 Rui-Sen Lu
Rui-Sen Lu Pan Li
Pan Li Ying-Xiong Qiu
Ying-Xiong Qiu- Key Laboratory of Conservation Biology for Endangered Wildlife of the Ministry of Education, College of Life Sciences, Zhejiang University, Hangzhou, China
The genus Cardiocrinum (Endlicher) Lindley (Liliaceae) comprises three herbaceous perennial species that are distributed in East Asian temperate-deciduous forests. Although all three Cardiocrinum species have horticultural and medical uses, studies related to species identification and molecular phylogenetic analysis of this genus have not been reported. Here, we report the complete chloroplast (cp) sequences of each Cardiocrinum species using Illumina paired-end sequencing technology. The cp genomes of C. giganteum, C. cathayanum, and C. cordatum were found to be 152,653, 152,415, and 152,410 bp in length, respectively, including a pair of inverted repeat (IR) regions (26,364–26,500 bp) separated by a large single-copy (LSC) region (82,186–82,368 bp) and a small single-copy (SSC) region (17,309–17,344 bp). Each cp genome contained the same 112 unique genes consisting of 30 transfer RNA genes, 4 ribosomal RNA genes, and 78 protein-coding genes. Gene content, gene order, AT content, and IR/SC boundary structures were almost the same among the three Cardiocrinum cp genomes, yet their lengths varied due to contraction/expansion of the IR/SC borders. Simple sequence repeat (SSR) analysis further indicated the richest SSRs in these cp genomes to be A/T mononucleotides. A total of 45, 57, and 45 repeats were identified in C. giganteum, C. cathayanum, and C. cordatum, respectively. Six cpDNA markers (rps19, rpoC2-rpoC1, trnS-psbZ, trnM-atpE, psaC-ndhE, ycf15-ycf1) with the percentage of variable sites higher than 0.95% were identified. Phylogenomic analyses of the complete cp genomes and 74 protein-coding genes strongly supported the monophyly of Cardiocrinum and a sister relationship between C. cathayanum and C. cordatum. The availability of these cp genomes provides valuable genetic information for further population genetics and phylogeography studies on Cardiocrinum.
Introduction
The tribe Lilieae sensu Tamura (1998) belongs to Liliaceae sensu APG III (Angiosperm Phylogeny Group, 2009), and contains five genera: Lilium L., Nomocharis Franch., Fritillaria L., Notholirion Wallich ex Boissier, and Cardiocrinum (Endlicher) Lindley (Gao et al., 2012). This tribe is characterized by papillose tepals (except Fritillaria) and numerous fleshy bulb-scales, as well as a morphologically distinct karyotype (Tamura, 1998). Among the five genera, Cardiocrinum, the subject of our study, is a small genus of bulbous plants, comprising three species: C. giganteum (Wall.) Makino, C. cathayanum (E. H. Wilson) Stearn, and C. cordatum (Thunb.) Makino. These species are long-lived, monocarpic, perennial herbs of East Asian temperate broad-leaved deciduous forests, and mainly differ in individual height, manner of flowering, floral characteristics (e.g., flower number/size/shape, bracts caducous vs. persistent) and geographic distribution (Ohara et al., 2006). Two of them, C. giganteum and C. cathayanum, form a parapatric species pair with abutting ranges in central China. The former is scattered in isolated patches across the Himalaya—Hengduan Mountains (including Bhutan, northeast India, Myanmar, Nepal, Sikkim), Southwest, and Central China (Phartyal et al., 2012), whereas C. cathayanum mainly occurs in isolated stands of montane deciduous forests in Southeast China. By contrast, C. cordatum is native to Japan and certain islands in the Russian Far East (Sakhalin, Kuriles; Araki et al., 2010). All three species of Cardiocrinum have self-compatible, visually showy flowers, and are insect (many bumblebee species) pollinated flowers that mature into capsules containing several 100 seeds with thin filmy wings (Ohara et al., 2006). Despite taxonomic recognition of three distinct species within the genus, the possibility of hybridization has long been suspected from morphological and/or distributional considerations, especially between the parapatric species pair C. giganteum and C. cathayanum with abutting ranges in Central China. In addition, although recent molecular phylogenetic studies supported the monophyly of Lilieae and recovered Cardiocrinum spp. as one of the early diverging lineages (Hayashi and Kawano, 2000; Patterson and Givnish, 2002; Gao et al., 2012; Kim et al., 2013), species relationships within Cardiocrinum largely remained unclear because usually only C. giganteum was included in all previous studies. Therefore, it is necessary to construct a robust phylogenetic tree of Cardiocrinum to facilitate a better understanding of the speciation, diversification, and biogeography of the genus in East Asia.
Cardiocrinum species are widely grown as ornamental plants in temperate regions of the Northern Hemisphere for their large and gorgeous flowers (Phartyal et al., 2012). On the other hand, they are known to contain bioactive compounds, such as isopimarane-type diterpenoids (Liu, 1984) and inhibitors of 5-lipoxygenase activation, as well as high levels of various trace elements, such as Ca, Mg, Fe, and Zn (Wang et al., 2007). In China, Cardiocrinum species are locally used as medicinal plants and food sources. For example, Cardiocrinum seeds have been proven to be a potential herbal replacement for Aristolochia fruits in treating cough (Li et al., 2010); and the starchy bulbs of C. giganteum are the staple food of local people in Guangxi and Yunnan (Li, 1997). The great economic value of Cardiocrinum species has brought about overexploitation and habitat fragmentation/isolation of their natural populations (Li et al., 2012), which might decrease not only population size but also genetic diversity. Despite of its ecological and economic importance, molecular research of Cardiocrinum has lagged far behind. So far, only a few microsatellite loci have been developed for C. cordatum and C. giganteum (Abdoullaye et al., 2010; Li et al., 2012). Evidently more effective molecular markers are needed to foster efforts regarding the identification, conservation, utilization, and breeding of Cardiocrinum species in the context of phylogeographic and population genetic analyses.
Chloroplasts, derived from photosynthetic bacteria, have their own genomes encoding an array of proteins in relation to photosynthesis, nitrogen fixation and biosynthesis of starch, pigments, fatty acids, and amino acids (Neuhaus and Emes, 2000; Howe et al., 2003; Liu et al., 2012). In contrast to nuclear genomes, plant chloroplast genomes show high copy numbers per cell and a much smaller size for complete sequencing (McNeal et al., 2006). The chloroplast genomes in angiosperms usually have a circular structure ranging from 115 to 165 kb in length and consist of two copies of a large inverted repeat (IR) region separated by a large single-copy (LSC) region and a small single-copy (SSC) region (Raubeson and Jansen, 2005; Wicke et al., 2011; Shetty et al., 2016). Due to the lack of recombination, low rates of nucleotide substitutions, and usually uniparental inheritance, chloroplast DNA sequences are a primary source of data for inferring plant phylogenies (Shaw et al., 2005). With the development of next-generation sequencing (NGS) technology, it is now more convenient to obtain complete chloroplast genome sequences and promptly extend gene-based phylogenetics to phylogenomics. Whole chloroplast genomes are increasingly being used for phylogenetic analyses and have proven to be effective in resolving evolutionary relationships, especially at lower taxonomic levels where recent divergence, and rapid radiations have resulted in limited sequence variation by using traditional methods (Cai et al., 2015; Ruhsam et al., 2015).
