 Xiang Lu
Xiang Lu Xuan Zhou1
Xuan Zhou1 Shan Liang
Shan Liang Chengwei Yang
Chengwei Yang- 1Guangdong Provincial Key Laboratory of Biotechnology for Plant Development, School of Life Sciences, South China Normal University, Guangzhou, China
- 2Tasmanian Institute of Agriculture, University of Tasmania, Kings Meadows, TAS, Australia
- 3College of Pratacultural Science, Gansu Agriculture University, Lanzhou, China
- 4Dongli Planting and Farming Industrial Co., LTD, Lianzhou, China
The annual Zea mays ssp. mexicana L. is a member of teosinte, a wild relative of the Zea mays spp. mays L. This subspecies has strong growth and regeneration ability, high tiller numbers, high protein and lysine content as well as resistance to many fungal diseases, and it can be effectively used in maize improvement. In this study, we reported a Zea mays ssp. mexicana L. transcriptome by merging data from untreated control (CK), cold (4°C) and drought (PEG2000, 20%) treated plant samples. A total of 251,145 transcripts (N50 = 1,269 bp) and 184,280 unigenes (N50 = 923 bp) were predicted, which code for homologs of near 47% of the published maize proteome. Under cold conditions, 2,232 and 817 genes were up-regulated and down-regulated, respectively, while fewer genes were up-regulated (532) and down-regulated (82) under drought stress, indicating that Zea mays ssp. mexicana L. is more sensitive to the applied cold rather than to the applied drought stresses. Functional enrichment analyses identified many common or specific biological processes and gene sets in response to drought and cold stresses. The ABA dependent pathway, trehalose synthetic pathway and the ICE1-CBF pathway were up-regulated by both stresses. GA associated genes have been shown to differentially regulate the responses to cold in close subspecies in Zea mays. These findings and the identified functional genes can provide useful clues for improving abiotic stress tolerance of maize.
Introduction
In order to survive in nature through evolution, plants developed multiple strategies to adjust to various abiotic stresses by promoting a series of physiological and metabolic processes such as stomatal closure, repression of cell growth and photosynthesis, and activation of respiration (Mishra et al., 2014). Stress induced damages are reduced or removed by improving sugar content, increasing antioxidants, taking mechanical action and inducing molecular chaperone chemicals in plants. The gene regulation networks involved are reviewed elsewhere (Zhu, 2016).
Low temperature and drought stresses are major environmental factors that impact on the geographical distribution and composition of plant species. They can also lead to a decrease in crop quality and productivity (Viswanathan and Zhu, 2002). A number of genes and alternative spliced isoforms responding to these stresses at the transcriptional level have been reported (Leyva et al., 1995; Shinozaki et al., 2003; Thatcher et al., 2016). Transcription factors make up of a large group which extensively involve in these processes (Zhu, 2016). Among them a class of DREB/CBF transcription factors which bind to the DRE/CRT element (A/GCCGAC) play roles in the regulation of the expression of target genes in response to cold and drought in Arabidopsis (Nakashima et al., 2009). DREB/CBF-like genes were significantly up-regulated by both stresses of cold and drought (Agarwal et al., 2006). AaDREB1 from Adonis amurensis, as an example, was capable to enhance tolerance to salt, drought, and low temperature in transgenic Arabidopsis and rice (Zong et al., 2016). Transcription factors including NAC, bZIP, MYB, and MYC also play important roles in cold and drought stresses. Overexpression of MlNAC5 (Miscanthus lutarioriparius), SlNAC1 (Suaeda liaotungensis K.), and VaCBF4 (Vitis amurensis) enhanced drought and cold stress tolerance of Arabidopsis, respectively (Li et al., 2013; Yang et al., 2015; Zong et al., 2016). MAPK related genes that are involved in signal transduction are significantly induced by both cold and drought (Pitzschke et al., 2009). A MAPK gene (GRMZM2G174170_T01) from maize, for example, was activated under cold and drought stresses and was suggested to contribute to stress tolerance in maize (Shan et al., 2013). The expression levels of most ZmVQ genes (41 out of 61 members) encoding VQ motif-containing proteins were changed by the drought stress, and half of ZmVQ genes were co-expressed with ZmWRKY genes in maize (Song et al., 2015).
Phytohormones such as abscisic acid (ABA), ethylene, cytokinin (CK), auxin (IAA), gibberellin (GA), and jasmonate (JA) play important roles in regulating plant growth and development and also in the responses to various biotic and abiotic stresses (Peleg and Blumwald, 2011). ABA synthesis is one of the fastest responses of plants to abiotic stress, triggering ABA-inducible gene expression and causing stomatal closure, thereby reducing water loss via transpiration and eventually restricting cellular growth. 9-cis-epoxycarotenoid dioxygenase (NCED) is a key enzyme in the ABA biosynthetic pathway in several plants which cleaves carotenoids to form the phytohormone ABA (Riahi et al., 2013). Cold or drought stress can induce ABA biosynthesis and exogenous ABA can improve cold or drought tolerance in tomato, Arabidopsis and tobacco (Thompson et al., 2000; Wan and Li, 2006; Zhang et al., 2008). In maize, thousands of genes are thought to be involved in abiotic stress. In particular, the gibberellin (GA) metabolic genes. Thus, understanding their expression changes could help to detect molecular mechanisms of the GA pathway under stress conditions (Colebrook et al., 2014). The response of GA metabolic genes to abiotic stress has been investigated and these genes participate in the CBF1-mediated stress-response pathway (Niu et al., 2014) and the GA20ox gene that is responsible for GA biosynthesis is down-regulated by cold treatment in Zea maize (Shan et al., 2013).
Zea mays ssp. mexicana L. is a member of teosintes and a close wild relative of cultivated maize (Almeida et al., 2011). Although teosintes have not yet been widely used in maize breeding, the high genetic diversity shows that Zea mays ssp. mexicana L. is a genetic reservoir for the improvement of agronomic characteristics of cultivated maize and teosintes (Wang et al., 2008). Since its introduction to China from Japan in 1979, the planting areas of teosinte have increased extensively in southern China, including Guangdong, Fujian, Guangxi, and Sichuan Provinces (Song et al., 2005). As an important forage or silage source, teosinte has a larger plant biomass, higher tiller number and better resistances to various stresses than cultivated maize (Niazi et al., 2015). Similar to maize, most lines of Zea mays ssp. mexicana L. originate from high altitude in northern and central Mexico and can adapt to acid soil with the best growing temperatures ranging from 25° to 35°C (Fukunaga et al., 2005). It is considered to be a very cold-sensitive crop, especially during the germination and early autotrophic growth stages, despite it having originated at altitudes of about 1,000–2,000 m (Hincha and Zuther, 2014).
RNA-seq (RNA sequencing) has become the most convenient and cost effective tool for understanding gene structure and quantitative transcriptome profiling. It does not depend on the existence of known genomic sequences, and shows a great ability for detecting differentially expressed genes with a broader dynamic range of expression levels (Wang et al., 2009). To discover genetic bases and molecular mechanisms in response to cold or drought in Zea mays ssp. mexicana L., the RNA-seq technique was employed in this study and the transcriptome of Zea mays ssp. mexicana L. was presented for the first time. In total, 414,232,462 high quality clean reads were obtained by RNA-seq and were used for de novo assembly and annotation of genes from Zea mays ssp. mexicana L. Differentially expressed genes (DEGs) under cold or drought were also identified. These data will be valuable for the exploration of genetic and molecular mechanisms in response to stresses in Zea mays ssp. mexicana L. and provide gene resources for breeding programs.
Materials and Methods
Plant Materials and Growth Conditions
Plump seeds of Zea mays ssp. mexicana L. variety “8493” were used in this study. Seeds were washed 3 times with distilled water, then soaked in 75% ethanol and 2% sodium hypochlorite for 10 min and 3 min, respectively, and washed 3 times with distilled water before planting in plastic boxes (54 × 28 × 7 cm) containing soil substrates (Jiffy, Netherlands, http://www.jiffygroup.com/jiffy_product_category/substrates/). The boxes were transferred into a climate control box (RXZ 500-C, JIANGNAN Instrument), allowing the seeds to germinate and grow under a photoperiod of 10-h light/14-h dark and a humidity of 60% at 25°C. Thirteen-day-old seedlings were assigned into 3 groups for different treatments. For cold treatment (cold), seedlings were treated for 12 h at 4°C under 10-h light/14-h dark while for drought treatment (drought), seedlings were treated with Hoagland solution containing PEG2000 (20%) for 3 h under the same light condition. Seedlings of the control group (CK) grew under the same photoperiod and were assigned neither of the above treatments. After treatments, fresh tissues (roots, stems and leaves) were sampled and pooled, followed by quick-freezing with liquid nitrogen and storing at −80°C for further analysis. Two replicates for each treatment were applied and plant materials from at least three seedlings were pooled for each sample.
RNA Extraction
Total RNA was extracted using TRIzol® reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. RNA was treated with RNase-free DNase I (Takara, Japan) to remove any possible DNA. The integrity was checked by gel electrophoresis and the Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Concentration of total RNA was determined using NanoDrop 8000 spectrophotometer (NanoDrop, Wilmington, DE). Total RNA with RIN values ≥7.3 and 28S:18S ratios ≥1.2 were accepted for the following RNA-seq analysis.
RNA-seq Analysis
The Illumina HiSeq2500 platform was applied for RNA-seq analysis. To obtain a comprehensive overview of the Zea mays spp. mexicana L. transcriptome and the transcript profiles under cold and drought, six libraries were constructed and paired-end sequencing was carried out according to the manufacturer's instructions (Illumina, San Diego, CA). In brief, poly(A)-tailed mRNA was enriched using biotin-Oligo (dT) magnetic beads and fragmented into short fragments of 200–700 bp, followed by converting into double-stranded cDNA. The cDNA fragments were then purified with a QiaQuick PCR extraction kit and adapters were added to both ends of the short fragments. cDNA fragment pools were loaded to Illumina HiSeq2500 platform for sequencing. The generated raw data were processed for further analysis. Library construction and RNA-seq were carried out by Novogene Bioinformatics Technology Co. Ltd, China.
