 Xianjun Liu1,2†
Xianjun Liu1,2† Ying Lu1†Mingli Yan3Donghong Sun1Xuefang HuShuyan Liu1Sheyuan Chen1Chunyun Guan1
Ying Lu1†Mingli Yan3Donghong Sun1Xuefang HuShuyan Liu1Sheyuan Chen1Chunyun Guan1 Zhongsong Liu1*
Zhongsong Liu1*- 1Oilseed Crops Institute, Hunan Agricultural University, Changsha, Hunan, China
- 2College of Life Sciences, Resources and Environment Sciences, Yichun University, Yichun, China
- 3School of Biology, Hunan University of Science and Technology, Xiangtan, China
Proanthocyanidins (PA) is a type of prominent flavonoid compound deposited in seed coats which controls the pigmentation in all Brassica species. Annotation of Brassica juncea genome survey sequences showed 72 PA genes; however, a functional description of these genes, especially how their interactions regulate seed pigmentation, remains elusive. In the present study, we designed 19 primer pairs to screen a bacterial artificial chromosome (BAC) library of B. juncea. A total of 284 BAC clones were identified and sequenced. Alignment of the sequences confirmed that 55 genes were cloned, with every Arabidopsis PA gene having 2–7 homologs in B. juncea. BLAST analysis using the recently released B. rapa or B. napus genome database identified 31 and 58 homologous genes, respectively. Mapping and phylogenetic analysis indicated that 30 B. juncea PA genes are located in the A-genome chromosomes except A04, whereas the remaining 25 genes are mapped to the B-genome chromosomes except B05 and B07. RNA-seq data and Fragments Per Kilobase of a transcript per Million mapped reads (FPKM) analysis showed that most of the PA genes were expressed in the seed coat of B. juncea and B. napus, and that BjuTT3, BjuTT18, BjuANR, BjuTT4-2, BjuTT4-3, BjuTT19-1, and BjuTT19-3 are transcriptionally regulated, and not expressed or downregulated in yellow-seeded testa. Importantly, our study facilitates in better understanding of the molecular mechanism underlying Brassica PA profiles and accumulation, as well as in further characterization of PA genes.
Introduction
In oilseed brassicas, a yellow-seeded form is preferred over a black- or brown-seeded counterpart mainly because of a thinner seed coat and higher oil content (Friedt and Snowdon, 2009; Velasco and Ferna'ndez-Martı'nez, 2009). Importantly, proanthocyanidins (PAs) play a critical role in this differential pigmentation process (Auger et al., 2010; Fang et al., 2012; Lu et al., 2012).
Proanthocyanidins (PAs) are end-products of a well-studied branch of the flavonoid biosynthetic pathway in higher plants (Winkel-Shirley, 2001; Lepiniec et al., 2006; Saito et al., 2013). In Arabidopsis, a close relative of the Brassica species, 19 single-copy genes have been associated with PA (Appelhagen et al., 2014, 2015; Ichino et al., 2014). These genes can be divided into three classes based on their functions: structural, transcriptionally regulatory, or genes responsible for PA modification, transport, and oxidation. PA genes have also been cloned from a dozen other plant species (Hichri et al., 2011; Falcone Ferreyra et al., 2012) such as maize, and soybean (Yang et al., 2010; Senda et al., 2012). In contrast to single-copy genes in Arabidopsis, several plant species have multiple homologs for a given PA gene. For example, there are nine CHS homologs in soybean (Yi et al., 2010).
In Brassica species homologous cloning is used to isolate PA genes by such as DFR/TT3 (Yan et al., 2008; Akhov et al., 2009), ANS/TT18 (Yan et al., 2011), ANR/BAN (Nesi et al., 2009), TT10 (Zhang et al., 2013), TT2 (Wei et al., 2007), TT8 (Padmaja et al., 2014), TT12 (Chai et al., 2009), TT16 (Deng et al., 2012; Chen et al., 2013), TTG1 (Zhang et al., 2009; Yan et al., 2014) and TTG2 (Li et al., 2015). However, homologous cloning has drawbacks. It needs prior knowledge of sequences of homologous gene, and is slow and difficult to amplify all members of a gene family, particularly in polyploid species, e.g., Brassica juncea, an allotetraploid species. To address these limitations, next-generation sequencing has been widely adopted. Up to date the genomes of over 100 plant species, including B. rapa (Wang et al., 2011), B. olearcea (Liu et al., 2014), and B. napus (Chalhoub et al., 2014) have been sequenced. Very recently, the genome sequence of B. nigra has also been released (http://www.ncbi.nlm.nih.gov/genome/10988). Whole-genome sequence annotation facilitates in genome-wide identification of PA genes (Velasco et al., 2007; Guo et al., 2014). However, the PA genes of Brassica species have not been analyzed in great detail. Furthermore, the complete genome sequencing of Brassica juncea has not been achieved to date. Yang et al. (2014) has conducted a survey of genome sequences in B. juncea. Genome survey sequencing (GSS) can provide information about gene content, functional elements and molecular markers (Jiao et al., 2012; Hirakawa et al., 2015), as well as compare genes of related species for the phylogenetic reconstruction of other non-model species.
Reverse transcription-polymerase chain reaction (RT-PCR), real-time fluorescent quantitative PCR, and transcriptome sequencing (RNA-seq) can analyze the spatial and temporal expression pattern, functions and interactions among various genes (Agarwal et al., 2014). RNA-seq is widely used to estimate transcript amounts and to obtain a quantitative account of transcript amounts in organisms, organs, tissues, or specific cell types, frequently comparing transcript amounts among different samples (Martin et al., 2013; Weber, 2015).
In the present study, GSS was conducted on the inbred line of B. juncea var. Purple-leaf Mustard (PM), and a total of 69,193 coding genes, including 72 PA genes, were predicted by annotation of GSS. Approximately 19 primer pairs specific for PA genes were then designed to screen a bacterial artificial chromosome (BAC) library of B. juncea, which was constructed from the same inbred line. In total, 284 BAC clones were identified and 55 B. juncea PA genes were confirmed by sequencing of fragments amplified from representative BAC clones. Its genomic or chromosomal positions were predicted by mapping to the sequenced B. rapa, B. nigra, or B. napus genomes, which was used as reference genomes to perform phylogenetic analysis on the full-length gene sequences and the end sequences of gene-carrying BACs. The expression level of PA genes were estimated in the seed coat and compared between the yellow- and brown-seed coat by fragments per kilobase of exon model per million mapped reads (FPKM) analysis of RNA-seq data in B. juncea and B. napus. Identification, mapping, and expression analysis of the PA genes in the present study may facilitate in better understanding the genetic mechanism underlying proanthocyanidin biosynthesis, profile, and accumulation in various Brassica species.
Materials and Methods
Plant Accessions
The inbred line of B. juncea var. PM was used for GSS and construction of the BAC library. RNA was extracted from the seed coat of the inbred line of B. juncea var. Sichuan Yellow (SY, yellow-seeded) and its brown-seeded near-isogenic lines (NILA and NILB), the black-seeded B. napus cv. Xiangyou 15 and two of its F7 recombinant inbred linesRIL52 and RIL55 15 days after pollination (DAP, torpedo to late torpedo stage) (Liu et al., 2009; Nesi et al., 2009). The plants were grown in a greenhouse under a photoperiod of 16 h/8 h (day/night cycle) at 22°C.
Genome Sequencing, Sequence Assembly, Gene Prediction, and Annotation
Paired-end (PE) libraries were prepared using total DNA from PM, which were then constructed according to the instructions provided by Illumina (San Diego, CA, USA) with a 500-bp insert size and 125-bp read length. Sequence analyses were conducted using the Illumina HiSeq 2000 platform.
