 Sajjad Asaf1†
Sajjad Asaf1† Muhammad Waqas1,2†
Muhammad Waqas1,2† Abdul L. Khan3
Abdul L. Khan3 Muhammad A. Khan1Sang-Mo Kang1
Muhammad A. Khan1Sang-Mo Kang1 Qari M. Imran1Raheem Shahzad1
Qari M. Imran1Raheem Shahzad1 Saqib Bilal1
Saqib Bilal1 Byung-Wook Yun1
Byung-Wook Yun1 In-Jung Lee1*
In-Jung Lee1*- 1School of Applied Biosciences, Kyungpook National University, Daegu, South Korea
- 2Department of Agriculture, Abdul Wali Khan University Mardan, Mardan, Pakistan
- 3Chair of Oman's Medicinal Plants and Marine Natural Products, University of Nizwa, Nizwa, Oman
Oryza minuta, a tetraploid wild relative of cultivated rice (family Poaceae), possesses a BBCC genome and contains genes that confer resistance to bacterial blight (BB) and white-backed (WBPH) and brown (BPH) plant hoppers. Based on the importance of this wild species, this study aimed to understand the phylogenetic relationships of O. minuta with other Oryza species through an in-depth analysis of the composition and diversity of the chloroplast (cp) genome. The analysis revealed a cp genome size of 135,094 bp with a typical quadripartite structure and consisting of a pair of inverted repeats separated by small and large single copies, 139 representative genes, and 419 randomly distributed microsatellites. The genomic organization, gene order, GC content and codon usage are similar to those of typical angiosperm cp genomes. Approximately 30 forward, 28 tandem and 20 palindromic repeats were detected in the O. minuta cp genome. Comparison of the complete O. minuta cp genome with another eleven Oryza species showed a high degree of sequence similarity and relatively high divergence of intergenic spacers. Phylogenetic analyses were conducted based on the complete genome sequence, 65 shared genes and matK gene showed same topologies and O. minuta forms a single clade with parental O. punctata. Thus, the complete O. minuta cp genome provides interesting insights and valuable information that can be used to identify related species and reconstruct its phylogeny.
Introduction
The angiosperm chloroplast (cp) is a uniparentally inherited and stable structure. Accordingly, it is considered to be an informative and valuable resource for phylogenetic analysis in plants at multiple taxonomic levels (Nadachowska-Brzyska et al., 2015) compared to mitochondrial genomes (Timmis et al., 2004). Most cp genomes range from 120 to 210 kb and have a quadripartite structure that is typically composed of a small single-copy region (SSC), a large single-copy region (LSC) and a pair of inverted repeats (IRs) (Yurina and Odintsova, 1998; Wang et al., 2015). In most cases, differences in the length of the IRs determine length differences of the cp genome (Chang et al., 2006; Guisinger et al., 2011).
Previously, phylogenetic analyses have been based on sequencing one or a few loci from the chloroplast. Due to the availability of complete chloroplast sequences in public databases and advances in next-generation sequencing techniques, analyses based on the entire chloroplast genome are achievable and yield higher quality and more valuable information, which could reveal detailed insight into genomic organization (Martin et al., 2005). Indeed, examining the entire cp genome can resolve previously ambiguous phylogenetic relationships among species (Jansen et al., 2007; Moore et al., 2010). Due to availability of high-throughput sequencing technology as well as the comparatively small size and structural similarity of cp genomes, hundreds of sequencing projects in terrestrial plants have recently been reported (Wu, 2016b).
Rice is an important cereal crop that provides essential food and energy for more than half of the world's population. In addition, rice is considered a model crop for studies on cereal genomics. Two species of the genus Oryza (O. sativa, and O. glaberrima) are cultivated, though there are more than 20 wild species (Evenson and Gollin, 1997; Sang and Ge, 2007). Different species are categorized into 10 genome types, six are diploid (AA, BB, CC, EE, FF, and GG) (2n = 2x = 24) and the other four are allelotetraploid (BBCC, CCDD, HHJJ, and HHKK) (2n = 4x = 28) (Ge et al., 1999). About one half of the species in Oryza genus are allotetraploids that originated through interspecific hyberdization and genome doubling (Vaughan, 1989; Bao and Ge, 2008; Jacquemin et al., 2013). Rice (O. sativa) with an AA genome type, is one of the most important species, and it is further divided into the subspecies japonica and indica, which are distributed globally (Chang, 1976; Wambugu et al., 2015).
Because of the importance of Oryza as a major food crop, great attention has been given to understanding the genetic makeup and phylogeny of this genus, both within the genus and species (Guo and Ge, 2005). In plants, sequencing functional genes in cpDNA (chloroplast DNA) is helpful for resolving issues related to molecular taxonomy and phylogenetic reconstruction (Jansen et al., 2007; Moore et al., 2010; Wu and Ge, 2012), and such approaches can yield vast benefits in plant breeding and conservation strategies. Currently, 10 cp genomes belonging to Oryzeae have been published (Waters et al., 2012; Brozynska et al., 2014). Some wild Oryza species are better able than cultivated Oryza species to resist biotic and abiotic stresses and attack from insect pests. Thus, cultivated species can be improved through introgression of resistance genes from wild species (Heinrichs et al., 1985). For example, resistance traits from wild O. minuta, a tetraploid wild relative of cultivated rice, have been reported. O. minuta has a BBCC genome type and exhibits significant potential to resist blast blight, bacterial blight (BB), and white-backed plant hopper (WBPH) and brown plant hopper (BPH) diseases (Vaughan, 1994). Such diseases are damaging to the growth and yield of cultivated rice. In addition, stress tolerance genes from O. minuta have been successfully transferred to cultivated rice through introgression (Amante-Bordeos et al., 1992; Rahman et al., 2009). Overall, wild species such as O. minuta possess valuable genetic diversity that can contribute greatly to improving the growth and yield of various crops (Amante-Bordeos et al., 1992). To identify desirable genes and ensure effective conservation, it is essential to analyze phylogenetic and evolutionary relationships among species (Guo et al., 2013). Previously, it was reported that O. minuta was originated from allopolyploidization of O. officinalis (paternal) and O. punctate (meternal) (Ammiraju et al., 2010; Zou et al., 2015).
In this study, we assembled for the first time the complete chloroplast genome sequence of O. minuta, and performed detailed phylogenetic analyses on the basis of complete cp genome and 65 shared genes. The complete cp genome of O. minuta, in conjunction with previously reported cp genome sequences, will improve our understanding of O. minuta and the evolutionary history of genus Oryza. Hence, we analyzed the fully assembled cp genome of O. minuta and compared it to eleven closely related species: O. australiensis EE, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis CC, and O. punctata BB.
Materials and Methods
In this study, a standard protocol for DNA extraction was used as described in detailed by Sierro et al. (2014). The extracted DNA was sequenced using an Illumina HiSeq-2000 (Illumina, San Diego, CA, USA) platform at Macrogen (Macrogen, Seoul, Korea), and the O. minuta cp genome was obtained by de novo assembly of the entire genome sequence via a bioinformatics pipeline (http://phyzen.com). A 400-bp paired-end library was produced according to the Illumina PE standard protocol, generating 28,110,596 bp of total reads with a 120-bp average read length. Raw reads with Phred scores of 20 or less were removed from the total PE reads using the CLC-quality trim tool, and de novo assembly was conducted on trimmed reads using CLC Genomics Workbench v7.0 (CLC Bio, Aarhus, Denmark) with parameters of minimum (200 to 600 bp) autonomously controlled overlap size. All contigs were then mapped and assembled against the reference cp genomes of O. officinalis and O. punctata by following a previously described method (Wu, 2016a,b). Primers were designed (Table S1) to test for correct sequence assembly. PCR amplification was performed in a total volume of 20 μl containing 1 × reaction buffer, 0.4 μl dNTPs (10 mM), 0.1 μl Taq (Solg™ h-Taq DNA Polymerase), 1 μl (10 pm/μl) primers, and 1 μl (10 ng/μl) DNA. The PCR program consisted of initial denaturation at 95°C for 5 min followed by 35 cycles of 95°C for 30 s, 65°C for 20 s and 72°C for 30 s, with a final extension step at 72°C for 5 min. After incorporation of the sequencing results, the finished cp genome was applied as a reference to map previously obtained short reads to refine the assembly based on maximum sequence coverage.
