 Mehak Taneja
Mehak Taneja Shivi Tyagi1
Shivi Tyagi1 Santosh Kumar Upadhyay
Santosh Kumar Upadhyay- 1Department of Botany, Panjab University, Chandigarh, India
- 2National Agri-Food Biotechnology Institute, Mohali, India
The Ca2+/cation antiporters (CaCA) superfamily proteins play vital function in Ca2+ ion homeostasis, which is an important event during development and defense response. Molecular characterization of these proteins has been performed in certain plants, but they are still not characterized in Triticum aestivum (bread wheat). Herein, we identified 34 TaCaCA superfamily proteins, which were classified into TaCAX, TaCCX, TaNCL, and TaMHX protein families based on their structural organization and evolutionary relation with earlier reported proteins. Since the T. aestivum comprises an allohexaploid genome, TaCaCA genes were derived from each A, B, and D subgenome and homeologous chromosome (HC), except chromosome-group 1. Majority of genes were derived from more than one HCs in each family that were considered as homeologous genes (HGs) due to their high similarity with each other. These HGs showed comparable gene and protein structures in terms of exon/intron organization and domain architecture. Majority of TaCaCA proteins comprised two Na_Ca_ex domains. However, TaNCLs consisted of an additional EF-hand domain with calcium binding motifs. Each TaCaCA protein family consisted of about 10 transmembrane and two α-repeat regions with specifically conserved signature motifs except TaNCL, which had single α-repeat. Variable expression of most of the TaCaCA genes during various developmental stages suggested their specified role in development. However, constitutively high expression of a few genes like TaCAX1-A and TaNCL1-B indicated their role throughout the plant growth and development. The modulated expression of certain genes during biotic (fungal infections) and abiotic stresses (heat, drought, salt) suggested their role in stress response. Majority of TaCCX and TaNCL family genes were found highly affected during various abiotic stresses. However, the role of individual gene needs to be established. The present study unfolded the opportunity for detail functional characterization of TaCaCA proteins and their utilization in future crop improvement programs.
Introduction
Calcium ion (Ca2+) is a vital element in plants due to its roles as an essential nutrient and secondary messenger (Hirschi, 2004; Case et al., 2007). It is involved in signal transduction in response to various internal and/or external stimuli (Poovaiah and Reddy, 1993; Dodd et al., 2010; Spalding and Harper, 2011). The concentration of Ca2+ increases during perturbation of stimuli, which get recognized by calcium binding proteins or sensor proteins. These proteins further transfer the signal downstream to start phosphorylation cascade that ultimately lead to the regulation of gene expression (Tuteja and Mahajan, 2007). The elevation in Ca2+ concentration inside the cell is also reported in response to various growth regulators, plant nutrients, pathogens and abiotic stresses, which established the role of calcium signaling during development and stress responses in plants (Tuteja and Sopory, 2008; Kader and Lindberg, 2010; Zhang et al., 2014a).
The modulation in Ca2+ concentration across the cell membrane is basically mediated by three classes of transporters- Ca2+-ATPases (PMCAs), Ca2+ permeable channels, and Ca2+/cation antiporters (CaCAs), which function in combination of each other (Haug-Collet et al., 1999; Sanders et al., 1999; Axelsen and Palmgren, 2001). The CaCA superfamily proteins are reported in diverse group of organisms from bacteria to higher plants and animals as well. These proteins usually facilitate the efflux of Ca2+ against concentration gradient across the membrane, and influx of monovalent cations like H+, Na+, or K+ in exchange (Saier et al., 1999; Cai and Lytton, 2004a; Emery et al., 2012; Pittman and Hirschi, 2016a). CaCA proteins basically form a composite superfamily, which consists of five different families of exchanger proteins- YRBG, Na+/Ca2+ exchanger (NCX), Na+/Ca2+, K+ exchanger (NCKX), cation/Ca2+ exchanger (CCX), and H+/cation exchanger (CAX), which are classified on the basis of their function and evolutionary relationship (Cai and Lytton, 2004a; Emery et al., 2012; Pittman and Hirschi, 2016a). Recently, Singh et al. (2015) proposed the classification of all the CaCA superfamily proteins as NCX family due to the occurrence of Na_Ca_ex (PF01699) domain and tight evolutionary relationship between them. However, Pittman and Hirschi (2016a) by phylogenetic analysis and structure modeling further established that the CaCA superfamily members exhibit diverse structural and functional characteristics, and all of them cannot be classified as NCX proteins.
The CaCA superfamily proteins consist of comparable topological structure with an average of 10 transmembrane (TM) domains and two α-repeats region inside the TM 2-3 and 7-8. These repeats play vital role during ion-selectivity, binding and transportation by various groups of CaCA proteins (Kamiya and Maeshima, 2004; Ottolia et al., 2005; Shigaki et al., 2005; Nicoll et al., 2007). Recent crystallography analyses of certain CaCA proteins from archaea, bacteria and yeast provided detail insight into the structure (Liao et al., 2012; Nishizawa et al., 2013; Waight et al., 2013; Wu et al., 2013). The length of CaCA superfamily proteins varies from 300 to 1000 amino acid (AA) residues with almost similar topological structure. A typical CaCA protein consists of clusters of five TM helices in each half, separated by a cytosolic loop, with certain exceptions. For instance, the CCX proteins contain additional TM helix in C-terminal half (Cai and Lytton, 2004b). Further, the length of N-terminal region and cytosolic loop also varies in various groups of CaCA superfamily proteins (Emery et al., 2012).
Large scale genome analysis of numerous archaea, bacteria, fungi, algae, land plants, and animals established that- (1) the YBRG proteins are exclusively present in prokaryotes, (2) the CAX proteins are present in all organisms, (3) CCXs are present in all eukaryotes but not in prokaryotes, while (4) NCX and NCKX proteins are present in animal cells and algae, but are lacking in higher plants (Shigaki et al., 2006; Lytton, 2007; Emery et al., 2012; Khananshvili, 2013, 2014; Pittman and Hirschi, 2016a). However, land plants have evolved two additional groups of CaCA proteins- Mg2+/H+ exchanger (MHX) proteins and EF-hand domain containing CAX (EF-CAX) proteins (Emery et al., 2012; Gaash et al., 2013). The EF-CAX was also found in algae (Emery et al., 2012) and later named as NCX like protein (NCL) due to their functional divergence from CAX proteins (Wang et al., 2012; Li et al., 2016).
A plant MHX was first identified in Arabidopsis thaliana (AtMHX) as a member of NCX family, which enhanced Mg2+ concentration in vacuole without Na+/Ca2+ exchange activity (Shaul et al., 1999). Recently, the MHX proteins were identified from numerous land plants, which formed a distinct group with tight phylogenetic clustering (Emery et al., 2012; Gaash et al., 2013; Pittman and Hirschi, 2016a). The plant MHX proteins also differ from NCX proteins in certain key amino acid residues in α1 and α2 repeat regions responsible for Na+/Ca2+ exchange activity (Nicoll et al., 1996a; Iwamoto et al., 2000; Philipson and Nicoll, 2000; Ottolia et al., 2005; Emery et al., 2012). Furthermore, the MHX proteins consist of smaller central loop without Ca2+ binding domain (Lytton, 2007; Gaash et al., 2013).
The CAX proteins comprised the largest proportion of CaCA superfamily, and present in each group of organism (Shigaki et al., 2006; Manohar et al., 2011; Emery et al., 2012; Pittman and Hirschi, 2016b), except mammals and insects. These are broadly classified into type 1, type 2, and type 3 categories on the basis of phylogenetic clustering (Shigaki et al., 2006). Plants generally consist of type 1 CAX proteins, which are further divided into type 1A and type 1B groups (Shigaki et al., 2006; Emery et al., 2012). The CAX proteins usually exist as multi-gene families in higher plants. Generally, 5-6 CAX proteins are identified in various monocot and dicot plants, with a maximum of 14 CAX proteins in Glycine max, about half of them belonging to each type 1A and 1B groups (Emery et al., 2012; Pittman and Hirschi, 2016a). It has been suggested that both the groups differ in their ion selectivity; type 1B members like AtCAX2 and AtCAX5 facilitate the transportation of several ions such as Ca2+, Cd2+, and Mn2+, while type 1A proteins like AtCAX1 and AtCAX3 are thought to be specific for Ca2+ homeostasis (Hirschi et al., 2000; Shigaki et al., 2003; Edmond et al., 2009; Conn et al., 2011). However, in-planta and heterologous expression of type 1A CAX proteins in yeast facilitate transport of multiple ions (Kamiya et al., 2005; Shigaki et al., 2005, 2010; Koren'kov et al., 2007; Mei et al., 2009). Hence, CAX family proteins are recently referred as H+/cation exchangers.
The NCL proteins are reported as a new member of CaCA superfamily (Wang et al., 2012), separated from CAX family due to the presence of long cytoplasmic loop with EF-hand domains (Emery et al., 2012; Pittman and Hirschi, 2016a). These are identified in numerous land plants, and AtNCL is reported to perform Na+/Ca2+exchange activity (Emery et al., 2012; Li et al., 2016). However, tight phylogenetic clustering in conserved α2-repeat tree established that these proteins are evolutionary more closer to CAX proteins than NCX proteins (Emery et al., 2012; Pittman and Hirschi, 2016a).
The CCX family of proteins were identified from diverse groups of organisms including protozoa, invertebrate, and vertebrate animals, fungi and plants (Cai and Lytton, 2004a; Emery et al., 2012; Pittman and Hirschi, 2016a). Functional characterization revealed Na+(Li+)/Ca2+ exchanger activity in a mammalian CCX protein, earlier named as NCKX6 (Palty et al., 2004; Cai and Lytton, 2004b). However, CCX3 of Arabidopsis showed H+/K+ exchanger activity and it could also transport Na+ and Mn2+ but not Ca2+ (Morris et al., 2008). Phylogenetic analysis classified CCX proteins from various organisms into three subgroups. These results indicated functional variation in CCX family of proteins (Emery et al., 2012; Pittman and Hirschi, 2016a). Normally 3-6 CCX proteins are reported in land plants with a maximum of 8 in Glycine max. These are also found highly conserved across the organisms especially in α1 and α2 -repeat regions (Emery et al., 2012).
The CaCA superfamily proteins are functionally characterized in several plant species, which established their basic role in cation transport and homeostasis. Modulated expression of these proteins have also been observed in various plants during numerous abiotic stresses, which indicated their role in stress response (Hirschi et al., 1996, 2000; Shaul et al., 1999; Kamiya et al., 2005; Morris et al., 2008; Wang et al., 2012; Singh et al., 2015; Li et al., 2016). Functional characterization of certain CaCA proteins like AtNCL, AtCAX1, OsCAX4, SsCAX1 and others further established their role in various kinds of abiotic stress response (Mei et al., 2007; Han et al., 2012; Wang et al., 2012; Yamada et al., 2014; Li et al., 2016; Pittman and Hirschi, 2016b; Zhang et al., 2016).
Despite being very important proteins, CaCA superfamily has not been identified and characterized in Triticum aestivum (bread wheat), a staple food crop of about one-third population of world. T. aestivum consist of an allohexaploid genome (2n = 6x = 42), evolved by hybridization events of three sub-genomes A, B, and D (Marcussen et al., 2014). The unavailability of genomic information, composite nature of genome and complexity in application of modern functional genomics tools could be the probable reason for the same. In recent years, the genome sequence and numerous development and stress related high throughput RNA sequence data have been available from T. aestivum (IWGSC, 2014; Zhang et al., 2014b, 2016; Liu Z. et al., 2015; Pingault et al., 2015), which enabled the genome wide characterization of various gene families (Shumayla et al., 2016a,b; Zeng et al., 2016). Since the CaCA proteins are involved in several vital functions from development to stress response, there characterization becomes essential in an important food crop like T. aestivum.
Herein, we have performed the genome wide identification and comprehensive characterization of CaCA proteins (TaCaCA) in T. aestivum genome. They were further characterized for gene and protein structural organization, and phylogenetic relationship. Furthermore, the expression analysis of individual TaCaCA genes was carried out during various tissues developmental stages, and in biotic and abiotic stress conditions as well. The present study provided inclusive information about TaCaCA superfamily proteins that would be very useful in future crop development programs.