The obtained reads were subjected to quality control as follows: bases with quality scores <10 were filtered out by FastQC-0.11.3 (Schmieder and Edwards, 2011). Adaptor sequences in the reads were trimmed using fastx clipper of the FASTX-Toolkit 0.0.13 (http://hannonlab.cshl.edu/fastx_toolkit). After trimming, reads including N nucleotide lengths of <100 bases were excluded, and the remaining high-quality data was used for de novo sequence assembly by SOAP (Schmieder and Edwards, 2011). Protein-encoding sequences in the assembled genomic sequences of PM were predicted by Augustus 2.7 (Stanke and Waack, 2003) using the A. thaliana training set under the default parameters. Reciprocal best-hit analysis (Moreno-Hagelsieb and Latimer, 2008) was performed to compare the results of the prediction by using B. rapa training sets.
Construction, Pooling, and Screening of the BAC Library
The B. juncea BAC library named ZBjuH was constructed from the inbred line of the PM that were treated with the restriction endonuclease HindIII (Luo and Wing, 2003). This library consists of 71,808 clones with an average insert size of 126 kb genomic DNA, and an estimated 10.8-fold coverage of the B. juncea genome. The clones were arranged in 187 384-well plates. The clones were organized into three-dimensional BAC pools of plates, rows, and columns. The superplate consisted of 19 DNA samples, each representing 10 BAC plates, except for superplate 19, which only consists of 7 384-well plates. The first dimension consisted of the BAC clone plate of 187 DNA samples. The second and third dimensions consisted of 8 and 12 DNA samples, respectively, for the pooled 16 rows and 24 columns of the BAC clones. Screening of single BAC clones was performed in a five-step PCR process (Figure S1). The PCR primers were designed according to the conserved sequences of the PA genes that were annotated from the B. juncea GSS (Table S1). PCR reactions were performed in a total volume of 10 μL with a reaction mixture as follows: 10 × PCR buffer (1.0 μL), dNTP mix (10 mM each, 0.15 μL), 1 U Taq DNA polymerase (Takara, Japan), 1 μL template, 10 mM forward primer (0.5 μL), 10 mM reverse primer (0.5 μL) and ddH2O up to 10 μL. A “touchdown” PCR amplification program is used as follows: 94°C for 5 min; 6 cycles of 30 s at 94°C, 40 s at 62°C with a 1°C decrease in the annealing temperature per cycle, and 1 min at 72°C; 30 cycles of 30 s at 94°C, 45 s at 56°C, and 1 min at 72°C; and a final extension at 72°C for 10 min. The PCR products were observed by electrophoresis on 1.5% agarose gels using ethidium bromide and UV visualization. The BAC clones from which the fragment of expected size was amplified were considered positive BAC clones.
Grouping and Sequencing for Full-Length Gene of Positive BAC Clones
Gene fragments amplified from the positive BAC clones were sequenced and aligned with annotated PA genes using DNAMAN4.0 (LynnonBiosoft, USA) to confirm whether the cloned and the annotated gene were the same copy. When a cloned gene harbored a single nucleotide difference (SNP) and/or insertion or deletion (Indels) in its sequence from the corresponding annotated gene, the cloned and the annotated genes are considered different. For each PA gene, one or two BAC clones were selected for sequencing of the full-length genes by the high-quality, longer read Sanger method (Life Technologies, Shanghai).
Identification and Phylogenetic Analysis of PA Genes in B. napus, B. nigra, and B. rapa
The sequences of cloned B. juncea PA genes were mapped to the released B. napus (http://www.genoscope.cns.fr/blat-server/cgi-bin/colza/webBlat), B. nigra (http://www.ncbi.nlm.nih.gov/genome/10988), or B. rapa (http://brassicadb.org/brad/blastPage.php) reference genome to search for homologous B. napus, B. nigra or B. rapa PA genes with an identity ≥90%. Phylogenetic analysis of homologous PA genes in B. juncea, B. rapa, B. napus, and Arabidopsis was performed by using neighbor-joining (NJ) method as provided in MEGA 5.2 (Tamura et al., 2011), and the reliability of the phylogenetic trees was evaluated by the bootstrap method, with 1000 replications. The B. juncea PA genes on the same branch (clade) of the phylogenetic tree were classified into a homologous group.
Sequencing and Mapping of BAC Ends
The BACs used for full-length sequencing of the gene were also sequenced for end-sequencing on an ABI 3730X DNA analyzer (Life Technologies, Shanghai). The sequencing primers were modified pIndigoBAC536 cloning vector-derived sequencing primers M13R (5′-CAGGAAACAGCTAT-GACC-3′) and S2 (5′-CGAATTCGAGCTCGGTACCC-3′). The sequence obtained by using the primer M13R was designated as left end (L) of the BAC clone, whereas the sequence by S2 was considered the right end (R). BAC end-sequences (BESs) were also mapped to the recently sequenced B. napus (http://www.genoscope.cns.fr/blat-server/cgi-bin/colza), B. nigra (http://www.ncbi.nlm.nih.gov/genome/10988) or B. rapa (http://brassicadb.org) reference genome to assign a genomic location when at least 100 bp aligned to the reference genome, with at least 75% identity. If hits were obtained at multiple locations in any one of the reference genomes, then a BES was assigned to the position of the hit with the highest identity. The position of a BES was indicated by the first and the last assigned nucleotide (nt) on each reference genome.
Expression Analysis of PA Genes in Seed Coat
Isolation, reverse transcription and RNA-seq analysis of RNA from fresh seed coats were performed as described by Liu et al. (2013). The expression level of every PA gene in the seed coat was calculated using the FPKM method (Mortazavi et al., 2008). To compare transcript abundance of cloned PA genes in seed coat between the yellow-seeded inbred SY and its brown-seeded near-isogenic lines (NILA and NILB), the respective mapped reads from the SY/NILA and the SY/NILB pairs for each gene were counted using TopHat v2.0.9 (Kim et al., 2013). Fold changes for each gene between NILs and SY were computed as the ratio of the FPKM values. When the FPKM value of NILs or SY was 0, the substitute 0.001 was used for estimation of fold change. To display changes of PA gene expression in seed coat, the heatmap was constructed by using Heml software (“Normalization:” Logarithmic Base 2, “DEMO:” Canvas) (Deng et al., 2014).
The primers used in RT-PCR expression analysis are listed in Table S2. The following cycling parameters were used for amplification of the PA genes: 1 cycle of 4 min at 94°C; 38 cycles of 50 s at 94°C, 50 s at 58°C, 1 min at 72°C; one cycle of 6 min at 72°C. The PCR products were verified by gel electrophoresis as earlier described.
Results
Identification and Cloning of PA Genes in B. juncea
A total of 56.2 Gb high-quality sequencing data were assembled into 835 Mb of genomic sequence, with contig and scaffold N50 sizes of 2584 bp and 16,777 bp in B. juncea (Table S3). A total of 233,309 coding genes were predicted by Augustus 2.7 (Table S3) and annotated by alignment of the deduced amino acid sequence to B. rapa genes (http://brassicadb.org/brad/). Approximately 69,193 records were screened out, with sequence identity greater than 70% and alignment length greater than 100 amino acids, which correspond to 32,798 B. rapa genes (Table S4). For a B. rapa gene, an averaged 2.1 homologs, at most 11 homologs, were detected in the B. juncea genome. Among the 69,193 predicted B. juncea genes, 72 were identified as PA genes (Table S5). The number of B. juncea genes that were homologous to a given Arabidopsis PA gene varied from two (DFR, TT1, TT2, TT8, TTG1, and TT12) to six (TT4, TT6, and ANR) (Table S5). Furthermore, two annotated B. juncea genes of TT6 and TT7 were located within the same scaffold (Table S5).
A total of 284 positive BAC clones were identified using 19 PA gene-specific primer pairs from ZBjuH BAC library (Table 1). The amplified fragments were sequenced, and 284 clean sequences with sizes between 192 and 1487 bp were obtained. Alignment showed that these fragments represented 55 B. juncea PA genes, corresponding to 16 Arabidopsis PA genes, with each Arabidopsis PA gene having 2–7 B. juncea homologs (Table 1). All cloned B. juncea PA genes, except for BjuTT4-2, BjuTT4-7, and BjuTT16-6, showed genomic sequences that were similar to the corresponding predicted PA genes. These amplified sequences were not evenly distributed among genes. For 6 genes, only one sequence was each identified, whereas at least 10 sequences were detected for 7 other genes. The remaining 42 genes were each carried by 2–9 BAC clones (Table 1), which is consistent with coverage of the genome by the BAC library used. No BAC clones were identified for six the annotated genes (TT4_g135394, TT5_g158015, ANR_g228640, ANR_g226654, TT19_g144296, and TT19_g167454) (Figure S3).
