 Zeeshan Nasim
Zeeshan Nasim Muhammad Fahim
Muhammad Fahim Ji Hoon Ahn
Ji Hoon Ahn- 1Creative Research Initiatives, Department of Life Sciences, Korea University, Seoul, South Korea
- 2Genetic Resources Conservation Lab, Institute of Biotechnology and Genetic Engineering, University of Agriculture, Peshawar, Pakistan
Eukaryotic cells use nonsense-mediated mRNA decay (NMD) to clear aberrant mRNAs from the cell, thus preventing the accumulation of truncated proteins. In Arabidopsis, two UP-Frameshift (UPF) proteins, UPF1 and UPF3, play a critical role in NMD. Although deficiency of UPF1 and UPF3 leads to various developmental defects, little is known about the mechanism underlying the regulation of flowering time by NMD. Here, we showed that the upf1-5 and upf3-1 mutants had a late-flowering phenotype under long-day conditions and the upf1-5 upf3-1 double mutants had an additive effect in delaying flowering time. RNA sequencing of the upf mutants revealed that UPF3 exerted a stronger effect than UPF1 in the UPF-mediated regulation of flowering time. Among genes known to regulate flowering time, FLOWERING LOCUS C (FLC) mRNA levels increased (up to 8-fold) in upf mutants, as confirmed by qPCR. The upf1-5, upf3-1, and upf1-5 upf3-1 mutants responded to vernalization, suggesting a role of FLC in delayed flowering of upf mutants. Consistent with the high FLC transcript levels and delayed flowering in upf mutants, levels of FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) mRNAs were reduced in the upf mutants. However, RNA-seq did not identify an aberrant FLC transcript containing a premature termination codon (PTC), suggesting that FLC is not a direct target in the regulation of flowering time by NMD. Among flowering time regulators that act in an FLC-dependent manner, we found that MAF3, NF-YA2, NF-YA5, and TAF14 showed increased transcript levels in upf mutants. We also found that BBX19 and ATC, which act in an FLC-independent manner, showed increased transcript levels in upf mutants. An aberrant transcript containing a PTC was identified from MAF3 and BBX19 and the levels of the aberrant transcripts increased in upf mutants. Taking these results together, we propose that the late-flowering phenotype of upf mutants is mediated by at least two different pathways, namely, by MAF3 in an FLC-dependent manner and by BBX19 in an FLC-independent manner.
Introduction
The survival of plant species largely depends on successful seed production via the formation of flowers; therefore, plants have evolved a complex mechanism to ensure successful reproduction (Huijser and Schmid, 2011), including a complicated genetic network that integrates endogenous and environmental cues to modulate the timing of the floral transition. At least 306 genes and eight genetic pathways affect flowering, including the photoperiod, autonomous, vernalization, ambient temperature, and gibberellic acid-dependent pathways (Bernier and Périlleux, 2005; Bouche et al., 2016).
FLOWERING LOCUS C (FLC) encodes a MADS-box transcription factor that binds to over 500 target sites in Arabidopsis and regulates genes involved in different developmental processes (Deng et al., 2011). FLC negatively regulates flowering (Michaels and Amasino, 1999) by repressing the expression of FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1; Searle et al., 2006), thereby preventing precocious flowering. FLC mRNA levels inversely correlate with the timing of flowering (Michaels and Amasino, 1999; Sheldon et al., 1999). The flc mutants show early flowering, whereas FLC overexpression causes very late flowering (Hepworth et al., 2002). Several pathways affect FLC expression, including the FRIGIDA (FRI) pathway, which activates FLC expression, the autonomous pathway, which negatively regulates FLC, and the vernalization pathway, which epigenetically silences FLC in response to prolonged cold. In vernalization, the expression of FLC is silenced through histone methylation and chromatin modification (Bastow et al., 2004; Michaels, 2009). FLC expression is regulated by various classes of proteins. For example, MADS AFFECTING FLOWERING 3 (MAF3), a close homolog of FLC, directly interacts with FLC (Gu et al., 2013) and negatively regulates flowering at low temperatures (Suter et al., 2014). MAF3 also represses FT and SOC1 expression through direct binding to their genomic loci (Gu et al., 2013). FLC is positively regulated by the members of the NUCLEAR FACTOR Y family, including NF-YA2 and NF-YA5 (Xu et al., 2013), which encode CCAAT-binding transcription factors.
In the regulation of flowering, important downstream targets of FLC include FT and SOC1. FT is considered the long-sought florigen (Zeevaart, 2008) and promotes the floral transition by directly interacting with FD to up-regulate the flower meristem identity gene APETALA1 (AP1) to specify the fate of floral cells (Abe et al., 2005). SOC1 is a floral activator that encodes a conserved MADS box protein (Lee et al., 2000). SOC1 mainly regulates LEAFY (LFY), another flower meristem identify gene, to induce floral initiation (Lee et al., 2000). The ft and soc1 mutants exhibit a delayed flowering phenotype (Kobayashi et al., 1999; Borner et al., 2000), indicating their importance in the determination of the timing of flowering. Among several regulators that regulate FT expression, B-BOX DOMAIN PROTEIN 19 (BBX19) indirectly regulates FT expression by interacting with CONSTANS (CO) and preventing CO from inducing FT expression (Wang et al., 2014). Although bbx19 mutation causes very weak early flowering, overexpression of BBX19 causes a delayed flowering phenotype (Wang et al., 2014), suggesting that BBX19 acts as a floral repressor. Arabidopsis thaliana CENTRORADIALIS homolog (ATC) is a floral regulator that represses flowering upon overexpression (Mimida et al., 2001). ATC and FT can interact with FD and affect the expression of AP1 (Huang et al., 2012). ATC overexpression causes late flowering in plants grown under both long day (LD) and short day (SD) conditions (Mimida et al., 2001). However, atc mutants exhibit an early flowering phenotype only under SD conditions (Mimida et al., 2001; Huang et al., 2012), suggesting the enhanced ability of ATC to compete with FT for FD interaction, when FT expression levels are low.
Alternative splicing is an important mechanism for controlling gene expression, since it adds proteomic complexity from the limited number of genes encoded in the genome. However, alternative splicing can generate a wide variety of unproductive isoforms carrying a premature termination codon (PTC; Filichkin and Mockler, 2012). To avoid accumulation of potentially harmful truncated proteins, RNA surveillance mechanisms clear these isoforms from the cell (Hori and Watanabe, 2005; Kertesz et al., 2006; Yoine et al., 2006; Riehs et al., 2008; Kurihara et al., 2009; Dai et al., 2016). One of these mechanisms, nonsense-mediated mRNA decay (NMD) clears PTC-containing aberrant transcripts from cells in eukaryotes (Maquat, 2005). About 1–10% of genes are regulated by NMD in different organisms such as yeast, flies, mammals, and plants (He et al., 2003; Mendell et al., 2004; Rehwinkel et al., 2005; Kurihara et al., 2009). In plants, the NMD is triggered by the presence of PTCs in a transcript, long 3′ untranslated regions, or intron-containing 3′ untranslated regions (Kertesz et al., 2006; Kerényi et al., 2008; Nyikó et al., 2013). A study of 270 Arabidopsis genes revealed that among these NMD-triggering features in plants, the presence of a PTC is the most frequent target of NMD (Kalyna et al., 2012).
In Arabidopsis, homologs of UPF1 (Arciga-Reyes et al., 2006), UPF2 (Kerényi et al., 2008), UPF3 (Hori and Watanabe, 2005), and SMG7 (Riehs et al., 2008) play important roles in NMD. Arabidopsis UPF1 shares a 50–75% amino acid sequence similarity with its homologs in yeasts, humans, Caenorhabditis elegans, and Drosophila, suggesting their functional similarities (Arciga-Reyes et al., 2006). UPF1 is required for the rapid degradation of mRNAs containing both spliced and unspliced PTCs. UPF3 is also required for the suppression of aberrant mRNAs originating from alternative splicing, emphasizing the important role of UPF3 in plant NMD (Hori and Watanabe, 2005). Collectively, these findings indicate that UPF1 and UPF3 are indispensable for the decay of abnormal transcripts in plants (Arciga-Reyes et al., 2006). Previous studies reported that the upf1 and upf3 mutations cause various developmental defects, including late flowering (Arciga-Reyes et al., 2006), but little is known about the mechanism of the regulation of flowering time in NMD-deficient mutants in Arabidopsis.
Here, we report that the upf1 and upf3 mutations led to delayed flowering and their double mutants showed an additive effect in delaying flowering time. RNA-seq analysis showed that FLC mRNA levels increased in upf mutants. Consistent with this, FT and SOC1 mRNA levels decreased in upf mutants, suggesting that FLC functions as a main component of the late-flowering phenotype of upf mutants. However, in upf mutants, our RNA-seq analysis did not find a PTC-containing transcript from FLC. Among the genes that regulate FLC, the transcript levels of MAF3, NF-YA2, NF-YA5, and TAF14 increased in upf mutants and among the genes that regulate flowering time in an FLC-independent manner, the transcript levels of ATC and BBX19 increased in upf mutants. Also, we found aberrant transcripts that contain a PTC from MAF3 and BBX19. Therefore, we propose that at least two pathways mediate the late-flowering phenotype of NMD-deficient mutants: a pathway in which MAF3 acts in an FLC-dependent manner and one in which BBX19 acts in an FLC-independent manner.