Genome Annotation and Sequence Architecture
The program DOGMA was used to annotate the O. minuta cp genome (Wyman et al., 2004). The annotation results were checked manually, and codon positions were adjusted by comparison to homologs from the cp genomes of O. australiensis and O. sativa ssp. indica in the database. All transfer RNA sequences were verified using tRNAscan-SE version 1.21 (Schattner et al., 2005) with the default settings. OGDRAW (Lohse et al., 2007) was applied to illustrate the structural features of the O. minuta cp genome. To examine deviations in synonymous codon usage by avoiding the influence of amino acid composition, the relative synonymous codon usage (RSCU) was determined using MEGA 6 software (Kumar et al., 2008). mVISTA software was used in the Shuffle-LAGAN mode to compare the complete variation in the O. minuta cp genome with eleven other cp genomes using the O. minuta annotation as a reference (Frazer et al., 2004).
Characterization of Repeat Sequences and SSRs
We employed REPuter to identify repeat sequences, including palindromic, reverse, and direct repeats, within the cp genome (Kurtz et al., 2001). The following settings for repeat identification were used: (1) Hamming distance of 3; (2) 90% or greater sequence identity; (3) a minimum repeat size of 30 bp. Phobos version 3.3.12 (Leese et al., 2008) was used to detect (SSRs) within the cp genome, with the search parameters set at ten repeat units ≥10 for mononucleotides, eight repeat units ≥8 for dinucleotides, four repeat units ≥4 for trinucleotides and tetranucleotides, and three repeat units ≥3 for pentanucleotide and hexanucleotide SSRs. Tandem repeats in the O. minuta cp genome were identified using Tandem Repeats Finder version 4.07 b (Benson, 1999) with the default settings.
Sequence Divergence and Phylogenetic Analysis
Complete cp genomes as well as a separate partition using only 65 shared genes were employed to analyze the average pairwise sequence divergence for 11 Oryza species: O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis, and O. punctata. Missing and ambiguous gene annotations were confirmed by comparative sequence analysis after a multiple sequence alignment and gene order comparison. These regions were aligned using MAFFT (version 7.222) (Katoh and Standley, 2013) with the default parameters. Kimura's two-parameter (K2P) model was selected to calculate pairwise sequence divergences (Kimura, 1980). To resolve the O. minuta phylogenetic position within the rice tribe (Oryzeae), 13 published cp genomes were downloaded from the NCBI database for analyses. First, multiple alignments were performed using the complete cp genomes based on the conserved structure and gene order of the chloroplast genomes (Wicke et al., 2011). Four methods were employed to construct phylogenetic trees, including Bayesian inference (BI) implemented with MrBayes 3.12 (Ronquist and Huelsenbeck, 2003), maximum parsimony (MP) with PAUP 4.0 (Swofford, 1993), and maximum likelihood (ML) and neighbor-joining (NJ) with MEGA 6 (Kumar et al., 2008) using described settings (Wu et al., 2015; Asaf et al., 2016a). In the second phylogenetic analysis, 65 shared genes from the cp genomes of 12 Oryza species and two Zizania outgroup species were aligned in ClustalX using the default settings, followed by manual adjustment to preserve reading frames. The above four phylogenetic-inference methods were used to infer trees from the 65 concatenated genes using the same settings (Wu et al., 2015; Asaf et al., 2016a).
Results and Discussion
Chloroplast Genome Organization of O. minuta
The O. minuta cp genome was assembled by mapping all Illumina reads to the draft cp genome sequence using CLC Genomics Workbench v7.0. A total of 1,577,251 reads were obtained, with an average length of 120 bp, for 504.211X coverage of the cp genome. The consensus sequence for a specific position was generated by assembling reads mapped with at least 875 reads per position and was used to construct the complete sequence of the O. minuta cp genome. The complete O. minuta cp genome is 135,094 bp in size (GenBank: KU179220), which is similar to the already reported cp genome sizes of related Oryza species and is within the range of other angiosperms (Yang et al., 2010). The cp genome possesses a typical quadripartite structure, which includes a pair of inverted repeats (IRa and IRb 20,836 bp) and separate SSC (12,446 bp) and LSC (80,974 bp) regions (Table 1, Figure 1). The GC content (39%) of the O. minuta cp genome is very similar to that of other Oryza species cp genomes (Table 1) (Wu et al., 2015). However, the GC content is unequally distributed in the O. minuta cp genome: it is highest in the IR regions (44.3%), moderate in the LSC regions (37.1%) and lowest in the SSC regions (33.3%). This high IR GC percentage is due to the presence of eight ribosomal RNA (rRNA) sequences in these regions. These results are similar to a previously reported high GC percentage in IR regions (Qian et al., 2013).
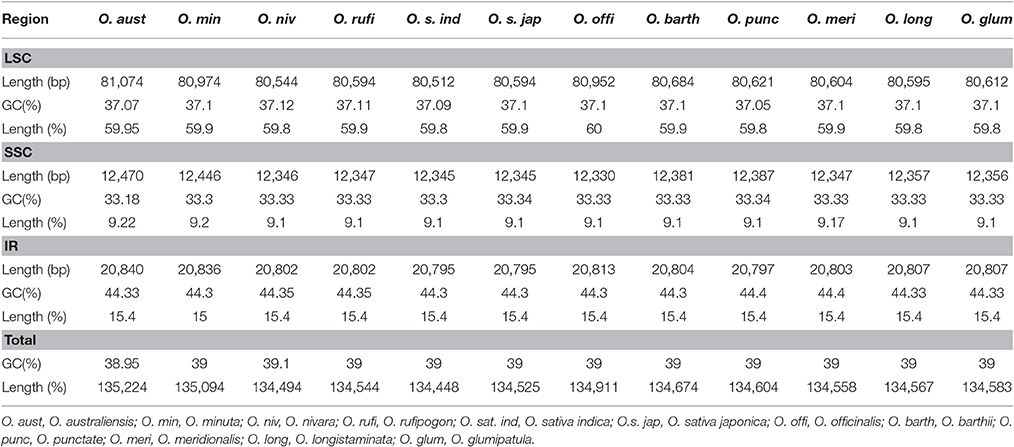
Table 1. Summary of complete chloroplast genomes for twelve Oryza species.
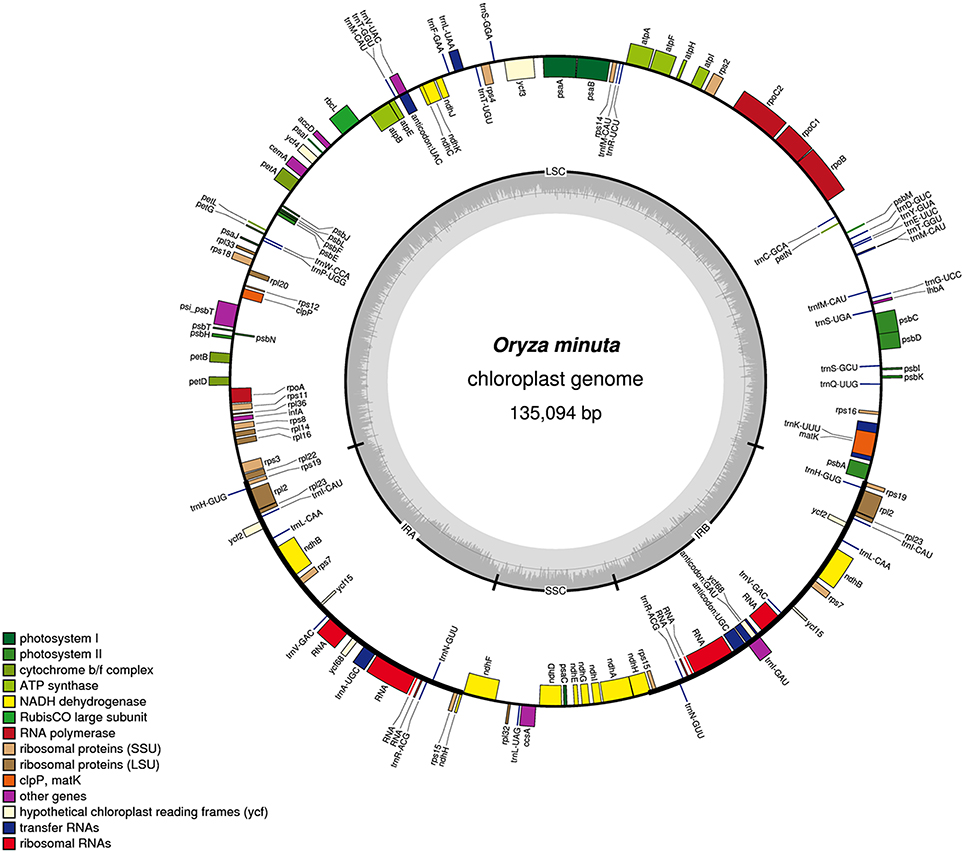
Figure 1. Gene map of the O. minuta chloroplast genome. Genes drawn inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. Genes belonging to different functional groups are color coded. The darker gray color in the inner circle corresponds to the GC content, and the lighter gray color corresponds to the AT content.
