 Zhongying Ren1,2†
Zhongying Ren1,2† Daoqian Yu1,2†Zhaoen Yang1,2Changfeng Li2,3Ghulam Qanmber2Yi Li2Jie Li2Zhao Liu2Lili Lu2Lingling Wang2Hua Zhang1Quanjia Chen1Fuguang Li1,2*
Daoqian Yu1,2†Zhaoen Yang1,2Changfeng Li2,3Ghulam Qanmber2Yi Li2Jie Li2Zhao Liu2Lili Lu2Lingling Wang2Hua Zhang1Quanjia Chen1Fuguang Li1,2* Zuoren Yang1,2*
Zuoren Yang1,2*- 1Xinjiang Research Base, State Key Laboratory of Cotton Biology, Xinjiang Agriculture University, Urumqi, China
- 2Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China
- 3Cotton Research Institute, Anhui Academy of Agricultural Sciences, Hefei, China
Cotton is one of the major world oil crops. Cottonseed oil meets the increasing demand of fried food, ruminant feed, and renewable bio-fuels. MADS intervening keratin-like and C-terminal (MIKC)-type MADS-box genes encode transcription factors that have crucial roles in various plant developmental processes. Nevertheless, this gene family has not been characterized, nor its functions investigated, in cotton. Here, we performed a comprehensive analysis of MIKC-type MADS genes in the tetraploid Gossypium hirsutum L., which is the most widely cultivated cotton species. In total, 110 GhMIKC genes were identified and phylogenetically classified into 13 subfamilies. The Flowering locus C (FLC) subfamily was absent in the Gossypium hirsutum L. genome but is found in Arabidopsis and Vitis vinifera L. Among the genes, 108 were distributed across the 13 A and 12 of the D genome's chromosomes, while two were located in scaffolds. GhMIKCs within subfamilies displayed similar exon/intron characteristics and conserved motif compositions. According to RNA-sequencing, most MIKC genes exhibited high flowering-associated expression profiles. A quantitative real-time PCR analysis revealed that some crucial MIKC genes determined the identities of the five flower organs. Furthermore, the overexpression of GhAGL17.9 in Arabidopsis caused an early flowering phenotype. Meanwhile, the expression levels of the flowering-related genes CONSTANS (CO), LEAFY (LFY) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 (SOC1) were significantly increased in these lines. These results provide useful information for future studies of GhMIKCs' regulation of cotton flowering.
Introduction
Transcription factors play an indispensable role in growth and development, and MADS transcription factor family members have been detected in the genomes of plants, animals, and fungi (Becker et al., 2000; Becker and Theissen, 2003; Messenguy and Dubois, 2003). In monophyletic evolution, they are divided into two classes: type I and type II (Alvarez-Buylla et al., 2000). The type I MADS-box genes are serum response factor-like genes in animals and fungi, while they are M-type genes in plants. They are characterized by a highly conserved MADS domain of the 58–60 amino acids, located in the N-terminal region of the proteins, which are involved in DNA binding and dimerization. Functional investigations have been mainly restricted to Arabidopsis (Parenicová et al., 2003). The type II family plays a significant role in regulating flowering during plant development (Mondragon-Palomino, 2013). Type II genes are closely related to the myocyte enhancer factor-2-like genes of animals and yeast. However, MADS intervening keratin-like and C-terminal (MIKC)-type MADS-box genes are found only in plants.
The MIKC-type plant genes contain three additional domains other than the MADS (M): Intervening (I), Keratin (K), and the C-terminal (C) domains (Theißen et al., 1996; Kaufmann et al., 2005). The I domain forms DNA-binding dimers, which is less conserved (Riechmann et al., 1996). The K domain, which consists of ~70 amino acids, is mainly responsible for dimerization by a coiled-coil structure (Ma et al., 1991; Fan et al., 1997). The C domain exhibits transactivation and mediates protein–protein interactions (Kramer and Irish, 1999; Honma and Goto, 2001). Based on structure divergence at the I domain, the MIKC-type genes are classified into two subgroups, MIKCC and MIKC*. Earlier investigations found 39 and 37 MIKCC genes in Arabidopsis and O. sativa, respectively (Parenicová et al., 2003; Arora et al., 2007). The MIKCC type plays a crucial role in the flowering time, floral organ identity determination and fruit ripening in plant growth and development (Theissen, 2001; Becker and Theissen, 2003; Theissen and Melzer, 2007; Li et al., 2016).
The genetic floral organ model is derived from the analysis of homeotic floral mutants. The ABC model was named after three classes of genes (A, B, and C) (Coen and Meyerowitz, 1991), and has developed into the more exact ABCDE model. MIKCC family genes have combined and determined the identities of the floral organs: sepals (A + E), petals (A + B + E), stamens (B + C + E), carpels (C + E), and ovules (D + E) (Bowman et al., 1991; Coen and Meyerowitz, 1991; Ma and Depamphilis, 2000; Zahn et al., 2006; Silva et al., 2015). In Arabidopsis, the functional genes were divided into five classes: Class A: APETALA1 (AP1); Class B: PISTILATA (PI) and AP3; Class C: AGAMOUS (AG) (Acri-Nunes-Miranda and Mondragón-Palomino, 2014); Class D: SEEDSTICK/AGAMOUS-LIKE11 (STK/AGL11); and Class E: SEPALLATA (SEP1, SEP2, SEP3, and SEP4) (Ferrándiz et al., 2000; Pinyopich et al., 2003). Other MIKCC genes that regulated flowering time and flower initiation have been identified as follows: Suppressor of Overexpression Of Constans1 (SOC1) (Lee et al., 2000; Hepworth et al., 2002); Flowering Locus c (FLC) (Michaels and Amasino, 1999; Searle et al., 2006; Reeves et al., 2007); AGAMOUSLIKE GENE 24 (AGL24) (Michaels et al., 2003; Liu et al., 2008) and Short Vegetative Phase (SVP) (Hartmann et al., 2000; Michaels et al., 2003; Lee et al., 2007). Others are involved in fruit ripening, such as SHATTERPROOF 1–2 and FUL (Ferrándiz et al., 2000; Liljegren et al., 2000), in seed pigmentation and endothelium development, such as TRANSPARENT TESTA16 (Nesi et al., 2002), and in root development such as AGL12 and AGL17 (Rounsley et al., 1995; Tapia-López et al., 2008). Studies of the evolutionary history of MIKC genes have explored the internal mechanisms behind their functional diversification in plant growth and development.
Cotton is not only the most important source of natural fiber for textile industry (Pang et al., 2010), but also a major contributor in world oilseed economy. The extracted cottonseed oil has long been considered to be a good vegetable oil (Michaelk et al., 2010; Sawan, 2014; Zhang et al., 2014). Simultaneously, as an alternative and sustainable oil source, cottonseed oil has been developed into biodiesel and used as substitutes for petroleum (Carlsson, 2009; Alhassan et al., 2014). As the top five oil crops in the world (Wang et al., 2016), cottonseed oil occupies about 21% of the cottonseed production (Malik and Ahsan, 2016; Wang et al., 2016; Yang and Zheng, 2016). The formation of cotton seed originates from ovule which is an important part of floral organs. G. hirsutum's MIKC functions are highly significant in plant developmental processes. Especially, a number of genes could involve in the development of flower morphology (Honma and Goto, 2001; Messenguy and Dubois, 2003). For example, GhMADS3, a homolog of Arabidopsis AG and putative C function gene, overexpression can improve sepal-to-carpel and petal-to-stamen transformations in transgenic tobacco (Guo et al., 2007). GhMADS13, a high homolog of Arabidopsis AGL6, overexpression significantly promotes flower buds in cotton (Wu et al., 2009), and GhMADS14 is enhanced gradually during the early stages of fiber elongation (Zhou et al., 2014). In previous study, 53 members of the G. hirsutum MIKCC gene family were identified based on the G. raimondii genome (Jiang et al., 2014). However, owing to the lack of G. hirsutum genome sequences, a comprehensive analysis of MIKC-type MADS genes in G. hirsutum has not yet been reported.
Recently, the G. hirsutum genome was sequenced. To systematically analyze the MIKC-Type MADS family genes in G. hirsutum, 92 MIKCC, and 18 MIKC* members of the MIKC family were identified from the whole G. hirsutum genome. Phylogeny, structures, locations and expression patterns were comprehensively analyzed. AGL17 is the biggest subgroup, and the involvement of GhAGL17 subfamily gene in regulating flowering was confirmed by ectopic expression in Arabidopsis. Our findings provide a foundation for the genetic improvement of cotton flowering.
Materials and Methods
Identification of MIKC Genes in Gossypium hirsutum L
To identify members of the MIKC gene family in G. hirsutum, Arabidopsis MIKC sequences were obtained from the TAIR database (http://www.arabidopsis.org) and used as queries for a BLASTP algorithm-based against the G. hirsutum genome database (https://www.cottongen.org/species/Gossypium_hirsutum/nbi-AD1_genome_v1.1) (Zhang et al., 2015). The MIKC protein domain was analyzed using the Hidden Markov Model (HMM) from the Pfam database (http://pfam.xfam.org/). The SRF-TF and K-box domains were confirmed by Pfam accessions (PF00319 and PF01486, respectively). All of the candidate proteins were manually checked using the above described methods to remove the redundant sequences.
Phylogenetic Tree Construction
To construct a MIKC-protein phylogenetic tree using MEGA 6.06, MIKC proteins from four plant species, Arabidopsis, O. sativa, V. vinifera, and G. hirsutum, were employed. The neighbor-joining method with amino acid p-distance was applied to construct the tree (Tamura et al., 2011), and the reliability was obtained by bootstrapping with 1,000 replicates.