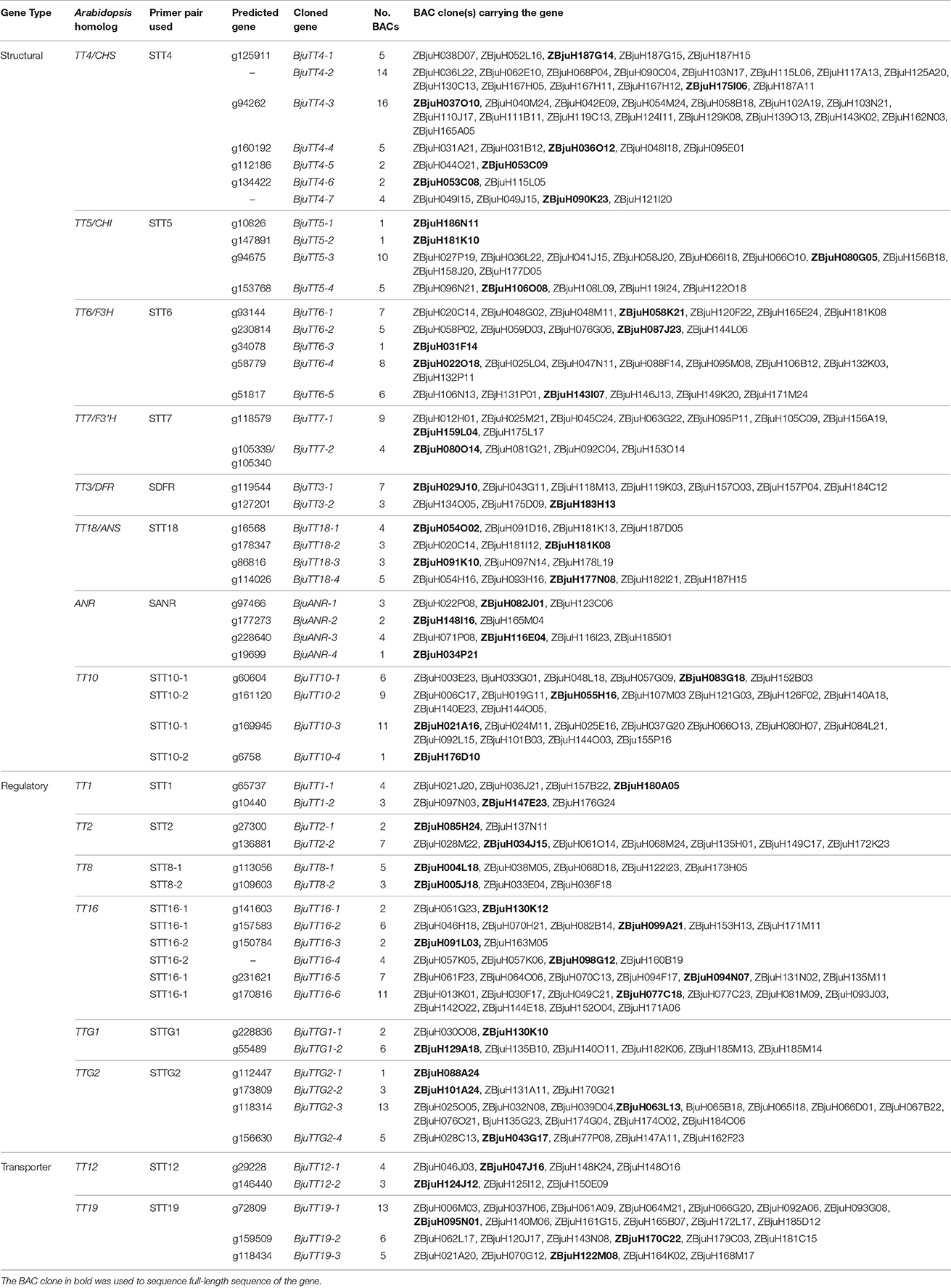
Table 1. Grouping of the PA gene carrier BAC clones screened by PCR from Brassica juncea.
One or two BAC clones were chosen for each of the above mentioned PA gene groups of BAC clones and sequenced by walking to obtain full-length gene sequence. Alignment of the resultant full-length gene with its respective GSS sequence indicated that two predicted genes was in fact from the same gene because each of them was only a portion of the same gene (Table S6). Finally, 55 PA genes were confirmed in B. juncea by BAC sequencing (Table 2).
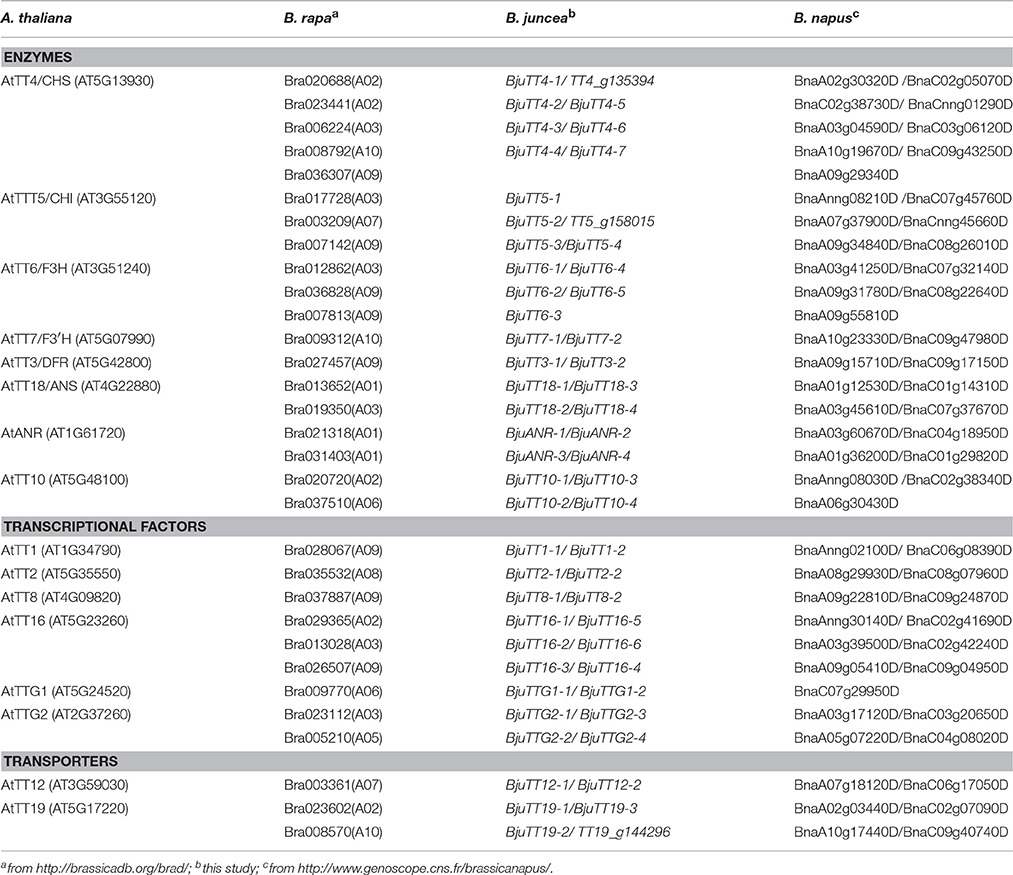
Table 2. Proanthocyanidins-associated genes identified in B. rapa, B. juncea, and B. napus.
Genomic Locations of PA Genes in Brassica Species
BLAST of these cloned 55 B. juncea PA genes against the B. rapa or B. napus reference genome identified 31 and 58 homologous genes in B. rapa and B. napus, respectively (Table 2). The neighbor-joining tree of the PA genes from B. juncea, B. rapa, B. napus, and Arabidopsis showed that TT4 genes were clustered into five homologous groups, TT5, TT6, and TT16 each into three groups; TT10, TT18, TTG2, and TT19 each into two groups; and the remaining TT3, TT7, ANR, TT1, TT2, TT8, TTG1, and TT12 genes were clustered into only one homologous group, indicating that these genes were highly conserved in terms of genomic sequence (Figure S2).