Here, we present the complete and annotated DNA sequences for the cp genomes of the three Cardiocrinum species. Our study aims were as follows: (1) to investigate global structural patterns of Cardiocrinum cp genomes; (2) examine variations of simple sequence repeats (SSRs) and repeat sequences among the three Cardiocrinum cp genomes; (3) to evaluate the morphology-based classification of Cardiocrinum species and resolve their phylogenetic relationships using the chloroplast genome sequence data; and (4) to screen fast evolving DNA regions among the three chloroplast genomes. The results will provide abundant information for the identification as well as phylogenetic, phylogeographic and population genetic studies of Cardiocrinum species, and aid in the conservation and utilization of their genetic resources.
Materials and Methods
Plant Material and DNA Extraction
Fresh leaves of C. giganteum from Sichuan Province (China), C. cathayanum from Zhejiang Province (China), and C. cordatum from the Miyazaki Prefecture (Japan) were sampled and dried with silica gel. Voucher specimens were deposited in the Herbarium of Zhejiang University (HZU). Genomic DNA was extracted from approximately 3 mg of the silica-dried leaf tissue using DNA Plantzol Reagent (Invitrogen) according to the manufacturer's protocol. The quality and concentration of the DNA products were assessed using agarose gel electrophoresis and an Agilent BioAnalyzer 2100 (Agilent Technologies).
DNA Sequencing and Genome Assembly
Purified DNA was used to generate short-insert (500 bp) paired-end sequencing libraries according to the Illumina standard protocol. Genomic DNA from each species was indexed by tags and pooled together in one lane of an HiSeq™ 2000 (Illumina, San Diego, California, USA) for sequencing at Beijing Genomics Institute (BGI, Shenzhen, China). For each species, approximately 2.0 Gb of raw data were generated with pair-end 125 bp read length. The raw reads were assembled into whole chloroplast genomes in a multi-step approach employing a modified pipeline that involved a combination of both reference guided and de novo assembly approaches (Cronn et al., 2008). First, paired-end sequence reads were trimmed to remove low-quality bases (Q < 20, 0.01 probability error) and adapter sequences using CLC-quality trim tool (quality_trim software included in CLC ASSEMBLY CELL package, http://www.clcbio.com/products/clc-assembly-cell/) before undertaking sequence assembly. Second, the contigs were assembled using CLC de novo assembler with the following optimized parameters: bubble size of 98, minimum contig length of 200, mismatch cost of 2, deletion and insertion costs of 3, length fraction of 0.9, and similarity fraction of 0.8. Third, all the contigs were aligned to the reference chloroplast genome of L. longiflorum (KC968977) using BLAST (http://blast.ncbi.nlm.nih.gov/), and aligned contigs (≥90% similarity and query coverage) were ordered according to the reference chloroplast genome. Then, contigs were aligned with the reference genome to construct the draft chloroplast genome of each species in Geneious 9.0.5 software (http://www.geneious.com). Finally, clean reads were remapped to the draft genome sequences and yield the complete chloroplast genome sequences.
Genome Annotation and Whole Genome Comparison
The chloroplast genomes were annotated by using the program DOGMA (Dual Organellar GenoMe Annotator; Wyman et al., 2004), coupled with manual corrections for start and stop codons. Protein-coding genes were identified by using the plastid/bacterial genetic code. Intron/exon boundaries were further determined using MAFFT v7 (Katoh and Standley, 2013) with those of the chloroplast genomes of L. longiflorum and Fritillaria hupehensis Hsiao et K. C. Hsia (NC024736) as references. We also used the program tRNAscan-SE (Schattner et al., 2005) with default settings to verify tRNA boundaries identified by DOGMA. The graphical maps of the Cardiocrinum chloroplast genomes were drawn using the OrganellarGenome DRAW tool (ORDRAW; Lohse et al., 2007), with subsequent manual editing.
The mVISTA program (http://genome.lbl.gov/vista/mvista/submit.shtml) was used to compare the complete plastid genome of C. giganteum with those of C. cathayanum and C. cordatum, taking the annotation of the chloroplast genome of Lilium longiflorum as a reference. Default parameters were utilized to align the chloroplast genomes in Shuffle-LAGAN mode and a sequence conservation profile was visualized in an mVISTA plot (Frazer et al., 2004). To explore the divergence hotspot regions in Cardiocrinum and facilitate its utilization in identification, all the regions, including coding regions, introns and intergenic spacers, were sequentially extracted under the following two criteria: (a) total number of mutation (Eta) > 0; and (b) an aligned length >200 bp. The nucleotide variability was calculated with DnaSP 5.10 (Librado and Rozas, 2009). Any large structural events, such as gene order rearrangements and IR expansions/contractions, were recorded.
Characterization of Repeat Sequences and SSRs
Size and location of repeat sequences, including direct (forward), inverted (palindromic), complement, and reverse repeats in the Cardiocrinum chloroplast genomes were identified by running REPuter (Kurtz and Schleiermacher, 1999). For all the repeat types, the constraint set in REPuter was 90% or greater sequence identity with hamming distance equal to 3. Simple sequence repeats (SSRs) were detected using MISA perl script (Thiel et al., 2003) with thresholds of 10 repeat units for mononucleotide SSRs, 5 repeat units for dinucleotide SSRs, 4 repeat units for trinucleotide SSRs, and 3 repeat units for tetra-, penta-, and hexa-nucleotide SSRs.
Phylogenetic Analysis
Altogether the complete chloroplast genome sequences of 12 species from Liliaceae were used for phylogenetic analysis (Table S1), including four Fritillaria species, four Lilium species, Erythronium sibiricum (Fisch. & C.A.Mey.) Krylov (P. Li, unpublished data) and the three Cardiocrinum species sequenced here (Table S1). Because of the close relationship of Liliaceae and Smilacaceae, Smilax china L. of Smilacaceae (Liu et al., 2012) was included as outgroup (Table S1). The sequences were aligned using MAFFT v7 (Katoh and Standley, 2013) and manually edited where necessary. The unambiguously aligned DNA sequences were used for phylogenetic tree construction. In order to examine the phylogenetic utility of different regions, phylogenetic analyses were performed using Maximum likelihood (ML) and Bayesian inference (BI) methods based on the following two data sets: (1) the complete chloroplast genome sequences; and (2) a set of 74 protein-coding genes shared by the chloroplast genomes of the13 species (Table S2). In both analyses, all the gaps were excluded after alignment.