Exon/Intron Structure, Motif and Chromosomal Location Analyses
The exon/intron structures of MIKC genes were retrieved by the alignment of predicted coding sequences with corresponding genomic sequences using the gene structure display server (GSDS) program (http://gsds.cbi.pku.edu.cn/).
The online program MEME (http://meme-suite.org/) was employed to determine the conserved motifs in GhMIKCs with the following optimum parameters: a motif width of 8–200 amino acids and a maximum of 13 motifs. The identified motifs were annotated using the program InterProScan (Quevillon et al., 2005).
The chromosomal distributions of MIKC genes were obtained based on genome annotation data. The MapInspect software was applied to draw images of their physical locations in G. hirsutum.
Gene Expression Analysis
The expression of MIKC family genes were measured using RNA-sequencing method. The raw RNA-sequencing data of G. hirsutum TM-1 seven different tissues (root, stem, leaf, flower, ovule, seed, and fiber) was downloaded from the NCBI Gene Expression repository under the accession number PRJNA248163 (Table S4) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA248163/). The relative data were normalized to calculate the expression levels. Hierarchical clustering was performed using Genesis 1.7.7 (Sturn et al., 2002).
RNA Isolation and the qRT-PCR Analysis
Gossypium hirsutum L. (cv CCRI24) was cultivated in the field in Zhengzhou, China. Five different tissue parts of flower: sepal, petal, stamen, carpel, and ovule were sampled, respectively at full bloom stage. Arabidopsis (Columbia-0) was used as wild type; the leaves of wild type and transgenic lines grown for 25 days were harvested. All samples were frozen immediately in liquid nitrogen and kept at −80°C for total RNA extraction. Total RNA was extracted from each sample using the TRIzol reagent (TIANGEN, Beijing, China) and treated with RNase-free DNase I. Gel electrophoresis and a Nanodrop2000 nucleic acid analyzer were employed to detect the quality of RNA. The first cDNA strand was synthesized from 1 μg total RNA using the Transcriptor First Strand cDNA Synthesis Kit version DRR047A (TaKaRa, Dalian, China). The cDNA was diluted five times for the next experiments.
The gene-specific primers used for qRT-PCR were listed in Supplementary Table S2 and S3. The G. hirsutum His3 gene and Arabidopsis Actin2 gene were used as an internal control respectively. The qRT-PCR was performed using SYBR Green (Roche) on a LightCycler480 system (Roche). Each reaction was conducted in a 96-well plate with a volume of 20 μl. The PCR cycling parameters were as follows: 95°C for 5 min, 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s, followed by an increase from 60 to 95°C. The relative expression levels were analyzed using the LightCycler® 480 gene scanning software. Three biological replicates were measured and each biological replicate was run three times.
Isolation of GhAGL17.9 and Transformation of Arabidopsis
We amplified GhAGL17.9 using cDNA templates from the mix of CCRI24 root, stem, leaf and flower. The amplified product was cloned into vector pCambia2301 (CAMBIA) containing the CAULIFLOWER MOSAIC VIRUS (CaMV) 35S constitutive promoter, and then, the constructed vector was introduced into Agrobacterium tumefaciens GV3101 (Clough and Bent, 1998). Floral dip method was used for Agrobacterium-mediated transformation of Arabidopsis. Positive transgenic lines were selected on MS medium containing kanamycin. To grow the transgenic lines, seedlings were sown in plastic pots filled with a nutrient soil and vermiculite mix. Then, they were grown in a culturing room at 22°C under a 16-h light/8-h dark cycle for 1 month.
Results
Identification of MIKC Genes in Gossypium hirsutum L
HMMER and BLASTP algorithm-based searches were used to identify MIKC protein HMM profiles based on the highly conserved MADS and K-box domains. To identify the maximum number of MIKC genes in G. hirsutum, HMMs of SRF-TF, and K-box domains (PF00319 and PF01486, respectively) were extracted from Pfam database to use as queries against protein sequences from the G. hirsutum genome (https://www.cottongen.org/species/Gossypium_hirsutum/nbi-AD1_genome_v1.1). A total of 145 putative MIKC proteins were identified. To verify the results, we conducted a multiple sequence alignment and removed 35 redundant sequences. Finally, 110 MIKC protein sequences were identified by confirming their conserved domains using the Pfam web server (Figure S3). From the sequences, 92 MIKCC genes and 18 MIKC* genes were identified. Thus, 84% of the MIKC genes were MIKCC in G. hirsutum (Figure 1A). The identified MIKC genes were listed with their corresponding locus tag (Table 1). We named the MIKCC genes on the basis of their assignment to the 13 previously classified Arabidopsis, O. sativa and P. tremula subfamilies (Parenicová et al., 2003; Leseberg et al., 2006; Arora et al., 2007). Subgroup AGL17 had the greatest (13%) number of GhMIKCC genes; however, subgroups TM8 and AGL12 had the lowest (2%) number of GhMIKCC genes (Figure 1B). The GhMIKCs' encoding amino acids were relatively conserved, and MIKCC proteins were highly conserved, ranging from 200 to 300 amino acids in most cases. MIKC* proteins generally possessed more than 300 amino acids. The chromosomal locations of the 108 GhMIKCs were distributed in different subgroups of the A and D genomes, while GhAGL17.12 and GhAP3.10 were located on scaffolds.
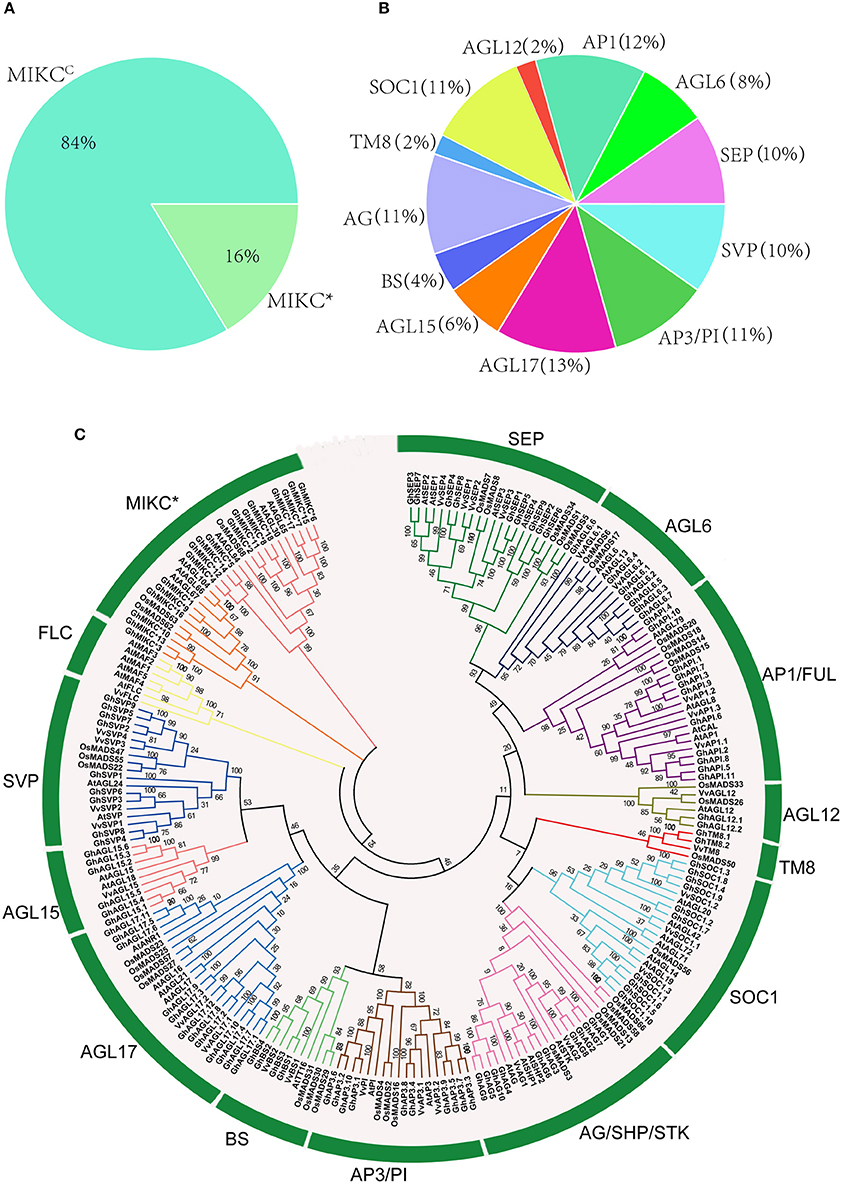
Figure 1. (A,B). Classification and percentage of GhMIKC genes. (C). Neighbor-joining phylogenetic tree using MIKC proteins of Gossypium hirsutum L., Arabidopsis, Oryza sativa L. and Vitis vinifera L. Full-length protein sequences were aligned using the MEGA 6.06 program with 1,000 bootstrap replicates. The numbers of the MIKC proteins are listed in Supplementary S1.