Mapping of these cloned 55 B. juncea PA genes to the B. rapa, B. nigra, or B. napus reference genome indicated that 30 and 29 PA genes were homologous to the genes located in A-genome chromosomes except A04 of B. rapa and B. napus, respectively, whereas 23 of the other 25 genes were located in the B-genome chromosomes except B05 and B07 of B. nigra, the remaining two gene (BjuTT5-4 and BjuTT2-2) were anchored on scaffold_30.1 and scaffold_500.1 of B. nigra, respectively, which have not yet been mapped onto a chromosome (Table 3, Figure 1). These PA genes have >95 identity (Table 3). Moreover, 23 of these A-genome PA genes were, respectively, located on the same chromosomes in B. rapa and B. napus, but additional genes may be located in either the same or different A-genome chromosomes or C-genome chromosomes because their positions have not been mapped to the B. napus reference genome (Table 3). The B-genome and the C-genome contributed 25 and 29 PA genes to B. juncea and B. napus genome, respectively, which is approximately equal to the number of PA genes from the A-genome.
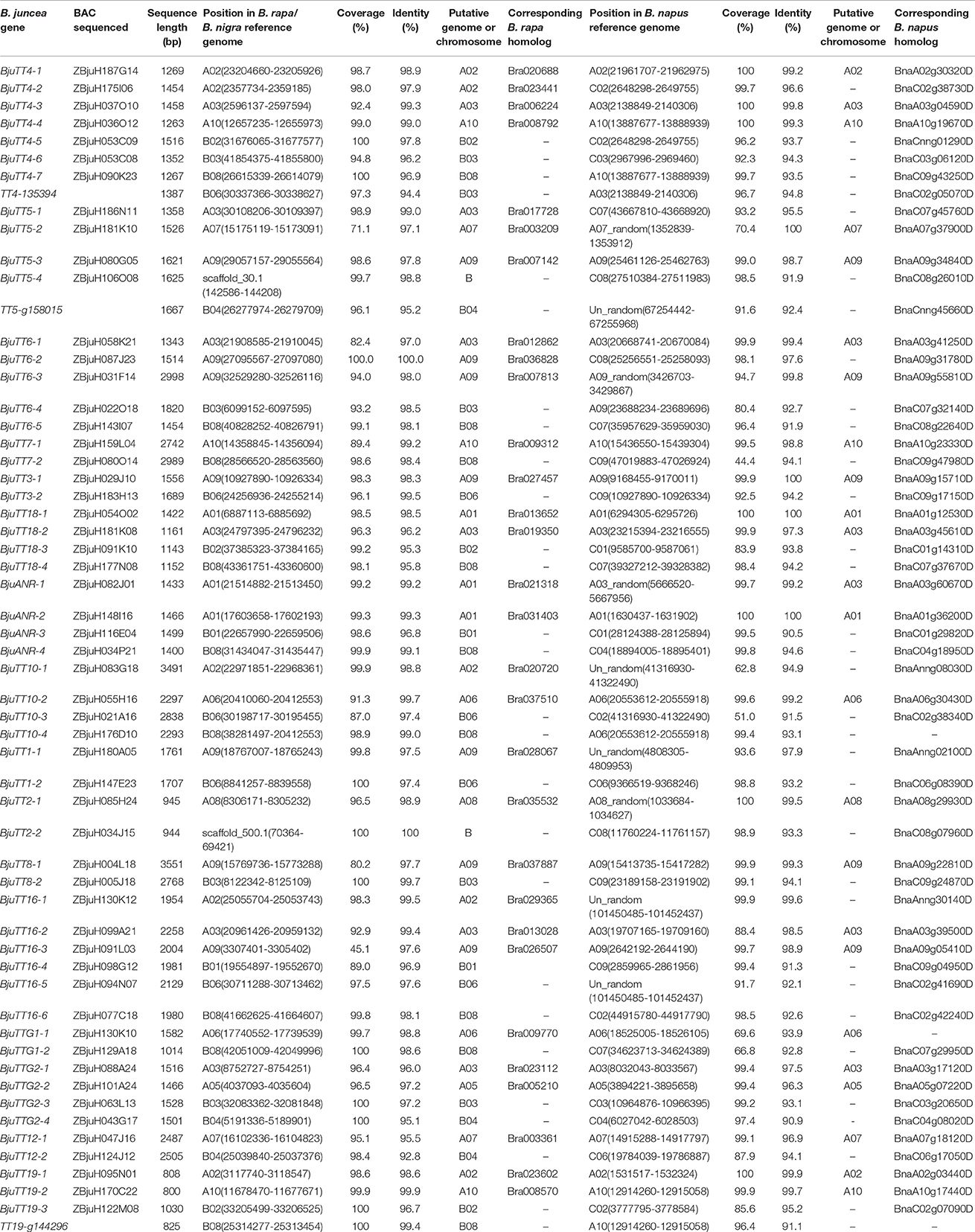
Table 3. Mapping to the Brassica rapa, B. nigra, or B. napus reference genome of full-length sequences of the B. juncea PA genes cloned in this study.
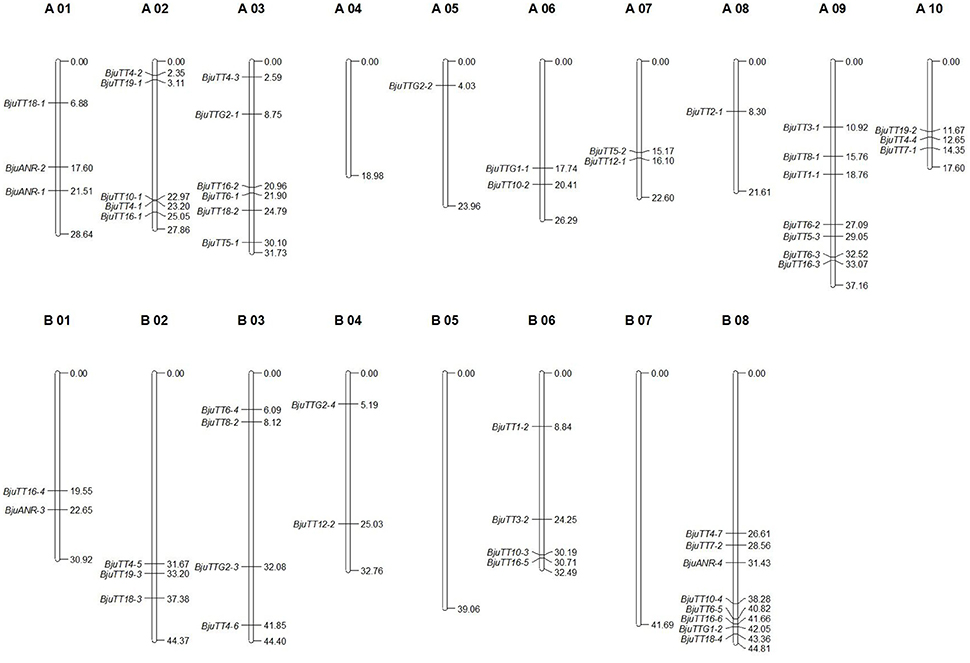
Figure 1. Putative chromosomal positions of cloned proanthocyanidins-associated genes in Brassica juncea. BjuTT5-4 and BjuTT2-2 were not located on the chromosome in B. juncea.