A total of 139 genes were found in the O. minuta cp genome, of which 110 are unique, including 91 protein-coding genes, 40 tRNA genes, and 8 rRNA genes (Figure 1, Table 2). Of these, 11 protein-coding, four rRNA, and eight tRNA genes are duplicated in the IR regions. The LSC region comprises 62 protein-coding and 24 tRNA genes, whereas the SSC region comprises 11 protein-coding genes and one tRNA gene. The protein-coding genes present in the O. minuta cp genome include nine genes encoding large ribosomal proteins (rpl2, 14, 16, 20, 22, 23, 32, 33, 36), 12 genes encoding small ribosomal proteins (rps2, 3, 4, 7, 8, 11, 12, 14, 15, 16, 18, 19), five genes encoding photosystem I components (psaA, B, C, I, J), 10 genes related to photosystem II (Table 2), and six genes (atpA, B, E, F, H, I) encoding ATP synthase and electron transport chain components (Table 2). A similar pattern of protein-coding genes is also present in O. sativa (Zhang et al., 2012) and O. glaberrima (Wambugu et al., 2015). There are 11 intron-containing genes, 10 of which contain one intron, with only ycf3 genes having two introns (Table S2). The ndhA gene has the longest intron (965 bp).
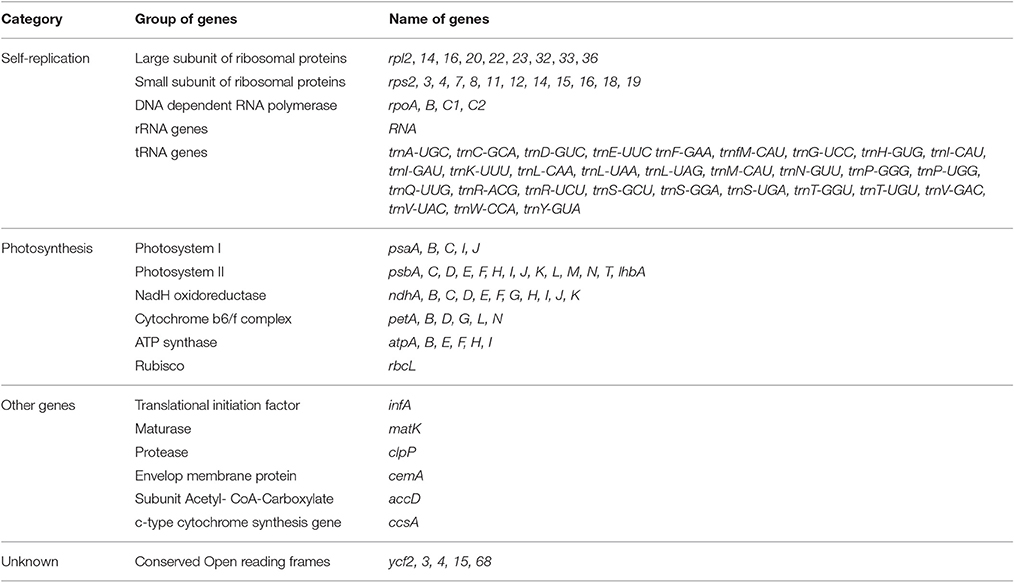
Table 2. Genes in the sequenced O. minuta chloroplast genome.
Protein, rRNAs, and tRNAs are encoded by 45.1, 6.83, and 2.2% of the entire cp genome, respectively, and the remaining 45.8% is composed of non-coding regions (Table 3). The total protein-coding sequences (CDSs) are 60,948 bp in length and consist of 91 genes encoding 20,354 codons (Tables 1, 4). The O. minuta cp genome codon usage frequency was determined based on tRNA and protein-coding gene sequences (Table 5). Leucine (10.7%) and cysteine (1.2%) are the maximum and minimum commonly encoded amino acids, and isoleucine, serine, glycine, arginine and alanine are encoded by 7.9, 7.5, 7.4, 6.5, and 6.1% of CDSs, respectively (Figure S1). Similar ratios for amino acids are present in previously reported cp genomes (Qian et al., 2013; Chen et al., 2015).
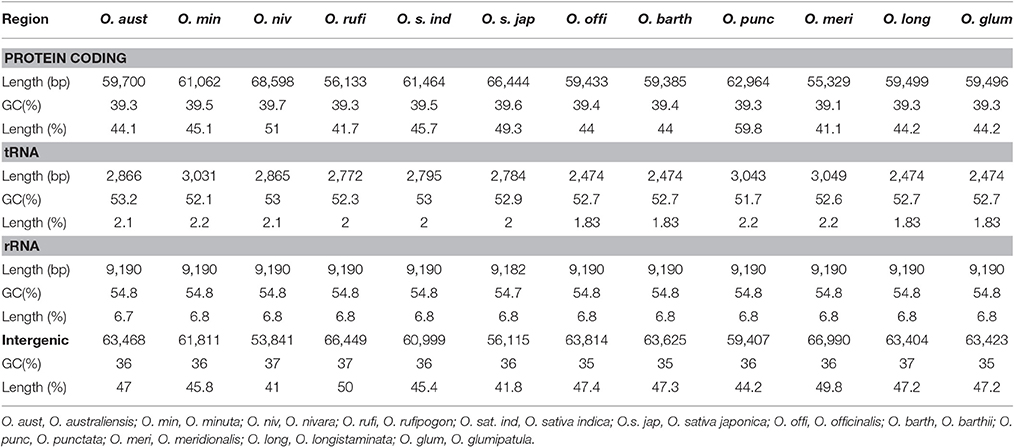
Table 3. Comparison of coding and non-coding region sizes among twelve Oryza species.
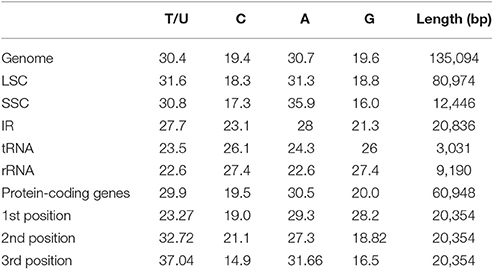
Table 4. Base compositions in the O. minuta cp genome.
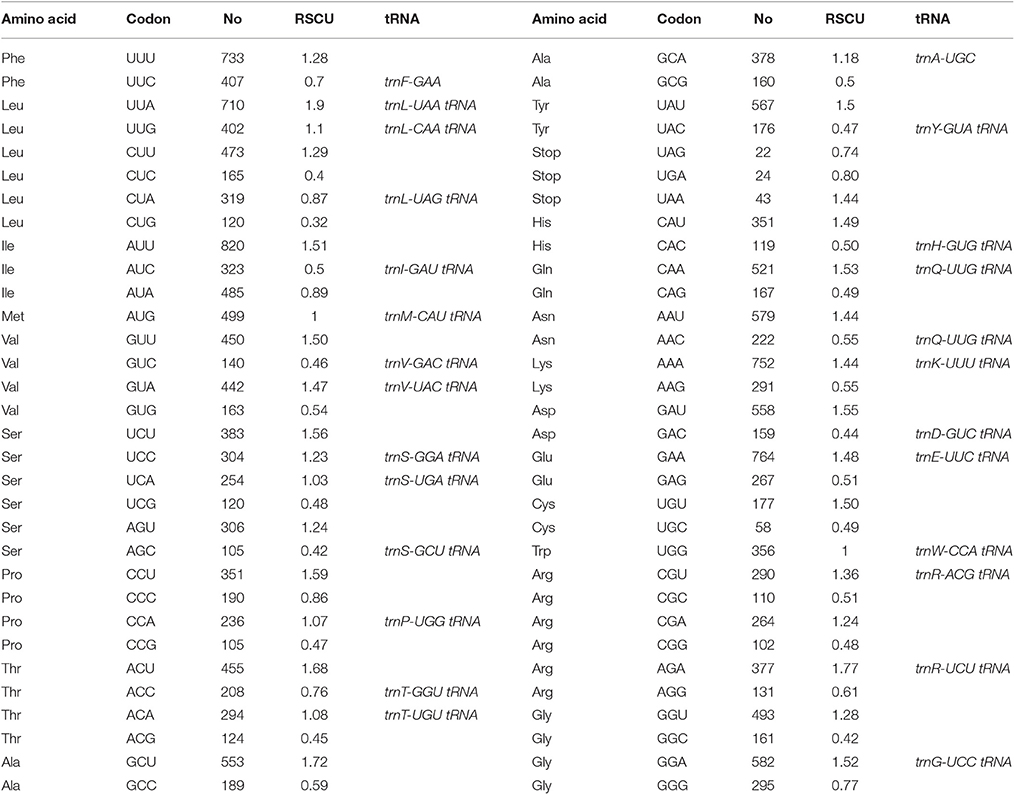
Table 5. The codon–anticodon recognition pattern and codon usage for the O. minuta chloroplast genome.