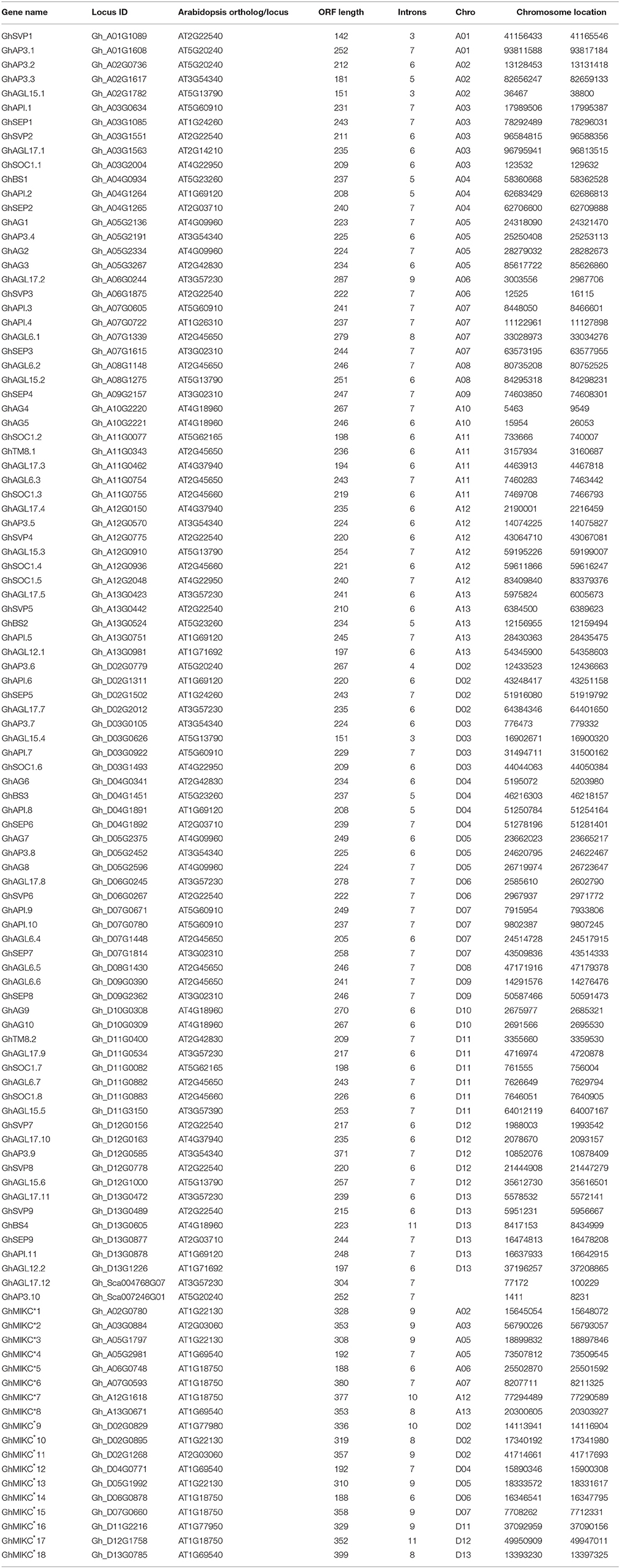
Table 1. MIKC genes identified in Gossypium hirsutum L.
Phylogenetic Analysis of the MIKC Gene Family
To examine the phylogenetic relationships among G. hirsutum MIKC proteins and to categorize them within the established subfamilies from other plants, we performed a multiple alignment analysis using the neighbor-joining method of 110 full-length MIKC proteins from G. hirsutum, 44 MIKC proteins from V. vinifera, 46 MIKC proteins from Arabidopsis, and 41 MIKC proteins from O. sativa (Table S1). The MIKCC proteins were divided into 13 subfamilies (SVP, BS, AGL17, AGL15, AP3-PI, AGL12, SOC1, AG/SHP/STK, AP1/FUL, AGL6, SEP, TM8, and FLC; Figure 1C). The AGL17 subgroup was the largest, and the FLC subgroup was absent in the G. hirsutum genome. Additionally, no TM8 family members were found in Arabidopsis. TM8 constituted the smallest clade, having only four members, including two GhMIKCs, GhTM8.1, and GhTM8.2. The MIKC* proteins were divided into two subfamilies.
Gene Structure and Protein Motif Analysis
A phylogenetic analysis revealed that our tree corresponded to those reported recently in V. vinifera and C. sativus (Díaz-Riquelme et al., 2009; Hu and Liu, 2012). The structures of the MIKC genes also helped to determine phylogenetic relationships (Figure 2). Most members had significant sequence identities in the same subfamily and similar exon-intron structures, indicating close evolutionary relationships. The most important differences were in the exon-intron lengths (Figure 2B). In general, most members contained eight exons in the SEP, AGL6, and AP1 gene families (except GhAGL6.1, GhAGL6.4, GhAP1.2, GhAP1.6, and GhAPI.8). The SVP (other than SVP1) and AGL12 subgroups had seven exons, whereas GhAGL15.1 and GhAGL15.4 of the AGL15 subgroup had four exons, which was consistent with GhSVP1 of the SVP subgroup. The AGL17 genes displayed relatively longer lengths compared with other subgroup genes. Additionally, GhBS4 had 11 introns and the first exon was meaningfully shorter, while in GhSOC1.5, the second of seven introns was longer than the others. The MIKC* had much shorter gene lengths and more introns than the MIKCC. GhMIKC*12 had the fourth longest intron, which distinguished it from other members of the MIKC* family.
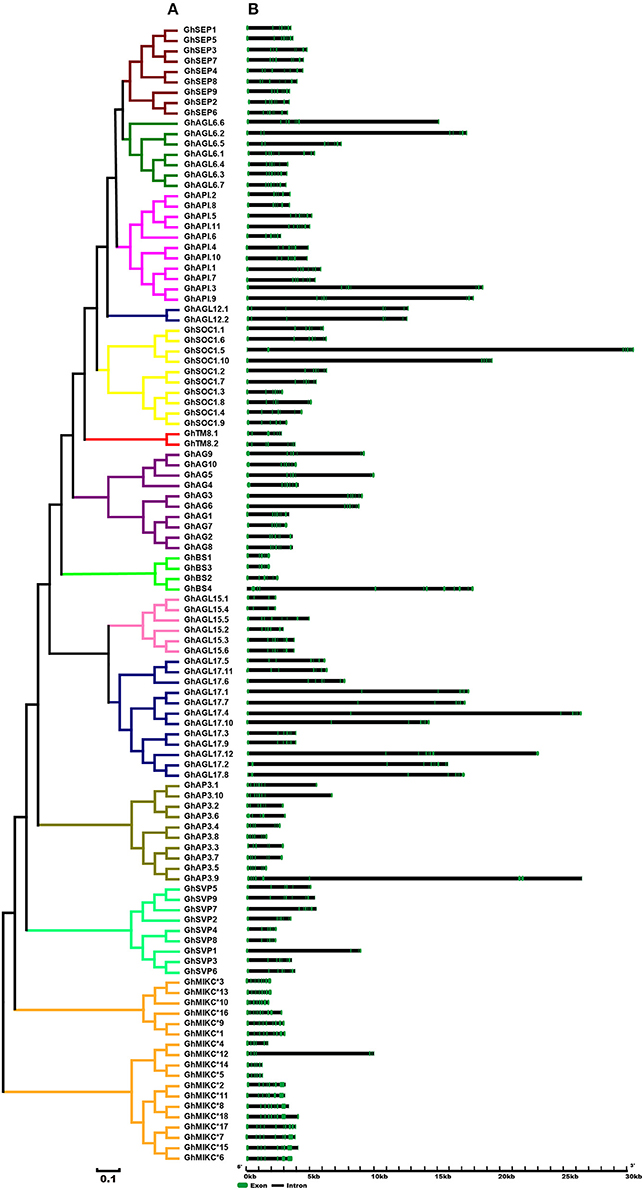
Figure 2. (A). Phylogenetic relationships and (B). Gene structure of MIKC genes in Gossypium hirsutum L. The neighbor-joining tree was constructed with MEGA v6.06. The 13 subfamilies are marked with different colored lines. Exons and introns are represented by green and black lines.
We used MEME to analyze MIKC proteins, and 13 conserved motifs were identified (Figure 3B). Most of the closely related MIKC proteins had similar motif type distributions in the same subfamily (Figure 3A). The most striking divergence among the subgroups was in the composition of the C-terminal domains. Motif 1 contained the MADS domain in all of the MIKC families, except GhSEP8. The highly conserved sequence logs were showed in Figure S2. The differences between I regions and K-box domains were distinctly shown in the MIKCC and MIKC* proteins (Figure 3B). The K-box domain contained three motifs, 2, 4, and 9, in GhMIKCC. However, motif 4, 5, 8, and 9 were present in the GhMIKC* K-box domain, depending on the lengths. The I region in the MIKCC subfamily contained Motifs 3 and 6, while members of the MIKC* contained motifs 6 and 11, which resulted in a longer I region.
