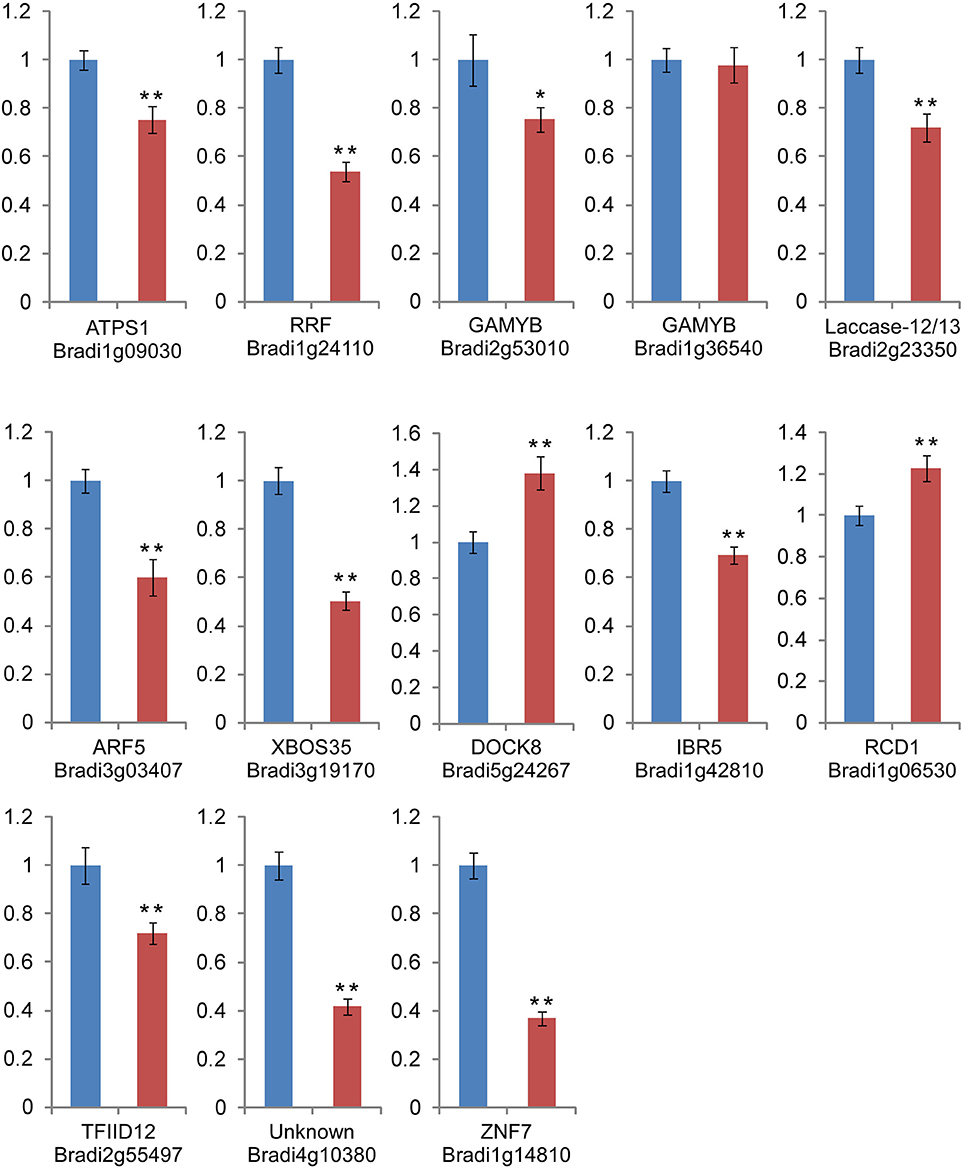
Figure 6. RT-qPCR analysis of the miRNA target genes. The Actin and SamDC genes were served as the endogenous controls. Blue bar: relative gene expression level in the control library. Red bar: relative gene expression level in the H2O2 stress library. The data are derived from three biological repeats and represent mean ± standard deviation (n = 3). “*” and “**” indicate significant difference at P < 0.05 and 0.01 level, respectively.
Verification of miRNA-Guided Cleavage of Target mRNAs in B. distachyon
To verify the miRNA-guided target cleavage, RLM-5′-RACE experiment was performed to detect cleavage product of 3 known (bdi-miR160b-5p, bdi-miR159b-3p.1, and bdi-miR397a) and two novel bdi-miRNAs (novel_mir_152 and novel_mir_222). As shown in Figure 7, all five of the B. distachyon miRNAs guided the target cleavage, often at the tenth nucleotide (Figure 7).
