 Pan Li1†
Pan Li1† Rui-Sen Lu1†
Rui-Sen Lu1† Wu-Qin Xu1
Wu-Qin Xu1 Tetsuo Ohi-Toma2
Tetsuo Ohi-Toma2 Min-Qi Cai1
Min-Qi Cai1 Ying-Xiong Qiu1*
Ying-Xiong Qiu1* Kenneth M. Cameron3
Kenneth M. Cameron3 Cheng-Xin Fu1
Cheng-Xin Fu1- 1Key Laboratory of Conservation Biology for Endangered Wildlife of the Ministry of Education, and Laboratory of Systematic & Evolutionary Botany and Biodiversity, College of Life Sciences, Zhejiang University, Hangzhou, China
- 2Botanical Gardens, Graduate School of Science, University of Tokyo, Tokyo, Japan
- 3Department of Botany, University of Wisconsin, Madison, WI, USA
The genus Amana Honda (Liliaceae), when it is treated as separate from Tulipa, comprises six perennial herbaceous species that are restricted to China, Japan and the Korean Peninsula. Although all six Amana species have important medicinal and horticultural uses, studies focused on species identification and molecular phylogenetics are few. Here we report the nucleotide sequences of six complete Amana chloroplast (cp) genomes. The cp genomes of Amana range from 150,613 bp to 151,136 bp in length, all including a pair of inverted repeats (25,629–25,859 bp) separated by the large single-copy (81,482–82,218 bp) and small single-copy (17,366–17,465 bp) regions. Each cp genome equivalently contains 112 unique genes consisting of 30 transfer RNA genes, four ribosomal RNA genes, and 78 protein coding genes. Gene content, gene order, AT content, and IR/SC boundary structure are nearly identical among all Amana cp genomes. However, the relative contraction and expansion of the IR/SC borders among the six Amana cp genomes results in length variation among them. Simple sequence repeat (SSR) analyses of these Amana cp genomes indicate that the richest SSRs are A/T mononucleotides. The number of repeats among the six Amana species varies from 54 (A. anhuiensis) to 69 (Amana kuocangshanica) with palindromic (28–35) and forward repeats (23–30) as the most common types. Phylogenomic analyses based on these complete cp genomes and 74 common protein-coding genes strongly support the monophyly of the genus, and a sister relationship between Amana and Erythronium, rather than a shared common ancestor with Tulipa. Nine DNA markers (rps15–ycf1, accD–psaI, petA–psbJ, rpl32–trnL, atpH–atpI, petD–rpoA, trnS–trnG, psbM–trnD, and ycf4–cemA) with number of variable sites greater than 0.9% were identified, and these may be useful for future population genetic and phylogeographic studies of Amana species.
Introduction
Tulips (genus Tulipa sensu lato) are among the world's most well-known, beloved, and economically important flowering plants. Their horticultural popularity, especially in Europe during the mid-seventeenth Century, led to bulbs being infamously traded in Holland as a form of speculative currency during a period that came to be known by historians as “tulip mania.” Although, there has been considerable research into the biology of tulips native to the Middle East and North Africa (Eijk et al., 1991; van Tunen et al., 1993; Van Creij et al., 1997; van Rossum et al., 1998; Zonneveld, 2009), much less is known of the East Asian tulips (e.g., Tulipa edulis), a group of species that most botanists today recognize as a distinct genus Amana Honda (Liliaceae). Amana is comprised of ca. six species of geophytic, perennial, understory herbs that are endemic to temperate East Asia (Ohwi and Kitagawa, 1992; Chen and Mordak, 2000; Shen, 2001; Tan et al., 2007; Han