We analyzed the above two data matrices under both ML and BI frameworks using an unpartitioned strategy. In addition, we also conducted partitioned analyses for 74-gene data set using two model partitionin strategies: (1) partitioning by codon position (three partitions), and (2) partitioning by each gene (74 partitions). ML analysis was conducted using RAxML-HPC v8.2.8 with 1000 bootstrap replicates on the CIPRES Science Gateway website (Miller et al., 2010). Akaike Information Criterion (AIC) in jModelTest v2.1.4 (Posada, 2008) was used to determine the best-fitting models of nucleotide substitutions and a GTR + G + I substitution model was selected for both data sets. BI analyses were conducted in MrBayes v3.2 (Ronquist and Huelsenbeck, 2003). The Markov chain Monte Carlo (MCMC) algorithm was run for two million generations with trees sampled every 500 generations. The first 25% of generations were discarded as burn-in. A 50% majority-rule consensus tree was constructed from the remaining trees to estimate posterior probabilities (PPs).
Results and Discussion
Genome Organization and Features
Illumina paired-end (125 bp) sequencing produced 16,593,274, 17,071,940 and 16,590,680 clean reads for C. giganteum, C. cathayanum, C. cordatum, respectively. The de novo assembly generated 17,157 contigs with an N50 length of 351 bp and a total length of 6.38 Mb for C. cathayanum, 20,859 contigs with an N50 length of 366 bp and a total length of 8.25 Mb for C. cordatum, and 26,859 contigs with an N50 length of 391 bp and a total length of 11.55 Mb for C. giganteum (Table 1). Each draft chloroplast genome was generated from a combined product of four initial contigs, with no gaps and no Ns. The determined nucleotide sequences of the three Cardiocrinum chloroplast genomes ranged narrowly from 152,410 bp in C. cordatum to 152,653 bp in C. giganteum (Figure 1, Table 1). All three chloroplast genomes exhibited the general quadripartite structure typical of angiosperms, consisting of a pair of IRs (26,364–26,500 bp) separated by the LSC (82,186–82,368 bp) and SSC (17,309–17,319 bp) regions. The chloroplast genome sequences were deposited in GenBank (accession numbers, KX528334 for C. giganteum, KX575836 for C. cathayanum, and KX575837 for C. cordatum).
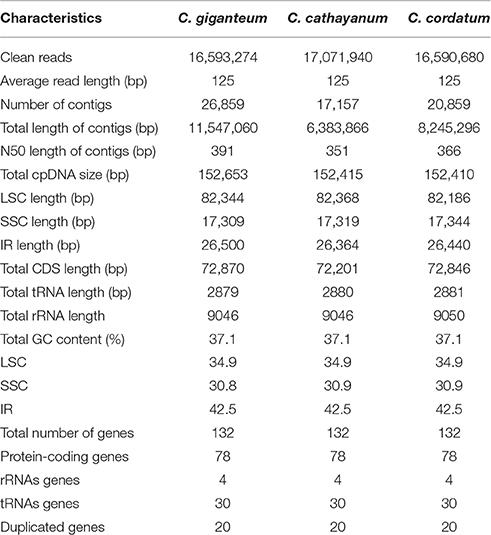
Table 1. The basic characteristics of three Cardiocrinum chloroplast genomes.
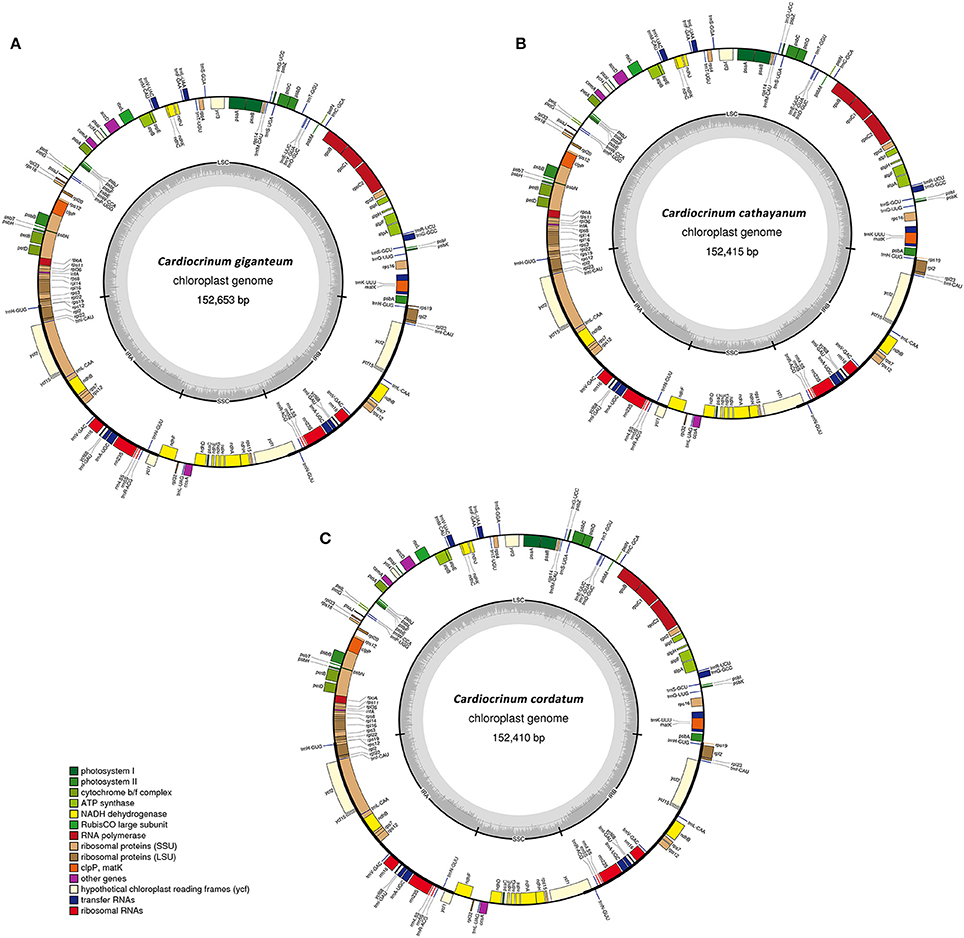
Figure 1. Gene maps of the three Cardiocrinum chloroplast genomes. (A) Cardiocrinum giganteum; (B) C. cathayanum; (C) C. cordatum. Genes shown on the outside of the circle are transcribed clockwise, and genes inside are transcribed counter-clockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner corresponds to GC content, and the lighter gray corresponds to AT content.
The three Cardiocrinum chloroplast genomes encoded an identical set of 132 genes, of which 112 were unique and 20 were duplicated in the IR regions (Table 2), and the arrangements of these 132 genes in them were totally collinear. The 112 unique genes included 78 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. Protein-coding regions accounted for 47.37–47.80% of the whole genome, while tRNA and rRNA regions accounted for 1.89 and 5.93–5.94%, respectively (Table 1). The remaining regions were non-coding sequences, including intergenic spacers, introns, and pseudogenes. The overall GC content was 37.1%, whereas the GC content in the LSC, SSC and IR regions were 34.9, 30.8–30.9, and 42.5%, respectively (Table 1), indicating nearly identical levels among the three Cardiocrinum chloroplast genomes. The GC content of the Cardiocrinum chloroplast genomes is close to that reported in other Liliales chloroplast genomes (Liu et al., 2012; Do et al., 2013; Kim and Kim, 2013).