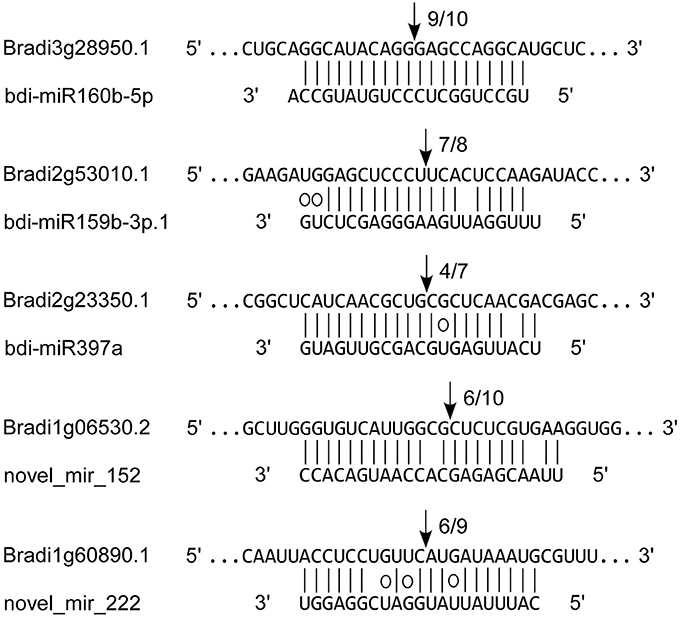
Figure 7. Validation of known and novel miRNAs by RLM-5′-RACE. Each top strand represents the miRNA complementary site on target mRNA, and each bottom strand represents the miRNA. Watson-Crick pairing (vertical dashes) and G:U wobble pairing (circles) are indicated. The arrows indicated the cleavage sites of target genes, and the numbers showed the frequency of cloned 5′ RACE products.
Discussion
To examine the H2O2-responsive miRNAs, two sRNA libraries were constructed from a mixture of 12-day old B. distachyon seedlings treated with 20 mM H2O2 and a control sample, and were then subjected to next-generation deep sequencing. We identified a set of known and novel H2O2-responsive miRNAs with notable expression pattern changes and we also provide a miRNA-mediated oxidative stress response PPI network. These results provide useful information for elucidating the response and defense mechanisms for H2O2 stress at the post-transcriptional level in plants.
Given that the expression of target genes is negatively regulated by miRNAs, the expression patterns of target genes always show an inverse correlation with those of miRNAs. However, our miRNA-target network showed that the regulatory mechanism of miRNA may be more complex. Some miRNAs can each regulate several target genes with the same function or different functions, and some genes can be regulated by several miRNAs. Among the known differential miRNAs in our study, some regulated two or more target genes with the same function, such as bdi-miR159a and bdi-miR169d/k, whereas more miRNAs, such as bdi-miR395, bdi-miR408, and bdi-miR528, regulated two or more target genes with different functions (Figure 4 and Table 1). Among the novel differential miRNAs, novel_mir_152 was downregulated (log2 fold change = −8.13) under H2O2 stress and mediated four target genes encoding a cell differentiation protein RCD1 homolog associated with RNA degradation and cell differentiation. The 20 potential genes regulated by novel_mir_120 are involved in various functions. Gene Bradi2g55497, which encodes transcription initiation factor TFIID subunit 12, was regulated by three novel miRNAs (novel_mir_98, novel_mir_120, and novel_mir_222). Two of them (novel_mir_98 and novel_mir_120) were downregulated under H2O2 stress, while novel_mir_222 was upregulated. Compared with novel_mir_98 and novel_mir_120, novel_mir_222 may have the opposite effect on the expression of Bradi2g55497. Furthermore, RT-qPCR analysis demonstrated that the final result of the antagonism is the downregulation of Bradi2g55497 (Figure 4), so novel_mir_222 may play a main role under H2O2 stress. Four of the target genes of downregulated novel_mir_120 and upregulated novel_mir_222 are the same, which seems to imply that they are a pair of miRNAs with opposing effects. Additionally, novel_mir_91 and novel_mir_197 also exhibited the same pattern. Previous studies also found similar phenomenon. For example, genes SPL and AP2, encoded DNA-binding transcription factors, can be regulated by miR156 and miR172, which showed opposite expression patterns (Ding et al., 2014).
