 Longming Zhu
Longming Zhu Weichen Ni
Weichen Ni Shuai Liu
Shuai Liu Binhua Cai
Binhua Cai Han Xing
Han Xing Sanhong Wang
Sanhong Wang- 1Department of Horticulture, Nanjing Agricultural University, Nanjing, China
- 2National Key Laboratory of Crop Genetics and Germplasm Enhancement, Department of Agricultural, Nanjing Agricultural University, Nanjing, China
Alternaria blotch disease of apple (Malus × domestica Borkh.), caused by the apple pathotype of Alternaria alternata, is one of the most serious fungal diseases to affect apples. To develop an understanding of how apples respond to A. alternata apple pathotype (AAAP) infection, we examined the host transcript accumulation over the period between 0 and 72 h post AAAP inoculation. Large-scale gene expression analysis was conducted of the compatible interaction between “Starking Delicious” apple cultivar and AAAP using RNA-Seq and digital gene expression (DGE) profiling methods. Our results show that a total of 9080 differentially expressed genes (DEGs) were detected (>two-fold and FDR < 0.001) by RNA-Seq. During the early phase of infection, 12 h post inoculation (HPI), AAAP exhibited limited fungal development and little change in the transcript accumulation status (950 DEGs). During the intermediate phase of infection, the period between 18 and 36 HPI, increased fungal development, active infection, and increased transcript accumulation were detected (4111 and 3838 DEGs detected at each time point, respectively). The majority of DEGs were detected by 72 HPI, suggesting that this is an important time point in the response of apples' AAAP infection. Subsequent gene ontology (GO) and pathway enrichment analyses showed that DEGs are predominately involved in biological processes and metabolic pathways; results showed that almost gene associated with photosynthesis, oxidation-reduction were down-regulated, while transcription factors (i.e., WRKY, MYB, NAC, and Hsf) and DEGs involved in cell wall modification, defense signaling, the synthesis of defense-related metabolites, including pathogenesis-related (PRs) genes and phenylpropanoid/cyanoamino acid /flavonoid biosynthesis, were activated during this process. Our study also suggested that the cell wall defensive vulnerability and the down-regulation of most PRs and HSP70s in “Starking Delicious” following AAAP infection might interpret its susceptible to AAAP.
Introduction
Alternaria blotch disease of apple, caused by the Alternaria alternata apple pathotype (AAAP), is one of the most serious fungal diseases affecting apples globally, especially in East Asia (Saito et al., 2001). This disease affects apple tree growth and production via the infection of leaves, young shoots, and fruits and leads to marked declines in tree vigor. Initial lesions appear first on apple leaves in late spring or early summer as small, round, black-colored spots that gradually enlarging to 2–5 mm in diameter and have a brownish purple border. These lesions may coalesce or undergo secondary enlargement and become irregular and much darker, acquiring a “frog-eye” appearance (Li et al., 2013). Severe AAAP infection can result in 60–80% defoliation (Filajdic and Sutton, 1991), leading to premature fruit drop. However, both the rate of occurrence and severity of Alternaria leaf blotch vary between apple cultivars. For example, “Jonathan,” “Jonagold,” and “Gala” are resistant to AAAP, while the “Starking Delicious” and “Indo” varieties are highly susceptible to infection (Abe et al., 2010). Thus, extensive breeding research has been carried out to understand genetics of the disease resistance as well as the underlying mechanism in order that new cultivars resistant to the pathogen can be produced. Little progress has been made in these areas, however, mainly because of our limited genetic understanding of response to the pathogen and the long apple breeding cycle. Application of fungicides is currently the main method to control AAAP; however, continuous and repetitive use of these chemicals can result in the individual selection of resistant pathogen and potentially pollutes the environment. Thus, an improved understanding of the defense mechanisms used by apples in response to AAAP will contribute to the design of new and safer control strategies, as well as aiding in the development of resistant cultivars.
Plants have evolved a number of strategies to effectively combat invasion of pathogens, involving a large number of physiological responses including hypersensitive reaction (HR), cell walls modifications, and the production of antimicrobial proteins and metabolites (Cohn et al., 2001). These physiological responses are associated with the reprogramming of numerous defense-related genes and transcription factors. Thus, as technology has advanced, approaches in comparative “omics” have greatly contributed to the effort of defining gene and protein functions and our understanding of their expression and changes in accumulation during plant-pathogen interactions (Mathioni et al., 2011; Zhao et al., 2014). Enormous datasets have been derived from model plant organisms, enabling the identification of defense-related genes and proteins (Thilmony et al., 2006; Mukherjee et al., 2010; Proietti et al., 2013). Apple is not just an agronomically important crop; it is also a model species within Rosaceace (Shulaev et al., 2008). Although, several studies to date have analyzed apple-pathogen interactions (Sarowar et al., 2011; Vilanova et al., 2014), this study is the first global transcription analysis of apple in response to AAAP.
In this study, we conducted a large-scale gene expression analysis of the “Starking Delicious” apple cultivar in response to AAAP infection using RNA-Seq and digital gene expression (DGE) methods. Use of these approaches enabled us to gain molecular insights into the changes in transcriptional expression that are associated with apple defense to the fungus, as well as the transcriptional response of “Starking Delicious” to AAAP infection, and the possible reasons that underlie its susceptibility to this pathogen.
Materials and Methods
Plant Materials, AAAP Culture, and Inoculation Method
Two-year-old “Starking Delicious” cultivar apple plants grafted on Malus robusta stocks were grown in a greenhouse at Nanjing Agricultural University, located in Nanjing, Jiangsu Province, China. The AAAP fungus was expanded on a potato dextrose agar (PDA; 200 g potato extract, 20 g dextrose, 20 g agar, 1 L water) medium for 5 days at 26°C under dark conditions. The inoculation method was carried out according to the protocol described previously (Abe et al., 2010); the fourth and fifth youngest opened leaves from shoot tips were collected from plants and inoculated with mycelia together with PDA medium punched using a hole punch (diameter = 5 mm). In each treatment, leaves were inoculated with six cakes of mycelium applied to both side of the midrib of the abaxial leaf surfaces; the mock-inoculation of leaves using PDA medium cakes instead of mycelia was also carried out as a control. Thus, five groups of leaves were inoculated with mycelia or PDA cakes at 36, 18, 6, and 4 h intervals and then incubated at 25°C under a 14 h light/10 h dark cycle in sterilized plastic chambers. The groups were then sampled simultaneously when the first set of inoculated leaves reached the 72 h post inoculation (HPI) time point; these five groups represent the five infection stage, 72, 36, 18, 12, and 8 HPI. Leaves sampled at 8 and 12 HPI were used to visualize new hypha development and necrotic tissue. Trypan blue stain is as previously described (Koch and Slusarenko, 1990). All microscopic observations were conducted using a compound microscope.
Leaf samples were placed rapidly into liquid nitrogen and then stored at −70°C for RNA extraction. Each stage contained three parallel leaves from three apple trees that represented three biological replicates.
Total RNA Extraction, Library Construction, and Sequencing
Total RNA was isolated using a cetyltrimethyl ammonium bromide method (Chang et al., 1993; Pavy et al., 2008). Equal quantities of RNA from three biological replications at each stage were pooled to construct a complementary DNA (cDNA) library. Oligo-(dT) magnetic beads were used to isolate poly-(A) messenger RNA (mRNA) from total RNA, and mRNA was fragmented in fragmentation buffer. Using these short fragments (≈200 bp) as templates, random hexamer-primers were then used to synthesize first-strand cDNA, while second-strand cDNA was synthesized using buffer, dNTPs, RNaseH, and DNA polymerase I. Short double-stranded cDNA fragments were purified using a QiaQuick PCR extraction kit (Qiagen, Venlo, Netherlands), resolved with elution buffer for end reparation and the addition of poly (A), before being ligated to sequencing adapters. Subsequent to purification using agarose gel electrophoresis, suitable fragments were enriched via PCR amplification, and libraries were sequenced on the Illumina HiSeq™2000 platform at the Beijing Genomics Institute (Shenzhen, China; http://www.genomics.cn/index), following the manufacturer's protocols.
RNA-Seq Data Analysis
Raw reads from the image data output from the sequencing machine were generated by Base Calling and saved in FASTQ format. Clean reads were generated by removing reads with adaptors, reads where the number of unknown bases was more than 10%, and low-quality reads (the percentage of the low-quality bases with which value ≤5 was more than 50% in one read).
Clean reads were then aligned to reference sequences using the software SOAPaligner/soap2 (Li et al., 2009) with no more than two mismatches were allowed in each alignment. The Malus × domestica Borkh. cv. “Golden Delicious” genome assemblies as well as related gene annotations were downloaded from the Phytozome V9.0 database (ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Mdomestica/assembly), (ftp://ftp.jgi-psf.org/pub/compgen/phytozome/v9.0/Mdomestica/annotation).
Gene expression levels were calculated using the “reads per kb per million reads” (RPKM) method (Mortazavi et al., 2008), while differential expressed genes (DEGs) were analyzed as described previously (Audic and Claverie, 1997). In addition, the false discovery rate (FDR) ≤ 0.001 and an absolute value of ≥ 1 were used as the thresholds to judge the significance of differences in gene expression.