Table 2. Gene composition of Cardiocrinum chloroplast genomes.
Nine of the protein-coding genes and six of the tRNA genes possessed a single intron, whereas three genes (rps12, clpP, and ycf3) contained two introns (Table 2). All the protein-coding genes had standard AUG as initiator codon. The gene rps12 was trans-spliced; the 5′ end exon was located in the LSC region and the 3′ exon and intron were duplicated and located in the IR regions. The infA region that contained several internal stop codons and is thus interpreted as pseudogenes. The pseudogenization of infA is also found in other angiosperm chloroplast genomes (Schmitz-Linneweber et al., 2001; Sloan et al., 2014; Raman and Park, 2015). Whether or not ycf68 and ycf15 occur as pseudogenes or protein-coding genes has already been discussed in previous studies (Raubeson et al., 2007). In general, based only on their sequence conservation over broad evolutionary distances and lack of internal stop codons, the two regions (ycf15 and ycf68) have been hypothesized to represent functional protein-coding genes (Raubeson et al., 2007). However, in the present study, they appear as pseudogenes because their coding sequences (CDS) contain several internal stop codons. Thus, the sequences of ycf15 and ycf68 are not annotated in the Cardiocrinum genomes. Furthermore, the rps19 gene, located in the boundary region between LSC and IRa, has apparently lost its protein-coding ability due to partial gene duplication. The same phenomenon was also found in the ycf1 gene at the SSC and IRb border (Figure 2).
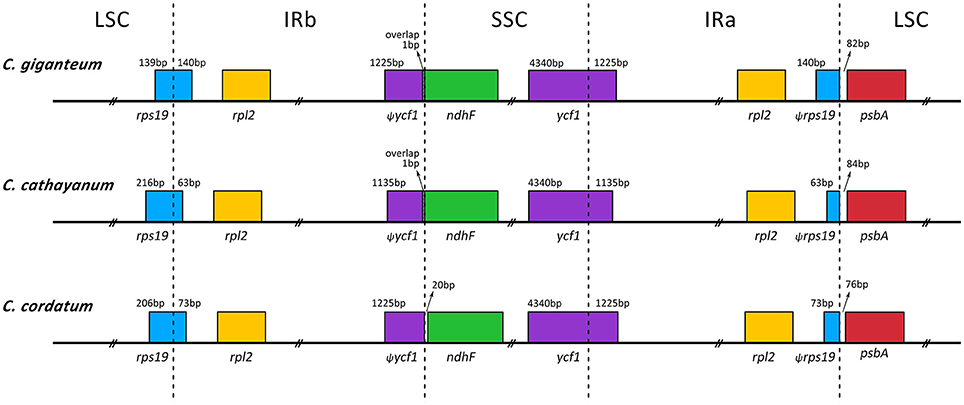
Figure 2. Comparison of LSC, IR, and SSC junction positions among the three Cardiocrinum chloroplast genomes.
Contraction and Expansion of Inverted Repeats (IRs)
Generally, the lengths of IR (IRa and IRb) regions differ among various plant species. The expansion and contraction of the IR regions and the single-copy (SC) boundary regions often results in length variation of angiosperm chloroplast genomes (Kim and Lee, 2004). We compared exact IR/SC border positions and their adjacent genes among the three Cardiocrinum chloroplast genomes (Figure 2). Although overall genomic structure including gene number and gene order was well-conserved, the three Cardiocrinum chloroplast genomes exhibited obvious differences at the IR/SC boundary regions (Figure 2). The IR region expanded into the rps19 gene, creating a pseudogene fragment ψrps19 at the IRa/LSC border with lengths of 63–140 bp (C. giganteum: 140 bp; C. cathayanum: 63 bp; C. cordatum: 73 bp). The ycf1 gene crossed the SSC/IRa region and the pseudogene fragment ψycf1 was located at the IRb region with 1135–1225 bp. For C. giganteum and C. cathayanum chloroplast genomes, the ndhF gene and the ψycf1 fragment overlapped by 1 bp at the junction of the IRa and SSC regions. However, for C. cordatum, the ndhF gene was entirely located in the SSC region and the distance between ndhF and ψycf1 was 20 bp (Figure 2).
Comparative Genomic Analysis of the Genus Cardiocrinum
Comparison of the sequences revealed several regions of high sequence length polymorphism (Figure 3). Being largely consistent with recent studies (Nazareno et al., 2015; Yao et al., 2015; Zhang et al., 2016), most of the sequence variations were found to be located in the LSC and SSC regions, while the IR regions exhibited comparatively fewer sequence variations. The lower sequence divergence observed in the IRs than SC regions for Cardiocrinum species and other angiosperms is likely due to copy correction between IR sequences by gene conversion (Khakhlova and Bock, 2006). We eventually identified 97 regions (43 coding regions, 42 intergenic spacers, and 12 introns) with more than 200 bp in length. Of these 97 regions, nucleotide variability (Pi) ranged from 0.0003 (ycf2) to 0.01927 (rpoC2-rpoC1) among the three Cardiocrinum species (Figure 4; Table S3). As found in most angiosperms (Zhang et al., 2011; Choi et al., 2016), sequence divergence in intergenic regions was higher than that in genic regions of these three chloroplast genomes. The mean value of Pi in non-coding regions was 0.42%, which was almost twice as much as in the coding regions (0.27% on average). Intergenic regions with a percentage of Pi exceeding 1% were rpoC2-rpoC1, trnS-psbZ, trnM-atpE, psaC-ndhE, and ycf15-ycf1. However, the highest proportion of variability in genic regions was 0.96% (rps19) (Figure 4; Table S3). Together, these six divergence hotspot regions should be useful for developing molecular markers for phylogenetic and phylogeographic analyses as well as plant identification of Cardiocrinum species.
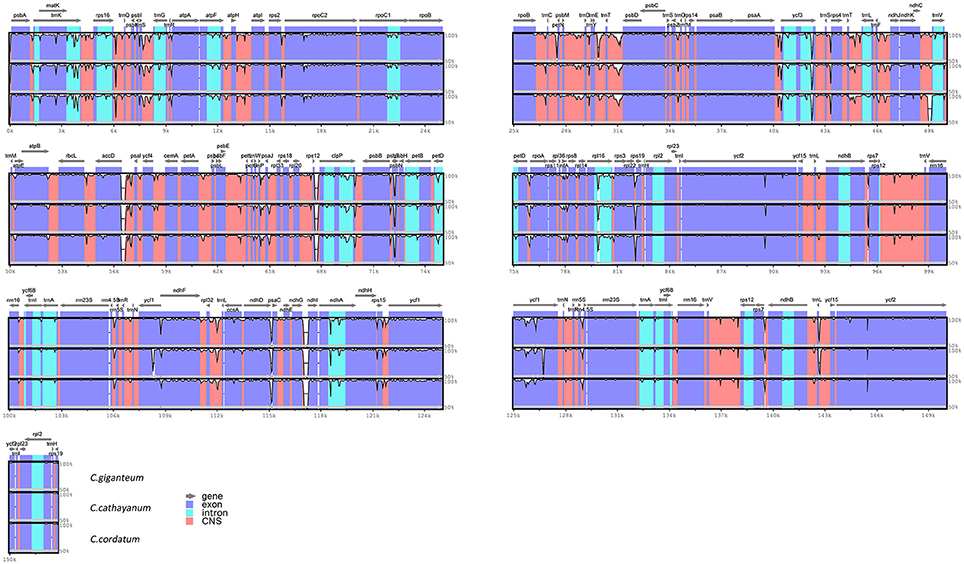
Figure 3. Sequence identity plots among the three Cardiocrinum chloroplast genomes, with Lilium longiflorum as a reference. Annotated genes are displayed along the top. The vertical scale represents the percent identity between 50 and 100%. Genome regions are color coded as exon, intron, and conserved non-coding sequences (CNS).
