 Pablo Peláez
Pablo Peláez Alejandrina Hernández-López
Alejandrina Hernández-López Georgina Estrada-Navarrete
Georgina Estrada-Navarrete Federico Sanchez1†
Federico Sanchez1†- 1Departamento de Biología Molecular de Plantas, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 2Laboratorio Nacional de Genómica para la Biodiversidad, Unidad de Genómica Avanzada del Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Irapuato, Mexico
Agrobacterium rhizogenes is a pathogenic bacteria that causes hairy root disease by transferring bacterial DNA into the plant genome. It is an essential tool for industry and research due to its capacity to produce genetically modified roots and whole organisms. Here, we identified and characterized small RNAs generated from the transfer DNA (T-DNA) of A. rhizogenes in hairy roots of common bean (Phaseolus vulgaris). Distinct abundant A. rhizogenes T-DNA-derived small RNAs (ArT-sRNAs) belonging to several oncogenes were detected in hairy roots using high-throughput sequencing. The most abundant and diverse species of ArT-sRNAs were those of 21- and 22-nucleotides in length. Many T-DNA encoded genes constituted phasiRNA producing loci (PHAS loci). Interestingly, degradome analysis revealed that ArT-sRNAs potentially target genes of P. vulgaris. In addition, we detected low levels of ArT-sRNAs in the A. rhizogenes-induced calli generated at the wound site before hairy root emergence. These results suggest that RNA silencing targets several genes from T-DNA of A. rhizogenes in hairy roots of common bean. Therefore, the role of RNA silencing observed in this study has implications in our understanding and usage of this unique plant-bacteria interaction.
Introduction
Agrobacterium rhizogenes and Agrobacterium tumefaciens are plant pathogenic bacteria capable of inducing uncontrolled cell proliferation and development of hairy roots and crown gall tumors, respectively. During the infection process, a transferred DNA from the bacteria root inducing (Ri) or tumor-inducing (Ti) plasmids is integrated into the plant cells genome (Tzfira and Citovsky, 2006). The genes encoded in the T-DNA (oncogenes) lead to abnormal proliferation of cells through changes in signal transduction pathways and transcription factors that affect the production of plant hormones (Britton et al., 2008). Transformed cells produce amino acid and sugar derivatives (opines) that are used by bacteria as nutrients and trigger conjugal transfer of T-DNA-carrying plasmids (Subramoni et al., 2014). The unique properties of these bacteria to perform cross-kingdom DNA transfer have been widely exploited to transform plants. A. tumefaciens strains with Ti plasmids lacking T-DNA oncogenes do not induce tumor growth and have been used as efficient delivery systems for plant transformation (Krenek et al., 2015). Hairy roots harboring the A. rhizogenes Ri plasmid T-DNA have also become an important tool for basic research and biotechnology (Georgiev et al., 2012). Transgenic roots emerge from totipotent calli cells that, subsequently, differentiate into highly branched ageotropic roots. Whole plants regenerated from hairy roots exhibit a particular phenotype (called hairy root phenotype) that comprises loss of apical dominance, wrinkled leaves, reduced fertility, shortened internodes, stunted growth, and abnormal organs (Tepfer, 1984). Several studies have attempted to determine the precise function of the T-DNA genes from A. rhizogenes strains in the infection process and in the hairy root phenotype (Britton et al., 2008).
RNA silencing constitutes a defense mechanism against foreign genetic material, such as transgenes or viral nucleic acids (Hamilton and Baulcombe, 1999). Small RNAs of 21–24 nucleotides (nt) in length trigger post-transcriptional gene silencing (PTGS) or transcriptional gene silencing (TGS) of endogenous or foreign genetic elements (Baulcombe, 2004). RNA silencing is initially triggered by double-stranded RNA (dsRNA), which is cleaved by Dicer-like proteins (DCLs) to form small RNAs (sRNAs). These sRNAs are then loaded into Argonaute proteins (AGOs) to direct RNA or DNA silencing via base pairing complementarity (Chapman and Carrington, 2007). Furthermore, RNA-dependent RNA polymerases (RDRs) produce secondary sRNAs that supports systemic spread of RNA silencing through long distances within plants (Willmann et al., 2011).
Transgenes incorporated by Agrobacterium-mediated transformation are frequently subject to RNA silencing. Dunoyer et al. (2006) proposed that transgene silencing could be a defense response typically conducted against T-DNA oncogenes of virulent bacteria similar to that occurring during viral infections. Analyzing the defensive role of RNA silencing in virulent A. tumefaciens infections, they found that mutants of dcl1 and plants deficient in miRNA functions became immune to A. tumefaciens. Accumulation and activity of microRNAs (miRNAs) in tumors were only moderately altered, highlighting the contribution of miRNA-regulated processes to tumor development. Besides, Arabidopsis thaliana rdr6 mutants and viral suppressor expressing plants were more susceptible to the infection. Surprisingly, RNA silencing against T-DNA genes happens at the early stages of the infection but is inhibited in tumors. Small RNAs of 21-nt in length from the tryptophan 2-monooxygenase (iaaM) and the agropine synthase (ags) T-DNA genes were detected at 3 days post-infiltration of virulent A. tumefaciens in leaves of Nicotiana benthamiana; however, no sRNAs were detected in tumors. The authors proposed that suppression of RNA silencing pathways in A. tumefaciens-induced tumors could be an event intrinsic to dedifferentiation and/or proliferation probably caused by phytohormones (Dunoyer et al., 2006). In this regard, the 6b protein from the T-DNA region of the Ti plasmid was reported to interact with AGO1 and the SERRATE (SE) protein involved in the biogenesis of miRNAs (Wang et al., 2011). The reduced accumulation of miRNAs observed in plants overexpressing the 6b protein suggests that it could act as a suppressor of RNA silencing and may contribute to the RNA silencing suppression state observed in tumors (Wang et al., 2011). Interestingly, the 6b protein is not present on the T-DNA of the Ri plasmid of A. rhizogenes (Britton et al., 2008). Recently, small RNAs from the T-DNA of A. rhizogenes were detected in Nicotiana tabacum plants that acquired the T-DNA naturally through horizontal gene transfer (Kovacova et al., 2014).
Hairy roots are differentiated roots that have been proven to be a powerful tool for loss-of-function analyses of genes in many species via RNA silencing. They have been generally employed in plants recalcitrant to A. tumefaciens-mediated transformation, such as common bean. Nevertheless, A. rhizogenes-induced hairy roots should not have the RNA silencing pathways suppressed like A. tumefaciens-induced tumors. Genes from the T-DNA of A. rhizogenes could be regulated by the RNA silencing mechanism in this biological system and small RNAs belonging to these genes could be detected. Herein we report A. rhizogenes T-DNA-derived small RNAs (ArT-sRNAs) in hairy roots of common bean. We characterized these T-DNA derived small RNAs and discussed their relevance to plant-bacteria interactions. Distinct abundant A. rhizogenes T-DNA-derived small RNAs belonging to several oncogenes and to the opine synthase gene were detected. Our data demonstrates that many T-DNA encoded genes constitute phasiRNA-producing loci probably subjected to PTGS triggered by dsRNA-derived ArT-sRNAs of 21- and 22-nt in length. We also found that these sRNAs generated from bacteria's genetic material may target host genes. Our results indicate that RNA silencing may regulate T-DNA genes of the Ri plasmid of A. rhizogenes in hairy roots of common bean, suggesting that RNA silencing may play an important role in this naturally occurring plant transformation.
Materials and Methods
Plant Material
Hairy roots and calli were obtained from Phaseolus vulgaris L. cv. Negro Jamapa adjusting the procedure described by Estrada-Navarrete et al. (2007). Ten days after infection with A. rhizogenes K599, the primary root was removed. Then plants were grown in hydroponics (B&D solution supplemented with 8 mM KNO3, pH 6.5) for 11 days in a chamber with 16 h of light and 8 h of dark at 28°C until hairy roots collection. This differs from Estrada-Navarrete et al. (2007) protocol (step 20). For 6 days old calli induction and collection, seeds were germinated as Estrada-Navarrete et al. (2007) protocol (steps 1–7). Then, after seedlings were grown in hydroponics for 2 days with B&D solution, bacteria (steps 9–10) was inoculated in the stem (not in the cotyledonary nodes). Seedlings were grown 6 days in hydroponics inside a chamber to preserve humidity. Common bean non-transgenic roots were obtained from germinated seeds (steps 1–7) grown for 4 days in hydroponics. All samples were frozen in liquid nitrogen and stored at −80°C until RNA extraction.