De novo Assembly and Sequence Clustering
The quality of raw data was controlled by using the FastQC tool (http://www.bioinformatics.babraham.ac.uk). Reads containing substantial unknown nucleotides (the proportion of N > 5%) and low-quality reads (>50% of the bases with a quality score ≤ 5) were discarded, and the first 10 bases of processed data were removed. The generated clean data from each sample were merged to perform de novo assembly using Trinity with default parameters and an optimized k-mer length of 25. A set of transcripts were generated and served as a reference transcriptome. Based on the shared sequence, transcripts from a gene were clustered into a subclass and the longest transcript was regarded as the unigene. The above processes were performed by Trinity software with default parameters (http://trinityrnaseq.sf.net, updated on 2014/07/17; Haas et al., 2013). To identify possible plant transcription factors (TFs), transcriptional regulators (TRs) and protein kinases (PKs), Plant Transcription factor and Protein Kinase Identifier and Classifier (iTAK) V1.5 was employed to analyse unigenes with the best match result.
Raw Illumina sequences and assembled sequences are available in the Gene Expression Omnibus (GEO) database of the National Center for Biotechnology Information (NCBI) (accession number: GSE76939).
Identification of Differentially Expressed Unigenes (DEGs)
The paired-reads from each sample were mapped to an assembled reference transcriptome by Bowtie software v1.1.1 with default parameters, and the number of mapped reads were calculated by RSEM (v 2.15) (Langmead et al., 2009; Li and Dewey, 2011). FPKM values were assigned to each unigene, representing normalized expression level by eliminating the influences from library construction and the length of genes (Trapnell et al., 2010). Unigenes with FPKM ≥ 1 were used for further analysis. Fold changes for each unigene under cold or drought condition were determined by comparing the FPKM value to that in control sample, and those unigenes with more than 2-fold change and adjusted P < 0.005 were identified as differentially expressed genes (DEGs) by DESeq 2 (V 1.2.10) package of the R statistical program (Love et al., 2014).
Annotation and Function Classification of Transcripts and Unigenes
Annotations were assigned to each unigene based on the top hit in BLASTX search against the protein databases, with the non-redundant (Nr) protein database at GeneBank (http://www.ncbi.nlm.nih.gov) as the highest priority, followed by Swiss-Prot (http://www.expasy.ch/sprot), KEGG (http://www.genome.jp/kegg) and eukaryotic KOG (http://www.ncbi.nlm.nih.gov/KOG) in that priority order. The significant thresholds of E-value were set at ≤ 10−6. CDS (5′–>3′) predictions of unigenes were confirmed with the priority result of BLAST searched protein.
The accession number and the GI code of the top hit at the BLASTX search against the Nr database were retrieved and ID mapping was performed to obtain GO annotations for the queried unigenes. To further annotate the unigenes, the Blast2GO (v2.5.0) program was employed to get GO annotations based on molecular function (MF), biological process (BP), and cellular component (CC) features (Conesa et al., 2005). GO enrichment analysis were carried out by AgriGO software with FDR < 0.05.
Quantitative Real-Time PCR Analysis
To further verify the expression profiles of genes in our Illumina RNA-seq data, total RNAs were extracted from three independent groups, which were different from those used for RNA-Seq. First-strand cDNA synthesis and qRT-PCR were carried out with the PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa) and TransStart® Tip Green qPCR SuperMix (Transgen), respectively. qRT-PCR was carried out in the BIO-RAD CFX96 sequence detection system according to the manufacturer's instructions. A total of 12 genes were randomly selected and gene-specific primers were designed with online Primer-blast of NCBI. The unigene annotated maize actin-2 was used as the endogenous control (GeneBank accession number NP_001146931.1). Each PCR reaction (20 μL) contained 10 μL 2 × Green qPCR SuperMix, 0.2 μM of each primer and appropriately diluted cDNA. The thermal cycling conditions were 94°C for 30 s, followed by 40 cycles of 5 s at 94°C and 30 s at 60°C then 72°C for 30 s. At the second dissociation stage, 95°C for 10 s, 65°–95°C with increment of 0.5°C for 0.05 s were used. All reactions were performed in triplicate, including the non-template controls. The relative expression level was calculated with the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Results
De novo Assembly and Functional Annotation of the Zea mays ssp. mexicana L. Transcriptome
To comprehensively understand the transcript profiles of Zea mays spp. mexicana L. under cold and drought, a total of six samples, including two biological repeats for each condition, were collected and two replicated runs of Illumine sequencing were performed for each sample. Raw reads of 153,219,582, 156,576,828 and 150,941,988 were generated from the control, cold and drought group, respectively. After quality check, the adapters, low-quality sequences and ambiguous reads were removed. A total of 136,233,336 (control), 141,732,126 (cold), and 136,267,000 (drought) clean paired-end reads, corresponding to 17.04 Gb (control), 17.72 Gb (cold), and 17.02 Gb (drought) clean bases, respectively, were used for future assembly. De novo assembly was carried out using the Trinity software and 251,145 transcripts and 184,280 unigenes with N50 values of 1,269 and 923 bp were obtained, respectively (Table 1). Approximately 47% of assembled transcripts and 35% of unigenes had a length of more than 500 bp (Supplemental File 1).
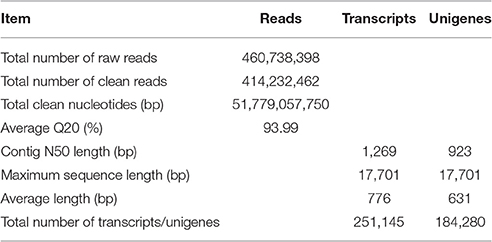
Table 1. Overview of the sequencing and assembly of Zea mays spp. mexicana L.
Annotations were assigned to each transcript and unigene by BlastX search against the plant protein collections of four public databases (NR, Swiss-Prot, KEGG, and KOG). There were124,297 (49.49%) transcripts and 68,177 (36.7%) unigenes that had at least one significant match (E < 1e-6) in one of these databases. Among all annotated transcripts/unigenes, more than 98.5% had at least one hit in the NR database at NCBI, which allowed retrieving GO annotations by the ID mapping method. A total of 28,382 (15.40%) unigenes were assigned to at least one GO term, among which 9769, 5404, and 13,209 unigenes were included into groups of “biological process,” “cellular component,” and “molecular function,” respectively (Figure 1). Among the “biological process” group, 9.96, 7.38, and 7.04% were annotated into “fatty acid biosynthetic process,” “pinoresinol biosynthetic process” and “(1–>3)-beta-D-glucan biosynthetic process,” respectively. In the “molecular function” group the subgroups of “nucleic acid binding” (9.68%), “RNA-directed DNA polymerase activity” (8.4%) and “zinc ion binding” (7.19%) ranked the highest. Additionally, 15.30, 7.49, and 7.48% of unigenes were annotated as cellular components of “integral component of membrane,” “cytoplasm,” and “ribosome,” respectively (Supplemental File 2).
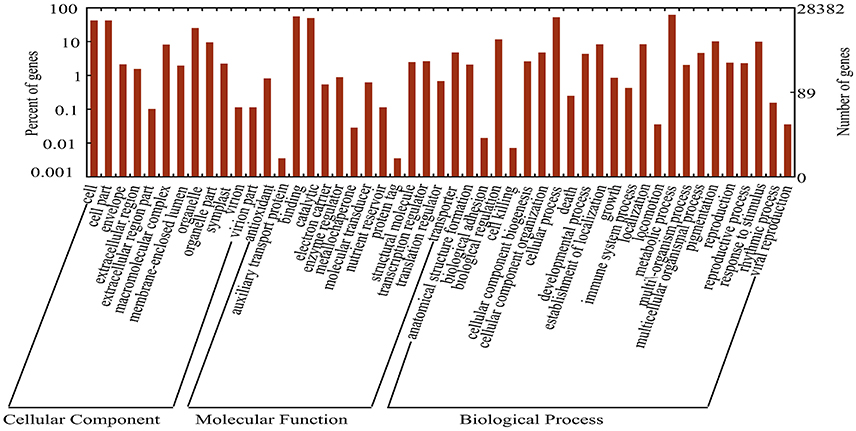
Figure 1. Functional classification of assembled unigenes. Functional classification of the assembled unigenes based on Gene Ontology (GO) categorization. GO annotations were assigned to unigenes based on the best BLAST hits, and a total of 98,947 GO terms were retrieved for 28,382 unigenes which were grouped into 49 categories. The left Y-axis represents the percentages of unigenes in each main category. The right Y-axis indicates the numbers of unigenes in each GO category.
Homologs were also identified by searching against the protein database of the most closed relative Zea mays L. and the model species, Arabidopsis thaliana. A total of 60,583 and 41,293 unigenes had at least one hit sequence from maize and Arabidopsis thaliana, respectively, with an E < 1e-6. After removing the replicate records, 27,379 and 15,643 homologs were obtained, representing 46.78 and 44.47% of the maize and Arabidopsis proteome, respectively (Supplemental File 3).
A total of 2,157 and 512 unigenes were identified as transcription factors (TF) or transcription regulators (TR) based on the Blast search against the Nr protein database at NCBI, covering 62 TF families and 25 TR families, respectively (Table 2). In addition, predicted protein kinases (PK) that play crucial roles during signal transduction and other biological processes were also obtained, including 1,598 unigenes covering 67 PK families (Table 3).