Materials and Methods
Identification, Classification and Chromosomal Distribution
To identify the CaCA superfamily proteins (TaCaCA) in the genome of T. aestivum, BLASTp search (e-value 0.00001) of known CaCA proteins sequences from rice and Arabidopsis (Emery et al., 2012; Gaash et al., 2013; Singh et al., 2015; Pittman and Hirschi, 2016a) was performed against the protein model sequences of T. aestivum obtained from IWGSC server (Nussbaumer et al., 2013; IWGSC, 2014) (http://www.wheatgenome.org/, http://wheat-urgi.versailles.inra.fr/Seq-Repository/Genes-annotations). A total of 6 CAX, 5 CCX and 1 each MHX and NCL sequences from Arabidopsis, and 6 CAX, 5 CCX, 2 MHX, and 2 NCL sequences from rice were used in BLAST search as described earlier (Pittman and Hirschi, 2016a). The putatively identified CaCA protein sequences were also searched against the newly reported protein model sequences (TGACv1) of T. aestivum generated by The Genome Analysis Centre (TGAC), available at Ensembl Plants (http://plants.ensembl.org/Triticum_aestivum/Info/Index) and other available sequences at NCBI database. The FGENESH pipeline (http://www.softberry.com/) was used for the prediction of coding sequence (Solovyev et al., 2006). The hydropathy analysis of putatively identified sequences was performed using TMHMMv2.01 (Krogh et al., 2001; Petersen et al., 2011) to further confirm their identity as CaCA proteins. The presence of EF-hand (PfamID: PF00036) and Na_Ca_ex (PfamID: PF01699) domains were confirmed by BLAST search against local pfam database (ftp://ftp.sanger.ac.uk/pub/databases/Pfam) following the method established in our laboratory (Finn et al., 2014; Shumayla et al., 2016a,b). CaCA superfamily proteins were also identified in T. urartu (TuCaCA) and Aegilops tauschii (AeCaCA) genomes, which are reported as progenitors of A and D subgenome of T. aestivum, respectively (Marcussen et al., 2014). The identified CaCA superfamily proteins were further classified into CAX, CCX, MHX, and NCL protein groups based on their relative sequence homology with known sequences from Arabidopsis and rice.
The chromosomal localization of TaCaCA genes was obtained by BLASTn search against available chromosome sequences of T. aestivum at URGI and Ensembl Plants (https://urgi.versailles.inra.fr/blast/, http://plants.ensembl.org/Triticum_aestivum/).
Homeologs, Orthologs and Duplication Events Prediction
Bi-directional best hit approach at e-value 10−10 was used for the prediction of homeologous and orthologous genes as reported in earlier studies (Shumayla et al., 2016a,b). The identification of homeologous TaCaCA genes was based on their chromosome group, chromosome arm location and high percent (≥90%) identity. The identified homeologous genes were further confirmed by clustering them with T. aestivum unigene clusters (http://www.ncbi.nlm.nih.gov/UniGene). Further, the known pericentromeric inversion of chromosome 4AS-4AL (Liu et al., 1992) and chromosomal translocation 4AL-5AL (Hernandez et al., 2012) were also considered during homeolog identification. A unique name was assigned to each TaCaCA gene on the basis of their homeologous group and subgenome following the rules recommended for gene symbolization in T. aestivum (http://wheat.pw.usda.gov/ggpages/wgc/98/Intro.htm).
The orthologous proteins were identified by BLASTp search of TaCaCA protein sequences (e-value 10−10) against the protein model sequences of T. urartu, Ae. tauschii, rice and Arabidopsis downloaded from their respective databases (http://plants.ensembl.org, http://rice.plantbiology.msu.edu/, http://www.arabidopsis.org). Prediction of paralogous genes originated by duplication events were carried out by BLASTn search (at e-value 10−10) with high sequence similarity than the usual similarity between TaCaCA genes from different homeologous groups.
Phylogenetic Analysis
The phylogenetic analysis of TaCaCA superfamily proteins with various orthologs was performed using full-length, core domain and α2-repeat region sequences as reported in earlier studies (Emery et al., 2012; Pittman and Hirschi, 2016a). The core domain of individual proteins was obtained by removing the non-conserved N- and C-terminus portions, and central loop region as well. However, the conserved α2-repeat region in each protein was determined by aligning them with previously determined consensus sequence of CaCA proteins (Cai and Lytton, 2004a; Emery et al., 2012; Pittman and Hirschi, 2016a). The sequences were aligned using muscle program (Edgar, 2004) and phylogenetic tree was constructed by maximum likelihood method using MEGA7 with 1000 bootstrap replicates (Kumar et al., 2016).
Sequence Alignment and Exon/Intron Organization
The multiple sequence alignments were carried out by using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/), MAFFT (Katoh et al., 2002), muscle (Edgar, 2004), and Multalin (Corpet, 1988) programs. The genomic sequence of T. aestivum was downloaded from URGI (https://urgi.versailles.inra.fr/) and Ensembl Plants (http://plants.ensembl.org/Triticum_aestivum/) databases. The number of exons and introns in each TaCaCA gene were determined by aligning the coding sequences with genomic sequences. The number was further confirmed by blast search against T. aestivum genome dataset of IWGSC available at Ensembl Plants. The exon/ intron organization was analyzed using GSDS 2.0 (gsds.cbi.pku.edu.cn/) server (Hu et al., 2015).
Protein Characterization
The trans-membrane (TM) regions in individual TaCaCA protein was predicted using five different programs- TMHMM v2.0 (http://www.cbs.dtu.dk/services/TMHMM/), TMMOD, Phobius, SosuiG v.1.1 and DAS-Tmfilter (Hirokawa et al., 1998; Krogh et al., 2001; Cserzo et al., 2004; Käll et al., 2004; Kahsay et al., 2005; Petersen et al., 2011). The PROTTER v1.0 (http://wlab.ethz.ch/protter/start/) was also used for the prediction of TM topology (Omasits et al., 2014). The domain composition was established using Prosite-scan (http://prosite.expasy.org/scanprosite/), InterProScan (https://www.ebi.ac.uk/interpro/search/sequence-search), SMART (http://smart.embl-heidelberg.de/) and NCBI conserved domain blast (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) servers (Jones et al., 2014; Letunic et al., 2014; Marchler et al., 2015). The domain architecture map was prepared using IBS (http://ibs.biocuckoo.org/online.php) server (Liu W. et al., 2015). The NCBI blast server (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used for similarity analysis with other sequences in the database. The localization of the protein within the cell was predicted using pSORT (http://wolfpsort.org/), CELLO v.2.5 (http://cello.life.nctu.edu.tw/), TargetP_v1, PredSL, ngLOC and ProtComp (http://www.softberry.com/berry.phtml) servers (Emanuelsson et al., 2000; Petsalaki et al., 2006; Yu et al., 2006; Horton et al., 2007; King et al., 2012). Occurrence of signal peptide was recognized using SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/). The ExPasy compute pI/MW tool (http://web.expasy.org/compute_pi/) was used to compute the isoelectric point and molecular weight (Gasteiger et al., 2005).
Expression Analysis
The expression profile of TaCaCA genes was performed in various tissue developmental stages, and biotic and abiotic stress conditions using the high throughput RNA seq data. The tissue specific expression was analyzed using the data generated from three developmental stages of each root, leaf, stem, spike and grain in duplicates (Pingault et al., 2015). The data is available for download at https://urgi.versailles.inra.fr/files/RNASeqWheat/. The expression value was calculated as fragments per kilobase of transcript per million fragments mapped (FPKM) using RNA-Seq by Expectation-Maximization (RSEM) protocol from Trinity package (Li and Dewey, 2011; Haas et al., 2013). The tissue specific expression of individual gene was also confirmed at WheatExp server (http://wheat.pw.usda.gov/WheatExp/#) using the data (accession number: ERP004714) developed from similar tissues and developmental stages (Choulet et al., 2014).
The effect of biotic stress on the expression of TaCaCA genes was analyzed using the RNA seq data (accession number PRJNA243835) developed from leaves after 24 h of inoculation of fungus Blumeria graminis f. sp. tritici (Bgt) and Puccinia striiformis f. sp. tritici (Pst), separately (Zhang et al., 2014b). The RNA seq data was generated in triplicates and expression was calculated using Trinity package as described (Haas et al., 2013).
The effect of heat (HS), drought (DS), and combination of these stresses (HD) was analyzed using RNA seq data developed in duplicates (accession number SRP045409) from leaf samples after one and 6 h of incubation (Liu Z. et al., 2015). The effect of salt (NaCl) stress was studied using RNA seq data produced in three biological replicates (accession number SRP062745) from root tissues after 6, 12, 24, and 48 h of treatment (Zhang et al., 2016). The fold expression change of each TaCaCA gene under various stresses was also calculated in comparison to their respective controls using Trinity (Haas et al., 2013).
The genes having less than 10 FPKM expression value in each stage of an experiment were not included for evaluation or fold change calculations. The TaCaCA genes showing more than 2-fold up or down regulation were considered as differentially expressed. The Hierarchical Clustering Explorer 3.5 (http://www.cs.umd.edu/hcil/hce/) was used for the generation of heat-maps from various expression data.
Results and Discussion
Identification, Classification and Genome Wide Distribution
A total of 34 TaCaCA superfamily proteins were identified in T. aestivum genome by extensive BLAST search of known sequences from rice and Arabidopsis (File S1). These were further confirmed by hydropathy analysis for the presence of TM domains, and conserved α1 and α2-repeat regions by aligning them with known sequences as suggested in earlier studies (Emery et al., 2012; Pittman and Hirschi, 2016a). The identified TaCaCA proteins were classified into 14 TaCAX, 10 TaCCX, 7 TaNCL, and 3 TaMHX proteins on the basis of their close similarity and tight phylogenetic clustering with respective known proteins from various plants (Figure S1; File S1). In other higher plants, a total of 14 (6 AtCAX, 5 AtCCX, 2 AtNCL, and 1 AtMHX), 15 (6 OsCAX, 5 OsCCX, 2 OsNCL, and 2 OsMHX), and 14 (6 ZmCAX, 6 ZmCCX, 1 ZmNCL, and 1 ZmMHX) CaCA protein are reported in Arabidopsis, rice, and maize, respectively (Emery et al., 2012; Singh et al., 2015; Pittman and Hirschi, 2016a). It was earlier established that T. aestivum consisted of higher number of genes in each gene family (Shumayla et al., 2016a,b) due to their large allohexaploid (2n = 6x = 42; AABBDD) nature of genome (Marcussen et al., 2014). Therefore, it was also expected in TaCaCA superfamily. Occurrence of 27 CaCA proteins (14 CAX, 8 CCX, 4 NCL, and 1 MHX) in a tetraploid soybean further established that the polyploid plants may contain more number of CaCA proteins than the diploid plants (Schmutz et al., 2010; Emery et al., 2012).
Genome-wide analysis established that the TaCaCA genes were derived from each subgenomes of T. aestivum as reported in case of other gene families (Shumayla et al., 2016a,b; Zeng et al., 2016). A total of 10 (5 TaCAX, 2 TaCCX, 2 TaNCL, and 1 TaMHX), 10 (4 TaCAX, 3 TaCCX, 2 TaNCL, and 1 TaMHX) and 14 (5 TaCAX, 5 TaCCX, 3 TaNCL, and 1 TaMHX) genes were located on A, B, and D subgenomes, respectively (Figure 1A). Since the A and D sub-genomes are derived from T. urartu and Ae. tauschii (Marcussen et al., 2014), we analyzed the occurrence of CaCA family genes in their recently reported genomic sequences (Jia et al., 2013; Ling et al., 2013) following the similar procedure. A total of 7 (3 TuCAX, 1 TuCCX, 2 TuNCL, and 1 TuMHX) and 11 (4 AeCAX, 4 AeCCX, 2 AeNCL, and 1 AeMHX) CaCA genes were found on T. urartu and Ae. tauschii genome, respectively (File S1), which was quite lower than genes detected on A and D sub-genomes of T. aestivum. The results indicated occurrence of duplication events during evolution of TaCaCA genes after hybridization of various sub-genomes. The detail about duplication is discussed in later section.
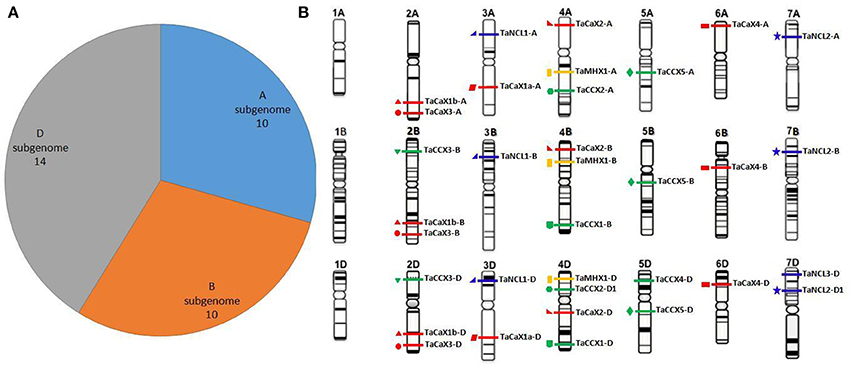
Figure 1. Distribution of the TaCaCA superfamily genes on A, B, and D subgenomes and chromosomes. Figure shows the frequency of TaCaCA genes on A, B, and D sub-genomes (A) and pictorial representation on various chromosomes (B). The figures of various chromosomes are adapted from the IWGSC website (www.wheatgenome.org). The TaCAX, TaCCX, TaNCL, and TaMHX genes are represented by red, green, blue and yellow bars, respectively. The homeologous genes in each family are shown by similar symbol.
The TaCaCA genes were distributed on each chromosome group of A, B, and D sub-genome, except chromosome group 1 (Figure 1B). A maximum of four genes were located on chromosome 4D. In total four (chromosomes 2B, 2D, 4A, and 4B), five (chromosomes 2A, 3A, 3D, 5D, and 7D) and eight (chromosomes 3B, 5A, 5B, 6A, 6B, 6D, 7A, and 7B) chromosomes comprised three, two and one genes, respectively. Further analysis indicated that the various families of CaCA superfamily were basically located on a particular group of chromosome. For instance, the TaCAXs were found on chromosome groups 2, 3, 4, and 6; TaCCXs on chromosome groups 2, 4, and 5; TaNCLs on chromosome groups 3 and 7; while TaMHXs were located on chromosome group 4. Occurrence of CaCA genes on various chromosomes has also been reported in Arabidopsis and rice, where these genes are distributed on four out of five, and eight out of twelve chromosomes, respectively. Five genes of Arabidopsis are located on chromosome I, while three genes are reported on each of chromosome II, III and XI of rice (Singh et al., 2015).