RNA Extraction and Library Sequencing
Total RNA was extracted from hairy roots and calli using the Trizol reagent (Invitrogen). Ten micrograms of each sample were prepared for deep sequencing of small RNA libraries. Sequences ranging from 18 to 30 nt were purified for the construction of the libraries. A small RNA library of hairy roots was prepared following Illumina's Small RNA alternative sample preparation protocol v1.5. The small RNA library of calli was constructed according to the Illumina's TruSeq Small RNA Sample Prep Kit. Libraries were Single Read-sequenced using the Genome Analyzer IIx (GAIIx) and the Illumina Cluster Station at the Instituto de Biotecnología (Universidad Nacional Autónoma de México). Degradome library construction for the hairy roots sample was performed as described by Ma et al. (2010) with some changes. Approximately 150 ng of poly(A)+ RNA was used to anneal with biotinylated random primers. Strapavidin capture of RNA fragments was performed using biotinylated random primers. A 5′ adaptor ligation was only performed to those RNAs containing 5′-monophosphates. The library was sequenced with the Illumina's Cluster Station and the GAIIx using the 5′ adapter only. This resulted in the sequencing of the first 36 nt of the inserts that represent the 5′ ends of the original RNAs (LC Sciences). All sequence data is available at the European Nucleotide Archive (EMBL-EBI) with the accession number PRJEB7993.
Bioinformatics and Data Analysis
Sequence length distribution and alignment (PatMaN; Prüfer et al., 2008) of small RNAs were performed using the UEA small RNA analysis toolkit (Version 2.5.0; Stocks et al., 2012). Small RNAs (18–26 nt) were aligned to the A. rhizogenes plasmid Ri T-DNA region without mismatches (GenBank accession number: EF433766). Whole T-DNA alignments were seen with the Integrative Genomics Viewer (IGV; Thorvaldsdóttir et al., 2013). RNA secondary structures of the T-DNA loci were predicted with the Vienna RNA secondary structure server using the minimum free energy algorithm (MFE; Hofacker, 2003). P. vulgaris genes targeted by T-DNA derived sRNAs were predicted in silico using the plant small RNA target analysis server psRNATarget (Dai and Zhao, 2011) with a maximum expectation threshold of 0–2. P. vulgaris (Phytozome v9.0) spliced mRNA transcripts without alternative splice variants were used for target prediction analysis. Multiple alignment of rolA sequences from different Ri plasmids (pRi1724/gb: AP002086, pRi8196/gb:M60490, pRiA4/gb:X12579) was performed using ClustalW2 with default settings (Larkin et al., 2007) and viewed with Jalview (Waterhouse et al., 2009). Previously reported microRNA sequences from P. vulgaris were used for the identification of microRNAs in the small RNA libraries and for the non-transgenic root miRNA accumulation analysis (Peláez et al., 2012). Normalized read frequencies (RPM) of miRNAs from the same family were added for miRNA accumulation analysis.
Analysis of PhasiRNAs
Two strategies were used for the identification of loci producing phasiRNAs of 21 nt in length (Chen et al., 2007; Axtell, 2010). The algorithm developed by Chen et al. (2007) was used through the UEA small RNA analysis toolkit implementation (Figures 5B,C). Only sRNAs derived from the T-DNA and the plasmid Ri T-DNA sequence (gb: EF433766) were used as input. Moreover, this algorithm was performed for the identification of phasing-generating loci from P. vulgaris used as controls (Supplementary Table 9). Combined abundances of two and five bins were used in the methodology developed by Axtell (2010) to calculate the phase ratio of rolB and CUS loci, respectively.
Northern Blot Analysis
Total RNA was extracted using Trizol reagent (Invitrogen). RNA (20 ug) was separated by 15% PAGE/8 M urea/ 1x TBE buffer. Gels were electro-blotted to a Hybond-N+ membrane (GE Healthcare) and UV cross-linked twice. Oligonucleotide probes (Supplementary Table 10) were labeled using [γ-32P]-ATP (Perkin-Elmer) and T4 PNK (New England Biolabs). Labeled probes were purified with the Quick spin-oligo columns (Roche) before hybridization. Hybridizations were performed at 42°C overnight in UltraHyb-oligo solution (Ambion). After hybridization, membranes were washed twice in 2 × SSC/0.1% SDS and exposed to the Phosphor Screen System (GE Healthcare). The screen was scanned in a Storm 860 Gel and Blot Imaging System (GE Healthcare). As loading control, an oligonucleotide probe complementary to the U6 small nuclear RNA was used. Signal intensities were quantified using the ImageQuant 5.2 software (Molecular Dynamics). The U6 signal intensity in each blot was used for normalization and calculation of expression ratios. The same loading control was employed for membranes used more than once.
Degradome Data Analysis
Perfect alignments of degradome sequences to the T-DNA were performed using PatMaN (Prüfer et al., 2008). The PAREsnip tool was used for the discovery of small RNA-guided cleavage sites with default parameters (Folkes et al., 2012). The small RNAs derived from the T-DNA, the T-DNA sequence (gb: EF433766) and the degradome sequences were used as input for the characterization of sRNA-mediated cleavages on the A. rhizogenes plasmid Ri T-DNA region. Primary transcripts (Phytozome v9.0) were used as input for the discovery of P. vulgaris targets cleaved by T-DNA derived sRNAs. Degradome sequences from seedlings of common bean were explored in the same way as input and used as control (Formey et al., 2015). Host targets were discarded if other sRNA from the library was also predicted to cleave the transcript and presented a better hit in at least one of the parameters evaluated such as alignment score, category, reads abundance, or p-value. Arabidopsis homolog targets were functionally grouped according to the Gene Ontology (GO) categories (Berardini et al., 2004). GO enrichment analysis was performed through the Plant GeneSet Enrichment Analysis Tool kit under default parameters (Yi et al., 2013). MicroRNAs (Supplementary Table 4) were used for analysis of validated target-sRNA interactions in the degradome library (Supplementary Table 5).
Quantitative PCR
Detection and quantification of transcripts accumulation in hairy roots was performed using the iQ5 Real-Time PCR Detection System and the iQ5 Optical System Software (Bio-Rad). First strand cDNA was synthesized from DNA-free RNA with the RevertAid H Minus First Strand cDNA Synthesis kit (Fermentas). The qRT-PCR reactions were carried out in triplicate for three biological replicates with the Maxima SYBR green-fluorescein qPCR master mix (Fermentas). The gene-specific oligonucleotides belonging to the T-DNA genes were tested also on non-transformed tissues as controls (Supplementary Table 11). The melting temperature was set at 60°C. Quantification was based on a cycle threshold (Ct) value and the genes expression levels were normalized with the elongation factor 1 (EF1) reference gene.