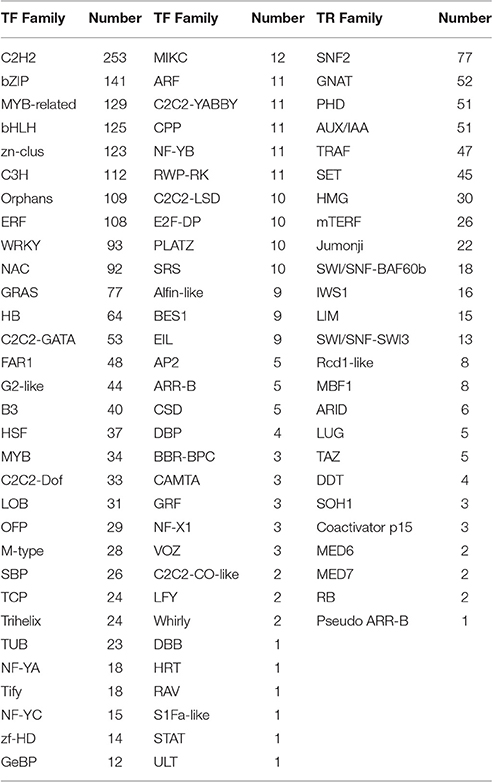
Table 2. Transcription factor (TF) and transcription regulators (TR) gene families identified from Zea mays spp. mexicana L.
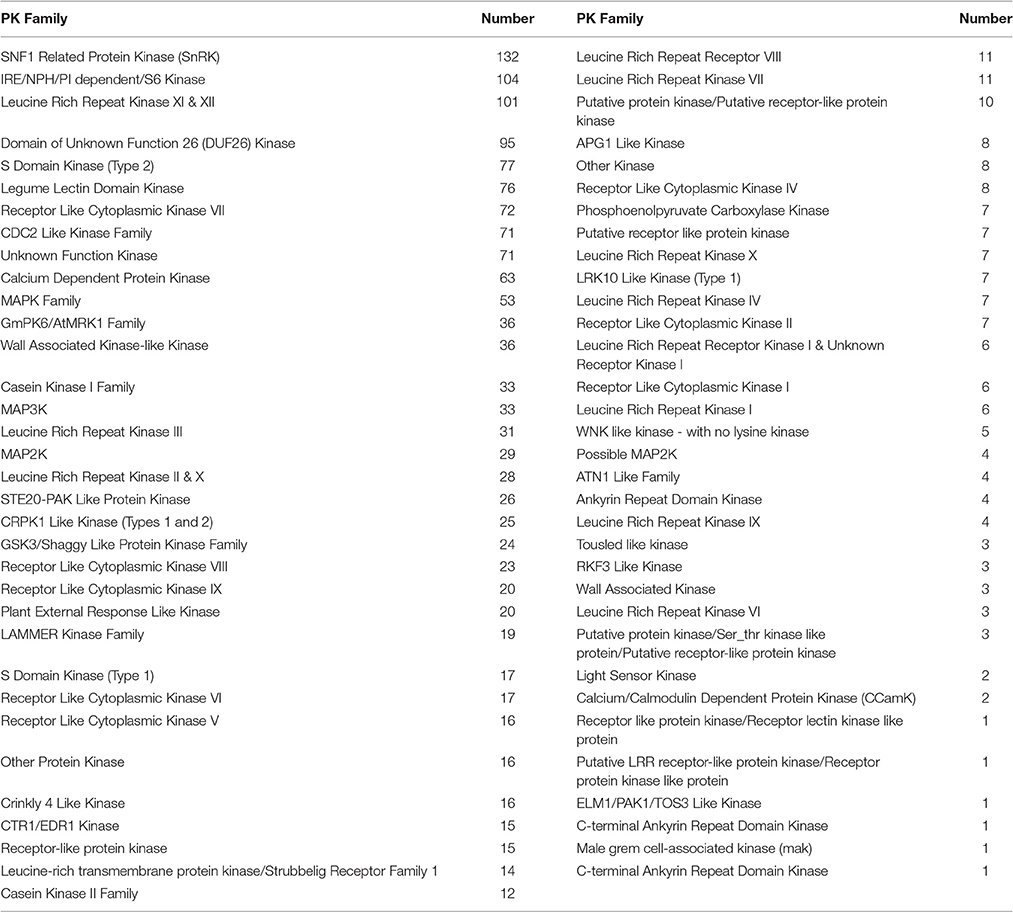
Table 3. Protein kinase (PK) gene families identified from the Zea mays ssp. mexicana L.
Expression Quantification of Unigenes and Identification of Differentially Expressed Genes (DEGs)
The expression levels of unigenes were evaluated using FPKM values. A total of 43,432 (23.57%), 42,849 (23.25%), 55,928 (30.35%), 48,945 (26.56%), 47,327 (25.68%), and 43,879 (23.81%) unigenes had FPKM ≥ 1 from the sample of control-1, control-2, cold-1, cold-2, drought-1, and drought-2, respectively. However, the percent of unigenes with FPKM ≥ 100 from each sample was less than 1%, ranging from 0.78 to 0.84% (Supplemental File 1). Compared with the control, the cold treatment at 4°C resulted in a larger number of genes with high FPKM values than did the drought treatment with PEG, indicating that the responses induced by cold was more dramatic than that by drought stress in Zea mays spp. mexicana L. at least with the relative levels of stress used in this trial.
To investigate differential responses of Zea mays spp. mexicana L. to cold or drought stress, data from replicated treatment groups were merged to perform pair-wise comparisons, and unigenes that matched the criteria of |log2Change|> = 2 and adjusted p < 0.005 were identified as differentially expressed genes (DEGs). Totally, 5,338 DEGs were obtained, with 3,049 and 614 genes differentially responding to cold and drought stresses, respectively. Among those cold-responsive DEGs, 2,232 were up-regulated and 817 were down-regulated (Figure 2C, Supplemental File 4). Notably, annotations for 41.1% of the up-regulated and 25.7% of the down-regulated DEGs were not retrieved from the public maize database (Supplemental File 4), suggesting some special regulation paths in Zea mays spp. mexicana L. For those DEGs which responded to drought, however, the number of DEGs was less, with only 532 and 82 genes being activated and inhibited by drought, respectively, of which 14.47 (77) and 4.2% (17) were unannotated (Supplemental File 4). The expression level of the whole set of DEGs in all six samples were also evaluated by plotting the Log10-transformed FPKM values in descending order (Figure 2A), and the distributions of DEGs' expression suggested activation was present in both cold- and drought-treated samples, with more dramatic transcriptional changes in the cold-treated samples. Taken together, these results indicated that Zea mays spp. mexicana L. had distinct gene networks in response to cold and drought.
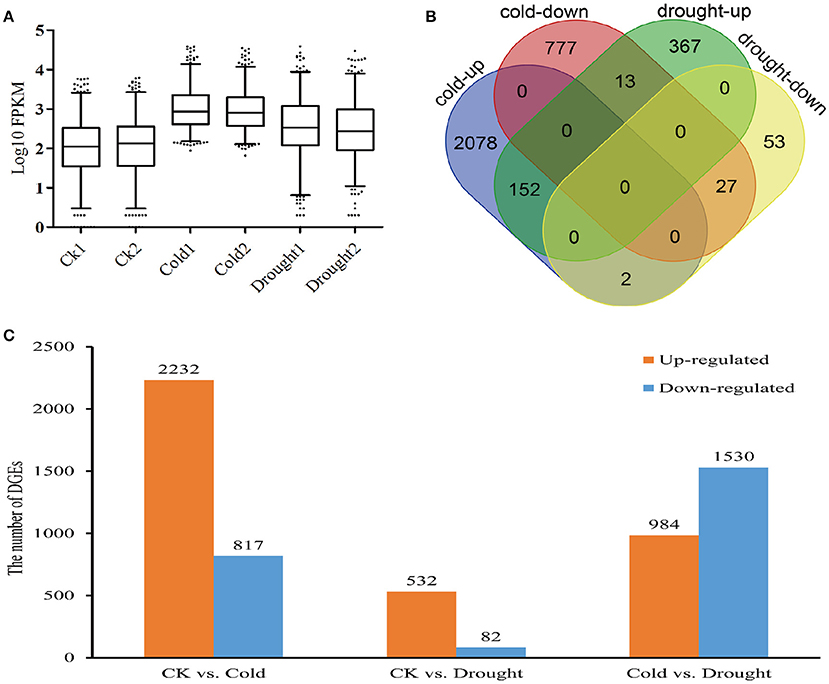
Figure 2. Gene expressions of differentially expressed genes (DEGs). (A) Distribution of Log10(read number) of DEGs in all tested samples. (B) Venn diagram for DEGs in response to cold and drought stress in Zea mays ssp. mexicana L. (C) The numbers of up- and down-regulated unigenes under cold and drought conditions.
Venn analysis indicated that 194 unigenes were regulated by both factors and 2,855 and 420 were specifically induced by either cold or drought (Figure 2B, Supplemental File 4).Of the 194 shared DEGs, 179 genes had similar response patterns to both cold and drought, including 152 up-regulated genes and 27 down-regulated genes. Two unigenes (c52482_g3 and c52482_g6) similar to protein phosphatase 2C3 and an unannotated unigene (c50928_g2) gave the greatest up-regulation, while the c29390_g1 (thioredoxin H4), c63024_g4 (hypothetical protein ZEAMMB73_373831) and an unannotated unigene (c55190_g2) comprised were the three most down-regulated. Fifteen genes showed the opposite response patterns under cold and drought stresses with 13 genes being down-regulated by cold but up-regulated by drought.
GO Enrichment of DEGs
GO enrichment analysis was performed to investigate over-represented biological processes or molecular functions involved in responses to cold or drought stress. A total of 45 and 89 GO terms were identified based on FDR < 0.05 as over-representations under cold and drought, respectively (Tables 4, 5 and Supplemental File 7).
