 Alicja Macko-Podgórni1
Alicja Macko-Podgórni1 Gabriela Machaj1
Gabriela Machaj1 Katarzyna Stelmach1
Katarzyna Stelmach1 Douglas Senalik2
Douglas Senalik2 Ewa Grzebelus1
Ewa Grzebelus1 Massimo Iorizzo3
Massimo Iorizzo3 Philipp W. Simon2
Philipp W. Simon2 Dariusz Grzebelus1*
Dariusz Grzebelus1*- 1Institute of Plant Biology and Biotechnology, Faculty of Biotechnology and Horticulture, University of Agriculture in Krakow, Krakow, Poland
- 2Vegetable Crops Research Unit, United States Department of Agriculture-Agricultural Research Service, Department of Horticulture, University of Wisconsin–Madison, Madison, WI, USA
- 3Plants for Human Health Institute, Department of Horticultural Science, North Carolina State University, Kannapolis, NC, USA
Carrot is one of the most important vegetables worldwide, owing to its capability to develop fleshy, highly nutritious storage roots. It was domesticated ca. 1,100 years ago in Central Asia. No systematic knowledge about the molecular mechanisms involved in the domestication syndrome in carrot are available, however, the ability to form a storage root is undoubtedly the essential transition from the wild Daucus carota to the cultivated carrot. Here, we expand on the results of a previous study which identified a polymorphism showing a significant signature for selection upon domestication. We mapped the region under selection to the distal portion of the long arm of carrot chromosome 2, confirmed that it had been selected, as reflected in both the lower nucleotide diversity in the cultivated gene pool, as compared to the wild (πw/πc = 7.4 vs. 1.06 for the whole genome), and the high FST (0.52 vs. 0.12 for the whole genome). We delimited the region to ca. 37 kb in length and identified a candidate domestication syndrome gene carrying three non-synonymous single nucleotide polymorphisms and one indel systematically differentiating the wild and the cultivated accessions. This gene, DcAHLc1, belongs to the AT-hook motif nuclear localized (AHL) family of plant regulatory genes which are involved in the regulation of organ development, including root tissue patterning. AHL genes work through direct interactions with other AHL family proteins and a range of other proteins that require intercellular protein movement. Based on QTL data on root thickening we speculate that DcAHLc1 might be involved in the development of the carrot storage root, as the localization of the gene overlapped with one of the QTLs. According to haplotype information we propose that the ‘cultivated’ variant of DcAHLc1 has been selected from wild Central Asian carrot populations upon domestication and it is highly predominant in the western cultivated carrot gene pool. However, some primitive eastern landraces and the derived B7262 purple inbred line still carry the ‘wild’ variant, reflecting a likely complexity of the genetic determination of the formation of carrot storage roots.
Introduction
Carrot is one of the most important root vegetable crops grown worldwide on ca. 1.2 million hectares (FAOSTAT, 2013). Its progenitor, wild Daucus carota L., is a weed commonly occurring across continents in the temperate climatic zone. Asia Minor and the inner Asiatic regions have been indicated as likely centers of origin of the cultivated carrot (Vavilov, 1992). As a storage root similar to modern carrots, it has been grown in those regions since the 10th century (Mackevic, 1932; Zagorodskikh, 1939). The first domesticated carrots produced purple or yellow roots (Banga, 1963), while orange carrots did not appear in Europe before the 15th century (Banga, 1957a,b; Stolarczyk and Janick, 2011). Recent molecular studies have provided answers to questions concerning carrot evolution and domestication and confirmed that domesticated carrots were derived from wild populations of Central Asian D. carota (Iorizzo et al., 2013). It was also clearly shown that the cultivated germplasm could be divided into two distinct groups: the eastern and the western gene pools (Baranski et al., 2012; Iorizzo et al., 2013; Grzebelus et al., 2014), in agreement with an earlier hypothesis on carrot evolution by Small (1978), based on morphological observations.
Nevertheless, little information has been available on the molecular basis of domestication traits in carrot. To date, research on crop domestication has focused on staple food crops with little attention toward root vegetables (Meyer et al., 2012). Thus, traits such as loss of seed shattering, dormancy, and branching, referred to as the domestication syndrome, have been extensively studied (Zohary and Hopf, 2000). However, with respect to carrot and other root crops, the list of traits important for primary domestication should include, among others, the ability to form fleshy roots, minimal lateral root branching and biennial growth habit. Of these traits, only the Vrn1 locus responsible for early flowering, which was an apparent target for selection in the course of carrot domestication, has been recently mapped on carrot chromosome 2 (Alessandro et al., 2013). However, the gene has not been characterized. After primary traits have been selected and fixed, the process of domestication has often directed more attention to quality traits such as color, shape, flavor, and physiological traits contributing to uniformity (Doebley et al., 2006). Further improvement of carrot required selection for a range of traits determining root quality, e.g., shape, color, smoothness, etc. To date, only the genetic factors underlying carrot root color have been extensively investigated, leading to the identification and mapping of Y and Y2 genes governing carotene accumulation (Bradeen and Simon, 1998) and P1, the gene involved in anthocyanin accumulation (Yildiz et al., 2013). Recently, a high quality assembly of the carrot genome has been reported, together with several resequenced genomes of wild and cultivated accessions of diverse origin (Iorizzo et al., 2016). This work allowed identification of the Y gene, which was shown to play a key regulatory role in carotenoid accumulation and shed light on the complexity of that process in the carrot storage root. Thus, Y can be considered as the first carrot domestication gene characterized in detail.
Previously, we reported on several Diversity Array Technology (DArT) polymorphisms showing signatures of selection upon domestication which were divided into three categories, i.e., those that were selected primarily in the cultivated carrots, representing primary domestication events, those under continuous selection from wild to eastern to western, and those differentiating western from both eastern and wild, representing secondary domestication events, likely related to traits differentiating eastern and western cultivated carrots (Grzebelus et al., 2014). One of those markers, crPt-895548, showing the most pronounced signature of selection was subsequently converted to a codominant cleaved amplified polymorphic site (CAPS) marker named cult (cultivated) which was shown to discriminate wild and domesticated D. carota gene pools (Macko-Podgórni et al., 2014). The polymorphism resulted from a 6 bp-long insertion in the cultivated carrot, comprising the PstI restriction site. Here, we characterized in detail the genomic region on carrot chromosome 2 flanking the cult polymorphism and being under selection in the cultivated carrot gene pool. Based on the observation that the systematic difference between the wild and the cultivated carrots was limited to a very narrow region comprising a putative regulatory gene belonging to the AT-hook motif nuclear localized (AHL) family, we speculated that it was involved in the development of the carrot storage root. The selection likely operated on the standing variation, as the variant selected for in the cultivated carrot had been present in wild carrots from Central Asia, but not in wild carrots outside that region.
Materials and Methods
Plant Materials
To analyze nucleotide diversity (π) and pairwise population differentiation (FST) we used sequencing data for a set of 29 resequenced genomes of D. carota (Iorizzo et al., 2016; sequence read archive accession SRP062070) comprising 11 wild, 9 eastern cultivated, and 9 western cultivated carrot accessions. For long-range PCR, we used eight plants representing wild and cultivated populations (Table 1) and four plants from an F2 (D. carota subsp. commutatus × line 2874B) preselected as homozygous ‘wild’ and ‘cultivated’ with respect to the cult marker. One hundred and eighty-three plants from the same F2 population were used to construct a genetic map and identify QTLs for root thickening. The plants were grown in the experimental field of the Institute of Plant Biology and Biotechnology at Prusy near Krakow, Poland. The seeds were sown on April 24, 2014 and roots were harvested on September 9, 2014. Head diameter and crown diameter were measured for each root immediately after harvest (Supplementary Figure 1A) and head to crown ratio was calculated. For RT-qPCR analysis, two orange-rooted cultivated (2874B and Kokubu Senko Oonaga) and two wild (D. carota subsp. commutatus and D. carota subsp. carota) accessions were grown in the greenhouse. The plants were harvested at three time-points, i.e., (1) 4-week-old seedlings, (2) 8- and (3) 20-week-old plants. In time point (1) whole plants were used, in time points (2) and (3), leaves, upper and lower portions of storage roots were collected separately. At each time point, samples were taken from three randomly selected plants of each accession, immediately frozen in liquid nitrogen and stored at -80°C.
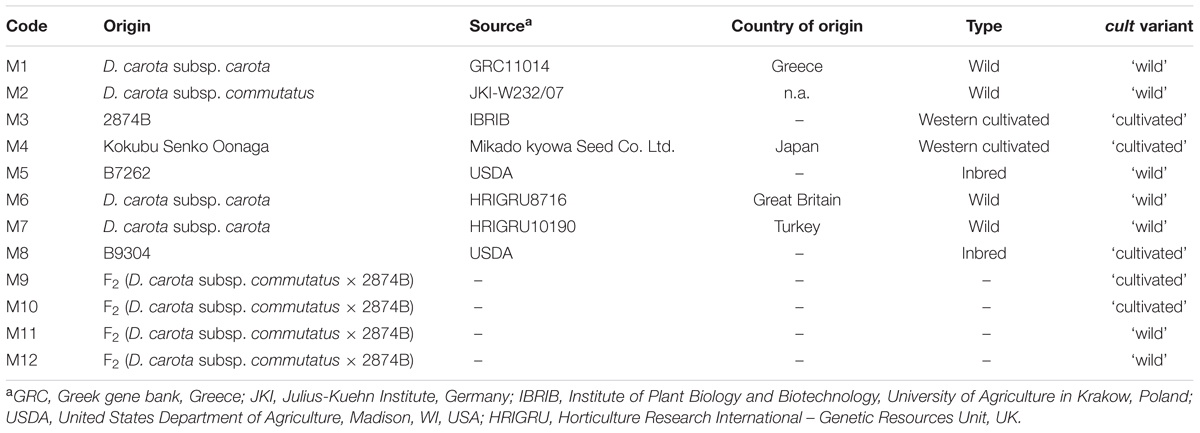
TABLE 1. List of accessions used for long-range PCR amplification and sequencing of the region spanning the cult polymorphism.