Among these, the maximum and minimum codons used are ATT (820), encoding isoleucine, and TTG and ATT (1, 1), encoding methionine. The AT content is 52.5, 60.0, and 68.7% at the 1st, 2nd, and 3rd codon positions, respectively, within CDS regions (Table 4). The preference for a high AT content at the 3rd codon position is similar to the A and T concentrations reported in various terrestrial plant cp genomes (Morton, 1998; Nie et al., 2012; Qian et al., 2013). In total, 42.65 and 57% of all types of preferred synonymous codons (RSCU>1) ending with A and U and C and G, respectively, were found. Non-preferred synonymous codons (RSCU <1) are 42.40 and 57.50% for C and G and A and U. Usage of the start codon AUG and UGG, the latter encoding tryptophan, has no bias (RSCU = 1) (Table 5).
Repeat Analysis
Repeat sequences, which play a role in genome rearrangements, are very helpful in phylogenetic studies (Cavalier-Smith, 2002; Nie et al., 2012). Furthermore, analyses of various cp genomes revealed that repeat sequences are essential to induce indels and substitutions (Yi et al., 2013). Repeat analysis of the O. minuta cp genome showed 20 palindromic repeats, 30 forward repeats, and 28 tandem repeats (Figure 2A). Among these, 17 forward repeats are 30–44 bp in length, with only three tandem repeats of the same length and 18 15–29 bp in length (Figures 2A–D). Similarly, 11 palindromic repeats are 30–44 bp, and 6 repeats are 45–59 bp in length (Figure 2B). Overall, 78 repeats were found in the O. minuta cp genome. Similarly, 73, 73, 76, 71 72, 78, 72, 71, 73, 77, and 74 repeat pairs were found in previously reported O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis and O. punctata genomes, respectively (Figure 2A). This suggests that O. minuta is more similar to O. barthii and O. officinalis in terms of repeats. Approximately 29.4% of these repeats are distributed in protein-coding regions. Previous reports suggest that sequence variation and genome rearrangement occur due to the slipped-strand mispairing and improper recombination of these repeat sequences (Cavalier-Smith, 2002; Asano et al., 2004; Timme et al., 2007). Furthermore, the presence of these repeats indicates that the locus is a crucial hotspot for genome reconfiguration (Gao et al., 2009; Nie et al., 2012). Additionally, these repeats are an informative source for developing genetic markers for phylogenetic and population studies (Nie et al., 2012).
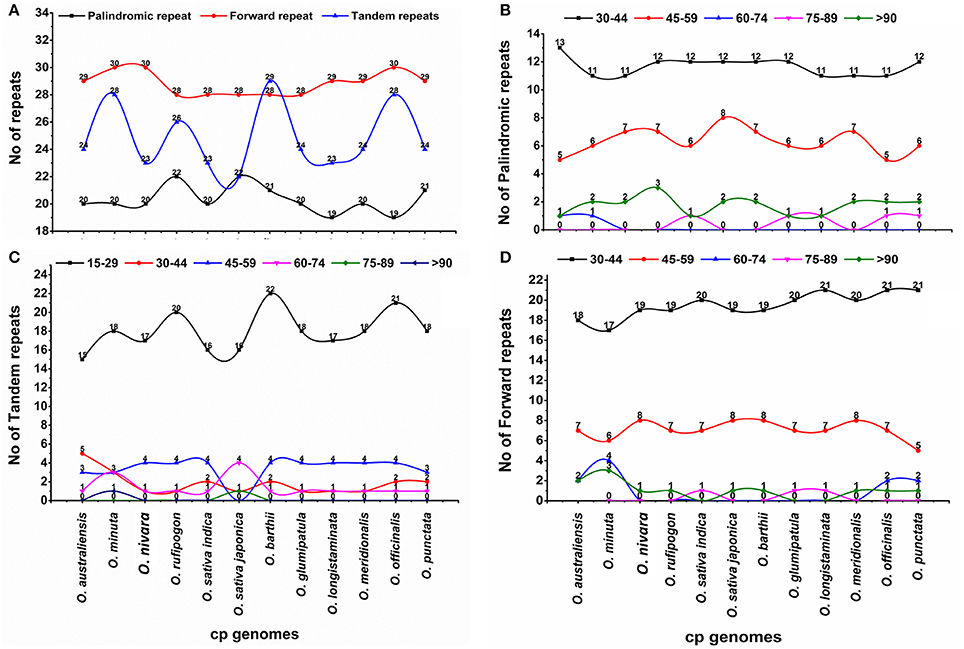
Figure 2. Analysis of repeated sequences in twelve Oryza chloroplast genomes. (A) Total of three repeat types; (B) frequency of the palindromic repeat by length; (C) frequency of the tandem repeat by length; (D) frequency of forward repeat by length.
SSR Analysis
Simple sequence repeats (SSRs), or microsatellites, are repeating sequences of typically 1–6 bp that are distributed throughout the genome. In this study, we detected perfect SSRs in O. minuta together with 11 other Oryza species cp genomes (Figure 3A). Certain parameters were set because SSRs of 10 bp or longer are prone to slipped-strand mispairing, which is believed to be the main mechanism for SSR polymorphisms (Rose and Falush, 1998; Raubeson et al., 2007; Huotari and Korpelainen, 2012). A total of 419 perfect microsatellites were found in the O. minuta cp genome (Figure 3A). Similarly, 418, 413, 416, 416, 419, 420, 419, 419, 421, 429, and 422 SSRs were detected in O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis and O. punctata, respectively (Figure 3A). The majority of SSRs in these cp genomes possess a dinucleotide repeat motif, varying in quantity from 269 in O. sativa ssp. indica to 276 in O. officinalis. Mononucleotide SSRs are the second most common, ranging from 92 in O. nivara to 100 in O. officinalis. Using our search criterion, only one pentanucleotide SSR was found in O. nivara, O. rufipogon, O. indica and O. officinalis (Figure 3A). In O. minuta, most mononucleotide SSRs are A (97%) and T (2.12.30%) motifs, with the majority of dinucleotide SSRs being A/G (47.05%) and A/T (38.60%) motifs (Figure 3B). Approximately 62% of SSRs are located in non-coding regions; approximately 4.3% are present in rRNA sequences and 2.3% in tRNA genes (Figure 3C). Further analysis revealed that approximately 66.82% of SSRs occur in the LSC region, whereas 24.34 and 8.83% were found in IR and SSC regions, respectively (Figure 3D). These results are similar to previous reports that SSRs are unevenly distributed in cp genomes, and the findings might provide more information for selecting effective molecular markers for detecting intra- and interspecific polymorphisms (Powell et al., 1995a,b; Provan et al., 1997; Pauwels et al., 2012). Furthermore, most mononucleotides and dinucleotides are composed of A and T, which may contribute to bias in base composition, consistent with other cp genomes (Li et al., 2013). Our findings are comparable to previous reports that SSRs found in cp genome are generally composed of polythymine (polyT) or polyadenine (polyA) repeats and infrequently contain tandem cytosine (C) and guanine (G) repeats (Kuang et al., 2011). Therefore, these SSRs identified contribute to the AT richness of the O. minuta cp genome, as previously reported for various species (Kuang et al., 2011; Chen et al., 2015).