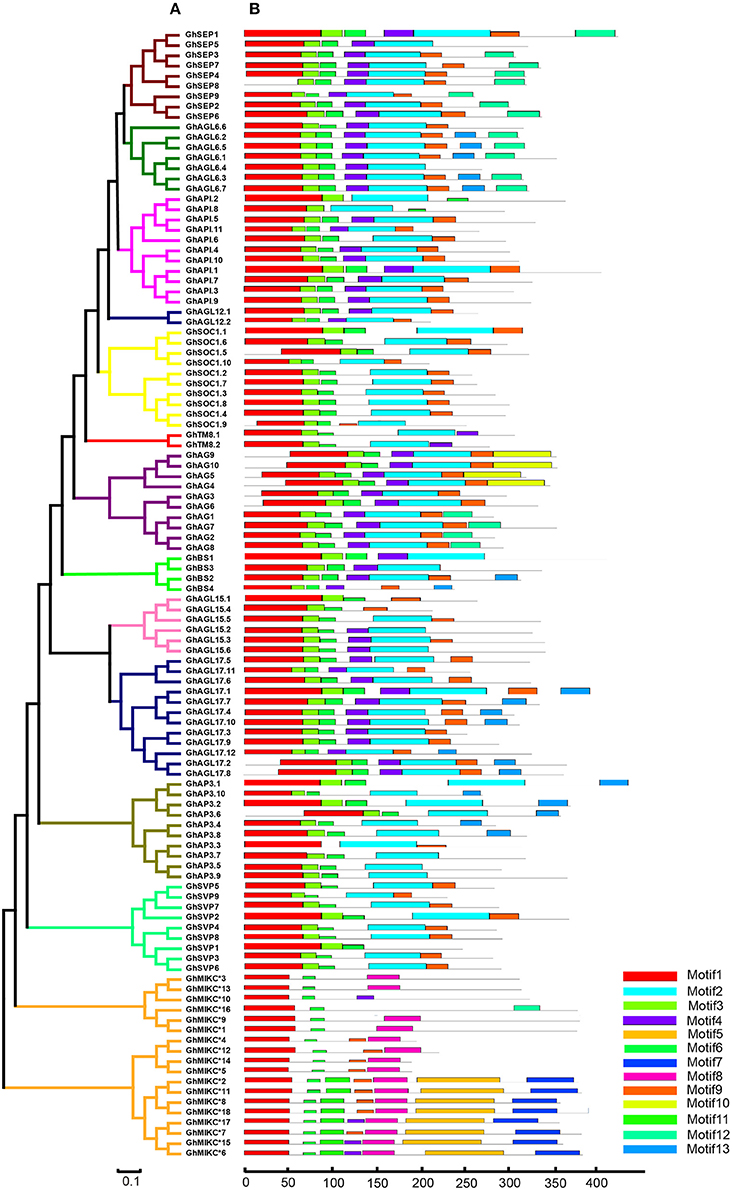
Figure 3. (A). Phylogenetic relationships and (B). Conserved motifs of GhMIKC proteins. The motif compositions were determined using MEME. Motif 1 contains MADS domain, Motifs 2, 4, 5, 8, and 9 contain K-box domains.
Chromosomes Distributions of GhMIKC Genes
Among the 25 G. hirsutum chromosomes, MIKC genes were physically located on all of the 13 A chromosomes and on 12 of the 13 D chromosomes (Figure 4). Among the 110 MIKC genes, two genes, GhAP3.10 and GhAGL17.12, could not be distributed on the G. hirsutum chromosomes, but were located on unmapped scaffolds (7,246 and 4,768, respectively). The greatest numbers of genes were located on Dt-chr12 (eight genes), followed by Dt-chr2, At-chr12, At-chr13, Dt-chr11, and Dt-chr13 (seven genes on each). In contrast, two genes were located on chromosomes At-chr1, At-chr8, At-chr10, Dt-chr9, and Dt-chr10. Only one gene was mapped on At-chr9 and Dt-chr8, and no genes were located on Dt-chr1.
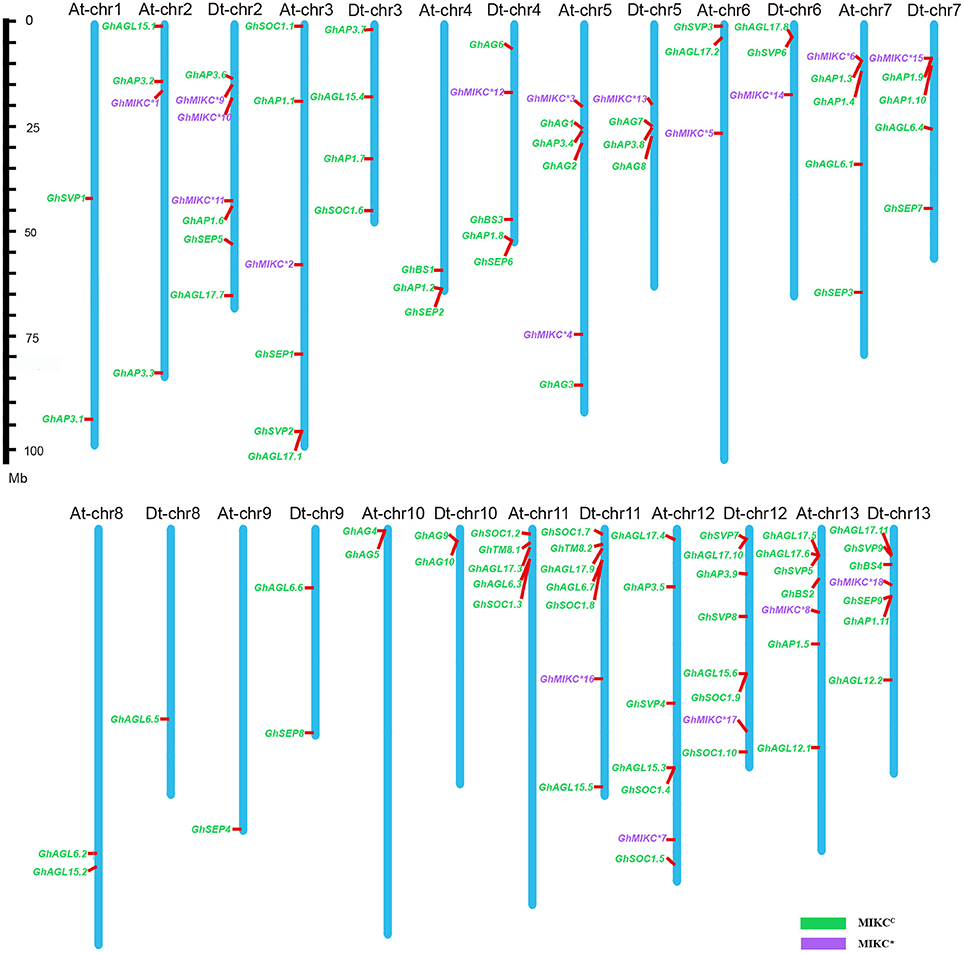
Figure 4. Chromosomal distribution of Gossypium hirsutum L. MIKC genes. The scale represents megabases (Mbs). The chromosome numbers are shown above each vertical blue bar. Two genes (GhAP3.10 and GhAGL17.12 were found on unassembled scaffolds 7,246 and 4,768, respectively) could not be anchored on a specific chromosome. MIKCC and MIKC* genes are shown in different colors.
Expression Pattern Analyses of MIKC Genes
To explore the expression patterns of the MIKC family genes in G. hirsutum- specific developmental processes, the 110 genes' expression profiles were detected in seven different tissues (root, stem, leaf, flower, ovule, seed, and fiber) by transcriptome sequencing (Figure 5). A heat map showed that different genes shared similar expression patterns within subfamilies. For example, the SEP, AG, AP1, AP3/PI, TM8, and AGL6 subgroups were preferentially expressed in flowers. Similarly, the SVP subfamily was expressed especially in flowers. Additionally, some SEP members (GhSEP1, GhSEP3, GhSEP4, GhSEP7, and GhSEP8), and the AG and BS subgroups, were highly expressed in reproductive organs (ovules and fibers). Simultaneously, SEP, AP1 and five of the AGL6 genes (GhAGL6.1, GhAGL6.2, GhAGL6.5, GhAGL6.6, and GhAGL6.7) were also detected in roots. Interestingly, the SOC family displayed diverse expression profiles. GhSOC1.3 and GhSOC1.7 had high expression levels in roots. In addition, GhSOC1.8 was mainly expressed in flower, while GhSOC1.1, GhSOC1.4 and GhSOC1.9 were highly expressed in leaves. GhSOC1.6 and GhSOC1.10 were exclusively and highly expressed in stems. The GhAGL15 (GhAGL15.2, GhAGL15.4, and GhAGL15.5) and GhAGL17 (GhAGL17.7, GhAGL17.9, and GhAGL17.11) subfamilies were relatively highly expressed in roots, and GhAGL17.2, GhAGL17.8, and GhAGL17.12 were expressed in flowers. Four of the GhMIKC* genes (GhMIKC*6, GhMIKC*7, GhMIKC*17, and GhMIKC*18) had high expression levels in flowers, and GhMIKC*7 and GhMIKC*17 were also highly expressed in seeds.
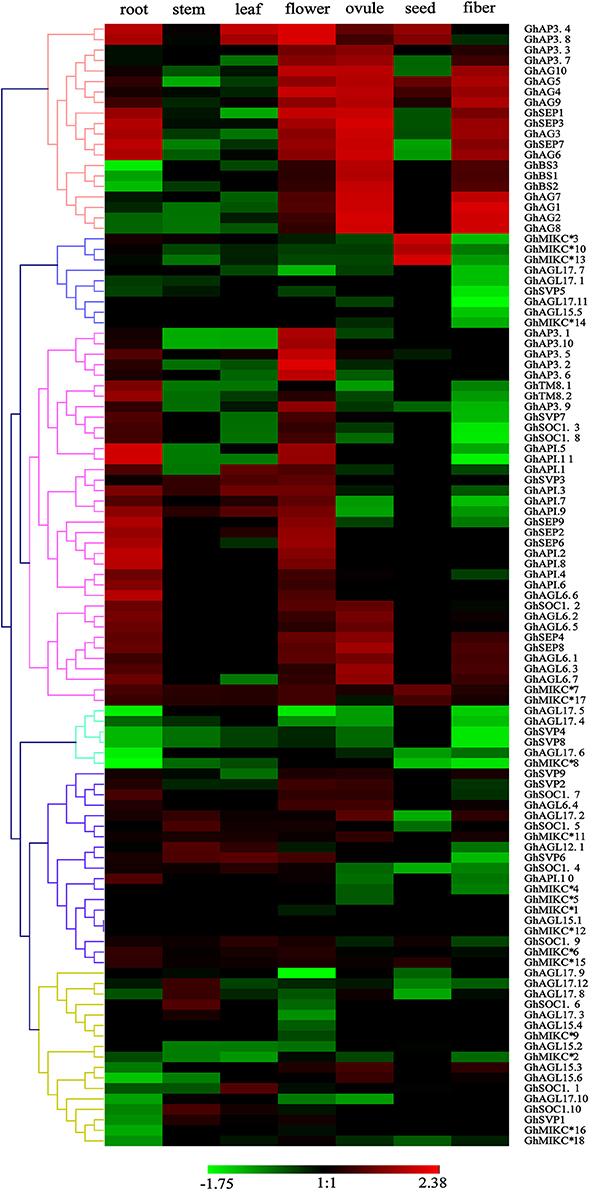
Figure 5. Heat map showing the hierarchical clustering of expression levels of Gossypium hirsutum L. MIKC genes in seven different tissues. The relative gene expression data was normalized. Gene names are displayed to the right of each row. Cluster analyses of gene expression levels with different color scales are displayed at the bottom.