The upregulation of a miRNA always results in the aggravated degradation of its target gene in plants, and vice versa. These target genes of upregulated miRNAs may be associated with stress response, whereas the target genes of downregulated miRNAs are always involved in stress resistance. miR528, a monocot-specific miRNA, was upregulated under H2O2 stress (log2 fold change = 2.88). bdi-miR528 is also upregulated in Bd21 under drought stress (Bertolini et al., 2013) and in rice under H2O2 stress (Li et al., 2010). Bradi3g19170, the target gene of bdi-miR528, codes the E3 ubiquitin-protein ligase XBOS35, which may be involved in ubiquitination for proteasomal degradation and the ethylene (ET) signaling pathway during abiotic stress response (Sobeih et al., 2004; Prasad et al., 2010). Thus, miR528 may be a vital miRNA involved in water and oxidative stress response in monocots. As the largest MIR gene family in B. distachyon, the MIR395 gene family includes 15 members. In our study, 13 members of the MIR395 gene family were overexpressed under H2O2 stress and their target genes were Bradi1g09030 (encoded ATP-sulfurylase 1), Bradi1g24110 (encoded Ribosome-recycling factor), and Bradi2g52840 (encoded disease resistance protein RGA4), which may be involved in oxidative stress response. Similar to our results, all of the identified MIR395 gene family members are also upregulated under drought stress in B. distachyon (Bertolini et al., 2013). All of the target genes of bdi-miR397a, except Bradi3g03407, encode laccase, which plays an important role during the formation of lignin in the cell wall (Mayer and Staples, 2002); a previous study also confirmed that miR397 is a negative regulator of laccase genes (Lu et al., 2013). bdi-miR827-3p was upregulated under H2O2 stress, and a previous study (Jeong et al., 2013) found that this miRNA was also upregulated under phosphate starvation conditions in shoots. In contrast to our results, a previous study showed that miR397 and miR827 were downregulated in rice by H2O2 treatment, which indicates that the miRNA response to the same abiotic stress occurs in a genotype/species-dependent manner (Zhang, 2015).
In plants, miRNAs participate in the response to abiotic stress by mediating key components of complex gene networks (Zhang, 2015). Many of the target genes of miRNAs identified in our study are TFs, which is consistent with previous studies (Jones-Rhoades et al., 2006; Baev et al., 2011; Zhang, 2015). Thus, a miRNA can indirectly regulate the expression of downstream target genes through regulation of the expression of TFs. Previous studies revealed that the mechanism of the MYB TF involved in gibberellic acid (GA) signal transduction was regulated by miR159 (Achard et al., 2004; Alonso-Peral et al., 2010). In our study, bdi-miR159a/b (target genes Bradi2g53010 and Bradi1g36540, two MYB genes) was upregulated under H2O2 stress, which is consistent with the results in rice and B. distachyon under drought stress (Zhou et al., 2010; Bertolini et al., 2013). Auxin response factors (ARFs) are a class of auxin-responsive TFs mediated and regulated by miR160 (Mallory et al., 2005). miR160 regulates the expression of ARF genes by combining with the complementary sites of a non-coding region of ARF genes (Rhoades et al., 2002). In our study, gene ARF22 was targeted by four downregulated members of the bdi-MIR160 family (Table 1). Interestingly, gene ARF5 was targeted by the upregulated bdi-miR397a, instead of bdi-miR160. In addition, the expression of some miRNAs can also be regulated by TFs through specific binding to the promoter region of the miRNA. For example, miR160 can be transcriptionally regulated by proteins ARF6 and ARF17 in Arabidopsis (Gutierrez et al., 2009). Thus, there are fine tuning mechanisms in plants to modulate gene expression through miRNA-TF-mediated feedback loops (Meng et al., 2011; Iyer et al., 2012). bdi-miR169 was significantly downregulated under H2O2 stress in our study and a previous study in rice (Li et al., 2010); its target gene encodes a nuclear TF Y subunit A-2 (NF-YA-2) or NF-YA-4, which belongs to the CCAAT binding TF family. In Arabidopsis, the upregulation of NF-YA gene depends on the regulation of miR169 under drought stress (Li et al., 2008). Thön et al. (2010) found that CCAAT-binding TFs are involved in the response to oxidative stress. Therefore, miR169 may play a crucial role in the process of resistance to oxidative stress in plants. The network analysis showed that NF-YA could interact with V-type proton ATPase subunit d (encoded by Bradi2g42100), transcription initiation factor TFIID subunit 12 (encoded by Bradi2g55497), 2-oxoglutarate-dependent dioxygenase DAO (encoded by Bradi2g24715), and TF MYB (encoded by Bradi2g53010 and Bradi1g36540). One of the target genes of the upregulated bdi-miR408-5p encodes TF TCP15, whose activity is inhibited by oxidative stress (Viola et al., 2013). Overexpression of miR408 in Arabidopsis can improve its tolerance to oxidative stress (Ma et al., 2015).