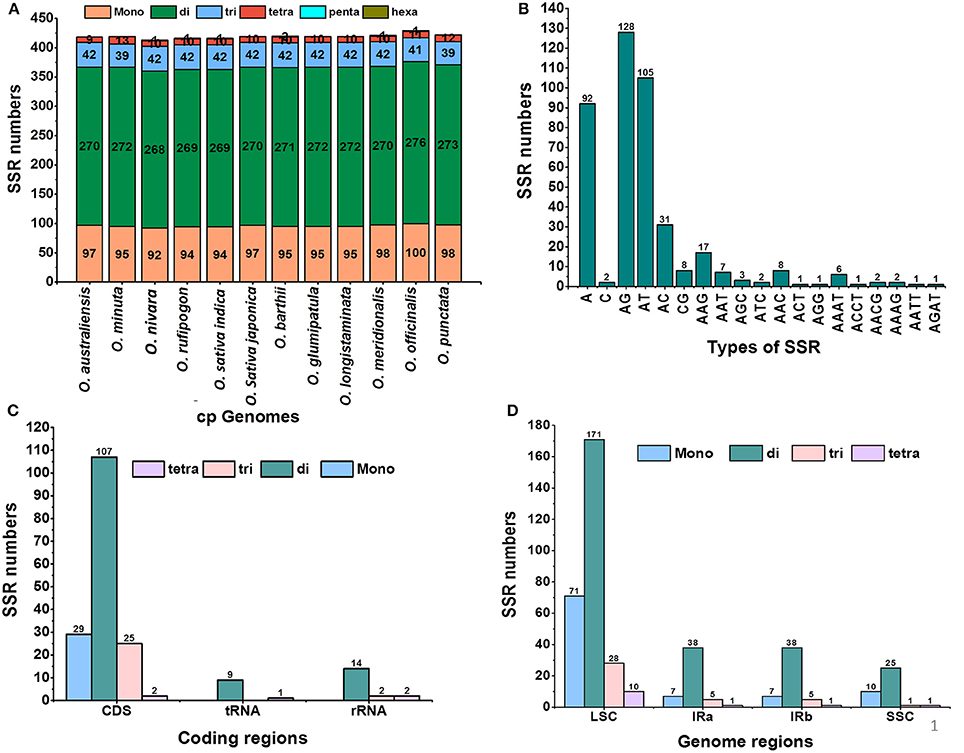
Figure 3. Analysis of simple sequence repeats (SSRs) in twelve Oryza chloroplast genomes. (A) Number of different SSR types detected in twelve genomes; (B) frequency of identified SSR motifs in different repeat class types; (C) frequency of identified SSRs in coding regions; (D) frequency of identified SSRs in LSC, SSC and IR regions.
Structural and Sequence Comparisons of cp Genomes in Oryza
Eleven complete cp genomes within the Oryza genus (O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis, and O. punctata) were selected for comparison with that of O. minuta (135,094 bp). O. australiensis has the largest genome, and this difference is mostly attributed to variation in the length of the LSC region (Table 1). Analysis of genes with known functions showed that O. minuta shares 65 protein-coding genes with eleven other Oryza species. The number of unique genes found in O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis, and O. punctata was 110, 100, 101, 108, 80, 104, 104, 104, 100, 104 and 114, respectively (Table S3). Furthermore, the O. minuta cp genome has a gene content and organization that are similar to other Oryza species and members of Poaceae (Wicke et al., 2011); however, as for other grasses, it lacks a ycf1 gene, and the accD gene is a truncated pseudogene. Because these genes are essential for the survival of photosynthetic plants (Drescher et al., 2000; Kode et al., 2005), they were most likely functionally transferred to the nucleus or functionally replaced by a eukaryotic gene, as observed for the accD plastid gene in other plant families (Babiychuk et al., 2011; Rousseau-Gueutin et al., 2011).
Pairwise cp genomic alignment between O. minuta and the 11 other genomes showed a high degree of synteny. The O. minuta cp genome annotation was used as a reference for plotting the overall sequence identity of the cp genomes of the 11 Oryza species in mVISTA (Figure 4), and the results revealed high sequence identity with all 11 Oryza species. However, except for O. australiensis, relatively lower identity was also observed with these species in various comparable genomic regions, particularly the rps3, rpl22, rpl23, rpl2, and rps19 regions (Figure 4). In addition, the LSC and SSC regions show less similarity than the two IR regions in all Oryza species. In addition, non-coding regions exhibit greater divergence than coding regions. These highly divergent regions include rbcL, rps16-trnQ, trnfM-trnM, psbM-petN, rpoC2, atpI-atpH, ndhA rpl33, petA-psbJ, ccsA, ndhF-rpl32, and ycf3. Similar results related to these genes were also reported by Qian et al. (2013). Our results also confirm similar differences among various coding regions in the analyzed species, as suggested by Kumar et al. (2009).
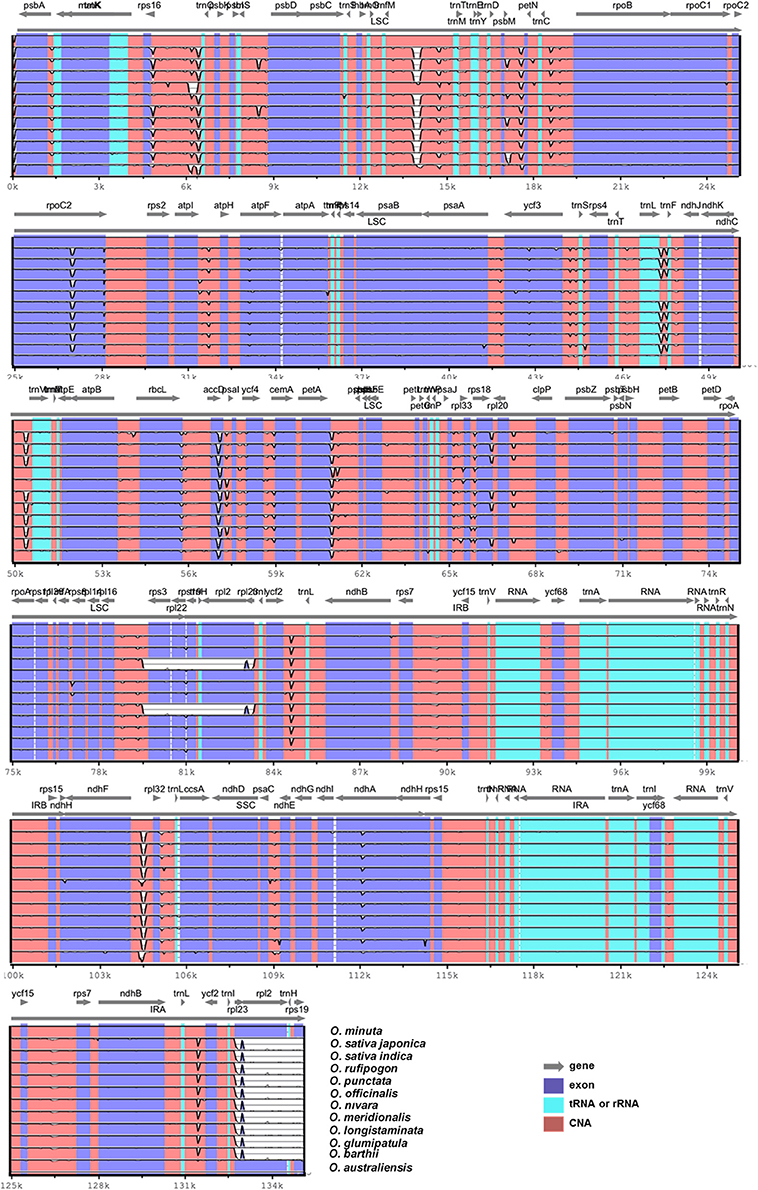
Figure 4. Alignment of twelve chloroplast genome sequences. VISTA-based identity plot showing sequence identity among twelve Oryza species using O. minuta as a reference. The thick black line shows the inverted repeats (IRs) in the chloroplast genomes.