Materials and Methods
Plant Materials, Growth Conditions, and Flowering Time Measurement
The upf1-5 (SALK_112922) and upf3-1 (SALK_025175) mutants used in this study were previously described (Hori and Watanabe, 2005; Arciga-Reyes et al., 2006). We generated upf1-5 upf3-1 double mutants by crossing upf1-5 and upf3-1 mutants and confirmed their homozygosity by PCR-based genotyping using AtUPF1-1F and AtUPF1-2R primers for upf1-5 and AtUPF3-4F and AtUPF3-6R primers for upf3-1 (Hori and Watanabe, 2005; Riehs-Kearnan et al., 2012; Supplementary Table 4). For RNA-seq and qPCR analyses, 8-day-old wild-type (WT) Columbia-0 (Col-0), upf1-5, upf3-1, and upf1-5 upf3-1 double mutants grown at 23°C under standard LD conditions (16:8 h light: dark) were used. Total leaf numbers and days to flowering were used for the measurement of flowering time. Total leaf numbers were counted when the primary inflorescences reached about 5 cm. Flowering time data are presented as a box plot (Spitzer et al., 2014). In our box plots, the center lines show the medians and plus signs (+) show the mean value; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range (IQR) from the 25th and 75th percentiles, and outliers that exceeded the 1.5X IQR are represented by ovals. The number of plants measured is shown above each genotype in the box plot.
Vernalization Treatment
The upf1-5 and upf3-1 single mutants and upf1-5 upf3-1 double mutants were vernalized by incubating their imbibed seeds at 4°C for 4 weeks, prior to sowing. For a non-vernalized control, the upf mutants and WT Col-0 seeds were imbibed at 4°C for 4 days. The vernalization response was recorded as differences in flowering time between vernalized and non-vernalized mutants. The flowering time difference in response to vernalization was statistically assessed using Student's t-test.
RNA Sequencing (RNA-seq)
Plants were grown for 8 days on MS plates at 23°C under standard LD conditions (16:8 h light: dark). About 50–100 seedlings were harvested at Zeitgeber Time 16 (ZT16) and pooled for RNA extraction, which was done using the Plant RNA Purification Reagent (Invitrogen). RNA sequencing was performed with a single biological replication for each sample. Because we did a single RNA-seq experiment, we verified the results via qPCR by using the same growth conditions. For RNA sequencing, library preparation was performed with an Illumina TruSeq Stranded Total RNA Sample Prep kit (Illumina), according to the manufacturer's protocols. Briefly, after removal of rRNA from the total RNA (~700 ng) using the rRNA removal kit, the RNA was cleaned using RNA purification beads. After the rRNA was removed, the remaining RNA was fragmented using fragment mix (EPH) at 94°C for 6 min. The fragmented RNA was primed with random hexamers and transcribed to first-strand cDNA using reverse transcriptase and random primers at 25°C for 10 min, 42°C for 15 min, and then 70°C for 15 min. Then a replacement strand was synthesized by incorporating dUTP in place of dTTP to generate double-stranded cDNA using polymerase at 16°C for 1 h. After cleanup of cDNA using sample purification beads, a single “A” nucleotide was added to the 3′ ends of the blunt fragments using A-tailing mix reagent by incubating at 37°C for 30 min and then at 70°C for 5 min. Indexing adapters were ligated to the ends of the DNA fragments using ligation mix two reagent at 30°C for 10 min. After washing with sample purification beads twice, PCR was performed to enrich DNA fragments having adapter molecules on both ends. Thermocycler conditions were as follows: 95°C for 3 min, 8 cycles of: 98°C for 20 s, 60°C for 15 s, and 72°C for 30 min, with a final extension at 72°C for 5 min. Finally, quality and band size of libraries were assessed using Agilent 2100 Bioanalyzer (Agilent). Libraries were quantified by qPCR using CFX96 Real Time System (Bio-Rad). After normalization, sequencing of the prepared library was conducted as paired-end reads on an Illumina HiSeq2000 sequencer. The resulting raw RNA-seq data are available at NCBI Gene Expression Omnibus (GEO) database under the accession number GSE87851.
Data Analysis
The raw sequence reads were processed for removal of adapter sequences, followed by qualitative analysis of raw reads using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The high-quality reads were then mapped against the Arabidopsis thaliana reference genome TAIR10 (downloaded from http://arabidopsis.org) using TopHat 2.0.6 (https://ccb.jhu.edu/software/tophat/index.shtml) and Bowtie 2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) using default parameters.
Identification of Differentially Expressed Genes (DEGs)
The resulting mapped reads were then assembled and normalized transcript abundances were determined using Cufflinks version 2.2.1 (Trapnell et al., 2012; http://cole-trapnell-lab.github.io/cufflinks) with default settings. Cuffdiff version 2.2.1 was used for the identification of DEGs between WT plants and upf mutants with default parameters, including a false discovery rate of 5% (Trapnell et al., 2012). The output of Cuffdiff was further analyzed using R package “CummeRbund” (Goff et al., 2012). Genes with fold-change (FC) values of two or more and FPKM (fragments per kilobase of transcript per million mapped reads) values of one or more were considered as differentially expressed genes.
Selection of Flowering Time Regulators and FLC Regulators
To get insight into the flowering time changes seen in upf mutants, we studied expression of 306 flowering time regulator genes acquired from the Flowering Interactive Database (FLOR-ID; Bouche et al., 2016). For the analysis of genes that regulate FLC, we collected a total of 58 genes that potentially regulate FLC expression (Xu et al., 2013; Bouche et al., 2016; Supplementary Table 2) and analyzed their expression patterns. For representation of differential gene expression in upf mutants, three heatmaps were generated: one for overall DEGs found in upf mutants, a second for expression of all 306 flowering time regulators, and a third map for 58 genes that were identified as FLC regulators.
Quantitative Real-Time PCR (qPCR) Analysis
Quantitative real-time PCR (qPCR) was employed to validate the transcriptome data of the floral integrators and their regulators. Total RNA was extracted from 8-day-old Arabidopsis seedlings sampled at ZT16, using Plant RNA purification reagent (Invitrogen). The extracted RNA (~2 μg) was reverse transcribed into cDNA using the Transcriptor First Strand cDNA Synthesis kit (Roche). Expression analysis was performed using SYBR Green I Master mix (Roche) in a LightCycler 480 (Roche). The data were normalized against two stable reference genes, PP2AA3 (AT1G13320) and a SAND family gene (AT2G28390) (Hong et al., 2010). All the qPCR data are presented as the mean of two biological replicates with three technical replicates each and the error bars indicate the standard deviation. Statistical significance of differences of gene expression levels between the samples was assessed by using Student's t-test. P < 0.05 were considered as significant. Information on the primers that were used for qPCR in this study is shown in Supplementary Table 4.
Identification of Potential Target Genes of NMD
For detection of potential NMD targets, we used the CummeRbund package to individually analyze each flowering time gene to identify genes that show accumulation of aberrant transcripts in NMD-deficient mutants. The shortlisted potential target transcripts were then detected using RT-PCR using PP2AA3 as an internal control (Hong et al., 2010). Sequences of normal and aberrant transcripts from RNA-seq data were extracted and analyzed for the presence of PTCs between the normal and aberrant transcripts. Putative cDNA sequences of both normal and aberrant transcripts were translated in silico to determine the effect of the PTCs on the translated protein.
Results
The upf Single and Double Mutants Showed Late Flowering
To investigate the effect of the loss of NMD function on flowering time, we measured flowering time of upf1-5 and upf3-1 single mutants and the upf1-5 upf3-1 double mutants at 23°C under standard LD conditions. Both upf1-5 and upf3-1 single mutants flowered later than wild-type plants (Figures 1A,B), such that upf1-5 and upf3-1 mutants flowered with 22 ± 1.6 and 23 ± 2.3 leaves, respectively, consistent with a previous study that showed upf1-5 and upf3-1 mutants flowered late under both LD and SD conditions and had narrow, jagged rosette leaves (Arciga-Reyes et al., 2006). A previous study also reported that upf3-1 mutants showed a more severe phenotype and grew more slowly than upf1-5 mutants (Shi et al., 2012). We also generated upf1-5 upf3-1 double mutants and measured their flowering time to investigate whether UPF1 and UPF3 act redundantly in flowering time. The upf1-5 upf3-1 double mutants exhibited a severe bushy phenotype with no well-defined leaves in the vegetative phase (Figure 1C). Because multiple inflorescences emerged almost simultaneously from the axillary meristems of upf1-5 upf3-1 double mutants after the transition to the reproductive phase (Figure 1C), we were not able to count the number of leaves of the primary inflorescence of upf1-5 upf3-1 mutants; instead, we measured days to flowering. By this measure, the upf1-5 and upf3-1 mutants flowered later than wild-type plants under LD conditions. The upf1-5 and upf3-1 mutants flowered 33.5 ± 3.8 and 35.25 ± 3.6 days after germination (Figure 1D), respectively, whereas wild-type plants flowered 23.4 ± 1.7 days after germination. The upf1-5 upf3-1 double mutants flowered 40.6 ± 2.4 days after germination, indicating that the double mutation had an additive effect in delaying flowering time. This flowering time analysis indicated that the upf1-5 and upf3-1 mutants are late flowering and their combined mutations have an additive effect on flowering.