Gene ontology (GO) categories were assigned to all genes via a BLASTX hit using the Blast2GO software. DEGs were first mapped to GO terms using a standard database (http://www.geneontology.org/), gene numbers for each term were calculated, and GO terms significantly enriched in DEGs compared to the background genome were determine with a hypergeometric test. All calculated P-values were then subjected to Bonferroni Correction, using a corrected P ≤ 0.05 as the threshold. GO terms that fulfilled this criterion were defined as significantly enriched in DEGs. On the basis of the Kyoto Encyclopedia of Gene and Genome (KEGG), pathway enrichment was then analyzed using the same method, while the WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) software was used for functional classification of GO terms following DEG GO annotation.
qRT-PCR Verification
Gene-specific primers for qRT-PCR were designed using the Beacon Designer 7.0 program (Premier Biosoft International, California, USA) and are listed in Table S1. Template cDNAs were synthesized using M-MLV reverse transcriptase (Promega, USA) from 1.0 μg of total RNAs following the manufacturer's instructions. 2 × SYBR-Green I RT-PCR Master Mix (Takara, Japan) was used as the labeling agent, while tubulin from M. domestica served as the internal reference gene. These reactions were performed on an Applied biosystems 7300 Real Time PCR System, with the reaction mixture (20 μL) containing 10 μL 2 × Master Mix, 10 μmol·L−1 forward and reverse primers (0.4 μL each), and 1 μL template cDNA. The PCR program in this case was 95°C for 1 min, followed by 40 cycles at 95°C for 20 s, 60°C for 20 s and 72°C for 40 s. Three independent biological replicates were performed for each sample, while the relative expression levels of the selected unigenes were calculated using the relative 2−ΔCT method (Livak and Schmittgen, 2001). The results represented in this case represent mean standard deviations for the three experimental replicates.
Results and Discussion
Changes in Symptoms Following AAAP Infection
We conducted an initial microscope study to determine an appropriate initial sampling time, staining inoculated leaves with trypan blue. The observations made during this study show that while some spores developed new hyphae by 8 HPI, no necrotic tissue formed by this time point. However, because necrotic tissue was present by 12 HPI, obviously infected by new hyphae (Figure 1A), this point was used as our first sampling time.
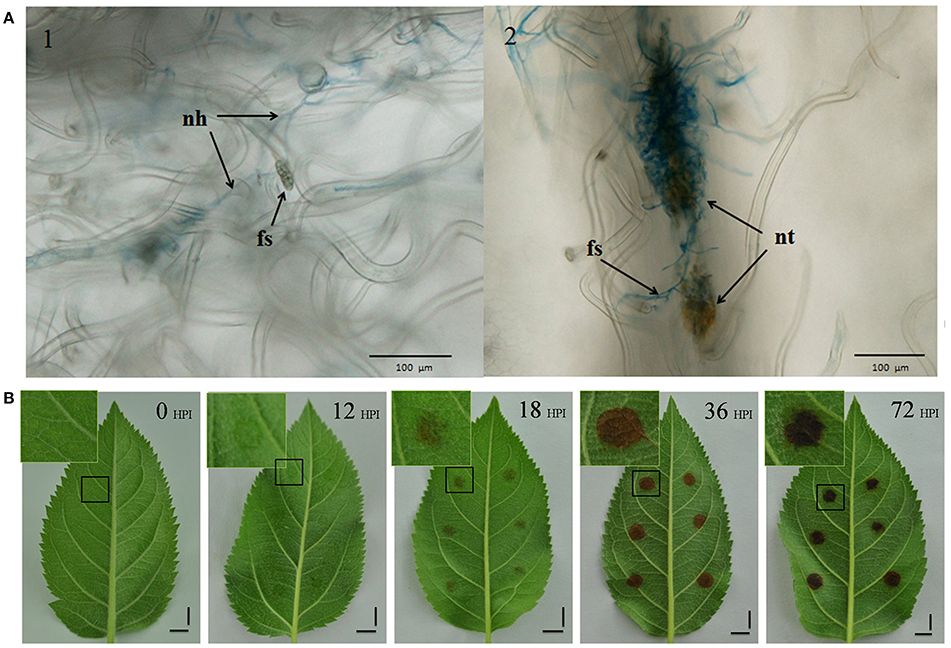
Figure 1. Symptoms of AAAP infection. (A) Infected leaves selected from 8 HPI (Panel 1) and 12 HPI (Panel 2) stained with trypan blue and imaged with optical microscopy. nh, new hyphae; fs, fungal spores; nt, necrotic tissue. Scale bar = 100 μm. (B) Changes in symptoms in “Starking Delicious” apple leaves after infection with A. alternata AP. Images show the alternaria blotch at 0, 12, 18, 36, and 72 HPI, with enlarged views of portions of damaged tissues in insets. Scale bar = 1 cm.
We recorded a series of macroscopic observations marking the progress of disease on the inoculated leaves (Figure 1B); leaf areas containing mycelial “cakes” appeared to be water soaked by 12 HPI even though the disease symptoms were not obvious, while blotches comprising damaged tissues appeared to be weakly brown by 18 HPI. Observation confirm that blotch size increased and deepened in color over the subsequent 18 h; following this, there was no further increase in blotch size but color deepened from brown to black.
RNA-Seq Data and Apple DGE Profiles in Response to AAAP Infection
According to the macroscopic and microscopic observation, we constructed four RNA libraries, i.e., 12, 18, 36, and 72 HPI, representative of four treatments of leaves inoculated by AAAP at 12, 18, 36, and 72 h. In addition, we mixed the mock-inoculated leaves of four time points into one sample as control (0 HPI). Five RNA libraries were analyzed using RNA-Seq method in combination with comparative DGE profiling. The sequencing raw data set has been deposited in National Center for Biotechnology Information Sequence Read Archive database (accession number accession number SRP091754). Approximately 11.96~12.61 million of raw RNA-Seq reads were produced for each sample. After filtering the dirty reads, 10.03~10.70 million and 7.83~8.36 million were mapped, of which 6.40~6.90 million and 5.38~5.61 million reads were uniquely mapped to reference genomes (Table S2-1) and specific genes (Table S2-2), respectively. On the basis of these mapped reads, a total of 41,022 gene expression levels were calculated using the RPKM method (Figure 2A, Excel S1).
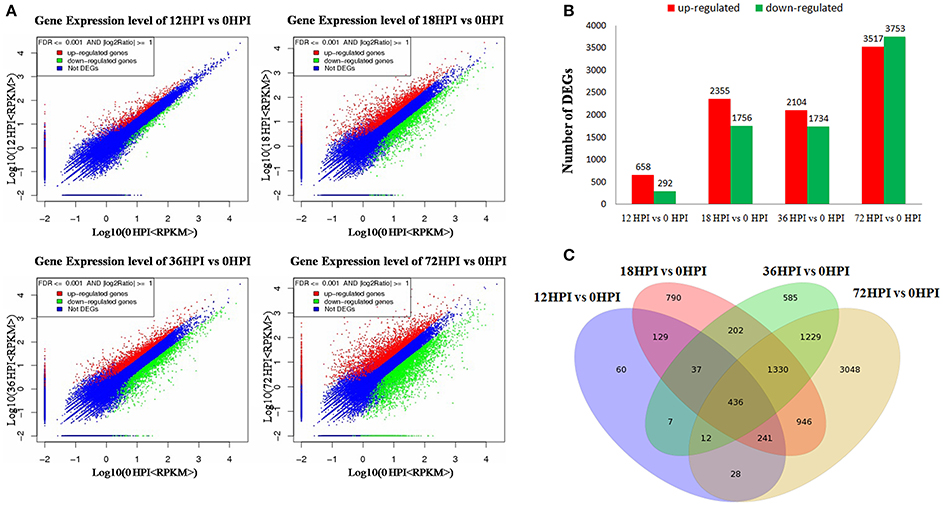
Figure 2. DEGs between samples. (A) Scattered plot of differential expression. (B) Numbers of DEGs compared between two samples (i.e., 12 vs. 0 HPI, 18 vs. 0 HPI, 36 vs. 0 HPI, 72 vs. 0 HPI, and with 0 HPI as the control). DEGs are shown in red (up-regulated) and green (down-regulated). (C) Venn diagram analysis of the DEGs in “Starking Delicious” apple leaves after inoculation with A. alternata AP.
Gene expression comparisons were performed between the AAAP-infected and control samples; compared with 12 and 0 HPI libraries (12 vs. 0 HPI), 950 DEGs were identified, of which 658 were up-regulated and 292 were down-regulated. In contrast, when the 18 and 0 HPI libraries were compared (18 vs. 0 HPI), 4111 DEGs were generated with 2355 up-regulated and 1756 down-regulated, respectively, while 3838 DEGs were identified when the 36 and 0 HPI libraries were compared (36 vs. 0 HPI), of which 2104 were up-regulated and 1734 were down-regulated, respectively. Out of all comparisons, however, the largest number of DEGs, 7270, were found when the 72 with 0 HPI libraries were compared (72 vs. 0 HPI) of which 3517 were up-regulated and 3753 were down-regulated, respectively (Figure 2B, Excel S2). Results showed that these transcriptomic changes do exhibit some shared characteristics when compared with fungal development and symptom phenotypes, including limited fungal development and little change in the transcript accumulation by 12 HPI. At intermediate stage, 18 and 36 HPI, increases in the level of fungal development, active infection, and increased transcript accumulation are seen, while the majority of DEGs were detected by 72 HPI which suggests that this time, in particular, is important for the apple in response to AAAP infection.