Among the differentially expressed novel miRNAs, novel_mir_160, a potential new member of the bdiMIR171 gene family, was downregulated (log2 fold change = −2.12) under H2O2 stress. miR171 is also significantly downregulated under drought stress in rice and potato (Zhou et al., 2010; Hwang et al., 2011). Although we did not identify the target genes of this potential novel bdiMIR171 gene family member, a previous study in potato showed that GRAS family TF, involved in development and stress responses, such as drought stress (Ma et al., 2010), is the putative target gene of miR171 (Hwang et al., 2011). The pre-miRNA of novel_mir_162 was the same as the precursor of bdi-miR395c, which may be a novel member of the bdiMIR395 family. Similar to other known bdi-miR395 family members, this miRNA was also upregulated (Table 2). In addition, novel_mir_40 was identified as a novel member of the bdiMIR169 family, which was also downregulated (log2 fold change = −10.28), as were the five known differentially expressed bdiMIR169 family members (Table 1). In contrast to typical members of the bdiMIR169 family, whose target genes always encode NF-YA TFs, the target gene of novel_mir_40 is Bradi1g25517, which encodes glucan endo-1,3-beta-glucosidase 3.
H2O2 can trigger various phytohormone signaling pathways involved in abiotic and biotic stress responses and there are complex crosstalks among different phytohormone signaling pathways (Harrison, 2012; Saxena et al., 2016). Recent study found out some immune hormone marker genes involved in salicylic acid (SA), jasmonic acid (JA) and ET signaling pathways in B. distachyon (Kouzai et al., 2016). In our study, we also found some genes related with phytohormone signaling pathways were the target of miRNAs. For example, the two MYB genes involved in GA pathway were regulated by bdi-miR159a/b, ARF22, and ARF5 genes involved in auxin pathway were the target of bdi-miR160 and bdi-miR397a, respectively, and the E3 ubiquitin-protein ligase XBOS35 and ethylene-responsive transcription factor RAP2-3 genes related with ET signaling pathways were mediated by bdi-miR528 and novel_mir_120/novel_mir_222, respectively.
Conclusion
In this study, we sequenced and analyzed the sRNA of model plant Bd21 seedlings under H2O2 stress and normal growth conditions using large-scale sequencing technology. Finally, we identified 39 known and 221 novel miRNAs, of which 31 known miRNAs and 30 novel miRNAs were involved in H2O2-stress response and resistance. Moreover, RT-qPCR analysis of several representative miRNAs and their target genes and cleavage site analysis through 5′ RACE validated our sequencing and bioinformatic results. The PPI network mediated by miRNAs revealed the regulation mechanism of signal transduction and oxidative stress resistance under H2O2 stress. Further analysis of the differentially expressed miRNAs and their target genes will help us understand the mechanism of oxidative stress response and tolerance in plants.
Author Contributions
DL carried out all experiments, data analysis and wrote the manuscript. SZ contributed the RLM-5′-RACE experiment. GZ, YB, and GC performed the RNA extraction and RT-qPCR, CH and ZY conducted GO annotation and enrichment analyses. DL and YY conceived the study, participated in the design and coordination, and in interpretation of the dataset. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was financially supported by grants from the National Natural Science Foundation of China (31471485), Natural Science Foundation of Beijing City and the Key Developmental Project of Science Technology, Beijing Municipal Commission of Education (KZ201410028031).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01567
Figure S1. First base bias analysis of the novel miRNAs. (A) First base bias of all novel Bd-miRNAs in the two libraries (CS and TS); (B) First base bias of 20-, 21-, 22-, and 23-nt novel Bd-miRNAs in the two libraries (CS and TS).
Figure S2. GO annotation of all potential miRNA target genes. Biological process, molecular function, and cellular component categories were displayed.
Table S1. All the RT-qPCR and RACE primers used in this study.
