 Rui Liu
Rui Liu Biao Lai
Biao Lai Bing Hu
Bing Hu Yonghua Qin
Yonghua Qin Guibing Hu
Guibing Hu Jietang Zhao
Jietang Zhao- 1State Key Laboratory for Conservation and Utilization of Subtropical Agro-Bioresources, South China Agricultural University, Guangzhou, China
- 2Key Laboratory of Biology and Genetic Improvement of Horticultural Crops-South China of Ministry of Agriculture, College of Horticulture, South China Agricultural University, Guangzhou, China
Litchi (Litchi chinensis Sonn.) is an important subtropical fruit in southern China and the fruit pericarp has attractive red skin at maturity, which is provided by anthocyanins accumulation. To understand the anthocyanin biosynthesis at post-transcriptional level, we investigated the roles of microRNAs (miRNAs) during fruit coloring. In the present study, four small RNA libraries and a mixed degradome library from pericarps of ‘Feizixiao’ litchi at different developmental phases were constructed and sequenced by Solexa technology. A total of 78 conserved miRNAs belonging to 35 miRNA families and 41 novel miRNAs were identified via high-throughput sequencing, and 129 genes were identified as their targets by the recently developed degradome sequencing. miR156a and a novel microRNA (NEW41) were found to be differentially expressed during fruit coloring, indicating they might affect anthocyanin biosynthesis through their target genes in litchi. qRT-PCR analysis confirmed the expression changes of miR156a and the novel microRNA (NEW41) were inversely correlated with the expression profiles of their target genes LcSPL1/2 and LcCHI, respectively, suggesting regulatory roles of these miRNAs during anthocyanin biosynthesis. The target genes of miR156a, LcSPL1/2, encode transcription factors, as evidenced by a localization in the nucleus, that might play roles in the regulation of transcription. To further explore the relationship of LcSPL1/2 with the anthocyanin regulatory genes, yeast two-hybrid and BiFC analyses showed that LcSPL1 proteins could interact with LcMYB1, which is the key regulatory gene in anthocyanin biosynthesis in litchi. This study represents a comprehensive expression profiling of miRNAs in anthocyanin biosynthesis during litchi fruit maturity and confirmed that the miR156- SPLs module was conserved in anthocyanin biosynthesis in litchi.
Introduction
Anthocyanins, synthesized by a specific branch of the flavonoid pathway, which represent a large class of secondary metabolites, have a variety of functions within plants (Winkel-Shirley, 2001). Athocyanins biosynthesis is regulated by enzyme-coding structural genes, including chalcone synthase (CHS), chalcone isomerase (CHI), flavonoid-3′-hydroxylase (F3’H), flavonoid-3′,5′-hydroxylase (F3′5′H), flavanone 3-hydroxylase (F3H), dihydroflavonol 4-reductase (DFR), anthocyanidin synthase (ANS), UDP-glucose: flavonoid 3-O-glucosyltransferase (UFGT), and is also orchestrated at the transcriptional level by MBW transcription factor (TF) complexes consisting of MYB, bHLH (basic helix-loop-helix) and WD40 (Koes et al., 2005; Saito et al., 2013; Xu et al., 2015). Recent researches have demonstrated that the MBW complex regulates the genes that encode enzymes specifically at the late steps of the anthocyanin pathway, and the MYB TF s have been identified to be the major determinant regulatory genes in the biosynthetic steps of anthocyanin metabolism (Gonzalez et al., 2008; Lai et al., 2014; Shen et al., 2014). AtPAP1 (AtMYB75), the known anthocyanin regulator originally defined in Arabidopsis, has been shown to effectively induce anthocyanin accumulation in various plant species (Zuluaga et al., 2008; Rowan et al., 2009; Li X. et al., 2010; Qiu et al., 2014). And now the PAP1 orthologs have been identified in many horticultural crops (Cutanda-Perez et al., 2009; Chagne et al., 2012; Schaart et al., 2013; Albert et al., 2014; Jin et al., 2016). Recently, our studies suggest that the structural gene LcUFGT and its regulator LcMYB1 play major roles in anthocyanin accumulation in litchi (Lai et al., 2014, 2015). In addition to MBW complex, additional TFs and regulatory genes have also been reported to affect anthocyanin biosynthesis, such as COP1 (Maier et al., 2013), JASMONATE ZIM-domain (JAZ) genes (Qi et al., 2011), the SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) gene (Gou et al., 2011), and NAC (Zhou et al., 2015). These genes show an interaction with the MBW complex forming a regulatory network that modulates the production of anthocyanins.
Mature microRNAs (miRNAs) are a group of 20–24 nt non-coding small RNAs with important roles in plant growth, development, metabolism, and responses to stress by repressing their targets at post-transcriptional level (Voinnet, 2009; Iwakawa and Tomari, 2015). In plants, miRNAs are generally processed from longer single-stranded RNA hairpin precursors by Dicer-like proteins to produce a double-stranded RNA duplex, and then incorporated into the RNA-induced silencing complex where they negatively regulate their target mRNA through imperfect sequence complementarity (Rogers and Chen, 2013; Li et al., 2016). To date, a large number of miRNAs have been identified in different plant species and deposited in miRBase1. Acting as powerful endogenous regulators, the identification of the miRNAs and their target mRNAs could help in better understanding of the biological roles of each miRNAs on the regulatory mechanism under various plant growth and development processes. Recently, a genome-wide 5′ RACE approach, known as degradome sequencing or PARE (parallel analysis of RNA ends), has been successfully adapted to screen miRNA targets in Arabidopsis, rice, and soybean (Addo-Quaye et al., 2008; German et al., 2008; Li Y.F. et al., 2010; Song et al., 2011).
Functional analysis of miRNAs has revealed that miRNAs regulate various aspects in plant development, including leaf morphogenesis, the differentiation and development of a flower, root development, the transition from vegetative growth to reproductive growth (Jones-Rhoades et al., 2006). For example, the target genes of miR160, miR167 and miR393 regulate auxin signaling, and the regulation of AUXIN RESPONSE FACTORs (ARFs) by miR160 focuses on many aspects of shoot and root development, while miR167 appears to be important in the regulation of flowers and fruit development (Mallory et al., 2004). Besides their roles in plant development, miRNAs are also important to cope with biotic and abiotic stresses, as well as in primary and secondary metabolism (Gou et al., 2011; Li et al., 2016). Evidences suggest that miRNAs are involved in anthocyanin biosynthesis through their target genes, while only few miRNAs have been reported to be responsible for the biosynthesis of anthocyanins (Gou et al., 2011; Xia et al., 2012). In Arabidopsis, miR828 negatively controls anthocyanin accumulation by repressing the expression of MYB75, MYB90, and MYB113, which are involved in anthocyanin biosynthesis (Luo et al., 2012). miR858 has been identified in Arabidopsis, apple, cotton and tomato (Xia et al., 2012; Guan et al., 2014; Jia et al., 2015). miR858 is predicted to target up to 66 MYB factors in apple (Xia et al., 2012). Small tandem target mimic-mediated blockage of miR858 (STTM858) results in anthocyanin accumulation in tomato (Jia et al., 2015). It is well documented that miR156 negatively regulates anthocyanin accumulation by targeting the TF, SPL9 (Gou et al., 2011).
In plants, miR156 targeting a subset of SPLs is highly conserved and plays important role in plant development (Nodine and Bartel, 2010). The miR156/SPLs module also participate in the biosynthesis of phenylpropanoids, as SPL9 was reported to repress anthocyanin accumulation by destabilizing the MBW complex, and directly preventing expression of anthocyanin biosynthetic genes in Arabidopsis (Gou et al., 2011). Recently, the miR156-targeted SPL has been connected to the spatiotemporal regulation of sesquiterpene biosynthesis in Arabidopsis and patchouli (Yu et al., 2015).
Litchi (Litchi chinensis Sonn.) is one of the most popular members of the Sapindaceae fruit with sweet, juicy flesh. The pericarp of litchi is mostly red at maturity, which is provided by anthocyanins accumulation. Apart from the delicious taste, attractive red skin of litchi is another important aspect of the fruit quality. Recently, the gene involved in anthocyanin biosynthesis and sequestration from litchi has been reported (Wei et al., 2011; Lai et al., 2014, 2015, 2016; Li et al., 2015; Hu et al., 2016). However, the mechanism of miRNAs regulating anthocyanin biosynthesis in litchi has not been reported. In the present study, we identified miRNAs in litchi pericarps during fruit ripening by using Solexa sequencing, and then analyzed the miR156/SPL module and its regulatory roles in the anthocyanin biosynthesis in litchi.
Materials and Methods
Plant Materials
Litchi (L. chinesis Sonn.) cv. ‘Feizixiao’ (FZX) was collected from the Yongfa Fruit Farm, Haikou, China. Pericarp disks of six different developmental phases (Sa and Sb, green; Sc, breaker; Sd, half red and half yellow; Se and Sf, red) were collected between May 4th and May 29th, 2013 at 5 days intervals. All samples were immediately frozen in liquid nitrogen and stored at -80°C until use.
Nicotiana benthamiana plants used for subcellular localization and bimolecular fluorescence complementation (BiFC) assays were grown in greenhouses with condition of 16 h/8 h day/night at 25°C.
Anthocyanin Analysis
The total anthocyanin content was determined according to the method published by Wrolstad et al. (1982) with some modifications. Samples were added into 6 ml of HCl/methanol (v/v) leaching solution for extraction of anthocyanin (for 5 h at 25°C in darkness). Each extraction solution diluted with pH 1.0 and pH 4.5 buffers was measured by ultraviolet scenery at 530 nm. Anthocyanin contents were calculated using a molar absorbance coefficient of 29, 600 (cyaniding-3-glucoside). Each sample was replicated three times.