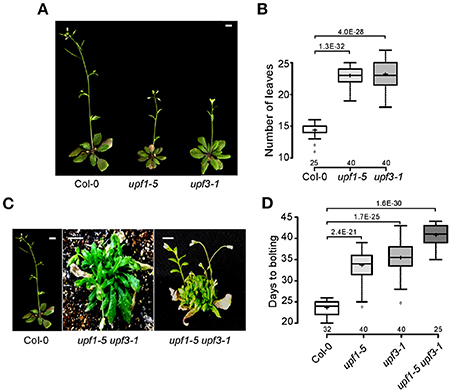
Figure 1. Late flowering of upf1-5, upf3-1, and upf1-5 upf3-1 mutants under LD conditions. (A) Late-flowering phenotypes of upf1-5 and upf3-1 mutants grown at 23°C under LD conditions. Scale bar = 2 cm (B) Total leaf numbers of upf1-5 and upf3-1 mutants presented as a box plot (see Section Materials and Methods for further information on box plots). (C) The severely bushy phenotype of upf1-5 upf3-1 double mutants with no well-defined leaves in the vegetative phase (middle) and multiple inflorescences generated from the axillary meristems in the reproductive phase (right). Scale bar = 2 cm (due to the late flowering of upf1-5 upf3-1 mutants, pictures of wild-type Col-0 and double mutants were taken at different times. Col-0 plants were photographed at 5 weeks old and upf1-5 upf3-1 double mutants were photographed at 8 weeks old). (D) Days to flowering of upf1-5, upf3-1, and upf1-5 upf3-1 mutants presented as a box plot. A t-test was used to assess the statistical significance of flowering time between wild-type and upf mutants (p-values are mentioned for each group).
Differentially Expressed Genes (DEGs) in upf Mutants
To gain insights on the transcript levels of flowering time genes in upf mutants, we performed RNA sequencing (RNA-seq) using upf1-5, upf3-1, and upf1-5 upf3-1 mutants with wild-type plants as a control (see Supplementary Table 1 for expression levels of all genes). Heat map analysis was performed to identify differentially expressed genes between upf mutants and wild-type plants (Figure 2A). Based on their expression patterns, we divided the DEGs into five major groups (Figure 2A). Group I included DEGs whose expression levels increased in both upf1-5 and upf3-1 mutants, but further increased in upf1-5 upf3-1 mutants. Group II included DEGs whose expression levels increased in upf3-1 mutants and upf1-5 upf3-1 mutants. Group III included DEGs whose expression levels increased only in upf1-5 upf3-1 mutants. Group IV included DEGs whose expression levels decreased in upf3-1 mutants and further decreased in upf1-5 upf3-1 mutants. The genes whose expression level changes were difficult to classify were placed in group V (Figure 2A). This heatmap analysis suggested that the expression patterns of DEGs in upf3-1 mutants were more similar to upf1-5 upf3-1 mutants than upf1-5 mutants. Furthermore, scatter plot distribution of gene expression also revealed the similarities in DEGs between upf3-1 single and upf1-5 upf3-1 double mutants (Supplementary Figure 1).
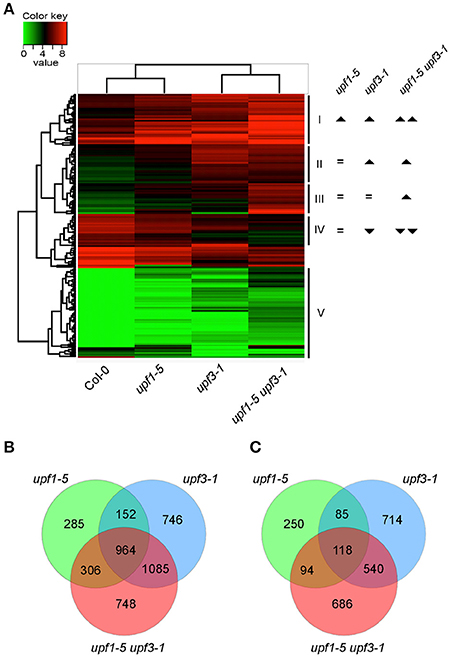
Figure 2. Differentially expressed genes in upf1-5, upf3-1, and upf1-5 upf3-1 mutants. (A) A heatmap showing differentially expressed genes among upf1-5, upf3-1, upf1-5 upf3-1, and wild-type plants. Based on the differential gene expression, upf1-5 mutants and wild-type plants grouped together, whereas upf3-1 and upf1-5 upf3-1 double mutants grouped together, indicating the high similarity between them. Upright triangle: up-regulation; inverted triangle: down-regulation. (B) Venn diagram of common and unique DEGs that were up-regulated in upf1-5, upf3-1, and upf1-5 upf3-1 mutants as compared to wild-type plants. (C) Venn diagram of common and unique DEGs that were down-regulated in upf1-5, upf3-1, and upf1-5 upf3-1 mutants as compared to wild-type plants. Note that the number of up-regulated genes was higher than down-regulated genes, suggesting the possible accumulation of NMD substrates in upf mutants.
We then selected individual DEGs after applying specific filters (genes with at least 2-fold difference in expression levels and FPKM value of 1 or more) from our RNA-seq data. The results showed that 2,254 genes were differentially expressed in upf1-5 mutants (1,707 were up-regulated and 547 were down-regulated), 4,404 genes were differentially expressed in upf3-1 mutants (2,947 were up-regulated and 1,457 were down-regulated; Figures 2B,C). In upf1-5 upf3-1 mutants, 4,541 genes were differentially expressed (3,103 up-regulated and 1,438 down-regulated). A total of 964 genes were commonly up-regulated among upf single mutants and upf1-5 upf3-1 double mutants. upf1-5 and upf1-5 upf3-1 double mutants shared 1,270 up-regulated genes, whereas upf3-1 and upf1-5 upf3-1 mutants shared 2,049 up-regulated genes (Figure 2B). In contrast, the upf1-5 and upf1-5 upf3-1 double mutants shared 212 down-regulated genes, whereas upf3-1 and upf1-5 upf3-1 mutants shared 658 down-regulated genes (Figure 2C). These indicated that in terms of number of DEGs, the upf3-1 mutants were closer to the upf1-5 upf3-1 mutants, compared with the upf1-5 mutants. Considering that the upf1-5 upf3-1 mutants showed an additive delayed flowering phenotype, these overall similarities in the number of DEGs between upf3-1 mutants and upf1-5 upf3-1 double mutants suggested that UPF3 exerted a stronger effect than UPF1 in the regulation of UPF-mediated flowering time.
To examine the biological functions of the DEGs, we performed Gene Ontology (GO) analysis using the ClueGO Cytoscape plug-in (Bindea et al., 2009). The “GO term fusion” option of ClueGO was used to reduce redundancy in GO terms. As expected, a variety of biological functions were affected by the NMD deficiency in upf mutants (Supplementary Figure 2). Most of the DEGs showed high enrichment for cellular metabolism of different biomolecules, metabolic process, gene expression regulation, transportation, protein localization, and response to different stimuli (Supplementary Figure 2), consistent with a previous finding (Rayson et al., 2012).
Up-Regulation of FLC in upf Mutants
To identify potential candidate genes responsible for the delayed flowering of upf mutants, we looked for DEGs related to the regulation of flowering time. Among such genes (Supplementary Figure 3), we found that the mRNA levels of FLOWERING LOCUS C (FLC), a floral repressor that is important for the vernalization response (Michaels and Amasino, 1999), increased in upf1-5, upf3-1, and upf1-5 upf3-1 mutants (Figure 3A, Supplementary Table 2). Our RNA-seq data showed that FLC mRNA levels increased by 2.92-fold in upf1-5 mutants and by 2.45-fold in upf3-1 mutants, whereas FLC mRNA levels increased by more than 5-fold in upf1-5 upf3-1 double mutants, compared to wild-type plants, indicating that UPF1 and UPF3 have an additive effect in regulating FLC expression (Figure 3A, Supplementary Table 2).
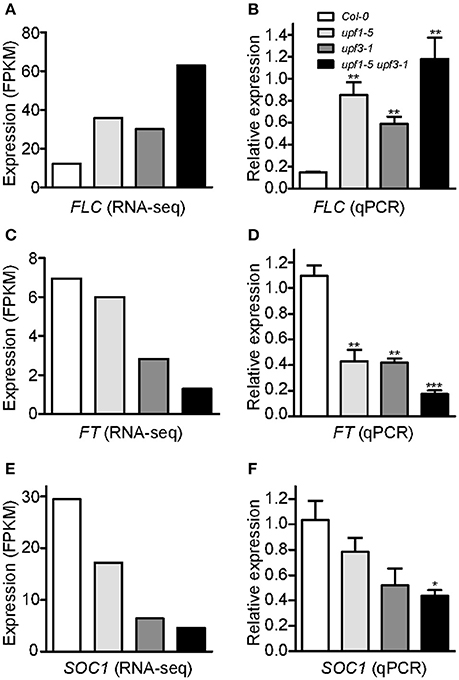
Figure 3. An increase in FLC mRNA levels and a decrease of FT and SOC1 mRNA levels in upf mutants. (A,B) FPKM levels determined by RNA-seq (A) and relative expression levels determined by qPCR (B) of FLC in upf1-5, upf3-1, upf1-5 upf3-1, and wild-type plants. (C,D) FPKM levels determined by RNA-seq (C) and relative expression levels determined by qPCR (D) of FT in upf1-5, upf3-1, upf1-5 upf3-1, and wild-type plants. (E,F) FPKM levels determined by RNA-seq (E) and relative expression levels determined by qPCR (F) of SOC1 in upf1-5, upf3-1, upf1-5 upf3-1, and wild-type plants.