Table S2. The distribution of sRNAs in each class.
Table S3. The known B. distachyon miRNAs identified in this study.
Table S4. The novel B. distachyon miRNAs identified in this study.
Table S5. List of known and novel miRNAs and their target genes. (A) List of known miRNAs and their target genes; (B) List of novel miRNAs and their target genes.
Abbreviations
miRNA, microRNA; RT-qPCR, Reverse transcription-quantitative polymerase chain reaction; ROS, Reactive oxygen species; H2O2, Hydrogen peroxide; SOD, Superoxide dismutase; TF, Transcription factor; RT-qPCR, Reverse transcription quantitative polymerase chain reaction; sRNA, Small-RNA; MFE, Minimum of free energy; GO, Gene Ontology; FDR, False discovery rate; PPI, Protein-protein interaction; CS, Control seedlings; TS, H2O2-treated seedlings; ARF, Auxin response factor; ET, Ethylene; SA, Salicylic acid; JA, Jasmonic acid.
References
Achard, P., Herr, A., Baulcombe, D. C., and Harberd, N. P. (2004). Modulation of floral development by a gibberellin-regulated microRNA. Development 131, 3357–3365. doi: 10.1242/dev.01206
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., and Axtell, M. J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18, 758–762. doi: 10.1016/j.cub.2008.04.042
Allen, E., Xie, Z., Gustafson, A. M., and Carrington, J. C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221. doi: 10.1016/j.cell.2005.04.004
Alonso-Peral, M. M., Li, J., Li, Y., Allen, R. S., Schnippenkoetter, W., Ohms, S., et al. (2010). The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol. 154, 757–771. doi: 10.1104/pp.110.160630
Apel, K., and Hirt, H. (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. doi: 10.1146/annurev.arplant.55.031903.141701
Audic, S., and Claverie, J.-M. (1997). The significance of digital gene expression profiles. Genome Res. 7, 986–995.
Baev, V., Milev, I., Naydenov, M., Apostolova, E., Minkov, G., Minkov, I., et al. (2011). Implementation of a de novo genome-wide computational approach for updating Brachypodium miRNAs. Genomics 97, 282–293. doi: 10.1016/j.ygeno.2011.02.008
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Bertolini, E., Verelst, W., Horner, D. S., Gianfranceschi, L., Piccolo, V., Inzé, D., et al. (2013). Addressing the role of microRNAs in reprogramming leaf growth during drought stress in Brachypodium distachyon. Mol. Plant 6, 423–443. doi: 10.1093/mp/sss160
Bian, Y. W., Lv, D. W., Cheng, Z. W., Gu, A. Q., Cao, H., and Yan, Y. M. (2015). Integrative proteome analysis of Brachypodium distachyon roots and leaves reveals a synergetic responsive network under H2O2 stress. J. Proteomics 128, 388–402. doi: 10.1016/j.jprot.2015.08.020
Budak, H., and Akpinar, A. (2011). Dehydration stress-responsive miRNA in Brachypodium distachyon: evident by genome-wide screening of microRNAs expression. OMICS 15, 791–799. doi: 10.1089/omi.2011.0073
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Ding, Q., Zeng, J., and He, X.-Q. (2014). Deep sequencing on a genome-wide scale reveals diverse stage-specific microRNAs in cambium during dormancy-release induced by chilling in poplar. BMC Plant Biol. 14:267. doi: 10.1186/s12870-014-0267-6
de Azevedo Neto, A. D., Prisco, J. T., Enéas-Filho, J., Medeiros, J. V., and Gomes-Filho, E. (2005). Hydrogen peroxide pre-treatment induces salt-stress acclimation in maize plants. J. Plant Physiol. 162, 1114–1122. doi: 10.1016/j.jplph.2005.01.007
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO. a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Foyer, C. H., Lopez-Delgado, H., Dat, J. F., and Scott, I. M. (1997). Hydrogen peroxide- and glutathione- associated mechanisms of acclimatory stress tolerance and signalling. Physiol. Plant 100, 241–254. doi: 10.1111/j.1399-3054.1997.tb04780.x
Friedman, R. C., Farh, K. K.-H., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. doi: 10.1101/gr.082701.108
Gutierrez, L., Bussell, J. D., Păcurar, D. I., Schwambach, J., Păcurar, M., and Bellini, C. (2009). Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 21, 3119–3132. doi: 10.1105/tpc.108.064758
Harrison, M. A. (2012). “Cross-talk between phytohormone signaling pathways under both optimal and stressful environmental conditions,” in Phytohormones and Abiotic Stress Tolerance in Plants, eds N. A. Khan, R. Nazar, N. Iqbal, and N. A. Anjum (Berlin; Heidelberg: Springer), 49–76.