Results
Small RNAs in the Hairy Root Disease
In order to identify small RNAs derived from the T-DNA of A. rhizogenes in the hairy root disease, small RNA libraries from hairy roots, and calli (formed before the emergence of hairy roots at the wound site) were generated. The presence and expression of several genes from the T-DNA in hairy roots (Supplementary Figure 1) were confirmed. Small RNA sequencing yielded 12,396,242 and 27,272,479 total raw reads for the hairy roots and calli samples, respectively. Small RNA sequences (18–26 nt) in hairy roots showed that the 21-nt (11%) and the 24-nt (32%) classes were the most abundant (redundant sequences). The 21-nt class was the second most diverse (unique sequences) in hairy roots and the fourth in A. rhizogenes-induced calli (Figure 1A). Perfect sequence alignment of small RNAs against the T-DNA of A. rhizogenes revealed the presence of several distinct and abundant ArT-sRNAs (18–26 nt) in hairy roots (Figure 1B). In hairy roots, 3176 distinct ArT-sRNAs corresponding to 17,000 small RNA reads per million (RPM) were detected. In contrast, only 563 ArT-sRNAs (unique sequences) corresponding to an abundance of 90 RPM were derived from T-DNA in calli (Figure 1C; Supplementary Table 1; De Paoli et al., 2009). Differences in the amount of ArT-sRNAs detected between hairy roots and calli may be a consequence of the amount of transformed cells in each sample. Most of the ArT-sRNAs observed in hairy roots aligned with transcriptional units of the T-DNA belonging to genes, such as ORF2, ORF8, rolA, rolB, ORF13, ORF14, and the opine synthase gene CUS (Figure 1B; Table 1), with the oncogene rolA being the major source of ArT-sRNAs in hairy roots. Interestingly, virtually no ArT-sRNAs derived from the oncogene rolC were detected in hairy roots. Also, nothing but 134 unique ArT-sRNAs were shared between hairy roots and calli.
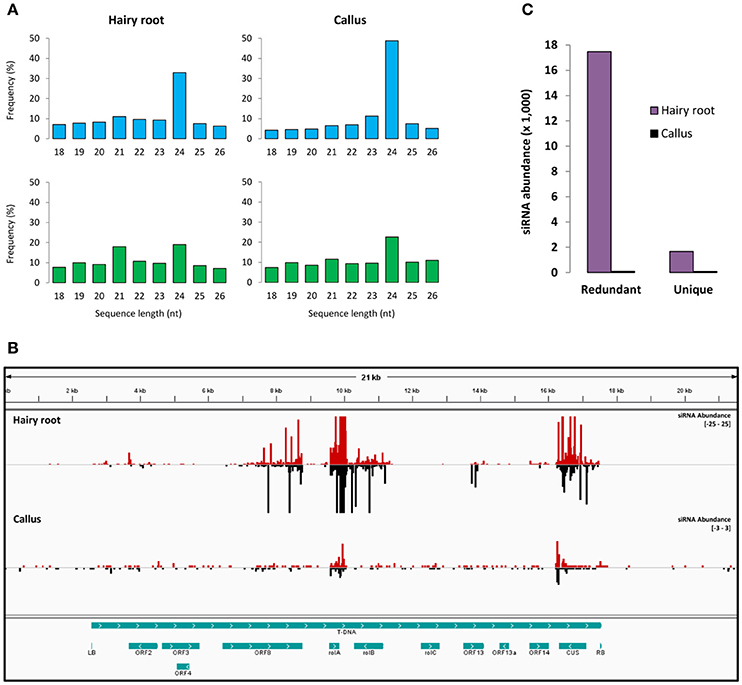
Figure 1. Small RNAs in hairy roots and A. rhizogenes-induced calli. (A) Sequence length distribution of small RNAs. Average percentage (Y-axis) of unique (blue bars) and redundant (green bars) sequences of 18–26 nt length (X-axis) for the hairy roots and the A. rhizogenes-induced calli libraries. (B) Abundance and distribution of ArT-sRNAs in the T-DNA. The small RNAs that aligned to the plus strand (red bars) and to the minus strand (black bars) of the T-DNA of A. rhizogenes (greenish blue long bar) are indicated for the hairy roots (upper part) and calli (lower part) libraries. The abundance scale (right) differs between the libraries to show ArT-sRNAs in calli. In the bottom, the coding regions and the borders from the T-DNA are represented (greenish blue bars). (C) Comparison of the number of ArT-sRNAs detected in hairy roots and calli. Abundance levels were graphed as small RNA reads per million.
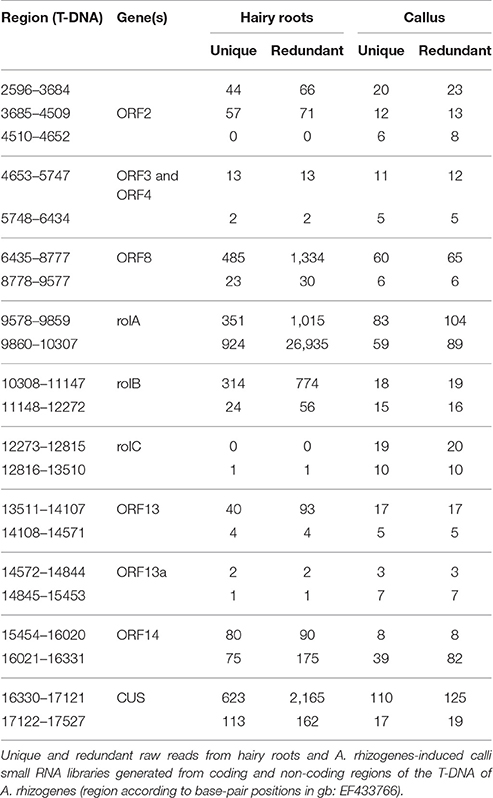
Table 1. Number and location of raw small RNA reads derived from the T-DNA.
Moreover, the impact of ArT-sRNAs production on the accumulation of microRNAs in hairy roots and calli was measured. Members of conserved miRNA families, such as miR156, miR157, miR159, miR160, miR162, miR164, miR166, miR167, miR168, miR169, miR319, miR390, miR393, miR394, miR396, and miR408 were considered. Production of miRNAs in hairy roots was similar compared with non-transgenic roots according to northern blot analysis for eight miRNAs (Figure 2A). Relative miRNA expression estimation using the frequencies of miRNAs detected in the hairy roots and the calli libraries showed that the majority of miRNAs analyzed accumulated less in calli than in hairy roots. Substantial differences in accumulation were detected in these two samples for microRNAs miR394, miR408, and miR319 (Figure 2B). These differences in accumulation were also observed for these three families of microRNAs between the read frequencies of calli and a previously reported small RNA library from non-transgenic roots (Supplementary Figure 2; Peláez et al., 2012). MicroRNAs miR408 and miR394 were up-regulated in hairy roots and non-transgenic roots compared with calli, whereas miR319 was down-regulated in both type of roots. Also, miR393, a microRNA involved in plant defenses and auxin signaling, was more abundant in calli than in hairy roots (Figure 2B; Peláez and Sanchez, 2013). Interestingly, miR319 was the most abundant miRNA detected in calli. This was not the case for small RNA libraries of common bean from roots, leaves, flowers, and seedlings (Peláez et al., 2012).
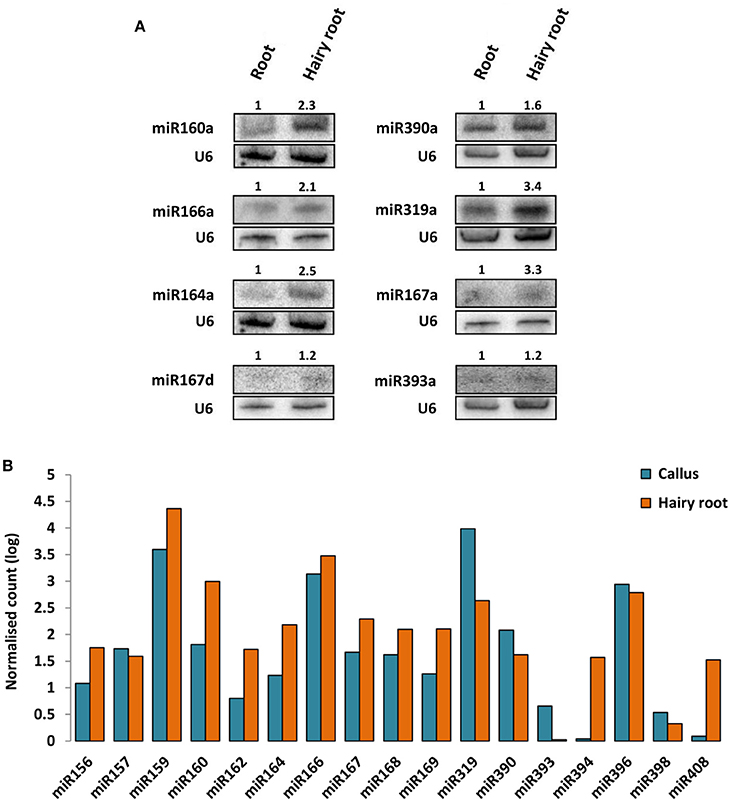
Figure 2. Accumulation of microRNAs in non-transgenic root, hairy roots and A. rhizogenes-induced calli. (A) Northern blot analysis of microRNAs in hairy roots and non-transgenic roots of common bean. The signal of the U6 small nuclear RNA was used as loading control. The accumulation ratios between non-transgenic roots and hairy roots were calculated (upper numbers in gels). (B) Differential accumulation analysis of microRNAs from hairy roots and calli libraries. Frequencies of reads were normalized (RPM) and represented in logarithmic scale (Y-axis).