We compared the cp genomes and calculated the average pairwise sequence divergence among the 12 species (Table S4). Of these, the O. minuta genome has 0.005 average sequence divergence, and high divergence was found for O. australiensis (0.00725); O. officinalis has the lowest average sequence divergence (0.0044). Furthermore, the twelve most divergent genes among these genomes are petG, matK, infA, ccsA, rpoC2, clcP, psbE, rbcL, psbN, rps18, rpl36, and ndhF. The highest average sequence distance was found for rpoC2 (0.01983), followed by petG (0.0154) (Figure 5). Both these genes are located in LSC regions and display a trend toward more rapid evolution.
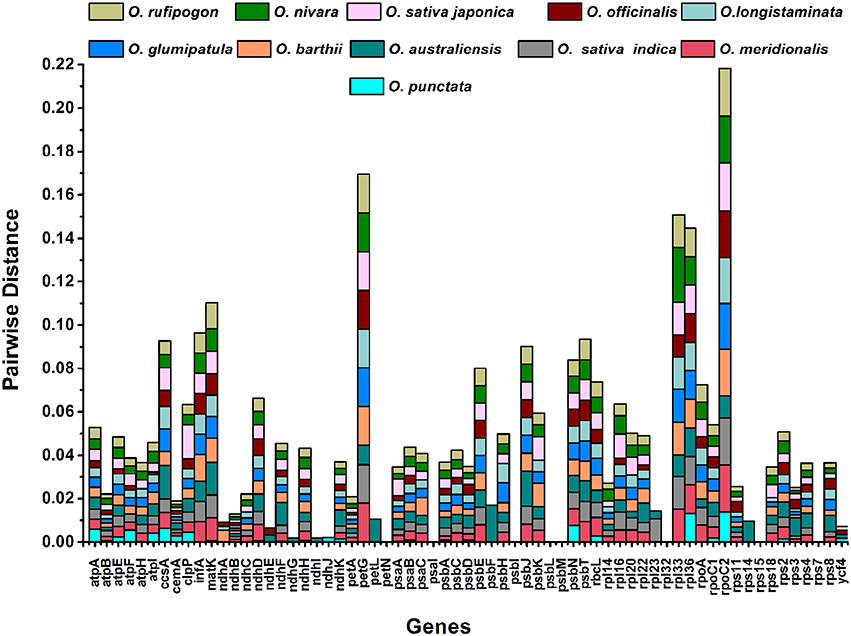
Figure 5. Pairwise sequence distances of Oryza minuta genes with O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis, and O. punctata.
IR Contraction and Expansion
Expansion and contraction at the borders of IR regions are the main reason for size variations in the cp genome and play a vital role in its evolution (Raubeson et al., 2007; Wang et al., 2008; Yang et al., 2010, 2014). A detailed comparison on four junctions (JLA, JLB, JSA, and JSB) between the two IRs (IRa and IRb) and the two single-copy regions (LSC and SSC) was performed among O. australiensis, O. nivara, O. rufipogon, O. sativa L. ssp. indica, O. sativa L. ssp. japonica, O. barthii, O. glumipatula, O. longistaminata, O. meridionalis, O. officinalis and O. punctata with regard to O. minuta by carefully analyzing the exact IR border positions and adjacent genes (Figure 6). Despite the similar length of the O. minuta IR region with the other eleven Oryza species, from 20,836 bp to 20,840 bp, some IR expansion and contraction was observed. JLA is located between rps19 and psbA, and variation in distances between rps19 and JLA range from 40 to 49 bp across all species; the distance in O. minuta is 46 bp. The distance between psbA and JLA is 81 bp in O. minuta, which is similar to the other genomes (81 bp). The distance between rpl22 and JLB varies from 23 bp to 29 bp. In O. minuta, 1-bp variations exist in the JSA border region compared to the other cp genomes. The ndhH gene traverses the SSC and IRa regions, with approximately 164 bp located in the IR region for O. minuta. Furthermore, there are 16-bp variations observed compared with O. officinalis for ndhF, ndhH and rps15 in the SSC and IRb regions, located 41 bp, 164 bp and 302 bp from the JSB and JSA border regions, respectively.

Figure 6. Comparison of border distances between adjacent genes and junctions of LSC, SSC, and two IR regions among chloroplast genomes of twelve Oryza species. Boxes above or below the main line indicate the adjacent border genes. The figure is not to scale with regard to sequence length and only shows relative changes at or near IR/SC borders.
Phylogenetic Analysis
The Oryza genus is composed of 23 species distributed in different regions of America, Africa, Asia, and Australia (Ge et al., 1999). Continued efforts have expanded our ability to differentiate among and to understand the genomic structure and phylogenetic relationships of rice species (Khush, 1997). Taxonomy and phylogeny of the rice genus have been extensively investigated at genus level (Ge et al., 1999; Zhu and Ge, 2005; Jacquemin et al., 2013). Previous evolutionary relationships among different rice genomes and species were estimated by nuclear and chloroplast DNA restriction fragment-length polymorphisms (Ge et al., 1999; Zou et al., 2015), but complete genome sequencing provides more detailed insight (Wambugu et al., 2015; Wu et al., 2015; Asaf et al., 2016b). In this regard, O. minuta has been poorly investigated. In this study, the phylogenetic position of O. minuta within Oryza was established by utilizing complete cp genomes and 65 shared genes among 12 Oryza members (Figures 7A,B). Two species, Zizania aquatic and Zizania latifolia were set as outgroups. Phylogenetic analysis using Bayesian inference (BI), maximum parsimony (MP), maximum likelihood (ML) and neighbor-joining (NJ) methods were performed. The results showed same phylogenetic signals for the complete cp genomes and 65 shared genes of O. minuta. The complete genome sequences (Table S5) and 65 shared genes (Tables S3, S6) from all species generated phylogenetic trees with same topologies (Figures 7A,B). In these phylogenetic trees based on the entire genome data set and 65 shared genes, O. minuta formed a single clade with O. punctata, with high BI and bootstrap support using four different methods (Figures 7A,B). Furthermore, the tree topology confirmed the relationship inferred from the phylogenetic work conducted by Ge et al. (1999) and Zou et al. (2015). This position of O. minuta confirms the previously published phylogeny described by Ge et al. (1999). Ge et al. (1999) reported that O. minuta BBCC shares a clade with O. punctata BB with regard to Adh1, whereas it forms a clade with O. officinalis CC in the Adh2 phylogenetic analysis. Similar resuls was suggested by Zou et al. (2015), whereby phylogenetic analysis of the four nuclear loci and three meternally interited chloroplast fragments from different Oryza species grouped O. minuta in a clade with maternal parent O. punctata BB (Zou et al., 2015). As the phylogenetic tree based on the matK gene represents the maternal genealogy of rice species, which can offer an opportunity to identify maternal parents of allotetraploid species, we performed an additional phylogenetic analysis of O. minuta using the matK gene from related species (Figure S2). The results revealed a single clade for O. minuta with parental O. punctata. Similar results was also suggested by Ge et al. (1999), whereby phylogenetic analysis of the matK gene from different Oryza species grouped O. minuta in a clade with the maternal parent O. punctata BB instead of O. officinalis CC. Furthermore, the result suggests that there is no conflict between the entire genome data set and 65 shared genes of these cp genomes.
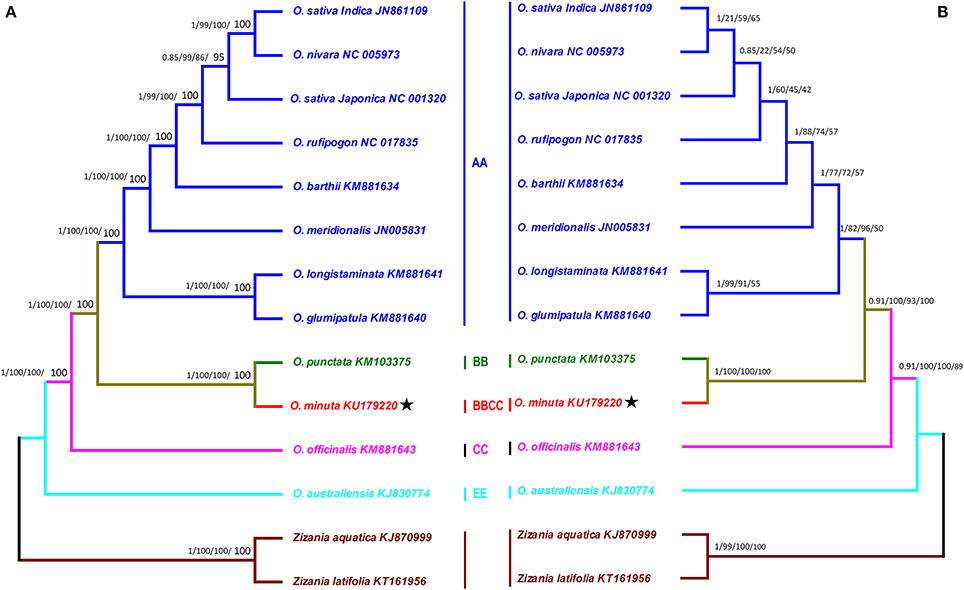
Figure 7. Phylogenetic trees were constructed for 14 species from the rice tribe using different methods, and two Bayesian trees are shown for data sets of the entire genome sequence and 65 shared genes. (A) The entire genome sequence data set (B). The data set of 65 shared genes. Each data set was used with four different methods, Bayesian inference (BI), maximum parsimony (MP), maximum likelihood (ML) and neighbor-joining (NJ). Numbers above the branches are the posterior probabilities of BI and bootstrap values of MP, ML, and NJ, respectively. Stars represent positions for O. minuta (KU179220) in the two trees.