As our global scale RNA-seq analysis revealed the elevated levels of FLC in the upf mutants that we tested (Figure 3A, Supplementary Table 2), we performed qPCR analyses to confirm the finding. FLC mRNA expression levels increased in upf1-5 and upf3-1 mutants (5.8- and 4-fold, respectively; Figure 3B). This suggested a role of FLC in delayed flowering of NMD-deficient mutants. Consistent with the further delayed flowering seen in upf1-5 upf3-1 mutants (Figure 1), FLC mRNA levels further increased in upf1-5 upf3-1 mutants (8.1-fold; Figure 3B).
Consistent with the increased levels of FLC, we also found that the mRNA levels of FLOWERING LOCUS T (FT; Kardailsky et al., 1999) and SOC1 (Lee et al., 2000), which were reported as major targets of FLC (Helliwell et al., 2006), decreased (Supplementary Table 2). Our RNA-seq analysis showed down-regulation of FT in upf1-5 and upf3-1 mutants (1.1 and 2.63-fold, respectively). In the upf1-5 upf3-1 double mutants, the mRNA levels of FT decreased by 5.1-fold, suggesting that the upf1 and upf3 mutations showed an additive effect (Figure 3C, Supplementary Table 2). Similar to FT, the mRNA levels of SOC1, another important floral integrator that is repressed directly by FLC (Hepworth et al., 2002), were reduced in upf1-5 and upf3-1 single mutants (1.7- and 4.5-fold, respectively), whereas the upf1-5 upf3-1 double mutants showed a 6.3-fold decrease (Figure 3E, Supplementary Table 2). Our qPCR results were consistent with our RNA-seq data that showed decreased levels of FT (Figure 3D) and SOC1 mRNAs (Figure 3F) in upf mutants. We also analyzed SHORT VEGETATIVE PHASE (SVP) expression levels, as FLC interacts with SVP to synergistically repress flowering (Li et al., 2008). However, we did not find apparent changes in SVP mRNA levels in upf mutants (Supplementary Figure 4). These results showed that FLC mRNA levels increased, whereas FT and SOC1 mRNA levels decreased in upf mutants, raising the possibility that FLC up-regulation represses FT and SOC1 expression in upf mutants, which eventually causes late flowering in upf mutants.
upf Mutants Were Vernalization-Responsive
After confirming the elevated levels of FLC in upf mutants, we next investigated whether FLC mediates the late-flowering phenotype of upf mutants. For this purpose, we analyzed whether vernalization treatment accelerated flowering of upf mutants, as FLC-mediated repression of flowering can be recovered by prolonged cold treatment (Michaels and Amasino, 1999; Sheldon et al., 1999). We transferred upf mutant seeds to 4°C for 4 weeks for vernalization treatment. Flowering time measurement indicated that vernalization accelerated the flowering of upf1-5 and upf3-1 mutants. Vernalized upf1-5 mutants flowered with 15.0 ± 1.5 leaves, whereas non-vernalized upf1-5 mutants flowered with 22.7 ± 2.2 leaves (Figures 4A,C). Similarly, vernalized upf3-1 mutants flowered with 16.3 ± 1.0 leaves, whereas non-vernalized upf3-1 mutants flowered with 23.3 ± 2.1 leaves (Figures 4B,C). The non-vernalized Col-0 plants flowered with 14.3 ± 1.3 leaves and the vernalized Col-0 plants flowered with 12.4 ± 1.2 leaves (Figure 4C). These observations confirmed that wild-type Col-0 plants, which contain a non-functional FRI allele (Lee and Amasino, 1995), showed weak responsiveness to vernalization treatment. As leaf numbers could not be counted for upf1-5 upf3-1 mutants, we also measured days to flowering of vernalized upf1-5 upf3-1 mutants under LD conditions to determine the vernalized response of upf1-5 upf3-1 mutants. The vernalized wild-type plants flowered 22.4 ± 2.3 days after germination, compared to the non-vernalized wild-type plants, which flowered 23.7 ± 1.6 days after germination. Vernalized upf1-5 mutants flowered 24.7 ± 3.5 days after germination, whereas non-vernalized upf1-5 mutants flowered 33.4 ± 3.8 days after germination (Figure 4E). Similarly, vernalized upf3-1 mutants flowered 28.1 ± 3.4 days after germination, whereas non-vernalized upf3-1 mutants flowered 35.6 ± 3.6 days after germination. In addition, vernalized upf1-5 upf3-1 mutants flowered 33 ± 4.3 days after germination, whereas non-vernalized upf1-5 upf3-1 mutants flowered 40.7 ± 2.4 days after germination (Figures 4D,E). These analyses indicated that vernalized upf1-5, upf3-1, and upf1-5 upf3-1 mutants flowered ~9, 7.5, and 7.7 days earlier than non-vernalized mutants, respectively. These observations suggested that FLC repression by vernalization likely accelerated flowering in upf mutants, suggesting a key role of FLC in the late flowering of upf mutants.
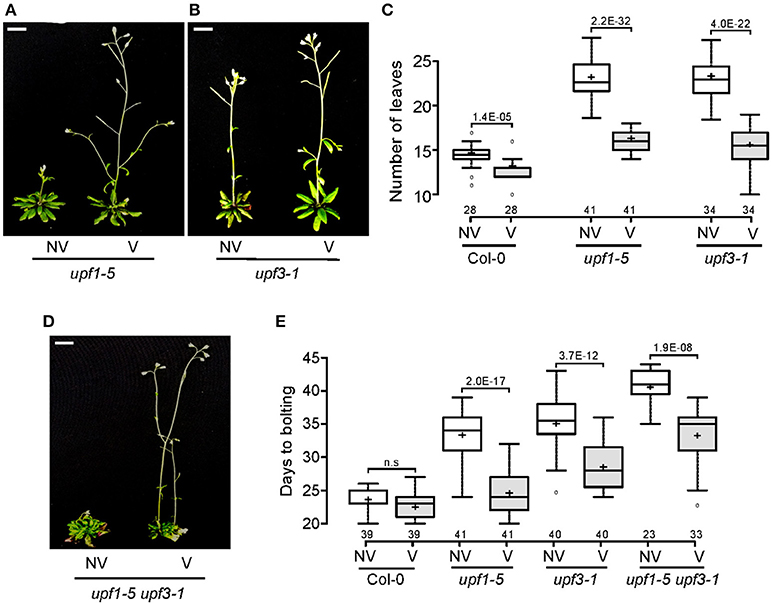
Figure 4. Vernalization response of upf1-5, upf3-1, and upf1-5 upf3-1 mutants. (A,B) Acceleration of flowering by vernalization in upf1-5 (A) and upf3-1 mutants (B). NV: non-vernalized, V: vernalized. Scale bar = 2 cm (C) Box plot showing total leaf numbers of vernalized/non-vernalized upf1-5 and upf3-1 mutants. Both upf1-5 and upf3-1 mutants flowered earlier in response to vernalization treatment. (D) Vernalization response of upf1-5 upf3-1 double mutants. NV: non-vernalized, V: vernalized. (E) Box plot showing days to flowering of vernalized/non-vernalized upf1-5, upf3-1, and upf1-5 upf3-1 mutants. A t-test was used to assess the statistical significance of flowering time difference in response to vernalization (p-value s mentioned for each group).
UPF-Mediated Regulation of Flowering Time May Target FLC Indirectly
Although it seemed that the late-flowering phenotype of upf mutants was largely mediated by FLC, we next looked for an aberrant FLC transcript that contains a PTC. In addition to AT5G10140.1, the predominant transcript that produces functional FLC protein, we found four alternatively spliced isoforms of FLC in our RNA-seq (Supplementary Figure 5A). Among them, AT5G10140.2, AT5G10140.3, and AT5G10140.4 were already reported. We found an unreported alternatively spliced isoform and named it AT5G10140.5. AT5G10140.5 had a similar structure to the fourth splice variant of FLC (i.e., AT5G10140.4), except for its untranslated regions (UTRs), as the novel AT5G10140.5 transcript had longer 3′- and 5′-UTRs. We then measured the transcript levels of each alternatively spliced isoform. The abundance of these four isoforms of FLC was not apparently altered in upf mutants (Supplementary Figure 5B). Moreover, we could not find a PTC from any of the alternative spliced isoforms. These results suggested that FLC is not a direct target of NMD in the regulation of flowering time in upf mutants.