Agrobacterium rhizogenes T-DNA-Derived Small RNAs
To unravel the probable biogenesis and action of common bean ArT-sRNAs, their size, strand origin, and phased patterns were analyzed. It was found that ArT-sRNAs were largely small RNAs of 21-nt in length in hairy roots and calli (Figure 3A). In hairy roots, 52.2% of the ArT-sRNAs (unique sequences), representing 67.9% of the total sequences that aligned to the T-DNA, were of 21-nt in length. Only 5% of the total sequences corresponded to the 24-nt class that is involved in transcriptional regulation. The 22-nt class of ArT-sRNAs was the second most diverse (18%) and abundant (19.7%). A similar pattern of accumulation was observed for the ArT-sRNAs detected in calli, which indicates that silencing of T-DNA transcripts may occur at the post-transcriptional level. Also, ArT-sRNAs were produced from the sense and antisense strands, suggesting that ArT-sRNAs may be generated from perfect long double stranded RNAs (Figure 3B). In hairy roots, 57% of redundant sequences of the ArT-sRNAs found aligned to the sense strand while 43% aligned to the antisense strand. Taking a closer look to particular genes encoded in the sense or antisense strand, the proportion of ArT-sRNAs that aligned to either strand was variable (Figure 4). Furthermore, phased secondary 21-nt small interfering RNAs characteristic of PHAS loci were identified for ORF8, rolA, rolB, ORF13, ORF14 and CUS genes (Figures 5A–C). Genes like rolA, ORF8, ORF13, and ORF14 exhibited one sRNA cluster and one predominant phase, whereas rolB and CUS presented two clusters without one main phase. Some phased small RNAs of the rolA transcript were among the most abundant sRNAs detected (Figure 5A).
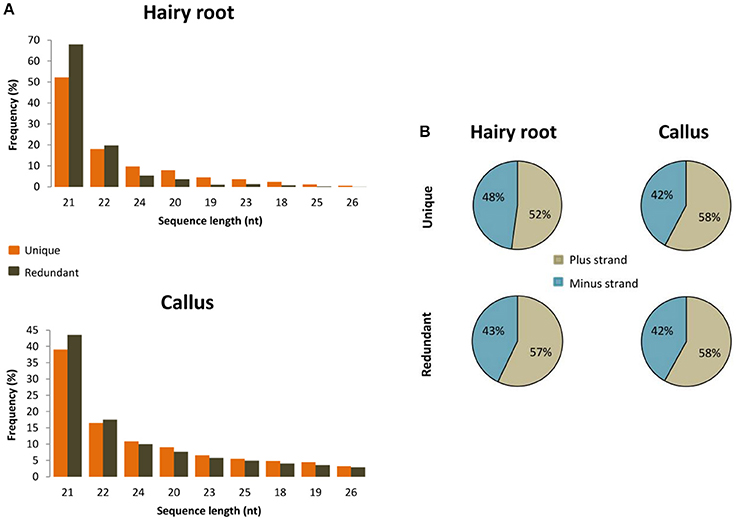
Figure 3. Size and orientation of ArT-sRNAs. (A) Sequence length distribution of ArT-sRNAs found in the hairy roots and A. rhizogenes-induced calli libraries that aligned to the T-DNA. (B) Average percentage of unique and redundant sequences that aligned with the plus strand (gray) and the minus strand (blue) of the T-DNA for each of the small RNA libraries.
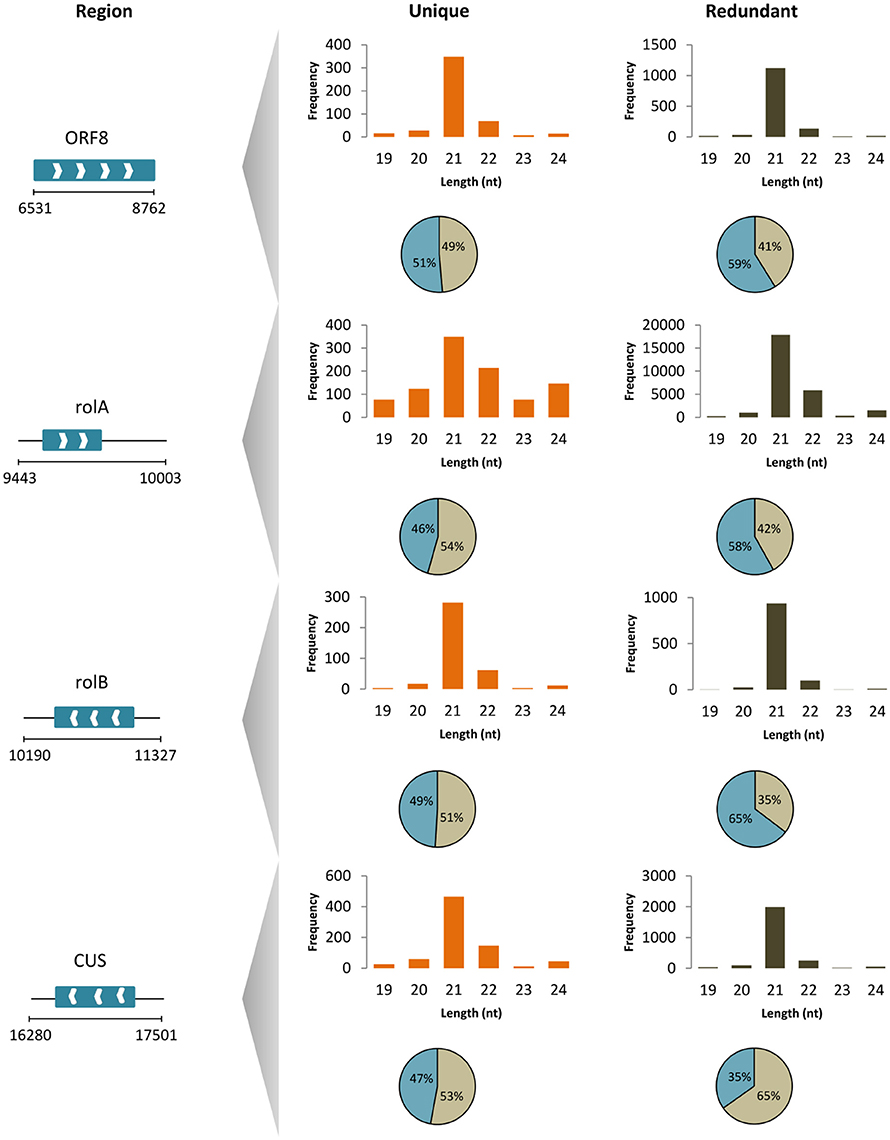
Figure 4. ArT-sRNAs from hairy roots mapped to T-DNA genes. Length distribution (column plots) and strand origin (circular plots; positive strand gray, negative strand blue) of unique and redundant reads of ArT-sRNAs for particular T-DNA genes (region according to base-pair positions in gb: EF433766). Genes encoded in the sense (white right arrows) and antisense (white left arrows) strand are shown.