To confirm the above genomic locations, the BAC clones used for sequencing full-length genes were also sequenced for BESs. The resulting BESs between 587 and 1233 bp in length were also mapped in a similar way. Mapping of the BESs to the B. rapa reference genome showed that both BESs of 23 A-genome B. juncea PA genes were mapped around the genomic position as mapped by the full-length sequence of the corresponding genes. However, one BES of the BACs carrying two A-genome genes, i.e., BjuTT2-1 and BjuTTG1-1 was mapped to an unfixed scaffold, whereas one BES of the BACs carrying the remaining five A-genome genes, i.e., BjuTT5-2, BjuTT6-1, BjuANR-2, BjuTT10-1, and BjuTTG2-1 was mapped to an unexpected genomic position (Table 4). Mapping of the BESs to the B. napus reference genome generated a more complicated picture. For only 15 A-genome B. juncea PA genes, both BESs were mapped around the genomic position as mapped by the full-length sequence of the corresponding genes. One or both BESs of the BACs carrying 7 A-genome genes, i.e., BjuTT5-1, BjuTT6-2, BjuTT7-1, BjuTT16-2, BjuTT1-1, BjuTT2-1, and BjuTTG1-1 were mapped to an unfixed scaffold, whereas one or both BESs of the BACs carrying the remaining 8 A-genome genes were mapped to an unexpected A-genome chromosome, or a C-genome chromosome in B. napus reference genome (Table 4). Mapping of the BESs to the B. nigra reference genome showed that both BESs of 19 B-genome B. juncea PA genes were mapped around the genomic position as mapped by the full-length sequence of the corresponding genes, one BES of the BACs carrying three B-genome genes, i.e., BjuTT4-6, BjuTT18-4, and BjuTT7-2 was mapped to an unexpected genomic position in the B. nigra reference genome, and then one BES of the BACs carrying the remaining three B-genome genes, i.e., BjuTT5-4, BjuTT1-2, and BjuTT2-2 was mapped to an unfixed scaffold (Table 4).
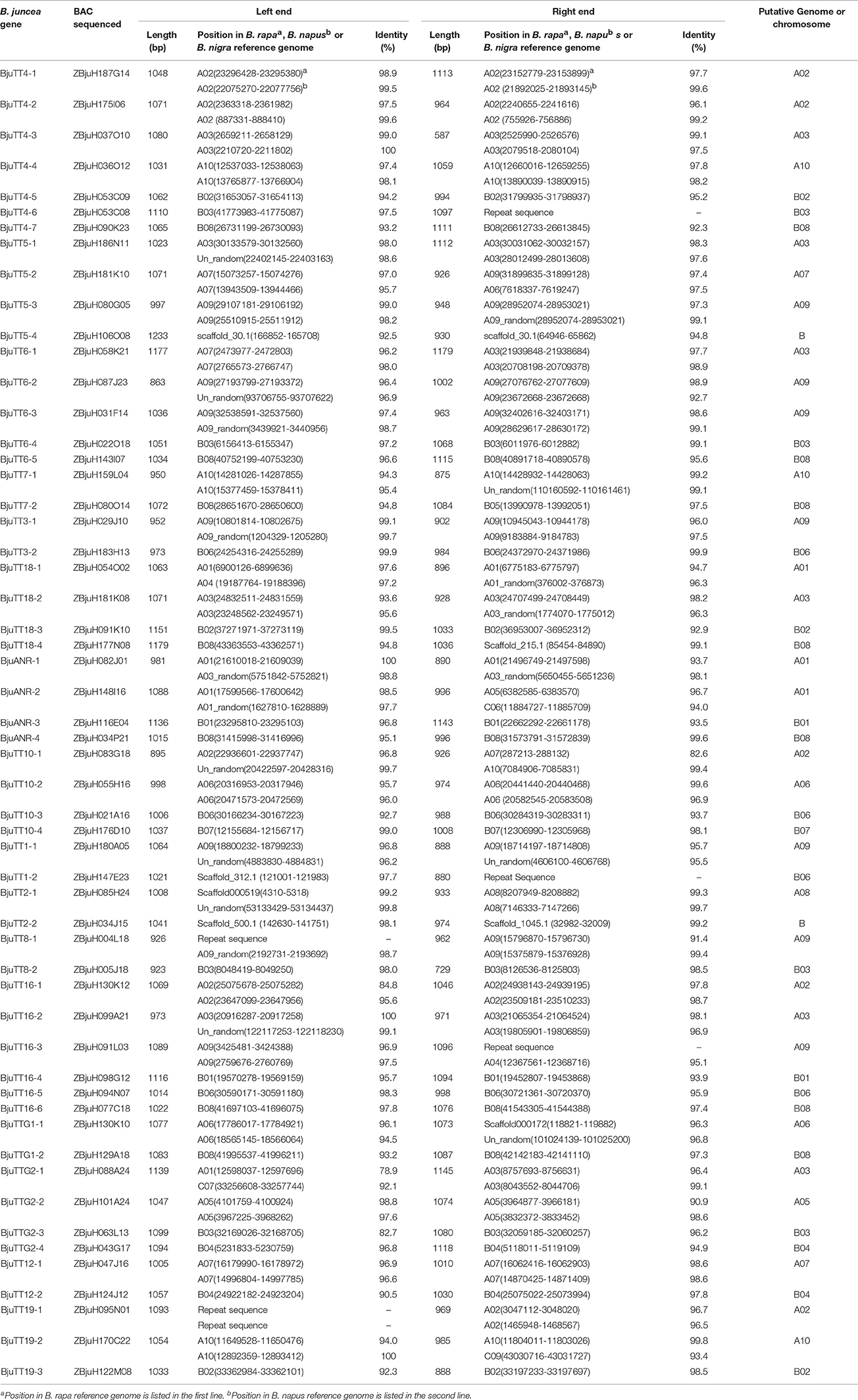
Table 4. Mapping to the B. rapa, B. nigra, or B. napus reference genome of end sequences of the PA gene carrier BACs from B. juncea.
Expression of PA Genes in Seed Coat of B. juncea and B. napus
Fragments Per Kilobase of a transcript per Million (FPKM) analysis indicated that 55 annotated B. napus PA genes (excluding BnaCnng01290D and BnaA09g29340D), and all cloned B. juncea PA genes except BjuTT5-1 and BjuTT5-4 were expressed in seed coat (Figure 2, Table S7). However, transcript abundance significantly varied among PA genes, as well as accessions. In general, the expression level of structural and transporter genes were higher than that of transcriptional factor genes in black- and brown-seeded accessions analyzed. No transcripts of BjuTT3, BjuANR, BjuTT18-1, BjuTT19-1, and BjuTT19-3 were detected in the seed coat of yellow-seeded SY. In addition, a 7-fold or greater difference in expression level of BjuTT3, BjuTT18, BjuANR, and BjuTT19 as well as BjuTT4-2, BjuTT4-3, BjuTT4-4, and BjuTT5-3 were found between SY and its brown-seeded near-isogenic lines (Figure 2, Table S7), implying that these differentially expressed genes are involved in seed pigmentation. Moreover, six additional genes, i.e., BjuTT4-5, BjuTT6-1, BjuTT6-4, BjuTT8-1, BjuTT16-3, and BjuTT16-6, were upregulated by at least 2-fold in seed coat of NILA, whereas four other genes (BjuTT4-5, TT4_g135394, BjuTT6-4, and BjuTT8-2) were upregulated by at least 2-fold in seed coat of NILB compared with SY (Figure 2, Table S7). RT-PCR analysis confirmed the differential expression profile of BjuTT3, BjuTT18, BjuANR, and BjuTT19 that was carried out using FPKM analysis (Figure 3).