ABCDE model genes regulate the formation of five floral organs in Arabidopsis (Sánchez-Fernández et al., 2001; Dietrich et al., 2009; Kuromori, 2010). To validate the participation of MIKC genes in regulating flowering, we selected 16 of ABCDE model orthologous genes to test their expression in five parts of floral organs (sepal, petal, stamen, carpel, and ovule) by qRT-PCR in G. hirsutum (Figure 6). GhAP1.4 and GhAP1.11 (A class) showed high expression levels in sepals, petals, and carpel. Differently, GhAP1.8 was preferentially expressed in sepal. GhAP3.5, GhAP3.6, and GhAP3.8 of the AP3 subfamily, belonging to B class, were expressed in petals and stamens. GhAG4 of the C class displayed the highest expression level in stamen. GhAG7 and GhAG8 of the D class had higher expression levels in carpel and ovules. GhSEP1, GhSEP4, and GhSEP6 (E class) were expressed in four different floral organs. GhBS2 and GhBS3 (B sister class) were mainly expressed in carpel and ovules. SOC1 accelerates the flowering time, and thus, it is involved in the promotion of floral organ formation. Therefore, high expression levels of GhSOC1.2 and GhSOC1.8 were detected in sepals, stamens and carpel. These results were consistent with the ABCDE model.
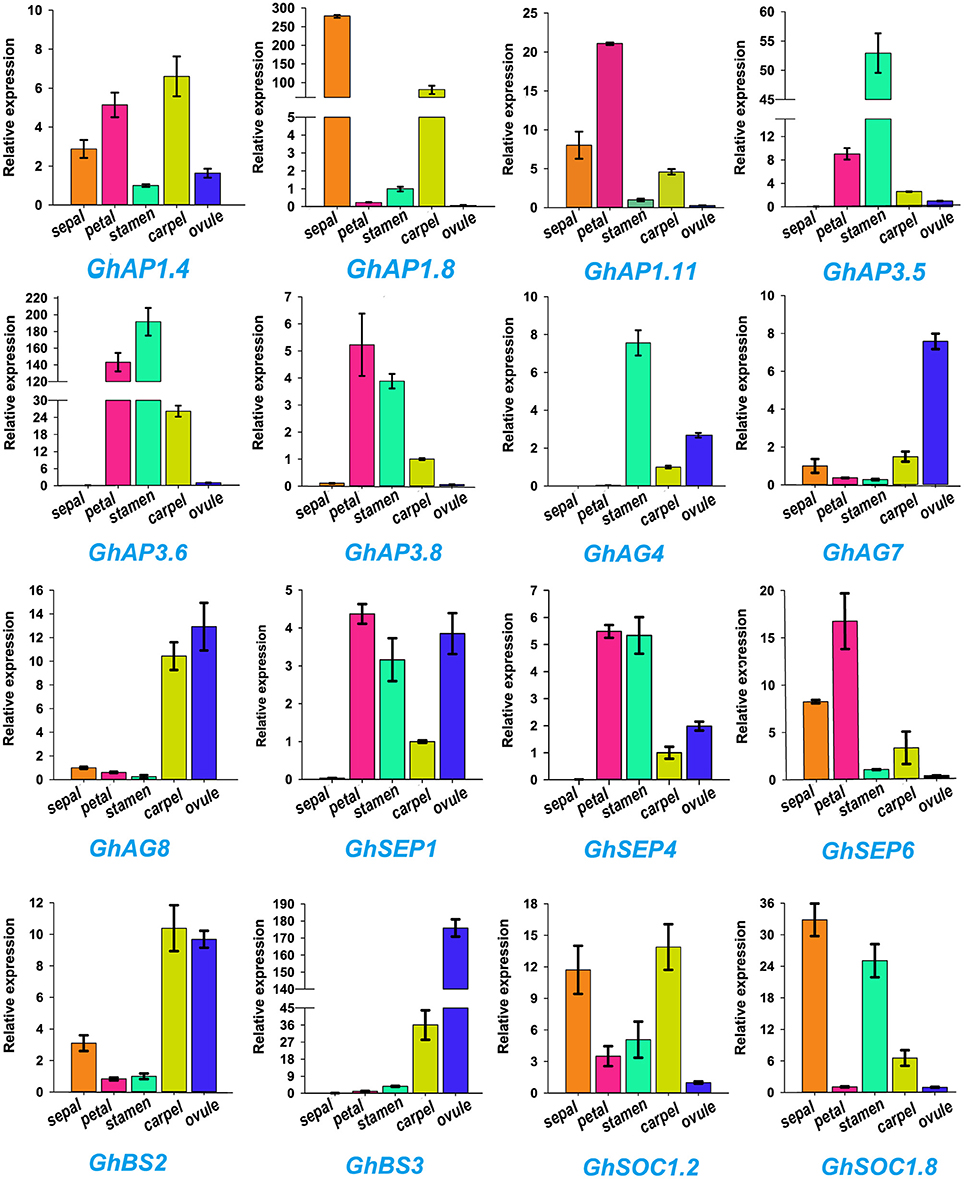
Figure 6. Expression profiles of 16 Gossypium hirsutum L. MIKC genes in five different tissues (sepal, petal, stamen, carpel, and ovule) as determined by qRT-PCR. The relative expression levels are shown against the reference gene His3. Error bars represent the standard deviations of three independent experiments.
Overexpression of the GhAGL17.9 Gene in Arabidopsis
AGL17 is the biggest subgroup (Figure 1B). To further investigate the role of the GhAGL17 subfamily in plant growth and development, we transformed GhAGL17.9 into Arabidopsis (Columbia-0) driven by the CAULIFLOWER MOSAIC VIRUS (CaMV) 35S promoter. We identified 12 T3 generation transgentic lines that showed an early flowering phenotype. QRT-PCR results confirmed that GhAGL17.9 was overexpressed in transgenic lines L1 and L3 (Figure 7). Meanwhile, the numbers of rosette leaves were significantly decreased compared with WT (Table 2). To explore the molecular mechanisms that impact the flowering time in transgentic lines, qRT-PCR was used to detect the expression of flowering-related genes in transgentic lines. LFY is a flowering integration promoting factor, AGL17 can positively regulate the expression of LFY gene (Han et al., 2008), and CO is a photoperiod pathway regulator, AGL17 acts downstream of CO (Han et al., 2008). As shown in Figure 7, the expression levels of LFY gene in lines 35S-L1 and 35S-L3 were three times higher than in the wild type. CO gene expression was not significantly increased. SOC1 is a flowering promoter that regulates different signals of the flowering pathways (Lee and Lee, 2010; Ding et al., 2013). The up-regulation of SOC1 activates downstream targets, including LFY and promotes flowering in Arabidopsis (Schönrock et al., 2006; Lee et al., 2008). Approximate four-fold increases in SOC1 expression levels were observed in two transgenic lines.
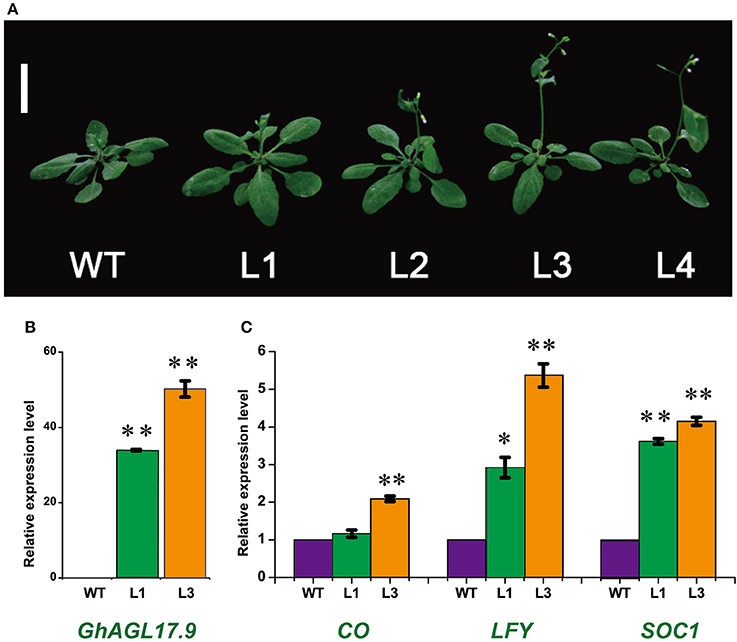
Figure 7. Phenotypes of transgenic Arabidopsis plants overexpressing GhAGL17.9 under the Cauliflower mosaic virus (CaMV) 35S promoter. (A). Morphology of wild type (WT) and transgenic seedlings after 22 days of growth. Bar = 2 cm. (B). A qRT-PCR analysis of GhAGL17.9 overexpression in WT and transgenic Arabidopsis. Significant differences compared with WT (t-test):**, P < 0.01. (C). Expression levels of SOC1, CO, and LFY as determined by qRT-PCR in WT and GhAGL17.9-overexpression plants. Actin2 was used as the internal control. Error bars represent the standard deviations of three independent experiments. Significant differences compared with WT (t-test):*, P < 0.05;**, P < 0.01.