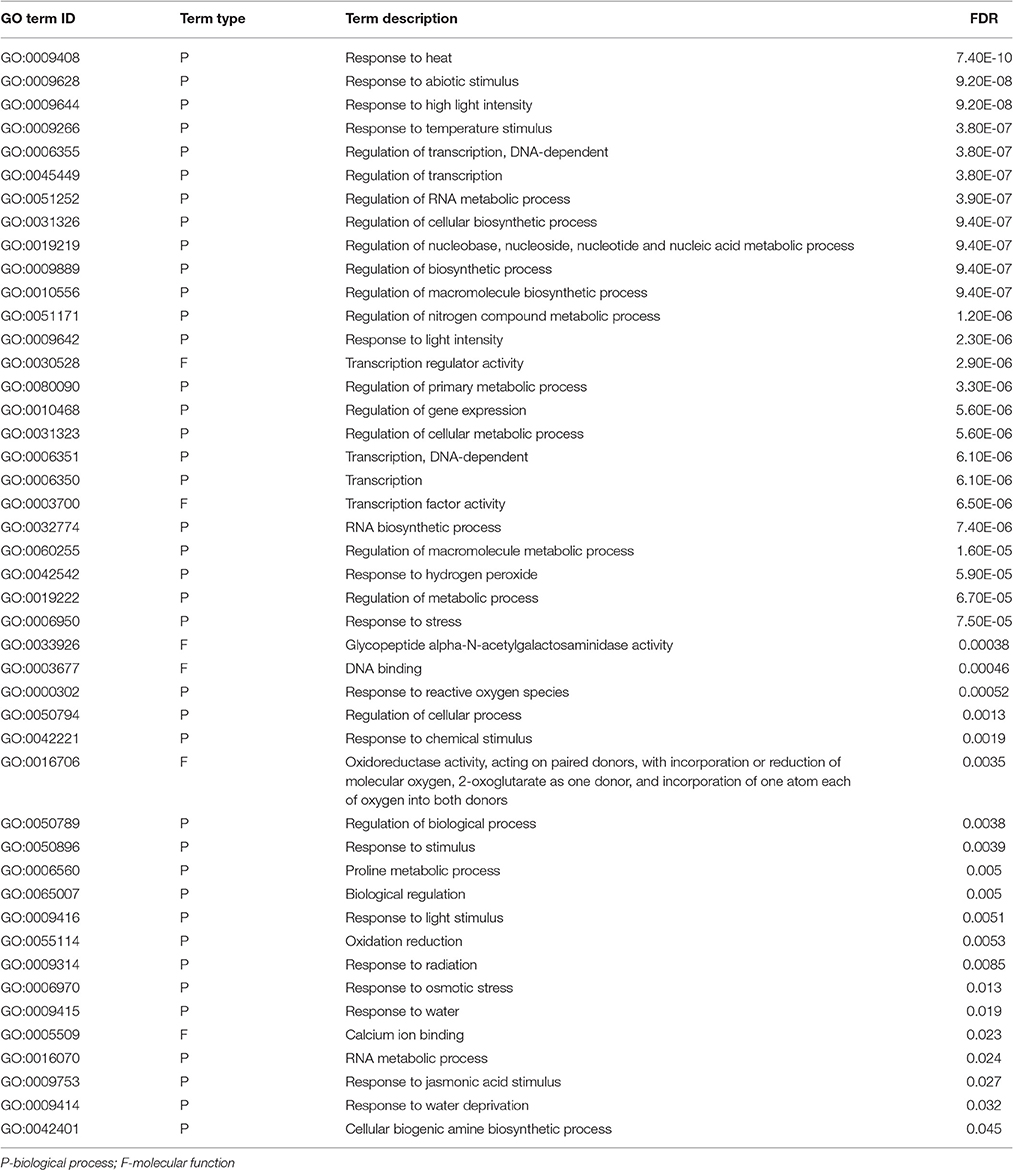
Table 4. Enriched GO terms corresponding to biological processes and molecular functions under cold stress.
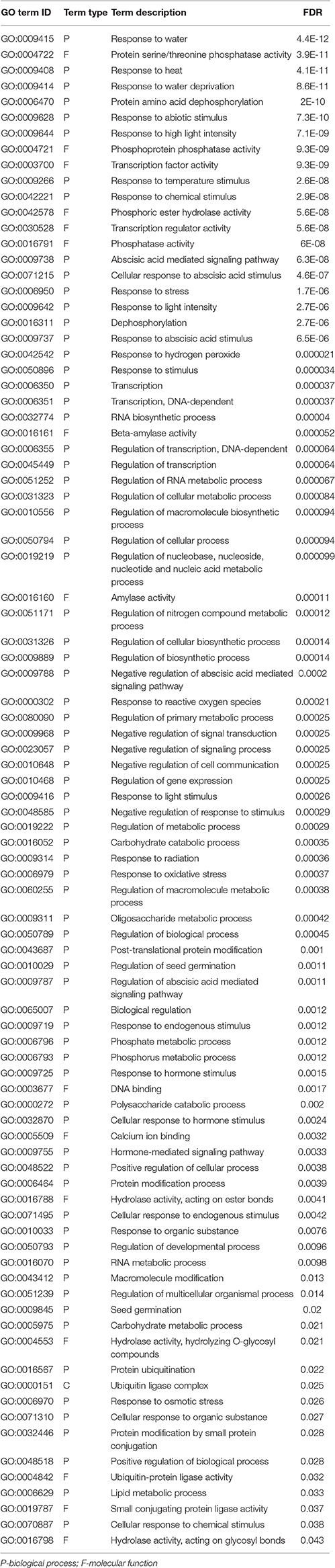
Table 5. Enriched GO terms corresponding to biological processes and molecular functions under drought stress.
Apparently, terms related to the response to abiotic stimuli, especially those to temperature and light, were significantly enriched in cold-treated Zea mays spp. mexicana L. seedlings. Genes for heat shock factor proteins (HSF) such as hsf1 (c59237_g1), hsp3 (c60986_g3), hsf4 (c46749_g1), hsf7 (c54684_g2), hsf26 (c46143_g1), hsp81 (c56395_g1), and hsp101 (c58173_g1) were down-regulated under cold stress. With respect to light responsive genes those for phosphatase 2C50 (c57868_g1) and chloroplastic-like stress response protein (c62415_g1) were up-regulated under cold stress. Additionally, terms related to the regulation of gene expression, mostly at the level of transcription, were over-represented. Several genes for encoding transcription factors, AP2/EREBP (c48755_g1 and c45827_g1), NAC (c60421_g1 and c47289_g2), bZIP (c61581_g3 and c59299_g1), WRKY (c59902_g1, c38115_g1 and c63357_g2 as examples) and ethylene-responsive transcription factor (c56755_g1 and c63899_g2), were up-regulated under cold stress. Together, these observations emphasized the roles of transcriptional regulation of cold-related responses in this species.
For the drought associated responses, however, there were a few differences. Under drought stress, DEGs were enriched for a total of 89 processes. Responses to stimuli, especially those to water deprivation, and regulation at the level of transcription were the most dramatic. Genes for the response to water deprivation were up-regulated under drought. These genes include dehydration-responsive element-binding protein 1D (c49699_g2 and c55715_g7), galactinol synthase gene (c60364_g1) and aquaporin PIP1-5 (c56750_g3 and c35748_g1). Genes involved in abscisic acid responses, such as EID1-like F-box protein (c57232_g1, c57232_g2 and c57232_g3), NAC domain transcription factor (c52045_g1), protein phosphatase 2C (c57868_g1, c50507_g1 and c60116_g4), and stachyose synthase (c55229_g2), were up-regulated under drought.
To further identify the differences between responses induced by cold and drought in Zea mays spp. mexicana L., a direct comparison was carried out between cold and drought-treated samples. A total of 2,514 assembled unigenes were identified by |log2change|> = 2 and adjusted p < 0.005 (Figure 2C). GO enrichment analysis for these DEGs indicated that the differences between cold and drought induced responses were in relation to abiotic stresses of temperature and water, transcriptional regulation and protein phosphorylation processes (Supplemental File 5). The FDR value in relation to the abscisic acid related signaling pathway was 0.011 which was very close to the criteria of 0.01 (Table 5), suggesting that this pathway was also a major difference between the two datasets. Together, the enrichment patterns identified by directly comparing responses to cold and drought were in agreement with the above results obtained from comparing each in turn with the untreated control.
qRT-PCR Verification
In order to validate the RNA-seq results, 12 DEGs with different expression patterns were selected for qRT-PCR analysis using gene-specific primers (Supplemental File 6). Among these DEGs, nine expressed under both cold and drought stresses, one regulated by cold only and two regulated by drought only were included. Eight of 10 cold-responsive DEGs and night of 11 drought-responsive DEGs were confirmed by qPCR (Figure 3).