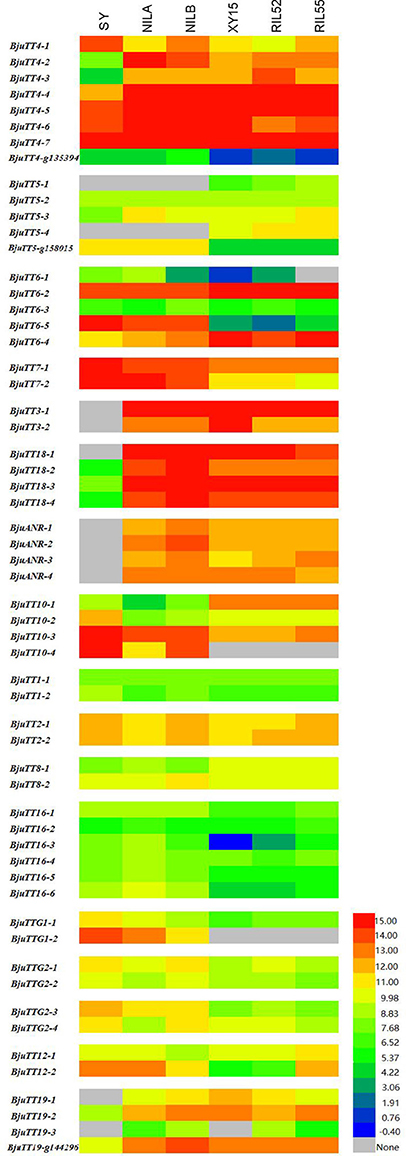
Figure 2. Expession heatmap of gene expression based on FPKM data. NILA, NILB, SY represent the seed coat of B.juncea, and XY15, RIL52, RIL55 represent the seed coat of B.nupus. The color key represents FPKM normalized log2 transformed counts.

Figure 3. RT-PCR analysis of genes for proanthocyanidin biosynthesis in the seed coats of Brassica juncea. SY, Sichuan Yellow; NILA and NILB, Near-Isogenic Lines A and B. Seed coats were separated from seeds at 15 days after pollination.
Discussion
In the present study, we identified 55, 58, and 31 PA genes in B. juncea, B. napus, and B. rapa through a combination of experimental and bioinformatics approaches, analyzed their phylogenetic relationship and genomic locations in Brassica, and detected and compared their expression in seed coats of different accessions by RNA-seq. Cloning of these genes not only lays a foundation for the elucidation of the molecular mechanism underlying PA accumulation/profile and seed pigmentation in Brassica species, but also facilitates in the functional characterization of each PA gene.
The PA genes in Arabidopsis (16) were almost doubled in B. rapa (31) and nearly quadrupled in B. juncea (55) and B. napus (58). The ancestral A, B, and C genomes of the Brassica species contributed a comparable number of PA genes. These findings are consistent with mesopolyploid nature of B. rapa and the allopolyploid nature of B. juncea and B. napus, implying that polyploidization plays an important role in expansion of PA genes. However, the number of PA genes in allopolyploid B. juncea and B. napus does not amount to the sum of PA genes from both ancestral species due to gene loss by genomic fractionation during allopolyploidization. Bra036307 and Bra009770 might have been lost in B. juncea and B. napus, respectively.
Phylogenetic analysis and genomic localization of B. juncea PA genes indicated that 30 and 29 B. juncea PA genes were homologous to genes located in the A-genome chromosomes of B. rapa and B. napus, respectively (Figure S2, Table 3). However, both BESs of 23 and 15 A-genome B. juncea PA genes were mapped around the B. rapa and B. napus genomic position, as mapped by the full-length sequence of the corresponding genes, respectively (Table 4). The other BESs were mapped to other chromosomes or not detected in the B. rapa and B. napus reference genome. These findings indicate that although B. rapa, B. juncea, and B. napus have the common A-genome, the chromosomes of each of these species do not harbor the same structure (Zou et al., 2016). On the other hand, assembly of the present reference genomes of Brassica species need improving.
For 6 of the annotated PA genes in B. juncea GSS, no BAC clones were identified. Sequence analysis revealed that the annotated genes ANR_g228640, ANR_g226654, and TT19_g167454 were false genes or artifacts that arose by misassembled sequences because these annotated genes only contain a part of the protein domains of the corresponding genes and its alignment ratios were significantly lower than other predicted genes (Table S5). No BACs carrying the annotated gene TT4_g135394, TT5_g158015, or TT19_g144296 were detected, most probably because the sequenced fragments amplified from positive BACs were too short to distinguish different members of a gene family (Table 1), or maybe because the primers used in screening the BAC library were not appropriate. In contrast, the cloned BjuTT4-1, BjuTT4-7, and BjuTT16-5 genes were not predicted from our GSS dataset, illustrating that these genes were missed in our genome sequence survey of B. juncea genome, most probably because of insufficient sequencing depth or assembly errors.
In Arabidopsis, three additional PA genes TT15 (DeBolt et al., 2009), TT9 (Ichino et al., 2014), and TT13/aha10 (Appelhagen et al., 2015) have recently been cloned. Their Brassica homologs were not investigated in the present study. In our next study, we will clone and analyze these genes to complete the set of PA genes in Brassica spp. Initial screening of our BAC library identified seven BAC clones for each of these three genes. Sequencing of the fragments amplified from these BACs is underway.
RNA-seq and FPKM analyses showed that BnaCnng01290D, BnaA09g29340D, BjuTT5-1 and BjuTT5-4 were not expressed in the seed coat, indicating that these genes might not be involved in seed pigmentation. Interestingly, the BjuTT3, BjuTT18, and BjuANR genes were not expressed in yellow-seeded testa, but expressed very high in brown-seeded testa of B. juncea (Figure 2, Table S7), which is consistent with previous results (Yan et al., 2008, 2011; Akhov et al., 2009; Liu et al., 2009, 2013; Jiang et al., 2013), suggesting that seed color is determined by expression of genes that encode enzymes that catalyze PA biosynthesis. Concomitant with the absence of expression of these enzyme-encoding genes in yellow-seeded testa, the early stage genes, BjuTT4-2 and BjuTT4-3, which encode chalcone synthase, and transporter genes, BjuTT19-1 and BjuTT19-3, which encode glutathione transferase, were remarkably downregulated or not expressed in yellow-seeded testa (Table S7). These findings illustrate that these genes are co-regulated with BjuTT3, BjuTT18, and BjuANR, and their expression is not essential to the production of biosynthetic substrates and epicatechin transport in yellow-seeded testa. Other BjuTT19 and BjuTT4 genes did not show differential expression between yellow- and brown-seeded testa (Figure 2, Table S7), implying that these genes are not involved in seed pigmentation and that their biological roles require further investigation.
To answer the questions why all the BjuTT3, BjuTT18, and BjuANR genes are not fully expressed in yellow-seeded testa and why these genes are mutated, transcriptionally regulated, or both, we also cloned full-length genomic sequences of these genes from SY and compared them with the corresponding sequences from PM. Comparative analysis showed no differences, except for a 33-bp and 2-bp difference in BjuTT18-2 and BjuTT3-1. In Arabidopsis, the genes TT3, TT18, and ANR are transcriptionally regulated by TT2-TT8-TTG1 complex (Xu et al., 2013). Comparison between SY and PM uncovered a 1275-bp insertion in exon 7 of BjuTT8-1 and a C-T transition in exon 7 of BjuTT8-2 of SY, which is almost in agreement with findings from Padmaja et al. (2014) who speculated that the TT8 gene controls seed pigmentation in B. juncea.
Conclusions
A total of 55 genes homologous to 16 Arabidopsis proanthocyandin-associated genes were identified and cloned from B. juncea. Approximately 58 and 31 PA genes were detected in B. napus and B. rapa genome databases. Around 30 of these cloned B. juncea genes were located in the A-genome chromosomes, except A04, whereas the remaining 25 were mapped to the B-genome chromosomes, except B05 and B07. A majority of these genes were expressed in the seed coat of B.juncea and B. napus. Tissue-specific expression of the TT4, TT5, and TT19 genes were observed in B. juncea and B. napus. BjuTT3, BjuTT18, BjuANR, BjuTT4-2, BjuTT4-3, BjuTT19-1, and BjuTT19-3 were transcriptionally regulated in the seed coat and not expressed or downregulated in yellow-seeded testa. In summary, the present study facilitates in better understanding the molecular mechanism underlying PA accumulation/profile and seed pigmentation, as well as in further characterization of the structure, variations, and functions of PA genes in Brassica spp.