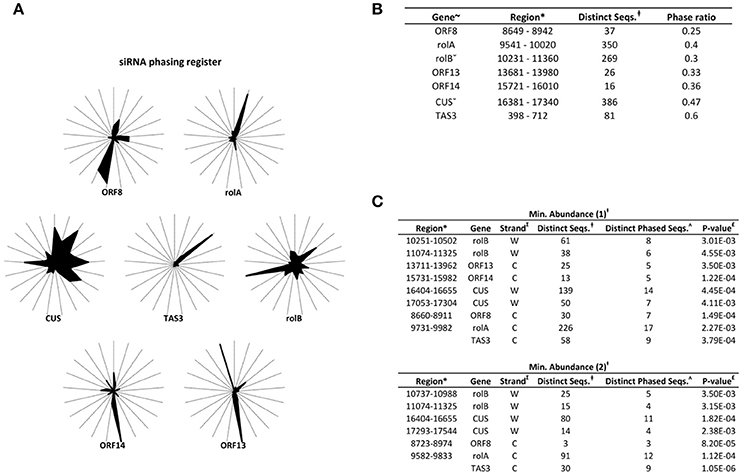
Figure 5. PhasiRNA-producing loci from the T-DNA. (A) Radial graphs representing the phasing register of ArT-sRNAs from hairy roots as performed by Axtell (2010). The 21 spokes of each graph correspond to the possible phasing registers. (B) Analysis of phasiRNA-producing loci represented in radial graphs. The genes (~), the region considered in the analysis (*) and the distinct small RNAs that mapped the region are indicated (‡). The region considered for the rolB and CUS genes is greater (ˇ). Loci with phase ratios of <0.25 were discarded. The TAS3 transcript that is targeted by miR390 to generate ta-siRNAs was used as control. (C) Identification of phasiRNAs as described by Chen et al. (2007) taking into account ArT-sRNAs with one (upper part) or more than two (lower part) absolute reads (Ɨ). The strand of the loci (∑), the number of phased ArT-sRNAs (ʌ) and the P-value (£) are indicated.
ArT-sRNAs-Mediated Cleavage of T-DNA Transcripts
Forty percent of the ArT-sRNAs of 21-nt in length that were detected in this analysis had a 5′ terminal uracil (Supplementary Figure 3). Small RNAs with this characteristic are recruited by AGO1. To gain insight into the cleavage of T-DNA transcripts through AGOs/ArT-sRNAs associations, a degradome (also called parallel analysis of RNA ends, PARE) library from hairy roots was deep-sequenced and analyzed. Sequence alignment resulted in the identification of 446 distinct sequences that aligned perfectly to the T-DNA of A. rhizogenes. Degradome sequences corresponding to all of the coding sequences of the T-DNA genes except for ORF13 were detected (Supplementary Figures 4, 5). Most of the reads detected had the sequence of the rolA and CUS genes. Discovery analysis of sRNA/target interactions using degradome sequences and ArT-sRNAs from hairy roots exposed 180 interactions between ArT-sRNAs and the transcripts of the T-DNA with significant p-values at 73 unique cleavage positions (Supplementary Table 2). According to degradome read abundances (Addo-Quaye et al., 2009), 5 interactions fell into category 0 (highly likely to occur), 116 into category 2, and 59 into category 4. From almost all of the interactions found, around 95.5%, pointed out to cleavage sites of the rolA transcript, mainly from the 3′ untranslated region (Figure 6A). These results strongly suggest that AGOs/ArT-sRNAs associations occur. Furthermore, one of the ArT-sRNAs that fell into category 0, and thus cleaved the most abundant fragment in the transcript of rolA, was indeed the most abundant ArT-sRNA in the library. This ArT-sRNA of 22-nt in length with 3304 absolute read counts in the library presented another cleavage position including two mismatches (Figure 6A). This cleavage position in the transcript of rolA was consistent with the phasing register of the detected rolA phase (Figure 5A). In addition, the sequence of this ArT-sRNA was found to be highly conserved among rolA sequences of different Ri plasmids (pRi8196, pRi1724, pRiA4, and pRi2659), which suggests functional roles for this small RNA (Figure 6B).
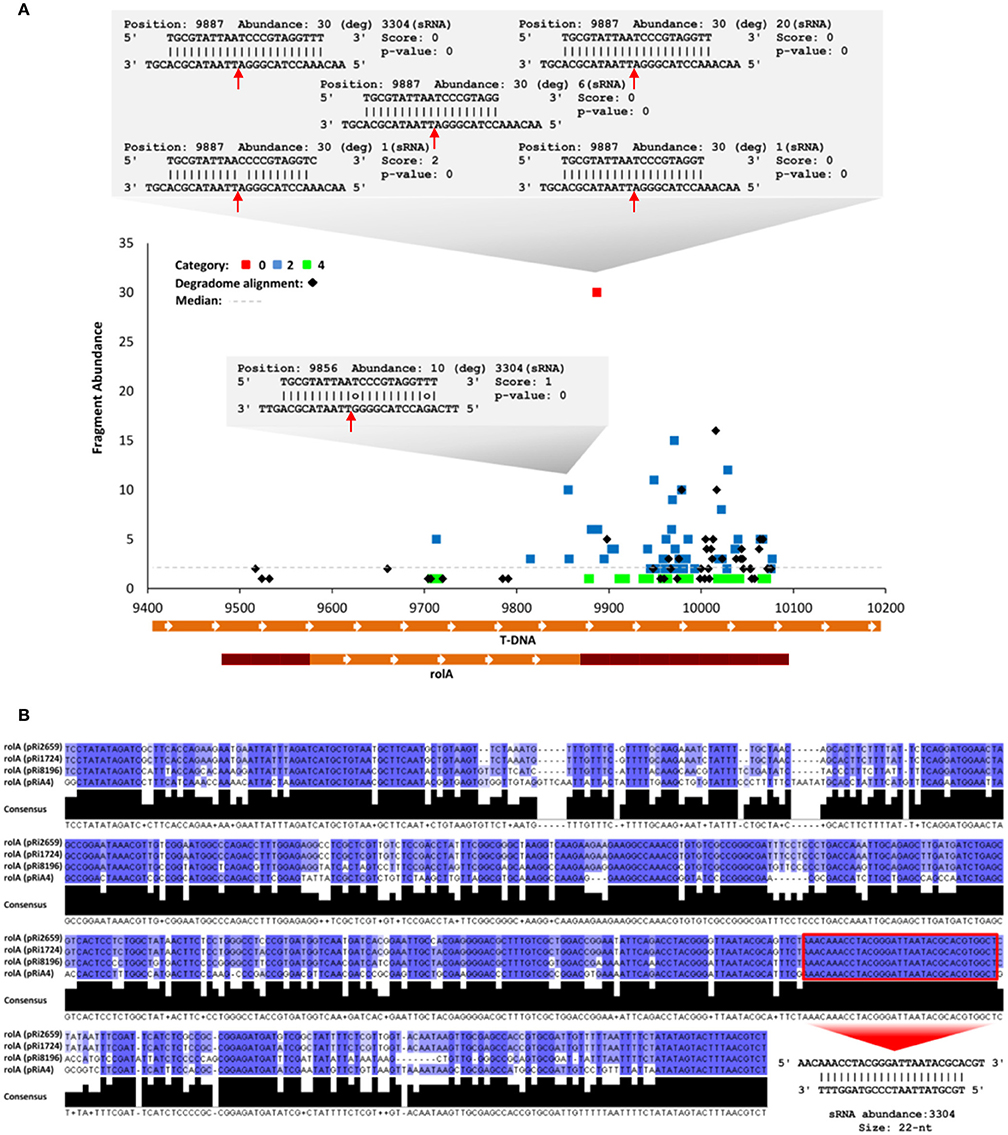
Figure 6. Cleavage analysis of rolA by ArT-sRNAs. (A) T-plot showing different categorized interactions (cleavage sites) between rolA and ArT-sRNAs. The interactions between rolA and the most abundant ArT-sRNA of 22-nt and its variants in length are shown (gray boxes). Cleavage sites for alignments are indicated (red arrows). In the T-DNA (X-axis), the coding region for rolA is indicated at the bottom (orange box) and the 5′ and 3′ untranslated regions (crimson red boxes). (B) Multiple alignment of rolA sequences from different Ri plasmids. The ArT-sRNA of 22-nt is indicated (box and red arrow).