Expression of MAF3, TAF14, and NF-YA2/5, Positive Regulators of FLC, Was Up-Regulated in upf Mutants
As FLC may not be the direct target of UPF-mediated regulation of flowering time, we analyzed expression of genes that act upstream of FLC. For this purpose, we measured the transcript levels of 58 genes that regulate flowering time (Bouche et al., 2016) from our RNA-seq data (Figure 5A), to understand the positive regulation of FLC in the upf mutants. Detailed expression data for of all FLC regulators can be found in Supplementary Table 3. Among 58 genes that potentially regulate FLC expression, we found a few genes whose expression patterns in the mutants were similar to that of FLC (Figures 5B–I). They were: (1) MADS AFFECTING FLOWERING 3 (MAF3), a homolog of FLC that inhibits flowering by up-regulating FLC expression and repressing FT and SOC1 expression (Gu et al., 2013; Suter et al., 2014); (2) TBP-ASSOCIATED FACTOR 14 (TAF14), which is also known as HOMOLOG OF YEAST YAF9 B (YAF9B) and promotes FLC expression through chromatin modification together with AtYAF9A (Zacharaki et al., 2012; Bieluszewski et al., 2015); and (3) NF-YA2 and NF-YA5, which encode factors that may have dual functions (as a floral promoter and a floral repressor) in the regulation of flowering time (Xu et al., 2013; Hou et al., 2014).
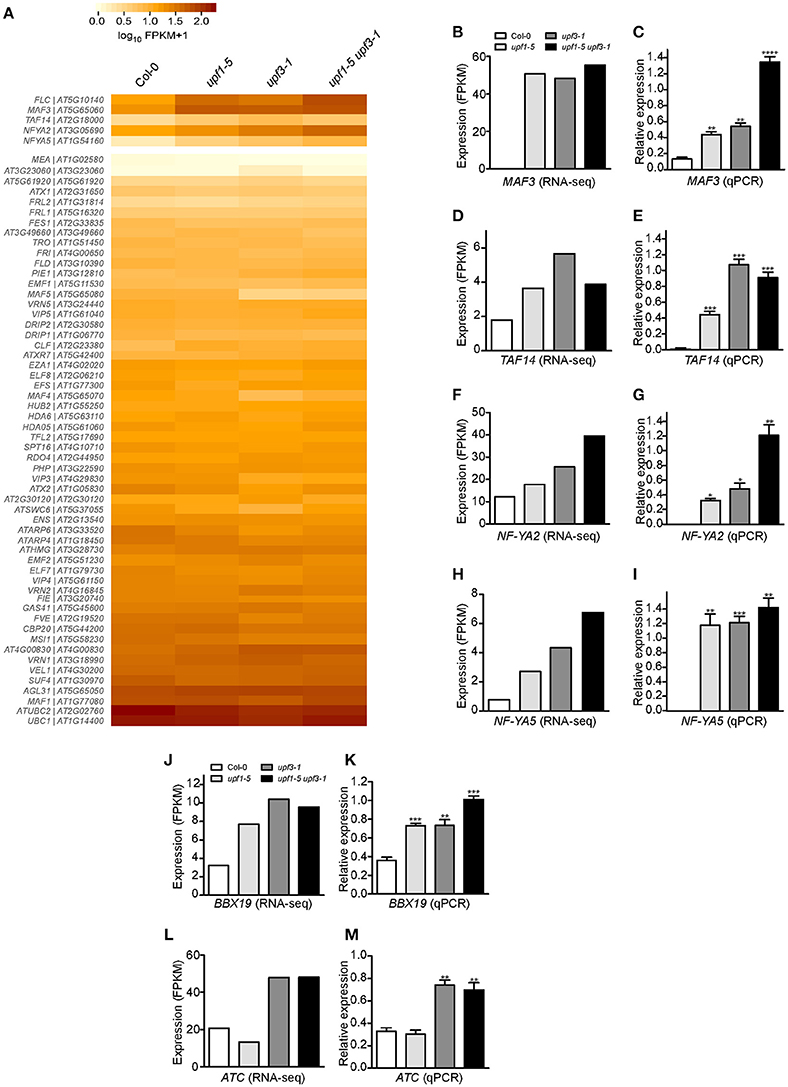
Figure 5. Expression of FLC and its regulators in upf mutants. (A) Heatmap shows the expression of 58 genes that possibly regulate FLC expression. MAF3, TAF14, NF-YA2, and NF-YA5, which showed expression patterns similar to that of FLC, are shown separately. (B,C) FPKM levels determined by RNA-seq (B) and relative expression levels determined by qPCR (C) of MAF3 in upf1-5, upf3-1, upf1-5 upf3-1, and wild-type plants. (D,E) FPKM levels determined by RNA-seq (D) and relative expression levels determined by qPCR (E) of TAF14 in upf mutants. TAF14 mRNA levels increased in upf single and double mutants, as detected by RNA-seq (D) and confirmed by qPCR (E). (F,G) FPKM levels determined by RNA-seq (F) and relative expression levels determined by qPCR (G) of NF-YA2 in upf mutants. (H,I) FPKM levels determined by RNA-seq (H) and relative expression levels determined by qPCR (I) of NF-YA5 in upf mutants. (J,K) FPKM levels determined by RNA-seq (J) and relative expression levels determined by qPCR (K) of BBX19 in upf mutants. (L,M) FPKM levels determined by RNA-seq (L) and relative expression levels determined by qPCR (M) of ATC in upf mutants.
RNA-seq analysis showed that mRNA levels of MAF3 were high in upf1-5 (2.9-fold that of wild type) and upf3-1 (2.8-fold) mutants. The mRNA level of MAF3 increased by 3.3-fold in upf1-5 upf3-1 double mutants (Figure 5B, Supplementary Table 3). Similarly, qPCR analysis showed increased transcript levels of MAF3 in upf1-5 (3.3-fold) and upf3-1 mutants (~4-fold; Figure 5C). The upf1-5 upf3-1 double mutants showed a 10.2-fold increase, suggesting that both upf mutations had an additive effect on MAF3 expression (Figure 5C). The mRNA levels of TAF14 also increased in upf1-5 (3.2-fold) and upf3-1 (2.0-fold) mutants, as well as in upf1-5 upf3-1 (2.1-fold) double mutants, (Figure 5D, Supplementary Table 3). The increased mRNA levels of TAF14 revealed by qPCR (Figure 5E) were consistent with RNA-seq data. The RNA-seq data showed that the mRNA level of NF-YA2 increased by 1.5- and 2.1-fold in upf1-5 and upf3-1 mutants, respectively, and was 3.3-fold higher in upf1-5 upf3-1 double mutants (Figure 5F, Supplementary Table 3). The qPCR results agreed with the RNA-seq data of elevated NF-YA2 mRNA levels (Figure 5G). The upf1-5 and upf3-1 mutants showed high levels of NF-YA5 mRNA (3.5- and 5.6-fold increase, respectively), and upf1-5 upf3-1 mutants showed an 8.7-fold increase in NF-YA5 mRNA (Figure 5H, Supplementary Table 3), suggesting that upf1 and upf3 mutations contribute additively to NF-YA5 mRNA levels in upf1-5 upf3-1 double mutants. The qPCR analysis of NF-YA2 also showed a similar expression pattern (Figure 5I).
Expression of BBX19 and ATC, Genes That Regulate Flowering Time in an FLC-Independent Manner, Was Up-Regulated in upf Mutants
We also analyzed the expression of genes that regulate flowering time in an FLC-independent manner to identify possible regulators of flowering time in NMD-deficient mutants. From our RNA-seq data, we found two floral repressors that showed consistent expression changes in upf mutants: B-BOX DOMAIN PROTEIN 19 (BBX19), which encodes a floral repressor that interacts with CO, thereby preventing CO from inducing FT expression (Wang et al., 2014), and ATC, which encodes a floral repressor that interacts with FD to affect the expression levels of the floral meristem identity gene AP1 (Mimida et al., 2001; Huang et al., 2012; Figures 5J,L). qPCR analysis revealed that BBX19 expression was significantly up-regulated with a fold change of ~2 in upf1-5 and upf3-1 mutants and ~3-fold in upf1-5 upf3-1 double mutants (Figure 5K). The transcript levels of ATC were similar between wild-type plants and upf1-5 mutants (Figure 5M); however, the ATC expression levels were elevated over 2-fold in upf3-1 and upf1-5 upf3-1 double mutants (Figure 5M). Thus, the elevated levels of BBX19 and ATC in upf mutants suggested an FLC-independent contribution to the delayed flowering of upf mutants.
MAF3 and BBX19 Are Potential Targets of NMD in the Determination of Flowering Time
To identify the direct targets of NMD in the regulation of flowering time, we analyzed the isoforms of all flowering time regulators. Among these genes, we found that MAF3 and BBX19 produced aberrantly spliced transcripts (Figures 6A,B). As these transcripts were not listed in TAIR, we then named them AT5G65060.3 (from MAF3) and AT4G38960.4 (from BBX19). The AT5G65060.3 transcript differed from AT5G65060.1, the predominant MAF3 transcript, by the presence of longer 3′- and 5′-UTRs and the absence of 38 nucleotides within the 4th exon. The 38-nucleotide deletion caused a predicted out-of-frame splicing event, which created a PTC in the same exon, leading to formation of a truncated MAF3 protein (Figure 6A). The full-length alignment of the transcripts and the proteins can be found in Supplementary Figures 6, 7. The AT4G38960.4 transcript generated from BBX19 had a long 5′-UTR, skipped the first two BBX19 exons, and retained part of the third intron, like the AT4G38960.2 transcript, a splice variant of BBX19. Importantly, seven nucleotides were absent in the 6th exon of the AT4G38960.4 transcript, compared to AT4G38960.3, the normal functional transcript of BBX19. The 7-nucleotide deletion likely created a PTC in the 5th exon to generate truncated BBX19 proteins (Figure 6B, Supplementary Figure 9). Considering that the AT5G65060.3 and AT4G38960.4 transcripts had a PTC and long UTRs (Figures 6A,B, Supplementary Figures 6, 8), a characteristic of aberrant transcripts that are destined for NMD under normal circumstances, these AT5G65060.3 and AT4G38960.4 transcripts could be potential targets of UPF-mediated regulation of flowering time.