Fluorescence In situ Hybridization
To obtain meiotic preparations, immature umbels of the DH1 carrot reference line (NCBI biosample accession SAMN03216637) were collected from flowering plants and fixed in Carnoy’s solution (ethanol:glacial acetic acid – 3:1). Prior to slide preparations, the umbels were washed from fixative solutions in distilled water (three times, 5 min each washing). Anthers isolated under a stereomicroscope were macerated in the enzyme mixture consisting of 4% (w/v) cellulose Onozuka R10 (Duchefa Biochemie, Haarlem, The Netherlands), 2% (w/v) pectolyase Y23 (Duchefa) and 0.04% (w/v) pectinase (Sigma), in 0.01 M citrate buffer, pH 4.8 for 40 min at 37°C. After digestion, one anther was transferred to a glass slide and preparation was performed as described by Iovene et al. (2011). Two FISH probes were used: a BAC probe DHBAC.b0022A10 specific to carrot chromosome 2 (Iorizzo et al., 2016) and a probe specific to a 37 kb-long region from 41,842,867 to 41,880,732 nt on chromosome 2 spanning the cult site. To prepare the cult probe, long PCR derived fragments (see below) covering 97% of that region were used for labeling. Both probes were labeled with either biotin-16-dUTP or digoxigenin-11-dUTP using nick translation mix (Roche Diagnostic, Mannheim, Germany) following the manufacturer’s protocol until the length of the probe fragments averaged about 100–500 bp. Labeled DNA was purified with Quick Spin G-50 Sephadex Columns (Roche Diagnostics) following the manufacturer’s protocol. FISH was carried out according to published protocols (Dong et al., 2000; Iovene et al., 2008). Carrot genomic DNA sheared up to 500 bp fragment size was used as blocking DNA in the hybridization mixture. To reduce the background signal of applied probes, 500× excess of blocking DNA was required. Biotin- and digoxigenin-labeled probes were immuno-detected with 10 μg/ml of Alexa Fluor 488-conjugated streptavidin antibody (Life Technologies) and 2 μg/ml rhodamine-conjugated anti-digoxigenin antibody (Roche Diagnostics), respectively. Chromosomes were counterstained with 1 μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) in Prolong Gold antifade solution (Invitrogen, Carlsbad, CA, USA). The slides were examined with AxioImager M2 Zeiss microscope. All images were captured digitally using BV MV System (Applied Spectral Imaging) and Case Data Manager 4.0 software (ASI).
Long-Range PCR
Amplification was carried out in 50 μl total volume containing 250 ng of genomic DNA, 15 μM of each primer, 25 mM of dNTP (Thermo Fisher Scientific), 3.5 U Expand Long Template Enzyme Mix (Roche) and 1× Expand Long Template Buffer 3 (Roche). PCR amplifications were performed in an Eppendorf Master Cycler Gradient using the following thermal conditions: 94°C (2 min), 10 cycles of 94°C (10 s), 57°C or 58°C (30 s), 68°C (6 min), 20 cycles of 94°C (15 s), 57°C or 58°C (30 s), 68°C (6 min + 20 s elongation for each successive cycle) and final elongation of 68°C (10 min). Each template was amplified using four to six primer pairs anchored in the exons of predicted genes in the investigated region (Supplementary Table 1). PCR products were purified using Agencourt AMPure XP purification system (Beckman Coulter) and pooled in equimolar amounts. Pooled amplicons were fragmented with NEBNext® dsDNA Fragmentase® (NEB) and used for library preparation (NEBNext® DNA Library Prep Master Mix Set for Illumina®) (NEB). All samples were sequenced on one lane of MiSeq (Illumina) and assembled by a commercial service provider Genomed SA, Warsaw, Poland. Polymorphisms between cultivated and wild carrot in the coding regions were identified upon mapping to reference transcript sequences (Iorizzo et al., 2016). A codon-based test of purifying selection (Nei and Gojobori, 1986) was run using MEGA6 (Tamura et al., 2013).
RT-qPCR
All steps of analysis followed the MIQE guidelines (Bustin et al., 2009). From each sample, total RNA was extracted using the TRIzol Plus RNA Purification Kit (Thermo Fisher Scientific) and DNA was removed with the Turbo DNA-free kit (Thermo Fisher Scientific) following the manufacturer’s protocol. Quality and quantity of RNA was determined using a NanoDrop 2000c (Thermo Scientific) and gel electrophoresis. cDNA was synthesized from 1 μg of RNA using iScript kit (Bio-Rad) and stored at -20°C. Primers for DCAR_008402 (F: CTCTATGTATCTTGTCCGCC, R: GAGATATTATGCTTGTCTGGTTC) were designed using Primer-BLAST (Ye et al., 2012) and checked with OligoAnalyzer (IDT). We used carrot actin (GenBank no. X17526.1) and ubiquitin (GenBank no. U68751.1) as reference genes, as proposed by Bowman et al. (2014). Primer efficiencies and RT-qPCR were performed as described by Bowman et al. (2014) using the StepOnePlus Real-Time PCR (Thermo Fisher Scientific). Relative expression ratios (RERs) were calculated using the ΔΔCt method (Livak and Schmittgen, 2001). ANOVA and post hoc Tukey HSD test were performed with R (Faraway, 2002).
Bioinformatic Analyses
Raw reads of 29 resequenced D. carota accessions (Iorizzo et al., 2016) comprising 11 wild and 18 cultivated carrot accessions were cleaned by trimming low quality reads and clipping adapters using Trimmomatic 0.35 (Bolger et al., 2014) with parameters minqual = 28, minlen = 50, LEADING:28, TRAILING:28, SLIDINGWINDOW:10:28, MINLEN:50 and mapped to the reference DH1 genome (Iorizzo et al., 2016; NCBI accession LNRQ01000000) using BWA-MEM 0.7.12 (Li and Durbin, 2009). Subsequently, duplicates were marked with Samblaster 0.1.22 (Faust and Hall, 2014) and files were converted into BAM and sorted using SAMtools 1.2 (Li et al., 2009). SNPs were called using Freebayes 1.0.1 (Garrison and Marth, 2012) and filtered using vcffilter1 with parameters -f “QUAL > 20 and QUAL/AO > 10 and SAF > 0 and SAR > 0 and RPR > 1 and RPL > 1.” A 1 Mb sequence centered around the cult site (41,361,527–42,364,686) was extracted and polymorphisms were additionally filtered with parameters –max-alleles 2 –maf 0.1 –max-missing-count 0 –remove-indels, and divided into cultivated and wild populations files using VCFTools 0.1.14 (Danecek et al., 2011). Nucleotide diversity (π) and pairwise population differentiation FST were calculated using VCFTools. Linkage disequilibrium (LD) was calculated and plotted using Haploview 4.2 (Barrett et al., 2005). The vcf file was also used to identify polymorphisms (synonymous and non-synonymous SNPs present in transcribed regions) differentiating cultivated and wild carrots.
Genotyping, Genetic Mapping, and QTL Analysis
DNA from 183 plants from the F2 (D. carota subsp. commutatus × line 2874B) population was extracted using a modified CTAB method (Murray and Thompson, 1980). Quality and quantity of DNA was tested using 1% agarose electrophoresis with a dilution series of λ phage DNA (10, 20, 40, 60, 80, 100, 140, and 180 ng) as a standard. Also, 200–300 ng of DNA from 20 randomly selected samples was digested with HindIII and separated using 1% agarose gel. Extracted DNA was freeze-dried and shipped to the University of Wisconsin Biotech Center, Madison (WI, USA) to perform genotyping-by-sequencing (GBS). Libraries were prepared according to Elshire et al. (2011) and run on one lane of HiSeq 2000 (Illumina) at the UW Biotech Center. The data were analyzed using TASSEL 4.3.11 essentially as described by Iorizzo et al. (2016) and filtered with plugin = GBSHapMapFiltersPlugin (mnTCov = “0.1”, mnSCov = “0.1”, mnMAF = “0.1”, mxMAF = “0.5”, mnR2 = “0.1”, mnBonP = “0.01”). The obtained vcf file was additionally filtered using VCFTools with parameters: –max-alleles 2 –min-alleles 2 –max-missing-count 10 –thin 100000 and converted into the format required for mapping using a custom Perl script. The polymorphism originally identified with the DArT marker crPt-895548 (Grzebelus et al., 2014), later referred to as cult, was genotyped in the codominant fashion according to the protocol developed by Macko-Podgórni et al. (2014). Mapping was performed in JoinMap 4.0 (Van Ooijen, 2006) using the regression mapping algorithm and the Haldane mapping function. SNPs showing segregation distortion (p < 0.001) were removed. Maps generated in round 2 were used for QTL analysis performed using Windows QTL Cartographer v.2.5 (Wang et al., 2012). The genetic map and root head diameter to crown diameter ratios of were used as input data. Composite interval mapping (CIM) with five control markers, window size of 10 cM and backward regression method was used for QTL identification. Empirical thresholds obtained from permutation tests (permutation times = 500, significance level = 0.05) were used to determine QTL significance. A putative QTL was declared significant when the LOD score was >2.5.