Small RNA and Degradome Libraries Construction
Total RNAs were extracted from litchi pericarps using the TRK-1001 total RNA purification kit (LC Science, Houston, TX, USA) according to the manufacturer’s instructions. Equal volumes of RNA extracts from Sa and Sb or Se and Sf were pooled for use as S1 or S4 for sRNA library construction and sequencing. Four libraries were denoted S1 (Sa and Sb), S2 (Sc), S3 (Sd), S4 (Se and Sf). For sRNA library construction, the sRNA fractions with the length of 10–40 nt were purified from 15% denaturing polyacrylamide gel. The purified sRNA was ligated to a 5′ adaptor and a 3′ adaptor sequentially by T4 RNA ligase and then transcribed into cDNA by RT-PCR following the Illumina protocol. These libraries were used for 50 bp single end sequencing using Illumina GAIIx (San Diego, CA, USA) at the LC-BIO (Hangzhou, China).
The method used for degradome library construction was previously described by German et al. (2008) with some modifications. Poly (A)+ RNA was isolated and annealed with Biotinylated Random Primers. The annealed products containing 5′-monophosphates were ligated to a 5′ adaptor and used to generate first-strand cDNA. And then we performed the single-end sequencing (36 bp) using the 5′ adapter only on an Illumina Hiseq2500 at the LC-BIO (Hangzhou, China).
Bioinformatic Analysis
Small RNA reads were processed following the procedures as described by Yin et al. (2008). Raw reads obtained using Illumina’s Sequencing Control Studio software version 2.8 (SCS v2.8, San Diego, CA, USA) were filtered to remove low quality reads and adapters using ACGT101-miR (LC Sciences, Houston, TX, USA). After rigorous screening, all clean reads of 18–25 nt with three or more copies in frequency were mapped to specific species precursors in miRBase 20.0 by BLAST search to identify known miRNAs and novel 3 p- and 5 p- derived miRNAs.
For the degradome sequencing data, raw reads were obtained using Illumina’s Pipeline v1.5 software to remove adaptor sequences and low quality sequencing reads. CleaveLand3.0 and the ACGT301-DGEv1.0 program were used for subsequent analysis. The degradome data were mapped to the transcriptome of pericarp during litchi maturation (Lai et al., 2015). All targets were selected and categorized as I, II, III, or IV based on the abundance of the resulting mRNA tag relative to the overall profile of degradome reads that matched the target as described by Addo-Quaye et al. (2008). In addition, t-plots were built to analyze the miRNA targets and RNA degradation patterns according to the distribution of signatures (and abundances) along these transcripts. To uncover the miRNA-gene regulatory network on the basis of biological process and molecular function, the gene ontology (GO) analysis of the candidate target transcripts was performed using the AgriGO toolkit (Du et al., 2010).
miRNAs and Target Gene Expression Analysis
The miRNA expression was analyzed by qRT-PCR using a published method with some modifications (Chen et al., 2005). Specific stem loop RT primer 5′-GTCGTATCCAGTGCAGGG
TCCGAGGTATTCGCACTGGATACGACGTGCTC-3′ and 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGC
ACTGGATACGACGACAGA-3′ was designed for miRNA156a and a new microRNA (NEW41), respectively. Total RNA was treated with RNase-free DNaseI (TaKaRa, Dalian, China) to remove genomic DNA. For miRNA expression analysis, RNA was reverse transcribed into cDNA using the MMLV-reverse transcriptase (Invitrogen) with miRNA specific stem loop primer. For miRNA target gene expression, 1 μg of total RNA was used for the synthesizing of cDNA, incubated with oligo (dT) primer by MMLV-reverse transcriptase (Invitrogen) following the supplier’s manual. A miRNA-specific primer and a universal primer (provided in the miScript SYBR Green PCR Kit) were used for miRNA expression analysis. And two specific primers were used to amplify each miRNA target gene. All primers were listed in Supplementary Table S1. All the qRT-PCR was carried out using the miScript SYBR Green PCR Kit (TOYOBO) and performed on the ABI 7500 Real-Time PCR Systems (Applied Biosystems, USA) according to the standard protocol. The relative expression changes were calculated using the 2-ΔΔCt method (Livak and Schmittgen, 2001) with Lcactin (HQ615689) as internal control gene. Each reaction was carried out in triplicate with different cDNA synthesized from three biological replicates.
Gene Cloning and Sequence Analysis
Based on the Litchi Genome Sequence Resource (unpublished) and the degradome sequencing results, specific primers (Supplementary Table S2) were designed to obtain the sequence of LcSPL1 and LcSPL2. The cDNA was synthesized from the total RNA of the mature pericarp of cultivar ‘FZX’ and used as the PCR templates. The PCR products were cloned into T/A cloning vector pMDH20-T (TaKaRa, Dalian, China) and transformed into Escherichia coli DH5α cells, the ligated vector DNAs were sequenced by Taihe Biotechnology Institute. Multiple sequence alignment was performed using ClustalX 1.832 and MEGA 5 (Tamura et al., 2011).
Subcellular Localization Analysis
The 35S::LcSPL1-GFP and 35S::LcSPL2-GFP vectors were constructed and transiently expressed in the leaves of N. benthamiana to observe the subcellular localization of LcSPL1 and LcSPL2. Briefly, the coding sequences of LcSPL1 and LcSPL2 without the stop codon were amplified by PCR (Primers were listed in Supplementary Table S2) and then subcloned into the pEAQ-HT-GFP vector using Age I in frame with the green fluorescent protein (GFP) sequence (Sainsbury et al., 2009). Then the fusion constructs and the pEAQ-HT-GFP vector as control were transformed into Agrobacterium tumefaciens strain GV3101 using freeze-thaw method. Agrobacterium cultures containing the 35S::LcSPL1-GFP, 35S::LcSPL2-GFP and pEAQ-HT-GFP constructs were infiltrated into the leaves of N. benthamiana. Two days after infiltration, leaf protoplasts were isolated and observed with a fluorescence microscope (Zeiss Axio Observer D1). All transient expression assays were repeated at least three times.
Transcriptional Activation Analysis in Yeast Cells
The full-length coding regions of LcSPL1/2 were amplified and fused to the GAL4 DBD to generate pGBKT7-LcSPL1 and pGBKT7-LcSPL1 constructs. The fusion plasmids together with the positive control (p-53+T-antigen) and negative control (pGBKT7) were transformed into the Y2H Gold yeast strain using the PEG/LiAc method. The transformed yeast cultures were dropped onto plates of minimal medium without Trp (SD/–Trp) and SD/–Trp–His–Ade plates at 30°C for 3 days, and their growth status and the α-galactosidase activity were tested to further determine the transcriptional activation of each protein. For the protein showing autoactivation in yeast cells, the C-terminus deletion and N-terminus deletion constructs were constructed and transformed into the yeast cells for transcription activation activity as described above. The primers used for transcriptional activation analysis were listed in Supplementary Table S2.
Yeast Two-Hybrid (Y2H) Assay
Yeast two-hybrid assays were performed to study the interaction of LcSPL1/2 with LcMYB1 or LcbHLH1/2/3 using the MatchmakerTM Gold Y2H system (Clontech) according to the manufacturer’s instructions. Full length of LcSPL1 showed autoactivation in yeast cells. The C-terminus deletion construct, LcSPL1-N were created in bait vector for Y2H analysis. LcSPL1-N and LcSPL2 was fused to the DNA-binding domain in the bait plasmid pGBKT7, respectively, and LcMYB1 or LcbHLH1/2/3 was independently cloned into activation domain in the prey vector pGADT7. Primers were shown in Supplementary Table S2. Different pairs of bait and prey constructs were then co-transformed into the yeast strain Y2HGold using the PEG/LiAc method, and the yeast cells were grown on SD medium without leucine and tryptophan (SD/-Leu/-Trp). Transformed colonies were screened for growth on quadruple dropout SD medium lacking adenine, histidine, leucine and tryptophan (SD/-Ade/-His/-Leu/-Trp), with X-α-Gal and Aureobasidin A (AbA) background. X-α-Gal was used to assay for α-galactosidase activity to confirm the positive interactions.
BiFC Assay
The entire coding sequences of LcSPL1/2 and LcMYB1 (without their stop codons) were amplified and subcloned into the pEAQ-CYFP and pEAQ-NYFP vectors described by Lai et al. (2016). The expression of target genes alone was used as negative controls. All constructed vectors were transformed into A. tumefaciens strain GV3101 and transiently expressed in leaves of N. benthamiana. The transformed leaf protoplasts were isolated and used for YFP fluorescence observation. All the transient expression assays were repeated at least three times. The primers used in the BiFC assay were listed in Supplementary Table S2.
Results
High-Throughput Sequencing of sRNAs
To identify miRNAs involved in the biosynthesis of anthocyanins in litchi, four sRNA libraries were constructed from the litchi pericarps at different developmental phases, and sequenced by the Solexa technology. After removing of low quality tags and contaminated adapter sequences, a total of 3036722 (S1), 1583580 (S2), 2802666 (S3), and 2836881 (S4) clean reads were obtained, respectively (Table 1). The size distribution of the sRNAs was summarized in Figure 1. The majority of sRNAs were 24 and 21 nt, with 24 nt having the highest abundance, and most conserved miRNAs belonging to the group of 21 nt sRNAs.