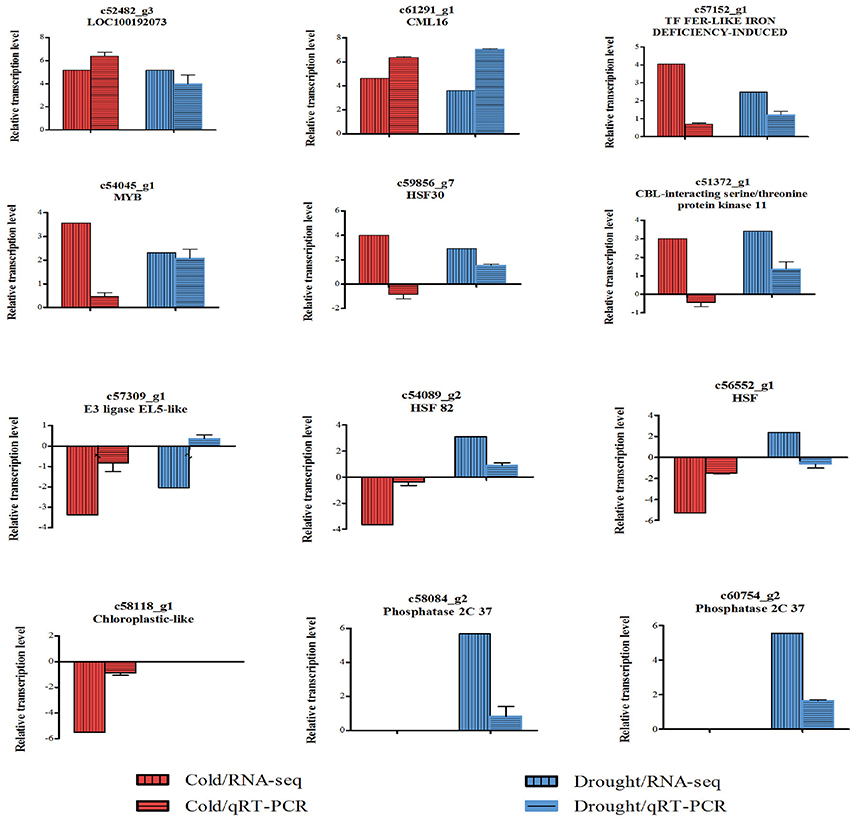
Figure 3. Verification for expressions of 12 selected DEGs. Relative expressions of the 12 genes in cold- or drought-treated Zea mays spp. mexicana L. seedlings were tested by qRT-PCR. Transcription levels were normalized to that of actin and compared to the untreated control. Values of fold change are showed in mean ± SEM. Three independent replicates were performed for each gene. Fold changes of these genes detected in RNA-Seq are also showed for reference. Relative transcription levels were calculated by log2−ΔΔCT method. The names of the closest putative orthologs are indicated for each gene, and the annotations are as following: c52482_g3, uncharacterized protein LOC_100192073; c61291_g1, calcium-binding protein CML16; c57152_g1, transcription factor FER-LIKE IRON DEFICIENCY-INDUCED; c54045_g1, MYB DNA-binding domain superfamily protein isoform X1; c59856_g7, heat shock factor protein HSF30; c51372_g1, CBL-interacting serine/threonine-protein kinase 11 isoform X1; c57309_g1, E3 ubiquitin-protein ligase EL5-like; c54089_g2, heat shock protein 82; c56552_g1, 17.0 kDa class II heat shock protein; c58118_g1, photosystem I reaction center subunit N, chloroplastic-like; c58084_g2, protein phosphatase 2C37; c60754_g2, homeobox-leucine zipper protein ATHB-6.
Discussion
RNA-Seq technology has been successfully used at the level of transcriptomes or whole genomes analysis in many plants and crops (Lu et al., 2010; Xia et al., 2011; Xu et al., 2012, 2014; Zhao et al., 2012; Garcia-Seco et al., 2015). Zea mays ssp. mexicana L. is a subspecies of Zea mays, and it has a genome size of 2.58 Gb (2n = 20) which is more than 20 times greater than the Arabidopsis thaliana (0.115 Gb) genome, 6 times greater than the rice (0.43 Gb) genome and equivalent to the Zea mays ssp. mays genome (2.73 Gb) (Hufford et al., 2012). The variety 8493, which serves as a representative of Zea mays spp. mexicana L., is reported to grow well under 25°C and can adapt to semi-arid and semi-humid environments (Huang et al., 1992). In this study, RNA-seq analyses were performed for de novo assembly of the transcriptome of Zea mays spp. mexicana L. and identification of the differentially expressed genes (DEGs) in response to cold or drought stress.
An Overview of the Zea mays ssp. mexicana L. Transcriptome
By pooling the clean reads from all samples, we obtained a limited transcriptome which provided a profile the transcript dynamics under cold and drought stresses. The transcriptome identified 184,280 unigenes, and 251,145 transcripts which Blast analysis divided in protein coding and other genes. When growing under cold (4°C) condition, the change of gene expression was dramatic at the global level of transcriptome (Figures 2A,C), indicating that cold induces an extensive activation of transcription. Moreover, GO analysis (Table 4) suggested there was an activation of gene networks involving in cold/heat-induced responses. The drought stress used here, however, induced fewer transcriptional changes, (only 15% as many) than cold (Figure 2C). These observations conformed the geographic distribution and the ideal growth conditions of Zea mays ssp. mexicana L. (Huang et al., 1992; Hufford et al., 2012). Similar observations have also been found in the elite maize inbred line Zheng 58 of Zea mays in which it was reported that the number of differentially expressed genes under drought stress was approximate 42% of that under cold (Shan et al., 2013), suggesting that the more sensitive response to cold rather than drought would be a conserved mechanism in Zea.
It is well known that gene expression in response to cold stress is somewhat different from that to drought stress (Shinozaki et al., 2003). In this study, only 194 DEGs are shared in stressed Zea mays ssp. mexicana L., (6.4 and 31.1% of cold- and drought-related DEGs, respectively). Among these, nearly 90% are regulated in a similar manner by both stresses, while only 15 DEGs are oppositely regulated (Supplemental File 4), indicating that there is a shared network to regulate the cold and drought induced responses in Zea mays ssp. mexicana L. On the other hand, specific regulations in response to cold or drought stress are also clearly visible in Zea mays ssp. mexicana L. The DEGs in response to cold are mostly enriched in processes of “Response to abiotic stimulus (GO:0009628),” especially the “Response to heat (GO:0009408),” and “Response to high light intensity (GO:0009644)” (Table 4), while the processes of “Response to water (GO:0009415),” “Response to water deprivation (GO:0009414),” and “protein amino acid dephosphorylation (GO:0004722)” are dominant in the collection of drought-induced DEGs (Table 5).
Transcriptional Regulatory Networks and Signaling Pathways involved in Cold and Drought Induced Responses in Zea mays ssp. mexicana L.
Transcription factors play crucial roles in the regulation of target gene expression by specifically binding to cis-acting elements in the promoter regions (Agarwal et al., 2006; Van Buskirk and Thomashow, 2006). In this study, WRKY, CBL, MYB, NAC, bHLH, bZIP, and AP2/EREBP families that are known to be involved in stress tolerance in plants were identified. Members of WRKY (c56800_g2 and c56800_g1), CBL (c51372_g1 and c49176_g1), MYB (c54045_g1), bHLH (c48928_g1), bZIP (c59299_g1), AP2/EREBP (c51992_g7 and c48755_g1) family are up-regulated by both stresses of cold and drought (Table 2), suggesting shared upstream pathways for signal transduction and regulation under these stimuli. From those down-regulated TF proteins, only one NAC (c57823_g1) protein is consistently down-regulated by both stresses, which is similar to the findings in Arabidopsis and rice (Lu et al., 2007; Takasaki et al., 2010) and suggests a negative regulation mechanism in Zea mays ssp. mexicana L. It is notable that some previously uncharacterized TFs are significantly up- or down-regulated in response to cold or drought only. For example, a member of TUB family (c61146_g4), was found in cold-induced DEGs set but not in the drought-induced set. This is similar to what has been observed in V. amurensis (Xu et al., 2014). In addition, some members in the ULT (c53036_g1), BIM2 (c64573_g2), and DBP (c51832_g3) families are found to be up-regulated by cold only (Table 2). The AP2/ERF transcription factor family comprises four major subfamilies, namely AP2, ERF, RAV, and dehydration-responsive element binding proteins (DREB/CBF) (Shinozaki et al., 2003). The roles of DREBs/CBFs in stresses induced response have been well established in numerous plants including A. thaliana (Ma et al., 2014), rice (Challam et al., 2015), maize (Liu et al., 2013), and soybean (Kidokoro et al., 2015). The DREB1s/CBFs can activate the downstream cold-responsive genes via specific binding to the DRE/CRT cis-acting element in their promoters, and expressions of these genes are positively correlated with cold tolerance (Mizoi et al., 2012). In Zea mays ssp. mexicana L., three DREB genes (c51992_g7, c49699_g1 and c49699_g2) were found to be significantly induced by both cold and drought stresses, suggesting that these proteins might functionally act in Zea mays ssp. mexicana L. during cold and drought stresses.
Signaling components such as kinases participate in numerous processes, including cell division, developmental programs, hormonal responses, and drought, salinity and reactive oxygen species (ROS) signaling responses (Wang et al., 2015). In particular, the MAPK cascade, serving as a signaling joint for various abiotic stress, positively regulates cold acclimation in plants (Chinnusamy et al., 2010). In Arabidopsis, the AtMEKK1/ANP1 (MAPKKK)-AtMKK2 (MAPKK)-AtMPK4/6 (MAPK) mitogen-activated protein kinase cascade is necessary for cold acclimation, and AtMKK2 (MAPKK) was identified as a key signal transducer. In addition, overexpression of AtMKK2 induces the cold responsive genes CBF2 and CBF3 and improves cold tolerance in Arabidopsis (Teige et al., 2004). In this study, one unigene coding for a MAP kinase (c14147_g1) and another one for a MAPKK protein (c62016_g1) were up-regulated by cold stress only, suggesting that the MAPK cascade may be involved in response to cold in Zea mays ssp. mexicana L.