Author Contributions
ZL and CG designed the research. XL, YL, MY, and DS performed the research and analyzed the data. XH took part in screening of the BAC library. SL and SC provided the genes primers and assisted with sequencing of the BAC clones. ZL and XL wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No.31101176 and No.31271762).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Meizhong Luo in Huazhong Agricultural University for constructing the B. juncea BAC library.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01831/full#supplementary-material
Figure S1. The PCR screening systems and process of BAC library. Screening of a specific clone by five-step PCR is shown: Step 1, screening of 19 superplates: A positive signal was detected in the superplate3 (plates SP 021–030). Steps 2–4, screening against superplate3 by 3D-PCR: Positive signals were identified in 1D, 2D, and 3D; these consisted of Plate034, C15&16, and RI&J, respectively, indicating that Plate034, column 15/16, and row I/J contained the specific BAC DNA. Step 5, screening of four candidate BACs: A positive signal was detected in the one of the four candidate BACs (ZBjuH034I15, ZBjuH034I16, ZBjuH034J15, and ZBjuH034J16). Consequently, a BAC clone containing the specific sequence was identified as the clone of ZBjuH034J15.
Figure S2. Phylogenetic trees of proanthocyanidin-associated genes from Brassica juncea, B. rapa, B. napus, and Arabidopsis thaliana. Phylogenetic reconstruction of proanthocyanidin biosynthetic genes from Brassica juncea, Arabidopsis thaliana, B. rapa, and B. napus. Phylogenetic trees were constructed from genomic sequences of PA genes using Neighbor-Joining (NJ) algorithm and 1000 bootstrap replications provided in MEGA5.2. (a) TT4; (b) TT5; (c) TT6; (d) TT7; (e) TT3; (f) TT18; (g) ANR; (h) TT10; (i) TT1; (j) TT2; (k) TT8; (l) TT16; (m) TTG1; (n) TTG2; (o) TT12; (p) TT19.
Figure S3. Sequences of annotated but unidentified proanthocyanidins-associated genes in Brassica juncea.
Table S1. Sequences of the primer pairs used in screening for proanthocyanidin-associated genes of Brassica juncea BAC library.
Table S2. Sequences of the primer pairs used in expression analysis of proanthocyanidin-associated genes of Brassica juncea.
Table S3. Global statistics of the genomic assembly of Brassica juncea.
Table S4. Annotated genes of Brassica juncea genome survey sequences (xls).
Table S5. Annotated proanthocyanidin-associated genes from Brassica juncea genome survey sequences (xls).
Table S6. Mapping to the GSS sequence of full-length sequences of Brassica juncea PA genes cloned in this study (xls).
Table S7. Transcript abundance of proanthocyanidin-associated genes in the transcriptome of Brassica napus and B. juncea seed coats.
References
Agarwal, P., Parida, S. K., Mahto, A., Das, S., Mathew, I. E., Malik, N., et al. (2014). Expanding frontiers in plant transcriptomics in aid of functional genomics and molecular breeding. Biotechnol. J. 9, 1480–1492. doi: 10.1002/biot.201400063
Akhov, L. A. L., Ashe, P. A. P., Tan, Y. T. Y., Datla, R. D. R., and Selvaraj, G. S. G. (2009). Proanthocyanidin biosynthesis in the seed coat of yellow-seeded, canola quality Brassica napus YN01-429 is constrained at the committed step catalyzed by dihydroflavonol 4-reductase. Botany 87, 616–625. doi: 10.1139/B09-036
Appelhagen, I., Nordholt, N., Seidel, T., Spelt, K., Koes, R., Quattrochio, F., et al. (2015). TRANSPARENT TESTA 13 is a tonoplast P3A -ATPase required for vacuolar deposition of proanthocyanidins in Arabidopsis thaliana seeds. Plant J. 82, 840–849. doi: 10.1111/tpj.12854
Appelhagen, I., Thiedig, K., Nordholt, N., Schmidt, N., Huep, G., Sagasser, M., et al. (2014). Update on transparent testa mutants from Arabidopsis thaliana: characterisation of new alleles from an isogenic collection. Planta 240, 955–970. doi: 10.1007/s00425-014-2088-0
Auger, B., Marnet, N., Gautier, V., Maia Grondard, A., Leprince, F., Renard, M., et al. (2010). A detailed survey of seed coat flavonoids in developing seeds of Brassica napus L. J. Agric. Food Chem. 58, 6246–6256. doi: 10.1021/jf903619v
Chai, Y. R., Lei, B., Huang, H. L., Li, J. N., Yin, J. M., Tang, Z. L., et al. (2009). TRANSPARENT TESTA 12 genes from Brassica napus and parental species: cloning, evolution, and differential involvement in yellow seed trait. Mol. Genet. Genomics 281, 109–123. doi: 10.1007/s00438-008-0399-1
Chalhoub, B., Denoeud, F., Liu, S., Parkin, I. A., Tang, H., Wang, X., et al. (2014). Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. doi: 10.1126/science.1253435
Chen, G., Deng, W., Peng, F., Truksa, M., Singer, S., Snyder, C. L., et al. (2013). Brassica napus TT16 homologs with different genomic origins and expression levels encode proteins that regulate a broad range of endothelium-associated genes at the transcriptional level. Plant J. 74, 663–677. doi: 10.1111/tpj.12151
DeBolt, S., Scheible, W. R., Schrick, K., Auer, M., Beisson, F., Bischoff, V., et al. (2009). Mutations in UDP-Glucose:sterol glucosyltransferase in Arabidopsis cause transparent testa phenotype and suberization defect in seeds. Plant Physiol. 151, 78–87. doi: 10.1104/pp.109.140582
Deng, W., Chen, G., Peng, F., Truksa, M., Snyder, C. L., and Weselake, R. J. (2012). Transparent testa16 plays multiple roles in plant development and is involved in lipid synthesis and embryo development in canola. Plant Physiol. 160, 978–989. doi: 10.1104/pp.112.198713
Deng, W., Wang, Y., Liu, Z., Cheng, H., and Xue, Y. (2014). HemI: a toolkit for illustrating heatmaps. PLoS ONE 9:e111988. doi: 10.1371/journal.pone.0111988
Falcone Ferreyra, M. L., Rius, S. P., and Casati, P. (2012). Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front. Plant Sci. 3:222. doi: 10.3389/fpls.2012.00222
Fang, J., Reichelt, M., Hidalgo, W., Agnolet, S., and Schneider, B. (2012). Tissue-specific distribution of secondary metabolites in Rapeseed (Brassica napus L.). PLoS ONE 7:e48006. doi: 10.1371/journal.pone.0048006
Friedt, W., and Snowdon, R. (2009). “Oilseed rape,” in Oil Crops, Handbook of Plant Breeding, Vol. 4, eds J. Vollmann and I. Rajcan (New York, NY: Springer), 91–126.