Results
Genomic Localization of the cult Region
We mapped the sequence of the cult amplicon to the high-quality carrot genome assembly (Iorizzo et al., 2016; NCBI accession LNRQ01000000). It mapped unambiguously to the long arm of chromosome 2, position 41,862,153–41,862,461, spanning a portion of intron 1 of the gene DCAR_008402. We applied fluorescence in situ hybridization, with long-PCR products used as probes, as a means to confirm that it was a single copy region. The single FISH signal was present in a distal region of the long arm of chromosome 2, as expected (Figure 1).
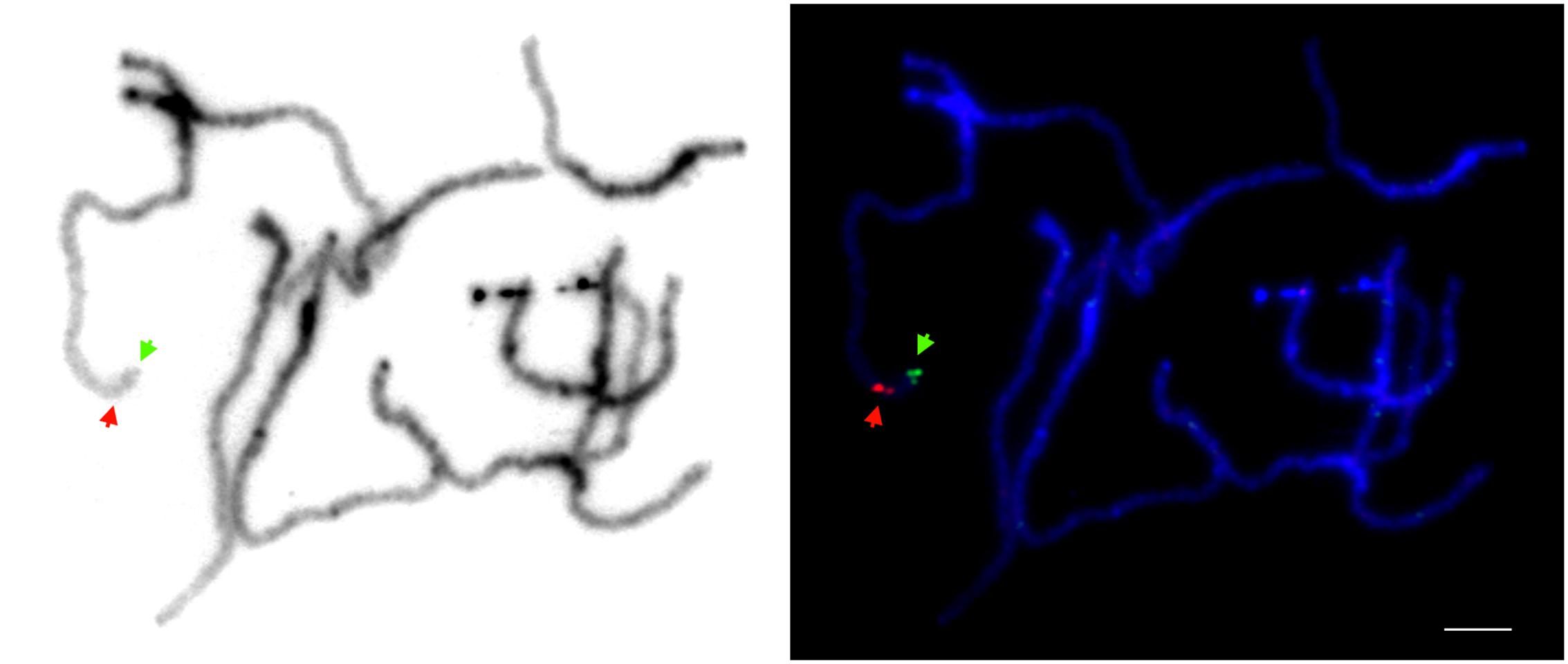
FIGURE 1. Physical localization of the region comprising the cult polymorphic site on carrot chromosomes analyzed by fluorescence in situ hybridization. Pachytene chromosomes were stained with DAPI (blue). Green and red signals represent the physical positions of DHBAC.b0022A10 (mapping to a distal position on the long arm of chromosome 2; Iorizzo et al., 2016) and long-range PCR amplicons spanning the cult region, respectively. Bar = 5 μm.
Delimitation of the Selective Sweep Range around the cult Polymorphic Site
We used previously reported genomic sequencing reads of wild and cultivated accessions of different origin (Iorizzo et al., 2016) to calculate nucleotide diversity of the wild (πw) and cultivated (πc) gene pools and the πw/πc ratio within the 1 Mb region centered around the cult polymorphic site. The πw/πc peaked around 41,858,000–41,866,000, where 7-fold decrease in nucleotide diversity was observed in the cultivated group (πw/πc = 7.4), confirming the previously reported selective sweep around the cult polymorphism (Grzebelus et al., 2014). For comparison, the genomic πw/πc ratio was 1.06, indicating a similar level of diversity in both gene pools. We also calculated pairwise population differentiation levels (FST) of wild and cultivated accessions in the same region. The highest FST values were observed in regions between 41,890,000–41,900,000 (peak at 0.52) and 41,854,000 and 41,868,000 (peak at 0.48) (Figure 2), while the FST value estimated for the whole genome was 0.12. Furthermore, we observed elevated LD in that region in the cultivated genomes, as compared to their wild relatives (Figure 3). Integration of these data allowed more precise delimitation of the region of nearly 37 Kb (41,844,694–41,881,540) comprising six genes (labeled 10 to 15 on Figure 3), which was likely under selection upon domestication. Within this region, six genes were predicted and annotated and at least three genes encode proteins that could provide regulatory functions potentially important with respect to domestication, i.e., two protein kinases (genes DCAR_008400 and DCAR_008405) and an AHL protein (gene DCAR_008402) (Table 2).
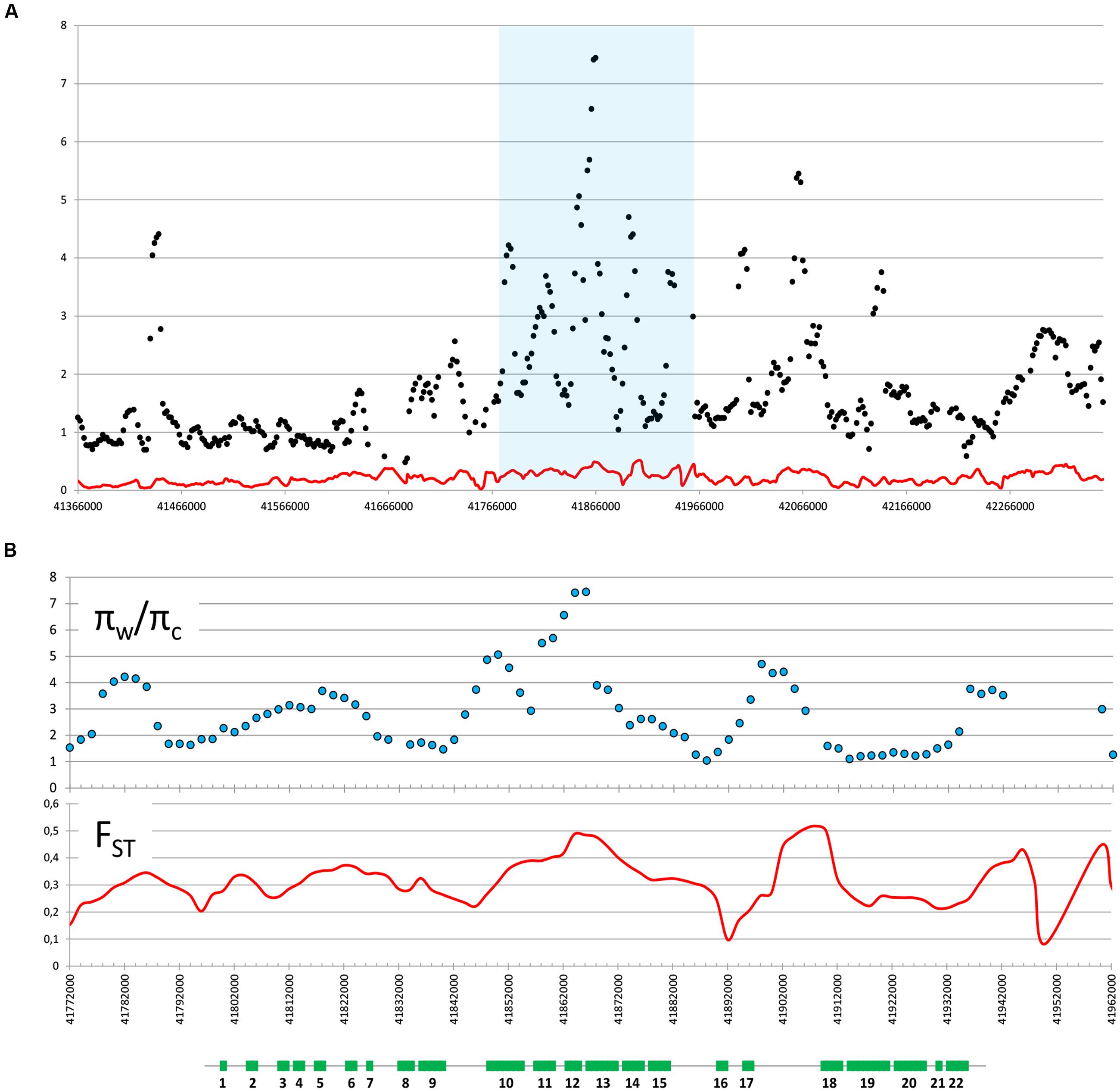
FIGURE 2. Wild/cultivated nucleotide diversity ratio (πw/πc) and pairwise population differentiation (FST). πw/πc (dots) and FST (red line) are graphed on carrot chromosome 2 centered around the cult polymorphic site in a 1 Mb (A) or 200 Kb (B) region (highlighted blue in A). Both parameters were calculated as average for sliding windows of size = 10 Kb and shift = 2 Kb. Schematic representation of genes (green rectangles) in the analyzed region is shown below graphs in panel B. Genes are numbered as in Table 2.