When these comparative results are illustrated as Venn diagram, it is clear that both unique and shared DEGs occur between, and among, pairs (Figure 2C). For example, 890 DEGs (93.68% of the total) in 12 HPI vs. 0 HPI, 3321 DEGs (81.79% of the total) in 18 vs. 0 HPI, 3253 DEGs (84.76% of the total) in 36 vs. 0 HPI and 4222 DEGs (59.07% of the total) in 72 vs. 0 HPI shared with the other compared libraries respectively, moreover, 436 DEGs are shared across all comparisons. These results suggest that as the pathogen infection progresses, more genes become involved in defense response, and a large number of genes that respond early to pathogen infection also function subsequently.
Functional Annotation of Differentially Expressed Genes (DEGs)
GO categories were developed using the Blast2GO program (http://www.blast2go.com/) in order to evaluate potential DEG functions. DEGs were classified into 50 functional categories including biological process (23), cellular component (15), and molecular function (12) (Figure S1). Results showed that 73 GO terms were enriched as a result of GO enrichment analysis, with the most significant biological process enrichments in the “Metabolic process,” “Single-organism metabolic process,” and “Organonitrogen compound metabolic process.” In contrast, the most significant cellular component enrichments occurred in the “Plastid,” and “Cytoplasmic part and organelle part,” while molecular function enrichments were in “Catalytic activity” and “Oxidoreductase activity.” The results of this analysis also demonstrate that GO terms were enriched differently at each of the four time points. For example, “monooxygenase activity,” “CoA-ligase activity,” and “prenyltransferase activity” as well as “response to stimulus” were particularly enriched by 12 HPI, while “kinase activity,” and “transferase activity” as well as “hexose metabolic process” were particularly enriched by 18 HPI. In addition, “RNA binding,” “structural molecule activity,” and “cis-trans isomerase activity” as well as “ribonucleoprotein complex biogenesis” were particularly enriched by 36 HPI, while “single-organism biosynthetic process,” “plastid organization,” “response to abiotic stimulus,” and “fatty acid biosynthetic process” were particularly enriched by 72 HPI (Figure 3). In addition, KEGG pathway mapping was also carried out based on orthology (KO) terms for assignments; results show that 45 KEGG pathways were significantly enriched. Of these, 17, 30, 22, and 32 pathways were enriched at 12, 18, 36, and 72 HPI, respectively, while nine were enriched at all measured time intervals (Table 1). Maps with highest DEGs representation those for the metabolic pathways (i.e., KO 01100 with 8688 DEGs), followed by those for the biosynthesis of secondary metabolites (i.e., KO 01110 with 5111 DEGs), plant-pathogen interactions (i.e., KO 04626 with 3658 DEGs), and plant hormone signal transduction (i.e., KO 04075 with 2107 DEGs). Taken together, these results suggest that apple have evolved a range of different molecular defense strategies depending on the infection stages of the pathogen.
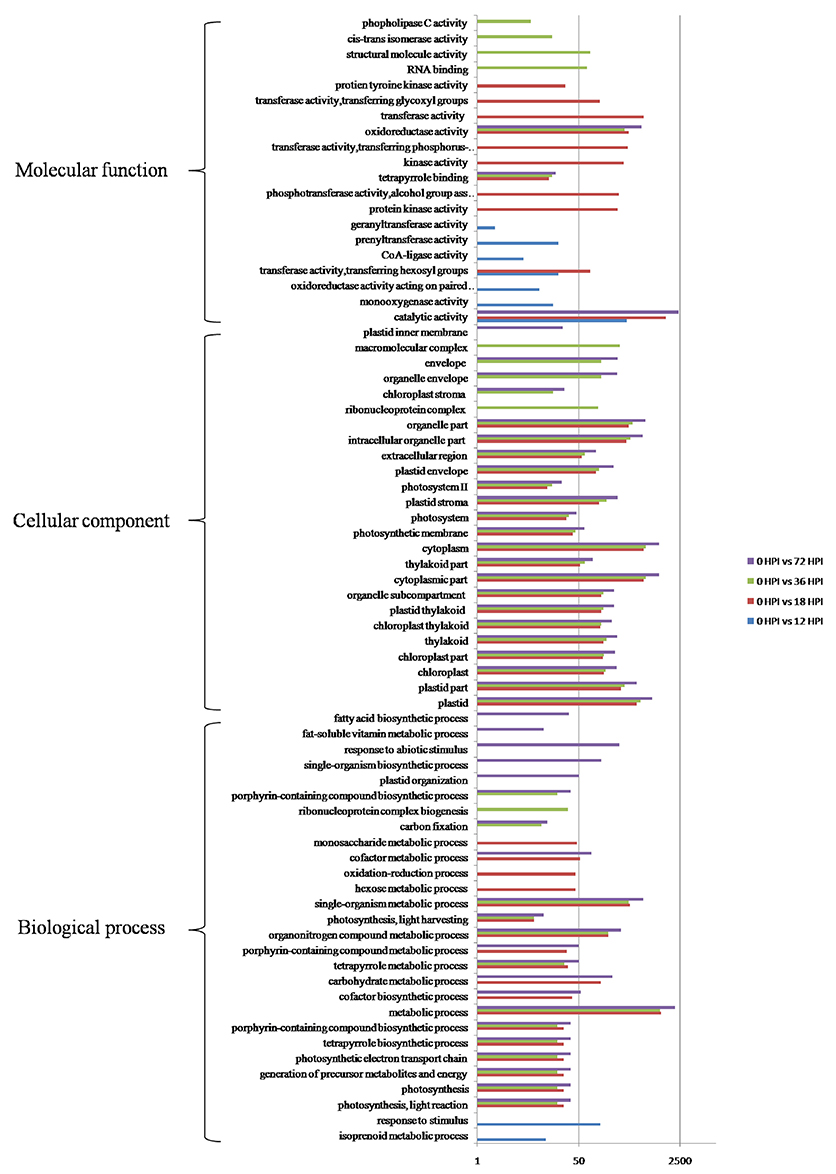
Figure 3. GO functional enrichment analyzes of DEGs.
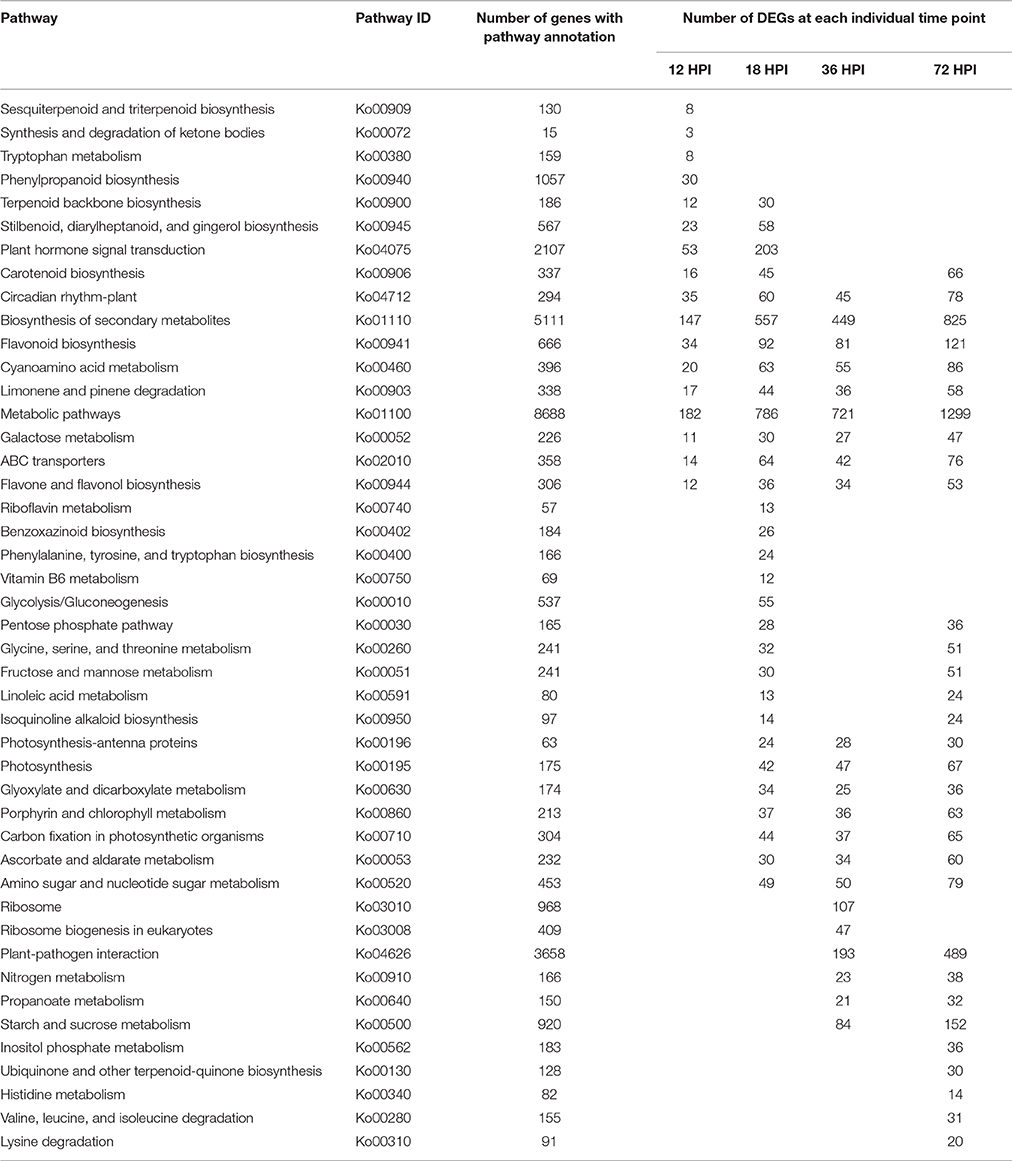
Table 1. Results of KEGG pathway enrichment analysis.