Potential Host Gene Silencing through ArT-sRNAs
ArT-sRNAs may present the required complementarity base pairing with the transcripts of P. vulgaris to cleave them. To search for complementarity base pairing between ArT-sRNAs and host transcripts (P. vulgaris), a target prediction analysis was performed using the plant small RNA target analysis server psRNATarget (Dai and Zhao, 2011). The identification of 399 unique ArT-sRNAs/target interactions using a stringent cut-off threshold of the complementarity score (Supplementary Table 3) was determined. The interactions detected were composed by 299 distinct transcripts and 326 distinct ArT-sRNAs. To provide further evidence of ArT-sRNAs-mediated cleavage of host gene transcripts, the degradome library obtained from hairy roots was analyzed in pursuit of putative interactions. MicroRNAs (Supplementary Table 4) were used for analysis of conserved validated target-sRNA interactions in the degradome library as controls. Most of the microRNA-target interactions found corresponded to conserved validated target-microRNA interactions (Supplementary Table 5). In total, 402 interactions between host targets and ArT-sRNAs were detected using PAREsnip with default parameters (Supplementary Table 6). The total number of unique host transcripts predicted to be cleaved by 346 unique ArT-sRNAs was 228. No interactions were found between ArT-sRNAs and these 228 transcripts using the degradome sequencing of non-transformed common bean seedlings (Formey et al., 2015). Most of the ArT-sRNAs that define these interactions were of 21-nt in length (53.4%). Taking into account all the interactions, 58 fell into category 0, 50 into category 1, 155 into category 2, 3 into category 3, and 136 into category 4. The host transcript targets identified correspond to a diverse set of protein families (Supplementary Table 7). Gene Ontology (GO) enrichment analysis was performed for the 98 of the unique host targets (Supplementary Table 8). The two most enriched gene sets within the cellular component category were cellular (GO: 0044464, p-value = 1.91E-09, FDR = 5.97E-07) and intracellular components (GO: 0044424, p-value = 2.97E-07, FDR = 6.96-05). Metabolic (GO: 0008152, p-value = 8.45E-12, FDR = 7.58E-08) and cellular processes (GO: 0009987, p-value = 7.68E-11, FDR = 3.44E-07) were the most enriched gene sets observed from the biological process category. The response to stress category was also detected by the GO enrichment analysis (GO: 0006950, p-value = 2.49E-08, FDR = 4.31E-05) suggesting probable regulation of host transcripts by ArT-sRNAs as a counter-defensive strategy. Given that most of the interactions found by PAREsnip were validated interactions (Folkes et al., 2012) and that we found several interactions between ArT-sRNAs and P. vulgaris transcripts, our findings suggest that ArT-sRNAs could target common bean transcripts.
Discussion
A. rhizogenes pathogenicity is characterized by the insertion of the T-DNA into the plants' nuclear genome. For this reason, it represents a powerful system to transform plants (Tzfira and Citovsky, 2006; Georgiev et al., 2012). Also, it is capable to transforming plants that are recalcitrant to A. tumefaciens infection (Taylor et al., 2006). Recent progress on the understanding of the induction and regeneration of hairy roots has increased its usage in the production of recombinant proteins, metabolic engineering, and phytoremediation (Georgiev et al., 2012). In contrast to A. tumefaciens, A. rhizogenes is frequently used with the root-inducing plasmid “armed,” carrying the T-DNA oncogenes (Taylor et al., 2006). For years, A. rhizogenes oncogenes have been considered modulators of neoplastic growth and differentiation (Britton et al., 2008). Recently, they have been also identified as triggers of secondary metabolism (Bulgakov et al., 2016). The biochemical and molecular characterization of the T-DNA genes from A. rhizogenes is not completely understood (Britton et al., 2008). In several studies, conflicting results regarding their functions have been reported, suggesting that new meristem emergence and subsequent differentiation of transformed plant cells may be conducted through complex molecular mechanisms (Britton et al., 2008). In recent years, small RNAs have emerged as important regulators of gene expression during different biological processes (Chapman and Carrington, 2007). Although, RNA silencing mechanism has been reported to regulate transgenes, viral nucleic acids and genes from the T-DNA of A. tumefaciens, its regulatory activity against the T-DNA of A. rhizogenes in hairy roots had not been analyzed (Blevins et al., 2006; Deleris et al., 2006; Dunoyer et al., 2006).
In this study, several abundant small RNAs generated from the T-DNA transcripts of A. rhizogenes in hairy roots were detected. Interestingly, miR319 was the most abundant miRNA family in the library of calli. MiR159 was the most abundant miRNA in roots, flowers and seedlings of P. vulgaris (Peláez et al., 2012). The miR319 family targets TCP (TEOSINTE BRANCHED-CYCLOIDEA/PROLIFERATING CELL FACTORS) genes involved in hormone and signaling pathways, proliferation, and differentiation (Schommer et al., 2012). Reduced activity of TCP genes caused by the increase of miR319 accumulation intensifies cellular proliferation (Schommer et al., 2012). Based on the findings above, this miRNA could be crucial for calli formation. MicroRNAs miR408 and miR394 were up-regulated in hairy roots and non-transgenic roots compared with calli. MiR408 targets genes of the phytocyanin family (cupredoxin, plnatacyanin, and uclacyanin) and a laccase (Abdel-Ghany and Pilon, 2008; Zhang and Li, 2013). MiR408 has been reported to play a role in abiotic stress responses and in vegetative development in Arabidopsis (Zhang and Li, 2013; Ma et al., 2015). Arabidopsis plants overexpressing miR408 presented enhanced root development under salinity stress and increased tolerance to oxidative stress (Ma et al., 2015). Oxidative and salinity stress tolerance have been reported for hairy roots (Bulgakov et al., 2013). Probably accumulation of miR408 contributes to these abiotic stress responses in hairy roots. MiR394 targets the gene LCR (LEAF CURLING RESPONSIVENESS), an F-box protein (SKP1-Cullin/CDC53-F-box). Roles described by Knauer et al. (2013) for miR394 makes it a highly attractive microRNA to study in Agrobacterium-plant interactions. MiR394 was found to be essential for shoot meristem formation. Interestingly, stem cells differentiate in the miR394 mutant plants (Knauer et al., 2013). In this study, miR394 was poorly detected in calli, suggesting a role for this microRNA in hairy root emergence. MiR393, a microRNA well known to be involved in plant defense responses to bacterial infection, was upregulated in calli when compared with hairy roots. MiR393 was almost undetectable in both roots (Figure 2A). Induced accumulation of miR393 was observed during A. tumefaciens infiltration at an early stage of infection (Pruss et al., 2008). MiR393 targets three auxin receptors: TIR1, AFB2, and AFB3. Besides its roles in plant defense responses, we consider that miR393 may play an important role during the hairy root disease because of auxin relevance in calli formation and organ induction (Ikeuchi et al., 2013).
Among the T-DNA transcripts producing ArT-sRNAs, the rolA transcript stands out as a major source of small RNAs. The precise function of the rolA protein is still unknown (Britton et al., 2008). Poor expression of rolA has been reported in roots and in leaves (Carneiro and Vilaine, 1993). Tobacco plants expressing rolA present wrinkled leaves, shortened internodes, and abnormal flowers (Carneiro and Vilaine, 1993). Local expression of rolA in the vascular bundles reduces the size of the surrounding parenchyma cells causing the wrinkled leaf phenotype. For this reason, and based on reciprocal grafting studies involving rolA, it has been proposed that rolA generates a diffusible factor (Guivarc'h et al., 1996; Britton et al., 2008). Given their nature, ArT-sRNAs derived from rolA may constitute this diffusible factor. ArT-sRNAs could also indirectly impact on plant phenotype through altering endogenous sRNA-regulated processes as described during PTGS of foreign genetic material (Martínez de Alba et al., 2011). Intriguingly, no ArT-sRNAs were derived from the rolC gene in hairy roots, one of the T-DNA genes widely studied as well. Strong and stable expression of rolC in roots of transformed plants previously reported together with the null detection of ArT-sRNAs generated from this gene, suggests that rolC is poorly or not at all subjected to silencing in hairy roots (Sugaya et al., 1989).