ICE1-CBF pathway Genes Involved in Cold and Drought Stresses
Cold induced responses come down to different metabolic pathways, gene regulations and cell compartments. The ICE1-CBF-COR transcriptional cascade is a highly conserved module which is activated in response to cold stress (Zhu et al., 2007; Lissarre et al., 2010), and has been demonstrated to have essential roles in response to cold, drought, and other abiotic stresses (Zhou et al., 2011; Peng et al., 2015). ICE1 is a MYC-type transcription factor that binds to the MYC recognition cis-elements (CANNTG) in the promoter of CBF3/DREB1A protein to activate the expression of CBF3/DREB1A, which consequently regulates 40% of COR genes and 46% of cold-regulated transcription factor genes (Lee et al., 2005). In Arabidopsis, three CBF proteins, namely CBF1, 2, and 3, are induced by low temperature but not by abscisic acid (ABA) or dehydration (Medina et al., 1999). CBF1 and CBF3 can respond to cold at the same time, but the response of the CBF2 lags behind. CBF2 negatively regulates the expression of CBF1 and CBF3 during cold acclimation, while CBF1 and CBF3 are not involved in regulation of other CBF genes. CBF1/CBF3 have lower cold acclimation ability and play a subsidiary function in the CBF/DREB1 pathway and cold acclimation (Gilmour et al., 2004; Novillo et al., 2007; Kim et al., 2015). Differences in the functions of CBFs in mangrove Avicennia marina, European bilberry (Vaccinium myrtillus) and Brassica rapa have been observed. For example, CBF4 is regulated by drought and ABA but not by cold (Haake et al., 2002; Lee et al., 2012; Oakenfull et al., 2013; Peng et al., 2013). In this study, two putative ICE1 homologs (c60792_g1, c13919_g1) were identified in this subspecies. However, expressions of these genes were not affected by cold. A possible explanation is that the ICE1 homolog may be induced rapidly and transiently, and is attenuated in a short time, making it undetectable. In addition, CBF6 has been found to play a role in freezing tolerance in Festuca pratensis (Ergon et al., 2016). In our study, a CBF6 (c57018_g4) homologous gene was found to be up-regulated in response to cold and drought stresses, suggesting that this CBF6 homologous gene might play roles in stress resistance in Zea mays ssp. mexicana L.
Trehalose May be a Common Regulator of the Response to Cold and Drought Stresses in Zea mays ssp. mexicana L.
Trehalose is widely found in various organisms and is a non-reducing disaccharide formed by linking two glucose units with an a,a-1,1-glycosidic linkage (Wang et al., 2005). The synthetization of trehalose involves two-steps. The first step is the formation of trehalose-6-phosphate (T6P) from UDP-glucose and glucose-6-phosphate, which is catalyzed by trehalose-6-phosphate synthase (TPS). The second step is the generation of trehalose through dephosphorylation which is catalyzed by trehalose-6-phosphate phosphatase (TPP) (Zang et al., 2011). The genes coding for the TPS and TPP enzymes are thus important for the regulation of trehalose production (Ilhan et al., 2015). Transgenic plants harboring TPS or TPP genes have enhanced tolerance to abiotic stresses by stabilizing dehydrated enzymes, as well as by protecting protein and cellular membranes structures (Nuccio et al., 2015). In our study, a key trehalose-6-phosphate phosphatase (TPP) gene (c45624_g2) was also found to be up-regulated under both cold and drought stresses in Zea mays ssp. mexicana L. This is in agreement with the positive roles of trehalose in tolerating low temperature and drought stresses.
Phytohormone Dependent Pathways in Zea mays ssp. mexicana L.
Plant hormones play vital roles in adaption to stressful environments by regulating growth, development, nutrient allocation and source/sink transitions. Previous studies showed that different types of plant hormones, including abscisic acid (ABA), cytokinins (CKs), jasmonic acid (JA), gibberellins (GAs), brassinosteroids (BRs), ethylene (ETH), and auxin, have functions in regulating freezing tolerance by CBF-dependent or -independent pathways (Shi et al., 2015). ABA is the key phytohormone used in plants for regulation of the responses to abiotic stresses. As an example, the secondary hyperosmotic stress derived from drought or salt stress causes the accumulation of ABA which in turn rapidly elicits downstream responses in plants (Zhu, 2016). ABA accumulation can cause stomatal closure, thereby reducing water loss and eventually restrict cellular growth. At the molecular level, ABA regulates gene transcription, protein synthesis, signaling pathways, ion transport (and the transport of other organic molecules) and the production of important protectants against dehydration and photoinhibition (Riahi et al., 2013). Cold stress is also reported to induce ABA biosynthesis, and the exogenous application of ABA improves the cold tolerance of plants (De Zelicourt et al., 2016). ABA biosynthesis is one of the important steps used by plants in responding to stresses via the ABA-dependent pathway. The 9-cis epoxycarotenoid dioxygenase 1 (NCED1) is regarded as a key rate-limiting enzyme in ABA biosynthesis. It was originally identified from maize viviparous 14 mutants, and is responsible for cleavage of the ABA precursor C40-cis-epoxycarotenoids, to either 9-cis-neoxanthin, 9-cis-violaxanthin or both to produce xanthoxin, the direct C15 precursor of ABA (Raghavendra et al., 2010; Boursiac et al., 2013). In Zea mays ssp. mexicana L., a homolog of NCED1 (c63716_g5) gene was identified. It was up-regulated by drought (Supplemental File 4), which is consistent with results reported previously in Arabidopsis (Behnam et al., 2013) and Phaseolus vulgaris L. (Qin and Jan, 1999), suggesting conserved regulation of this drought induced response in Zea mays ssp. mexicana L. It is notable that this NCED1 gene was also activated by cold stress (Supplemental File 4), which is similar to recent observations for OsNCED1 and OsNCED3 (Maruyama et al., 2014), suggesting that the ABA-dependent pathway plays roles in response to cold and drought in Zea mays ssp. mexicana L.
PP2C proteins are also reported to be involved in ABA-dependent regulations of stress induced responses, such as the 76 phosphatase 2C (PP2C-type) proteins which have been identified from Arabidopsis. Most of these (PP2C-type) proteins fall into 10 groups (A–J) (Schweighofer et al., 2004). ABA signaling can be mediated by type 2C protein phosphatases (PP2Cs) such as HAB1 and ABI2, which inhibit stress-activated SnRK2 kinases (Nakashima et al., 2009). When directly comparing the expression levels under cold with those under drought conditions in Zea mays ssp. mexicana L., four candidate PP2C genes, including c52482_g3, c52482_g6, c62830_g2, and c57868_g1, showed up-regulated patterns. A putative protein kinase SnRK1 (c57228_g1), however, was down-regulated only under cold stress. These results taken together suggest that PP2C protein is involved in ABA-dependent signaling and affects the regulation of cold induced responses in Zea mays ssp. mexicana L.
Previous studies on Zea mays ssp. maize L. transcript profile under cold, salt, and drought stresses indicate that the abundance of bioactive GAs was reduced and that the negative regulatory factor DELLAs were accumulated (Shan et al., 2013), suggesting a negative effects of gibberellin in stress induced responses. This is in disagreement with what we observed in cold-stressed Zea mays ssp. mexicana L. Genes for the GAs receptor GID1L2 precursor (c47232_g1), the GA 2-beta-dioxygenase (c57017_g1, c33506_g1 and c57017_g2) and the GA20 oxidase (c63822_g4, c49254_g1 and c56760_g2) that are involved in GAs synthesis or signaling were up-regulated by cold, suggesting a positive role of gibberellin in this subspecies in response to cold stress. Thus, GAs could differentially regulate the responses to cold in these two close Zea mays subspecies. Future comparative study will provide better understanding of the molecular basis of the GA associated response to cold in Zea mays.
In summary, this study provided an overview of the transcriptome of Zea mays ssp. mexicana L., and the changes of transcript abundance in response to cold or drought stress. Less than a half of the reported Zea mays proteome have homologs in Zea mays ssp. mexicana L., indicating that specific regulation networks or mechanisms exist in these close relatives. DEGs responsive to cold or drought conditions serve as candidates for further study on tolerance related gene networks in Zea mays ssp. mexicana L. The effects of drought and cold reported here have arisen from a limited range of potential types and severities of stress. Thus, it should be noted that a greater range of treatments for (e.g., timing, severity, frequency, source) need to be examined in future studies to provide more clues for understanding the adaptation and tolerance mechanisms in this species.
Author Contributions
XL conducted the bulk of experiments, data analysis, and wrote the paper draft. XZ and YC participated in the treatments of experimental materials. MZ and DM contributed to data interpretation and writing. CY and SL were responsible for experimental design, data interpretation and writing.
Funding
This research was supported by the Guangdong Science and Technology Department (2015A020209155) and the National Natural Science Foundation of China (U1201212).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the technical support of Qi Zhao and Yue-yuan Zheng from Sun Yat-Sen University, Hao-ran Shu from Guangzhou Magigen Biotechnology Corporation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00136/full#supplementary-material
Supplemental File 1. Distribution of the length of transcripts and unigenes.
Supplemental File 2. GO classification of annotated unigenes.
Supplemental File 3. Blast against maize and Arabidopsis protein database.
Supplemental File 4. Differentially expressed genes in different samples.
Supplemental File 5. GO enrichment analysis by direct comparing cold with drought stresses.
Supplemental File 6. Primers used in qRT-PCR.
Supplemental File 7. GO enrichment analysis and classify of DEGs.