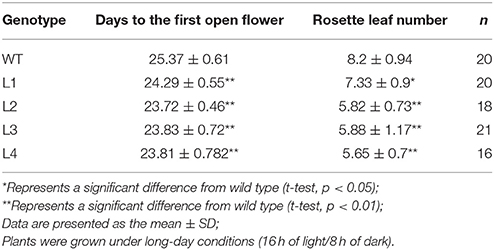
Table 2. Flowering time and the leaf numbers of rosette in WT and p35S::GhAGL17.9 plants.
Discussion
Cotton, as an oil crop, plays an important role in agriculture and industry all around the world (Houhoula et al., 2003; Waheed et al., 2010; Mujeli et al., 2016). Floral organs developments affect the yield and quality of cotton seed. The MIKC family members are plant-specific transcription factors containing MADS and K-box domains, and play crucial roles in plant seed development and floral identity (Nesi et al., 2002; De Folter et al., 2006; Mondragon-Palomino and Theissen, 2011). Many MIKC homologs have been analyzed in many plants, including Arabidopsis, O. sativa, P. tremula, Z. mays, S. bicolor, B. rapa, and R. sativus (Parenicová et al., 2003; Leseberg et al., 2006; Arora et al., 2007; Zhao et al., 2010; Duan et al., 2015; Li et al., 2016). However, the characterization and functional analysis of the MIKC family has not been performed in G. hirsutum, an allotetraploid species. In this study, we performed a comprehensive analysis of GhMIKCs, which included investigating chromosomal locations, phylogenetic relationships, gene structures, conserved motifs, and expression profiles in different tissues.
Overall Summary of the MIKC Family in Gossypium hirsutum L
In total, 110 MIKC genes were identified based on G. hirsutum genome sequences. Based on phylogenetic relationships with Arabidopsis and O. sativa orthologs (Figure S1), the G. hirsutum type II MADS family (MIKCC) was divided into 13 subfamilies (Figure 1C). Interestingly, an FLC subfamily was not identified in the G. hirsutum genome. Similar results were found in O. sativa, C. sativus, Z. mays, and S. bicolor genomes as well (Arora et al., 2007; Zhao et al., 2010; Hu and Liu, 2012). The FLC genes are involved in controlling flowering time through the vernalization and autonomous pathways (Helliwell et al., 2006, 2011; Greb et al., 2007). Vernalization is not required for flowering in O. sativa, C. sativus, Z. mays, and S. bicolor (Arora et al., 2007; Zhao et al., 2010; Hu and Liu, 2012). Thus, vernalization might not be essential for cotton flowering as well. In addition, we found that in most subgroups, the numbers of proteins in G. hirsutum were not doubled, compared with in the diploids Arabidopsis and O. sativa. This implied that gene duplication could give rise to the amplification of MIKC subfamily genes in a variety of forms (Flagel et al., 2008; Hargreaves et al., 2014). As previously reported, multiple duplications and diversifications in the different clades of different species cause different evolutionary constraints (Lynch and Conery, 2000; Flagel and Wendel, 2009; Airoldi and Davies, 2012).
Chromosomal assignments indicated that the gene locations were equally divided among four pairs of chromosomes (At-chr6 and Dt-chr6, At-chr7 and Dt-chr7, At-chr10 and Dt-chr10, and At-chr13 and Dt-chr13) in A as well as in D genome (Figure 4). However, five D-genome chromosomes (Dt-chr2, Dt-chr4, Dt-chr9, Dt-chr11, and Dt-chr12) contained more genes compared with the corresponding A-genome chromosomes (At-chr2, At-chr4, At-chr9, At-chr11, and At-chr12). Additionally, large numbers of MIKC genes were located on the last three chromosomes (chr11, chr12, and chr13) of both genomes. This could indicate that the current phenomena were derived from differential rates of genomic evolution and inter-genomic hereditary information transfer (Paterson et al., 2000; Wendel and Cronn, 2003).
Expression Profiles of MIKC Genes in Gossypium hirsutum L
Global expression patterns analyses in seven different tissues showed that the API, AP3, AG, SEP, and BS subfamilies were almost all expressed in the flower development stage (Figure 6). Floral organ identities and flower meristem are regulated by five kinds of genetic functional genes (A-B-C-D-E) during flower development, from sepals to ovules (Díaz-Riquelme et al., 2009; Na et al., 2014). A qRT-PCR analysis showed the expression patterns of the orthologous genes of the ABCDE model in flower organogenesis (Figure 6), which were consistent with previous findings in Arabidopsis (Ó'Maoiléidigh et al., 2014; Xie et al., 2015). Further, the API subgroup of A class genes were not only expressed in sepals and petals, but also exhibited carpel expression profiles. Before and after pollination, the API-like gene may aid in the carpel development in Orchidaceae, which triggered ovary development (Mondragon-Palomino and Theissen, 2011; Acri-Nunes-Miranda and Mondragón-Palomino, 2014). Thus, AP1 subgroup genes may have similar expression patterns in Orchidaceae and allotetraploid cotton. A few GhMIKC* genes were highly expressed in flowers and seeds, which was in accordance with previous results in Arabidopsis (Verelst et al., 2007) and O. sativa (Liu et al., 2013). These results indicated that the expression profiles of MIKC* genes were involved in functional redundancy and conservation in the process of G. hirsutum evolution.
Role of the GhAGL17 Gene in Flowering
In Arabidopsis, AGL17 acts as a novel flowering promoter, which is involved in the photoperiod pathway. Under long-day conditions, the overexpression of AtAGL17 causes early flowering (Han et al., 2008). As the largest subgroup of the GhMIKCC family, one member of the AGL17s, GhAGL17.9, was overexpressed in Arabidopsis to explore its biological functions. The transgenic lines displayed earlier flowering than wild type (Figure 7). The expression levels of the related positive marker genes, especially LFY and SOC1, which are involved in regulating the flowering process, were higher in p35S::GhAGL17.9 lines than in wild type. LFY overexpression can prematurely cause plant development and accelerate blossoming processes (Nilsson et al., 1998; Dornelas and Amaral, 2004). AGL17 targets LFY to promote flowering (Han et al., 2008). SOC1 encodes a MIKC protein, a floral pathway integrator, which is regulated by a variety of flower signaling pathways (Lee et al., 2000; Wang et al., 2009; Ding et al., 2013). However, the relationship between AGL17 and SOC1 in flowering is not clear, which remains to be functionally explored further in the future.
Conclusions
In this study, 110 MIKC genes were first identified in the G. hirsutum genome. The family was divided into 13 subgroups based on a phylogenetic tree, exon/intron structures, and the distributions of conserved motifs. Chromosomal locations of MIKC gene family members were also determined. Finally, the expression patterns of GhMIKCs were explored using transcriptome sequencing and qRT-PCR, which revealed the expression levels at different developmental stages. Most MIKCC genes were highly expressed in the floral organs, which was consistent with the ABCDE model. The overexpression of GhAGL17.9 in Arabidopsis resulted in early flowering through the upregulated expression of SOC1, CO, and LFY, which suggested that GhMIKCs play vital roles in cotton flowering. Our work provides functional insights into the roles of GhMIKC genes in cotton flowering.
Author Contributions
ZuY and FL conceived and designed the experiments. ZR and DY performed the experiments. ZhY conducted the phylogeny analysis. CL and LL prepared the materials. HZ and QC analyzed the data. ZR and ZuY wrote the paper. GQ, YL, JL, ZL, and LW helped to revise the paper. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31501345), Zhengzhou Science and Technology Program (153PXXCY180) and Young Elite Scientist Sponsorship Program by CAST. We thank Peng Huo (Zhengzhou Research Center, Institute of Cotton Research of CAAS, Zhengzhou) for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00384/full#supplementary-material
References
Acri-Nunes-Miranda, R., and Mondragón-Palomino, M. (2014). Expression of paralogous SEP-, FUL-, AG- and STK-like MADS-box genes in wild-type and peloric Phalaenopsis flowers. Front. Plant Sci. 5:76, doi: 10.3389/fpls.2014.00076
Airoldi, C. A., and Davies, B. (2012). Gene Duplication and the evolution of plant MADS-box transcription factors. J. Genet. Genomics 39, 157–165. doi: 10.1016/j.jgg.2012.02.008
Alhassan, Y., Kumar, N., Bugaje, I. M., Pali, H. S., and Kathkar, P. (2014). Co-solvents transesterification of cotton seed oil into biodiesel: effects of reaction conditions on quality of fatty acids methyl esters. Energy Conversion Manage. 84, 640–648. doi: 10.1016/j.enconman.2014.04.080
Alvarez-Buylla, E. R., Pelaz S., Liljegren, S. J., Gold, S. E., Burgeff, C., Ditta, G. S., et al. (2000). An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. U.S.A. 97, 5328–5333. doi: 10.1073/pnas.97.10.5328
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8:242. doi: 10.1186/1471-2164-8-242
Becker, A., and Theissen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489. doi: 10.1016/S1055-7903(03)00207-0
Becker, A., Winter, K. U., Meyer, B., Saedler, H., and Theissen, G. (2000). MADS-box gene diversity in seed plants 300 million years ago. Mol. Biol. Evol. 17, 1425–1434. doi: 10.1093/oxfordjournals.molbev.a026243
Bowman, J. L., Smyth, D. R., and Meyerowitz, E. M. (1991). Genetic interactions among floral homeotic genes of Arabidopsis. Development 112, 1–20.
Carlsson, A. S. (2009). Plant oils as feedstock alternatives to petroleum - A short survey of potential oil crop platforms. Biochimie 91, 665. doi: 10.1016/j.biochi.2009.03.021
Clough, S. J., and Bent, A. F. (1998). Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. Cell Mol. Biol. 16, 735–743.