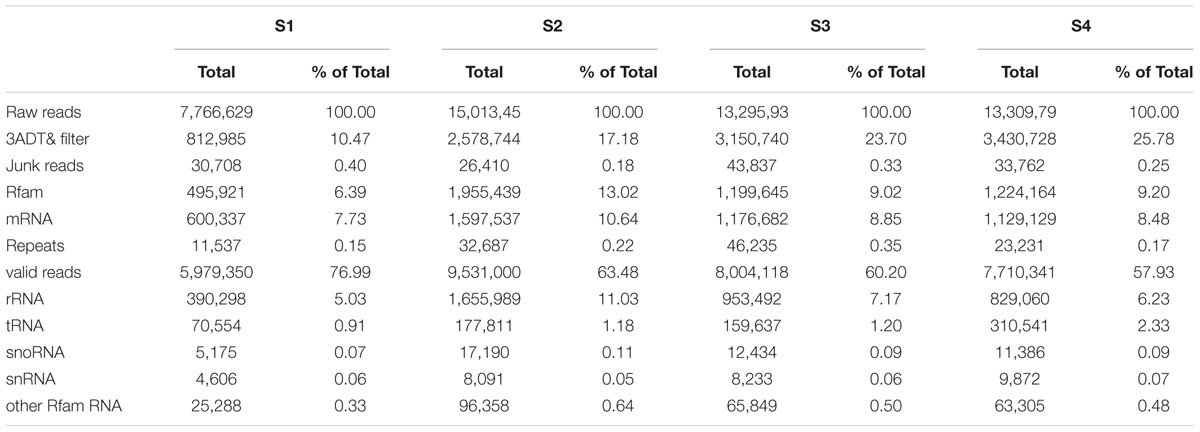
TABLE 1. Data summary of four sRNA sequencing from litchi pericarp.
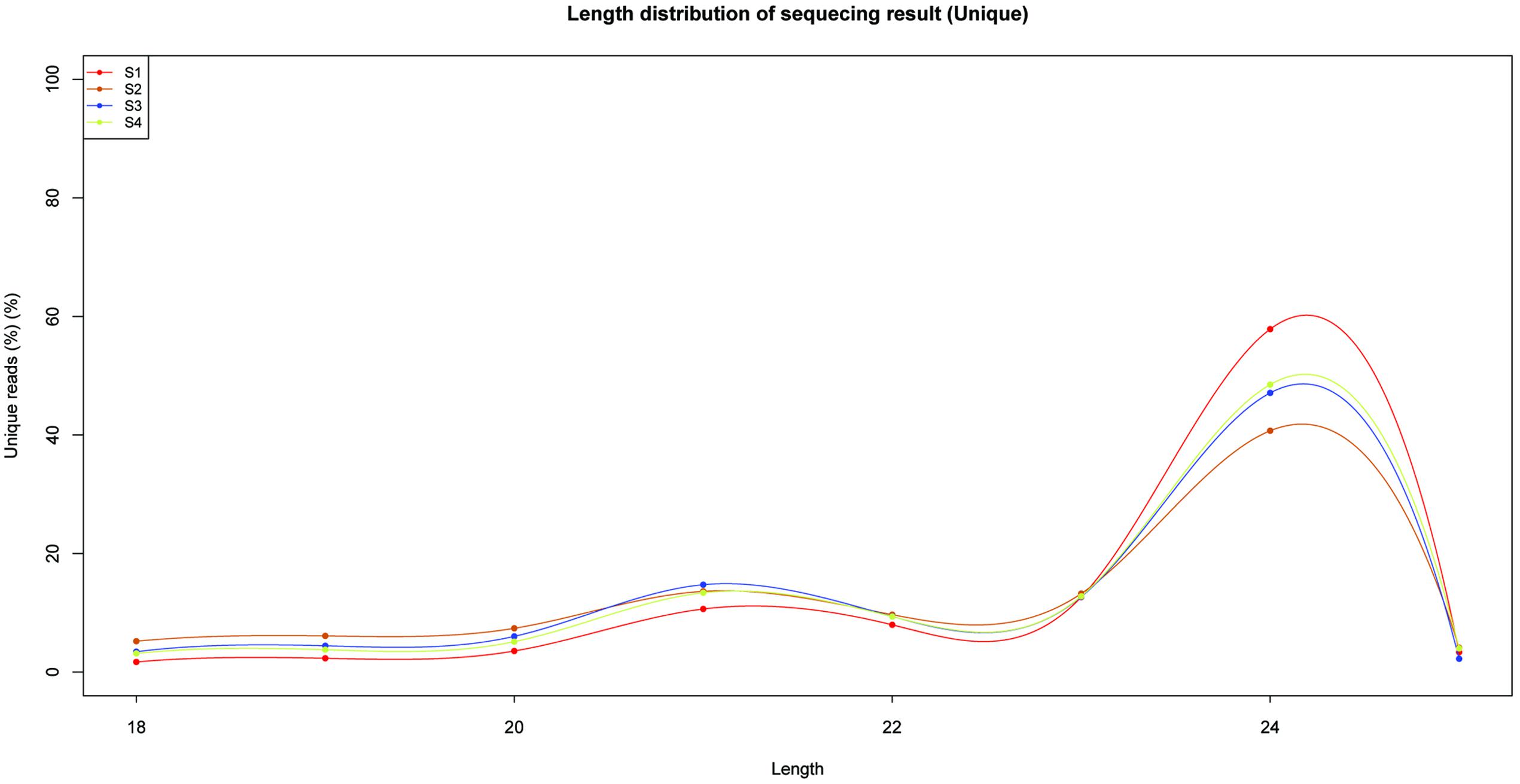
FIGURE 1. Length and size distribution of unique sequences in four sRNA libraries in litchi.
Identification of Conserved miRNAs in Litchi Pericarp
To identify the conserved miRNAs in litchi pericarp, the clean reads were mapped onto the genome of litchi, and then all the retained sequences were compared with known miRNA precursor or mature miRNA sequences in miRBase 20.0 allowing no more than two mismatches. The unique sequences mapping to specific species mature miRNAs in hairpin arms were identified as conserved miRNAs. A total of 78 conserved miRNAs were identified in the four sRNA libraries (Supplementary Table S3). Conserved miRNAs were found to be important in plant development, growth, and many other biological processes (Jones-Rhoades et al., 2006). In the present study, 35 highly conserved miRNA families were identified. According to the sequencing results, the expression levels of the conserved miRNAs were highly variable. Among them, lch-miR156a, lch-miR159, lch-miR166a, lch-miR403a, and lch-miR396b had relatively high number of reads, whereas lch-miR169 family members expressed at a lower level (Supplementary Table S3).
Identification of Novel miRNAs in Litchi Pericarp
The primary criterion for identification of novel miRNA was the ability of its sequence to fold-back into a stable hairpin structure. Novel miRNAs were identified by analyzing the precursors using the Mfold web server (Zuker, 2003). All the results were filtered using the strict criteria defined by Meyers et al. (2008). In total, 41 novel miRNAs were predicted (Supplementary Table S4). The 34 novel miRNAs were 18–25 nt long, and most of them were 24 nt long (Figure 1). Finally, 119 miRNAs, including conserved and new candidates, were identified in litchi pericarps of ‘FZX’ during fruit ripening, from the four libraries (S1/S2/S3/S4).
Target Genes Identification for miRNAs in Litchi by Degradome Sequencing
In the present study, degradome sequencing technology was performed to identify miRNA targets for both conserved and new miRNAs (Addo-Quaye et al., 2008). Two degradome libraries (S1S4 and S2S3) were constructed and sequenced using the Solexa Analyzer. A total of 8127257 and 7112553 raw reads were yielded, respectively. After removing the low quality and repeat sequences, 3551258 and 3818329 unique reads were obtained. The remaining sequences were mapped to the litchi pericarp transcriptome (Lai et al., 2015) (Supplementary Table S5). Finally, 3524213 and 3788804 sequences could be mapped perfectly onto the reference database. All these mapped sequences were analyzed to identify candidate target gene for miRNAs.
The sliced target transcripts were categorized into five groups according to the relative abundance of the tags at the target mRNA sites. In total, 129 targets were identified for conserved and novel miRNAs (Supplementary Table S6). These targets included ARFs, NAC domain TF, GRAS family TF, MYB TF, SPLs, AP2-like factors, SCLs and MADS-box factors (Supplementary Table S6), which were reported to play important roles in the plant growth and development. For example, miR166 targeted class III homeodomain leucine zipper (HD-ZIPIII) protein has been shown to be involved in the asymmetry development of leaves in maize (Juarez et al., 2004). The TF ARF10 was targeted by miR160, and ARF10 could alter hormone sensitivity during seed germination and seedling growth (Liu et al., 2007).
Identification of miRNAs Target Related to Anthocyanins Biosynthesis in Litchi Pericarp
Based on the results of miRNA identification and its target prediction (Supplementary Table S6), two miRNAs, miR156a and a novel miRNA-NEW41, were found to be related to anthocyanin biosynthesis in litchi pericarp. miR156a was found to target SPL TF (LcSPL1 and LcSPL2) (Figure 2), and miRNA-NEW41 was predicted to target LcCHI. SPL TF has been reported to be involved in anthocyanin biosynthesis in Arabidopsis (Gou et al., 2011), whereas LcCHI is an important structural gene in the biosynthesis of anthocyanins in litchi pericarp (Wei et al., 2011).
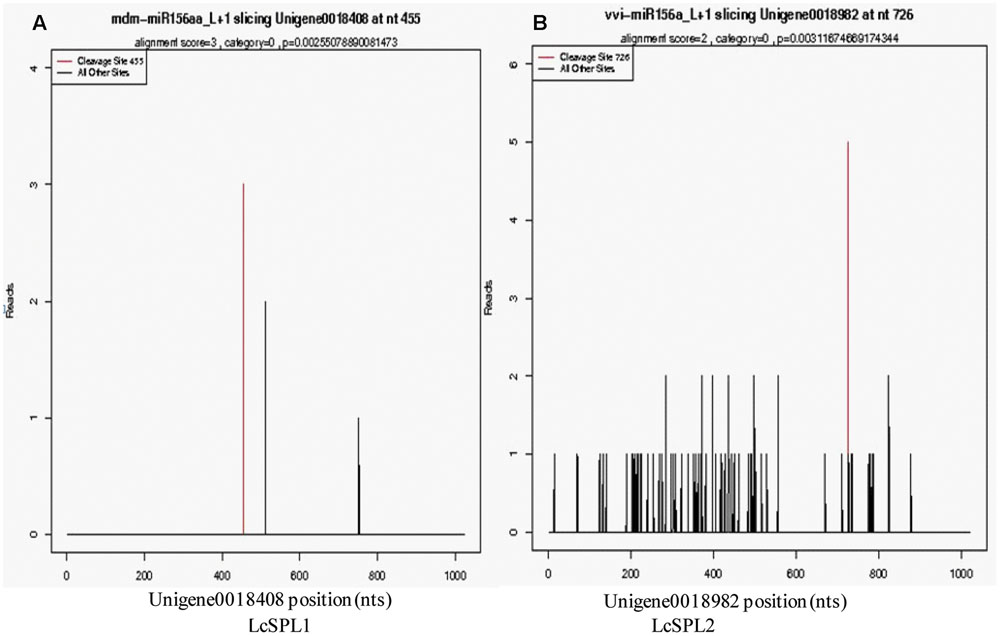
FIGURE 2. Target plots of the targets cleaved by the miR156a. The T-plots show the distribution of the degradome tags along the full-length of the target mRNA sequence. The red line represents the predicted cleavage site of the corresponding miRNAs. (A) Cleavage features in LcSPL1 mRNA by miR156a from the degradome library. (B) Cleavage features in LcSPL2 mRNA by miR156a from the degradome library.