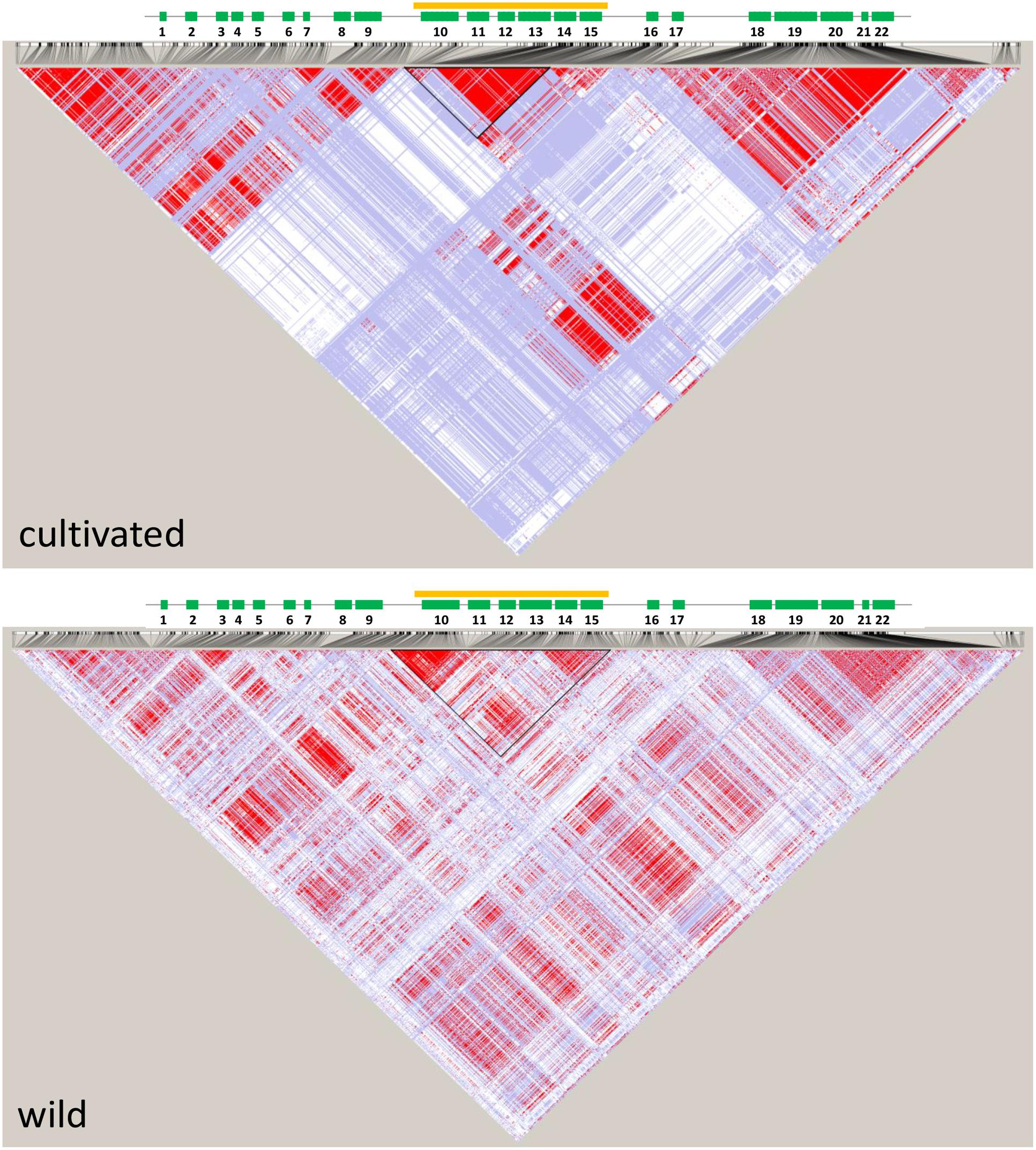
FIGURE 3. Linkage disequilibrium (LD) plots of a 200 Kb region of carrot chromosome 2 centered around the cult polymorphic site for the cultivated and wild genomes. Localization of genes is shown above (green rectangles), genes are numbered as in Table 2. The orange line and outlined triangle shows the region under selection.
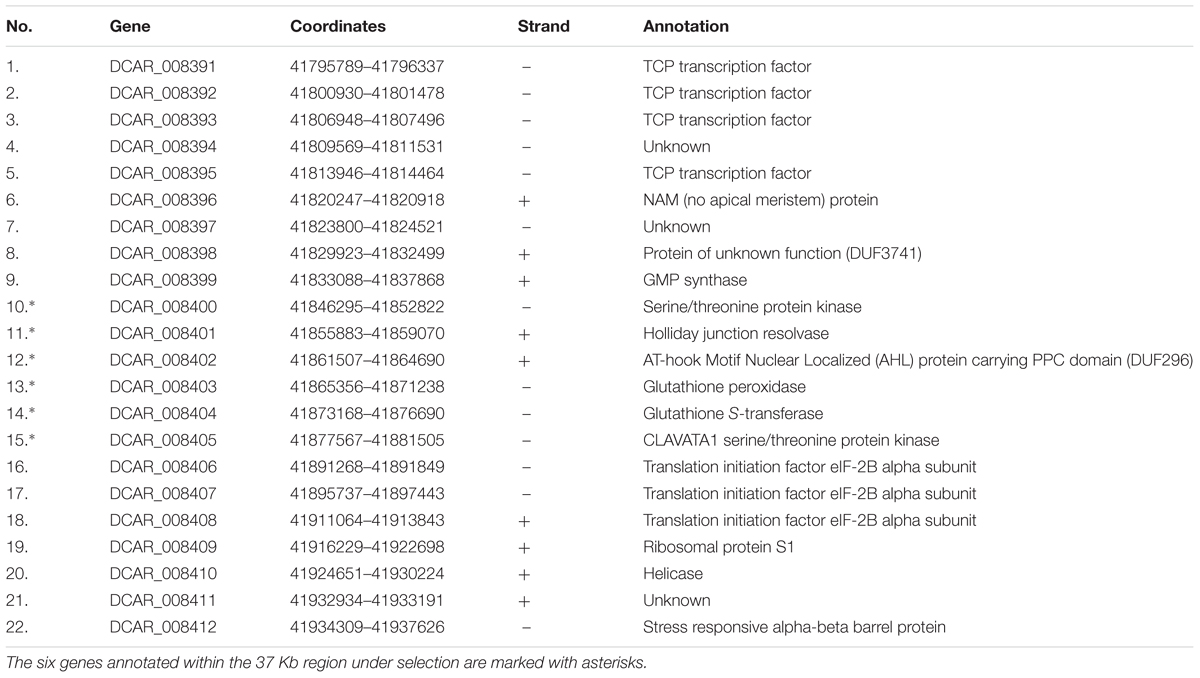
TABLE 2. Gene predictions within the 150 Kb region on carrot chromosome 2 spanning the cult polymorphic site differentiating wild and cultivated gene pools.
Structural Analysis of the Region under Selection
We used long-PCR to amplify and sequence the region spanning genes DCAR_008400 to DCAR_008405 in 12 plants representing wild and cultivated types (Table 1), including four F2 plants from the cross between the wild D. carota subsp. commutatus and the cultivated carrot line 2874B (M9 to M12 in Table 1). Two of the F2 plants were preselected as homozygous ‘wild’ and the other two as homozygous ‘cultivated’ with respect to the cult marker (Supplementary Figure 1B). As we did not observe recombination in any of the four F2 plants throughout the whole long-range PCR amplified region, we were able to characterize the two haplotypes corresponding to the wild and the cultivated parent and use them to search for putative functional polymorphisms. For the F2 plants and their parents, we observed 75 single nucleotide substitutions in the six coding regions. The two haplotypes derived from the wild and the cultivated parents were compared to SNP variants present in the remaining unrelated accessions. SNPs were present in six genes. For only one of these genes, DCAR_008402, we observed SNPs systematically differentiating the wild and the cultivated groups. Three of those substitutions were non-synonymous (Table 3). The inbred line B7262 carried ‘wild’ variants of these SNPs. In addition to these substitutions, the previously described insertion in intron 1 (Macko-Podgórni et al., 2014) was present in all cultivated accessions except B7262. To further investigate that relationship, we evaluated SNP variants in the three polymorphic sites of DCAR_008402, as well as the intronic indel, in the 29 resequenced genomes of D. carota. Again, 17 cultivated carrots both of eastern and western type carried variants attributed to the cultivated gene pool, while B7262 was confirmed to carry ‘wild’ variants. Interestingly, two Asian wild carrot accessions carried ‘cultivated’ variants in all four sites, the other three Asian wild accessions being mostly heterozygous, while no ‘cultivated’ variant was attributed to the six wild accessions of European origin (Supplementary Table 2).