Validation of RNA-Seq Expression Levels by qRT-PCR
Total RNA at four time points during the pathogen infection provided templates for qRT-PCR validation. We randomly selected 12 DEGs to validate our RNA-Seq results. qRT-PCR data for these genes were consistent with the RNA-Seq results from the four samples, which indicated a high degree of reproducibility between transcript abundances assayed using RNA-Seq and the expression profiles revealed by qRT-PCR data (Figure 4). However, the fact that quantities were not highly consistent suggests sensitivity differences between the two methods.
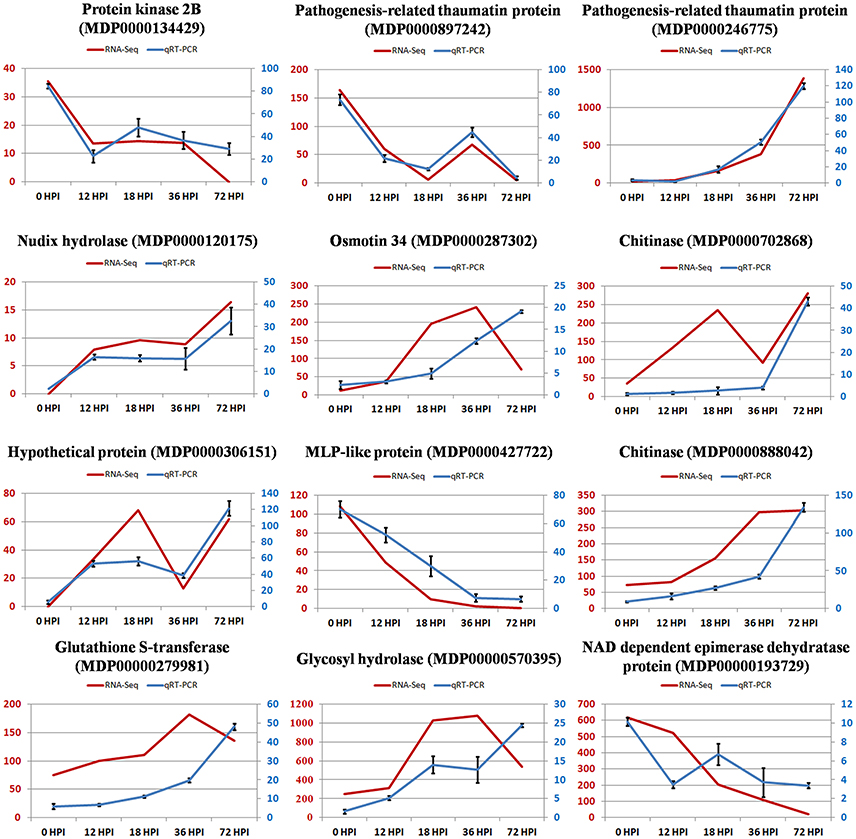
Figure 4. The relative expression level change of 12 selected genes from DEGs by quantitative real-time PCR. Left vertical coordinate is RPKM of RNA-Seq; right vertical coordinate is relative expression level of qRT-PCR.
DEGs Involved in Cell Wall Reinforcement or Disassembly
Plant cell wall forms a dynamic physical barrier that protects cell from microbial infection. Prospective plant pathogens must overcome the physical barrier presented by cuticle and plant cell wall. The cuticle is the first cell wall layer encountered by a pathogen, and its biosynthesis involves a number of genes, including WSD (wax-ester synthase/diacylglycerol O-acyltransferase; Li et al., 2008), CYP77A6 (cytochrome P450; Yeats et al., 2010), CYP94A1 (fatty acid omega-hydroxylase; Pinot et al., 1999; Pinot and Beisson, 2011), and HTH (HOTHEAD, fatty acid omega-hydroxy dehydrogenase; Kurdyukov et al., 2006). Our results show that the homologs of these genes in apple, WSD (MDP0000253706), HTH (MDP0000210966), CYP77A6 (MDP0000874252), and CYP94A1 (MDP0000166404), were all down-regulated during the infection process (Figure 5A), which implies that the biosynthesis of cutin, suberine, and wax was impaired in infected leaves making it easier for AAAP to penetrate. We also speculate that the weakness of this physical barrier may be one of the reasons why “Starking Delicious” apples are so susceptible to AAAP.

Figure 5. Heatmaps of DEGs involved in cell wall reinforcement or disassembly. The was colored using Cluster 3.0 (i.e., red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right. (A) Genes related with cuticle. (B) Genes related to cell walls.
Fungal necrotrophs degrade the cuticle via the action of cutinases and lipases (Bellincampi et al., 2015). These cuticular breakdown products can be sensed by plants, initiating activation of a series of genes associated with cell wall reinforcement and modification (Schweizer et al., 1996). Calloses, produced by callose synthase (CALS), are thought to reinforce the cell wall at sites of fungal penetration in an effort to impede infections (Underwood, 2012), while PME (pectin methylesterase), XTH (xyloglucan endotransglycosylase/hydrolase) are both genes that encode enzymes involved in cell wall degradation or modification (Albert et al., 2004; Raiola et al., 2011). The results of this study demonstrate that CALS1 (MDP0000177875) is up-regulated while PME (MDP0000276688) and XTH (MDP0000233177) are down-regulated, suggesting that apples are able to sense the signals of pathogen infection and activated relevant defense mechanisms. Nevertheless, we also found that paralogous genes of PME (MDP0000276688) and XTH (MDP0000233177) were up-regulated in the early stages of infection (Figure 5B); in Arabidopsis, the AtPME3 acts as a susceptibility factor, required by necrotrophic pathogens for the initial colonization of host tissue (Raiola et al., 2011). We hypothesize that the up-regulation of PME in the early stages of infection might provide the conditions for the initial colonization of apple leaves by AAAP.
DEGs Involved in Phytohormone Signaling
Phytohormones, including salicylic acid (SA), ethylene (ET), brassinosteroids (BR), abscisic acid (ABA), and jasmonic acid (JA), are critical regulators of plant-pathogen interactions (De Vleesschauwer et al., 2013). In the experiment, most of the genes involved ET (e.g., ERF, EIN, and ETR) and SA signaling (e.g., NPR1 and TGA) were up-regulated when the “Starking Delicious” cultivar was challenged by AAAP, while differential expression of the NPR1 gene, the key regulator of SA-mediated transcriptional reprogramming and immunity (Pajerowska-Mukhtar et al., 2013), was up-regulated more than four-fold (Figure 6, Excel S3). Traditionally it has been thought that SA mediates defense signaling against biotrophic and hemibiotrophic pathogens, while JA and ET are associated with defense responses to necrotrophs (Kunkel and Brooks, 2002; Glazebrook, 2005). Our results suggest that SA might also play a role in local immunity against necrotrophic pathogens.

Figure 6. Heatmaps of DEGs involved in phytohormone signaling pathways, including JA, SA, ET, BR, and ABA signaling pathways. The was colored using Cluster 3.0 (red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right. Abbreviations for genes on the left of the heatmap are defined in the text.
It has also been shown that ABA is a crucial regulator of plant-microbe interactions (Asselbergh et al., 2008; Cao et al., 2011). It has been repeatedly shown to paralyze plant defenses by antagonizing the SA pathway in Arabidopsis, thus it predominantly behaves as a negative regulator of immunity (Koga et al., 2004; De Vleesschauwer et al., 2013; Xu et al., 2013). Indeed, in our experiments, the genes involved in ABA signal perception and transduction pathways, for example PYR/PYL (pyrabactin resistance1/PYR1-like), ABF (ABA binding factor) were all up-regulated (Figure 6, Excel S3). Finally, BRs comprise a unique class of growth-promoting steroid hormones that have also been shown to be key regulators of plant immunity (Wang, 2012). Intensive research has deciphered the complex positive and negative roles of BRs and their signaling in innate immunity, the genes encoding BR signaling cascades, including BAK1 (BRI-associated receptor kinase 1; Chinchilla et al., 2009; Choudhary et al., 2012), BRI1 (brassinosteroid insensitive1), BSK (brassinosteroid signaling kinases), and BZR (brassinazole-resistant transcription factor), were up-regulated as a result of “Starking Delicious” apple in response to AAAP infection (Figure 5, Excel S3).
Interestingly, in our experiments, most of the genes involved in ET and BR biosynthesis and signaling cascades were differentially expressed by 12 HPI, while those involved in JA, SA, and ABA biosynthesis and signaling were not differentially expressed until 18 HPI. These results therefore indicate that ET and BR signaling cascades were triggered earlier by the pathogen than the JA, SA, and ABA signaling cascades.
The involvement and characteristics of differential expression of multiple phytohormone signaling genes suggests that these signals are not just simple linear and isolated cascades, but that they “crosstalk” with each other. Thus, overlaps in defense and the differences in efficacy could also be triggered by divergent pathways at the different stages of AAAP infection.
Transcription Factors (TFs)
TFs are key regulatory proteins, essential for the regulation of gene expression. In plants, WRKY (Pandey and Somssich, 2009), Hsf (Pajerowska-Mukhtar et al., 2012), zinc finger protein (Maldonado-Bonilla et al., 2013), ZIP (Dezar et al., 2011), and NAC (Sun et al., 2013) are all important regulators of plant defense responses. The results of our experiment show that almost all WRKYs, ZIPs, Hsfs, and NACs, as well as most MYBs and a portion of the genes encoding for zinc finger proteins were significantly up-regulated. The expression of related TFs in apple induced by AAAP can be seen in Figure 7 and Excel S3.