In S1, S2, S3, and S4 library, miR156a and miRNA-NEW41 were found to be differentially expressed (Supplementary Tables S3 and S4). Compared with the expression levels in S1 (green pericarp), the expression levels of both miR156a and miRNA-NEW41 were up-regulated and then down-regulated in the S4 (red pericarp). These results indicated that miR156a and miRNA-NEW41 might be involved in the anthocyanins biosynthesis in litchi.
Expression Analysis of miRNAs and Their Targets by qRT-PCR
To validate the results obtained by high-throughput sequencing, miR156a and miRNA-NEW41 were selected for further confirmation by qRT-PCR. The total anthocyanin content showed a trend of increasing in litchi during fruit maturation (Figure 3A). The expression levels of miRNA-NEW41 was slightly decreased and reached the lowest at sample Se, while its corresponding target gene LcCHI showed increased expression (Figure 3B). In parallel with the accumulation of anthocyanins, the expression patterns of miR156a were enhanced as the fruit developed toward full maturity, while its target genes LcSPL1 and LcSPL2 were reduced (Figures 3C,D). The expression patterns between miR156a and its targets (LcSPL1 and LcSPL2) were complementary, which further validated the regulatory role of miR156a on its targets LcSPL1 and LcSPL2. Together, the above results indicated the involvement of miR156a in the accumulation of anthocyanins in litchi, and was selected for further analysis.
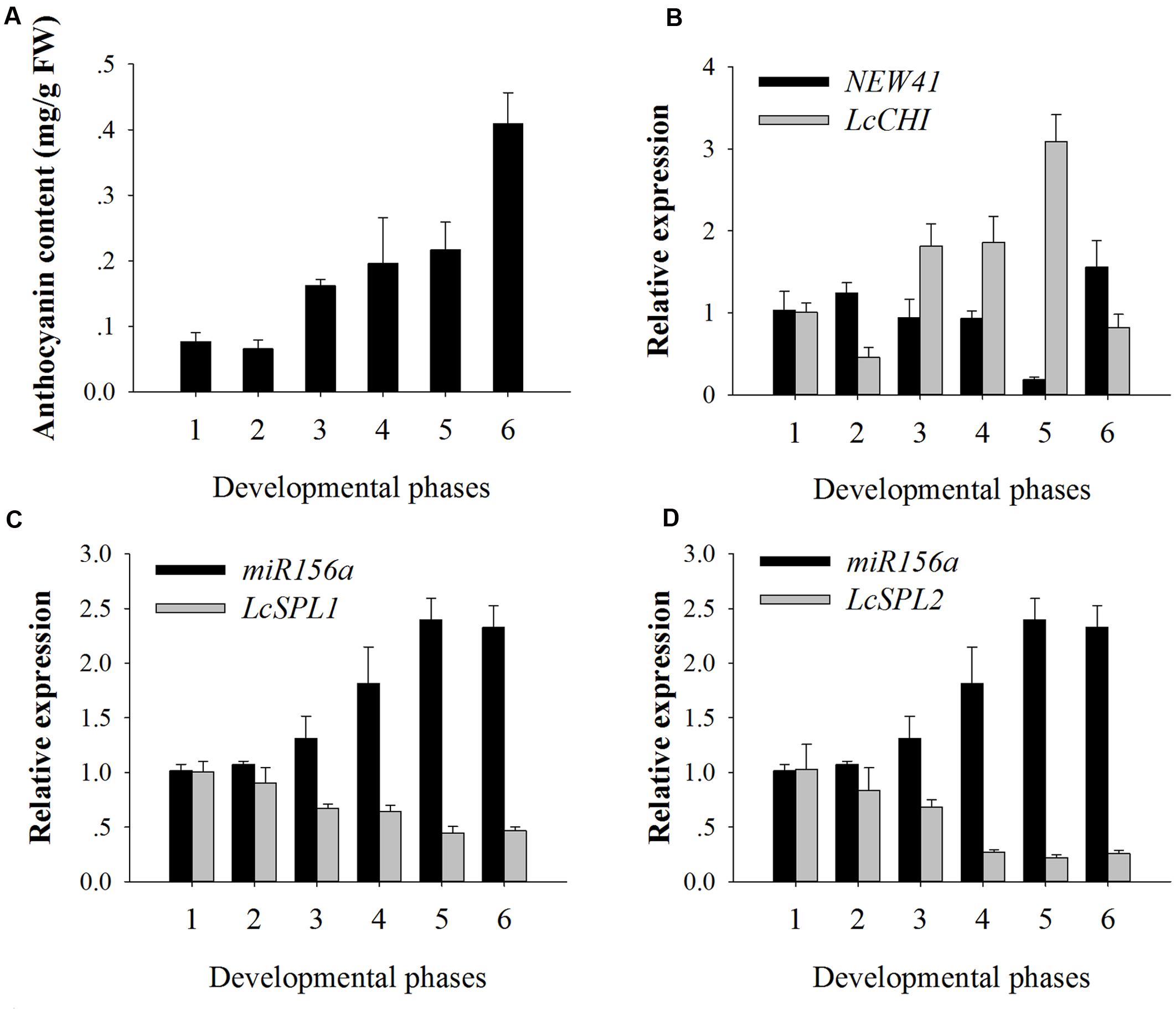
FIGURE 3. Anthocyanin contents and relative expressions of miRNAs and their target genes. (A) Anthocyanin contents of ‘FZX’ litchi during fruit maturation. (B) Expression analysis of miRNA-NEW41 and its target gene LcCHI in the pericarp of ‘FZX’ litchi. (C) Expression analysis of miR156a and its target genes LcSPL1 in the pericarp of ‘FZX’ litchi. (D) Expression analysis of miR156a and its target genes LcSPL2 in the pericarp of ‘FZX’ litchi. The reference gene, Lcactin, was used to normalize gene expression levels under identical conditions. The vertical bars represent the standard error of triplicate experiments.
Isolation and Subcellular Localization of LcSPL1/2
In Arabidopsis, miR156 and its target SPL9 negatively regulates anthocyanin accumulation (Gou et al., 2011). In the present study, two SPL TFs, LcSPL1 and LcSPL2 were isolated and characterized. The full length 915 bp LcSPL1 (GenBank accession number KY305107) and 552 bp LcSPL2 (accession no. KY305108) were isolated using the cDNA samples from red pericarp of ‘FZX’. The LcSPL1 gene encoded a putative protein of 304 amino acids with a predicted molecular weight of 34.435 KDa, and the isoelectric point was 7.23. The predicted molecular weight of LcSPL2 (183 amino acids) was 22.376 kD, and the isoelectric point was 8.34. Both LcSPL1 and LcSPL2 belong to the SBP family based on the protein functional domain analysis (data not shown).
SQUAMOSA PROMOTER BINDING PROTEIN-LIKEs, as TF, are expected to be localized in the nucleus. To determine the subcellular localization of LcSPL1 and LcSPL2, a 35S-LcSPL1-GFP and 35S-LcSPL1-GFP fusion protein was analyzed. Fluorescence from 35S-GFP was discerned in both cytoplasm and nucleus, whereas fluorescence from the 35S-LcSPL1-GFP and 35S-LcSPL1-GFP fusion was detected only in the nucleus, demonstrating that LcSPL1 and LcSPL2 are localized in the nucleus (Figure 4). The results indicated that LcSPL1 and LcSPL2 were both nuclear-localized proteins.
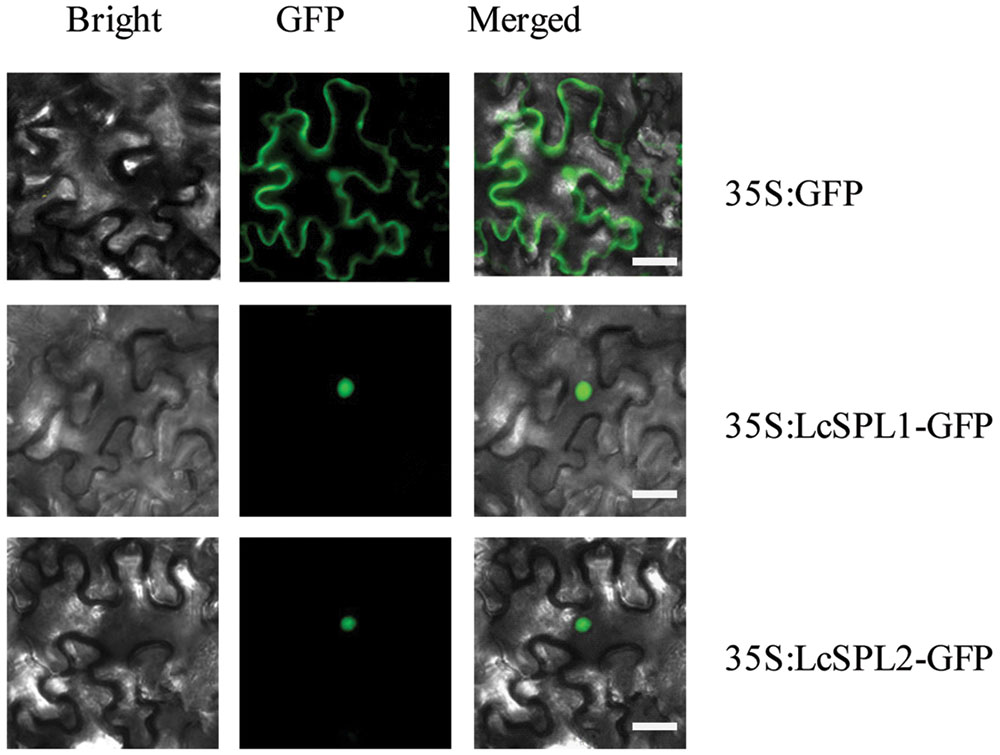
FIGURE 4. Subcellular localization of LcSPL1 and LcSPL2 as revealed using GFP fusion proteins. Bar = 20 μm.