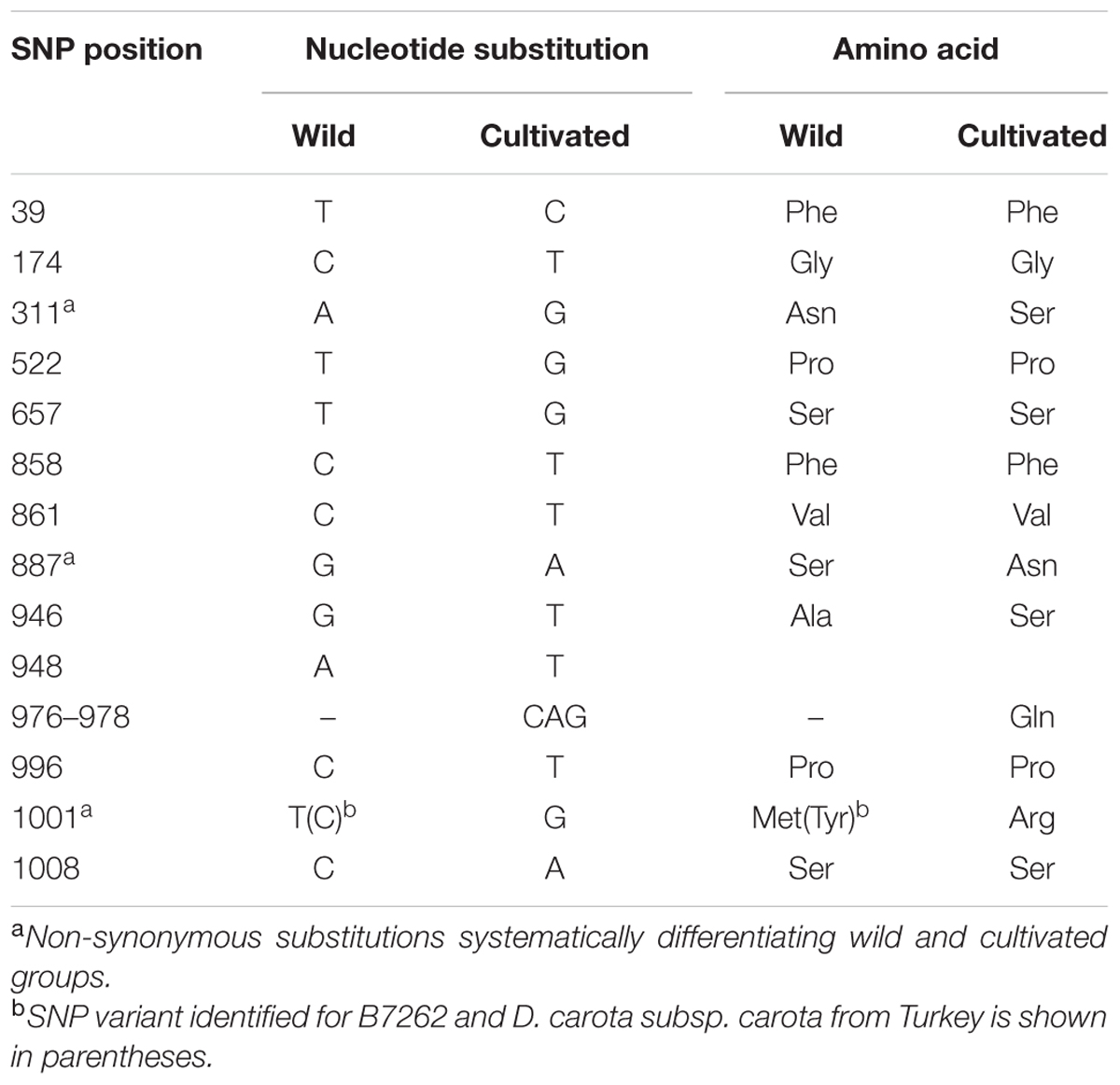
TABLE 3. Sequence variants in the DCAR_008402 coding sequence differentiating wild and cultivated accessions.
Structural and Functional Characteristics of a Candidate Domestication Gene
Following the results presented above, we carried out a more detailed analysis of DCAR_008402, a candidate domestication gene. It encodes a protein belonging to the AHL family of land plant specific transcription factors (Zhao et al., 2014). It can be classified into subfamily B2 of the AHL gene family (Zhao et al., 2014) and it clusters together with AtAHL5 and AtAHL12 from Arabidopsis thaliana (Supplementary Figure 2). Thus, it encodes a Type-II AHL protein containing two AT-hook motifs and Type-B PPC/DUF296 domain (Figure 4). Subsequently, we use the name DcAHLc1 to denote DCAR_008402.
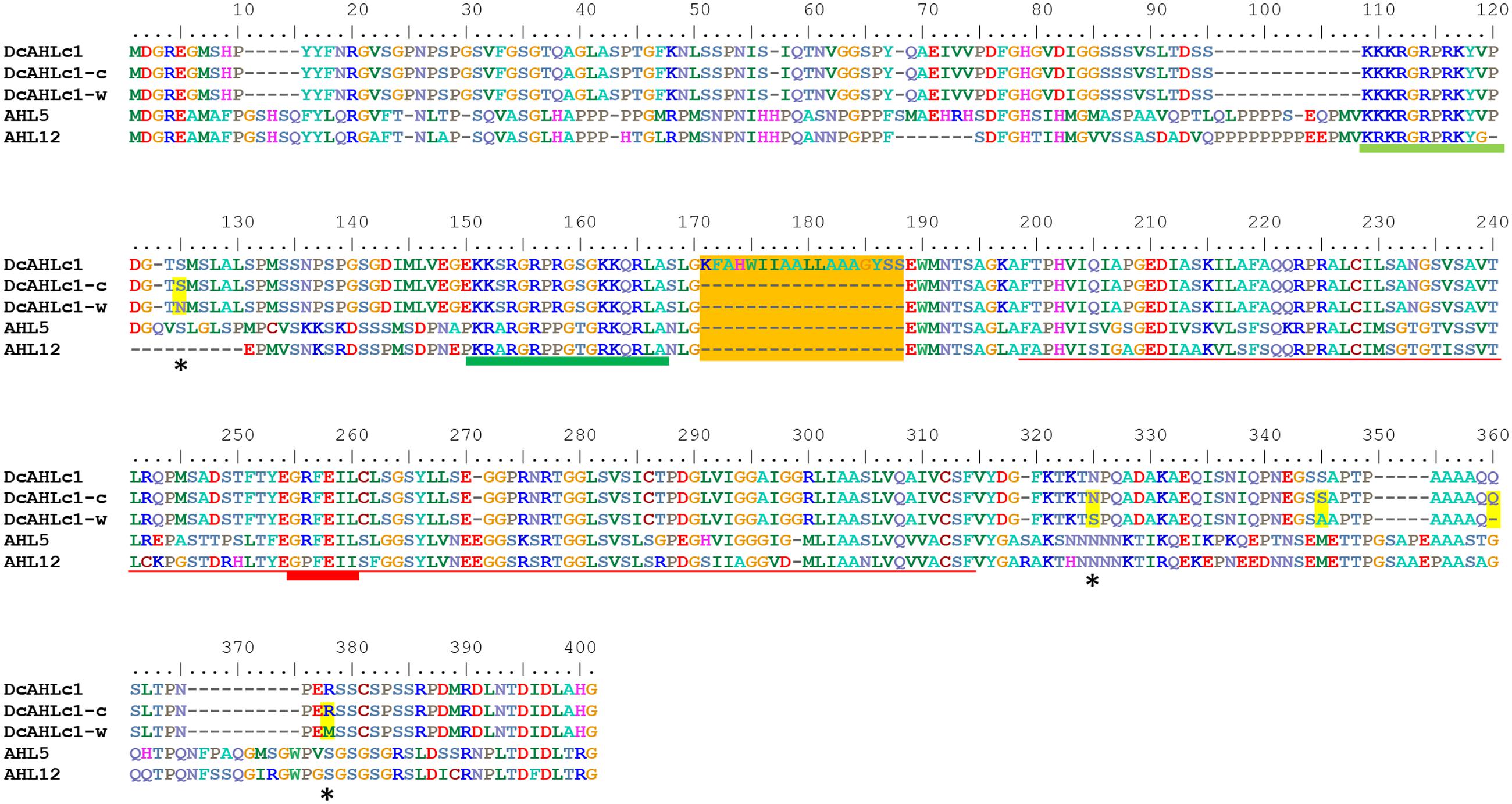
FIGURE 4. Alignment of proteins encoded by carrot DcAHLc1 gene and its homologs from A. thaliana. Alignment of three variants showing DcAHLc1 according to the gene model, major isoforms of the cultivated and the wild type (DcAHLc1-c and DcAHLc1-w, respectively) and the two A. thaliana homologs, AHL5 and AHL12. Amino acid substitutions in wild and cultivated variants are indicated by stars and yellow shading, and the putative additional exon is shaded orange. The dark green and light green lines show positions of AT-hook Type-I and Type-II, respectively (Zhao et al., 2014). The red line shows the position of the PPC/DUF296 domain (Fujimoto et al., 2004) with the GRFEIL conserved motif underlined with a thicker line.
A codon-based test for purifying selection (dN/dS) confirmed that DcAHLc1 was likely under selection in the cultivated gene pool (Supplementary Table 3). In addition to the three non-synonymous single nucleotide substitutions, an insertion in intron 1 common in the cultivated carrot was observed, as reported previously (Macko-Podgórni et al., 2014). This insertion produced an additional exon, likely resulting in an alternatively spliced isoform. Indeed, transcriptome data indicated that the major DcAHLc1 isoform did not include the additional exon, however, there was a minor isoform comprising this exon in the cultivated carrot (Figure 5). This exon was not present in any of the two A. thaliana homologs (Figure 4).