Prediction of Homeologs and Duplication Events
Due to the composite allohexaploid (AABBDD) nature of genome, each progenitor diploid subgenome has contributed in the composition of various gene families in T. aestivum (Shumayla et al., 2016a,b; Zeng et al., 2016). Majority of genes are derived from homeologous chromosome groups from A, B, and D subgenomes, and those are considered as homeologous genes in various studies. These genes mostly share high (≥90%) sequence homology. The homeologous relation of T. aestivum genes has also been considered in gene symbolization and nomenclature (http://wheat.pw.usda.gov/ggpages/wgc/98/Intro.htm). We had also identified the homeologous TaCaCA gene on the basis of similarity. A total of 14 distinct clusters (5 TaCAX, 5 TaCCX, 3 TaNCL, and 1 TaMHX) of homeologous genes were identified, majority of them consisted of at least one gene from each subgenome (File S2). However, a few homeologous genes were localized on either one or two subgenome. For instance, homeologous groups TaCCX1 and TaCCX3 were located on two (B and D) subgenomes, while TaCCX4 and TaNCL3 were found on only D subgenome (File S2). The number of distinct homeologous groups of TaCaCA genes was comparable to the total number of CaCA genes reported in diploid plant genomes like Arabidopsis and rice (Emery et al., 2012; Pittman and Hirschi, 2016a).
Gene duplication is earlier reported as a vital source for gene evolution (Magadum et al., 2013), which basically occurs due to retro-transpositions, imbalanced crossing over, and duplication of genome or chromosome. Occurrence of ~80% repeat sequences and various transposable elements in T. aestivum genome highly favors duplication as an important event during evolution (Marcussen et al., 2014; Glover et al., 2015). Several duplication events (DEs) are earlier predicted in various gene families of T. aestivum on the basis of high (≥80%) similarity (Shumayla et al., 2016a,b), but it was difficult in case of TaCaCA superfamily genes because of high divergence among various families of genes (File S3). The genes found on same subgenome and showed relatively higher similarity than the other genes were considered as putatively duplicated genes. Further, the information about CaCA genes was merely available from A (T. urartu) and D (Ae. tauschii) subgenome progenitors, therefore the DEs in TaCaCA genes were only predicted on these subgenomes.
The A subgenome of T. aestivum consisted of five TaCAX and two TaCCX genes, while their progenitor T. urartu had three TuCAX and one TuCCX. This indicated occurrence of at least two and one DEs during evolution of TaCAX and TaCCX genes, respectively. Probably, TaCAX3-D and TaCAX4-D were evolved by putative duplication of TaCAX2-D (File S3). In case of TaCCX, though both the genes (TaCCX2-A and TaCCX5-A) shared less than 50% sequence similarity, still these might be putative paralogous genes. The chromosomal translocation event between 4AL-5AL (Hernandez et al., 2012) also supports the same. Similarly, one DE was predicted in each TaCAX, TaCCX, and TaNCL gene family on D subgenome, because the progenitor Ae. tauschii was short of one gene in these families. The gene pairs TaCAX1a-D and TaCAX1b-D, TaCCX2-D and TaCCX4-D, TaNCL1-D and TaNCL3-D could be predicted as putative duplicated genes in D subgenome. Role of duplication events in evolvement of CaCA superfamily genes has also been earlier reported from both plant and animal species (On et al., 2008; Emery et al., 2012).
Phylogenetic Analysis
In earlier studies, usually three phylogenetic trees have been constructed using full length, core domain and α2-repeat motif sequences to get better separation of various members of CaCA superfamily proteins. It is due to the poor alignment of full length CaCA protein sequences especially in N- and C-terminus regions, variation in length of cytoplasmic loop and lack of α1-repeat motif in NCL proteins (Emery et al., 2012; Wang et al., 2012; Pittman and Hirschi, 2016a). Similar phylogenetic trees were constructed in the present study using CaCA protein sequences from various plant species. Four distinct clades of CAX, CCX, NCL, and MHX family proteins were found in each tree, which clearly depicted high homology within each family, and evident distinction between different families (Figure 2, Figure S1). Though, the NCL and MHX proteins formed distinct clades but showed tight phylogenetic relation with CAX and CCX proteins, respectively. Similar clustering is reported in earlier studies (Emery et al., 2012; Pittman and Hirschi, 2016a).
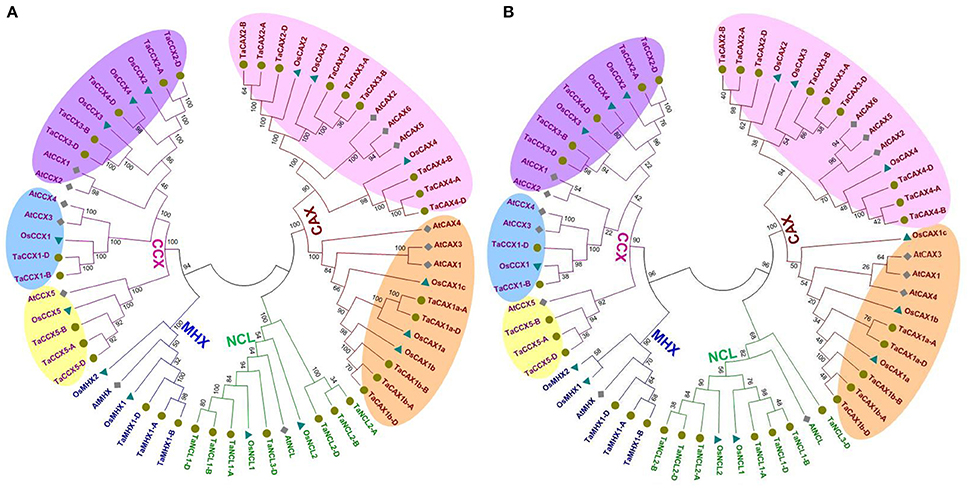
Figure 2. Phylogenetic analysis of the CaCA superfamily proteins. The evolutionary relationship between CaCA proteins of T. aestivum, Arabidopsis, and rice was analyzed using conserved core domain (A) and α2-repeat motif (B). Figure shows tight clustering of the CAX, CCX, NCL, and MHX family proteins from various plants, which are shown with red, purple, green, and blue color lines and fonts, respectively. The CAX and CCX family proteins are further classified into two (Type 1A and Type 1B) and three (group 1, 2, and 3) categories, respectively. Orange and pink shaded regions show Type 1A and Type 1B CAX proteins, while yellow, blue and purple shaded regions represent group 1, 2, and 3 CCX proteins, respectively.
The identified homeologous TaCaCA proteins from A, B, and D subgenomes of T. aestivum were closely clustered due to the high homology between them. The CaCA proteins identified from T. urartu and Ae. tauschii genomes were located in proximity to the related proteins from A and D subgenome of T. aestivum, respectively (Figure S1). It was probably due to their origin from respective subgenomes (Marcussen et al., 2014). For instance, TaNCL2-A and TaNCL2-D were proximally clustered with TuNCL2 and AeNCL2, respectively. Most of the predicted orthologous sequences from Arabidopsis and rice were clustered together with their respective TaCaCA proteins in various clades, such as CCX5 proteins from Arabidopsis, rice and T. aestivum genome were grouped together (Figure 2A). The evolutionary relation between orthologous CaCA proteins across the various plant species has also been observed earlier (Emery et al., 2012; Pittman and Hirschi, 2016a). Since the land plants are evolved from a common ancestor (McClung, 2010), they are found to be evolutionary related in the form of orthologous genes. The orthology of various other gene families of T. aestivum has also been earlier established with several land plants (Zeng et al., 2016; Shumayla et al., 2016a,b).
Since the CAX and CCX proteins are further classified into various groups on the basis of significant variation in their sequences and functional divergence (Cai and Lytton, 2004b; Palty et al., 2004; Shigaki et al., 2006; Morris et al., 2008; Pittman et al., 2009; Manohar et al., 2010; Emery et al., 2012), the phylogenetic trees developed from core domain and α2-repeat motif were also analyzed in terms of separation of those groups (Figures 2A,B). The OsCCX5 sequence did not contain α2-repeat motif, therefore it was not included in α2-repeat motif phylogeny. In plants, CAX proteins are basically divided into Type1A and Type1B groups, while CCXs comprised group 1, 2 and 3 (Emery et al., 2012). Type1A and Type1B CAX groups were clearly separated in both the trees, which indicated evolutionary divergence in TaCAX proteins, as earlier reported in other plants (Emery et al., 2012). In case of CCX, the phylogenetic tree developed from core domain sequences showed distinct separation of group 1, 2, and 3 proteins than the α2-repeat motif phylogeny (Figures 2A,B). For instance, AtCCX1 and AtCCX2 were clustered with group 3 CCX proteins in core domain phylogeny (Figure 2B) as reported in earlier study (Emery et al., 2012), while they were found closer to group 2 in α2-repeat motif phylogeny (Figure 2A). It is probably due to the conserved nature of this motif.
Gene and Protein Characterization
Several standard methods are reported for the characterization and comparison of different features of various genes in a gene family (Singh et al., 2015; Shumayla et al., 2016a,b; Zhou et al., 2016). The TaCaCA superfamily genes and proteins were analyzed in terms of various characteristic features like exon/intron organization, intron phase, length, molecular weight, pI, transmembrane, cytoplasmic loop size, cellular localization and domain architecture (Figure 3; Table 1; Files S4–S8). Out of 34 TaCaCA genes, 31 consisted of one or more introns, while three genes (TaCCX1-B, TaCCX2-A, and TaCCX4-D) were intron less. The average number of introns present in TaCAX, TaCCX, TaNCL, and TaMHX gene families were 10, 0.8, 5.5, and 7, respectively. A maximum of 12 introns were found in each TaCAX4-A and TaCAX4-D gene. The identified homeologus genes consisted of almost similar pattern of exon/ intron distribution (Figure 3A; Table 1; Files S4). The analysis of intronic phase indicated occurrence of 62, 23 and 14% introns in 0, 1, and 2 phase, respectively (Figure 3A). Similar distribution of these intron phases has been earlier reported in various organisms, which suggested the conserved nature of evolution of eukaryotic gene structure (Fedorov et al., 1992).
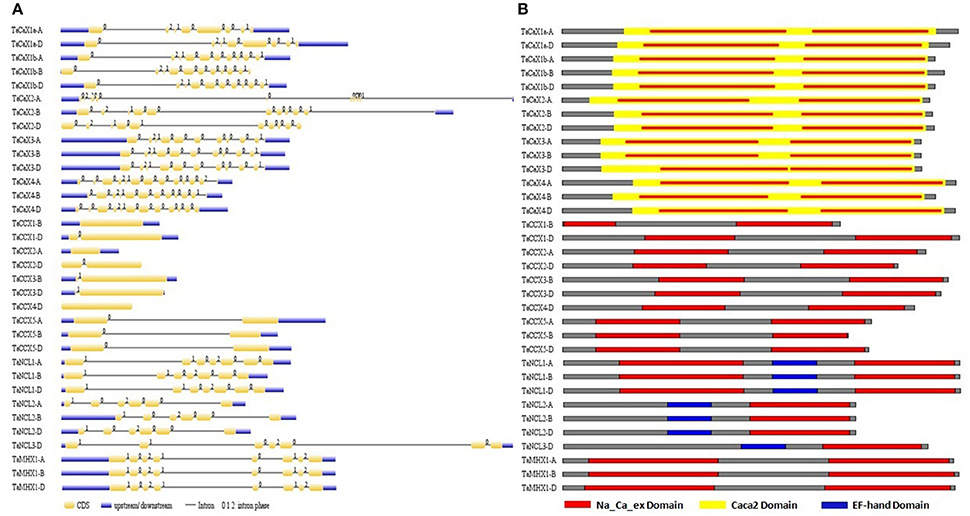
Figure 3. Gene and protein structure analysis. (A) Intron/exon configuration of TaCaCA superfamily genes. Exons and introns are shown as yellow boxes and black lines, respectively. Un-translated regions (UTRs) are shown as blue boxes. Various intron phases are represented as- 0; intron phase 0, 1; intron phase 1, 2; intron phase 2. (B) Figure shows domain architecture of TaCaCA proteins. The red, yellow and blue colored boxes represent Na_Ca_ex, Caca2 and EF-hand domains, respectively.
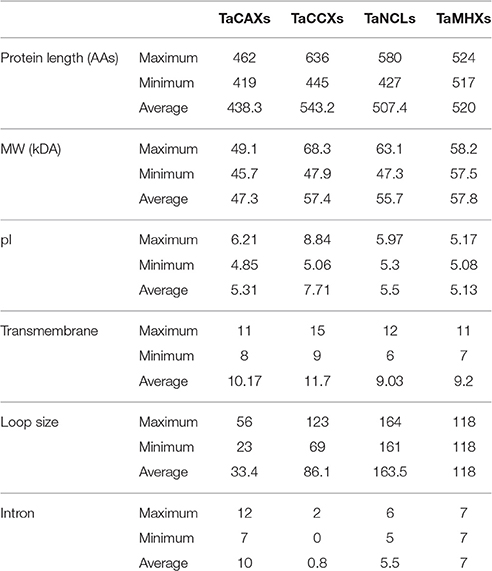
Table 1. General characteristic features of TaCaCA superfamily proteins.