Transcriptional Activation Ability of LcSPL1/2 in Yeast
To identify the transcriptional activation abilities of LcSPL1 and LcSPL2, a GAL4-responsive reporter system in yeast cells was performed. The yeast cells with vector pGBKT7 (empty) and cells that have been successfully transfected with the vector construct containing DBD-LcSPL2 could not grow on media minus Trp, Ade and His with Aureobasidin A background (Figure 5). While the yeast cells containing DBD-LcSPL1 and DBD-P53+T-antigen (positive control) grew well on the same media and showed α-galactosidase activity (Figure 5). These results confirmed that the DBD-LcSPL1 could act as a transcriptional activator and LcSPL2 had no transcriptional activation. To further identify the transcriptional activation domains of LcSPL1, the coding region of LcSPL1 was separated into two fragments, which were the N-and C-terminal regions of LcSPL1, and fused with GAL4-BD, and then transformed into yeast strain Y2H Gold. As shown in Figure 5, yeast cells harboring DBD-LcSPL1-C showed α-galactosidase activity. The result indicated that the activation domain of LcSPL1 was localized at C-terminus, and the N-terminal was used for protein interaction in Y2H.
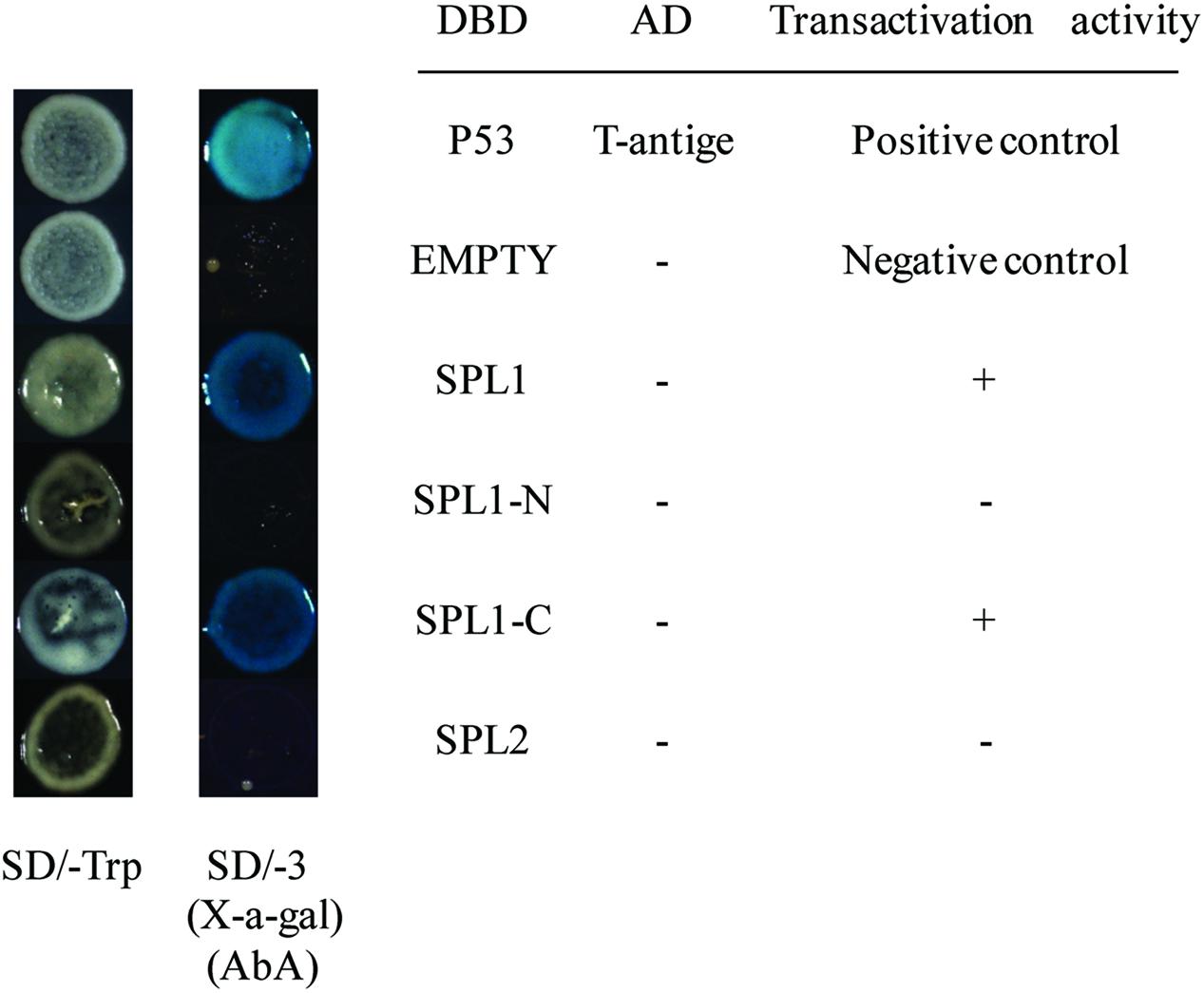
FIGURE 5. Analysis of the transcriptional activation ability of LcSPL1/2 in yeast cells. The full length construct of LcSPL1/2 and the N-and C-terminal derivatives of LcSPL1 were cloned into the pGBKT7 vector to create the DBD-LcSPL1/2, DBD-LcSPL1-N, and DBD-LcSPL1-C constructs, respectively, and then expressed in the yeast strain Y2H Gold. The plasmids pGBKT7-53+pGADT7-T and pGBKT7 were used as positive and negative controls, respectively. Yeast clones transformed with all the constructs above were grown on SD media minus Trp, Ade and His (-TAH), with X-a-Gal and Aureobasidin A (AbA) background for 3 days at 30°C.
The Interaction of LcSPL1/2 with LcMYB1 or LcbHLH 1/2/3
Recent report has suggested that SPLs, the targets of the miR156, are involved in anthocyanin biosynthesis pathways through directly preventing expression of anthocyanin biosynthetic genes by destabilizing the MBW complex (Gou et al., 2011). Based on our previously published results, LcMYB1 might play major roles in the MBW complex determining anthocyanin accumulation in litchi (Lai et al., 2014). Recently, two LcbHLH TFs interacting with LcMYB1 in regulating late structural genes of anthocyanin biosynthesis in litchi during anthocyanin accumulation has been reported (Lai et al., 2016). In the present study, to explore interaction of LcSPL1/2 with LcMYB1 or LcbHLH1/2/3, the Y2H has been employed. The co-transformed colonies were selected on SD medium lacking leucine and tryptophan (SD/-Leu/-Trp), and screened for growth on another SD medium without adenine, histidine, leucine, and tryptophan (SD/-Ade/-His/-Leu/-Trp) supplement with X-α-Gal and Aureobasidin A (AbA) background. As shown in Figure 6, yeast cells co-transformed with the positive control (pGBKT7-53+pGADT7-T) and LcSPL1 with LcMYB1, could grow on selective medium (synthetic medium lacking tryptophan, leucine, histidine, and adenine) supplement with X-α-Gal and AbA background, and turned blue at the presence of X-α-Gal. In contrast, the yeasts co-transformed with the control and the yeast cells harboring LcSPL2 with LcMYB1 combinations could not (Figure 6). These results suggested that LcMYB1 was able to form complex with LcSPL1, but not with LcSPL2.