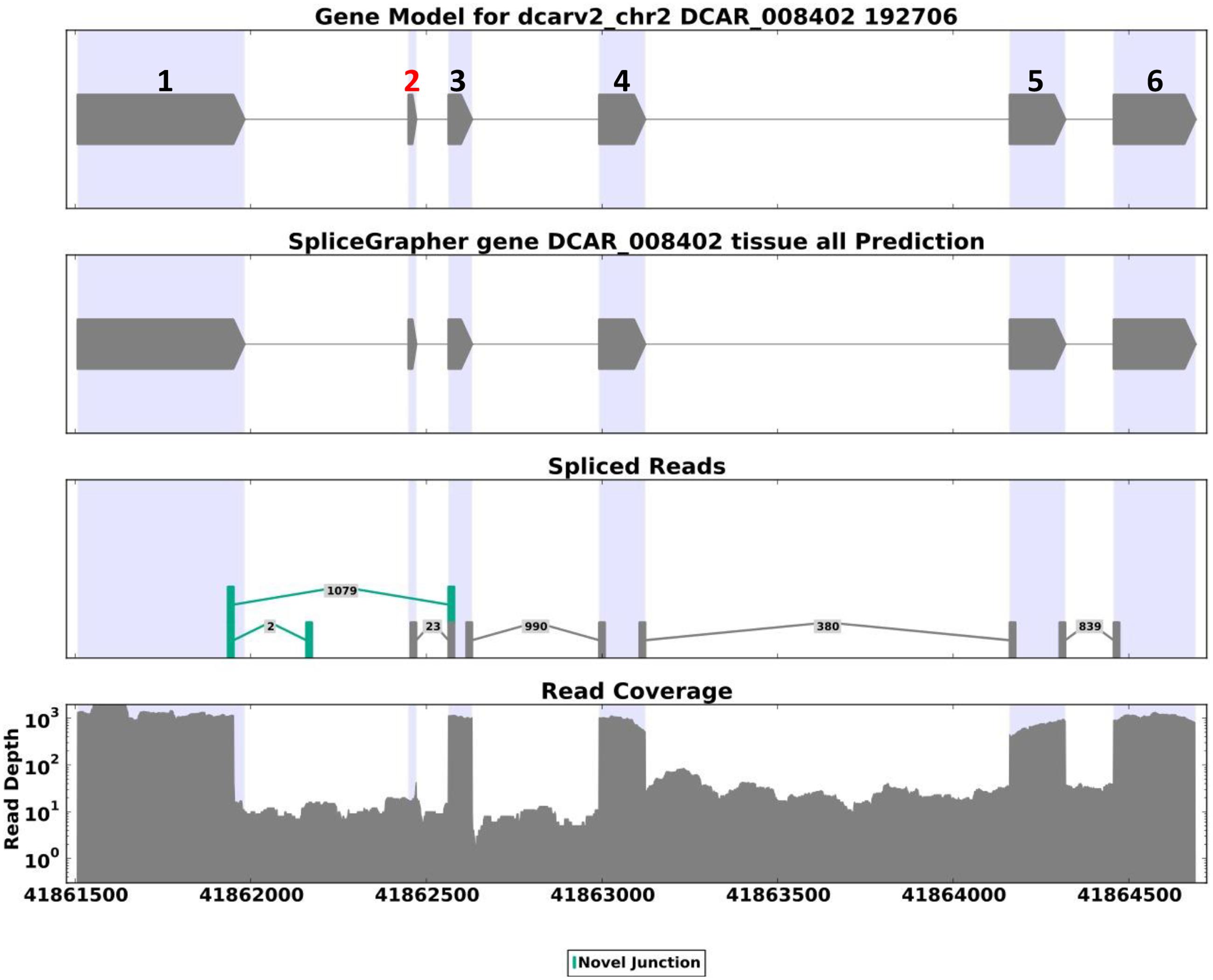
FIGURE 5. SpliceGrapher (Rogers et al., 2012) diagram showing the structure and abundance of alternatively spliced DCAR_008402 transcripts based on the cultivated carrot transcriptome reported by Iorizzo et al. (2016). The major isoform comprises a 3′ end-truncated exon 1 and no exon 2. There are two minor isoforms, one comprising exon 1 and a short fragment of intron 1 (supported by two reads), and the other one identical to the proposed gene model, comprising exon 2 derived from insertion in the cultivated carrot, marked red in the upper panel (supported by 23 reads).
RNAseq data (NCBI BioProject PRJNA291977) indicated that in the cultivated carrot, DcAHLc1 was expressed in all analyzed tissues, including fibrous and storage roots (hypocotyl, phloem, and xylem), buds and open flower, leaf and petiole, callus and germinating seeds (Supplementary Table 4). qRT-PCR indicated that no significant differences were observed in the total expression levels of DcAHLc1 in seedlings, developing or mature roots and leaves of wild and cultivated D. carota (Supplementary Figure 3). This suggests that structural differences between the wild and the cultivated variant of the gene might play a role on the functionality of this gene, rather than contrasting quantitative expression patterns.
A QTL for Root Thickening Overlaps with the cult Site
We performed a QTL analysis for root thickening measured by the root head diameter to crown diameter ratio in a field-grown wild × cultivated F2 population. Five QTLs were revealed on chromosomes 2, 3, 4, 5, and 8, jointly explaining over 62% of the total variation (Table 4; Supplementary Figure 4). The QTL on chromosome 2 overlapped the cult site (Figure 6), indicating that DcAHLc1 may possibly be involved in the development of the carrot storage root. However, QTLs on chromosomes 3, 4, and 5 had greater effect on root thickening than that on chromosome 2 (Table 4), likely reflecting the complexity of storage root developmental regulation.

TABLE 4. Chromosomal location and characteristics of QTLs for root thickening in D. carota subsp. commutatus × 2874B F2 population.
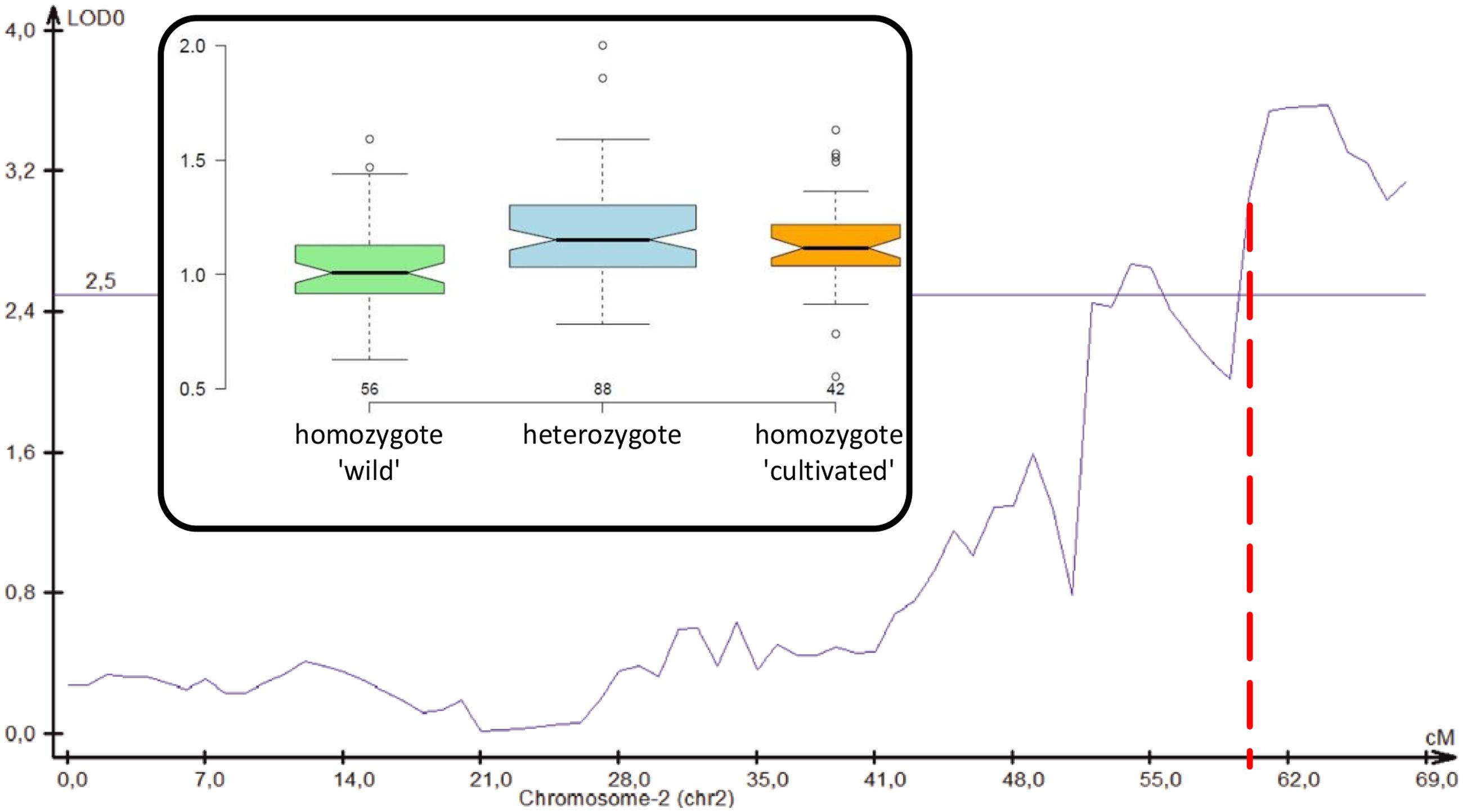
FIGURE 6. Position of a QTL for root thickening on carrot chromosome 2. Genetic distance is shown on the X-axis, LOD is on the Y-axis, thin horizontal line shows the significance threshold, red dashed vertical line shows the position of DcAHLc1. Inset: a boxplot presenting arm to crown ratio distributions in plants with respect to the cult marker variant.