Hwang, E.-W., Shin, S.-J., Yu, B.-K., Byun, M.-O., and Kwon, H.-B. (2011). miR171 family members are involved in drought response in Solanum tuberosum. J. Plant Biol. 54, 43–48. doi: 10.1007/s12374-010-9141-8
Iyer, N. J., Jia, X., Sunkar, R., Tang, G., and Mahalingam, R. (2012). microRNAs responsive to ozone-induced oxidative stress in Arabidopsis thaliana. Plant Signal. Behav. 7, 484–491. doi: 10.4161/psb.19337
Jeong, D.-H., Schmidt, S. A., Rymarquis, L. A., Park, S., Ganssmann, M., German, M. A., et al. (2013). Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol. 14:R145. doi: 10.1186/gb-2013-14-12-r145
Jia, X., Ren, L., Chen, Q.-J., Li, R., and Tang, G. (2009). UV-B-responsive microRNAs in Populus tremula. J. Plant Physiol. 166, 2046–2057. doi: 10.1016/j.jplph.2009.06.011
Jones-Rhoades, M. W., and Bartel, D. P. (2004). Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 14, 787–799. doi: 10.1016/j.molcel.2004.05.027
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Khraiwesh, B., Zhu, J.-K., and Zhu, J. (2012). Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim. Biophys. Acta 1819, 137–148. doi: 10.1016/j.bbagrm.2011.05.001
Kouzai, Y., Kimura, M., Yamanaka, Y., Watanabe, M., Matsui, H., Yamamoto, M., et al. (2016). Expression profiling of marker genes responsive to the defence-associated phytohormones salicylic acid, jasmonic acid and ethylene in Brachypodium distachyon. BMC Plant Biol. 16:59. doi: 10.1186/s12870-016-0749-9
Kozomara, A., and Griffiths-Jones, S. (2011). miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157 doi: 10.1093/nar/gkq1027
Li, R., Yu, C., Li, Y., Lam, T.-W., Yiu, S.-M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, T., Li, H., Zhang, Y.-X., and Liu, J.-Y. (2011). Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica). Nucleic Acids Res. 39, 2821–2833. doi: 10.1093/nar/gkq1047
Li, W.-X., Oono, Y., Zhu, J., He, X.-J., Wu, J.-M., Iida, K., et al. (2008). The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20, 2238–2251. doi: 10.1105/tpc.108.059444
Li, Y. F., Zheng, Y., Addo-Quaye, C., Zhang, L., Saini, A., Jagadeeswaran, G., et al. (2010). Transcriptome-wide identification of microRNA targets in rice. Plant J. 62, 742–759. doi: 10.1111/j.1365-313X.2010.04187.x
Li, Y., Zhang, Z., Liu, F., Vongsangnak, W., Jing, Q., and Shen, B. (2012). Performance comparison and evaluation of software tools for microRNA deep-sequencing data analysis. Nucleic Acids Res. 40, 4298–4305. doi: 10.1093/nar/gks043
Lu, S., Li, Q., Wei, H., Chang, M.-J., Tunlaya-Anukit, S., Kim, H., et al. (2013). Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc. Natl. Aca. Sci. U.S.A. 110, 10848–10853. doi: 10.1073/pnas.1308936110
Lv, D.-W., Li, X., Zhang, M., Gu, A.-Q., Zhen, S.-M., Wang, C., et al. (2014a). Large-scale phosphoproteome analysis in seedling leaves of Brachypodium distachyon L. BMC Genomics 15:375. doi: 10.1186/1471-2164-15-375
Lv, D.-W., Subburaj, S., Cao, M., Yan, X., Li, X., Appels, R., et al. (2014b). Proteome and phosphoproteome characterization reveals new response and defense mechanisms of Brachypodium distachyon leaves under salt stress. Mol. Cell. Proteomics 13, 632–652. doi: 10.1074/mcp.M113.030171
Ma, C., Burd, S., and Lers, A. (2015). miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 84, 169–187. doi: 10.1111/tpj.12999
Ma, H.-S., Liang, D., Shuai, P., Xia, X.-L., and Yin, W.-L. (2010). The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J. Exp. Bot. 6, 4011–4019. doi: 10.1093/jxb/erq217
Mallory, A. C., Bartel, D. P., and Bartel, B. (2005). MicroRNA-directed regulation of Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes. Plant Cell 17, 1360–1375. doi: 10.1105/tpc.105.031716
Mayer, A. M., and Staples, R. C. (2002). Laccase: new functions for an old enzyme. Phytochemistry 60, 551–565. doi: 10.1016/S0031-9422(02)00171-1