Figure 7. Heatmaps of DEGs encoding transcriptional factors including WRKYs, HSFs, NACs, MYBs, zinc finger proteins, and homebox-leucine zipper proteins. The was colored using Cluster 3.0 (i.e., red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right.
There are more than 100 WRKY family-members within the apple genome (http://planttfdb.cbi.pku.edu.cn/family.php?sp=Mdo&fam=WRKY), 38 of which were induced by AAAP infection on the basis of our experiments. Results show that most were up regulated, with the exception of MDP0000811478 and MDP0000228304; indeed, some WRKY genes were significantly up-regulated throughout the whole experimental period, for example MDP0000154734, MDP0000792088, and MDP0000161881. In particular, MDP0000154734, which shows high similarity with AtWRKY75, were up-regulated almost 30-fold by 12 HPI, increasing to 180-fold by 72 HPI (Figure 7). Previously work has shown that in Arabidopsis, AtWRKY75 acts as a positive regulator of JA- or SA-mediated defense signaling responses to the necrotrophic pathogen Pectobacterium carotovorum ssp. carotovorum (Choi et al., 2014). These results also suggest that the gene homologous to AtWRKY75 in apple may also play an important role in responses to AAAP infection.
An increasing body of evidence has implicated NAC genes in the defense responses of plant to pathogen infections and environmental stimuli (Xia et al., 2010; Nuruzzaman et al., 2015). It is thought that NAC genes positively regulate plant defense responses by activating PR genes, inducing a hypersensitive response (HR) and cell death at the site of infection, or negatively regulate plant basal pathogen resistance by suppressing PR1 expression (Jensen et al., 2007; Kaneda et al., 2009; Seo et al., 2010; Kim et al., 2012). In our experiment, 15 NAC genes exhibited differential expression out of a total of 180 in the apple genome (Figure 7; Su et al., 2013). Results show that most of these were up-regulated, with the exception of MDP0000291272 and MDP0000250487, while in Arabidopsis over-expression of ATAF2, a NAC transcription factor gene, which has a high degree of similarity with MDP0000291272, resulted in the repression of a number of PR proteins and higher susceptibility to the soil-borne fungal pathogen fusarium oxysporum (Delessert et al., 2005). We therefore hypothesize that up-regulation of most NAC genes combined with down-regulation of gene MDP0000291272 in the early stage of infection may play a positive role in the defense of the “Starking Delicious” apple cultivar against AAAP.
The MYB gene family is extraordinarily large in apple (Cao et al., 2013). These genes are involved in a variety of functions, including the anthocyanin biosynthetic pathway (Espley et al., 2007), morphogenesis (Vimolmangkang et al., 2013), and abiotic stress responses (Wang et al., 2014). There have been relatively few reports dealing with the responses of MYBs in biotic stress, although it is known that the R2R3 MYB transcription factor in wheat mediates host resistance to the Bipolaris sorokiniana pathogen via regulation of SA-signaling pathways and defense-related genes (Zhang et al., 2012). A total of 12 apple MYB genes were either up- or down- regulated by AAAP infection in our experiment (Figure 7), which suggests that these genes might also play a regulatory role in responses of the “Starking Delicious” cultivar against AAAP attack. However, this hypothesis requires confirmation by additional research.
Heat shock transcription factors (Hsfs) are also thought to be involved in a range of pathological conditions, cellular responses to oxidative stress, as well as certain developmental and differentiation processes (Morimoto, 1998; Hahn et al., 2004). There are 25 members of the Hsf family in apple genome (Giorno et al., 2012), of which five were induced by AAAP infection and were up-regulated in our experiments (Figure 7). Three of these can be classified as class B-Hsf, and MDP0000527802 and MDP00006225590 in particular significantly increased in expression following AAAP infection. These two genes exhibit a high degree of similarity with AtHsfB2b and AtHsfB1, respectively, while the double knockout hsfb1 and hsfb2b genes in Arabidopsis significantly improved disease resistance following A. brassicicola infection via strong up-regulation of the basal mRNA-levels in the defensin genes Pdf1.2a/b (Kumar et al., 2009). We therefore speculate that the up-regulation of class B-Hsfs genes in apple is counterproductive to disease resistance.
In addition, results show that a number of homeobox-leucine zipper proteins and zinc finger proteins were also induced by AAAP infection (Figure 7). However, unlike the other TFs discussed above, most zinc finger proteins were down-regulated in our experiments.
Defense-Related Proteins
There are 17 families of PR proteins, normally expressed at nearly undetectable levels in healthy tissues, but which rapidly accumulate to significant amounts in response to biotic or abiotic stress (Van Loon and Van Strien, 1999; van Loon et al., 2006). The accumulation of PR proteins is usually associated with systemic acquired resistance to a wide range of pathogens (Ward et al., 1991; Durrant and Dong, 2004). Apple PR genes were induced in response to AAAP infection in our experiment (Figure 8A); the expression of PR-1, most PR-2s, PR thaumatin-like proteins (PR-5s), and defense-regulated genes were all associated with SA-regulated defense responses (Ward et al., 1991; Yang et al., 1997) and gradually increased subsequent to 18 HPI (Figure 8A). Results also show similar expression patterns in SA receptor genes, NPR1s and TGAs (Figure 6), while chitinases, JA-regulated defense genes, responded more quickly to AAAP infection than SA-regulated counterparts. Our results thus demonstrate that JA signal transduction is dominant at early infection stages, and that most other PR genes, including PR-9s (peroxidases), PR-10s (ribonucleases), PR-12s (defensins), and PR-14s (lipid-transfer proteins), were down-regulated subsequent to 18 HPI. Overall, PR genes, with the exception of chitinases, fail to respond to AAAP infection timely in the “Starking Delicious” apple cultivar. It was known that the timing of PR gene expression was a crucial determinant of pathogenesis: Response at an early stage of infection was favorable to plant prevention of violation by the pathogen (Kaur et al., 2011). Down-regulation and lagged expression of most PR genes might imply that the “Starking Delicious” apple cultivar is sensitive to AAAP infection.
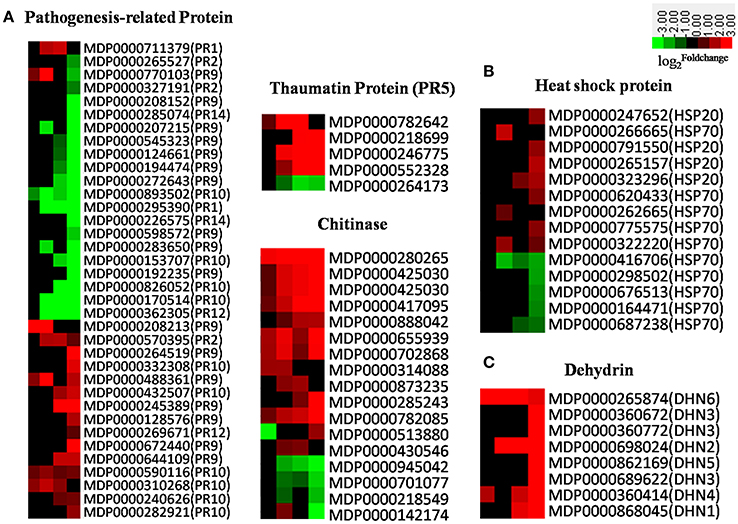
Figure 8. (A) Heatmaps of DEGs encoding pathogenesis-related (PR) proteins; (B) Heatmap of DEGs encoding heat shock proteins; (C) Heatmap of DEGs encoding dehydrin. The was colored using Cluster 3.0 (i.e., red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right.
Heat shock proteins (HSPs) are a subset of molecular chaperones, best known because they are rapidly induced in large numbers by stress (Neumann et al., 1994; Wang et al., 2004; Scarpeci et al., 2008). These proteins act as molecular chaperones to stabilize, reduce misfolding, and facilitate the refolding of proteins denatured as a result of stresses. Our results show that HSPs in the “Starking Delicious” cultivar exhibited a range of responses to AAAP. HSP20s, for example, did not respond at early stages of infection but slightly increased at later stages. In contrast, many of HSP70 transcripts were down-regulated (Figure 8B), a result confirmed by a proteomics study of apple-AAAP interactions that showed that these proteins accumulate in resistance as opposed to susceptible leaves (Zhang et al., 2015). Additional research has shown that HSP70s are induced by both P. expansum and P. digitatum pathogens in “Ultima Gala” apple cultivar (Spadoni et al., 2015), thus expression of these proteins is an important fact of apple disease-resistance. Down-regulation of HSP70s in the apple leaves suggests that the defense signal transduction pathway mediated by HSP70s may be compromised in the “Starking Delicious” cultivar, leading to AAAP susceptibility.
Apple dehydrins (DHNs) are another family of proteins that protect cells from damage caused by a variety of abiotic stresses, including drought, salt, and low temperature (Liang et al., 2012). A number of studies have confirmed that DHNs are also involved in biotic stress; for example, over-expression of wheat DHN5 in Arabidopsis induced the up-regulation of PR genes and led to increased resistance to fungal infections caused by Botrytis cinerea and Alternaria solani (Brini et al., 2011; Drira et al., 2016). Other research has shown that the grapevine DHN1 protein was up-regulated in Vitis yeshanensis in response to Eryiphe necator infection (Yang et al., 2012), while in our experiments, DHN genes in apple, including DHN1-6, were up-regulated following AAAP infection, with DHN4 (MDP0000360414) and DHN6 (MDP0000265874) responding particularly rapidly (Figure 8C). These results suggest that DHNs may play a protective role when plants respond to biotic stresses.