et al., 2014). The genus does, indeed, share many morphological characters with Tulipa L. (tulips), which is why most taxonomists until recently have classified it within Tulipa sensu lato (Sealy, 1957; Mao, 1980; Ohwi and Kitagawa, 1992; Liang, 1995; Tamura, 1998; Shen, 2001). However, Amana differs from Tulipa sensu stricto in having 2–3(–4) opposite or verticillate bracts in the upper part of the flowering stem and a longer style that is as long as the ovary (Tan et al., 2005). In many features it also resembles the genus Erythronium L. (trout lilies) from North America and Eurasia. At present, Amana is generally accepted as a separate genus (Tan et al., 2005, 2007; Christenhusz et al., 2013; Han et al., 2014). Recent molecular phylogenetic studies based on a few plastid regions and nuclear ribosomal ITS sequences (Hayashi and Kawano, 2000; Allen et al., 2003; Rønsted et al., 2005; Zarrei et al., 2009; Clennett et al., 2012; Christenhusz et al., 2013; Kim et al., 2013) have generally supported this separation. Amana, Erythronium, and Tulipa were strongly supported to be a monophyletic group in all of these studies, but the precise sister relationships among them has remained controversial. For example, some studies clustered Amana and Tulipa together (Hayashi and Kawano, 2000; Zarrei et al., 2009, ITS), whereas others supported a sister relationship between Erythronium and Tulipa (Allen et al., 2003; Christenhusz et al., 2013). Still others found that Amana is most closely related to Erythronium (Rønsted et al., 2005; Zarrei et al., 2009, five plastid regions combined; Clennett et al., 2012; Kim et al., 2013). All previous studies appear to have been based on insufficient information and thus could not fully resolve the phylogenetic relationships among these taxa.
The six currently recognized species of Amana occur in temperate deciduous or subtropical evergreen broad-leaved/mixed forests (Table 1). Within the genus, Amana edulis (Miq.) Honda is the most common and widely distributed species, ranging from China (central, eastern and northeastern provinces) to Japan (Honshu, Kyushu, and Shikoku) and the Korean peninsula (Ohwi and Kitagawa, 1992; Chen and Mordak, 2000; Park, 2007). The other five species are narrow endemics with non-overlapping areas among them, but all are broadly sympatric with A. edulis (Figure 1). However, these narrow endemic species rarely co-occur with the widespread A. edulis in intermixed populations due to the different altitudes of their natural habitats (A. edulis: 0–400 m, rarely to 850 m; other species: 600–1,400 m). Specifically, A. latifolia (Makino) Honda is restricted to a few sites in Honshu, Japan (Ohwi and Kitagawa, 1992); A. erythronioides (Baker) D. Y. Tan and D. Y. Hong and A. kuocangshanica D. Y. Tan and D. Y. Hong are confined to a few mountains near the coast of the East China Sea (Chen and Mordak, 2000; Tan et al., 2007), whereas A. anhuiensis (X. S. Shen) D. Y. Tan and D. Y. Hong and A. wanzhensis L. Q. Huang, B. X. Han and K. Zhang have more interior distributions in eastern China (Shen, 2001; Tan et al., 2008; Han et al., 2014; P. Li, pers. obs.). Despite the taxonomic recognition of six Amana species, the evolutionary history and interspecific relationships in this genus are still unclear because most species of Amana were missing from previous studies.
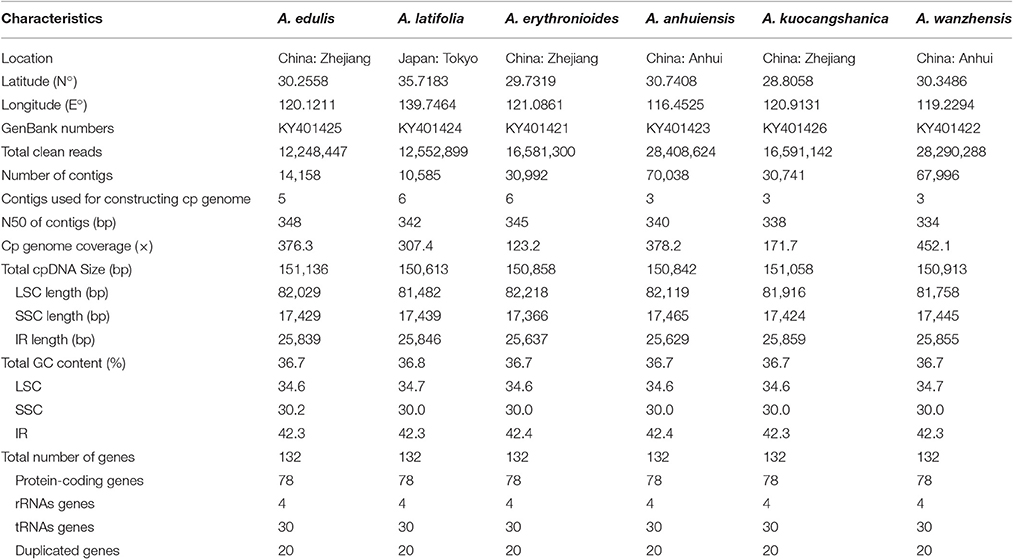
Table 1. The basic characteristics of six Amana species chloroplast (cp) genomes.