Meng, Y., Shao, C., and Chen, M. (2011). Toward microRNA-mediated gene regulatory networks in plants. Brief Bioinform. 12, 645–659. doi: 10.1093/bib/bbq091
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant MicroRNAs. Plant Cell 20, 3186–3190. doi: 10.1105/tpc.108.064311
Mittler, R. (2002). Oxidative stress, antioxidants and stress tolerance. Trends Plant Sci. 7, 405–410. doi: 10.1016/s1360-1385(02)02312-9
Mittler, R., Vanderauwera, S., Gollery, M., and Van Breusegem, F. (2004). Reactive oxygen gene network of plants. Trends Plant Sci. 9, 490–498. doi: 10.1016/j.tplants.2004.08.009
Neill, S., Desikan, R., and Hancock, J. (2002). Hydrogen peroxide signalling. Curr. Opin. Plant Biol. 5, 388–395. doi: 10.1016/S1369-5266(02)00282-0
Neill, S. J., Desikan, R., Clarke, A., Hurst, R. D., and Hancock, J. T. (2002). Hydrogen peroxide and nitric oxide as signalling molecules in plants. J. Exp. Bot. 53, 1237–1247. doi: 10.1093/jexbot/53.372.1237
Pastori, G. M., and Foyer, C. H. (2002). Common components, networks, and pathways of cross-tolerance to stress. The central role of “redox” and abscisic acid-mediated controls. Plant Physiol. 129, 460–468. doi: 10.1104/pp.011021
Prasad, M. E., Schofield, A., Lyzenga, W., Liu, H., and Stone, S. L. (2010). Arabidopsis RING E3 ligase XBAT32 regulates lateral root production through its role in ethylene biosynthesis. Plant Physiol. 153, 1587–1596. doi: 10.1104/pp.110.156976
Priest, H. D., Fox, S. E., Rowley, E. R., Murray, J. R., Michael, T. P., and Mockler, T. C. (2014). Analysis of global gene expression in Brachypodium distachyon reveals extensive network plasticity in response to abiotic stress. PLoS ONE 9:e87499. doi: 10.1371/journal.pone.0087499
Quan, L. J., Zhang, B., Shi, W. W., and Li, H. Y. (2008). Hydrogen peroxide in plants: a versatile molecule of the reactive oxygen species network. J. Integr. Plant Biol. 50, 2–18. doi: 10.1111/j.1744-7909.2007.00599.x
Rentel, M. C., and Knight, M. R. (2004). Oxidative stress-induced calcium signaling in Arabidopsis. Plant Physiol. 135, 1471–1479. doi: 10.1104/pp.104.042663
Rhoades, M. W., Reinhart, B. J., Lim, L. P., Burge, C. B., Bartel, B., and Bartel, D. P. (2002). Prediction of plant microRNA targets. Cell 110, 513–520. doi: 10.1016/S0092-8674(02)00863-2
Saxena, I., Srikanth, S., and Chen, Z. (2016). Cross talk between H2O2 and interacting signal molecules under plant stress response. Front Plant Sci. 7:570. doi: 10.3389/fpls.2016.00570
Schwab, R., Palatnik, J. F., Riester, M., Schommer, C., Schmid, M., and Weigel, D. (2005). Specific effects of microRNAs on the plant transcriptome. Dev. Cell 8, 517–527. doi: 10.1016/j.devcel.2005.01.018
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sobeih, W. Y., Dodd, I. C., Bacon, M. A., Grierson, D., and Davies, W. J. (2004). Long-distance signals regulating stomatal conductance and leaf growth in tomato (Lycopersicon esculentum) plants subjected to partial root-zone drying. J. Exp. Bot. 55, 2353–2363. doi: 10.1093/jxb/erh204
Sunkar, R., Kapoor, A., and Zhu, J.-K. (2006). Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18, 2051–2065. doi: 10.1105/tpc.106.041673
Szklarczyk, D., Franceschini, A., Kuhn, M., Simonovic, M., Roth, A., Minguez, P., et al. (2011). The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568. doi: 10.1093/nar/gkq973
Thön, M., Al Abdallah, Q., Hortschansky, P., Scharf, D. H., Eisendle, M., Haas, H., et al. (2010). The CCAAT-binding complex coordinates the oxidative stress response in eukaryotes. Nucleic Acids Res. 38, 1098–1113. doi: 10.1093/nar/gkp1091
Uchida, A., Jagendorf, A. T., Hibino, T., Takabe, T., and Takabe, T. (2002). Effects of hydrogen peroxide and nitric oxide on both salt and heat stress tolerance in rice. Plant Sci. 163, 515–523. doi: 10.1016/S0168-9452(02)00159-0
Upadhyaya, H., Khan, M., and Panda, S. (2007). Hydrogen peroxide induces oxidative stress in detached leaves of Oryza sativa L. Gen. Appl. Plant Physiol. 33, 83–95.