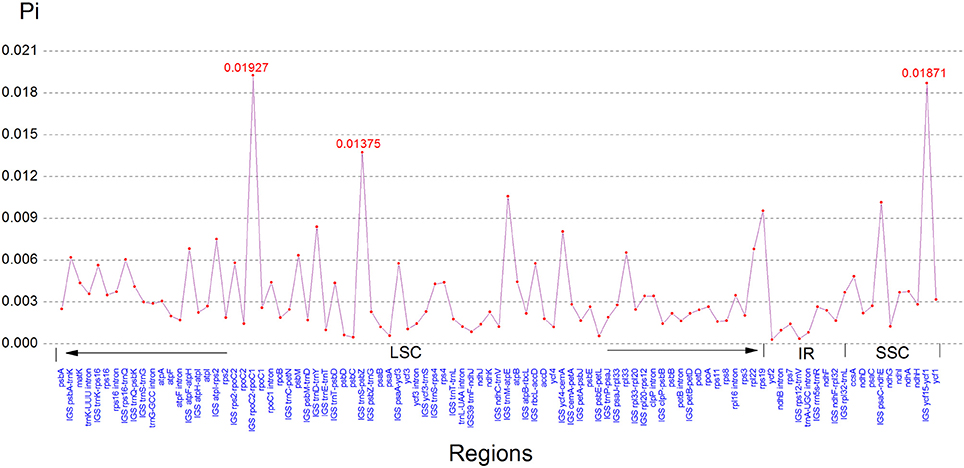
Figure 4. The nucleotide variability (Pi) values were compared among C. giganteum, C. cathayanum and C. cordatum.
Repeat Structure and SSR Analysis
A total of 147 repeats, including forward, palindromic and complement repeats, were detected in the three Cardiocrinum chloroplast genomes using REPuter (Kurtz and Schleiermacher, 1999; Figure 5A; Table S4). Cardiocrinum cathayanum contained the most repeats (57) comprising of 28 forward repeats, 28 palindromic repeats, and 1 complement repeat (Table S4). The other two species identically possessed 45 repeats (C. cordatum: 21 forward repeats, 23 palindromic repeats and 1 complement repeat; C. giganteum: 22 forward repeats, 22 palindromic repeats, and 1 complement repeat). The majority of repeats (88.9%) ranged from 30 to 40 bp in size (Figure 5B; Table S4). Under the criterion with identical lengths located in homologous regions as shared repeats, we investigated those repeats shared among the three Cardiocrinum chloroplast genomes. There were 38 repeats shared by the three Cardiocrinum chloroplast genomes, 3 repeats shared by C. giganteum and C. catahayanum; 2 repeats shared by C. giganteum and C. cordatum and 1 repeat shared by C. cathayanum and C. cordatum. Additionally, C. cathayanum owned the most unique repeats (15) while C. cordatum and C. giganteum had only four and two unique repeats, respectively (Figure 5C; Table S4). Repeats located in gene ycf2 occupied 47.7% (31 repeats) of total distinct repeats and 32.3% (21 repeats) were located in non-coding regions, while some were found in genes such as psaB, rps16, ycf3, and trnS-GCU (Table S4).
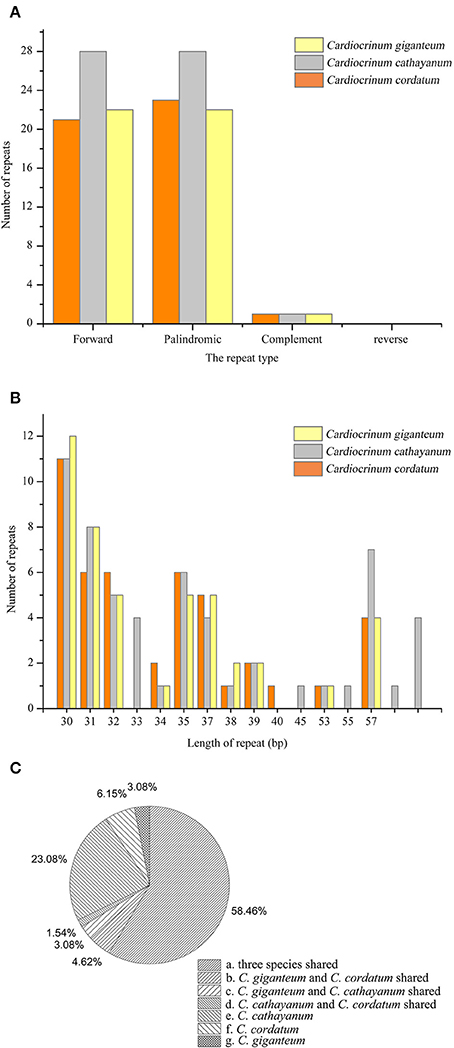
Figure 5. Analysis of repeated sequences in the three Cardiocrinum chloroplast genomes. (A) Frequency of repeats by length; (B) Frequency of repeat types; (C) Summary of the shared repeats among the Cardiocrinum cp genomes.
SSRs or microsatellites in the chloroplast genome present high diversity in copy numbers, and are important molecular markers for plant population genetics and evolutionary studies (Bodin et al., 2013; Zhao et al., 2015; Zhang et al., 2016). With MISA analysis, each Cardiocrinum chloroplast genome was found to contain 63–71 SSRs (C. giganteum: 71; C. cathayanum: 64; C. cordatum: 63) (Figure 6A; Table S5), of which 33 SSRs were the same for the three chloroplast genomes (similar repeat units located in similar genomic regions; Table S6), and the numbers of polymorphic SSRs ranged from 30 to 38. Among these SSRs, the mononucleotide A/T repeat units occupied the highest proportion with 59.2% in C. giganteum, 59.4% in C. cathayanum and 55.3% in C. cordatum (Figure 6A; Table S5). These numbers are slightly lower than those reported in previous studies on asterids (68%) and monocots (76%) (Huotari and Korpelainen, 2012; Qian et al., 2013). Among the total 198 SSRs, most loci were located in intergenic spacer (IGS) regions (60.3%), followed by CDS (23.1%) and introns (16.6%) (Figure 6B). This may be due to the fact there is a higher mutation rate in the IGS regions than the coding regions. We observed that 15 different SSRs were located in 9 protein-coding genes [ycf1 (× 5), cemA, rpoC2 (× 3), ycf2 (× 2), ndhH, rpl22, ndhD, ndhE, cemA] of the three Cardiocrinum chloroplast genomes. In general, the SSRs of these chloroplast genomes showed abundant variation, and can therefore be used in future population genetic studies of Cardiocrinum species.