Conclusion
This study reports the first complete chloroplast genome sequence of O. minuta (135,094 bp). The structure and organization of this genome is very similar to previously reported cp genomes from the tribe Oryzeae. The location and distribution of repeat sequences was detected, and sequence divergences among cp genomes and 65 shared genes were identified with related species. No major structural rearrangement of Oryza species cp genomes was observed. Phylogenetic analyses showed that data sets based on the entire genome and 65 shared genes generate trees with same topologies regarding the placement of O. minuta. These findings provide a valuable analysis of the complete cp genome of O. minuta, which can be used to identify species and clarify taxonomic questions.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
All the research work was financially supported by National Research Foundation of Korea (NRF), Ministry of Science, ICT and Future-Planning through Basic-Science Research Program (2014R1A1A1004918).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00304/full#supplementary-material
Table S1. Primers used for gap closure in O. minuta.
Table S2. Genes with introns in the O. minuta chloroplast genome and the lengths of exons and introns.
Table S3. List of shared and unique genes among 12 Oryza cp genomes.
Table S4. Average pairwise sequence distance of O. minuta with the cp genome of 11 Oryza species.
Table S5. The full alignment of complete cp genomes from the 14 species examined (NEXUS format).
Table S6. The alignment of 65 shared genes from the 14 species cp genomes examined (NEXUS format).
Figure S1. Amino acid frequencies in O. minuta cp protein-coding sequences. The frequencies of amino acids were calculated for all 97 protein-coding genes from start to stop codon.
Figure S2. A phylogenetic tree was constructed based on 14 species from the rice tribe using different methods. matK gene sequence data were used with maximum parsimony (MP), maximum likelihood (ML) and neighbor-joining (NJ) approaches. Numbers above the branches are the bootstrap values of MP, ML, and NJ, respectively. Stars represent position for O. minuta (KU179220).
References
Amante-Bordeos, A., Sitch, L. A., Nelson, R., Dalmacio, R. D., Oliva, N. P., Aswidinnoor, H., et al. (1992). Transfer of bacterial blight and blast resistance from the tetraploid wild rice Oryza minuta to cultivated rice, Oryza sativa. Theor. Appl. Genet. 84, 345–354. doi: 10.1007/bf00229493
Ammiraju, J. S. S., Fan, C. Z., Yu, Y. S., Song, X. A., Cranston, K. A., Pontaroli, A. C., et al. (2010). Spatio-temporal patterns of genome evolution in allotetraploid species of the genus Oryza. Plant J. 63, 430–442. doi: 10.1111/j.1365-313X.2010.04251.x
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S.-M., Khan, M. A., et al. (2016a). Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 7:843. doi: 10.3389/fpls.2016.00843
Asaf, S., Khan, A. L., Khan, A. R., Waqas, M., Kang, S. M., Khan, M. A., et al. (2016b). Mitochondrial genome analysis of wild rice (Oryza minuta) and its comparison with other related species. PLoS ONE 11:e0152937. doi: 10.1371/journal.pone.0152937
Asano, T., Tsudzuki, T., Takahashi, S., Shimada, H., and Kadowaki, K. (2004). Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11, 93–99. doi: 10.1093/dnares/11.2.93
Babiychuk, E., Vandepoele, K., Wissing, J., Garcia-Diaz, M., De Rycke, R., Akbari, H., et al. (2011). Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc. Natl. Acad. Sci. U.S.A. 108, 6674–6679. doi: 10.1073/pnas.1103442108
Bao, Y., and Ge, S. (2008). Historical retrospect and the perplexity on the studies of the Oryza polyploids. J. Syst. Evol. 46, 3–12. doi: 10.3724/SP.J.1002.2008.07069
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Brozynska, M., Furtado, A., and Henry, R. J. (2014). Direct chloroplast sequencing: comparison of sequencing platforms and analysis tools for whole chloroplast barcoding. PLoS ONE 9:e110387. doi: 10.1371/journal.pone.0110387
Cavalier-Smith, T. (2002). Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr. Biol. 12, R62–R64. doi: 10.1016/S0960-9822(01)00675-3
Chang, C. C., Lin, H. C., Lin, I. P., Chow, T. Y., Chen, H. H., Chen, W. H., et al. (2006). The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 23, 279–291. doi: 10.1093/molbev/msj029
Chang, T.-T. (1976). The origin, evolution, cultivation, dissemination, and diversification of Asian and African rices. Euphytica 25, 425–441. doi: 10.1007/BF00041576
Chen, J., Hao, Z., Xu, H., Yang, L., Liu, G., and Sheng, Y. (2015). The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 6:447. doi: 10.3389/fpls.2015.00447
Drescher, A., Ruf, S., Calsa, T., Carrer, H., and Bock, R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Evenson, R. E., and Gollin, D. (1997). Genetic resources, international organizations, and improvement in rice varieties*. Econ. Dev. Cult. Change 45, 471–500. doi: 10.1086/452288
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Gao, L., Yi, X., Yang, Y. X., Su, Y. J., and Wang, T. (2009). Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 9:130. doi: 10.1186/1471-2148-9-130
Ge, S., Sang, T., Lu, B. R., and Hong, D. Y. (1999). Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc. Natl. Acad. Sci. U.S.A. 96, 14400–14405. doi: 10.1073/pnas.96.25.14400
Guisinger, M. M., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2011). Extreme reconfiguration of plastid genomes in the angiosperm family geraniaceae: rearrangements, repeats, and codon usage (vol 28, pg 583, 2011). Mol. Biol. Evol. 28, 1543–1543. doi: 10.1093/molbev/msq229
Guo, S. B., Wei, Y., Li, X.-Q., Liu, K.-Q., Huang, F.-K., Chen, C.-Q., et al. (2013). Development and identification of introgression lines from cross of Oryza sativa and Oryza minuta. Rice Sci. 20, 95–102. doi: 10.1016/S1672-6308(13)60111-0
Guo, Y. L., and Ge, S. (2005). Molecular phylogeny of Oryzeae (Poaceae) based on DNA sequences from chloroplast, mitochondrial, and nuclear genomes. Am. J. Bot. 92, 1548–1558. doi: 10.3732/ajb.92.9.1548
Heinrichs, E. A., Medrano, F. G., and Rapusas, H. R., and International Rice Research Institute (1985). Genetic Evaluation for Insect Resistance in Rice. Manila: International Rice Research Institute.