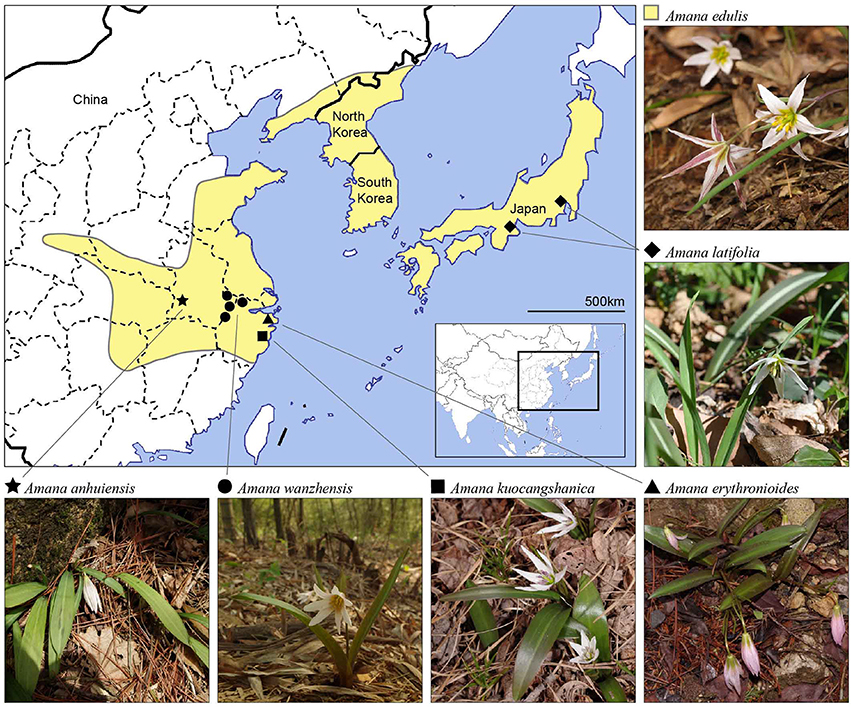
Figure 1. Distribution map of all six currently recognized Amana species. One of the species, A. edulis, is widespread and found primarily at low elevations (yellow-shaded areas). The other five species are narrow endemics restricted to disjunct montane habitats (filled symbols).
Not only are these plants valuable to humans as ornamentals, but they have considerable ethnobotanical uses as well. The bulb of Amana edulis is edible and commonly used as herbal medicine or starch source in China (Chen and Mordak, 2000). It has been used in traditional Chinese medicine (TCM) under the common name “Guangcigu” to treat sore throats, scrofula, ulcers and postpartum blood stasis (Chinese Herbalism Editorial Board, 1999). Other species in the genus Amana can be found as adulterants of Guangcigu, and these may result in different pharmacological actions, but such adulterants are often misidentified due to the similarity in their appearance with A. edulis (Ma H. L. et al., 2014). The increasing demand for wild-collected material of these economically important plants has brought about overexploitation of the natural populations in some regions. Therefore, a rapid and accurate method for species identification of Amana species is needed not only to facilitate proper medicinal uses, but also to aid conservation management.