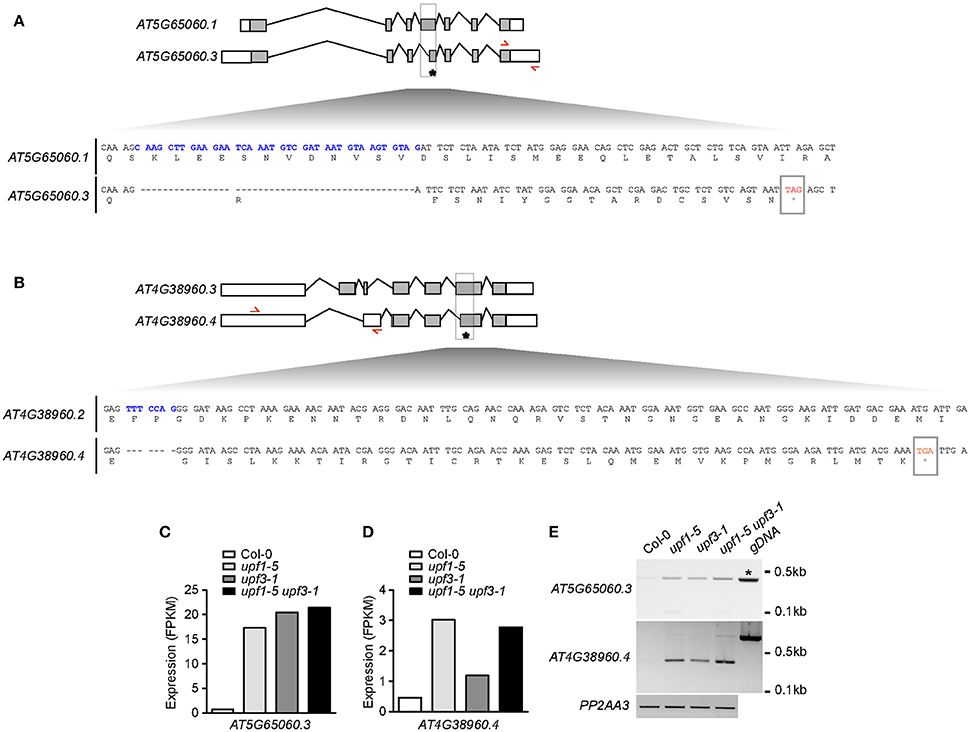
Figure 6. Production of aberrant MAF3 and BBX19 transcripts containing PTCs in upf mutants. (A,B) Comparison of the structures of a normal transcript (AT5G65060.1) and an aberrant transcript (AT5G65060.3) from MAF3 (A) and comparison of the structures of a normal transcript (AT4G38950.3) and an aberrant transcript (AT4G38950.4) that we identified from BBX19 (B) from our RNA-seq analysis. Each bottom panel shows deduced amino acid sequences from the normal transcript and the aberrant transcript containing a PTC, which is indicated by an asterisk. Sequences in blue indicate the cDNA fragment that is absent in the aberrant transcript. Red arrows indicate the binding sites of primers used to detect aberrant transcripts from MAF3 and BBX19. Gray boxes indicate coding regions. (C,D) FPKM levels of the aberrant transcript of MAF3 (AT5G65060.3) (C) and BBX19 (AT4G38960.4) (D) in upf mutants. (E) Detection of AT5G65060.3 and AT4G38960.4 transcripts via RT-PCR. Note that an amplicon with the same size was amplified from genomic DNA (gDNA) from MAF3 (asterisk). PP2AA3 was used as an internal control.
RNA-seq analysis showed that FPKM levels of AT5G65060.3 and AT4G38960.4 transcripts increased in upf mutants. The FPKM levels of the AT5G65060.3 transcript increased by 20.3, 23.9, and 25.9-fold in upf1-5, upf3-1, and upf1-5 upf3-1 mutants, respectively (Figure 6C). The FPKM levels of the AT4G38950.4 transcript increased by 6.5, 2.6, and 5.8-fold in upf1-5, upf3-1, and upf1-5 upf3-1 mutants, respectively (Figure 6D). We then carried out conventional RT-PCR to determine whether we could detect the aberrant transcripts identified in our RNA-seq analysis. We successfully detected amplicons using primers to specifically amplify the aberrant AT5G65060.3 and AT4G38960.4 transcripts (Figure 6E). Furthermore, the levels of AT5G65060.3 and AT4G38960.4 apparently increased in upf mutants, compared to wild-type plants, suggesting that AT5G65060.3 and AT4G38960.4 transcripts were likely substrates of NMD. These analyses suggested that MAF3 and BBX19 are potential targets of NMD in the regulation of flowering time in upf mutants.
Discussion
This study aimed to explain the late-flowering phenotype of NMD-deficient mutants (upf1-5, upf3-1, and upf1-5 upf3-1) using next-generation RNA sequencing and qPCR analyses. We found that FLC mRNA levels were high in upf mutants and FLC likely mediates late flowering of upf mutants via possible transcriptional repression of FT and SOC1. However, FLC might not be the direct target of NMD, suggesting that upstream regulators of FLC may be responsible for the UPF-mediated flowering time change. Indeed, we found that the transcript levels of MAF3 and BBX19 increased in upf mutants and that they produced aberrant transcripts containing a PTC.
NMD is a conserved eukaryotic surveillance mechanism that protects cells from potential harmful effects of truncated proteins as a result of faulty transcripts with PTCs. Early transcript profiling experiments in Arabidopsis showed that transcript levels of only ~0.5% of protein-coding genes were elevated in NMD-impaired protoplasts (Kurihara et al., 2009), which suggests the involvement of NMD in global-scale regulation of Arabidopsis genes, apart from degrading aberrant transcripts. With the development of more robust sequencing technologies, especially RNA sequencing (RNA-seq), further insight into the mechanism of NMD and its targets has been achieved over the past few years (Filichkin et al., 2010; Drechsel et al., 2013). By using paired-end RNA-seq, we were able to find a large number of protein coding genes that were differentially expressed in upf mutants (2,254 genes in upf1-5 mutants, 4,404 in upf3-1 mutants, and 4,541 in upf1-5 upf3-1 double mutants; Figures 2B,C). We found an additive effect of the upf1-5 and upf3-1 mutations on differentially expressed genes, which is consistent with the further delayed flowering seen in upf1-5 upf3-1 double mutants (Arciga-Reyes et al., 2006; Drechsel et al., 2013). Our data suggested that the upf3 mutation made a strong contribution to the differential expression of genes in the double mutants.
The transition from vegetative growth to flowering is a major developmental event in the plant life cycle. The precisely regulated timing of flowering has decisive consequences for the successful completion of the plant life cycle (Huijser and Schmid, 2011). Although a previous work suggested that NMD regulates SOC1 transcript levels in the presence of EARLY FLOWERING 9 (ELF9) protein (Song et al., 2009), the molecular mechanism governing the late-flowering phenotype of NMD-deficient mutants has remained an unsolved puzzle. The upf1-5 and upf3-1 mutants have a delayed-flowering phenotype (Arciga-Reyes et al., 2006), but no mechanism explaining the late-flowering time phenotype was suggested. Here, we attempted to understand the factors responsible for the delayed flowering of upf1-5 and upf3-1 mutants, two major components in the NMD pathway in Arabidopsis, as well as in their double mutant plants. We found that FLC expression levels increased in upf mutants, which is consistent with a previous finding (Kurihara et al., 2009). In their report, the authors performed a whole-genome tiling array and found FLC as one of the 138 protein-coding transcripts whose expression levels were up-regulated (over 1.8-fold) in upf mutants. From our RNA-seq and qPCR analyses, we found consistent results showing that FLC mRNA levels increased in both upf1-5 and upf3-1 single mutants and in upf1-5 upf3-1 double mutants (Figures 3A,B), which is consistent with the late-flowering phenotypes of upf mutants. This strongly suggests that FLC mediates the delayed flowering of NMD-deficient mutants, as FLC expression quantitatively and inversely correlates with flowering time (Michaels and Amasino, 1999; Sheldon et al., 1999). This conclusion is further supported by the vernalization responsiveness of upf mutants (Figure 4). As vernalization results in acceleration of flowering time via the stable repression of FLC through histone methylation (Searle et al., 2006), the recovery of the late-flowering phenotype of upf mutants suggests that FLC plays an important role in the determination of flowering time in the upf mutants.