Coen, E. S., and Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37. doi: 10.1038/353031a0
De Folter, S., Shchennikova, A. V., Franken, J., Busscher, M., Baskar, R., Grossniklaus, U., et al. (2006). A B sister MADS-box gene involved in ovule and seed development in petunia and Arabidopsis. Plant J. 47, 934–946. doi: 10.1111/j.1365-313X.2006.02846.x
Díaz-Riquelme, J., Lijavetzky, D., Martínez-Zapater, J. M., and Carmona, M. J. (2009). Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiol. 149, 354–369. doi: 10.1104/pp.108.131052
Dietrich, D., Schmuths, H., De Marcos Lousa, C., Baldwin, J. M., Baldwin, S. A., Baker, A., et al. (2009). Mutations in the Arabidopsis Peroxisomal ABC transporter COMATOSE allow differentiation between multiple functions in planta: insights from an Allelic Series. Mol. Biol. Cell 20, 530–543. doi: 10.1091/mbc.E08-07-0745
Ding, L., Wang, Y., and Yu, H. (2013). Overexpression of DOSOC1, an ortholog of Arabidopsis SOC1, promotes flowering in the Orchid dendrobium chao parya smile. Plant Cell Physiol. 54, 595–608. doi: 10.1093/pcp/pct026
Dornelas, M. C., and Amaral, W. A. N. D. (2004). EgLFY, the Eucalyptus grandis homolog of the Arabidopsis gene LEAFY is expressed in reproductive and vegetative tissues. Braz. J. Plant Physiol. 16, 105–114. doi: 10.1590/S1677-04202004000200006
Duan, W., Song, X., Liu, T., Huang, Z., Ren, J., Hou, X., et al. (2015). Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genomics 290, 239–255. doi: 10.1007/s00438-014-0912-7
Fan, H. Y., Hu, Y., Tudor, M., and Ma, H. (1997). Specific interactions between the K domains of AG and AGLs, members of the MADS domain family of DNA binding proteins. Plant J. 12, 999–1010. doi: 10.1046/j.1365-313X.1997.12050999.x
Ferrándiz, C., Liljegren, S. J., and Yanofsky, M. F. (2000). Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289, 436–438. doi: 10.1126/science.289.5478.436
Flagel, L., Udall, J., Dan, N., and Wendel, J. (2008). Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 6:16. doi: 10.1186/1741-7007-6-16
Flagel, L. E., and Wendel, J. F. (2009). Gene duplication and evolutionary novelty in plants. New Phytol. 183, 557–564. doi: 10.1111/j.1469-8137.2009.02923.x
Greb, T., Mylne, J. S., Crevillen, P., Geraldo, N., An, H., Gendall, A. R., et al. (2007). The PHD finger protein VRN5 functions in the epigenetic silencing of Arabidopsis, FLC. Curr. Biol. 17, 73–78. doi: 10.1016/j.cub.2006.11.052
Guo, Y., Zhu, Q., Zheng, S., and Li, M. (2007). Cloning of a MADS Box Gene(GhMADS3)from cotton and analysis of its homeotic role in transgenic tobacco. Acta Genet. Sin. 34, 527–535. doi: 10.1016/s1673-8527(07)60058-7
Han, P., García-Ponce, B., Fonseca-Salazar, G., Alvarez-Buylla, E. R., and Yu, H. (2008). AGAMOUS-LIKE 17, a novel flowering promoter, acts in a FT-independent photoperiod pathway. Plant J. 55, 253–265. doi: 10.1111/j.1365-313X.2008.03499.x
Hargreaves, A. D., Swain, M. T., Hegarty, M. J., Logan, D. W., and Mulley, J. F. (2014). Restriction and recruitment—gene duplication and the origin and evolution of snake venom toxins. Genome Biol. Evol. 6, 2088. doi: 10.1093/gbe/evu166
Hartmann, U., Höhmann, S., Nettesheim, K., Wisman, E., Saedler, H., and Huijser, P. (2000). Molecular cloning of SVP: a negative regulator of the floral transition in Arabidopsis. Plant J. 21, 351–360. doi: 10.1046/j.1365-313x.2000.00682.x
Helliwell, C. A., Robertson, M., Finnegan, E. J., Buzas, D. M., and Dennis, E. S. (2011). Vernalization-repression of Arabidopsis FLC requires promoter sequences but not antisense transcripts. PLoS ONE 6, 240–247. doi: 10.1371/journal.pone.0021513
Helliwell, C. A., Wood, C. C., Robertson, M., Peacock, W., and Dennis, E. S. (2006). The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 46, 183–192. doi: 10.1111/j.1365-313X.2006.02686.x
Hepworth, S. R., Valverde, F., Ravenscroft, D., Mouradov, A., and Coupland, G. (2002). Antagonistic regulation of flowering-time gene SOC1 by CONSTANS and FLC via separate promoter motifs. Embo J. 21, 4327–4337. doi: 10.1093/emboj/cdf432
Honma, T., and Goto, K. (2001). Complexes of MADS-BOX proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. doi: 10.1038/35054083
Houhoula, D. P., Oreopoulou, V., and Tzia, C. (2003). The effect of process time and temperature on the accumulation of polar compounds in cottonseed oil during deep-fat frying. J. Sci. Food Agric. 83, 314–319. doi: 10.1002/jsfa.1314
Hu, L., and Liu, S. (2012). Genome-wide analysis of the MADS-box gene family in cucumber. Genome 55, 245–256. doi: 10.1139/g2012-009
Jiang, S.-C., Pang, C.-Y., Song, M.-Z., Wei, H.-L., Fan, S.-L., and Yu, S.-X. (2014). Analysis of MIKCC−Type MADS-box gene family in Gossypium hirsutum. J. Integr. Agric. 13, 1239–1249. doi: 10.1016/S2095-3119(13)60520-1
Kaufmann, K., Melzer, R., and Theissen, G. (2005). MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347, 183–198. doi: 10.1016/j.gene.2004.12.014
Kramer, E. M., and Irish, V. F. (1999). Evolution of genetic mechanisms controlling petal development. Nature 399, 144–148. doi: 10.1038/20172
Kuromori, T. (2010). Aba transport factors found in Arabidopsis ABC transporters. Plant Signal. Behav. 5, 1124–1126. doi: 10.4161/psb.5.9.12566
Lee, H., Suh, S. S., Park, E., Cho, E., Ahn, J. H., Kim, S. G., et al. (2000). The AGAMOUS-LIKE 20 MADS domain protein integrates floral inductive pathways in Arabidopsis. Genes Dev. 14, 2366–2376. doi: 10.1101/gad.813600
Lee, J. H., Yoo, S. J., Park, S. H., Hwang, I., Lee, J. S., and Ahn, J. H. (2007). Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 21, 397–402. doi: 10.1101/gad.1518407
Lee, J., and Lee, I. (2010). Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 61, 2247–2254. doi: 10.1093/jxb/erq098
Lee, J., Oh, M., Park, H., and Lee, I. (2008). SOC1 translocated to the nucleus by interaction with AGL24 directly regulates LEAFY. Plant J. Cell Mol. Biol. 55, 832–843. doi: 10.1111/j.1365-313X.2008.03552.x
Leseberg, C. H., Li, A., Kang, H., Duvall, M., and Mao, L. (2006). Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 378, 84–94. doi: 10.1016/j.gene.2006.05.022
Li, C., Wang, Y., Xu, L., Nie, S., Chen, Y., Liang, D., et al. (2016). Genome-wide characterization of the MADS-Box gene family in Radish (Raphanus sativus L.) and assessment of its roles in flowering and floral organogenesis. Front. Plant Sci. 7:1390. doi: 10.3389/fpls.2016.01390
Liljegren, S. J., Ditta, G. S., Eshed, Y., Savidge, B., Bowman, J. L., and Yanofsky, M. F. (2000). SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766–770. doi: 10.1038/35008089
Liu, C., Chen, H., Er, H. L., Soo, H. M., Kumar, P. P., Han, J. H., et al. (2008). Direct interaction of AGL24 and SOC1 integratesflowering signals in Arabidopsis. Development 135, 1481–1491. doi: 10.1242/dev.020255
Liu, Y., Cui, S., Wu, F., Yan, S., Lin, X., Du, X., et al. (2013). Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25, 1288–1303. doi: 10.1105/tpc.113.110049
Lynch, M., and Conery, J. S. (2000). The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Ma, H., and Depamphilis, C. (2000). The ABCs of floral evolution. Cell 101, 5–8. doi: 10.1016/S0092-8674(00)80618-2
Ma, H., Yanofsky, M. F., and Meyerowitz, E. M. (1991). AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 5, 484–495. doi: 10.1101/gad.5.3.484
Malik, T. H., and Ahsan, M. Z. (2016). Review of the Cotton Market in Pakistan and its future prospects. Oilseeds Fats Crops and Lipids 23:D606. doi: 10.1051/ocl/2016043
Messenguy, F., and Dubois, E. (2003). Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 316, 1–21. doi: 10.1016/S0378-1119(03)00747-9
Michaelk, D., Deborahl, B., Williamrjr, M., Btodd, C., Fredm, B., Johnr, G., et al. (2010). Fatty acid profiles of cottonseed genotypes from the national cotton variety trials. J. Cotton Sci. 14, 64–73.