In this study, we chose to analyze the complete chloroplast (cp) genomes of all six Amana species because of the plastome's conservative rate of evolution, absence of recombination, uniparental inheritance, and small effective population size (Birky et al., 1983). These are the same reasons that cpDNA sequences have been extensively used in studies of plant population genetics, phylogeography, phylogeny, and DNA barcoding (Jansen et al., 2007; Moore et al., 2010; Shaw et al., 2014). Compared with phylogenetic studies limited to a few cpDNA regions, cp phylogenomic studies involve many more informative sites for potentially greater resolution and support (Burke et al., 2012). With the rapid development of next-generation sequencing, cp genome-scale data have been increasingly employed to infer phylogenetic relationships at almost any taxonomic levels in the past decade (Jansen et al., 2007; Moore et al., 2007, 2010; Parks et al., 2009; Barrett et al., 2013; Ma P. F. et al., 2014; Carbonell-Caballero et al., 2015; Zhang et al., 2016). In addition, based on comparative genomic analyses, cp genomic hotspots can be identified as DNA barcodes in discriminating species, in terms of informative regions for a specific plant genus, tribe or family (Doorduin et al., 2011; Li et al., 2013, 2014). Our objectives are to: (1) characterize and compare the cp genomes of all six Amana in order to gain insights into their evolutionary patterns; (2) resolve the phylogenetic relationships among all Amana species and among closely related genera; (3) screen and identify the most rapidly evolving DNA regions of the Amana genome for species identification and future phylogeographic studies of the genus.
Materials and Methods
Plant Samples, DNA Extraction and Sequencing
Fresh leaf samples of six Amana species, five from China and one from Japan (Table 1), were field-collected and dried with silica gel. Voucher herbarium specimens were deposited at the Herbarium of Zhejiang University (HZU). We extracted total DNA from ca. 3 mg of the silica-gel dried leaf tissue for each species using DNA Plantzol Reagent (Invitrogen) and following the manufacturer's protocol. The qualities and quantities of genomic DNA were checked on an Agilent BioAnalyzer 2100 (Agilent Technologies). Short-insert (500 bp) paired-end libraries were generated by using Genomic DNA Sample Prep Kit (Illumina) according to the manufacturer's protocol. Genomic DNA of each species was indexed by barcode tags and then pooled together for sequencing in one lane of HiSeq™ 2500 (Illumina, San Diego, California, USA) at Beijing Genomics Institute (BGI, Shenzhen, China).
Genome Assembly and Annotation
For each Amana species, raw reads (125 bp read length) were firstly cleaned by removing low-quality reads with Phred scores of <20 using the CLC-quality trim tool (http://www.clcbio.com/products/clc-assembly-cell/). Secondly, we assembled the clean reads into contigs on the CLC de novo assembler (http://www.clcbio.com/products/clc-assembly-cell/), under the following settings: minimum contig length of 200 bp, mismatch cost of 2, deletion and insertion costs of 3, length fraction of 0.8, and similarity fraction of 0.8. Thirdly, all the contigs were aligned to the reference genome (Lilium longiflorum Thunb., KC968977) using BLAST (http://blast.ncbi.nlm.nih.gov/), and aligned contigs were oriented according to the reference genome. Then, contigs were aligned with the reference genome for constructing the draft chloroplast genome of each Amana species in GENEIOUS V9.0.5 (http://www.geneious.com). Finally, clean reads were re-mapped to the draft genome and yielded the complete chloroplast genome sequences.
Preliminary annotation of these Amana chloroplast genomes was conducted on the program Dual Organellar GenoMe Annotator (DOGMA; Wyman et al., 2004). DOGMA annotations were further corrected for the start/stop codons and intron/exon boundaries by comparison with homologous genes from L. longiflorum (GenBank accession no. KC968977) and Fritillaria hupehensis (GenBank accession no. KF712486) using MAFFT v7 (Katoh and Standley, 2013). In addition, tRNAscan-SE (Schattner et al., 2005) was used to verify the tRNA genes with default parameters, and the ultimately annotated chloroplast genomes were deposited in GenBank (accession numbers listed in Table 1). The cp genome maps were drawn in OrganellarGenome DRAW (Lohse et al., 2007). Codon usage, as well as relative synonymous codon usage (RSCU, Sharp and Li, 1987) value was estimated for all exons of protein-coding genes with the program CODONW V1.4.2 (http://codonw.sourceforge.net/).