How is UPF-mediated flowering time regulated? Although FLC mRNA levels increased in upf mutants, we could not find any evidence that FLC is a direct target of NMD (Supplementary Figure 5). Among upstream flowering time regulators that act in an either FLC-dependent or FLC-independent manners, however, we found that MAF3 and BBX19 mRNA levels increased in upf mutants (Figures 5B,C,J,K, respectively). Furthermore, we detected aberrant transcripts of MAF3 and BBX19 in upf mutants (Figure 6), suggesting that these two genes might be targets of the UPF-dependent regulation of flowering time. Thus we propose that the regulation of UPF-dependent flowering time involves at least two independent pathways. The first pathway is the MAF3-FLC pathway, in which MAF3 positively regulates FLC expression (Gu et al., 2013) and subsequent up-regulation of FLC causes late flowering in upf mutants. The second pathway is the BBX19-FT pathway, which acts independently of FLC. BBX19 physically interacts with CO and prevents it from binding to the FT promoter to induce FT expression (Wang et al., 2014). Thus, increased BBX19 expression likely leads to delayed flowering in upf mutants. The existence of multiple pathways is consistent with the flowering time changes induced by changes in MAF3 and BBX19 expression. Since flowering time changes caused by the alteration of expression of MAF3 or BBX19 were not very dramatic, either the MAF3-FLC pathway or the BBX19-FT pathway is not sufficient to explain the strong late flowering of upf mutants. Thus, we suggest that NMD affects multiple pathways to modulate flowering time and at least the MAF3-FLC and BBX19-FT modules play an important role in such regulation.
We propose that MAF3 and BBX19 function as major players in UPF-mediated delayed flowering in upf mutants, but it should be noted that the detection of PTCs and altered transcripts in upf mutants involves an element of random chance. Therefore, we cannot exclude the possibility that the FLC locus produced PTCs and altered transcripts, but we were not able to capture the event. Furthermore, considering that PTCs and altered transcripts can arise from every transcribed gene in the genome, given enough time, it is possible that the delayed flowering resulted from the combinatorial effects of many genes that may or may not be related to flowering. Given that disruption of NMD in the upf1-5 and upf3-1 mutants likely has broad effects on cellular metabolism, stress responses, and other aspects of plant cell biology, we consider that the observed delay in flowering time reflects only a small part of the disruptions occurring in these mutants. However, this phenotype provided a valuable indicator of the metabolic alterations occurring in the mutant cells and allowed us to probe the effects on the regulation of flowering time.
Based on our results, we constructed a working model to explain the delayed flowering of upf mutants (Figure 7). According to this model, the regulation of UPF-dependent flowering time is controlled by at least two independent pathways, namely, an FLC-dependent pathway and an FLC-independent pathway. In the FLC-dependent pathway, MAF3 is likely a direct substrate of NMD and aberrant MAF3 transcripts that are not cleared out by NMD affect normal MAF3 function, which affects FLC. The increase in FLC then causes the repression of flowering by direct repression of FT and SOC1 by FLC. In the FLC-independent pathway, BBX19 is likely a direct substrate of NMD and aberrant BBX19 transcripts that are not removed by NMD affect normal BBX19 function, which eventually affects FT expression by inhibiting binding of CO to the FT locus (Wang et al., 2014). Thus, reduced FT and SOC1 expression is not sufficient to induce the expression of floral identity genes and hence the upf mutants flower later than wild-type plants.
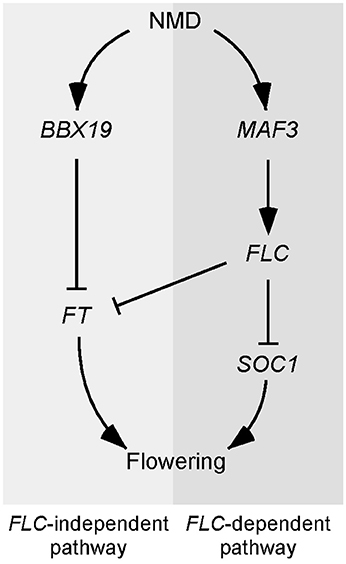
Figure 7. Model explaining the late-flowering phenotype of NMD-deficient mutants. Two independent pathways likely participate in the UPF-mediated regulation of flowering time. In the FLC-dependent pathway, MAF3 seems to be the direct target of NMD and the up-regulation of MAF3 in NMD-deficient mutants causes up-regulation of FLC, which in turn represses FT and SOC1 to delay flowering. In the FLC-independent pathway, BBX19 seems to be the direct target of NMD and its up-regulation suppresses FT expression, which eventually inhibits flowering.
Author Contributions
ZN analyzed the NGS data analysis, performed the experiments. MF performed the initial NGS data analysis. JA designed and supervised the study. ZN, MF, and JA wrote the manuscript.
Funding
Our work was supported by a National Research Foundation of Korea grant funded by the Korean government (Ministry of Science, ICT, and Future Planning; 2008-0061988) to JA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Y.J. Kim for her technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00191/full#supplementary-material
References
Abe, M., Kobayashi, Y., Yamamoto, S., Daimon, Y., Yamaguchi, A., Ikeda, Y., et al. (2005). FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309, 1052–1056. doi: 10.1126/science.1115983
Arciga-Reyes, L., Wootton, L., Kieffer, M., and Davies, B. (2006). UPF1 is required for nonsense-mediated mRNA decay (NMD) and RNAi in Arabidopsis. Plant J. 47, 480–489. doi: 10.1111/j.1365-313X.2006.02802.x
Bastow, R., Mylne, J. S., Lister, C., Lippman, Z., Martienssen, R. A., and Dean, C. (2004). Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427, 164–167. doi: 10.1038/nature02269
Bernier, G., and Périlleux, C. (2005). A physiological overview of the genetics of flowering time control. Plant Biotechnol. J. 3, 3–16. doi: 10.1111/j.1467-7652.2004.00114.x
Bieluszewski, T., Galganski, L., Sura, W., Bieluszewska, A., Abram, M., Ludwikow, A., et al. (2015). AtEAF1 is a potential platform protein for Arabidopsis NuA4 acetyltransferase complex. BMC Plant Biol. 15:1. doi: 10.1186/s12870-015-0461-1
Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., et al. (2009). ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. doi: 10.1093/bioinformatics/btp101
Borner, R., Kampmann, G., Chandler, J., Gleißner, R., Wisman, E., Apel, K., et al. (2000). A MADS domain gene involved in the transition to flowering in Arabidopsis. Plant J. 24, 591–599. doi: 10.1046/j.1365-313x.2000.00906.x
Bouche, F., Lobet, G., Tocquin, P., and Perilleux, C. (2016). FLOR-ID: an interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 44, D1167–D1171. doi: 10.1093/nar/gkv1054
Dai, Y., Li, W., and An, L. (2016). NMD mechanism and the functions of Upf proteins in plant. Plant Cell Rep. 35, 5–15. doi: 10.1007/s00299-015-1867-9
Deng, W., Ying, H., Helliwell, C. A., Taylor, J. M., Peacock, W. J., and Dennis, E. S. (2011). FLOWERING LOCUS C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 108, 6680–6685. doi: 10.1073/pnas.1103175108
Drechsel, G., Kahles, A., Kesarwani, A. K., Stauffer, E., Behr, J., Drewe, P., et al. (2013). Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell 25, 3726–3742. doi: 10.1105/tpc.113.115485
Filichkin, S. A., and Mockler, T. C. (2012). Unproductive alternative splicing and nonsense mRNAs: a widespread phenomenon among plant circadian clock genes. Biol. Direct 7:20. doi: 10.1186/1745-6150-7-20
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58. doi: 10.1101/gr.093302.109
Goff, L. A., Trapnell, C., and Kelley, D. (2012). CummeRbund: Visualization and Exploration of Cufflinks High-throughput Sequencing Data. R Package Version 2(0).