Huotari, T., and Korpelainen, H. (2012). Complete chloroplast genome sequence of Elodea canadensis and comparative analyses with other monocot plastid genomes. Gene 508, 96–105. doi: 10.1016/j.gene.2012.07.020
Jacquemin, J., Bhatia, D., Singh, K., and Wing, R. A. (2013). The international oryza map alignment project: development of a genus-wide comparative genomics platform to help solve the 9 billion-people question. Curr. Opin. Plant Biol. 16, 147–156. doi: 10.1016/j.pbi.2013.02.014
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., de Pamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Khush, G. S. (1997). Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 35, 25–34. doi: 10.1023/A:1005810616885
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kode, V., Mudd, E. A., Iamtham, S., and Day, A. (2005). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/g11-026
Kumar, S., Hahn, F. M., McMahan, C. M., Cornish, K., and Whalen, M. C. (2009). Comparative analysis of the complete sequence of the plastid genome of Parthenium argentatum and identification of DNA barcodes to differentiate Parthenium species and lines. BMC Plant Biol. 9:131. doi: 10.1186/1471-2229-9-131
Kumar, S., Nei, M., Dudley, J., and Tamura, K. (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinformatics 9, 299–306. doi: 10.1093/bib/bbn017
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Leese, F., Mayer, C., and Held, C. (2008). Isolation of microsatellites from unknown genomes using known genomes as enrichment templates. Limnol. Oceanogr. Methods 6, 412–426. doi: 10.4319/lom.2008.6.412
Li, X. W., Gao, H. H., Wang, Y. T., Song, J. Y., Henry, R., Wu, H. Z., et al. (2013). Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci. China Life Sci. 56, 189–198. doi: 10.1007/s11427-012-4430-8
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Martin, W., Deusch, O., Stawski, N., Grünheit, N., and Goremykin, V. (2005). Chloroplast genome phylogenetics: why we need independent approaches to plant molecular evolution. Trends Plant Sci. 10, 203–209. doi: 10.1016/j.tplants.2005.03.007
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Morton, B. R. (1998). Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 46, 449–459. doi: 10.1007/PL00006325
Nadachowska-Brzyska, K., Li, C., Smeds, L., Zhang, G. J., and Ellegren, H. (2015). Temporal dynamics of avian populations during pleistocene revealed by whole-genome sequences. Curr. Biol. 25, 1375–1380. doi: 10.1016/j.cub.2015.03.047
Nie, X. J., Lv, S. Z., Zhang, Y. X., Du, X. H., Wang, L., Biradar, S. S., et al. (2012). Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 7:e36869. doi: 10.1371/journal.pone.0036869
Pauwels, M., Vekemans, X., Gode, C., Frerot, H., Castric, V., and Saumitou-Laprade, P. (2012). Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 193, 916–928. doi: 10.1111/j.1469-8137.2011.04003.x
Powell, W., Morgante, M., Andre, C., McNicol, J. W., Machray, G. C., Doyle, J. J., et al. (1995a). hypervariable microsatellites provide a general source of polymorphic DNA markers for the chloroplast genome. Curr. Biol. 5, 1023–1029. doi: 10.1016/S0960-9822(95)00206-5
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995b). Polymorphic simple sequence repeat regions in chloroplast genomes - applications to the population-genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Provan, J., Corbett, G., McNicol, J. W., and Powell, W. (1997). Chloroplast DNA variability in wild and cultivated rice (Oryza spp.) revealed by polymorphic chloroplast simple sequence repeats. Genome 40, 104–110. doi: 10.1139/g97-014
Qian, J., Song, J., Gao, H., Zhu, Y., Xu, J., and Pang, X. (2013). The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8:e57607. doi: 10.1371/journal.pone.0057607
Rahman, M. L., Jiang, W., Chu, S. H., Qiao, Y., Ham, T. H., Woo, M. O., et al. (2009). High-resolution mapping of two rice brown planthopper resistance genes, Bph20(t) and Bph21(t), originating from Oryza minuta. Theor. Appl. Genet. 119, 1237–1246. doi: 10.1007/s00122-009-1125-z
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rose, O., and Falush, D. (1998). A threshold size for microsatellite expansion. Mol. Biol. Evol. 15, 613–615. doi: 10.1093/oxfordjournals.molbev.a025964
Rousseau-Gueutin, M., Ayliffe, M. A., and Timmis, J. N. (2011). Conservation of plastid sequences in the plant nuclear genome for millions of years facilitates endosymbiotic evolution. Plant Physiol. 157, 2181–2193. doi: 10.1104/pp.111.185074
Sang, T., and Ge, S. (2007). Genetics and phylogenetics of rice domestication. Curr. Opin. Genet. Dev. 17, 533–538. doi: 10.1016/j.gde.2007.09.005
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sierro, N., Battey, J. N. D., Ouadi, S., Bakaher, N., Bovet, L., Willig, A., et al. (2014). The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5:3833. doi: 10.1038/ncomms4833
Swofford, D. L. (1993). Paup - a computer-program for phylogenetic inference using maximum parsimony. J. Gen. Physiol. 102, A9.
Timme, R. E., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2007). A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am. J. Bot. 94, 302–312. doi: 10.3732/ajb.94.3.302
Timmis, J. N., Ayliffe, M. A., Huang, C. Y., and Martin, W. (2004). Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 5, 123–135. doi: 10.1038/nrg1271
Vaughan, D. A. (1994). The Wild Relatives of Rice: A Genetic Resources Handbook. Manila: International Rice Research Institute.
Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., and Henry, R. J. (2015). Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci. Rep. 5:13957. doi: 10.1038/srep13957
Wang, M. X., Cui, L. C., Feng, K. W., Deng, P. C., Du, X. H., Wan, F. H., et al. (2015). Comparative analysis of asteraceae chloroplast genomes: structural organization, RNA editing and evolution. Plant Mol. Biol. Rep. 33, 1526–1538. doi: 10.1007/s11105-015-0853-2
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., and Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
Waters, D. L., Nock, C. J., Ishikawa, R., Rice, N., and Henry, R. J. (2012). Chloroplast genome sequence confirms distinctness of Australian and Asian wild rice. Ecol. Evol. 2, 211–217. doi: 10.1002/ece3.66
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, Z. (2016a). The whole chloroplast genome of shrub willows (Salix suchowensis). Mitochondrial DNA A DNA Mapp Seq Anal. 27, 2153–2154. doi: 10.3109/19401736.2014.982602
Wu, Z. (2016b). The completed eight chloroplast genomes of tomato from Solanum genus. Mitochondrial DNA Part A 27, 4155–4157. doi: 10.3109/19401736.2014.1003890
Wu, Z. Q., and Ge, S. (2012). The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 62, 573–578. doi: 10.1016/j.ympev.2011.10.019
Wu, Z., Tembrock, L. R., and Ge, S. (2015). Are differences in genomic data sets due to true biological variants or errors in genome assembly: an example from two chloroplast genomes. PLoS ONE 10:e0118019. doi: 10.1371/journal.pone.0118019
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., and Yun, Q. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5:e12762. doi: 10.1371/journal.pone.0012762
Yang, Y., Dang, Y. Y., Li, Q., Lu, J. J., Li, X. W., and Wang, Y. T. (2014). Complete chloroplast genome sequence of poisonous and medicinal plant datura stramonium: organizations and implications for genetic engineering. PLoS ONE 9:e110656. doi: 10.1371/journal.pone.0110656
Yi, X., Gao, L., Wang, B., Su, Y. J., and Wang, T. (2013). The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol. Evol. 5, 688–698. doi: 10.1093/gbe/evt042
Yurina, N. P., and Odintsova, M. S. (1998). Comparative structural organization of plant chloroplast and mitochondrial genomes. Genetika 34, 5–22.
Zhang, T., Hu, S., Zhang, G., Pan, L., Zhang, X., Al-Mssallem, I. S., et al. (2012). The organelle genomes of Hassawi rice (Oryza sativa L.) and its hybrid in saudi arabia: genome variation, rearrangement, and origins. PLoS ONE 7:e42041. doi: 10.1371/journal.pone.0042041
Zhu, Q., and Ge, S. (2005). Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol. 167, 249–265. doi: 10.1111/j.1469-8137.2005.01406.x
Keywords: wild rice (Oryza minuta), cp genome, repeat analysis, codon usage, phylogeny, sequence divergence, SSRs
Citation: Asaf S, Waqas M, Khan AL, Khan MA, Kang S-M, Imran QM, Shahzad R, Bilal S, Yun B-W and Lee I-J (2017) The Complete Chloroplast Genome of Wild Rice (Oryza minuta) and Its Comparison to Related Species. Front. Plant Sci. 8:304. doi: 10.3389/fpls.2017.00304
Received: 02 December 2016; Accepted: 20 February 2017;
Published: 07 March 2017.
Edited by:
Tian Tang, Sun Yat-sen University, ChinaReviewed by:
Haipeng Li, Partner Institute for Computational Biology, ChinaJinfeng Chen, University of California, Riverside, USA
Copyright © 2017 Asaf, Waqas, Khan, Khan, Kang, Imran, Shahzad, Bilal, Yun and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: In-Jung Lee, aWpsZWVAa251LmFjLmty
†These authors have contributed equally to this work.