The average length of TaCAX, TaCCX, TaNCL, and TaMHX family proteins were 438, 543, 507, and 520 AA residues, respectively. However, the average molecular weight (MW) and pI of these families were 47, 57, 55, and 57 kDa, and 5.3, 7.7, 5.5, and 5.1, respectively (Table 1; File S4). The TaCCX1-D and TaCAX3-A with MW 68.3 and 45.7 kDa were the biggest and smallest TaCaCA proteins. Variation in length and size of various CaCA superfamily proteins has also been observed in earlier studies. The AtCCX4 (AT1G54115; 645 AAs) and OsCCX1 (LOC_Os03g08230; 639 AAs) are the largest, while AtCCX5 (AT1G08960; 416 AAs) and OsCCX5 (LOC_Os11g05070; 265 AAs) are the smallest CaCA proteins reported from Arabidopsis and rice, respectively (Emery et al., 2012; Singh et al., 2015).
Since the CaCA proteins are membrane bound in nature (Cai and Lytton, 2004a; Emery et al., 2012), the occurrence of TM regions and sub-cellular localization was also predicted using various tools. Further, the number of TMs predicted by various tools was varied in majority of proteins as reported in earlier studies (Emery et al., 2012; Pittman and Hirschi, 2016a), therefore the average occurrence was calculated (File S5). The average number of TM regions found in TaCAX, TaCCX, TaNCL, and TaMHX family proteins was 10, 11, 9, and 9, respectively. However, the average TMs in total TaCaCA proteins was 10 (Table 1; File S5). Similar occurrence of TMs is reported in various other plants species (Emery et al., 2012), which supported the accuracy of our finding. As reported in other organisms (Cai and Lytton, 2004a,b; Emery et al., 2012), the TM regions were found in two groups in N- and C-terminus half of each TaCaCA protein, which were separated by a cytoplamic loop (CL) (Figures S2–S6). The average N- and C-terminus TMs ratio was 6:5, 5:6, 6:5, and 5:5 in TaCAX, TaCCX, TaNCL, and TaMHX family proteins, respectively. The length of CL also varied in various families due to the presence and absence of their respective domains in this region (File S6). The TaNCL proteins consisted of longest CL (~163 AAs), which is due to the presence of EF-hand domain (Figure S4, File S8). The average size of CL in TaCAX, TaCCX, and TaMHX proteins was ~33, ~86 and ~118 AAs, respectively. About similar length of CL is reported in other plant species (Emery et al., 2012).
The sub-cellular localization analysis indicated large discrepancy in prediction of various tools (File S7). The CELLO server predicted localization of each protein in plasma membrane, while ProtComp predicted most of them as vacuolar. However, other tools showed assorted results. The incongruity in prediction of these servers has also been reported in earlier studies (Vaid et al., 2012). This might be due to the differences in the algorithm of prediction of each tool. To validate the prediction, we analyzed the localization known proteins like AtCAX1, AtCAX2, and AtMHX, which are established as vacuolar proteins (Shaul et al., 1999; Hirschi et al., 2000; Kamiya et al., 2006). We found ProtComp prediction was quite reasonable, which predicted vacuolar localization for the majority of proteins except TaCCX5 homeologs. However, it could not provide localization of NCLs. The occurrence of CaCA proteins in plasma membrane and vacuole has been earlier reported (Wang et al., 1994, 2012; Bickerton and Pittman, 2015; Singh et al., 2015), but the actual localization of each TaCaCA superfamily protein need to be established.
The domain composition and their organization play vital role in determining the fundamental function of a protein. Numerous in-silico methods and databases like SMART (Letunic et al., 2014), Scan-Prosite, InterProScan (Jones et al., 2014), NCBI-Conserved Domain Database (Marchler et al., 2015) are available to analyse the domain architecture of proteins. These have been used for the domain analysis of a variety of proteins including CaCAs from various plants and animal species (Upadhyay et al., 2013, 2016; Singh et al., 2015; Shumayla et al., 2016a,b). Each TaCaCA protein consisted of one or more Na_Ca_ex domain (PF01699) (Figure 3B; File S8), as reported in case of other plants (Singh et al., 2015). All the TaCAX, TaCCX, TaMHX, and TaNCL1 group proteins comprised two Na_Ca_ex domains, one in each N- and C terminus half (Figures 4–5; File S8; Figures S2–S6). However, the TaNCL2 and TaNCL3 proteins consisted of a single Na_Ca_ex domain, which was located in C-terminus half. Additionally, TaCAXs and TaNCLs consisted of caca2 (TIGR00846) and EF-hand (PF00036) domain, respectively. The caca2 domain was basically located throughout the core domain of each TaCAX protein, whereas the Ef-hand domain was positioned in CL region of TaNCLs (Figure 5B; File S8). Since each CaCA superfamily protein consisted of Na_Ca_ex domain, Singh et al. (2015) proposed their nomenclature as NCX proteins instead of classifying them in several families. However, the various families of CaCA superfamily differ significantly in their structure, conserved motifs and function (Cai and Lytton, 2004a; Emery et al., 2012). Further, these formed distinct phylogenetic groups in each organism, which suggested that their classification in different families is more meaningful rather than naming all of them as NCX proteins (Pittman and Hirschi, 2016a).

Figure 4. Multiple sequence alignment of CCX proteins. Figure shows alignment of CCX protein sequences of T. aestivum, Arabidopsis and rice. The group 1, 2, and 3 CCX sequences are separated by green lines. Conserved α1 and α2-repeat regions are shown in black colored boxes. The signature motifs “GNG(A/S)PD” in α1-repeat and “G(N/D)SxGD” in α2-repeat motif reported by Cai and Lytton (2004a) are shown in purple boxes. The predicted transmembrane (TM) spans are over-lined in blue color.

Figure 5. Multiple sequence alignments of NCL and MHX proteins. (A) Figure shows alignment of EF-hand domain and α-2 repeat region of NCL proteins from T. aestivum, Arabidopsis, and rice. Pink asterisks indicate calcium binding sites in EF-hand domain. (B) Alignment of α-1 and α-2 repeat regions of human NCXs (HsNCX1; NP_066920, HsNCX2; NP_055878 and HsNCX3; NP_891977) with MHX proteins from T. aestivum, Arabidopsis and rice. The signature motifs “GSSAPE” in α1-repeat and “GTSVPD” α2-repeat of human NCX protein are shown in pink coloured boxes. Residues involved in Na+/Ca2+ exchange in HsNCX1 are indicated by asterisks, while the red asterisks indicates the residues which are not conserved in the MHX proteins.
Since the numerous conserved motifs and AAs play significant role in functional divergence of various families of CaCA proteins (Cai and Lytton, 2004a; Shigaki et al., 2006; Emery et al., 2012; Gaash et al., 2013), their occurrence and conservation was also analyzed in TaCaCAs by multiple sequence alignments with the known proteins. As reported in other plant species, all the TaCAX proteins consisted of both the α-repeat motifs and various other conserved AA residues. The signature motif “GNxxE” was also found conserved in α-repeats of TaCAX proteins, which was followed by a conserved H and A residue in the α2-repeat region (Figure S2). Both the acidic E residues are reported as essential for ion transport. The conserved H residue is also found important, which could not be exchanged with R or A residue (Kamiya and Maeshima, 2004). Furthermore, we observed “GGLLNAT” and SLLGS(I/V)LSN(L/M)LLV(L/V)G in α1-repeat, and “FIS(I/V)IL(L/I)PIV” in α2-repeat as highly conserved motif in TaCAX protein along with the CAXs of Arabidopsis and rice. Besides above motifs, numerous conserved G residues were also found, which may provide conformational flexibility to the CAX proteins as reported in earlier studies (Yan and Sun, 1997; Shigaki et al., 2006). The TaCAX proteins also formed two distinct groups, which could be easily marked as Type 1A and Type 1B on the basis of their similarity with the known proteins from other plant species (Figure S2). Although, the α-repeats were quite conserved, but both group of proteins differ significantly in AAs composition especially between TM-4 and TM-8, and beyond TM-9. The Type 1A CAX appeared to be more hydrophilic along with the numerous acidic residues like E and D. Similar observation is earlier reported in other plants (Shigaki et al., 2006; Emery et al., 2012), however, role of these conserved motifs and residues needs to be established in future studies.
Similar to the CCX proteins in other organisms (Cai and Lytton, 2004a; Emery et al., 2012), the TaCCXs also comprised two α-repeat motifs around TM2-TM3 and TM9-TM10 (Figure 4; Figure S3). The signature motifs “GNG(A/S)PD” in α1-repeat and “G(N/D)SxGD” in α2-repeat were too found conserved in TaCCX proteins except TaCCX2-D, which lacked former one. Further, motifs “A(G/A)VTLL” and “LGxTVLAW” were highly conserved in proximity to the signature motifs in α1 and α2-repeats, respectively. The TaCCX proteins could be classified into three groups on the basis of their sequence similarity with known CCX proteins of rice and Arabidopsis. Despite of high similarity in α-repeat regions, the CCX sequences from various groups showed significant divergence, which may also be responsible for functional diversification as reported in earlier studies (Cai and Lytton, 2004a; Palty et al., 2004; Morris et al., 2008; Emery et al., 2012). Moreover, functional characterization of each group of CCX protein needs to be individually established especially for their substrate specificity.
Despite of similar 6:5 topology of TM domains, the NCL proteins are separated from CAXs due to the high dissimilarity in their sequences (Emery et al., 2012). Sequence alignment showed significant variation between TaCAX and TaNCL family proteins (Figure S5). The NCL proteins including TaNCLs merely comprised α2-repeat motif and lacked α1-repeat (Figure 5A; Figure S4-S5). It has been earlier demonstrated that the occurrence of two α-repeats are necessary to form a functional protein (Nicoll et al., 2007). The separated N- and C-terminus portions of AtCAX1 could not perform transportation (Zhao et al., 2009), but the mutated NCKX6 with single α-repeat motif appeared functional after the oligomerization of protein (Palty et al., 2006). Moreover, AtNCL is established as functional protein, and it is suggested that certain critical AA residues like E or D from α1 and α2-repeats, S or T from extracellular Na+ binding site, and T, S and N from intracellular Na+ region are conserved in this protein (Wang et al., 2012). However, the detailed functional and structural characterization of individual NCL proteins is still required. The NCL proteins varied from CAXs in signature sequence of α2-repeat motif as well (Figure S5). In contrary to the NCX proteins (Nicoll et al., 1990, 1996a,b), the TaNCL proteins lacked long (~230 AAs) calcium binding domain (CBD) in CL regions (Figure S6), but consisted a small (~65 AAs) EF-hand domain with calcium binding motifs (CBMs) (Figure 5A). The CBMs mostly comprised acidic and/or negatively charged amino acid residues such D, E, S, and N, which suggested their electrostatic interaction with positively charged Ca2+ ions. The binding of Ca2+ ions persuade conformational shifting in EF-hand domain, which is responsible for activation and inactivation of the target proteins (Ikura, 1996). The CBMs of EF-hand domain were also found conserved in NCL proteins reported from other plant species (Emery et al., 2012).
The MHX proteins are recently reported to be evolved as new family within the CaCA superfamily (Gaash et al., 2013). These are exclusively reported to be present in plants, which are probably evolved from NCX proteins after separation of the streptophytes and chlorophytes (Yoon et al., 2004; Gaash et al., 2013). Similar to the NCX proteins, MHXs also consisted of two α-repeats, but differed in signature motifs and lacked long cytoplasmic Ca2+ binding domain (Figure 5B, Figure S7). The MHX proteins comprised signature motifs “GTSFPQ” and “GTSWPD” in place of “GSSAPE” and “GTSVPD” in NCX at α1 and α2-repeats, respectively. Some of the key residues like E and D in α1-repeat, and D and F in α2-repeats are substituted with other AA residues in the MHXs (Emery et al., 2012; Gaash et al., 2013). Similar changes had been marked in case of TaMHXs. These residues play vital role in Ca2+ ion transport in NCX (Nicoll et al., 1996a). Mutation in these regions substantially reduced their affinity (Iwamoto et al., 2000), which explained the lack of Na+/Ca2+ exchange activity by MHX proteins. Moreover, the MHX transporters also lacked the Ca2+ binding domain in central loop region (Lytton, 2007). Certain other motifs like “ASKIA,” “TADSAI” and “ELGGP” reported as conserved in plant MHXs (Gaash et al., 2013) could also be found in TaMHXs (Figure S7). Since the AA residues in α-repeat regions play important role in ion selectivity (Shigekawa et al., 2002; Ottolia et al., 2005), sequence variation in these might be responsible for diversification in ion selectivity of various CaCA proteins. AtMHX is earlier reported to be involved in vacuolar transportation of Mg2+, Zn2+, Cd2+, and probably Fe2+ against protons (Shaul et al., 1999; Berezin et al., 2008). Since the plant MHXs share comparable structural organization with significant conservation, they may also be involved in analogous functions, but that needs to be established in individual proteins.