Gu, X., Le, C., Wang, Y., Li, Z., Jiang, D., Wang, Y., et al. (2013). Arabidopsis FLC clade members form flowering-repressor complexes coordinating responses to endogenous and environmental cues. Nat. Commun. 4:1947. doi: 10.1038/ncomms2947
He, F., Li, X., Spatrick, P., Casillo, R., Dong, S., and Jacobson, A. (2003). Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol. Cell 12, 1439–1452. doi: 10.1016/S1097-2765(03)00446-5
Helliwell, C. A., Wood, C. C., Robertson, M., James Peacock, W., and Dennis, E. S. (2006). The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 46, 183–192. doi: 10.1111/j.1365-313X.2006.02686.x
Hepworth, S. R., Valverde, F., Ravenscroft, D., Mouradov, A., and Coupland, G. (2002). Antagonistic regulation of flowering-time gene SOC1 by CONSTANS and FLC via separate promoter motifs. EMBO J. 21, 4327–4337. doi: 10.1093/emboj/cdf432
Hong, S. M., Bahn, S. C., Lyu, A., Jung, H. S., and Ahn, J. H. (2010). Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol. 51, 1694–1706. doi: 10.1093/pcp/pcq128
Hori, K., and Watanabe, Y. (2005). UPF3 suppresses aberrant spliced mRNA in Arabidopsis. Plant J. 43, 530–540. doi: 10.1111/j.1365-313X.2005.02473.x
Hou, X., Zhou, J., Liu, C., Liu, L., Shen, L., and Yu, H. (2014). Nuclear factor Y-mediated H3K27me3 demethylation of the SOC1 locus orchestrates flowering responses of Arabidopsis. Nat. Commun. 5:4601. doi: 10.1038/ncomms5601
Huang, N. C., Jane, W. N., Chen, J., and Yu, T. S. (2012). Arabidopsis thaliana CENTRORADIALIS homologue (ATC) acts systemically to inhibit floral initiation in Arabidopsis. Plant J. 72, 175–184. doi: 10.1111/j.1365-313X.2012.05076.x
Huijser, P., and Schmid, M. (2011). The control of developmental phase transitions in plants. Development 138, 4117–4129. doi: 10.1242/dev.063511
Kalyna, M., Simpson, C. G., Syed, N. H., Lewandowska, D., Marquez, Y., Kusenda, B., et al. (2012). Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic acids Res. 40, 2454–2469. doi: 10.1093/nar/gkr932
Kardailsky, I., Shukla, V. K., Ahn, J. H., Dagenais, N., Christensen, S. K., Nguyen, J. T., et al. (1999). Activation tagging of the floral inducer FT. Science 286, 1962–1965. doi: 10.1126/science.286.5446.1962
Kerényi, Z., Mérai, Z., Hiripi, L., Benkovics, A., Gyula, P., Lacomme, C., et al. (2008). Inter-kingdom conservation of mechanism of nonsense-mediated mRNA decay. EMBO J. 27, 1585–1595. doi: 10.1038/emboj.2008.88
Kertesz, S., Kerenyi, Z., Merai, Z., Bartos, I., Palfy, T., Barta, E., et al. (2006). Both introns and long 3′-UTRs operate as cis-acting elements to trigger nonsense-mediated decay in plants. Nucleic Acids Res. 34, 6147–6157. doi: 10.1093/nar/gkl737
Kobayashi, Y., Kaya, H., Goto, K., Iwabuchi, M., and Araki, T. (1999). A pair of related genes with antagonistic roles in mediating flowering signals. Science 286, 1960–1962. doi: 10.1126/science.286.5446.1960
Kurihara, Y., Matsui, A., Hanada, K., Kawashima, M., Ishida, J., Morosawa, T., et al. (2009). Genome-wide suppression of aberrant mRNA-like noncoding RNAs by NMD in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 106, 2453–2458. doi: 10.1073/pnas.0808902106
Lee, H., Suh, S.-S., Park, E., Cho, E., Ahn, J. H., Kim, S.-G., et al. (2000). The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev. 14, 2366–2376. doi: 10.1101/gad.813600
Lee, I., and Amasino, R. M. (1995). Effect of vernalization, photoperiod, and light quality on the flowering phenotype of Arabidopsis plants containing the FRIGIDA gene. Plant Physiol. 108, 157–162. doi: 10.1104/pp.108.1.157
Li, D., Liu, C., Shen, L., Wu, Y., Chen, H., Robertson, M., et al. (2008). A repressor complex governs the integration of flowering signals in Arabidopsis. Dev. Cell 15, 110–120. doi: 10.1016/j.devcel.2008.05.002
Maquat, L. E. (2005). Nonsense-mediated mRNA decay in mammals. J. Cell Sci. 118, 1773–1776. doi: 10.1242/jcs.01701
Mendell, J., Sharifi, N., Meyers, J., Martinez-Murillo, F., and Dietz, H. (2004). Erratum: nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 36, 1238–1238. doi: 10.1038/ng1104-1238c
Michaels, S. D. (2009). Flowering time regulation produces much fruit. Curr. Opin. Plant Biol. 12, 75–80. doi: 10.1016/j.pbi.2008.09.005
Michaels, S. D., and Amasino, R. M. (1999). FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. doi: 10.1105/tpc.11.5.949
Mimida, N., Goto, K., Kobayashi, Y., Araki, T., Ahn, J. H., Weigel, D., et al. (2001). Functional divergence of the TFL1-like gene family in Arabidopsis revealed by characterization of a novel homologue. Genes Cells 6, 327–336. doi: 10.1046/j.1365-2443.2001.00425.x
Nyikó, T., Kerényi, F., Szabadkai, L., Benkovics, A. H., Major, P., Sonkoly, B., et al. (2013). Plant nonsense-mediated mRNA decay is controlled by different autoregulatory circuits and can be induced by an EJC-like complex. Nucleic Acids Res. 41, 6715–6728. doi: 10.1093/nar/gkt366
Rayson, S., Arciga-Reyes, L., Wootton, L., Zabala, M. D. T., Truman, W., Graham, N., et al. (2012). A role for nonsense-mediated mRNA decay in plants: pathogen responses are induced in Arabidopsis thaliana NMD mutants. PLoS ONE 7:e31917. doi: 10.1371/journal.pone.0031917
Rehwinkel, J., Letunic, I., Raes, J., Bork, P., and Izaurralde, E. (2005). Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11, 1530–1544. doi: 10.1261/rna.2160905
Riehs, N., Akimcheva, S., Puizina, J., Bulankova, P., Idol, R. A., Siroky, J., et al. (2008). Arabidopsis SMG7 protein is required for exit from meiosis. J. Cell Sci. 121, 2208–2216. doi: 10.1242/jcs.027862
Riehs-Kearnan, N., Gloggnitzer, J., Dekrout, B., Jonak, C., and Riha, K. (2012). Aberrant growth and lethality of Arabidopsis deficient in nonsense-mediated RNA decay factors is caused by autoimmune-like response. Nucleic Acids Res. 40, 5615–5624. doi: 10.1093/nar/gks195
Searle, I., He, Y., Turck, F., Vincent, C., Fornara, F., Krober, S., et al. (2006). The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 20, 898–912. doi: 10.1101/gad.373506
Sheldon, C. C., Burn, J. E., Perez, P. P., Metzger, J., Edwards, J. A., Peacock, W. J., et al. (1999). The FLF MADS box gene: a repressor of flowering in Arabidopsis regulated by vernalization and methylation. Plant Cell 11, 445–458. doi: 10.1105/tpc.11.3.445
Shi, C., Baldwin, I. T., and Wu, J. (2012). Arabidopsis plants having defects in nonsense-mediated mRNA decay factors UPF1, UPF2, and UPF3 show photoperiod-dependent phenotypes in development and stress responses. J. Integr. Plant Biol. 54, 99–114. doi: 10.1111/j.1744-7909.2012.01093.x
Song, H.-R., Song, J.-D., Cho, J.-N., Amasino, R. M., Noh, B., and Noh, Y.-S. (2009). The RNA binding protein ELF9 directly reduces suppressor of overexpression of CO1 transcript levels in Arabidopsis, possibly via nonsense-mediated mRNA decay. Plant Cell 21, 1195–1211. doi: 10.1105/tpc.108.064774
Spitzer, M., Wildenhain, J., Rappsilber, J., and Tyers, M. (2014). BoxPlotR: a web tool for generation of box plots. Nat. Methods 11, 121–122. doi: 10.1038/nmeth.2811
Suter, L., Rüegg, M., Zemp, N., Hennig, L., and Widmer, A. (2014). Gene regulatory variation mediates flowering responses to vernalization along an altitudinal gradient in Arabidopsis. Plant Physiol. 166, 1928–1942. doi: 10.1104/pp.114.247346
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Wang, C.-Q., Guthrie, C., Sarmast, M. K., and Dehesh, K. (2014). BBX19 interacts with CONSTANS to repress FLOWERING LOCUS T transcription, defining a flowering time checkpoint in Arabidopsis. Plant Cell 26, 3589–3602. doi: 10.1105/tpc.114.130252
Xu, M. Y., Zhang, L., Li, W. W., Hu, X. L., Wang, M.-B., Fan, Y. L., et al. (2013). Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J. Exp. Bot. 65, 89–101. doi: 10.1093/jxb/ert353
Yoine, M., Nishii, T., and Nakamura, K. (2006). Arabidopsis UPF1 RNA helicase for nonsense-mediated mRNA decay is involved in seed size control and is essential for growth. Plant Cell Physiol. 47, 572–580. doi: 10.1093/pcp/pcj035
Zacharaki, V., Benhamed, M., Poulios, S., Latrasse, D., Papoutsoglou, P., Delarue, M., et al. (2012). The Arabidopsis ortholog of the YEATS domain containing protein YAF9a regulates flowering by controlling H4 acetylation levels at the FLC locus. Plant Sci. 196, 44–52. doi: 10.1016/j.plantsci.2012.07.010
Keywords: UPF1, UPF3, NMD, flowering, Arabidopsis
Citation: Nasim Z, Fahim M and Ahn JH (2017) Possible Role of MADS AFFECTING FLOWERING 3 and B-BOX DOMAIN PROTEIN 19 in Flowering Time Regulation of Arabidopsis Mutants with Defects in Nonsense-Mediated mRNA Decay. Front. Plant Sci. 8:191. doi: 10.3389/fpls.2017.00191
Received: 19 November 2016; Accepted: 30 January 2017;
Published: 14 February 2017.
Edited by:
Jill Christine Preston, University of Vermont, USAReviewed by:
Ben Holt, University of Oklahoma, USADaniel Paul Woods, University of Wisconsin-Madison, USA
Copyright © 2017 Nasim, Fahim and Ahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji Hoon Ahn, amFobkBrb3JlYS5hYy5rcg==