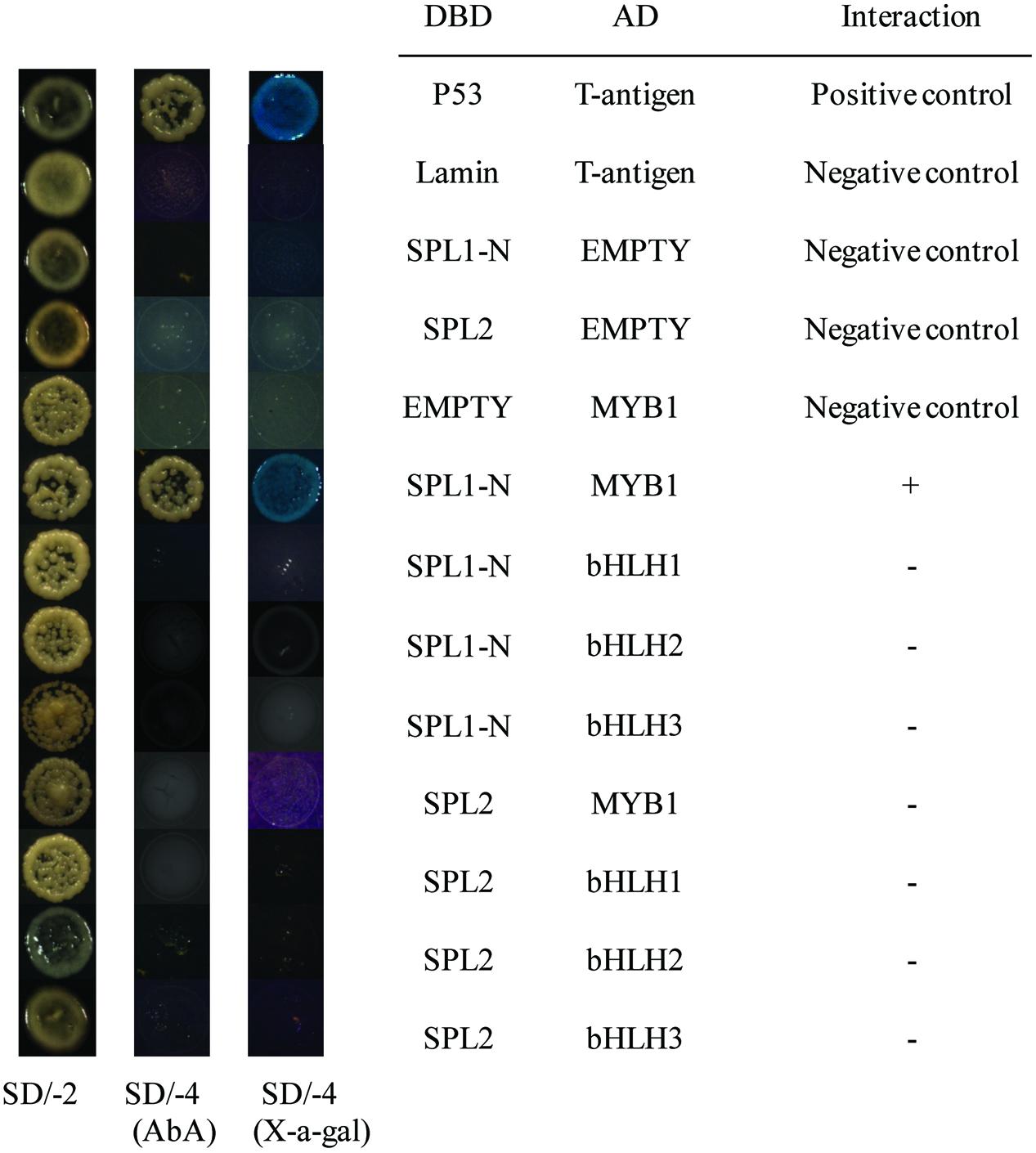
FIGURE 6. Interaction of LcSPL1/2 with LcMYB1 or LcbHLH1/2/3 detected in Y2H assays. The combinations of LcSPL1/2 and LcMYB1 or LcbHLH1/2/3 fused to pGBKT7 (BD) or pGADT7 (AD), respectively, was co-transformed to the Y2HGold yeast strain. All of the yeast clones were grown on appropriate media (SD/-Leu/-Trp and then SD/-Ade/-His/-Leu/-Trp) to test for activation of the reporter gene. The plasmids pGBKT7-53+pGADT7-T, pGBKT7-Lam+pGADT7-T, DBD–SPL1/2+pGADT7-T, pGBKT7+AD-MYB1 were used as positive and negative controls, respectively.
As shown in Figure 6, yeast cells co-transformed the positive control (pGBKT7-53+pGADT7-T) could grow on selective medium (synthetic medium lacking tryptophan, leucine, histidine, and adenine) supplemented with X-α-Gal and AbA background, and turned blue at the presence of X-α- Gal, but the yeasts co-transformed with the control and the yeast cells harboring LcSPL1/2 with LcbHLH1/2/3 combinations could not, indicating no direct interaction between LcSPL1/2 and LcbHLH1/2/3.
Subsequently, BiFC analysis was performed to further confirm the interactions between LcSPL1/2 and LcMYB1 observed in the Y2H assays (Figure 7). LcSPL1/2 tagged with pSPYNE (split YFP C-terminal fragment expression) and LcMYB1 tagged with pSPYCE (split YFP N-terminal fragment expression) were transiently co-expressed in N. benthamiana leaves by Agrobacterium. As shown in Figure 7, strong YFP fluorescent signal was detected in the nucleus of leaf epidermal cells expressing LcSPL1-cYFP and LcMYB1-nYFP, but not in the control or LcSPL2-cYFP and LcMYB1-nYFP. The BiFC assay further demonstrated the in vivo interaction between LcSPL1 and LcMYB1, which was consistent with the Y2H assays.
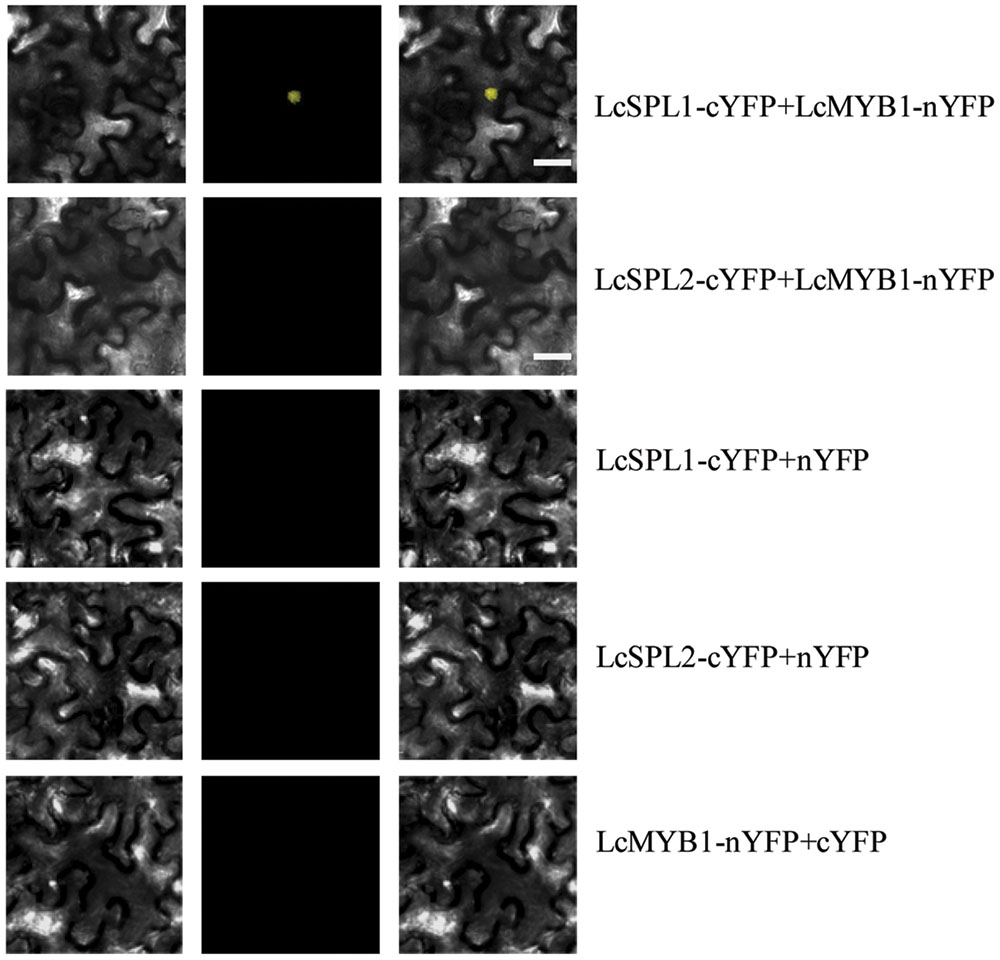
FIGURE 7. Bimolecular fluorescence complementation assay of the LcSPL1 and LcMYB1 interaction in Nicotiana benthamiana. Bright field, YFP and merged field were shown for each transformation combination. Bar = 20 μm.
Discussion
High-Throughput Sequencing
miRNAs play important roles in plant development, adaptation to biotic and abiotic stress, as well as in secondary metabolism (Jones-Rhoades et al., 2006; Gou et al., 2011; Li et al., 2016). Litchi miRNAs and targets had been identified in litchi fruit subjected to ambient storage and post-cold storage shelf life (Yao et al., 2015). However, whether miRNAs are involved in litchi anthocyanin biosynthesis is still unknown. Therefore, identification of litchi miRNAs and their target genes associated with anthocyanin biosynthesis was performed in the present study using the high-throughput deep sequencing technology. In this study, we constructed four sRNA libraries and two mixed degradome libraries from pericarps of ‘FZX’ litchi of different developmental phases. In general, 24 nt sRNAs were the most abundant, followed by 21 nt, which was consistent with the length distribution of sRNAs reported in apple and litchi (Xia et al., 2012; Yao et al., 2015). Finally, a total of 78 known miRNAs belonging to 35 highly conserved miRNA families as well as 41 new miRNA were identified in litchi (Supplementary Tables S3 and S4). Different miRNAs showed different expression levels during litchi fruit maturity, indicating miRNAs might be involved in litchi fruit development. Moreover, we identified a total of 129 potential targets of the conserved and new miRNAs by the recently developed degradome sequencing approach (Supplementary Table S6). The majority of these target genes are conserved in plant species, which had been reported to play important roles in plant development.
miRNAs and Their Targets Related to Litchi Anthocyanins Biosynthesis
Accumulating evidences suggest that miRNAs are involved in anthocyanin biosynthesis, such as miR156 and miR858 (Gou et al., 2011; Xia et al., 2012; Jia et al., 2015). In this study, miR156a and a novel miRNA-NEW41 were found to be related to the anthocyanin biosynthesis in litchi based on the sRNA and degradome sequencing. The target gene of miR156a is LcSPL, a family of plant-specific TFs. And miRNA-NEW41 target LcCHI, which is the important structural gene of anthocyanin biosynthesis in litchi. qRT-PCR confirmed that the expression of miRNA-NEW41 was essentially complementary to that of the target LcCHI (Figure 3B). As the miRNA targeted CHI in the regulation of anthocyanin biosynthesis has not been reported in plant, the roles of miRNA-NEW41 in litchi anthocyanin biosynthesis are deserved to be further studied.