DEGs Involved in Photosynthesis, Oxidation-Reduction, Amino Acid, and Secondary Metabolism
Our results confirm that almost all DEGs involved in photosynthesis were down-regulated as a result of pathogen infection (Figure S2). This indicates that the rate of photosynthesis gradually declined as disease progressed. Hundreds of transcripts associated with amino acid metabolism were differentially expressed in apple leaves attacked by AAAP, with some very significantly activated in later infection stages on the basis of KEGG pathway enrichment analysis (Table 1). Our results also demonstrated that a decrease in photosynthesis can be attributed to plants adjusting their metabolism to produce defense-related compounds (Walters and Boyle, 2005). Results also demonstrate that the genes encoding glutathione-S-transferases (GSTs), superoxide dismutases (SODs), catalases (CATs), L-ascorbate peroxidases (APXs), and monodehydroascorbate reductases (MDHARs), which all involved in the regulation of redox homeostasis, were all down-regulated as the result of AAAP infection (Figure 9).

Figure 9. Heatmaps of DEGs involved oxidation-reduction. The was colored using Cluster 3.0 (i.e., red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right.
Secondary metabolites function in biocidal defense and as regulatory signals in innate immunity (Dixon, 2001; Grayer and Kokubun, 2001). The differential expression of many genes encoding key enzymes in the lignin, phenylpropanoid, flavonoids, benzoxazinoids, cyanoanimo acid, alkaloid, and terpene biosynthetic pathways were detected in our experiment (Figure S3), of which β-glucosidases (BGLs) play an important defensive role by releasing glucosyl-blocking groups from metabolic intermediates and allowing for the modification of various phytoanticipins and phytotoxins (Morant et al., 2008; Cairns and Esen, 2010). The expression level of BGLs were drastically up- and down-regulated in our experiments (Figure 10A), and the presence of differentially-expressed isozyme genes suggested that these proteins have a range of different functions in apple in response to biotic stresses. In addition, oxylipins act as protective compounds and are potential signaling molecules (Blée, 2002), and lipoxygenases (LOXs) are key enzymes mediating their synthesis. The results of this study show that the majority of 9-LOXs were up-regulated in AAAP-infected leaves, while no difference in expression level was seen in the majority of 13-LOXs up to 72 HPI and the expression levels of 13-LOXs were then subsequently down-regulated by 72 HPI (Figure 10B). A body of research has demonstrated that the 9-LOX pathway is essential to plant defense against microbial pathogens (Rancé et al., 1998), and that 13-LOX-derived oxylipin biosynthesis is associated with HR induction in Arabidopsis during race-specific resistance to the Pseudomonas syringae pathogen (Veronico et al., 2006). Moreover, the first step of JA biosynthesis is catalyzed by a member of the 13-LOX family (Bannenberg et al., 2009). Therefore, we speculate that down-regulated expressions of these genes may impair the defense response of apple leaves.

Figure 10. Heatmaps of differentially expressed (A) BGLs and (B) LOXs. The was colored using Cluster 3.0 (i.e., red for up-regulated, green for down-regulated), each horizontal row represents a DEG with its gene ID, and the vertical columns represent 12, 18, 36, and 72 HPI from left to right.
Conclusions
In conclusion, transcriptomic analysis allowed us to detect a total of 9080 DEGs in the “Starking Delicious” apple cultivar in response to AAAP infection. GO and pathway enrichment analysis indicated that these DEGs were predominately involved in both biological processes and metabolic pathways. Almost all genes associated with photosynthesis and oxidation-reduction were down-regulated, and a large number of defense-related genes and transcription factors were activated as a result of the infection process. Transcriptomic data suggested that cell wall defensive vulnerability and the down-regulation of most PRs and HSP70s genes in the “Starking Delicious” cultivar following infection may present possible reasons for susceptibility to AAAP.
Author Contributions
SW was the recipient of funds; SW, LZ, and HX conceived the experiment. SL and BC prepared the plant materials and collected samples. LZ, WN, and BC undertook experiments and data analysis. SW, LZ, and HX prepared the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Science Foundation of China (31171935) and the Fundamental Research Funds for the Central Universities (KYZ201310). We are also grateful to Prof. Guo Yunzhong in Northwest A&F University, China for providing AAAP (Alternaria alternata apple pathotype) stain.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00022/full#supplementary-material
References
Abe, K., Iwanami, H., Kotoda, N., Moriya, S., and Takahashi, S. (2010). Evaluation of apple genotypes and Malus species for resistance to Alternaria blotch caused by Alternaria alternata apple pathotype using detached-leaf method. Plant Breed. 129, 208–218. doi: 10.1111/j.1439-0523.2009.01672.x
Albert, M., Werner, M., Proksch, P., Fry, S. C., and Kaldenhoff, R. (2004). The cell wall-modifying xyloglucan endotransglycosylase/hydrolase LeXTH1 is expressed during the defence reaction of tomato against the plant parasite Cuscuta reflexa. Plant Biol. 6, 402–407. doi: 10.1055/s-2004-817959
Asselbergh, B., De Vleesschauwer, D., and Hofte, M. (2008). Global switches and fine-tuning-ABA modulates plant pathogen defense. Mol. Plant Microbe Interact. 21, 709–719. doi: 10.1094/MPMI-21-6-0709
Audic, S., and Claverie, J. M. (1997). The significance of digital gene expression profiles. Genome Res. 7, 986–995.
Bannenberg, G., Mart Inez, M., Hamberg, M., and Castresana, C. (2009). Diversity of the enzymatic activity in the lipoxygenase gene family of Arabidopsis thaliana. Lipids 44, 85–95. doi: 10.1007/s11745-008-3245-7
Bellincampi, D., Cervone, F., and Lionetti, V. (2015). Plant cell wall dynamics and wall-related susceptibility in plant-pathogen interactions. Front. Plant Sci. 5:228. doi: 10.3389/fpls.2014.00228
Blée, E. (2002). Impact of phyto-oxylipins in plant defense. Trends Plant Sci. 7, 315–322. doi: 10.1016/S1360-1385(02)02290-2
Brini, F., Yamamoto, A., Jlaiel, L., Takeda, S., Hobo, T., Dinh, H. Q., et al. (2011). Pleiotropic effects of the wheat dehydrin DHN-5 on stress responses in Arabidopsis. Plant Cell Physiol. 52, 676–688. doi: 10.1093/pcp/pcr030
Cairns, J. R. K., and Esen, A. (2010). β-Glucosidases. Cell. Mol. Life Sci. 67, 3389–3405. doi: 10.1007/s00018-010-0399-2
Cao, F. Y., Yoshioka, K., and Desveaux, D. (2011). The roles of ABA in plant-pathogen interactions. J. Plant Res. 124, 489–499. doi: 10.1007/s10265-011-0409-y
Cao, Z. H., Zhang, S. Z., Wang, R. K., Zhang, R. F., and Hao, Y. J. (2013). Genome wide analysis of the apple MYB transcription factor family allows the identification of MdoMYB121 gene confering abiotic stress tolerance in plants. PLoS ONE 8:e69955. doi: 10.1371/journal.pone.0069955
Chang, S., Puryear, J., and Cairney, J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 11, 113–116. doi: 10.1007/BF02670468
Chinchilla, D., Shan, L., He, P., de Vries, S., and Kemmerling, B. (2009). One for all: the receptor-associated kinase BAK1. Trends Plant Sci. 14, 535–541. doi: 10.1016/j.tplants.2009.08.002
Choi, C., Park, Y. H., Kwon, S. I., Yun, C., Ahn, I., Park, S. R., et al. (2014). Identification of AtWRKY75 as a transcriptional regulator in the defense response to Pcc through the screening of Arabidopsis activation-tagged lines. Plant Biotechnol. Rep. 8, 183–192. doi: 10.1007/s11816-013-0308-x
Choudhary, S. P., Yu, J. Q., Yamaguchi-Shinozaki, K., Shinozaki, K., and Tran, L. S. (2012). Benefits of brassinosteroid crosstalk. Trends Plant Sci. 17, 594–605. doi: 10.1016/j.tplants.2012.05.012
Cohn, J., Sessa, G., and Martin, G. B. (2001). Innate immunity in plants. Curr. Opin. Immunol. 13, 55–62. doi: 10.1016/S0952-7915(00)00182-5
Delessert, C., Kazan, K., Wilson, I. W., Van Der Straeten, D., Manners, J., and Dennis, E. S. (2005). The transcription factor ATAF2 represses the expression of pathogenesis-related genes in Arabidopsis. Plant J. 43, 745–757. doi: 10.1111/j.1365-313X.2005.02488.x