Discussion
Detection of the Selective Sweep on Carrot Chromosome 2
In any crop, a set of traits differentiating the cultivated form from its wild progenitor, conferring adaptation to a cultivated environment and to consumer needs, can be defined. They are collectively called the domestication syndrome (Gepts, 2014). These traits have been under continuous selection in the course of the domestication process, resulting in local decrease of variability caused by selective sweeps in genomic regions overlapping with domestication syndrome genes in cultivated populations, as compared to their wild counterparts. Very often, domestication leads to overall decrease in genetic variability, referred to as a genetic bottleneck.
In carrot, reduction of lateral root branching and biennial growth habit were pointed out as the major primary domestication syndrome traits, the latter being crucial for the development of a non-woody storage root, while further improvements comprised root quality traits, such as pigment content and flavor. Unlike most other crops, no major decrease in overall genetic diversity was reported for carrot (Iorizzo et al., 2013, 2016; Grzebelus et al., 2014), likely resulting from its predominant outcrossing mating system and the constant bidirectional gene flow between wild and cultivated populations throughout the crop breeding history until very recently. Nevertheless, it should be possible to identify local reduction in diversity around domestication syndrome genes. In our previous study, we employed a low density screen for selective sweeps using a set of 900 DArT markers and identified 27 polymorphic sites showing signatures for selection (Grzebelus et al., 2014). It employed a relatively low number of polymorphisms, hence many domestication-associated regions remained unrecognized. Given the low marker density and a likely high rate of LD in carrot as an outcrossing species, the identification of the region on chromosome 2 can be viewed as a ‘needle in a haystack’ situation. Nevertheless, the data presented here confirm the presence of a selection sweep around the original DArT polymorphism. Using multiple lines of evidence, we revealed a 37 Kb-long genomic region on chromosome 2 under selection in the cultivated carrot. There were six genes annotated in that region, but only one of them, DCAR_008402, coding for a protein belonging to the AHL family, carried polymorphisms systematically differentiating wild and cultivated gene pools.
DcAHLc1 as a Domestication Syndrome Candidate Gene
The AHL gene family is widely distributed in land plants, with individual genomes carrying several copies. Twenty-nine AHL paralogs were identified in Arabidopsis thaliana (Fujimoto et al., 2004). Plant AHLs were further divided into two clades; intron-less Clade A and intron-containing Clade B, comprising AHLs of Type I, and Types II and III, respectively (Zhao et al., 2014). It has been reported that genes belonging to the AHL family regulate a range of processes related to plant growth and development, including not only hypocotyl growth (Street et al., 2008; Xiao et al., 2009; Zhao et al., 2013), root development (Zhou et al., 2013), floral development (Ng et al., 2009; Gallavotti et al., 2011; Jin et al., 2011; Yun et al., 2012), but also defense against pathogens (Lu et al., 2010; Yadeta et al., 2011). It has been shown that AHL3 and AHL4 proteins act jointly as transcription factors and are involved in the regulation of vascular tissue boundaries in A. thaliana determined by their intercellular trafficking (Zhou et al., 2013). AHL5 and AHL12, the closest homeologs to DcAHLc1, have not been functionally characterized in A. thaliana, as most other members of AHL Clade B. It seems conceivable that DcAHLc1 is a component of a regulatory complex involved in the development of the fleshy storage root typical for the cultivated carrot. However, the mechanism can be quite complex, as possibly it requires tissue- or even cell-specific expression (Zhou et al., 2013), intercellular movement (Jang and Lee, 2014), and direct interactions between AHLs via the PPC/DUF296 domain and with other proteins (Zhao et al., 2013). Our qPCR experiments failed to reveal any significant differences in expression of DcAHLc1, in part because of high differences among biological replicates, therefore we speculate that modifications of the interaction or movement capability resulting from changes in the gene sequence and/or structure could be responsible for the developmental shift, although the effects of tight cell-specific expression regulation which were difficult to detect using the applied methodology cannot be excluded.
While the ‘cultivated’ variant of DcAHLc1 was not observed in any of the European wild D. carota, it was present relatively frequently in the wild Asian populations. Notably, two of the wild Central Asian accessions, i.e., PI 274297 from Pakistan and PI 478369 from Xinjiang, China, were homozygous for the ‘cultivated’ variant. Convincing evidence was provided that carrot was domesticated in Central Asia (Iorizzo et al., 2013, 2016) which stands in line with reports that wild carrots from that region are capable of developing at least a primitive storage root (Vavilov, 1992). These observations imply that the mutation occurred in the wild D. carota from Central Asia, and selection upon domestication operated on the standing variation.
Most cultivated carrots evaluated carried the ‘cultivated’ variant of DcAHLc1, however, the ‘wild’ variant was present in the inbred line B7262. Previously, we observed the presence of the ‘wild’ variant mostly in primitive eastern landraces (Macko-Podgórni et al., 2014), from which the purple-rooted B7262 was derived (Simon et al., 1997). In our opinion, this observation further illustrates the complexity of the regulatory mechanism implying a possible compensatory effect, which might be provided by carrot AHL paralogs. Nevertheless, in advanced carrot cultivars of western type, the ‘cultivated’ variant is highly predominant. Thus, we conclude that DcAHLc1 is a candidate domestication gene in carrot. Given the high level of gene flow postulated between the wild and the cultivated carrot throughout most of the crop history which limited the expected genetic bottleneck (Iorizzo et al., 2013), the selective sweep around DcAHLc1 implies that it had to be under constant selection which underscores the significance of this gene for the cultivated carrot phenotype. On the basis of a QTL study, we tentatively postulate that the gene is involved in root thickening, however, the relationship has yet to be evaluated across multiple environments and populations and at a functional level. As DcAHLc1 is expressed in most developing tissues, pleiotropic effects of its action could be expected.
Conclusion
A DArT marker based low-density screen for selective sweeps inferred by the process of domestication across the genome of cultivated carrot revealed a highly significant polymorphism named cult. Here, we mapped the cult region to the distal portion of the long arm of chromosome 2, delimited the region under selection overlapping with the cult region to ca. 37 Kb and proposed a candidate domestication gene. The gene DcAHLc1 belongs to the AHL family of plant regulatory proteins. The gene variant predominant in the cultivated gene pool has been selected from the wild Central Asian D. carota. Following the preliminary evidence from a QTL study and knowledge about the mode of function of other AHLs related to root tissue patterning, we speculate that the DcAHLc1-encoded protein might be involved in the transition from the woody fibrous root of wild D. carota to the thick fleshy root typical for cultivated carrot. It provides an interesting example of the function of a gene belonging to a relatively poorly characterized family of plant transcription factors in determination of an agronomically important trait, i.e., the carrot storage root.
Author Contributions
AM-P and DG designed the study; GM and KS performed phenotyping; GM and KS performed genotyping; GM, MI, and DG performed mapping and QTL analyses; GM performed long-PCR, EG performed FISH; DS, MI, and PS provided resequencing data; AM-P, GM, DS, MI, and DG performed bioinformatic analyses; AM-P, EG, GM, KS, and DG drafted sections of the manuscript, MI and PS critically revised the manuscript, AM-P and DG prepared the final version of the paper. All authors read, reviewed, and approved the manuscript.
Funding
The research was funded by the Polish National Science Center, project no. 2012/05/B/NZ9/03401 and the statutory funds for science granted by the Polish Ministry of Science and Higher Education to the Faculty of Biotechnology and Horticulture, University of Agriculture in Krakow. MI was supported by the USDA National Institute of Food and Agriculture, Hatch project 1008691.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
We thank Ms. Emilia Morańska for her excellent assistance in FISH.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00012/full#supplementary-material
Footnotes
References
Alessandro, M. S., Galmarini, C. R., Iorizzo, M., and Simon, P. W. (2013). Molecular mapping of vernalization requirement and fertility restoration genes in carrot. Theor. Appl. Genet. 126, 415–423. doi: 10.1007/s00122-012-1989-1
Banga, O. (1957a). Origin of the European cultivated carrot. Euphytica 6, 54–63. doi: 10.1007/BF00179518
Banga, O. (1957b). The development of the original European carrot material. Euphytica 6, 64–76. doi: 10.1007/BF00179518
Banga, O. (1963). Origin and domestication of the western cultivated carrot. Genet. Agrar. 17, 357–370.
Baranski, R., Maksylewicz-Kaul, A., Nothnagel, T., Cavagnaro, P. F., Simon, P. W., and Grzebelus, D. (2012). Genetic diversity of carrot cultivars revealed by analysis of SSR loci. Genet. Resour. Crop Evol. 59, 163–170. doi: 10.1007/s10722-011-9777-3
Barrett, J. C., Fry, B., Maller, J., and Daly, M. J. (2005). Haploview, analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265. doi: 10.1093/bioinformatics/bth457
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic, a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowman, M. J., Willis, K. W., and Simon, P. W. (2014). Transcript abundance of Phytoene Synthase 1 and Phytoene Synthase 2 is associated with natural variation of storage root carotenoid pigmentation in carrot. J. Amer. Soc. Hort. Sci. 139, 63–68.
Bradeen, J. M., and Simon, P. W. (1998). Conversion of an AFLP fragment linked to the carrot Y2 locus to a simple, codominant, PCR-based marker form. Theor. Appl. Genet. 97, 960–967. doi: 10.1007/s001220050977
Broman, K. W., and Sen, Ś. (2009). A Guide to QTL Mapping with R/qtl. New York, NY: Springer-Verlag.
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines, Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Danecek, P., Auton, A., Abecasis, G., Albers, A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Doebley, J. F., Gaut, B. S., and Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Dong, F., Song, J., Naess, S. K., Helgeson, J. P., Gebhardt, G., and Jiang, J. (2000). Development and applications of a set of chromosome specific cytogenetic DNA markers in potato. Theor. Appl. Genet. 101, 1001–1007. doi: 10.1007/s001220051573
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
FAOSTAT (2013). The Statistics Division of FAO. Available at: http://faostat3.fao.org/ (accessed August 18, 2016).