Michaels, S. D., and Amasino, R. M. (1999). FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. doi: 10.1105/tpc.11.5.949
Michaels, S. D., Ditta, G., Gustafson-Brown, C., Pelaz, S., Yanofsky, M., and Amasino, R. M. (2003). AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. 33, 867–874. doi: 10.1046/j.1365-313X.2003.01671.x
Mondragón-Palomino, M. (2013). Perspectives on MADS-box expression during orchid flower evolution and development. Front. Plant Sci. 4:377. doi: 10.3389/fpls.2013.00377
Mondragon-Palomino, M., and Theissen, G. (2011). Conserved differential expression of paralogous DEFICIENS- and GLOBOSA-like MADS-box genes in the flowers of Orchidaceae: refining the “orchid code”. Plant J. 66, 1008–1019. doi: 10.1111/j.1365-313X.2011.04560.x
Mujeli, M., Kefas, H. M., Shitu, A., and Ayuba, I. (2016). Optimization of biodiesel production from crude cotton seed oil using central composite design. Am. J. Chem. Biochem. Eng. 1, 8–14. doi: 10.11648/j.ajcbe.20160101.12
Na, L., Liu, Y., Ming, Z., Min, J., and Li, H. (2014). Thinking out of the box: MADS-box genes and maize spikelet development. Afr. J. Biotechnol. 13, 4673–4679. doi: 10.5897/AJB11.3885
Nesi, N., Debeaujon, I., Jond, C., Stewart, A. J., Jenkins, G. I., Caboche, M., et al. (2002). The TRANSPARENT TESTA16 locus encodes the ARABIDOPSIS BSISTER MADS domain protein and is required for proper development and pigmentation of the seed coat. Plant Cell 14, 2463–2479. doi: 10.1105/tpc.004127
Nilsson, O., Lee, I., Blázquez, M. A., and Weigel, D. (1998). Flowering-time genes modulate the response to LEAFY activity. Genetics 150, 403–410.
Ó'Maoiléidigh, D. S., Graciet, E., and Wellmer, F. (2014). Gene networks controlling Arabidopsis thaliana flower development. New Phytol. 201, 16–30. doi: 10.1111/nph.12444
Pang, C. Y., Wang, H., Pang, Y., Xu, C., Jiao, Y., Qin, Y. M., et al. (2010). Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol. Cell. Proteomics 9, 2019. doi: 10.1074/mcp.M110.000349
Parenicová, L., de Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-Box transcription factor family in arabidopsis: new openings to the MADS world. Plant Cell Online 15, 1538–1551. doi: 10.1105/tpc.011544
Paterson, A. H., Bowers, J. E., Burow, M. D., Draye, X., Elsik, C. G., Jiang, C. X., et al. (2000). Comparative genomics of plant chromosomes. Plant Cell 12, 1523–1540. doi: 10.1105/tpc.12.9.1523
Pinyopich, A., Ditta, G. S., Savidge, B., Liljegren, S. J., Baumann, E., Wisman, E., et al. (2003). Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424, 85–88. doi: 10.1038/nature01741
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Reeves, P. A., He, Y., Schmitz, R. J., Amasino, R. M., Panella, L. W., and Richards, C. M. (2007). Evolutionary conservation of the FLOWERING LOCUS C-mediated vernalization response: evidence from the sugar beet (Beta vulgaris). Genetics 176, 295–307. doi: 10.1534/genetics.106.069336
Riechmann, J. L., Krizek, B. A., and Meyerowitz, E. M. (1996). Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. U.S.A. 93, 4793–4798. doi: 10.1073/pnas.93.10.4793
Rounsley, S. D., Ditta, G. S., and Yanofsky, M. F. (1995). Diverse roles for MADS box genes in Arabidopsis development. Plant Cell 7, 1259–1269. doi: 10.1105/tpc.7.8.1259
Sturn, A., Quackenbush, J., and Trajanoski, Z. (2002). Cluster analysis of microarray data. Bioinformatics 18, 207–208. doi: 10.1093/bioinformatics/18.1.207
Sánchez-Fernández, R., Davies, T. G., Coleman, J. O., and Rea, P. A. (2001). The Arabidopsis thaliana ABC protein superfamily, a complete inventory. J. Biol. Chem. 276, 30231–30244. doi: 10.1074/jbc.M103104200
Sawan, Z. M. (2014). Cottonseed yield and its quality as affected by mineral fertilizers and plant growth retardants. Agric. Sci. 05, 186–209. doi: 10.4236/as.2014.53023
Schönrock, N., Bouveret, R., Leroy, O., Borghi, L., Köhler, C., Gruissem, W., et al. (2006). Polycomb-group proteins repress the floral activator AGL19 in the FLC-independent vernalization pathway. Genes Dev. 20, 1667–1678. doi: 10.1101/gad.377206
Searle, I., He, Y., Turck, F., Vincent, C., Fornara, F., Kröber, S., et al. (2006). The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 20, 898–912. doi: 10.1101/gad.373506
Silva, C. S., Puranik, S., Round, A., Brennich, M., Jourdain, A., Parcy, F., et al. (2015). Evolution of the Plant Reproduction Master Regulators, L. F. Y., and the MADS Transcription Factors: The Role of Protein Structure in the Evolutionary Development of the Flower. Front. Plant Sci. 6:1193. doi: 10.3389/fpls.2015.01193
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tapia-López, R., García-Ponce, B., Dubrovsky, J. G., Garay-Arroyo, A., Pérez-Ruíz, R. V., Kim, S. H., et al. (2008). An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol. 146, 1182–1192. doi: 10.1104/pp.107.108647
Theissen, G. (2001). Development of floral organ identity: stories from the MADS house. Curr. Opin. Plant Biol. 4, 75–85. doi: 10.1016/S1369-5266(00)00139-4
Theißen, G., Kim, J. T., and Saedler, H. (1996). Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43, 484–516. doi: 10.1007/BF02337521
Theissen, G., and Melzer, R. (2007). Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann. Bot. 100, 603–619. doi: 10.1093/aob/mcm143
Verelst, W., Saedler, H. and Münster, T. (2007). MIKC* MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiol. 143, 447–460. doi: 10.1104/pp.106.089805
Waheed, A., Rasool, G., and Asghar, A. (2010). Effect of interesterified palm and cottonseed oil blends on cookie quality. Agric. Biol. J. North Am. 1, 402–406. doi: 10.5251/abjna.2010.1.3.402.406
Wang, J. W., Czech, B., and Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, Q., Zhu, Y., Sun, L., Li, L., Jin, S., and Zhang, X. (2016). Transgenic Bt cotton driven by the green tissue-specific promoter shows strong toxicity to lepidopteran pests and lower Bt toxin accumulation in seeds. Sci. China Life Sci. 59, 172–182. doi: 10.1007/s11427-015-4920-6
Wendel, J. F., and Cronn, R. C. (2003). Polyploidy and the evolutionary history of cotton. Adv. Agron. 78, 139–186. doi: 10.1016/S0065-2113(02)78004-8
Wu, D., Yu, S. X., Fan, S. L., Song, M. Z., and Wang, L. N. (2009). Cloning and expression analysis of a MADS-box protein gene (GhMADS-13) from upland cotton. Genomics Appl. Biol. 28, 223–228.
Xie, W., Huang, J., Liu, Y., Rao, J., Luo, D., He, M. (2015). Exploring potential new floral organ morphogenesis genes of Arabidopsis thaliana using systems biology approach. Front. Plant Sci. 6:829. doi: 10.3389/fpls.2015.00829
Yang, T., and Zheng, Y. (2016). State and trends of oil crops production in China. OCL 23, D603. doi: 10.1051/ocl/2016046
Zahn, L. M., Feng, B., and Ma, H. (2006). Beyond the ABC-Model: regulation of floral homeotic genes. Adv. Bot. Res. 44, 163–207. doi: 10.1016/S0065-2296(06)44004-0
Zhang, L., Luo, Y., Hou, Z., He, Z., and Eli, W. (2014). Synthesis of carbonated cotton seed oil and its application as lubricating base oil. J. Am. Oil Chemists' Soc. 91, 143–150. doi: 10.1007/s11746-013-2358-1
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhao, Y., Li, X., Chen, W., Peng, X., Cheng, X., Zhu, S., et al. (2010). Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tissue Organ Culture 105, 159–173. doi: 10.1007/s11240-010-9848-8
Zhou, Y., Li, B. Y., Li, M., Li, X. J., Zhang, Z. T., Li, Y., et al. (2014). A MADS-box gene is specifically expressed in fibers of cotton (Gossypium hirsutum) and influences plant growth of transgenic Arabidopsis in a GA-dependent manner. Plant Physiol. Biochem. 75, 70–79. doi: 10.1016/j.plaphy.2013.12.003
Keywords: Gossypium hirsutum L., GhMIKCs, phylogeny, structure, expression patterns, flower
Citation: Ren Z, Yu D, Yang Z, Li C, Qanmber G, Li Y, Li J, Liu Z, Lu L, Wang L, Zhang H, Chen Q, Li F and Yang Z (2017) Genome-Wide Identification of the MIKC-Type MADS-Box Gene Family in Gossypium hirsutum L. Unravels Their Roles in Flowering. Front. Plant Sci. 8:384. doi: 10.3389/fpls.2017.00384
Received: 25 December 2016; Accepted: 06 March 2017;
Published: 22 March 2017.
Edited by:
Leire Molinero-Ruiz, Instituto de Agricultura Sostenible (CSIC), SpainReviewed by:
Antonio José Monforte, Instituto de Biología Molecular y Celular de Plantas (CSIC), SpainAnkica Kondic-Spika, Institute of Field and Vegetable Crops, Serbia
Copyright © 2017 Ren, Yu, Yang, Li, Qanmber, Li, Li, Liu, Lu, Wang, Zhang, Chen, Li and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zuoren Yang, eWFuZ3p1b3JlbjQwMTJAMTYzLmNvbQ==
Fuguang Li, YXlsaWZ1Z0AxNjMuY29t
†These authors have contributed equally to this work.