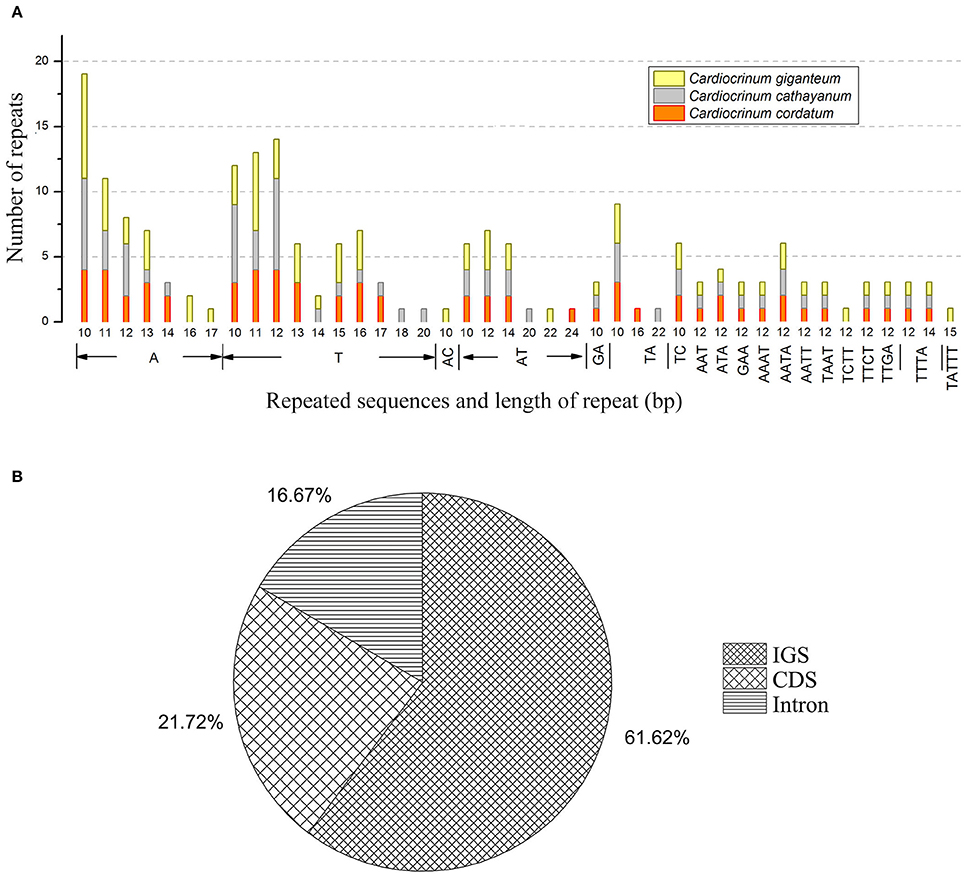
Figure 6. Simple sequence repeats (SSRs) in the three Cardiocrinum chloroplast genomes. (A) Numbers of SSRs by length; (B) Distribution of SSR loci. IGS: intergenic spacer region.
Phylogenetic Analysis
The whole chloroplast genomes and protein-coding genes have been successfully used to resolve phylogenetic relationships at almost any taxonomic level during the past decade (De Las Rivas et al., 2002; Moore et al., 2007; Zhang et al., 2016). In the present study, two data sets including 74 commonly present protein-coding genes (Table S2) and the complete chloroplast genome sequences of 12 species from Liliaceae were used to perform phylogenetic analysis, with Smilax china (Smilacaceae) used as outgroup (Table S1). The BI and ML analyses yielded nearly identical tree topologies across all analyses, with 100% bootstrap (BS) values and 1.0 Bayesian posterior probabilities (PP) at each node (Figure 7). Thus, only the phylogenetic trees based on complete genome sequences using no partitioning scheme are shown. All these phylogenetic trees identically supported the monophyly of Cardiocrinum, which in turn formed a sister clade to the Lilium+Fritillaria group. The phylogenetic trees in this study also indicate a sister relationship of Fritillaria to Lilium, which is consistent with a previous phylogenetic study based on four plastid loci (Kim et al., 2013). Within Cardiocrinum, C. giganteum from the HHM region/Southwest China was identified as sister to C. cathayanum (Southeast China)—C. cordatum (Japan, Russian Far East Islands; Figure 7). According to Wu and Wu (1998), the Sino-Japanese Floristic Region (SJFR) can be divided into two subkingdoms: the Sino-Himalayan and the Sino-Japanese Forest subkingdoms. The phylogenetic relationships in Cardiocrinum are found to be consistent with Wu and Wu (1998) floristic division. Major genetic subdivisions between the Sino-Himalayan and Sino-Japanese Forest subkingdoms have also been found in other plant taxa (e.g., Spiraea japonica complex: Zhang et al., 2006; Ainsliaea: Mitsui et al., 2008) and likewise across the East China Sea between Southeast China and Japan (e.g., Kalopanax septemlobus: Sakaguchi et al., 2012; Euptelea: Cao et al., 2016). However, considering that chloroplast genome is a haploid, uniparentally-inherited, single locus (Birky, 1995), comparative phylogenies and phylogeography between biparental (nuclear) and uniparental (chloroplast) markers are needed to elucidate the timing and processes underlying species diversification, hybridization and range evolution within Cardiocrinum. Overall, our phylogenomic analyses based on chloroplast genomes have provided the first successful attempt to clarify intrageneric relationships within Cardiocrinum. In addition, they also recovered phylogenetic relationships within the tribe Lilieae, which are consistent with previous phylogenetic results based on chloroplast and/or nuclear markers (Gao et al., 2012; Kim et al., 2013).
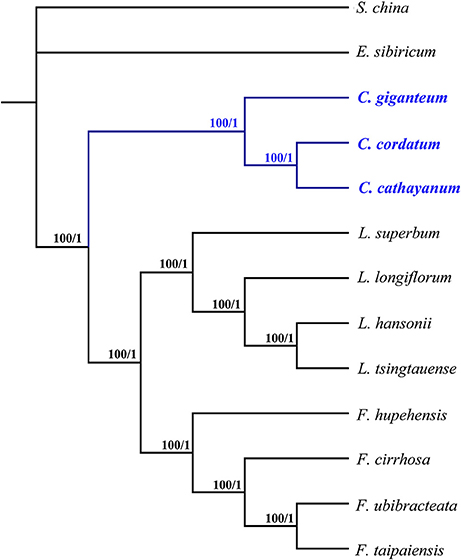
Figure 7. Phylogenetic relationships of the three Cardiocrinum species inferred from Maximum likelihood (ML) and Bayesian inference (BI) based on complete genome sequences using no partitioning scheme. Numbers above the lines represent ML bootstrap values and BI posterior probability. The phylogenetic tree based on 74 protein-coding genes is completely consistent with this topology.