References
Agarwal, P. K., Agarwal, P., Reddy, M., and Sopory, S. K. (2006). Role of DREB transcription factors in abiotic and biotic stress tolerance in plants. Plant Cell Rep. 25, 1263–1274. doi: 10.1007/s00299-006-0204-8
Almeida, C., Amorim, E. P., Barbosa Neto, J. F., Cardoso Filho, J. A., and Sereno, M. J. C. D.M. (2011). Genetic variability in populations of sweet corn, common corn and teosinte. Crop Breed. Appl. Biotechnol. 11, 64–69. doi: 10.1590/S1984-70332011000100009
Behnam, B., Fujita, M., Takasaki, H., Osakabe, Y., Shinozaki, K., Iuchi, S., et al. (2013). Characterization of the promoter region of an arabidopsis gene for 9-cis-epoxycarotenoid dioxygenase involved in dehydration-inducible transcription. DNA Res. 20, 315–324. doi: 10.1093/dnares/dst012
Boursiac, Y., Léran, S., Corratgé-Faillie, C., Gojon, A., Krouk, G., and Lacombe, B. (2013). Review: ABA transport and transporters. Trends Plant Sci. 18, 325–333. doi: 10.1016/j.tplants.2013.01.007
Challam, C., Ghosh, T., Rai, M., and Tyagi, W. (2015). Allele mining across DREB1A and DREB1B in diverse rice genotypes suggest a highly conserved pathway inducible by low temperature. J. Genet. 231–238. doi: 10.1007/s12041-015-0507-z
Chinnusamy, V., Zhu, J.-K., and Sunkar, R. (2010). Gene regulation during cold stress acclimation in plants. Methods Mol. Biol. 639, 39–55. doi: 10.1007/978-1-60761-702-0_3
Colebrook, E. H., Thomas, S. G., Phillips, A. L., and Hedden, P. (2014). The role of gibberellin signalling in plant responses to abiotic stress. J. Exp. Biol. 217, 67–75. doi: 10.1242/jeb.089938
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
De Zelicourt, A., Colcombet, J., and Hirt, H. (2016). Review: the role of MAPK Modules and ABA during Abiotic Stress Signaling. Trends Plant Sci. 21, 677–685. doi: 10.1016/j.tplants.2016.04.004
Ergon, Å., Melby, T. I., Rognli, O. A., and Höglind, M. (2016). Vernalization requirement and the Chromosomal VRN1-region can affect freezing tolerance and expression of cold-regulated genes in Festuca pratensis. Front. Plant Sci. 7, 207–221. doi: 10.3389/fpls.2016.00207
Fukunaga, K., Hill, J., Vigouroux, Y., Matsuoka, Y., Sanchez, J., Liu, K., et al. (2005). Genetic diversity and population structure of teosinte. Genetics 169, 2241–2254. doi: 10.1534/genetics.104.031393
Garcia-Seco, D., Zhang, Y., Gutierrez-Ma-ero, F., Martin, C., and Ramos-Solano, B. (2015). RNA-Seq analysis and transcriptome assembly for blackberry (Rubus sp. Var. Lochness) fruit. BMC Genomics 16:5. doi: 10.1186/s12864-014-1198-1
Gilmour, S. J., Fowler, S. G., and Thomashow, M. F. (2004). Arabidopsis transcriptional activators CBF1, CBF2, and CBF3 have matching functional activities. Plant Mol. Biol. 54, 767–781. doi: 10.1023/B:PLAN.0000040902.06881.d4
Haake, V., Cook, D., Riechmann, J., Pineda, O., Thomashow, M. F., and Zhang, J. Z. (2002). Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol. 130, 639–648. doi: 10.1104/pp.006478
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hincha, D., and Zuther, E. (2014). “Introduction: Plant Cold Acclimation and Freezing Tolerance,” in Plant Cold Acclimation, eds D. K. Hincha and E. Zuther (New York, NY: Springer), 1–6.
Huang, Z. C., Liang, C. Y., Peng, J. C., and Shi, A. J. (1992). “8493” Green forage maize hybrid breeding and application. China Herb. Sci. 29, 8–9.
Hufford, M. B., Bilinski, P., Pyhäjärvi, T., and Ross-Ibarra, J. (2012). Teosinte as a model system for population and ecological genomics. Trends Genet. 28, 606–615. doi: 10.1016/j.tig.2012.08.004
Ilhan, S., Ozdemir, F., and Bor, M. (2015). Contribution of trehalose biosynthetic pathway to drought stress tolerance of Capparis ovata Desf. Plant Biol. 17, 402–407. doi: 10.1111/plb.12227
Kidokoro, S., Watanabe, K., Ohori, T., Moriwaki, T., Mizoi, J., Sekita, S., et al. (2015). Soybean DREB1/CBF-type transcription factors function in heat and drought as well as cold stress-responsive gene expression. Plant J. 81, 505–518. doi: 10.1111/tpj.12746
Kim, Y. S., Lee, M., Lee, J. H., Lee, H. J., and Park, C. M. (2015). The unified ICE–CBF pathway provides a transcriptional feedback control of freezing tolerance during cold acclimation in Arabidopsis. Plant Mol. Biol. 89, 187–201. doi: 10.1007/s11103-015-0365-3
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. doi: 10.1186/gb-2009-10-3-r25
Lee, B.-H., Henderson, D. A., and Zhu, J.-K. (2005). The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17, 3155–3175. doi: 10.1105/tpc.105.035568
Lee, S. C., Lim, M. H., Yu, J. G., Park, B. S., and Yang, T. J. (2012). Genome-wide characterization of the CBF/DREB1 gene family in Brassica rapa. Plant Physiol. Biochem. 61, 142–152. doi: 10.1016/j.plaphy.2012.09.016
Leyva, A., Jarillo, J. A., Salinas, J., and Martinez-Zapater, J. M. (1995). Low temperature induces the accumulation of phenylalanine ammonia-lyase and chalcone synthase mRNAs of Arabidopsis thaliana in a light-dependent manner. Plant Physiol. 108, 39–46. doi: 10.1104/pp.108.1.39
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformat. 12:323. doi: 10.1186/1471-2105-12-323
Li, J., Wang, N., Xin, H., and Li, S. (2013). Overexpression of VaCBF4, a transcription factor from Vitis amurensis, improves cold tolerance accompanying increased resistance to drought and salinity in Arabidopsis. Plant Mol. Biol. Report. 31, 1518–1528. doi: 10.1007/s11105-013-0627-7
Lissarre, M., Ohta, M., Sato, A., and Miura, K. (2010). Cold-responsive gene regulation during cold acclimation in plants. Plant Signal. Beh. 5, 948–952. doi: 10.4161/psb.5.8.12135
Liu, S., Wang, X., Wang, H., Xin, H., Yang, X., Yan, J., et al. (2013). Genome-wide analysis of ZmDREB genes and their association with natural variation in drought tolerance at seedling stage of Zea mays L. PLoS Genet. 9, 1. doi: 10.1371/journal.pgen.1003790
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 15, 1–21. doi: 10.1186/s13059-014-0550-8
Lu, P.-L., Chen, N.-Z., An, R., Su, Z., Qi, B.-S., Ren, F., et al. (2007). A novel drought-inducible gene, ATAF1, encodes a NAC family protein that negatively regulates the expression of stress-responsive genes in Arabidopsis. Plant Mol. Biol. 63, 289–305. doi: 10.1007/s11103-006-9089-8
Lu, T., Lu, G., Fan, D., Zhu, C., Li, W., Zhao, Q., et al. (2010). Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 20, 1238–1249. doi: 10.1101/gr.106120.110
Ma, J. T., Wang, Z. L., Yin, C. C., Guo, Q. Q., Zhou, M. L., and Wu, Y. M. (2014). A novel DREB transcription factor from Halimodendron halodendron leads to enhance drought and salt tolerance in Arabidopsis. Biol. Plant. 59, 74–82. doi: 10.1007/s10535-014-0467-9
Maruyama, K., Urano, K., Yoshiwara, K., Morishita, Y., Sakurai, N., Suzuki, H., et al. (2014). Integrated analysis of the effects of cold and dehydration on rice metabolites, phytohormones, and gene transcripts. Plant Physiol. 164, 1759–1771. doi: 10.1104/pp.113.231720
Medina, J. N., Bargues, M., Terol, J., Pérez-Alonso, M., and Salinas, J. (1999). The Arabidopsis CBF gene family is composed of three genes encoding AP2 domain-containing proteins whose expression is regulated by low temperature but not by abscisic acid or dehydration. Plant Physiol. 119, 463–470. doi: 10.1104/pp.119.2.463
Mishra, A., Heyer, A. G., and Mishra, K. B. (2014). Chlorophyll fluorescence emission can screen cold tolerance of cold acclimated Arabidopsis thaliana accessions. Plant Methods 10, 1–10. doi: 10.1186/1746-4811-10-38
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Nakashima, K., Fujita, Y., Kanamori, N., Katagiri, T., Umezawa, T., Kidokoro, S., et al. (2009). Three Arabidopsis SnRK2 protein kinases, SRK2D/SnRK2. 2, SRK2E/SnRK2. 6/OST1 and SRK2I/SnRK2. 3, involved in ABA signaling are essential for the control of seed development and dormancy. Plant Cell Physiol. 50, 1345–1363. doi: 10.1093/pcp/pcp083
Niazi, I. A. K., Rauf, S., Teixeira Da Silva, J. A., and Munir, H. (2015). Comparison of teosinte (Zea mexicana L.) and inter-subspecific hybrids (Zea mays L. × Zea mexicana) for high forage yield under two sowing regimes. Crop Past. Sci. 66, 49–61. doi: 10.1071/cp14155
Niu, S. H., Gao, Q., Li, Z. X., Chen, X. Y., and Li, W. (2014). The Role of Gibberellin in the CBF1-Mediated stress-response pathway. Plant Mol. Biol. Report. 4, 852–863. doi: 10.1007/s11105-013-0693-x
Novillo, F., Medina, J., and Salinas, J. (2007). Arabidopsis CBF1 and CBF3 have a different function than CBF2 in cold acclimation and define different gene classes in the CBF regulon. Proc. Natl. Acad. Sci. U.S.A. 104, 21002–21007. doi: 10.1073/pnas.0705639105