Recently, Gou et al. (2011) reported that SPL9 competed with TT8, a bHLH, for binding to PAP1, and could suppress DFR expression by interfering with a MYB-bHLH-WD40 transcriptional activation complex, and then influence the anthocyanin biosynthesis in Arabidopsis. In this investigation, we analyzed the expression profile of miR156a and its corresponding targets LcSPL1 and LcSPL2 in the different development phases of litchi pericarp, and the results showed that the expression level of miR156a was in parallel with the accumulation of anthocyanins, whereas its targets LcSPL1/2 showed a reverse trend (Figures 3C,D). These results indicated that miR156a might also be related to the accumulation of anthocyanins in litchi. However, the functions of miR156a in litchi are unknown. Transgenic plants ectopically expressing the miR156a or its targets LcSPL1/2 in Arabidopsis might provide an efficient platform to study the function of miRNA. For example, Sun et al. (2013) reported that transgenic Arabidopsis plants ectopically expressed Md-miRNA156h gene exhibited late flowering and long vegetative growth.
Previous studies showed that miR858 participated in anthocyanin biosynthesis in Arabidopsis, apple and tomato (Luo et al., 2012; Xia et al., 2012; Jia et al., 2015). However, the expression of miR858a maintained a low level during litchi fruit maturity (Supplementary Table S3). Whether miR858 is responsible for anthocyanin biosynthesis in litchi is worth for further study.
LcSPL Genes Analysis
SQUAMOSA PROMOTER BINDING PROTEIN-LIKE is a family of plant-specific TFs that have been identified in various species, such as Arabidopsis (Unte et al., 2003), tomato (Salinas et al., 2012), populous (Li and Lu, 2014), and Citrus (Shalom et al., 2015). In plants, miR156 and its target gene SPL have previously been shown to play important roles in several plant developmental processes, such as flowering time and phase change, organ size, and fertility (Wang et al., 2008; Wang et al., 2009; Xing et al., 2010). Here, we described a critical role of miR156a/SPL in regulating the biosynthesis of anthocyanins in litchi. miR156a and two target genes, which were LcSPL1 and LcSPL2, were obtained from the sRNA and degradome sequencing. Subcellular localization indicated that LcSPL1 and LcSPL2 were all localized in the nucleus (Figure 4), where they participated in the regulation of anthocyanin biosynthesis. SPLs responsible for anthocyanin biosynthesis have also been reported in Arabidopsis (Gou et al., 2011).
LcSPL1 Interacting with LcMYB1 in Regulating Anthocyanin Biosynthesis
Anthocyanin biosynthesis is known to be controlled by both enzyme-coding structural genes and their regulated genes, which are influenced by hormones, light, temperature, and nutrition (Koes et al., 2005; Saito et al., 2013). The data presented here revealed a role for LcSPL1 in regulating the expression of LcMYB1, thereby controlling anthocyanin biosynthesis. In the present study, Y2H and BiFC assays confirmed that LcSPL1 could physically interact with LcMYB1 (Figures 6 and 7), which has been proved to play major roles in anthocyanin biosynthesis in litchi (Lai et al., 2014). However, there was no interaction between LcSPL1/2 and LcbHLL1/2/3 (Figure 6). Recently, LcbHLH1/3 interacting with LcMYB1 in regulating late structural genes of anthocyanin biosynthesis in Nicotiana and Litchi has been reported (Lai et al., 2016). In previous study, SPL9 was reported to destabilize the MBW complex to repress anthocyanin biosynthesis (Gou et al., 2011). However, the WD40 related to anthocyanin biosynthesis in litchi has not been identified. Therefore, whether the LcSPL1/2 could bind WD40 in litchi is worth for further research. Our results, together with previous reports, clearly suggest that LcSPL1 is involved in litchi anthocyanin biosynthesis through interaction with its regulatory gene LcMYB1.
Previous studies indicated that the expression of LcUFGT, LcF3H, LcDFR, LcGST, and their regulatory gene LcMYB1 were up-regulated as the fruit developed toward full maturity (Wei et al., 2011; Lai et al., 2014; Li et al., 2015; Hu et al., 2016). In the present study, the expression of miR156a was enhanced with the accumulation of anthocyanins, while its target genes LcSPL1/2 were opposite (Figures 3C,D). Taken together, these results suggest the up-regulation of miR156a lead to the down-regulation of LcSPL1, which negatively regulate anthocyanin biosynthesis via interaction with LcMYB1. To the best of our knowledge, this is the first report that miRNAs are involved in the anthocyanin biosynthesis in litchi, and our findings reveal a novel mechanism on regulation of anthocyanin biosynthesis in litchi. Here, we report that miR156a-targeted LcSPL1 functionally interfere with anthocyanin biosynthesis through interaction with LcMYB1.
Author Contributions
RL, GH, and JZ designed the research. RL, BL, and BH performed the experiments. RL, YQ, GH, and JZ analyzed the data and wrote the manuscript. All of the authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The project was supported by the China Litchi and Longan Industry Technology Research System (Project No. CARS-33) and Pearl River S&T Nova Program of Guangzhou (No. 201610010147).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.02059/full#supplementary-material
Footnotes
References
Addo-Quaye, C., Eshoo, T. W., Bartel, D. P., and Axtell, M. J. (2008). Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 18, 758–762. doi: 10.1016/j.cub.2008.04.042
Albert, N. W., Davies, K. M., Lewis, D. H., Zhang, H., Montefiori, M., Brendolise, C., et al. (2014). A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell 26, 962–980. doi: 10.1105/tpc.113.122069
Chagne, D., Lin-Wang, K., Espley, R. V., Volz, R. K., How, N. M., Rouse, S., et al. (2012). An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol. 161, 225–239. doi: 10.1104/pp.112.206771
Chen, C., Ridzon, D. A., Broomer, A. J., Zhou, Z., Lee, D. H., Nguyen, J. T., et al. (2005). Real-time quantification of miRNA by stem-loop RT-PCR. Nucleic Acids Res. 33, e179. doi: 10.1093/nar/gni178
Cutanda-Perez, M. C., Ageorges, A., Gomez, C., Vialet, S., Terrier, N., Romieu, C., et al. (2009). Ectopic expression of VlmybA1 in grapevine activates a narrow set of genes involved in anthocyanin synthesis and transport. Plant Mol. Biol. 69, 633–648. doi: 10.1007/s11103-008-9446-x
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). Agrigo: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, 64–70. doi: 10.1093/nar/gkq310
German, M. A., Pillay, M., Jeong, D. H., Hetawal, A., Luo, S., Janardhanan, P., et al. (2008). Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 26, 941–946. doi: 10.1038/nbt1417
Gonzalez, A., Zhao, M., Leavitt, J. M., and Lloyd, A. M. (2008). Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 53, 814–827. doi: 10.1111/j.1365-313X.2007.03373.x
Gou, J. Y., Felippes, F. F., Liu, C. J., Weigel, D., and Wang, J. W. (2011). Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 23, 1512–1522. doi: 10.1105/tpc.111.084525
Guan, X., Pang, M., Nah, G., Shi, X., Ye, W., Stelly, D. M., et al. (2014). miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 5:3050. doi: 10.1038/ncomms4050
Hu, B., Zhao, J., Lai, B., Qin, Y., Wang, H., and Hu, G. (2016). LcGST4 is an anthocyanin-related glutathione S-transferase gene in Litchi chinensis Sonn. Plant Cell Rep. 35, 831–843. doi: 10.1007/s00299-015-1924-4
Iwakawa, H. O., and Tomari, Y. (2015). The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 25, 651–665. doi: 10.1016/j.tcb.2015.07.011
Jia, X., Shen, J., Liu, H., Li, F., Ding, N., Gao, C., et al. (2015). Small tandem target mimic-mediated blockage of microRNA858 induces anthocyanin accumulation in tomato. Planta 242, 283–293. doi: 10.1007/s00425-015-2305-5
Jin, W., Wang, H., Li, M., Wang, J., Yang, Y., Zhang, X., et al. (2016). The R2R3 MYB transcription factor PavMYB10.1 involves in anthocyanin biosynthesis and determines fruit skin colour in sweet cherry (Prunus avium L.). Plant Biotechnol. J. 14, 2120–2133. doi: 10.1111/pbi.12568
Jones-Rhoades, M. W., Bartel, D. P., and Barte, L. B. (2006). MicroRNAS and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Juarez, M. T., Kui, J. S., Thomas, J., Heller, B. A., and Timmermans, M. C. P. (2004). microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428, 84–88. doi: 10.1038/nature02363
Koes, R., Verweij, W., and Quattrocchio, F. (2005). Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 10, 236–242. doi: 10.1016/j.tplants.2005.03.002
Lai, B., Du, L. N., Liu, R., Hu, B., Su, W. B., Qin, Y. H., et al. (2016). Two LcbHLH transcription factors interacting with LcMYB1 in regulating late structural genes of anthocyanin biosynthesis in Nicotiana and Litchi chinensis during anthocyanin accumulation. Front. Plant Sci. 7:166. doi: 10.3389/fpls.2016.00166
Lai, B., Hu, B., Qin, Y., Zhao, J., Wang, H., and Hu, G. (2015). Transcriptomic analysis of Litchi chinensis pericarp during maturation with a focus on chlorophyll degradation and flavonoid biosynthesis. BMC Genomics 16:225. doi: 10.1186/s12864-015-1433-4