Conclusions
In this study, the chloroplast genomes of the three species of Cardiocrinum are reported for the first time and their organization is described. These three chloroplast genomes exhibit typical quadripartite and circular structure that is rather conserved in genomic structure and the synteny of gene order. However, these chloroplast genomes show obvious variations at the boundaries of the four regions because of the expansion and contraction of the inverted repeat (IR) regions and the single-copy (SC) boundary regions. The six rapidly evolving regions and 147 repeat sequences identified in the Cardiocrinum chloroplast genome can be selected for future studies to develop markers and conduct phylogenetic analysis. In addition, the cp SSRs with abundant variation identified herein should be useful in characterizing the population genetic structure of Cardiocrinum species. Our phylogenomic analyses based on two data sets including 13 species from Liliaceae and Smilacaceae provided strong support for the monophyly of Cardiocrinum as sister to Fritillaria–Lilium within the tribe Lilieae. Furthermore, within Cardiocrinum, C. giganteum was identified as sister to C. cathayanum–C. cordatum, which thus reflects a biogeographically interesting phylogenetic tripartition of the genus across the SJFR. Overall, the data obtained in this study will be beneficial to expand our understanding of the evolutionary history of the tribe Lilieae in general, and the times and modes of Cardiocrinum diversification in particular.
Author Contributions
YQ conceived the ideas; RL and PL contributed to the sampling; RL performed the experiment and analyzed the data. The manuscript was written by RL and YQ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Yong-Hua Zhang, Shan-Shan Zhu and Shota Sakaguchi for their great help in collecting plant materials and Hans-Peter Comes for valuable comments on an earlier version of this manuscript. This research was supported by the National Natural Science Foundation of China (Grant Nos. 31370241, 31570214) and the International Cooperation and Exchange of the National Natural Science Foundation of China (Grant Nos. 31511140095, 31561143015).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.02054/full#supplementary-material
References
Abdoullaye, D., Acevedo, I., Adebayo, A. A., Behrmann-Godel, J., Benjamin, R. C., Bock, D. G., et al. (2010). Permanent genetic resources added to molecular ecology resources database 1 August 2009-30 September 2009. Mol. Ecol. Resour. 10, 232–236. doi: 10.1111/j.1755-0998.2009.02796.x
Angiosperm Phylogeny Group (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161, 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Araki, K., Shimatani, K., Nishizawa, M., Yoshizane, T., and Ohara, M. (2010). Growth and survival patterns of Cardiocrinum cordatum var. glehnii (Liliaceae) based on a 13-year monitoring study: Life history characteristics of a monocarpic perennial herb. Bot. Bot. 88, 745–752. doi: 10.1139/B10-041
Birky, C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. U.S.A. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Bodin, S. S., Kim, J. S., and Kim, J. H. (2013). Complete chloroplast genome of Chionographis japonica (Willd.) Maxim. (Melanthiaceae): comparative genomics and evaluation of universal primers for Liliales. Plant Mol. Biol. Rep. 31, 1407–1421. doi: 10.1007/s11105-013-0616-x
Cai, J., Ma, P. F., Li, H. T., and Li, D. Z. (2015). Complete plastid genome sequencing of four Tilia species (Malvaceae): a comparative analysis and phylogenetic implications. PLoS ONE 10:e0142705. doi: 10.1371/journal.pone.0142705
Cao, Y. N., Comes, H. P., Sakaguchi, S., Chen, L. Y., and Qiu, Y. X. (2016). Evolution of East Asia's Arcto-Tertiary relict Euptelea (Eupteleaceae) shaped by Late Neogene vicariance and Quaternary climate change. BMC Evol. Biol. 16:1. doi: 10.1186/s12862-016-0636-x
Choi, K. S., Chung, M. G., and Park, S. (2016). The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Front. Plant Sci. 7:355. doi: 10.3359/fpls.2016.00355
Cronn, R., Liston, A., Parks, M., Gernandt, D. S., Shen, R., and Mockler, T. (2008). Multiplex sequencing of plant chloroplast genomes using solexa sequencing-by-synthesis technology. Nucleic Acids Res. 36:e122. doi: 10.1093/nar/gkn502
De Las Rivas, J., Lozano, J. J., and Ortiz, A. R. (2002). Comparative analysis of chloroplast genomes: functional annotation, genome-based phylogeny, and deduced evolutionary patterns. Genome Res. 12, 567–583. doi: 10.1101/gr.209402
Do, H. D., Kim, J. S., and Kim, J. H. (2013). Comparative genomics of four Liliales families inferred from the complete chloroplast genome sequence of Veratrum patulum O. Loes. (Melanthiaceae). Gene 530, 229–235. doi: 10.1016/j.gene.2013.07.100
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, Y. D., Zhou, S. D., He, X. J., and Wan, J. (2012). Chromosome diversity and evolution in tribe Lilieae (Liliaceae) with emphasis on Chinese species. J. Plant Res. 125, 55–69. doi: 10.1007/s10265-011-0422-1
Hayashi, K., and Kawano, H. (2000). Molecular systematics of Lilium and allied genera (Liliaceae): phylogenetic relationships among Lilium and related genera based on the rbcL and matK gene sequence data. Plant Spec. Biol. 15, 73–93. doi: 10.1046/j.1442-1984.2000.00025.x
Howe, C. J., Barbrook, A. C., Koumandou, V. L., Nisbet, R. E., Symington, H. A., and Wightman, T. F. (2003). Evolution of the chloroplast genome. Philos. Trans. R. Soc. B 358, 99–106. doi: 10.1098/rstb.2002.1176
Huotari, T., and Korpelainen, H. (2012). Complete chloroplast genome sequence of Elodea canadensis and comparative analyses with other monocot plastid genomes. Gene 508, 96–105. doi: 10.1016/j.gene.2012.07.020
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software Version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j.1365-313X.2006.02673.x
Kim, J. S., Hong, J. K., Chase, M. W., Fay, M. F., and Kim, J. H. (2013). Familial relationships of the monocot order Liliales based on a molecular phylogenetic analysis using four plastid loci: matK, rbcL, atpB and atpF-H. Bot. J. Linn. Soc. 172, 5–21. doi: 10.1111/boj.12039
Kim, J. S., and Kim, J. H. (2013). Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea). PLoS ONE 8:e68180. doi: 10.1371/journal.pone.0068180
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kurtz, S., and Schleiermacher, C. (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15, 426–427. doi: 10.1093/bioinformatics/15.5.426
Li, M., Ling, K. H., Lam, H., Shaw, P. C., Cheng, L., Techen, N., et al. (2010). Cardiocrinum seeds as a replacement for Aristolochia fruits in treating cough. J. Ethnopharmacol. 130, 429–432. doi: 10.1016/j.jep.2010.04.040
Li, R., Yang, J., Yang, J., and Dao, Z. (2012). Isolation and characterization of 21 microsatellite loci in Cardiocrinum giganteum var. yunnanense (Liliaceae), an important economic plant in China. Int. J. Mol. Sci. 13, 1437–1443. doi: 10.3390/ijms13021437
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Liu, J., Qi, Z. C., Zhao, Y. P., Fu, C. X., and Xiang, Q. Y. (2012). Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales - influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 64, 545–562. doi: 10.1016/j.ympev.2012.05.010
Liu, R. M. (1984). The isopimarane-type diterpenoid from fruits of Cardiocrinum giganteum var. yunnanense. Acta Bot. Yunnanica 6, 219–222.