Expression Analysis in Tissue Developmental Stages
Calcium ion is an essential secondary messenger in plants which play vital roles in various developmental, biochemical, and physiological processes. The expression analysis is a method to predict the putative function of an unknown gene (Zeng et al., 2016). Further, CaCA superfamily genes are also reported to play vital role in plant development (Shaul et al., 1999; Cheng et al., 2005; Wang et al., 2012; Pittman and Hirschi, 2016b). Therefore, their expression was analyzed in various developmental stages of T. aestivum using high throughput transcriptome data (Pingault et al., 2015). Each TaCaCA gene showed significant but varied expression in different developmental stages of all the analyzed tissues. Expression of CAX genes was usually high in each developmental stage as compared to the other genes. Likewise high expression of CAX genes in different tissues has also been reported in Arabidopsis and rice (Cheng et al., 2002, 2005; Pittman et al., 2004; Kamiya et al., 2005, 2006, 2012; Mei et al., 2009; Wang et al., 2012; Bickerton and Pittman, 2015; Singh et al., 2015). The TaCAX1a-A, TaCAX1a-D, TaCAX1b-A, TaCAX1b-B, and TaCAX1b-D in CAX family, TaCCX1-B, TaCCX1-D, TaCCX2-A and TaCCX2-D in CCX family, TaNCL1-A, TaNCL1-B, and TaNCL1-D in NCL family, and TaMHX1-D in MHX family were relatively high expressing genes in one or more developmental stages (Figure 6; File S9). The majority of TaCAX genes were highly expressed in grain except TaCAX3 group genes, which were highly expressed in root and stem (Figure 6A). The high expression of CAX genes is also reported in seed of rice and Arabidopsis (Goel et al., 2012; Bickerton and Pittman, 2015).
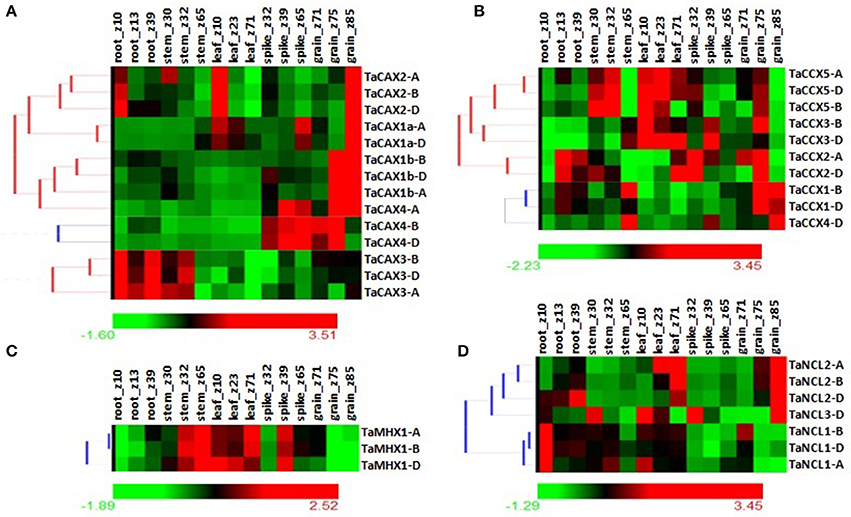
Figure 6. Relative expression profile of TaCaCA superfamily genes in various tissue developmental stages. Heat map shows relative expression profile of (A) TaCAX, (B) TaCCX, (C) TaMHX, and (D) TaNCL genes in three developmental stages of each root, leaf, stem, spike, and grain tissue. The developmental stages are shown in Zadoks scale.
In case of CCX family genes, TaCCX1 and TaCCX4 were highly expressed in grain, TaCCX5 in leaf and stem, TaCCX3 in leaf and grain, while TaCCX2 in root, spike and grain (Figure 6B). The orthologous CCX genes from rice and Arabidopsis also showed comparable expression pattern (Morris et al., 2008; Singh et al., 2015). The TaMHX genes were highly expressed in stem, leaf and spike up to certain extent (Figure 6C) as observed in case of other plants (Singh et al., 2015). Since MHX transporter is usually involved in Mg2+ transportation (Shaul et al., 1999), which plays vital role in chlorophyll synthesis, this might be the reason for their high expression in green tissues. Furthermore, we could not observe significant expression of TaMHX in grain and root developmental stages. The over expression of AtMHX caused reduced growth in transgenic tobacco and Arabidopsis plants, which indicated their role in development (Berezin et al., 2008). An interesting expression pattern of TaNCL genes was observed. The TaNCL1 group genes were high expressing in root, while TaNCL2 in grain and leaf. TaNCL3 showed assorted expression in various tissues (Figure 6D). Comparable expression of these genes is reported in other plants also (Singh et al., 2015). The variable expression of TaCaCA genes in different tissue developmental stages indicated their specific role during development. However, constitutively high expression of certain genes like TaCAX1-A and TaNCL1-B suggested their role throughout the various tissue developments.
Expression Analysis in Biotic and Abiotic Stresses
Various abiotic and biotic stresses affect numerous biochemical and physiological pathways in plants and ultimately responsible for yield loss (Munns and Tester, 2008; Prasad et al., 2011; Pradhan et al., 2012; Izadi et al., 2014). The calcium signaling has been reported as an important mechanism for these kinds of stress sensing in plants (Tuteja and Sopory, 2008; Kader and Lindberg, 2010; Zhang et al., 2014a). Modulation in expression of CaCA superfamily genes such as CAXs and NCL has been earlier reported during abiotic stresses in various plant species (Wang et al., 2012; Li et al., 2016; Pittman and Hirschi, 2016b). Therefore, it becomes necessary to study the impact of these stresses on expression pattern of TaCaCA genes. In the present study, we analyzed the effect of fungal pathogens (Blumeria graminis; Bgt, and Puccinia striiformis; Pst) under biotic stress, and heat, drought and salt under abiotic stress (Figure 7; Files S10–S12).
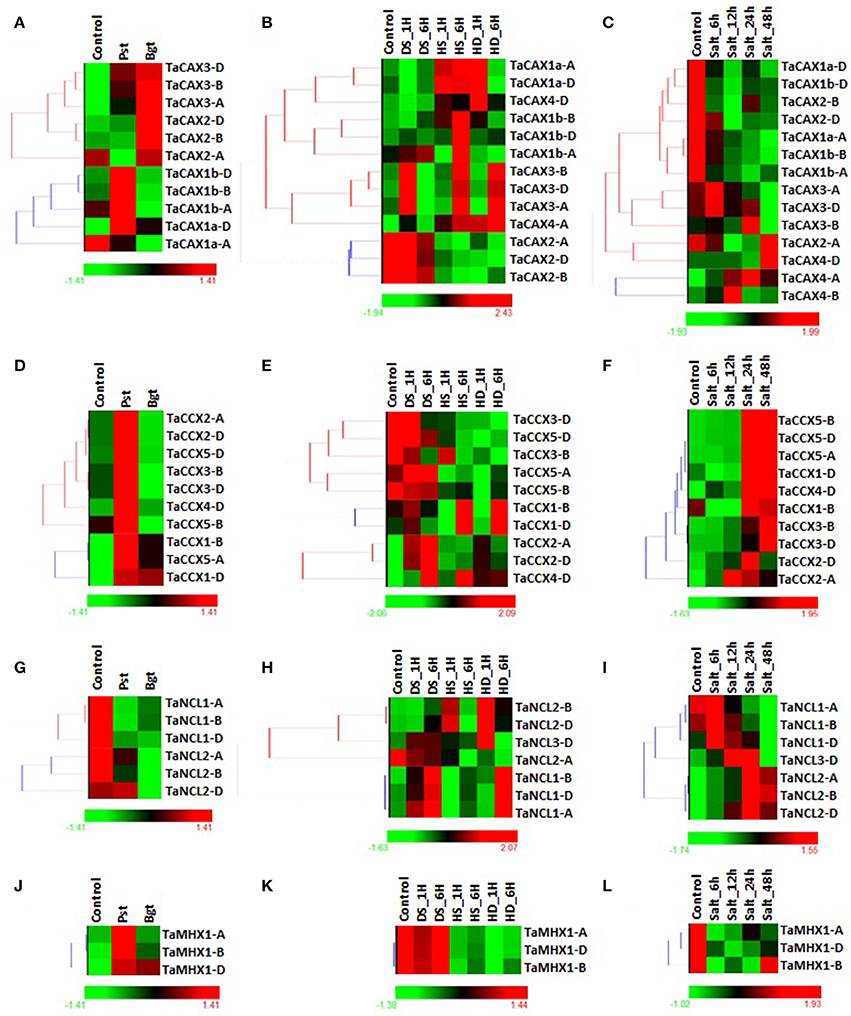
Figure 7. Differential expression analysis of TaCaCA superfamily genes under biotic and abiotic stresses. The heat map shows differential expression pattern of (1) TaCAX genes under (A) biotic, (B) heat/drought, and (C) salt stress, (2) TaCCX genes under (D) biotic, (E) heat/drought and (F) salt stress (F), (3) TaNCL genes under (G) biotic, (H) heat/drought and (I) salt stress, and (4) TaMHX genes under (J) biotic, (K) heat/drought stress and (L) salt stress. The symbols shown in figure indicate-HS; heat stress, DS; drought stress, HD; combination of heat and drought stress, Bgt; after Blumeria graminis infection, Pst; after Puccinia striiformis inoculation.
The expression of TaCAX genes were not much affected after fungal infestation. Only TaCAX2 group genes were slightly (~2–3-folds) up-regulated (Figure 7A; File S10). The results indicated that the CAX genes are probably not much involved in fungal stress response. The TaCAX4 group genes were found highlyS up-regulated during heat, drought and combination of these stresses, while TaCAX1b-B and TaCAX1b-D were up-regulated solely during heat stress (Figure 7B; File S11). Comparable expression of orthologous OsCAX4 (LOC_Os02g04630) and OsCAX1b (LOC_Os05g51610) genes of rice, and AtCAX5 (AT1G55730) and AtCAX2 (AT3G13320) genes of Arabidopsis has been observed during similar stresses (Singh et al., 2015). During salt stress, TaCAX1a-A, TaCAX1a-D, TaCAX1b-A, and TaCAX1b-D were highly down-regulated, while TaCAX4 group genes were up-regulated (Figure 7C; File S12). Modulated expression of numerous CAX genes has been reported in various plants under salt stress (Shigaki and Hirschi, 2006; Mei et al., 2007; Manohar et al., 2011; Han et al., 2012; Yamada et al., 2014). The expression pattern and role of CAX genes during different stresses has been recently listed from several plant species (Bickerton and Pittman, 2015), however the specific function of individual TaCAX genes is required to be established.
A total of six TaCCX genes showed differential expression during biotic stress. Majority of them were 2–3-folds up-regulated after Pst infestation, while two (TaCCX2-A and TaCCX3-D) were down regulated after Bgt infestation (Figure 7D; File S10). Likewise, the expression of orthologous CCX genes of Arabidopsis (AtCCX1; AT5G17860, AtCCX2; AT5G17850) are found up-regulated after wounding (Singh et al., 2015), which is usually considered as similar to pathogen infection. Heat, drought and their combination stresses modulated the expression of each TaCCX gene (Figure 7E; File S11), which indicated their role in these conditions. The TaCCX2 and TaCCX4 group genes were 2–18-folds up-regulated in different conditions. The TaCCX4-D was highly up-regulated in each stress. The TaCCX1, TaCCX3 and TaCCX5 group genes were 2–17-folds down-regulated in one or more stress condition. The TaCCX3-D was extremely down-regulated gene during all the stresses. Modulation of CCX genes expression during heat and drought stress is also reported in other plant species. For instance, AtCCX1 (AT5G17860) and AtCCX2 (AT5G17850) of Arabidopsis are up-regulated in similar stresses, while OsCCX4 (LOC_Os12g42910) is up-regulated in dehydration and down-regulated in heat stress (Singh et al., 2015). During exposure to the high salt conditions, majority of the TaCCX genes were 3–110-folds up-regulated. The TaCCX2-D (9–58-folds) and TaCCX3-D (11–110-folds) were highly affected genes at various stages of treatment (Figure 7F; File S12). The results indicated that the TaCCX genes were more affected during various abiotic stresses as compared to the other TaCaCA superfamily genes. The CCX genes are also found up-regulated during salt stress in rice and Arabidopsis (Morris et al., 2008; Singh et al., 2015).