Lai, B., Li, X. J., Hu, B., Qin, Y. H., Huang, X. M., Wang, H. C., et al. (2014). LcMYB1 is a key determinant of differential anthocyanin accumulation among genotypes, tissues, developmental phases and ABA and light stimuli in Litchi chinensis. PLoS ONE 9:e86293. doi: 10.1371/journal.pone.0086293
Li, C., and Lu, S. (2014). Molecular characterization of the SPL gene family in Populus trichocarpa. BMC Plant Biol. 14:131. doi: 10.1186/1471-2229-14-131
Li, L., Xue, M., and Yi, H. (2016). Uncovering microRNA-mediated response to SO2 stress in Arabidopsis thaliana by deep sequencing. J. Hazard. Mater. 316, 178–185. doi: 10.1016/j.jhazmat.2016.05.014
Li, X., Gao, M. J., Pan, H. Y., Cui, D. J., and Gruber, M. Y. (2010). Purple canola: Arabidopsis PAP1 increases antioxidants and phenolics in Brassica napus leaves. J. Agric. Food Chem. 58, 1639–1645. doi: 10.1021/jf903527y
Li, X. J., Zhang, J. Q., Wu, Z. C., Lai, B., Huang, X. M., Qin, Y. H., et al. (2015). Functional characterization of a glucosyltransferase gene, LcUFGT1, involved in the formation of cyanidin glucoside in the pericarp of Litchi chinensis. Physiol. Plant. 156, 139–149. doi: 10.1111/ppl.12391
Li, Y. F., Zheng, Y., Addo-Quaye, C., Zhang, L., Saini, A., Jagadeeswaran, G., et al. (2010). Transcriptome-wide identification of microRNA targets in rice. Plant J. 62, 742–759. doi: 10.1111/j.1365-313X.2010.04187.x
Liu, P. P., Montgomery, T. A., Fahlgren, N., Kasschau, K. D., Nonogaki, H., and Carrington, J. C. (2007). Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 52, 133–146. doi: 10.1111/j.1365-313X.2007.03218.x
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, Q. J., Mittal, A., Jia, F., and Rock, C. D. (2012). An autoregulatory feedback loop involving PAP1 and TAS4 in response to sugars in Arabidopsis. Plant Mol. Biol. 80, 117–129. doi: 10.1007/s11103-011-9778-9
Maier, A., Schrader, A., Kokkelink, L., Falke, C., Welter, B., Iniesto, E., et al. (2013). Light and the E3 ubiquitin ligase COP1/SPA control the protein stability of the MYB transcription factors PAP1 and PAP2 involved in anthocyanin accumulation in Arabidopsis. Plant J. 74, 638–651. doi: 10.1111/tpj.12153
Mallory, A. C., Reinhart, B. J., Jones-Rhoades, M. W., Tang, G., Zamore, P. D., Barton, M. K., et al. (2004). MicroRNA control of PHABULOSA in leaf development: importance of pairing to the microRNA 5’ region. EMBO J. 23, 3356–3364. doi: 10.1038/sj.emboj.7600340
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., Bowman, J. L., et al. (2008). Criteria for annotation of plant microRNAs. Plant Cell 20, 3186–3190. doi: 10.1105/tpc.108.064311
Nodine, M. D., and Bartel, D. P. (2010). MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 24, 2678–2692. doi: 10.1101/gad.1986710
Qi, T., Song, S., Ren, Q., Wu, D., Huang, H., Chen, Y., et al. (2011). The Jasmonate-ZIM-domain proteins interact with the WD-Repeat/bHLH/MYB complexes to regulate Jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 23, 1795–1814. doi: 10.1105/tpc.111.083261
Qiu, J., Sun, S., Luo, S., Zhang, J., Xiao, X., Zhang, L., et al. (2014). Arabidopsis AtPAP1 transcription factor induces anthocyanin production in transgenic Taraxacum brevicorniculatum. Plant Cell Rep. 33, 669–680. doi: 10.1007/s00299-014-1585-8
Rogers, K., and Chen, X. (2013). Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25, 2383–2399. doi: 10.1105/tpc.113.113159
Rowan, D. D., Cao, M., Lin-Wang, K., Cooney, J. M., Jensen, D. J., Austin, P. T., et al. (2009). Environmental regulation of leaf colour in red 35S:PAP1 Arabidopsis thaliana. New Phytol. 182, 102–115. doi: 10.1111/j.1469-8137.2008.02737.x
Sainsbury, F., Thuenemann, E. C., and Lomonossoff, G. P. (2009). pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol. J. 7, 682–693. doi: 10.1111/j.1467-7652.2009.00434.x
Saito, K., Yonekura-Sakakibara, K., Nakabayashi, R., Higashi, Y., Yamazaki, M., Tohge, T., et al. (2013). The flavonoid biosynthetic pathway in Arabidopsis: structural and genetic diversity. Plant Physiol. Biochem. 72, 21–34. doi: 10.1016/j.plaphy.2013.02.001
Salinas, M., Xing, S., Höhmann, S., Berndtgen, R., and Huijse, R. P. (2012). Genomic organization, phylogenetic comparison and differential expression of the SBP-box family of transcription factors in tomato. Planta 235, 1171–1184. doi: 10.1007/s00425-011-1565-y
Schaart, J. G., Dubos, C., Romero De La Fuente, I., van Houwelingen, A. M. M. L., de Vos, R. C. H., Jonker, H. H., et al. (2013). Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria x ananassa) fruits. New Phytol. 197, 454–467. doi: 10.1111/nph.12017
Shalom, L., Shlizerman, L., Zur, N., Doron-Faigenboim, A., Blumwald, E., and Sadka, A. (2015). Molecular characterization of SQUAMOSA PROMOTER BINDINGPROTEIN-LIKE (SPL) gene family from Citrus and the effect of fruit load on their expression. Front. Plant Sci. 6:389. doi: 10.3389/fpls.2015.00389
Shen, X., Zhao, K., Liu, L., Zhang, K., Yuan, H., Liao, X., et al. (2014). A Role for PacMYBA in ABA-regulated anthocyanin biosynthesis in red-colored sweet cherry cv. Hong Deng (Prunus avium L.). Plant Cell Physiol. 55, 862–880. doi: 10.1093/pcp/pcu013
Song, Q. X., Liu, Y. F., Hu, X. Y., Zhang, W. K., Ma, B., Chen, S. Y., et al. (2011). Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 11:5. doi: 10.1186/1471-2229-11-5
Sun, C., Zhao, Q., Liu, D. D., You, C. X., and Hao, Y. J. (2013). Ectopic expression of the apple Md-miRNA156h gene regulates flower and fruit development in Arabidopsis. Plant Cell Tissue Organ Cult. 112, 343–351. doi: 10.1007/s11240-012-0241-7
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Unte, U. S., Sorensen, A. M., Pesaresi, P., Gandikota, M., Leister, D., Saedler, H., et al. (2003). SPL8, an SBP-Box gene that affects pollen sac development in Arabidopsis. Plant Cell 15, 1009–1019. doi: 10.1105/tpc.010678
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell 136, 669–687. doi: 10.1016/j.cell.2009.01.046
Wang, J. W., Czech, B., and Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, J. W., Schwab, R., Czech, B., Mica, E., and Weigel, D. (2008). Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell 20, 1231–1243. doi: 10.1105/tpc.108.058180
Wei, Y. Z., Hu, F. C., Hu, G. B., Li, X. J., Huang, X. M., and Wang, H. C. (2011). Differential expression of anthocyanin biosynthetic genes in relation to anthocyanin accumulation in the pericarp of Litchi chinensis Sonn. PLoS ONE 6:e19455. doi: 10.1371/journal.pone.0019455
Winkel-Shirley, B. (2001). Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 126, 485–493. doi: 10.1104/pp.126.2.485
Wrolstad, R. E., Culbertson, J. D., Cornwell, C. J., and Mattick, L. R. (1982). Detection of adulteration in blackberry juice concentrates and wines. J. Assoc. Off. Anal. Chem. 65, 1417–1423.
Xia, R., Zhu, H., An, Y. Q., Beers, E. P., and Liu, Z. (2012). Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol. 13:R47. doi: 10.1186/gb-2012-13-6-r47
Xing, S., Salinas, M., Hohmann, S., Berndtgen, R., and Huijser, P. (2010). miR156-targeted and nontargeted SBP-Box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 22, 3935–3950. doi: 10.1105/tpc.110.079343
Xu, W., Dubos, C., and Lepiniec, L. (2015). Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 20, 176–185. doi: 10.1016/j.tplants.2014.12.001
Yao, F. R., Zhu, H., Yi, C., Qu, H. X., and Jiang, Y. M. (2015). MicroRNAs and targets in senescent litchi fruit during ambient storage and post-cold storage shelf life. BMC Plant Biol. 15:181. doi: 10.1186/s12870-015-0509-2
Yin, Z., Li, C., Han, X., and Shen, F. (2008). Identification of conserved microRNAs and their target genes in tomato (lycopersicon esculentum). Gene 414, 60–66. doi: 10.1016/j.gene.2008.02.007
Yu, Z. X., Wang, L. J., Zhao, B., Shan, C. M., Zhang, Y. H., Chen, D. F., et al. (2015). Progressive regulation of sesquiterpene biosynthesis in Arabidopsis and Patchouli (Pogostemon cablin) by the miR156-targeted SPL transcription factors. Mol. Plant 8, 98–110. doi: 10.1016/j.molp.2014.11.002
Zhou, H., Lin-Wang, K., Wang, H., Gu, C., Dare, A. P., Espley, R. V., et al. (2015). Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 82, 105–121. doi: 10.1111/tpj.12792
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. doi: 10.1093/nar/gkg595
Keywords: Litchi chinensis, anthocyanin, microRNA, high-throughput sequencing, miR156a, LcSPL1 LcMYB1
Citation: Liu R, Lai B, Hu B, Qin Y, Hu G and Zhao J (2017) Identification of MicroRNAs and Their Target Genes Related to the Accumulation of Anthocyanins in Litchi chinensis by High-Throughput Sequencing and Degradome Analysis. Front. Plant Sci. 7:2059. doi: 10.3389/fpls.2016.02059
Received: 07 November 2016; Accepted: 23 December 2016;
Published: 10 January 2017.
Edited by:
Gerrit TS. Beemster, University of Antwerp, BelgiumReviewed by:
Kunal Mukhopadhyay, Birla Institute of Technology, Mesra, IndiaChangjie Xu, Zhejiang University, China
Copyright © 2017 Liu, Lai, Hu, Qin, Hu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jietang Zhao, anR6aGFvQHNjYXUuZWR1LmNu