The detection of ArT-sRNAs, mainly of 21- and 22-nt in length, suggests that the T-DNA genes may be silenced at the post-transcriptional level and that their transcripts are probably processed by DCL4 and DCL2 as occurs during regulation of RNA virus (Deleris et al., 2006). Also, the identification of ArT-sRNAs as phasiRNAs indicates an active role of one or more RDRs during the regulation of these genes. DCL4 often produces phasiRNAs of 21-nt from RDR6-dependent dsRNA that can be loaded into AGO1 (Fei et al., 2013). Besides, cleavage analysis of the T-DNA transcripts by ArT-sRNAs indirectly shows that this small RNAs could be loaded into AGO protein-complexes. Most of them could be loaded onto AGO1, which has an important role in antiviral silencing (Peláez and Sanchez, 2013). The interactions detected for conserved microRNAs using the PAREsnip algorithm and the degradome data were mainly validated interactions. For this reason, the amount of interactions detected between ArT-sRNAs and host transcripts suggests that ArT-sRNAs may help shape hairy root phenotype through direct regulation of the host transcripts. Further experiments are required to truly validate this regulation. Although, its known that virus-derived small interfering RNAs and fungal small RNAs regulate host genes like endogenous sRNAs (Qi et al., 2009; Shimura et al., 2011; Smith et al., 2011; Weiberg et al., 2013).
RNA silencing of transcripts from the T-DNA of A. rhizogenes still has to be determined in hairy roots and calli because several factors could influence their accumulation levels in these two different samples. Transcripts from the T-DNA in hairy roots were detected by quantitative PCR (Supplementary Figure 1) but, the degree of silencing for these transcripts still has to be evaluated. Also, more experiments have to be done to define if poor accumulation of ArT-sRNAs and reduced accumulation of most miRNAs in calli are a consequence of the amount of transformed cells or a RNA silencing suppression state similar to A. tumefaciens-induced tumors. It is also possible that ArT-sRNAs detected in calli belong to cells that have begun the process of differentiation. Nevertheless, these results contribute to the understanding and usage of hairy roots as an experimental model. We propose that, if the T-DNA genes are silenced in the hairy root disease, one may consider the population of ArT-sRNAs when designing constructs for its overexpression in hairy roots to avoid possible silencing. Also, it would be interesting to determine if the role of RNA silencing against the T-DNA genes has an impact on the efficiency of constructs designed to silence genes. For this reason, studies have to take into account these observations when elucidating the function of several T-DNA genes, specially of rolA. Our results also suggest that additional genes of the T-DNA of A. tumefaciens could be silenced at the beginning of the infection (Dunoyer et al., 2006). Future studies may reveal if regulation of the T-DNA transcripts by RNA silencing contributes to hairy root emergence or phenotype.
Author Contributions
PP conceived the study. PP wrote the manuscript and carried out plant growth, RNA extraction, and preparation and the bioinformatic analyses. PP and AH carried out the quantitative PCR analysis. GE and FS provided intellectual suggestions. Authors read and approved the final manuscript.
Funding
Financial support for this research was provided by the Consejo Nacional de Ciencia y Tecnología (CONACYT) and Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-DGAPA-IN206415). PP was supported by a fellowship from CONACYT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Unidad Universitaria de Secuenciación Masiva de DNA (UUSMD-UNAM) for sequencing services. We are grateful to the Instituto de Biotecnología-UNAM for giving us access to its computer cluster and to Selene Fernández and Stewart Gillmor for fruitful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00096/full#supplementary-material
References
Abdel-Ghany, S. E., and Pilon, M. (2008). MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 283, 15932–15945. doi: 10.1074/jbc.M801406200
Addo-Quaye, C., Miller, W., and Axtell, M. J. (2009). CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 25, 130–131. doi: 10.1093/bioinformatics/btn604
Axtell, M. J. (2010). A method to discover phased siRNA loci. Methods Mol. Biol. 592, 59–70. doi: 10.1007/978-1-60327-005-2_5
Berardini, T. Z., Mundodi, S., Reiser, L., Huala, E., Garcia-Hernandez, M., Zhang, P., et al. (2004). Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 135, 745–755. doi: 10.1104/pp.104.040071
Blevins, T., Rajeswaran, R., Shivaprasad, P. V., Beknazariants, D., Si-Ammour, A., Park, H. S., et al. (2006). Four plant Dicers mediate viral small RNA biogenesis and DNA virus induced silencing. Nucleic Acids Res. 34, 6233–6246. doi: 10.1093/nar/gkl886
Britton, M. T., Escobar, M. A., and Dandekar, A. M. (2008). “The oncogenes of Agrobacterium tumefaciens and Agrobacterium rhizogenes,” in Agrobacterium: from Biology to Biotechnology, eds T. Tzfira and V. Citovsky (New York, NY: Springer), 523–563.
Bulgakov, V. P., Shkryl, Y. N., Veremeichik, G. N., Gorpenchenko, T. Y., and Vereshchagina, Y. V. (2013). Recent advances in the understanding of Agrobacterium rhizogenes-derived genes and their effects on stress resistance and plant metabolism. Adv. Biochem. Eng. Biotechnol. 134, 1–22. doi: 10.1007/10_2013_179
Bulgakov, V. P., Veremeichik, G. N., Grigorchuk, V. P., Rybin, V. G., and Shkryl, Y. N. (2016). The rolB gene activates secondary metabolism in Arabidopsis calli via selective activation of genes encoding MYB and bHLH transcription factors. Plant Physiol. Biochem. 102, 70–79. doi: 10.1016/j.plaphy.2016.02.015
Carneiro, M., and Vilaine, F. (1993). Differential expression of the rolA plant oncogene and its effect on tobacco development. Plant J. 3, 785–792. doi: 10.1111/j.1365-313X.1993.00785.x
Chapman, E. J., and Carrington, J. C. (2007). Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 8, 884–896. doi: 10.1038/nrg2179
Chen, H.-M., Li, Y.-H., and Wu, S.-H. (2007). Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 104, 3318–3323. doi: 10.1073/pnas.0611119104
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Deleris, A., Gallego-Bartolome, J., Bao, J., Kasschau, K. D., Carrington, J. C., and Voinnet, O. (2006). Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. Science 313, 68–71. doi: 10.1126/science.1128214
De Paoli, E., Dorantes-Acosta, A., Zhai, J., Accerbi, M., Jeong, D.-H., Park, S., et al. (2009). Distinct extremely abundant siRNAs associated with cosuppression in petunia. RNA 15, 1965–1970. doi: 10.1261/rna.1706109
Dunoyer, P., Himber, C., and Voinnet, O. (2006). Induction, suppression and requirement of RNA silencing pathways in virulent Agrobacterium tumefaciens infections. Nat. Genet. 38, 258–263. doi: 10.1038/ng1722
Estrada-Navarrete, G., Alvarado-Affantranger, X., Olivares, J.-E., Guillén, G., Díaz-Camino, C., Campos, F., et al. (2007). Fast, efficient and reproducible genetic transformation of Phaseolus spp. by Agrobacterium rhizogenes. Nat. Protoc. 2, 1819–1824. doi: 10.1038/nprot.2007.259
Fei, Q., Xia, R., and Meyers, B. C. (2013). Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 25, 2400–2415. doi: 10.1105/tpc.113.114652