Expression of TaNCL genes were also affected during various biotic and abiotic stresses as reported in other plant species (Wang et al., 2012; Singh et al., 2015). The TaNCL2 group genes were found 2–8-folds down-regulated during both Pst and Bgt infestation, while TaNCL1 and TaNCL3 were not much affected (Figure 7G; File S10). TaNCL2-A and TaNCL2-B were highly (~8-folds) down-regulated during Pst and Bgt invasion, respectively. Variation in expression of AtNCL (AT1G53210) is also observed during wounding (Singh et al., 2015), which indicated role of NCL genes in pathogen response. Both TaNCL1 and TaNCL2 group genes showed assorted expression during heat, drought and their combination stresses (Figure 7H; File S11). Such as TaNCL1 group genes were up-regulated during drought, while down-regulated during heat stress. The TaNCL2-A was down-regulated in most of the treatment, while TaNCL2-B and TaNCL2-D were found up-regulated. These results indicated functional divergence in TaNCL genes, despite of their similar structural organization. Moreover, the AtNCL (AT1G53210) and OsNCL1 (LOC_Os01g11414) genes were up-regulated during heat stress in Arabidopsis and rice, respectively (Singh et al., 2015). During salt stress, TaNCL2 group genes were (2–17-folds) up-regulated during all the treatments, in which TaNCL2-B was highly up-regulated in most of the treatments (Figure 7I; File S12). TaNCL3-D showed slight up-regulation till 24 h of treatment, while down-regulated at 48 h. Both TaNCL1-A and TaNCL1-B were ~3-folds down-regulated at 48 h of treatment. Increased expression of AtNCL and OsNCL1 genes has also been reported in earlier studies (Wang et al., 2012; Singh et al., 2015).
The expression of TaMHX genes was not significantly affected during biotic stress; but these were 2–4-folds down-regulated during heat, drought and salt stresses (Figure 7J–L; Files S10–S12). However, the up-regulation of orthologous OsMHX (LOC_Os11g43860) and AtMHX (AT2G47600) genes is reported during similar stress conditions (Singh et al., 2015). The results indicated that the similar genes from different plants might behave differently during comparable stress conditions.
Conclusions
Transporter proteins are an integral part of an organism to exchange the various mineral or metal ions. The Ca2+ ions play vital role in numerous development and defense related mechanisms in various plant species. The CaCA superfamily proteins are a kind of transporters involved in exchange of various metal ions including Ca2+ ion homeostasis. In the present study, we identified 34 TaCaCA superfamily proteins in T. aestivum, which were further classified into TaCAX (14 genes), TaCCX (10 genes), TaNCL (7 genes), and TaMHX (3 genes) families on the basis of their structural organization and phylogenetic relation with known proteins from other plant species. Similar to the other gene families in T. aestivum, the TaCaCA genes were also derived from each A, B, and D sub-genome and chromosome. The predicted homeologous genes showed similar gene and protein structure organization in each TaCaCA family. The hydropathy analysis indicated the occurrence of 10, 11, 9, and 9 TMs in TaCAX, TaCCX, TaNCL, and TaMHX proteins, respectively. Most of the TaCaCA proteins consisted of two Na_Ca_ex domains except TaNCL2 and TaNCL3 group proteins, which had single Na_Ca_ex domain. The TaNCL proteins comprised an additional EF-hand domain in CL region with conserved calcium binding motifs. As reported in other organisms, two α-repeats with specifically conserved signature motifs were also detected in each family of TaCaCA superfamily except TaNCL, which contained single α-repeat. The TaCAXs and TaCCXs were further classified into two (Type 1A and Type 1B) and three (group 1, 2, and 3) groups on the basis of similarity with the earlier known proteins, respectively. The significant expression of each TaCaCA gene during various developmental stages suggested their role in growth and development. The TaCAX1a and TaCAX1b group genes were specifically highly expressed in grain, while TaNCL1 in root developmental stages. Modulated expression of certain TaCaCA genes during various biotic and abiotic stresses revealed their role in stress response. The TaCCX and TaNCL family genes were found more responsive than TaCAXs and TaMHXs during various abiotic stresses. The present study enlightened various characteristic features of CaCA superfamily proteins in T. aestivum, however the function of individual gene needs to be investigated using functional genomics tools in future studies.
Author Contributions
SU conceived the idea and designed the experiments. MT, ST, and SS performed the experiments. MT and SU analyzed the data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are thankful to Panjab University, Chandigarh, India and National Agri-Food Biotechnology Institute, Mohali, India for research facilities. We are grateful to Department of Science and Technology, Government of India for financial support under Promotion of University Research and Scientific Excellence (PURSE) grant scheme.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01775/full#supplementary-material
Figure S1. Phylogenetic clustering of putatively identified CaCA superfamily proteins. The tree was constructed using full length sequences of putatively identified CaCA proteins from T. aestivum, T. urartu, and Aegilops tauschii with the reported sequences from rice and arabidopsis. The CAX, CCX, NCL, and MHX family proteins are shown with red, purple, green, and blue color lines and fonts, respectively. Tight phylogenetic clustering confirmed the identity of isolated CaCA proteins from these plants.
Figure S2. Multiple sequence alignment of CAX proteins. The amino acid sequences of CAX proteins from T. aestivum, Arabidopsis thaliana, and Oryza sativa are aligned. Type 1A and Type 1B group of CAX proteins are separated by green line. The α1 and α2-repeat regions are shown in black boxes. The reported signature motif (GNxxE) in α-repeat regions (Kamiya and Maeshima, 2004; Cai and Lytton, 2004a) is indicated by green shaded region. The predicted transmembrane (TM1 to TM11) spans are over-lined in blue. The other conserved residues reported in CaCA superfamily proteins (Cai and Lytton, 2004a) are indicated by asterisk. Hyphens indicate gaps introduced to maximize the alignment. The identified Na_Ca_ex domains are shown as red shaded regions in both N- and C-terminus half of CAX proteins.
Figure S3. Multiple sequence alignment of CCX proteins. The amino acid sequences of CCX proteins from T. aestivum, A. thaliana, and O. sativa are aligned. The group 1, 2, and 3 CCX sequences are separated by green line. Conserved α1 and α2-repeat regions are shown in black colored boxes. The signature motifs “GNG(A/S)PD” in α1-repeat and “G(N/D)SxGD” in α2-repeat motifs (Cai and Lytton, 2004a) are shown by green shaded region. The predicted transmembrane (TM) spans (TM1 to TM11) are over-lined in blue color. The identified Na_Ca_ex domains are shown as red shaded regions in both N- and C-terminus half of CCX proteins.
Figure S4. Multiple sequence alignment of NCL proteins. The amino acid sequences of NCL proteins from T. aestivum, A. thaliana, and O. sativa are aligned. The α-2 repeat region and EF-hand domain is shown in black and red boxes, respectively. Calcium binding sites within EF-hand domain are indicated by pink asterisks. The Na_Ca_ex domains are shaded red. The predicted transmembrane spans (TM1 to TM11) are over-lined in blue.
Figure S5. Multiple sequence alignment of CAX and NCL proteins. The amino acid sequences of CAX and NCL proteins from T. aestivum, A. thaliana, and O. sativa are aligned together. The α-1 and α-2 repeat regions of are boxed in black while the signature motif (GNxxE) in dark red. The EF-hand domain is shown green box. Figure shows that NCL proteins lack α-1 repeat region and signature motif of CAX proteins.
Figure S6. Multiple sequence alignment of NCL and NCX proteins. The amino acid sequences of T. aestivum NCL proteins and human NCX proteins are aligned. The calcium binding domain of HsNCX proteins is over-lined in yellow, EF-hand domain of TaNCLs is red under-lined. The calcium binding sites in EF-hand domain is shown by red asterisks. Figure shows large variation between long (~230 AAs) calcium binding domain of HsNCXs and short (~65 AAs) EF-hand domain of TaNCL proteins.
Figure S7. Multiple sequence alignment of MHX and NCX proteins. The amino acid sequences of MHX proteins from T. aestivum, A. thaliana, and O. sativa and NCX proteins from human are aligned. The α-1 and α-2 repeat regions are boxed in black. Te predicted transmembrane (TM1 to TM10) spans are over-lined in blue, while Calcium binding domain HsNCX is yellow over-lined. Residues involved in Na+/Ca2+ exchange in HsNCX1 are indicated by asterisks, while the red asterisks indicates the residues which are not conserved in the MHX proteins. The Na_Ca_ex domains are shown as red shaded regions in both N- and C-terminus half of proteins.
File S1. List of protein sequences identified as putative CaCA proteins from various databases. Green highlighted sequences showed comparable size and hydropathy to the reported sequences, therefore these were used during further characterization.
File S2. Clustering of TaCaCA superfamily genes into homeologous groups. The genes derived from homeologous chromosomes and showed ≥90% similarity are considered as homeologous genes.
File S3. Similarity matrix between various families of TaCaCA superfamily genes obtained by bi-directional best blast hit approach. The genes highlighted with same color are predicted as putatively duplicated genes in each gene family due to their localization on same subgenome and relatively higher similarity than other genes.
File S4. Analysis of chromosomal localization, gene structure and various other characteristic features of TaCaCA superfamily genes and proteins.
File S5. Number of transmembrane domains predicted by various tools in TaCaCA superfamily proteins.
File S6. Cytoplasmic loop sizes of various TaCaCA superfamily proteins.
File S7. Prediction of sub-cellular localization of TaCaCA superfamily proteins using various tools.
File S8. Domain organization in TaCaCA superfamily proteins. Each TaCaCA protein was individually search against Pfam, NCBI-conserved domain database, SMART, Prosite-Scan and InterProScan servers to analyse the domain architecture.
File S9. Expression analysis of TaCaCA genes in three developmental stages of various tissues. The top expressing genes from various families are highlighted in red boxes.
File S10. Fold expression change in TaCaCA superfamily genes during biotic stress of Blumeria graminis (Bgt) and Puccinia striiformis (Pst). Two-folds or more up and down regulated genes are shown in yellow and red color cells, respectively.
File S11. Fold expression change in TaCaCA superfamily genes during drought (DS), heat (HS) and their combination (HD) stress. Two-folds or more up and down regulated genes are shown in yellow and red color cells, respectively.
File S12. Fold expression change in TaCaCA superfamily genes during salt stress. Two-folds or more up and down regulated genes are shown in yellow and red color cells, respectively.
References
Axelsen, K. B., and Palmgren, M. G. (2001). Inventory of the superfamily of P-Type ion pumps in Arabidopsis. Plant Physiol. 126, 696–706. doi: 10.1104/pp.126.2.696
Berezin, I., Mizrachy-Dagry, T., Brook, E., Mizrahi, K., Elazar, M., Zhuo, S., et al. (2008). Overexpression of AtMHX in tobacco causes increased sensitivity to Mg2+, Zn2+, and Cd2+ ions, induction of V-ATPase expression, and a reduction in plant size. Plant Cell Rep. 27, 939–949. doi: 10.1007/s00299-007-0502-9
Bickerton, P. D., and Pittman, J. K. (2015). “Role of cation/proton exchangers in abiotic stress signaling and stress tolerance in plants,” in Elucidation of Abiotic Stress Signaling in Plants: Functional Genomics Perspectives, Vol. 1, ed K. G. Pandey (New York, NY: Springer), 95–117.