Faraway, J. J. (2002). Practical Regression and ANOVA Using R. Available at http://www.mathstat.ualberta.ca/wiens/stat568/misc\%20resources/Faraway-PRA.pdf
Faust, G. G., and Hall, I. M. (2014). SAMBLASTER, fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. doi: 10.1093/bioinformatics/btu314
Fujimoto, S., Matsunaga, S., Yonemura, M., Uchiyama, S., Azuma, T., and Fukui, K. (2004). Identification of a novel plant MAR DNA binding protein localized on chromosomal surfaces. Plant Mol. Biol. 56, 225–239. doi: 10.1007/s11103-004-3249-5
Gallavotti, A., Malcomber, S., Gaines, C., Stanfield, S., Whipple, C., Kellogg, E., et al. (2011). BARREN STALK FASTIGIATE1 is an AT-Hook protein required for the formation of maize ears. Plant Cell 23, 1756–1771. doi: 10.1105/tpc.111.084590
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv 1207, 3907.
Gepts, P. (2014). The contribution of genetic and genomic approaches to plant domestication studies. Curr. Opin. Plant Biol. 18, 51–59. doi: 10.1016/j.pbi.2014.02.001
Grzebelus, D., Iorizzo, M., Senalik, D., Ellison, S., Cavagnaro, P., Macko-Podgórni, A., et al. (2014). Diversity, genetic mapping, and signatures of domestication in the carrot (Daucus carota L.) genome, as revealed by Diversity Arrays Technology (DArT) markers. Mol. Breed. 33, 625–637. doi: 10.1007/s11032-013-9979-9
Iorizzo, M., Ellison, S., Senalik, D., Zeng, P., Satapoomin, P., Bowman, M., et al. (2016). A high-quality carrot genome assembly reveals new insights into carotenoid accumulation and Asterid genome evolution. Nat. Genet. 48, 657–666. doi: 10.1038/ng.3565
Iorizzo, M., Senalik, D., Ellison, S., Grzebelus, D., Cavagnaro, P. F., Allender, C., et al. (2013). Genetic structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am. J. Bot. 100, 930–938. doi: 10.3732/ajb.1300055
Iovene, M., Cavagnaro, P. F., Senalik, D., Buell, C. R., Jiang, J., and Simon, P. W. (2011). Comparative FISH mapping of Daucus species (Apiaceae family). Chromosome Res 19, 493–506. doi: 10.1007/s10577-011-9202-y
Iovene, M., Grzebelus, E., Carputo, D., Jiang, J., and Simon, P. W. (2008). Major cytogenetic landmarks and karyotype analysis in Daucus carota and other Apiaceae. Am. J. Bot. 95, 793–804. doi: 10.3732/ajb.0700007
Jang, G., and Lee, J.-Y. (2014). Intercellular trafficking of transcription factors in the vascular tissue patterning. Physiol. Plant. 151, 184–191. doi: 10.1111/ppl.12140
Jin, Y., Luo, Q., Tong, H., Wang, A., Cheng, Z., Tang, J., et al. (2011). An AT-hook gene is required for palea formation and floral organ number control in rice. Dev. Biol. 359, 277–288. doi: 10.1016/j.ydbio.2011.08.023
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Boinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and 2ΔΔC(T) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H., Zou, Y., and Feng, N. (2010). Overexpression of AHL20 negatively regulates defenses in Arabidopsis. J. Integr. Plant Biol. 52, 801–808. doi: 10.1111/j.1744-7909.2010.00969.x
Macko-Podgórni, A., Iorizzo, M., Smółka, K., Simon, P. W., and Grzebelus, D. (2014). Conversion of a diversity arrays technology marker differentiating wild and cultivated carrots to a co-dominant cleaved amplified polymorphic site marker. Acta Biochim. Pol. 61, 19–22.
Meyer, R. S., Duval, A. E., and Jensen, H. R. (2012). Patterns and processes in crop domestication, an historical review and quantitative analysis of 203 global food crops. New Phytol. 196, 29–48. doi: 10.1111/j.1469-8137.2012.04253.x
Murray, M. G., and Thompson, W. F. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4326. doi: 10.1093/nar/8.19.4321
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426.
Ng, K.-H., Yu, H., and Ito, T. (2009). AGAMOUS controls GIANT KILLER, a multifunctional chromatin modifier in reproductive organ patterning and differentiation. PLoS Biol. 7:e1000251. doi: 10.1371/journal.pbio.1000251
Rogers, M. F., Thomas, J., Reddy, A. S., and Ben-Hur, A. (2012). SpliceGrapher: detecting patterns of alternative splicing from RNA-Seq data in the context of gene models and EST data. Genome Biol. 13:R4. doi: 10.1186/gb-2012-13-1-r4
Simon, P. W., Rubatzky, V., Bassett, M. J., Strandberg, J. O., and White, J. M. (1997). B7262 purple carrot inbred. HortSci. 32, 146–147.
Small, E. (1978). A numerical taxonomic analysis of the Daucus carota complex. Can. J. Bot. 56, 248–276. doi: 10.1139/b78-033
Stolarczyk, J., and Janick, J. (2011). Carrot, history and iconography. Chron. Horticult. 51, 13–18.
Street, I. H., Shah, P. K., Smith, A. M., Avery, N., and Neff, M. M. (2008). The AT-hook-containing proteins SOB3/AHL29 and ESC/AHL27 are negative modulators of hypocotyl growth in Arabidopsis. Plant J. 54, 1–14. doi: 10.1111/j.1365-313X.2007.03393.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6, Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Van Ooijen, J. W. (2006). JoinMap 4, Software for the Calculation of Genetic Linkage Maps in Experimental Populations. Wageningen: Kyazma BV.
Vavilov, N. I. (1992). Origin and Geography of Cultivated Plants. New York, NY: Cambridge University Press.
Wang, S., Basten, C. J., and Zeng, Z.-B. (2012). Windows QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University.
Xiao, C., Chen, F., Yu, X., Lin, C., and Fu, Y.-F. (2009). Over-expression of an AT-hook gene, AHL22, delays flowering and inhibits the elongation of the hypocotyl in Arabidopsis thaliana. Plant Mol. Biol. 71, 39–50. doi: 10.1007/s11103-009-9507-9
Yadeta, K. A., Hanemian, M., Smit, P., Hiemstra, J. A., Pereira, A., Marco, Y., et al. (2011). The Arabidopsis thaliana DNA-binding protein AHL19 mediates Verticillium wilt resistance. Mol. Plant Microbe Interact. 24, 1582–1591. doi: 10.1094/MPMI-04-11-0090
Ye, J., Coulouris, G., Cutcutache, I., Rozen, S., and Madden, T. (2012). Primer-BLAST, a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:1. doi: 10.1186/1471-2105-13-134
Yildiz, M., Willis, D. K., Cavagnaro, P. F., Iorizzo, M., Abak, K., and Simon, P. W. (2013). Expression and mapping of anthocyanin biosynthesis genes in carrot. Theor. Appl. Genet. 126, 1689–1702. doi: 10.1007/s00122-013-2084-y
Yun, J., Kim, Y.-S., Jung, J.-H., Seo, P. J., and Park, C.-M. (2012). The AT-hook motif-containing protein AHL22 regulates flowering initiation by modifying FLOWERING LOCUS T chromatin in Arabidopsis. J. Biol. Chem. 287, 15307–15316. doi: 10.1074/jbc.M111.318477
Zagorodskikh, P. (1939). New data on the origin and taxonomy of the cultivated carrot. C. R. (Doklady) Acad. Sci. USSR 25, 522–525.
Zhao, J., Favero, D. S., Peng, H., and Neff, M. M. (2013). Arabidopsis thaliana AHL family modulates hypocotyl growth redundantly by interacting with each other via the PPC/DUF296 domain. Proc. Natl. Acad. Sci. U.S.A. 110, E4688–E4697. doi: 10.1073/pnas.1219277110
Zhao, J., Favero, D. S., Qiu, J., Roalson, E. H., and Neff, M. M. (2014). Insights into the evolution and diversification of the AT-hook motif nuclear localized gene family in land plants. BMC Plant Biol. 14:266. doi: 10.1186/s12870-014-0266-7
Zhou, J., Wang, X., Lee, J.-Y., and Lee, J.-Y. (2013). Cell-to-cell movement of two interacting AT-hook factors in Arabidopsis root vascular tissue patterning. Plant Cell 25, 187–201. doi: 10.1105/tpc.112.102210
Keywords: AT-hook motif nuclear localized (AHL), domestication syndrome, genotyping-by-sequencing, linkage disequilibrium, single nucleotide polymorphism, storage root
Citation: Macko-Podgórni A, Machaj G, Stelmach K, Senalik D, Grzebelus E, Iorizzo M, Simon PW and Grzebelus D (2017) Characterization of a Genomic Region under Selection in Cultivated Carrot (Daucus carota subsp. sativus) Reveals a Candidate Domestication Gene. Front. Plant Sci. 8:12. doi: 10.3389/fpls.2017.00012
Received: 25 October 2016; Accepted: 04 January 2017;
Published: 18 January 2017.
Edited by:
Henry T. Nguyen, University of Missouri, USAReviewed by:
Steven B. Cannon, Agricultural Research Service (USDA), USAClaudio R. Galmarini, National University of Cuyo, Argentina
Copyright © 2017 Macko-Podgórni, Machaj, Stelmach, Senalik, Grzebelus, Iorizzo, Simon and Grzebelus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dariusz Grzebelus, ZC5ncnplYmVsdXNAb2dyLnVyLmtyYWtvdy5wbA==