Folkes, L., Moxon, S., Woolfenden, H. C., Stocks, M. B., Szittya, G., Dalmay, T., et al. (2012). PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 40:e103. doi: 10.1093/nar/gks277
Formey, D., Iñiguez, L. P., Peláez, P., Li, Y.-F., Sunkar, R., Sánchez, F., et al. (2015). Genome-wide identification of the Phaseolus vulgaris sRNAome using small RNA and degradome sequencing. BMC Genomics 16:423. doi: 10.1186/s12864-015-1639-5
Georgiev, M. I., Agostini, E., Ludwig-Müller, J., and Xu, J. (2012). Genetically transformed roots: from plant disease to biotechnological resource. Trends Biotechnol. 30, 528–537. doi: 10.1016/j.tibtech.2012.07.001
Guivarc'h, A., Carneiro, M., Vilaine, F., Pautot, V., and Chriqui, D. (1996). Tissue-specific expression of the rolA gene mediates morphological changes in transgenic tobacco. Plant Mol. Biol. 30, 125–134. doi: 10.1007/BF00017807
Hamilton, A. J., and Baulcombe, D. C. (1999). A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286, 950–952. doi: 10.1126/science.286.5441.950
Hofacker, I. L. (2003). Vienna RNA secondary structure server. Nucleic Acids Res. 31, 3429–3431. doi: 10.1093/nar/gkg599
Ikeuchi, M., Sugimoto, K., and Iwase, A. (2013). Plant callus: mechanisms of induction and repression. Plant Cell 25, 3159–3173. doi: 10.1105/tpc.113.116053
Knauer, S., Holt, A. L., Rubio-Somoza, I., Tucker, E. J., Hinze, A., Pisch, M., et al. (2013). A protodermal miR394 signal defines a region of stem cell competence in the arabidopsis shoot meristem. Dev. Cell 24, 125–132. doi: 10.1016/j.devcel.2012.12.009
Kovacova, V., Zluvova, J., Janousek, B., Talianova, M., and Vyskot, B. (2014). The evolutionary fate of the horizontally transferred agrobacterial mikimopine synthase gene in the genera Nicotiana and Linaria. PLoS ONE 9:e113872. doi: 10.1371/journal.pone.0113872
Krenek, P., Samajova, O., Luptovciak, I., Doskocilova, A., Komis, G., and Samaj, J. (2015). Transient plant transformation mediated by Agrobacterium tumefaciens: principles, methods and applications. Biotechnol. Adv. 33, 1024–1042. doi: 10.1016/j.biotechadv.2015.03.012
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Ma, C., Burd, S., and Lers, A. (2015). miR408 is involved in abiotic stress responses in Arabidopsis. Plant J. 84, 169–187. doi: 10.1111/tpj.12999
Ma, Z., Coruh, C., and Axtell, M. J. (2010). Arabidopsis lyrata small RNAs: transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell 22, 1090–1103. doi: 10.1105/tpc.110.073882
Martínez De Alba, A. E., Jauvion, V., Mallory, A. C., Bouteiller, N., and Vaucheret, H. (2011). The miRNA pathway limits AGO1 availability during siRNA-mediated PTGS defense against exogenous RNA. Nucleic Acids Res. 39, 9339–9344. doi: 10.1093/nar/gkr590
Peláez, P., and Sanchez, F. (2013). Small RNAs in plant defense responses during viral and bacterial interactions: similarities and differences. Front. Plant Sci. 4:343. doi: 10.3389/fpls.2013.00343
Peláez, P., Trejo, M. S., Iñiguez, L. P., Estrada-Navarrete, G., Covarrubias, A. A., Reyes, J. L., et al. (2012). Identification and characterization of microRNAs in Phaseolus vulgaris by high-throughput sequencing. BMC Genomics 13:83. doi: 10.1186/1471-2164-13-83
Prüfer, K., Stenzel, U., Dannemann, M., Green, R. E., Lachmann, M., and Kelso, J. (2008). PatMaN: rapid alignment of short sequences to large databases. Bioinformatics 24, 1530–1531. doi: 10.1093/bioinformatics/btn223
Pruss, G. J., Nester, E. W., and Vance, V. (2008). Infiltration with Agrobacterium tumefaciens induces host defense and development-dependent responses in the infiltrated zone. Mol. Plant Microbe. Interact. 21, 1528–1538. doi: 10.1094/MPMI-21-12-1528
Qi, X., Bao, F. S., and Xie, Z. (2009). Small RNA deep sequencing reveals role for Arabidopsis thaliana RNA-dependent RNA polymerases in viral siRNA biogenesis. PLoS ONE 4:e4971. doi: 10.1371/journal.pone.0004971
Schommer, C., Bresso, E. G., Spinelli, S. V., and Palatnik, J. F. (2012). “Role of microRNA miR319 in plant develompent,” in MicroRNAs in Plant Development and Stress Responses, Signaling and Communication in Plants, ed R. Sunkar (Berlin; Heidelberg: Springer), 29–47.
Shimura, H., Pantaleo, V., Ishihara, T., Myojo, N., Inaba, J., Sueda, K., et al. (2011). A viral satellite RNA induces yellow symptoms on tobacco by targeting a gene involved in chlorophyll biosynthesis using the RNA silencing machinery. PLoS Pathog. 7:e1002021. doi: 10.1371/journal.ppat.1002021
Smith, N. A., Eamens, A. L., and Wang, M. B. (2011). Viral small interfering RNAs target host genes to mediate disease symptoms in plants. PLoS Pathog. 7:e1002022. doi: 10.1371/journal.ppat.1002022
Stocks, M. B., Moxon, S., Mapleson, D., Woolfenden, H. C., Mohorianu, I., Folkes, L., et al. (2012). The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics 28, 2059–2061. doi: 10.1093/bioinformatics/bts311
Subramoni, S., Nathoo, N., Klimov, E., and Yuan, Z. C. (2014). Agrobacterium tumefaciens responses to plant-derived signaling molecules. Front. Plant Sci. 5:322. doi: 10.3389/fpls.2014.00322
Sugaya, S., Hayakawa, K., Handa, T., and Uchimiya, H. (1989). Cell-specific expression of the rolC gene of the TL-DNA of Ri plasmid in transgenic tobacco plants. Plant Cell Physiol. 305, 649–653.
Taylor, C. G., Fuchs, B., Collier, R., and Lutke, W. K. (2006). Generation of composite plants using Agrobacterium rhizogenes. Methods Mol. Biol. 343, 155–167. doi: 10.1385/1-59745-130-4:155
Tepfer, D. (1984). Transformation of several species of higher plants by Agrobacterium rhizogenes: sexual transmission of the transformed genotype and phenotype. Cell 37, 959–967. doi: 10.1016/0092-8674(84)90430-6
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Tzfira, T., and Citovsky, V. (2006). Agrobacterium-mediated genetic transformation of plants: biology and biotechnology. Curr. Opin. Biotechnol. 17, 147–154. doi: 10.1016/j.copbio.2006.01.009
Wang, M., Soyano, T., Machida, S., Yang, J.-Y., Jung, C., Chua, N.-H., et al. (2011). Molecular insights into plant cell proliferation disturbance by Agrobacterium protein 6b. Genes Dev. 25, 64–76. doi: 10.1101/gad.1985511
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Weiberg, A., Wang, M., Lin, F.-M., Zhao, H., Zhang, Z., Kaloshian, I., et al. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123. doi: 10.1126/science.1239705
Willmann, M. R., Endres, M. W., Cook, R. T., and Gregory, B. D. (2011). The Functions of RNA-Dependent RNA Polymerases in Arabidopsis. Arabidopsis Book 9:e0146. doi: 10.1199/tab.0146
Yi, X., Du, Z., and Su, Z. (2013). PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 41, W98–W103. doi: 10.1093/nar/gkt281
Keywords: Agrobacterium rhizogenes, hairy roots, RNA silencing, small RNAs, T-DNA
Citation: Peláez P, Hernández-López A, Estrada-Navarrete G and Sanchez F (2017) Small RNAs Derived from the T-DNA of Agrobacterium rhizogenes in Hairy Roots of Phaseolus vulgaris. Front. Plant Sci. 8:96. doi: 10.3389/fpls.2017.00096
Received: 01 September 2016; Accepted: 17 January 2017;
Published: 01 February 2017.
Edited by:
Bernie Carroll, University of Queensland, AustraliaReviewed by:
Ming-Bo Wang, Commonwealth Scientific and Industrial Research Organisation (CSIRO), AustraliaChristopher Andrew Brosnan, ETH Zurich, Switzerland
Copyright © 2017 Peláez, Hernández-López, Estrada-Navarrete and Sanchez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo Peláez, cGFibG8ucGVsYWV6QGNpbnZlc3Rhdi5teA==
†In Memoriam: In memory of professor Federico Sanchez Ph.D.