Cai, X. J., and Lytton, J. (2004a). The cation/Ca2+ exchanger superfamily: phylogenetic analysis and structural implications. Mol. Biol. Evol. 21, 1692–1703. doi: 10.1093/molbev/msh177
Cai, X. J., and Lytton, J. (2004b). Molecular cloning of a sixth member of the K+-dependent Na+/Ca2+ exchanger gene family, NCKX6. J. Biol. Chem. 279, 5867–5876. doi: 10.1074/jbc.M310908200
Case, R. M., Eisner, D., Gurney, A., Jones, O., Muallem, S., and Verkhratsky, A. (2007). Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signaling system. Cell Calcium 42, 345–350. doi: 10.1016/j.ceca.2007.05.001
Cheng, N. H., Pittman, J. K., Shigaki, T., and Hirschi, K. D. (2002). Characterization of CAX4, an Arabidopsis H+/Cation antiporter. Plant Physiol. 128, 1245–1254. doi: 10.1104/pp.010857
Cheng, N. H., Pittman, J. K., Shigaki, T., Lachmansingh, J., LeClere, S., Lahner, B., et al. (2005). Functional association of Arabidopsis CAX1 and CAX3 is required for normal growth and ion homeostasis. Plant Physiol. 138, 2048–2060. doi: 10.1104/pp.105.061218
Choulet, F., Alberti, A., Theil, S., Glover, N., Barbe, V., Daron, J., et al. (2014). Structural and functional partitioning of bread wheat chromosome 3B. Science 345:1249721. doi: 10.1126/science.1249721
Conn, S. J., Gilliham, M., Athman, A., Schreiber, A. W., Baumann, U., Moller, I., et al. (2011). Cell-specific vacuolar calcium storage mediated by CAX1 regulates apoplastic calcium concentration, gas exchange, and plant productivity in Arabidopsis. Plant Cell 23, 240–257. doi: 10.1105/tpc.109.072769
Corpet, F. (1988). Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881–10890. doi: 10.1093/nar/16.22.10881
Cserzo, M., Eisenhaber, F., Eisenhaber, B., and Simon, I. (2004). TM or not TM: transmembrane protein prediction with low false positive rate using DAS-TM filter. Bioinformatics 20, 136–137. doi: 10.1093/bioinformatics/btg394
Dodd, A. N., Kudla, J., and Sanders, D. (2010). The language of calcium signaling. Annu. Rev. Plant Biol. 61, 593–620. doi: 10.1146/annurev-arplant-070109-104628
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edmond, C., Shigaki, T., Ewert, S., Nelson, M. D., Connorton, J. M., Chalova, V., et al. (2009). Comparative analysis of CAX2-like cation transporters indicates functional and regulatory diversity. Biochem. J. 418, 145–154. doi: 10.1042/BJ20081814
Emanuelsson, O., Nielsen, H., Brunak, S., and von Heijne, G. (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016. doi: 10.1006/jmbi.2000.3903
Emery, L., Whelan, S., Hirschi, K. D., and Pittman, J. K. (2012). Protein phylogenetic analysis of Ca2+/CA antiporters and insights into their evolution in plants. Front. Plant Sci. 3:1. doi: 10.3389/fpls.2012.00001
Fedorov, A., Suboch, G., Bujakov, M., and Fedorova, L. (1992). Analysis of nonuniformity in intron phase distribution. Nucleic Acids Res. 20, 2553–2557. doi: 10.1093/nar/20.10.2553
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Gaash, R., Elazar, M., Mizrahi, K., Avramov-Mor, M., Berezin, I., and Shaul, O. (2013). Phylogeny and a structural model of plant MHX transporters. BMC Plant Biol. 13:75. doi: 10.1186/1471-2229-13-75
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). Protein identification and analysis tools on the expasy server. Proteomics Protoc. Handb. 112, 571–607. doi: 10.1385/1-59259-890-0:571
Glover, N. M., Daron, J., Pingault, L., Vandepoele, K., Paux, E., Feuillet, C., et al. (2015). Small-scale gene duplications played a major role in the recent evolution of wheat chromosome 3B. Genome Biol. 16, 188. doi: 10.1186/s13059-015-0754-6
Goel, A., Gaur, V. S., Arora, S., Gupta, S., and Kumar, A. (2012). In silico analysis of expression data for identification of genes involved in spatial accumulation of calcium in developing seeds of rice. OMICS 16, 402–413. doi: 10.1089/omi.2012.0004
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Han, N., Lan, W. J., He, X., Shao, Q., Wang, B. S., and Zhao, X. J. (2012). Expression of a Suaeda salsavacuolar H+/Ca2+ transporter gene in Arabidopsis contributes to physiological changes in salinity. Plant Mol. Biol. Report. 30, 470–477. doi: 10.1007/s11105-011-0353-y
Haug-Collet, K., Pearson, B., Webel, B., Szerencsei, R. T., Winkfein, R. J., Schnetkamp, P. P. M., et al. (1999). Cloning and characterization of a potassium-dependent sodium/calcium exchanger in Drosophila. J. Cell Biol. 147, 659–669. doi: 10.1083/jcb.147.3.659
Hernandez, P., Martis, M., Dorado, G., Pfeifer, M., Gálvez, S., et al. (2012). Next-generation sequencing and syntenic integration of flow-sorted arms of wheat chromosome 4A exposes the chromosome structure and gene content. Plant J. 69, 377–386. doi: 10.1111/j.1365-313X.2011.04808.x
Hirokawa, T., S., Boon-Chieng, S., and Mitaku, S. (1998). SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14:378. doi: 10.1093/bioinformatics/14.4.378
Hirschi, K. D. (2004). The calcium conundrum. Both versatile nutrient and specific signal. Plant Physiol. 136, 2438–2442. doi: 10.1104/pp.104.046490
Hirschi, K. D., Korenkov, V. D., Wilganowski, N. L., and Wagner, G. J. (2000). Expression of Arabidopsis CAX2 in tobacco. Altered metal accumulation and increased manganese tolerance. Plant Physiol. 124, 125–133. doi: 10.1104/pp.124.1.125
Hirschi, K. D., Zhen, R. G., Cun-ningham, K. W., Rea, P. A., and Fink, G. R. (1996). CAX1, an H+/Ca2+ antiporter from Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 93, 8782–8786. doi: 10.1073/pnas.93.16.8782
Horton, P., Park, K. J., and Obayashi, T. (2007). WoLF PSORT: protein localization predictor. Nucl Acids Res. 35, W585–W587. doi: 10.1093/nar/gkm259
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Ikura, M. (1996). Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 21, 14–17. doi: 10.1016/S0968-0004(06)80021-6
Iwamoto, T., Uehara, A., Imanaga, I., and Shigekawa, M. (2000). The Na+/Ca2+ exchanger NCX1 has oppositely oriented re-entrant loop domains that contain conserved aspartic acids whose mutation alters its apparent Ca2+ affinity. J. Biol. Chem. 275, 38571–38580. doi: 10.1074/jbc.M003788200
Izadi, H. M., Rabbani, J., Emam, Y., Pessarakli, M., and Tahmasebi, A. (2014). Effects of salinity stress on physiological performance of various wheat and barley cultivars. J. Plant Nutr. 37, 520–531. doi: 10.1080/01904167.2013.867980
Jia, J., Zhao, S., Kong, X., Li, Y., Zhao, G., He, W., et al. (2013). Aegilopstauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496, 91–95. doi: 10.1038/nature12028
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kader, M. A., and Lindberg, S. (2010). Cytosolic calcium and pH signaling in plants under salinity stress. Plant Signal. Behav. 5, 233–238. doi: 10.4161/psb.5.3.10740
Kahsay, R. Y., Gao, G., and Liao, L. (2005). An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics 21, 1853–1858. doi: 10.1093/bioinformatics/bti303
Käll, L., Krogh, A., and Sonnhammer, E. L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036. doi: 10.1016/j.jmb.2004.03.016
Kamiya, T., Akahori, T., Ashikari, M., and Maeshima, M. (2006). Expression of the vacuolar Ca2+/H+ exchanger, OsCAX1a, in rice: cell and age specificity of expression, and enhancement by Ca2+. Plant Cell Physiol. 47, 96–106. doi: 10.1093/pcp/pci227
Kamiya, T., Akahori, T., and Maeshima, M. (2005). Expression profile of the genes for rice cation/H+ exchanger family and functional analysis in yeast. Plant Cell Physiol. 46, 1735–1740. doi: 10.1093/pcp/pci173
Kamiya, T., and Maeshima, M. (2004). Residues in internal repeats of the rice cation/H+ exchanger are involved in the transport and selection of cations. J. Biol. Chem. 279, 812–819. doi: 10.1074/jbc.M309726200
Kamiya, T., Yamagami, M., Hirai, M. Y., and Fujiwara, T. (2012). Establishment of an in-planta magnesium monitoring system using CAX3 promoter-luciferase in Arabidopsis. J. Exp. Bot. 63, 355–363. doi: 10.1093/jxb/err283
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Khananshvili, D. (2013). The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease. Mol. Aspects Med. 34, 220–235. doi: 10.1016/j.mam.2012.07.003
Khananshvili, D. (2014). Sodium-calcium exchangers (NCX): Molecular hallmarks underlying the tissue-specific and systemic functions. Plügers Arch. 466, 43–60. doi: 10.1007/s00424-013-1405-y
King, B. R., Vural, S., Pandey, S., Barteau, A., and Guda, C. (2012). ngLOC: software and web server for predicting protein subcellular localization in prokaryotes and eukaryotes. BMC Res Notes 5:351. doi: 10.1186/1756-0500-5-351
Koren'kov, V., Park, S., Cheng, N. H., Sreevidya, C., Lachmansingh, J., Morris, J., et al. (2007). Enhanced Cd2+- selective root-tonoplast-transport in tobaccos expressing Arabidopsis cation exchangers. Planta 225, 403–411. doi: 10.1007/s00425-006-0352-7
Krogh, A., Larsson, B., Heijne, G. V., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 70 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Letunic, I., Doerks, T., and Bork, P. (2014). SMART: recent updates new developments and status in 2015. Nucleic Acids Res. 43, D257–D260. doi: 10.1093/nar/gku949
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12:323. doi: 10.1186/1471-2105-12-323
Li, P., Zhang, G., Gonzales, N., Guo, Y., Hu, H., et al. (2016). Ca2+ and diurnal rhythm-regulated Na+/Ca2+ exchanger AtNCL affects flowering time and auxin signaling in Arabidopsis. Plant Cell Environ. 39, 377–392. doi: 10.1111/pce.12620
Liao, J., Li, H., Zeng, W., Sauer, D. B., Belmares, R., and Jiang, Y. (2012). Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335, 686–690. doi: 10.1126/science.1215759
Ling, H. Q., Zhao, S., Liu, D., Wang, J., Sun, H., Zhang, C., et al. (2013). Draft genome of the wheat A-genome progenitor Triticum Urartu. Nature 496, 87–90. doi: 10.1038/nature11997
Liu, C., Atkinson, M., Chinoy, C., Devos, K. M., and Gale, M. D. (1992). Non-homoeologous translocations between group 4, 5 and 7 chromosomes within wheat and rye. Theor. Appl. Genet. 83, 305–312. doi: 10.1007/BF00224276
Liu, W., Xie, Y., Ma, J., Luo, X., Nie, P., and Zuo, Z. (2015). IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361. doi: 10.1093/bioinformatics/btv362
Liu, Z., Xin, M., Qin, J., Peng, H., Ni, Z., Yao, Y., et al. (2015). Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L). BMC Plant Biol. 15:152. doi: 10.1186/s12870-015-0511-8
Lytton, J. (2007). Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem. J. 406, 365–382. doi: 10.1042/BJ20070619
Magadum, S., Banerjee, U., Murugan, P., Gangapur, D., and Ravikesavan, R. (2013). Gene duplication as a major force in evolution. J. Genet. 92, 155–161. doi: 10.1007/s12041-013-0212-8
Manohar, M., Mei, H., Franklin, A. J., Sweet, E. M., Shigaki, T., Riley, B. B., et al. (2010). Zebrafish (Danio rerio) endomembrane antiporter similar to a yeast cation/H+ transporter is required for neural crest development. Biochemistry 49, 6557–6566. doi: 10.1021/bi100362k
Manohar, M., Shigaki, T., and Hirschi, K. D. (2011). Plant cation/H+ exchangers (CAXs): biological functions and genetic manipulations. Plant Biol. 13, 561–569. doi: 10.1111/j.1438-8677.2011.00466.x
Marchler, A. B., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Marcussen, T., Sandve, S. R., Heier, L., Spannagl, M., Pfeifer, M., Jakobsen, K. S., et al. (2014). Ancient hybridizations among the ancestral genomes of bread wheat. Science 345:1250092. doi: 10.1126/science.1250092
McClung, C. R. (2010). A modern circadian clock in the common angiosperm ancestor of monocots and eudicots. BMC Biol. 8:55. doi: 10.1186/1741-7007-8-55
Mei, H., Cheng, N. H., Zhao, J., Park, P., Escareno, R. A., Pittman, J. K., et al. (2009). Root development under metal stress in Arabidopsis thaliana requires the H+/cation an antiporter CAX4. New Phytol. 183, 95–105. doi: 10.1111/j.1469-8137.2009.02831.x
Mei, H., Zhao, J., Pittman, J. K., Lachmansingh, J., Park, S., and Hirschi, K. D. (2007). In planta regulation of the Arabidopsis Ca2+/H+ antiporter CAX1. J. Exp. Bot. 58, 3419–3427. doi: 10.1093/jxb/erm190
Morris, J., Tian, H., Park, S., Sreevidya, C. S., Ward, J. M., and Hirschi, K. D. (2008). AtCCX3 is an Arabidopsis endomembrane H+-dependent K+ transporter. Plant Physiol. 148, 1474–1486. doi: 10.1104/pp.108.118810
Munns, R., and Tester, M. (2008). Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681. doi: 10.1146/annurev.arplant.59.032607.092911
Nicoll, D. A., Hryshko, L. V., Matsuoka, S., Frank, J. S., and Philipson, K. D. (1996a). Mutation of amino acid residues in the putative transmembrane segments of the cardiac sarcolemmal Na+-Ca2+exchanger. J. Biol. Chem. 271, 13385–11339. doi: 10.1074/jbc.271.23.13385
Nicoll, D. A., Longoni, S., and Philipson, K. D. (1990). Molecular cloning and functional expression of the cardiac sarcolemmal Na(+)-Ca2+ exchanger. Science 250, 562–565.
Nicoll, D. A., Quednau, B. D., Qui, Z., Xia, Y.-R., Lusis, A. J., and Philipson, K. D. (1996b). Cloning of a third mammalian Na+-Ca2+exchanger, NCX3. J. Biol. Chem. 271, 24914–24921.