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
McNeal, J. R., Leebens-Mack, J. H., Arumuganathan, K., Kuehl, J. V., Boore, J. L., and DePamphilis, C. W. (2006). Using partial genomic fosmid libraries for sequencing complete organellar genomes. Biotechniques 41, 69–73. doi: 10.2144/000112202
Miller, M., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of Gateway Computing Environments Workshop (GCE), (New Orleans, LA: IEEE), 1–8.
Mitsui, Y., Chen, S. T., Zhou, Z. K., Peng, C. I., Deng, Y. F., and Setoguchi, H. (2008). Phylogeny and biogeography of the genus Ainsliaea (Asteraceae) in the Sino-Japanese region based on nuclear rDNA and plastid DNA sequence data. Ann. Bot. 101, 111–124. doi: 10.1093/aob/mcm267
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Nazareno, A. G., Carlsen, M., and Lohmann, L. G. (2015). Complete chloroplast genome of Tanaecium tetragonolobum: the first Bignoniaceae plastome. PLoS ONE 10:e0129930. doi: 10.1371/journal.pone.0129930
Neuhaus, H. E., and Emes, M. J. (2000). Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Phys. 51, 111–140. doi: 10.1146/annurev.arplant.51.1.111
Ohara, M., Narumi, T., Yoshizane, T., Okayasu, T., Masuda, J., and Kawano, S. (2006). Life-history monographs of Japanese plants. 7: Cardiocrinum cordatum (Thunb.) Makino (Liliaceae). Plant Spec. Biol. 21, 201–207. doi: 10.1111/j.1442-1984.2006.00166.x
Patterson, T. B., and Givnish, T. J. (2002). Phylogeny, concerted convergence, and phylogenetic niche conservatism in the core Liliales: insights from rbcL and ndhF sequence data. Evolution 56, 233–252. doi: 10.1554/0014-3820(2002)056
Phartyal, S. S., Kondo, T., Baskin, C. C., and Baskin, J. M. (2012). Seed dormancy and germination in the giant Himalayan lily (Cardiocrinum giganteum var. giganteum): an assessment of its potential for naturalization in northern Japan. Ecol. Res. 27, 677–690. doi: 10.1007/s11284-012-0940-x
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., Pang, X., et al. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607. doi: 10.1371/journal.pone.0057607
Raman, G., and Park, S. (2015). Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. longicalyncinus, from a comparative genomics perspective. PLoS ONE 10:e0141329. doi: 10.1371/journal.pone.0141329
Raubeson, L. A., and Jansen, R. K. (2005). “Chloroplast genomes of plants,” in Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants, ed R. J. Henry (Cambridge, MA: CABI), 45–68.
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Ruhsam, M., Rai, H. S., Mathews, S., Ross, T. G., Graham, S. W., Raubeson, L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 15, 1067–1078. doi: 10.1111/1755-0998.12375
Sakaguchi, S., Qiu, Y. X., Liu, Y. H., Qi, X. S., Kim, S. H., Han, J., et al. (2012). Climate oscillation during the quaternary associated with landscape heterogeneity promoted allopatric lineage divergence of a temperate tree kalopanax septemlobus (araliaceae) in East Asia. Mol. Ecol. 21, 3823–3838. doi: 10.1111/j.1365-294X
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schmitz-Linneweber, C., Maier, R. M., Alcaraz, J. P., Cottet, A., Herrmann, R. G., and Mache, R. (2001). The plastid chromosome of spinach (Spinacia oleracea): complete nucleotide sequence and gene organization. Plant Mol. Biol. 45, 307–315. doi: 10.1023/A:1006478403810
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Shetty, S. M., Md Shah, M. U., Makale, K., Mohd-Yusuf, Y., Khalid, N., and Othman, R. Y. (2016). Complete chloroplast genome sequence of Musa balbisiana corroborates structural heterogeneity of inverted repeats in wild progenitors of cultivated bananas and plantains. Plant Genome 9. doi: 10.3835/plantgenome2015.09.0089
Sloan, D. B., Triant, D. A., Forrester, N. J., Bergner, L. M., Wu, M., and Taylor, D. R. (2014). A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol. Phylogenet. Evol. 72, 82–89. doi: 10.1016/j.ympev.2013.12.004
Tamura, M. N. (1998). “Liliaceae,” in The Families and Genera of Vascular Plants. III. Flowering Plants-Monocotyledons, Lilianae (Except Orchidaceae), ed K. Kubitzki (Berlin: Springer Press), 343–353.
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Wang, Y. Z., Li, S. B., Guo, H. C., Fan, H. C., Sha, B. C., Chen, F. S., et al. (2007). Determination of trace elements in Cardiocrinum giganteum by FAAS. Spectrosc. Spect. Anal. 27, 1854–1857.
Wicke, S., Schneeweiss, G. M., de Pamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, Z. Y., and Wu, S. S. (1998). Floristic Characteristics and Diversity of East Asian Plants. Beijing: China Higher Education Press. [In Chinese with English Summary].
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yao, X., Tang, P., Li, Z., Li, D., Liu, Y., and Huang, H. (2015). The first complete chloroplast genome sequences in Actinidiaceae: genome structure and comparative analysis. PLoS ONE 10:e0129347. doi: 10.1371/journal.pone.0129347
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhang, Y. J., Ma, P. F., and Li, D. Z. (2011). High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6:e20596. doi: 10.1371/journal.pone.0020596
Zhang, Z., Fan, L., Yang, J., Hao, X., and Gu, Z. (2006). Alkaloid polymorphism and its sequence variation in the Spiraea japonica complex (Rosaceae) in China: traces of the biological effects of the Himalaya-Tibet plateau uplift. Am. J. Bot. 93, 762–769. doi: 10.3732/ajb.93.5.762
Keywords: Liliaceae, Cardiocrinum, chloroplast genome, genomic structure, phylogenomics, taxonomic identification
Citation: Lu R-S, Li P and Qiu Y-X (2017) The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 7:2054. doi: 10.3389/fpls.2016.02054
Received: 04 November 2016; Accepted: 22 December 2016;
Published: 10 January 2017.
Edited by:
Renchao Zhou, Sun Yat-sen University, ChinaReviewed by:
Zhi-Yong Zhang, Jiangxi Agricultural University, ChinaGoro Kokubugata, National Museum of Nature and Science, Japan
Wei-Ning Bai, Beijing Normal University, China
Copyright © 2017 Lu, Li and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying-Xiong Qiu, cXl4aGVyb0B6anUuZWR1LmNu