Guo, N., Cheng, F., Wu, J., Liu, B., Zheng, S., Liang, J., et al. (2014). Anthocyanin biosynthetic genes in Brassica rapa. BMC Genomics 15:426. doi: 10.1186/1471-2164-15-426
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., and Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hirakawa, H., Okada, Y., Tabuchi, H., Shirasawa, K., Watanabe, A., Tsuruoka, H., et al. (2015). Survey of genome sequences in a wild sweet potato, Ipomoea trifida (H. B. K.) G. Don. DNA Res. 22, 171–179. doi: 10.1093/dnares/dsv002
Ichino, T., Fuji, K., Ueda, H., Takahashi, H., Koumoto, Y., Takagi, J., et al. (2014). GFS9/TT9 contributes to intracellular membrane trafficking and flavonoid accumulation in Arabidopsis thaliana. Plant J. 80, 410–423. doi: 10.1111/tpj.12637
Jiang, J., Shao, Y., Li, A., Lu, C., Zhang, Y., and Wang, Y. (2013). Phenolic composition analysis and gene expression in developing seeds of yellow- and black-seeded Brassica napus. J. Integr. Plant Biol. 55, 537–551. doi: 10.1111/jipb.12039
Jiao, Y., Jia, H. M., Li, X. W., Chai, M. L., Jia, H. J., Chen, Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13:201. doi: 10.1186/1471-2164-13-201
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Lepiniec, L., Debeaujon, I., Routaboul, J. M., Baudry, A., Pourcel, L., Nesi, N., et al. (2006). Genetics and biochemistry of seed flavonoids. Annu. Rev. Plant Biol. 57, 405–430. doi: 10.1146/annurev.arplant.57.032905.105252
Li, Q., Yin, M., Li, Y., Fan, C., Yang, Q., Wu, J., et al. (2015). Expression of Brassica napus TTG2, a regulator of trichome development, increases plant sensitivity to salt stress by suppressing the expression of auxin biosynthesis genes. J. Exp. Bot. 66, 5821–5836. doi: 10.1093/jxb/erv287
Liu, S., Liu, Y., Yang, X., Tong, C., Edwards, D., Parkin, I. A., et al. (2014). The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5:3930. doi: 10.1038/ncomms4930
Liu, X. J., Yuan, M. Z., Guan, C. Y., Chen, S. Y., and Liu, S. Y. (2009). Inheritance, Mapping, and Origin of Yellow-Seeded Trait in Brassica juncea. Acta Agron. Sin. 35, 839–847. doi: 10.3724/SP.J.1006.2009.00839
Liu, X., Lu, Y., Yuan, Y., Liu, S., Guan, C., Chen, S., et al. (2013). De novo transcriptome of Brassica juncea seed coat and identification of genes for the biosynthesis of flavonoids. PLoS ONE 8:e71110. doi: 10.1371/journal.pone.0071110
Lu, Y., Liu, X., Liu, S., Yue, Y., Guan, C., and Liu, Z. (2012). A simple and rapid procedure for identification of seed coat colour at the early developmental stage of Brassica juncea and Brassica napus seeds. Plant Breed. 131, 176–179. doi: 10.1111/j.1439-0523.2011.01914.x
Luo, M., and Wing, R. A. (2003). An improved method for plant BAC library construction. Methods Mol. Biol. 236, 3–20. doi: 10.1385/1-59259-413-1:3
Martin, L. B., Fei, Z., Giovannoni, J. J., and Rose, J. K. (2013). Catalyzing plant science research with RNA-seq. Front. Plant Sci. 4:66. doi: 10.3389/fpls.2013.00066
Moreno-Hagelsieb, G., and Latimer, K. (2008). Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics 24, 319–324. doi: 10.1093/bioinformatics/btm585
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nesi, N., Lucas, M. O., Auger, B., Baron, C., Lécureuil, A., Guerche, P., et al. (2009). The promoter of the Arabidopsis thaliana BAN gene is active in proanthocyanidin-accumulating cells of the Brassica napus seed coat. Plant Cell Rep. 28, 601–617. doi: 10.1007/s00299-008-0667-x
Padmaja, L. K., Agarwal, P., Gupta, V., Mukhopadhyay, A., Sodhi, Y. S., Pental, D., et al. (2014). Natural mutations in two homoeologous TT8 genes control yellow seed coat trait in allotetraploid Brassica juncea (AABB). Theor. Appl. Genet. 127, 339–347. doi: 10.1007/s00122-013-2222-6
Saito, K., Yonekura-Sakakibara, K., Nakabayashi, R., Higashi, Y., Yamazaki, M., Tohge, T., et al. (2013). The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. doi: 10.1016/j.plaphy.2013.02.001
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Senda, M., Kurauchi, T., Kasai, A., and Ohnishi, S. (2012). Suppressive mechanism of seed coat pigmentation in yellow soybean. Breed. Sci. 61, 523–530. doi: 10.1270/jsbbs.61.523
Stanke, M., and Waack, S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl. 2), ii215–ii225. doi: 10.1093/bioinformatics/btg1080
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Velasco, L., and Ferna‘ndez-Martı’nez, J. (2009). “Other brassicas,” in Oil Crops, Handbook of Plant Breeding, Vol. 4, eds J. Vollmann and I. Rajcan (New York, NY: Springer), 127–153.
Velasco, R., Zharkikh, A., Troggio, M., Cartwright, D. A., Cestaro, A., Pruss, D., et al. (2007). A high quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2:e1326. doi: 10.1371/journal.pone.0001326
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039. doi: 10.1038/ng.919
Weber, A. P. (2015). Discovering new biology through sequencing of RNA. Plant Physiol 169, 1524–1531. doi: 10.1104/pp.15.01081
Wei, Y. L., Li, J. N., Lu, J., Tang, Z. L., Pu, D. C., and Chai, Y. R. (2007). Molecular cloning of Brassica napus TRANSPARENT TESTA 2 gene family encoding potential MYB regulatory proteins of proanthocyanidin biosynthesis. Mol. Biol. Rep. 34, 105–120. doi: 10.1007/s11033-006-9024-8
Winkel-Shirley, B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. doi: 10.1104/pp.126.2.485
Xu, W., Grain, D., Le Gourrierec, J., Harscoet, E., Berger, A., Jauvion, V., et al. (2013). Regulation of flavonoid biosynthesis involves an unexpected complex transcriptional regulation of TT8 expression, in Arabidopsis. New Phytol. 198, 59–70. doi: 10.1111/nph.12142
Yan, M., Liu, X., Guan, C., Liu, L., Xiang, J., Lu, Y., et al. (2014). Cloning of TTG1 gene and PCR identification of genomes A, B and C in Brassica species. Genetica 142, 169–176. doi: 10.1007/s10709-014-9764-7
Yan, M. L., Liu, X. J., Guan, C. Y., Chen, X. B., and Liu, Z. S. (2011). Cloning and expression analysis of an anthocyanidin synthase gene homolog from Brassica juncea. Mol. Breed. 28, 313–322. doi: 10.1007/s11032-010-9483-4
Yan, M. L., Liu, X. J., Liu, Z. S., Guan, C. Y., Yuan, M. Z., and Xiong, X. H. (2008). Cloning and expression analysis of Dihydroflavonol 4-Reductase gene in Brassica juncea. Acta Agron. Sin. 34, 1–7. doi: 10.3724/SP.J.1006.2008.00001
Yang, J., Song, N., Zhao, X., Qi, X., Hu, Z., and Zhang, M. (2014). Genome survey sequencing provides clues into glucosinolate biosynthesis and flowering pathway evolution in allotetrapolyploid Brassica juncea. BMC Genomics 15:107. doi: 10.1186/1471-2164-15-107
Yang, K., Jeong, N., Moon, J. K., Lee, Y. H., Lee, S. H., Kim, H. M., et al. (2010). Genetic analysis of genes controlling natural variation of seed coat and flower colors in soybean. J. Hered. 101, 757–768. doi: 10.1093/jhered/esq078
Yi, J., Derynck, M. R., Chen, L., and Dhaubhadel, S. (2010). Differential expression of CHS7 and CHS8 genes in soybean. Planta 231, 741–753. doi: 10.1007/s00425-009-1079-z
Zhang, J., Lu, Y., Yuan, Y., Zhang, X., Geng, J., Chen, Y., et al. (2009). Map-based cloning and characterization of a gene controlling hairiness and seed coat color traits in Brassica rapa. Plant Mol. Biol. 69, 553–563. doi: 10.1007/s11103-008-9437-y
Zhang, K., Lu, K., Qu, C., Liang, Y., Wang, R., Chai, Y., et al. (2013). Gene silencing of BnTT10 family genes causes retarded pigmentation and lignin reduction in the seed coat of Brassica napus. PLoS ONE 8:e61247. doi: 10.1371/journal.pone.0061247
Keywords: Brassica spp., proanthocyanidin biosynthesis, gene cloning, BAC library, seed color
Citation: Liu X, Lu Y, Yan M, Sun D, Hu X, Liu S, Chen S, Guan C and Liu Z (2016) Genome-Wide Identification, Localization, and Expression Analysis of Proanthocyanidin-Associated Genes in Brassica. Front. Plant Sci. 7:1831. doi: 10.3389/fpls.2016.01831
Received: 13 August 2016; Accepted: 21 November 2016;
Published: 09 December 2016.
Edited by:
Maoteng Li, Huazhong University of Science and Technology, ChinaReviewed by:
Chunyu Zhang, Huazhong Agricultural University, ChinaQing-Yong Yang, Huazhong Agricultural University, China
Copyright © 2016 Liu, Lu, Yan, Sun, Hu, Liu, Chen, Guan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongsong Liu, enNsaXU0OEBzb2h1LmNvbQ==
†These authors have contributed equally to this work.