Nicoll, D. A., Ren, X. Y., Ottolia, M., Phillips, M., Paredes, A. R., Abramson, J., et al. (2007). What we know about the structure of NCX1 and how it relates to its function. Ann. N.Y. Acad. Sci. 1099, 1–610. doi: 10.1196/annals.1387.014
Nishizawa, T., Kita, S., Maturana, A. D., Furuya, N., Hirata, K., Kasuya, G., et al. (2013). Structural basis for the counter-transport mechanism of a H+/Ca2+ exchanger. Science 341, 168–172. doi: 10.1126/science.1239002
Nussbaumer, T., Martis, M. M., Roessner, S. K., Pfeifer, M., Bader, K. C., Sharma, S., et al. (2013). MIPS Plants DB: a database framework for comparative plant genome research. Nucleic Acids Res. 41, D1144–D1151. doi: 10.1093/nar/gks1153
Omasits, U., Ahrens, C. H., Müller, S., and Wollscheid, B. (2014). Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886. doi: 10.1093/bioinformatics/btt607
On, C., Marshall, C. R., Chen, N., Moyes, C. D., and Tibbits, G. F. (2008). Gene structure evolutionof the Na+-Ca2+ exchanger (NCX) family. BMC Evol. Biol. 8:127. doi: 10.1186/1471-2148-8-127
Ottolia, M., Nicoll, D. A., and Philipson, K. D. (2005). Mutational analysis of the alpha-1 repeat of the cardiac Na+-Ca2+ exchanger. J. Biol. Chem. 280, 1061–1069. doi: 10.1074/jbc.M411899200
Palty, R., Hershfinkel, M., Yagev, O., Saar, D., Barkalifa, R., Khananshvili, D., et al. (2006). Single R-domain constructs of the Na+/Ca2+ exchanger, NCLX, oligomerize to form a functional exchanger. Biochemistry 45, 11856–11866. doi: 10.1021/bi060633b
Palty, R., Ohana, E., Hershfinkel, M., Volokita, M., Elgazar, V., Beharier, O., et al. (2004). Lithium- calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 279, 25234–25240. doi: 10.1074/jbc.M401229200
Petersen, N. T., Brunak, S., Heijne, G. V., and Nielsen, H. (2011). SignalP 40: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Petsalaki, E. I., Bagos, P. G., Litou, Z. I., and Hamodrakas, S. J. (2006). PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Genomics Proteomic. Bioinform. 4, 48–55. doi: 10.1016/S1672-0229(06)60016-8
Philipson, K. D., and Nicoll, D. A. (2000). Sodium-calcium exchange: a molecular perspective. Annu. Rev. Physiol. 62, 111–133. doi: 10.1146/annurev.physiol.62.1.111
Pingault, L., Choulet, F., Alberti, A., Glover, N., Wincker, P., Feuillet, C., et al. (2015). Deep transcriptome sequencing provides new insights into the structural and functional organization of the wheat genome. Genome Biol. 16:29. doi: 10.1186/s13059-015-0601-9
Pittman, J. K., Edmond, C., Sunderland, P. A., and Bray, C. M. (2009). A cation-regulated and proton gradient-dependent cation transporter from Chlamydomonasreinhardtiihas a role in calcium and sodium homeostasis. J. Biol. Chem. 284, 525–533. doi: 10.1074/jbc.M807173200
Pittman, J. K., and Hirschi, K. D. (2016a). Phylogenetic analysis and protein structure modelling identifies distinct Ca2+/Cation antiporters and conservation of gene family structure within Arabidopsis and rice species. Rice 9, 3. doi: 10.1186/s12284-016-0075-8
Pittman, J. K., and Hirschi, K. D. (2016b). CAX-ing a wide net: cation/H (+) transporters in metal remediation and abiotic stress signaling. Plant Biol. 18, 741–749. doi: 10.1111/plb.12460
Pittman, J. K., Shigaki, T., Marshall, J. L., Morris, J. L., Cheng, N. H., and Hirschi, K. D. (2004). Functional and regulatory analysis of the Arabidopsis thaliana CAX2 cation transporter. Plant Mol. Biol. 56, 959–971. doi: 10.1007/s11103-004-6446-3
Poovaiah, B. W., and Reddy, A. S. (1993). Calcium and signal transduction in plants. CRC Crit. Rev. Plant Sci. 12, 185–211. doi: 10.1080/713608046
Pradhan, G. P., Prasad, P. V. V., Fritz, A. K., Kirkham, M. B., and Gill, B. S. (2012). Effects of drought 6 and high temperature stress on synthetic hexaploid wheat. Funct. Plant Biol. 39, 190–198. doi: 10.1071/FP11245
Prasad, P. V. V., Pisipati, S. R., Momcilovic, I., and Ristic, Z. (2011). Independent and combined effects of high temperature and drought stress during grain filling on plant yield and chloroplast EF-Tu expression in Spring Wheat. J. Agron. Crop Sci. 197, 430–441. doi: 10.1111/j.1439-037X.2011.00477.x
Saier, M. H. Jr., Eng, B. H., Fard, S., Garg, J., Haggerty, D. A., Hutchinson, W. J., et al. (1999). Phylogenetic characterization of novel transport protein families revealed by genome analyses. Biochim. Biophys. Acta 1422, 1–56. doi: 10.1016/S0304-4157(98)00023-9
Sanders, D., Brownlee, C., and Harper, J. F. (1999). Communicating with calcium. Plant Cell 11, 691–706. doi: 10.1105/tpc.11.4.691
Schmutz, J., Steven, B., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Shaul, O., Hilgemann, D. W., de-Almeida-Engler, J., Van Montagu, M., Inze, D., and Galili, G. (1999). Cloning and characterization of a novel Mg2+/H+ exchanger. EMBO J. 18, 3973–3980. doi: 10.1093/emboj/18.14.3973
Shigaki, T., Barkla, B. J., Miranda-Vergara, M. C., Zhao, J., Pantoja, O., and Hirschi, K. D. (2005). Identification of a crucial histidine involved in metal transport activity in the Arabidopsis cation/H+ exchanger CAX1. J. Biol. Chem. 280, 30136–30142. doi: 10.1074/jbc.M503610200
Shigaki, T., and Hirschi, K. D. (2006). Diverse functions and molecular properties emerging for CAX cation/H+ exchangers in plants. Plant Biol. 8, 419–429. doi: 10.1055/s-2006-923950
Shigaki, T., Mei, H., Marshall, J., Li, X., Manohar, M., and Hirschi, K. D. (2010). The expression of the open reading frame of Arabidopsis CAX1, but not its cDNA, confers metal tolerance in yeast. Plant Biol. 12, 935–939. doi: 10.1111/j.1438-8677.2010.00368.x
Shigaki, T., Pittman, J. K., and Hirschi, K. D. (2003). Manganese specificity determinants in the Arabidopsis metal/H+ antiporter CAX2. J. Biol. Chem. 278, 6610–6617. doi: 10.1074/jbc.M209952200
Shigaki, T., Rees, I., Nakhleh, L., and Hirschi, K. D. (2006). Identification of three distinct phylogenetic groups of CAX cation/proton antiporters. J. Mol. Evol. 63, 815–825. doi: 10.1007/s00239-006-0048-4
Shigekawa, M., Iwamoto, T., Uehara, A., and Kita, S. (2002). Probing ion binding sites in the Na+/Ca2+ exchanger. Ann. N.Y. Acad. Sci. 976, 19–30. doi: 10.1111/j.1749-6632.2002.tb04710.x
Shumayla, S. S., Sharma, S., Kumar, R., Mendu, V., Singh, K., and Upadhyay, S. K. (2016b). Genomic dissection and expression profiling revealed functional divergence in Triticum aestivum leucine rich repeat receptor like kinases (TaLRRKs). Front. Plant Sci. 7:1374. doi: 10.3389/fpls.2016.01374
Shumayla, S. S., Sharma, S., Pandey, A. K., Singh, K., and Upadhyay, S. K. (2016a). Molecular characterization and global expression analysis of lectin receptor kinases in bread wheat (Triticum aestivum). PLoS ONE 11:e0153925. doi: 10.1371/journal.pone.0153925
Singh, A. K., Kumar, R., Tripathi, A. K., Gupta, B. K., Pareek, A., and Pareek-Singla, S. L. (2015). Genome-wide investigation and expression analysis of Sodium/Calcium exchanger gene family in rice and Arabidopsis. Rice 8, 21. doi: 10.1186/s12284-015-0054-5
Solovyev, V., Kosarev, P., Seledsov, I., and Vorobyev, D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7:S10. doi: 10.1186/gb-2006-7-s1-s10
Spalding, E. P., and Harper, J. F. (2011). The ins and outs of cellular Ca(2+) transport. Curr. Opin. Plant Biol. 14, 715–720. doi: 10.1016/j.pbi.2011.08.001
The International Wheat Genome Sequencing Consortium (IWGSC) (2014). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788. doi: 10.1126/science.1251788
Tuteja, N., and Mahajan, S. (2007). Calcium signaling network in plants: an overview. Plant Signal. Behav. 2, 79–85. doi: 10.4161/psb.2.2.4176
Tuteja, N., and Sopory, S. K. (2008). Chemical signaling under abiotic stress environment in plants. Plant Signal. Behav. 3, 525–536. doi: 10.4161/psb.3.8.6186
Upadhyay, S. K., Dixit, S., Sharma, S., Singh, H., Kumar, J., Verma, P. C., et al. (2013). siRNA Machinery in Whitefly (Bemisia tabaci). PLoS ONE 8:e83692. doi: 10.1371/journal.pone.0083692
Upadhyay, S. K., Singh, H., Dixit, S., Mendu, V., and Verma, P. C. (2016). Molecular characterization of vitellogenin and vitellogenin receptor of Bemisia tabaci. PLoS ONE 11:e0155306. doi: 10.1371/journal.pone.0155306
Vaid, N., Pandey, P. K., and Tuteja, N. (2012). Genome-wide analysis of lectin receptor-like kinase family from Arabidopsis and rice. Plant Mol. Biol. 80, 365–388. doi: 10.1007/s11103-012-9952-8
Waight, A. B., Pedersen, B. P., Schlessinger, A., Bonomi, M., Chau, B. H., Roe-Zurz, Z., et al. (2013). Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499, 107–110. doi: 10.1038/nature12233
Wang, P., Li, Z., Wei, J., Zhao, Z., Sun, D., and Cui, S. (2012). A Na+/Ca2+ exchanger-like protein (AtNCL) involved in salt stress in Arabidopsis. J. Biol. Chem. 287, 44062–44070. doi: 10.1074/jbc.M112.351643
Wang, Z., Li, R. L., Xu, X. Z., and Zhu, W. C. (1994). Ca2+ transport across the tonoplast of wheat roots. J. Wuhan Univ. 5, 111–115.
Wu, M., Tong, S., Waltersperger, S., Diederichs, K., Wang, M., and Zheng, L. (2013). Crystal structure of Ca2+/H+ antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc. Natl. Acad. Sci. U.S.A. 110, 11367–11372. doi: 10.1073/pnas.1302515110
Yamada, N., Theerawitaya, C., Cha-um, S., Kirdmanee, C., and Takabe, T. (2014). Expression and functional analysis of putative vacuolar Ca2+-transporters (CAXs and ACAs) in roots of salt tolerant and sensitive rice cultivars. Protoplasma 251, 1067–1075. doi: 10.1007/s00709-014-0615-2
Yan, B. X., and Sun, Y. Q. (1997). Glycine residues provide flexibility for enzyme active sites. J. Biol. Chem. 272, 3190–3194. doi: 10.1074/jbc.272.6.3190
Yoon, H. S., Hackett, J. D., Ciniglia, C., Pinto, G., and Bhattacharya, D. (2004). A molecular timeline for the origin of photosynthetic eukaryotes. Mol. Biol. Evol. 21, 809–818. doi: 10.1093/molbev/msh075
Yu, C. S., Chen, Y. C., Lu, C. H., and Hwang, J. K. (2006). Prediction of protein subcellular localization. Proteins 64, 643–651. doi: 10.1002/prot.21018
Zeng, L., Deng, R., Guo, Z., Yang, S., and Deng, X. (2016). Genome-wide identification and characterization of Glyceraldehyde-3-phosphate dehydrogenase genes family in wheat (Triticum aestivum). BMC Genomics 17:240. doi: 10.1186/s12864-016-2527-3
Zhang, H., Yang, Y., and Wang, C. (2014b). Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genomics 15:898. doi: 10.1186/1471-2164-15-898
Zhang, L., Du, L., and Poovaiah, B. W. (2014a). Calcium signaling and biotic defense responses in plants. Plant Signal. Behav. 9:e973818. doi: 10.4161/15592324.2014.973818
Zhang, Y., Liu, Z., Khan, A. A., Lin, Q., Han, Y., Mu, P., et al. (2016). Expression partitioning of homeologs and tandem duplications contribute to salt tolerance in wheat (Triticum aestivum L). Sci. Rep. 6:21476. doi: 10.1038/srep21476
Zhao, J., Connorton, J. M., Guo, Y. Q., Li, X. K., Shigaki, T., Hirschi, K. D., et al. (2009). Functional studies of split Arabidopsis Ca2+/H+ exchangers. J. Biol. Chem. 284, 34075–34083. doi: 10.1074/jbc.M109.070235
Keywords: abiotic, biotic, Ca2+/cation antiporters, TaCAX, TaCCX, TaNCL, TaMHX, Triticum aestivum
Citation: Taneja M, Tyagi S, Sharma S and Upadhyay SK (2016) Ca2+/Cation Antiporters (CaCA): Identification, Characterization and Expression Profiling in Bread Wheat (Triticum aestivum L.). Front. Plant Sci. 7:1775. doi: 10.3389/fpls.2016.01775
Received: 24 August 2016; Accepted: 10 November 2016;
Published: 28 November 2016.
Edited by:
Pierre-Emmanuel Courty, University of Fribourg, SwitzerlandReviewed by:
Jon Pittman, University of Manchester, UKDaniel Khananshvili, Tel Aviv University, Israel
Copyright © 2016 Taneja, Tyagi, Sharma and Upadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Santosh Kumar Upadhyay, c2FudG9zaG5icmlAaG90bWFpbC5jb20=; c2t1cGFkaHlheUBwdS5hYy5pbg==