Nuccio, M. L., Zhou, H. P., Chen, X., Gao, Y., Haque, E., Basu, S. S., et al. (2015). Expression of trehalose-6-phosphate phosphatase in maize ears improves yield in well-watered and drought conditions. Nat. Biotechnol. 33, 862–869. doi: 10.1038/nbt.3277
Oakenfull, R. J., Baxter, R., and Knight, M. R. (2013). A C-repeat binding factor transcriptional activator (CBF/DREB1) from European bilberry (Vaccinium myrtillus) induces freezing tolerance when expressed in Arabidopsis thaliana. PLoS ONE 8:e54119. doi: 10.1371/journal.pone.0054119
Peleg, Z., and Blumwald, E. (2011). Hormone balance and abiotic stress tolerance in crop plants. Curr. Opin. Plant Biol. 14, 290–295. doi: 10.1016/j.pbi.2011.02.001
Peng, Y.-L., Wang, Y.-S., Cheng, H., Sun, C.-C., Wu, P., Wang, L.-Y., et al. (2013). Characterization and expression analysis of three CBF/DREB1 transcriptional factor genes from mangrove Avicennia marina. Aquat. Toxicol. 140, 68–76. doi: 10.1016/j.aquatox.2013.05.014
Peng, Y. L., Wang, Y. S., Cheng, H., and Wang, L. Y. (2015). Characterization and expression analysis of a gene encoding CBF/DREB1 transcription factor from mangrove Aegiceras corniculatum. Ecotoxicology 24, 1733–1743. doi: 10.1007/s10646-015-1485-x
Pitzschke, A., Schikora, A., and Hirt, H. (2009). MAPK cascade signalling networks in plant defence. Curr. Opin. Plant Biol. 12, 421–426. doi: 10.1016/j.pbi.2009.06.008
Qin, X., and Jan, A. D. Z. (1999). The 9-Cis-Epoxycarotenoid cleavage reaction is the key regulatory step of Abscisic acid biosynthesis in water-stressed bean. Plant Biol. 96, 15354–15361. doi: 10.1073/pnas.96.26.15354
Raghavendra, A. S., Gonugunta, V. K., Christmann, A., and Grill, E. (2010). ABA perception and signalling. Trends Plant Sci. 15, 395–401. doi: 10.1016/j.tplants.2010.04.006
Riahi, L., Zoghlami, N., Dereeper, A., Laucou, V., Mliki, A., and This, P. (2013). Molecular characterization and evolutionary pattern of the 9- cis -epoxycarotenoid dioxygenase NCED1 gene in grapevine. Mol. Breed. 32, 253–266. doi: 10.1007/s11032-013-9866-4
Schweighofer, A., Hirt, H., and Meskiene, I. (2004). Plant PP2C phosphatases: emerging functions in stress signaling. Trends Plant Sci. 9, 236–243. doi: 10.1016/j.tplants.2004.03.007
Shan, X., Li, Y., Jiang, Y., Jiang, Z., Hao, W., and Yuan, Y. (2013). Transcriptome profile analysis of maize seedlings in response to high-salinity, drought and cold stresses by deep sequencing. Plant Mol. Biol. Report. 31, 1485–1491. doi: 10.1007/s11105-013-0622-z
Shi, Y., Ding, Y., and Yang, S. (2015). Cold signal transduction and its interplay with phytohormones during cold acclimation. Plant Cell Physiol. 56, 7–15. doi: 10.1093/pcp/pcu115
Shinozaki, K., Yamaguchi-Shinozaki, K., and Seki, M. (2003). Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 6, 410–417. doi: 10.1016/S1369-5266(03)00092-X
Song, J.-C., Fan, L., Qiao, Y.-K., Fen, M.-S., Gao, S.-L., Yang, Z.-Z., et al. (2005). Research of the growth characters and nutritional contents of feed Euchlaena mexicana schred. Pratacult. Sci. 22, 53–56.
Song, W., Zhao, H., Zhang, X., Lei, L., and Lai, J. (2015). Genome-wide identification of VQ motif-containing proteins and their expression profiles under abiotic stresses in Maize. Front. Plant Sci. 6:1177. doi: 10.3389/fpls.2015.01177
Takasaki, H., Maruyama, K., Kidokoro, S., Ito, Y., Fujita, Y., Shinozaki, K., et al. (2010). The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol.Genet. Genomics 284, 173–183. doi: 10.1007/s00438-010-0557-0
Teige, M., Scheikl, E., Eulgem, T., Doczi, F., Ichimura, K., and Shinozaki, K. (2004). The MKK2 pathway mediates cold and salt stress signaling in Arabidopsis. Mol. Cell 15, 141–152. doi: 10.1016/j.molcel.2004.06.023
Thatcher, S. R., Danilevskaya, O. N., Meng, X., Beatty, M., Zastrow-Hayes, G., Harris, C., et al. (2016). Genome-wide analysis of alternative splicing during development and drought stress in maize. Plant Physiol. 170, 586–599. doi: 10.1104/pp.15.01267
Thompson, A. J., Jackson, A. C., Symonds, R. C., Mulholland, B. J., Dadswell, A. R., Blake, P. S., et al. (2000). Ectopic expression of a tomato 9-cis-epoxycarotenoid dioxygenase gene causes over-production of abscisic acid. Plant J. 23, 363–374. doi: 10.1046/j.1365-313x.2000.00789.x
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Van Buskirk, H. A., and Thomashow, M. F. (2006). Arabidopsis transcription factors regulating cold acclimation. Physiol. Plant. 126, 72–80. doi: 10.1111/j.1399-3054.2006.00625.x
Viswanathan, C., and Zhu, J.-K. (2002). Molecular genetic analysis of cold–regulated gene transcription. Philos. Trans. R. Soc. B Biol. Sci. 357, 877–886. doi: 10.1098/rstb.2002.1076
Wan, X.-R., and Li, L. (2006). Regulation of ABA level and water-stress tolerance of Arabidopsis by ectopic expression of a peanut 9-cis-epoxycarotenoid dioxygenase gene. Biochem. Biophys. Res. Commun. 347, 1030–1038. doi: 10.1016/j.bbrc.2006.07.026
Wang, J., Pan, C., Wang, Y., Ye, L., Wu, J., Chen, L., et al. (2015). Genome-wide identification of MAPK, MAPKK, and MAPKKK gene families and transcriptional profiling analysis during development and stress response in cucumber. BMC Genomics 16, 386. doi: 10.1186/s12864-015-1621-2
Wang, L., Yang, A., He, C., Qu, M., and Zhang, J. (2008). Creation of new maize germplasm using alien introgression from Zea mays ssp. mexicana. Euphytica 164, 789–801. doi: 10.1007/s10681-008-9730-5
Wang, Y.-J., Hao, Y.-J., Zhang, Z.-G., Chen, T., Zhang, J.-S., and Chen, S.-Y. (2005). Isolation of trehalose-6-phosphate phosphatase gene from tobacco and its functional analysis in yeast cells. J. Plant Physiol. 162, 215–223. doi: 10.1016/j.jplph.2004.06.014
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Xia, Z., Xu, H., Zhai, J., Li, D., Luo, H., He, C., et al. (2011). RNA-Seq analysis and de novo transcriptome assembly of Hevea brasiliensis. Plant Mol. Biol. 77, 299–308. doi: 10.1007/s11103-011-9811-z
Xu, D.-L., Long, H., Liang, J.-J., Zhang, J., Chen, X., Li, J.-L., et al. (2012). De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode. BMC Genomics 13:133. doi: 10.1186/1471-2164-13-133
Xu, W., Li, R., Zhang, N., Ma, F., Jiao, Y., and Wang, Z. (2014). Transcriptome profiling of Vitis amurensis, an extremely cold-tolerant Chinese wild Vitis species, reveals candidate genes and events that potentially connected to cold stress. Plant Mol. Biol. 86, 527–541. doi: 10.1007/s11103-014-0245-2
Yang, X., Wang, X., Ji, L., Yi, Z., Fu, C., Ran, J., et al. (2015). Overexpression of a Miscanthus lutarioriparius NAC gene MlNAC5 confers enhanced drought and cold tolerance in Arabidopsis. Plant Cell Rep. 34, 943–958. doi: 10.1007/s00299-015-1756-2
Zang, B., Deng, X. W., Li, H., Wang, X., and Li, W. (2011). Analysis of trehalose-6-phosphate synthase (TPS) gene family suggests the formation of TPS complexes in rice. Plant Mol. Biol. 76, 507–522. doi: 10.1007/s11103-011-9781-1
Zhang, Y., Yang, J., Lu, S., Cai, J., and Guo, Z. (2008). Overexpressing SgNCED1 in tobacco increases ABA level, antioxidant enzyme activities, and stress tolerance. J. Plant Growth Regul. 27, 151–158. doi: 10.1007/s00344-008-9041-z
Zhao, Z., Tan, L., Dang, C., Zhang, H., Wu, Q., and An, L. (2012). Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant, Chorispora bungeana. BMC Plant Biol. 12:222. doi: 10.1186/1471-2229-12-222
Zhou, M. Q., Shen, C., Wu, L. H., Tang, K. X., and Lin, J. (2011). CBF-dependent signaling pathway: a key responder to low temperature stress in plants. Crit. Rev. Biotechnol. 31, 186–192. doi: 10.3109/07388551.2010.505910
Zhu, H., Gao, W., Shi, Y. F., and Zhang, X. J. (2007). The CCAAT-binding factor CBF/NF-Y regulates the human acetylcholinesterase promoter activity during calcium ionophore A23187-induced cell apoptosis. Biochim. Biophys. Acta 1770, 1475–1482. doi: 10.1016/j.bbagen.2007.07.007
Zhu, J.-K. (2016). Review: abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Keywords: Zea mays ssp. mexicana L., cold tolerance, drought tolerance, transcriptome, differentially expressed genes
Citation: Lu X, Zhou X, Cao Y, Zhou M, McNeil D, Liang S and Yang C (2017) RNA-seq Analysis of Cold and Drought Responsive Transcriptomes of Zea mays ssp. mexicana L. Front. Plant Sci. 8:136. doi: 10.3389/fpls.2017.00136
Received: 18 November 2016; Accepted: 23 January 2017;
Published: 07 February 2017.
Edited by:
Bunyamin Tar'an, University of Saskatchewan, CanadaReviewed by:
Benedetto Ruperti, University of Padova, ItalyChuang Ma, Northwest Agricultural and Forestry University, China
Copyright © 2017 Lu, Zhou, Cao, Zhou, McNeil, Liang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shan Liang, bGlhbmdzaEBzY251LmVkdS5jbg==
Chengwei Yang, eWFuZ2Nod0BzY251LmVkdS5jbg==