De Vleesschauwer, D., Gheysen, G., and Hofte, M. (2013). Hormone defense networking in rice: tales from a different world. Trends Plant Sci. 18, 555–565. doi: 10.1016/j.tplants.2013.07.002
Dezar, C. A., Giacomelli, J. I., Manavella, P. A., Re, D. A., Alves-Ferreira, M., Baldwin, I. T., et al. (2011). HAHB10, a sunflower HD-Zip II transcription factor, participates in the induction of flowering and in the control of phytohormone-mediated responses to biotic stress. J. Exp. Bot. 62, 1061–1076. doi: 10.1093/jxb/erq339
Dixon, R. A. (2001). Natural products and plant disease resistance. Nature 411, 843–847. doi: 10.1038/35081178
Drira, M., Hanin, M., Masmoudi, K., and Brini, F. (2016). Comparision of full-length and conserved segments of wheat dehydrin DHN-5 overexpressing in Arabidopsis thaliana showed different responses to abiotic and biotic stress. Funct. Plant Biol. 43, 1048–1060. doi: 10.1071/FP16134
Durrant, W. E., and Dong, X. (2004). Systemic aquired resistance. Annu. Rev. Phytopathol. 42, 185–209. doi: 10.1146/annurev.phyto.42.040803.140421
Espley, R. V., Hellens, R. P., Putterill, J., Stevenson, D. E., Kutty Amma, S., and Allan, A. C. (2007). Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 49, 414–427. doi: 10.1111/j.1365-313X.2006.02964.x
Filajdic, N., and Sutton, T. B. (1991). Identification and distribution of Alternaria mali on apples in north carolina and susceptibility of different varieties of apples to Alternaria blotch. Plant Dis. 75, 1045–1048. doi: 10.1094/PD-75-1045
Giorno, F., Guerriero, G., Baric, S., and Mariani, C. (2012). Heat shock transcriptional factors in Malus domestica: identification, classification and expression analysis. BMC Genomics 13:1. doi: 10.1186/1471-2164-13-639
Glazebrook, J. (2005). Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 43, 205–227. doi: 10.1146/annurev.phyto.43.040204.135923
Grayer, R. E., and Kokubun, T. (2001). Plant-fungal interactions: the search for phytoalexins and other antifungal compounds from higher plants. Phytochemistry 56, 253–263. doi: 10.1016/S0031-9422(00)00450-7
Hahn, J. S., Hu, Z., Thiele, D. J., and Iyer, V. R. (2004). Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 24, 5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004
Jensen, M. K., Rung, J. H., Gregersen, P. L., Gjetting, T., Fuglsang, A. T., Hansen, M., et al. (2007). The HvNAC6 transcription factor: a positive regulator of penetration resistance in barley and Arabidopsis. Plant Mol. Biol. 65, 137–150. doi: 10.1007/s11103-007-9204-5
Kaneda, T., Taga, Y., Takai, R., Iwano, M., Matsui, H., Takayama, S., et al. (2009). The transcription factor OsNAC4 is a key positive regulator of plant hypersensitive cell death. EMBO J. 28, 926–936. doi: 10.1038/emboj.2009.39
Kaur, P., Jost, R., Sivasithamparam, K., and Barbetti, M. J. (2011). Proteome analysis of the Albugo candida-Brassica juncea pathosystem reveals that the timing of the expression of defence-related genes is a crucial determinant of pathogenesis. J. Exp. Bot. 62, 1285–1298. doi: 10.1093/jxb/erq365
Kim, H. S., Park, H. C., Kim, K. E., Jung, M. S., Han, H. J., Kim, S. H., et al. (2012). A NAC transcription factor and SNI1 cooperatively suppress basal pathogen resistance in Arabidopsis thaliana. Nucleic Acids Res. 40, 9182–9192. doi: 10.1093/nar/gks683
Koch, E., and Slusarenko, A. (1990). Arabidopsis is susceptible to infection by a downy mildew fungus. Plant Cell 2, 437–445. doi: 10.1105/tpc.2.5.437
Koga, H., Dohi, K., and Mori, M. (2004). Abscisic acid and low temperatures suppress the whole plant-specific resistance reaction of rice plants to the infection of Magnaporthe grisea. Physiol. Mol. Plant Pathol. 65, 3–9. doi: 10.1016/j.pmpp.2004.11.002
Kumar, M., Busch, W., Birke, H., Kemmerling, B., Nurnberger, T., and Schoffl, F. (2009). Heat shock factors HsfB1 and HsfB2b are involved in the regulation of Pdf1.2 expression and pathogen resistance in Arabidopsis. Mol. Plant 2, 152–165. doi: 10.1093/mp/ssn095
Kunkel, B. N., and Brooks, D. M. (2002). Cross talk between signaling pathways in pathogen defense. Curr. Opin. Plant Biol. 5, 325–331. doi: 10.1016/S1369-5266(02)00275-3
Kurdyukov, S., Faust, A., Trenkamp, S., Bär, S., Franke, R., Efremova, N., et al. (2006). Genetic and biochemical evidence for involvement of HOTHEAD in the biosynthesis of long-chain alpha-, omega-dicarboxylic fatty acids and formation of extracellular matrix. Planta 224, 315–329. doi: 10.1007/s00425-005-0215-7
Li, F., Wu, X., Lam, P., Bird, D., Zheng, H., Samuels, L., et al. (2008). Identification of the wax ester synthase/acyl-coenzyme A: diacylglycerol acyltransferase WSD1 required for stem wax ester biosynthesis in Arabidopsis. Plant Physiol. 148, 97–107. doi: 10.1104/pp.108.123471
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Li, Y., Aldwinckle, H. S., Sutton, T., Tsuge, T., Kang, G., Cong, P., et al. (2013). Interactions of apple and the Alternaria alternata apple pathotype. Crit. Rev. Plant Sci. 32, 141–150. doi: 10.1080/07352689.2012.722026
Liang, D., Xia, H., Wu, S., and Ma, F. (2012). Genome-wide identification and expression profiling of dehydrin gene family in Malus domestica. Mol. Biol. Rep. 39, 10759–10768. doi: 10.1007/s11033-012-1968-2
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Maldonado-Bonilla, L. D., Eschen-Lippold, L., Gago-Zachert, S., Tabassum, N., Bauer, N., Scheel, D., et al. (2013). The Arabidopsis tandem zinc finger 9 protein binds RNA and mediates pathogen-associated molecular pattern-triggered immune responses. Plant Cell Physiol. 55, 412–425. doi: 10.1093/pcp/pct175
Mathioni, S. M., Belo, A., Rizzo, C. J., Dean, R. A., and Donofrio, N. M. (2011). Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genomics 12:49. doi: 10.1186/1471-2164-12-49
Morant, A. V., Jorgensen, K., Jorgensen, C., Paquette, S. M., Sánchez-Pérez, R., Moller, B. L., et al. (2008). beta-Glucosidases as detonators of plant chemical defense. Phytochemistry 69, 1795–1813. doi: 10.1016/j.phytochem.2008.03.006
Morimoto, R. I. (1998). Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12, 3788–3796. doi: 10.1101/gad.12.24.3788
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mukherjee, A. K., Carp, M. J., Zuchman, R., Ziv, T., Horwitz, B. A., and Gepstein, S. (2010). Proteomics of the response of Arabidopsis thaliana to infection with Alternaria brassicicola. J. Proteomics 73, 709–720. doi: 10.1016/j.jprot.2009.10.005
Neumann, D., Lichtenberger, O., Günther, D., Tschiersch, K., and Nover, L. (1994). Heat-shock proteins induce heavy-metal tolerance in higher plants. Planta 194, 360–367. doi: 10.1007/BF00197536
Nuruzzaman, M., Sharoni, A. M., Satoh, K., Karim, M. R., Harikrishna, J. A., Shimizu, T., et al. (2015). NAC transcription factor family genes are differentially expressed in rice during infections with Rice dwarf virus, Rice black-streaked dwarf virus, Rice grassy stunt virus, Rice ragged stunt virus, and Rice transitory yellowing virus. Front Plant Sci. 6:676. doi: 10.3389/fpls.2015.00676
Pajerowska-Mukhtar, K. M., Emerine, D. K., and Mukhtar, M. S. (2013). Tell me more: roles of NPRs in plant immunity. Trends Plant Sci. 18, 402–411. doi: 10.1016/j.tplants.2013.04.004
Pajerowska-Mukhtar, K. M., Wang, W., Tada, Y., Oka, N., Tucker, C. L., Fonseca, J. P., et al. (2012). The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Current Biol. 22, 103–112. doi: 10.1016/j.cub.2011.12.015
Pandey, S. P., and Somssich, I. E. (2009). The role of WRKY transcription factors in plant immunity. Plant Physiol. 150, 1648–1655. doi: 10.1104/pp.109.138990
Pavy, N., Boyle, B., Nelson, C., Paule, C., Giguere, I., Caron, S., et al. (2008). Identification of conserved core xylem gene sets: conifer cDNA microarray development, transcript profiling and computational analyses. New Phytol. 180, 766–786. doi: 10.1111/j.1469-8137.2008.02615.x
Pinot, F., and Beisson, F. (2011). Cytochrome P450 metabolizing fatty acids in plants: characterization and physiological roles. FEBS J. 278, 195–205. doi: 10.1111/j.1742-4658.2010.07948.x