Vandenabeele, S., Van Der Kelen, K., Dat, J., Gadjev, I., Boonefaes, T., Morsa, S., et al. (2003). A comprehensive analysis of hydrogen peroxide-induced gene expression in tobacco. Proc. Natl. Aca. Sci. U.S.A. 100, 16113–16118. doi: 10.1073/pnas.2136610100
Viola, I. L., Güttlein, L. N., and Gonzalez, D. H. (2013). Redox modulation of plant developmental regulators from the class I TCP transcription factor family. Plant Physiol. 162, 1434–1447. doi: 10.1104/pp.113.216416
Vogel, J. P., Garvin, D. F., Mockler, T. C., Schmutz, J., Rokhsar, D., Bevan, M. W., et al. (2010). Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768. doi: 10.1038/nature08747
Vranová, E., Inzé, D., and Van Breusegem, F. (2002). Signal transduction during oxidative stress. J. Exp. Bot. 53, 1227–1236. doi: 10.1093/jexbot/53.372.1227
Wahid, A., Perveen, M., Gelani, S., and Basra, S. (2007). Pretreatment of seed with H2O2 improves salt tolerance of wheat seedlings by alleviation of oxidative damage and expression of stress proteins. J. Plant Physiol. 164, 283–294. doi: 10.1016/j.jplph.2006.01.005
Wang, B., Sun, Y., Song, N., Wang, X. J., Feng, H., Huang, Z. S., et al. (2013). Identification of UV-B-induced microRNAs in wheat. Genet. Mol. Res. 12, 4213–4221. doi: 10.4238/2013.October.7.7
Wu, J., Wang, D., Liu, Y., Wang, L., Qiao, X., and Zhang, S. (2014). Identification of miRNAs involved in pear fruit development and quality. BMC Genomics 15:953. doi: 10.1186/1471-2164-15-953
Zhang, B. (2015). MicroRNA: a new target for improving plant tolerance to abiotic stress. J. Exp. Bot. 66, 1749–1761. doi: 10.1093/jxb/erv013
Zhang, J., Xu, Y., Huan, Q., and Chong, K. (2009). Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics 10:449. doi: 10.1186/1471-2164-10-449
Zhou, L., Liu, Y., Liu, Z., Kong, D., Duan, M., and Luo, L. (2010). Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J. Exp. Bot. 61, 4157–4168. doi: 10.1093/jxb/erq237
Zhou, X., Wang, G., and Zhang, W. (2007). UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 3, 103. doi: 10.1038/msb4100143
Keywords: Bd21, H2O2 stress, microRNA, high-throughput sequencing, regulatory network
Citation: Lv D-W, Zhen S, Zhu G-R, Bian Y-W, Chen G-X, Han C-X, Yu Z-T and Yan Y-M (2016) High-Throughput Sequencing Reveals H2O2 Stress-Associated MicroRNAs and a Potential Regulatory Network in Brachypodium distachyon Seedlings. Front. Plant Sci. 7:1567. doi: 10.3389/fpls.2016.01567
Received: 29 April 2016; Accepted: 05 October 2016;
Published: 20 October 2016.
Edited by:
Bernie Carroll, University of Queensland, AustraliaReviewed by:
Mingming Xin, China Agricultural University, ChinaZhiyong Liu, Chinese Academy of Sciences, China
Copyright © 2016 Lv, Zhen, Zhu, Bian, Chen, Han, Yu and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-Ming Yan, eWFueW1AY251LmVkdS5jbg==
†These authors have contributed equally to this work.