Pinot, F., Benveniste, I., Salaun, J. P., Loreau, O., Noel, J. P., Schreiber, L., et al. (1999). Production in vitro by the cytochrome P450 CYP94A1 of major C18 cutin monomers and potential messengers in plant-pathogen interactions: enantioselectivity studies. Biochem. J. 342, 27–32. doi: 10.1042/0264-6021:3420027
Proietti, S., Bertini, L., Timperio, A. M., Zolla, L., Caporale, C., and Caruso, C. (2013). Crosstalk between salicylic acid and jasmonate in Arabidopsis investigated by an integrated proteomic and transcriptomic approach. Mol. Biosyst. 9, 1169–1187. doi: 10.1039/c3mb25569g
Raiola, A., Lionetti, V., and Elmaghraby, I. (2011). Pectin methylesterase is induced in Arabidopsis upon infection and is necessary for a successful colonization by necrotrophic pathogens. Mol. Plant Microbe Interact. 24, 432–440. doi: 10.1094/MPMI-07-10-0157
Rancé, I., Fournier, J., and Esquerré-Tugayé, M. T. (1998). The incompatible interaction between Phytophthora parasitica var. nicotianae race 0 and tobacco is suppressed in transgenic plants expressing antisense lipoxygenase sequences. Proc. Natl. Acad. Sci. U.S.A. 95, 6554–6559. doi: 10.1073/pnas.95.11.6554
Saito, A., Nakazawa, N., and Suzuki, M. (2001). Selection of mutants resistant to Alternaria blotch from in vitro-cultured apple shoots irradiated with X- and γ-rays. J. Plant Physiol. 158, 391–400. doi: 10.1078/0176-1617-00235
Sarowar, S., Zhao, Y., Soria-Guerra, R. E., Ali, S., Zheng, D., Wang, D., et al. (2011). Expression profiles of differentially regulated genes during the early stages of apple flower infection with Erwinia amylovora. J. Exp. Bot. 62, 4851–4861. doi: 10.1093/jxb/err147
Scarpeci, T. E., Zanor, M. I., and Valle, E. M. (2008). Investigating the role of plant heat shock proteins during oxidative stress. Plant Signal. Behav. 3, 856–857. doi: 10.4161/psb.3.10.6021
Schweizer, P., Felix, G., Buchala, A., Müller, C., and Métraux, J. P. (1996). Perception of free cutin monomers by plant cells. Plant J. 10, 331–341. doi: 10.1046/j.1365-313X.1996.10020331.x
Seo, P. J., Kim, M. J., Park, J. Y., Kim, S. Y., Jeon, J., Lee, Y. H., et al. (2010). Cold activation of a plasma membrane-tethered NAC transcription factor induces a pathogen resistance response in Arabidopsis. Plant J. 61, 661–671. doi: 10.1111/j.1365-313X.2009.04091.x
Shulaev, V., Korban, S. S., Sosinski, B., Abbott, A. G., Aldwinckle, H. S., Folta, K. M., et al. (2008). Multiple models for Rosaceae genomics. Plant Physiol. 147, 985–1003. doi: 10.1104/pp.107.115618
Spadoni, A., Guidarelli, M., Philips, J., Mari, M., and Wisniewshi, M. (2015). Transcritional profiling of apple fruit in response to heat treatment: involvement of a defense response during Penicillium expansum infection. Postharvest Biol. Technol. 101, 37–48. doi: 10.1016/j.postharvbio.2014.10.009
Su, H., Zhang, S., Yuan, X., Chen, C., Wang, X. F., and Hao, Y. J. (2013). Genome-wide analysis and identification of stress-responsive genes of the NAM-ATAF1, 2-CUC2 transcription factor family in apple. Plant Physiol. Biochem. 71, 11–21. doi: 10.1016/j.plaphy.2013.06.022
Sun, L., Zhang, H., Li, D., Huang, L., Hong, Y., Ding, X. S., et al. (2013). Functions of rice NAC transcriptional factors, ONAC122 and ONAC131, in defense responses against Magnaporthe grisea. Plant Mol. Biol. 81, 41–56. doi: 10.1007/s11103-012-9981-3
Thilmony, R., Underwood, W., and He, S. Y. (2006). Genome-wide transcriptional analysis of the Arabidopsis thaliana interaction with the plant pathogen Pseudomonas syringae pv.tomato DC3000 and the human pathogen Escherichia coli O157:H7. Plant J. 46, 34–53. doi: 10.1111/j.1365-313X.2006.02725.x
Underwood, W. (2012). The plant cell wall: a dynamic barrier against pathogen invasion. Front. Plant Sci. 3:85. doi: 10.3389/fpls.2012.00085
Van Loon, L. C., and Van Strien, E. A. (1999). The families of pathogenesis-related proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol. Mol. Plant Pathol. 55, 85–97. doi: 10.1006/pmpp.1999.0213
van Loon, L. C., Rep, M., and Pieterse, C. M. (2006). Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 44, 7.1–7.28. doi: 10.1146/annurev.phyto.44.070505.143425
Veronico, P., Giannino, D., Melillo, M. T., Leone, A., Reyes, A., Kennedy, M. W., et al. (2006). A novel lipoxygenase in pea roots. Its function in wounding and biotic stress. Plant Physiol. 141, 1045–1055. doi: 10.1104/pp.106.081679
Vilanova, L., Wisniewski, M., Norelli, J., Vinas, I., Torres, R., Usall, J., et al. (2014). Transcriptomic profiling of apple in response to inoculation with a pathogen (Penicillium expansum) and a non-pathogen (Penicillium digitatum). Plant Mol. Biol. Rep. 32, 566–583. doi: 10.1007/s11105-013-0676-y
Vimolmangkang, S., Han, Y., Wei, G., and Korban, S. S. (2013). An apple MYB transcription factor, MdMYB3, is involved in regulation of anthocyanin biosynthesis and flower development. BMC Plant Biol. 13:176. doi: 10.1186/1471-2229-13-176
Walters, D. R., and Boyle, C. (2005). Induced resistance and allocation costs: what is the impact of pathogen challenge? Physiol. Mol. Plant Pathol. 66, 40–44. doi: 10.1016/j.pmpp.2005.04.002
Wang, R. K., Cao, Z. H., and Hao, Y. J. (2014). Overexpression of a R2R3 MYB gene MdSIMYB1 increases tolerance to multiple stresses in transgenic tobacco and apples. Physiol. Plant. 150, 76–87. doi: 10.1111/ppl.12069
Wang, W. X., Vinocur, B., Shoseyov, O., and Altman, A. (2004). Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9, 244–252. doi: 10.1016/j.tplants.2004.03.006
Wang, Z. (2012). Brassinosteroids modulate plant immunity at multiple levels. Proc. Natl. Acad. Sci. U.S.A. 109, 7–8. doi: 10.1073/pnas.1118600109
Ward, E. R., Uknes, S. J., Williams, S. C., Dincher, S. S., Wiederhold, D. L., Alexander, D. C., et al. (1991). Coordinate gene activity in response to agents that induce systemic acquired resistance. Plant Cell 3, 1085–1094. doi: 10.1105/tpc.3.10.1085
Xia, N., Zhang, G., Liu, X. Y., Deng, L., Cai, G. L., Zhang, Y., et al. (2010). Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Mol. Biol. Rep. 37, 3703–3712. doi: 10.1007/s11033-010-0023-4
Xu, J., Audenaert, K., Hofte, M., and De Vleesschauwer, D. (2013). Abscisic acid promotes susceptibility to the rice leaf blight pathogen Xanthomonas oryzae pv oryzae by suppressing salicylic acid-mediated defenses. PLoS ONE 8:e67413. doi: 10.1371/journal.pone.0067413
Yang, Y., He, M., Zhu, Z., Li, S., Xu, Y., Zhang, C., et al. (2012). Identification of the dehydrin gene family from grapevine species and analysis of their responsiveness to various forms of abiotic and biotic stress. BMC Plant Biol. 12:1. doi: 10.1186/1471-2229-12-140
Yang, Y., Shah, J., and Klessig, D. F. (1997). Signal perception and transduction in plant defense responses. Genes Dev. 11, 1621–1639. doi: 10.1101/gad.11.13.1621
Yeats, T. H., Howe, K. J., Matas, A. J., Buda, G. J., Thannhauser, T. W., and Rose, J. K. (2010). Mining the surface proteome of tomato (Solanum lycopersicum) fruit for proteins associated with cuticle biogenesis. J. Exp. Bot. 61, 3759–3771. doi: 10.1093/jxb/erq194
Zhang, C. X., Tian, Y., and Cong, P. H. (2015). Proteome analysis of pathogen-responsive proteins from apple leaves induced by Alternaria Blotch Alternaria alternate. PLoS ONE 10:e0122233. doi: 10.1371/journal.pone.0122233
Zhang, Z., Liu, X., Wang, X., Zhou, M., Zhou, X., Ye, X., et al. (2012). An R2R3 MYB transcription factor in wheat, TaPIMP1, mediates host resistance to Bipolaris sorokiniana and drought though regulation of defense- and stress-related genes. New Phytol. 196, 1155–1170. doi: 10.1111/j.1469-8137.2012.04353.x
Keywords: transcriptomic, apple (Malus × domestica Borkh.cv. “Starking Delicious”), Alternaria alternata apple pathotype, infection, defense response
Citation: Zhu L, Ni W, Liu S, Cai B, Xing H and Wang S (2017) Transcriptomics Analysis of Apple Leaves in Response to Alternaria alternata Apple Pathotype Infection. Front. Plant Sci. 8:22. doi: 10.3389/fpls.2017.00022
Received: 12 April 2016; Accepted: 04 January 2017;
Published: 20 January 2017.
Edited by:
Riccardo Velasco, Fondazione Edmund Mach, ItalyReviewed by:
Vincenzo Lionetti, Sapienza University of Rome, ItalyShu Yuan, Sichuan Agricultural University, China
Copyright © 2017 Zhu, Ni, Liu, Cai, Xing and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanhong Wang, d3NoM3hnQGhvdG1haWwuY29t