Genome Comparative Analysis and Molecular Marker Identification
Chloroplast genome comparisons across the six Amana species was performed in Shuffle-LAGAN mode on the mVISTA program (genome.lbl.gov/vista/index. shtml, Frazer et al., 2004), using the annotation of A. kuocangshanica as a reference. To evaluate whether different cp genome regions underwent different evolution patterns in this genus, and to explore highly variable regions for future population genetic and species identification studies, we sequentially extracted both coding regions and noncoding regions (including intergenic spacers and introns) after alignment using MAFFT v7 under the two criteria that aligned length is >200 bp and at least one mutation site is present. After that, the nucleotide variability of these regions was evaluated with DNASP V5.10 (Librado and Rozas, 2009).
Identification of Repeat Sequences and Simple Sequence Repeats
REPUTER (Kurtz and Schleiermacher, 1999) was used to determine the size and position of repeat sequences, which included direct, inverted, complement and reverse repeats in the Amana chloroplast genomes. The minimum length of repeat size and sequence identity was set to 30 bp and >90%. MISA perl script (Thiel et al., 2003) was applied to detect the simple sequence repeats (SSRs) in the six Amana cp genomes with thresholds of 10, 5, 4, 3, 3, 3 repeat units for mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs, respectively.
Phylogenetic Analyses
Phylogenetic analyses were conducted on the six Amana species and one species each for Erythronium, Tulipa and Lloydia, using L. longiflorum (KC968977) and Fritillaria cirrhosa D. Don (KF769143) as outgroups based on previous studies (Rønsted et al., 2005; Kim et al., 2013). Chloroplast sequences of these 11 species were aligned using MAFFT v7. In order to evaluate possible alternative hypotheses of phylogeny, topologies were constructed by both maximum likelihood (ML) and Bayesian inference (BI) methods using not only the complete cp genome sequences (162,505 bp), but also the exons of protein-coding genes (78,815 bp). We also tried two different partitioning strategies for the second dataset: (1) separating each gene as a partition, (2) divided the data matrix into three partitions, corresponding to the first, second and third codon positions.
The best-fitting models of nucleotide substitutions were determined by the Akaike Information Criterion (AIC) in JMODELTEST V2.1.4 (Posada, 2008). The GTR+I+G model was most suitable for both datasets. Maximum likelihood analyses were conducted using RAXML-HPC v8.2.8 (Stamatakis, 2014) with 1000 bootstrap replicates at the CIPRES Science Gateway website (Miller et al., 2010). Bayesian inference (BI) analyses were performed in MRBAYES v3.2 (Ronquist and Huelsenbeck, 2003). Two independent Markov Chain Monte Carlo chains were calculated simultaneously for five million generations with trees sampled every 500 generations. The first 25% of calculated trees were discarded as burn-in, and a consensus tree was constructed from the remaining trees to estimate posterior probabilities (PPs).
Results
Genome Assembly and Structural Features
After filtering the low-quality reads and adaptor sequences, 12,248,447–28,408,770 clean reads (of 125 bp length) were obtained for the six Amana species. Through de novo assembly, 10,585 contigs (A. latifolia) to 70,038 contigs (A. anhuiensis) were assembled with N50 contigs varing from 338 to 348 bp (Table 1). Subsequently, three to six initial contigs which were found to be significantly homologous to the reference genome were combined to generate each chloroplast genome, with no gaps or missing nucleotides (Ns) found.
The full length of the six Amana chloroplast genomes ranged from 150,613 to 151,136 bp (Table 1; Figure 2). Akin to other angiosperms, chloroplast genomes of the six Amana species present a typical quadripartite structure, including a pair of inverted repeat regions (IR with 25,629–25,859 bp) separated by one large single-copy region (LSC with 81,482–82,218 bp) and one small single-copy region (SSC with 17,366–17,465 bp). All the complete cp genomes with annotation were deposited in GenBank (accession numbers listed in Table 1). The GC content in the LSC, SSC, and IR regions, and also in the whole genome sequences, were nearly identical among the six Amana species (Table 1).
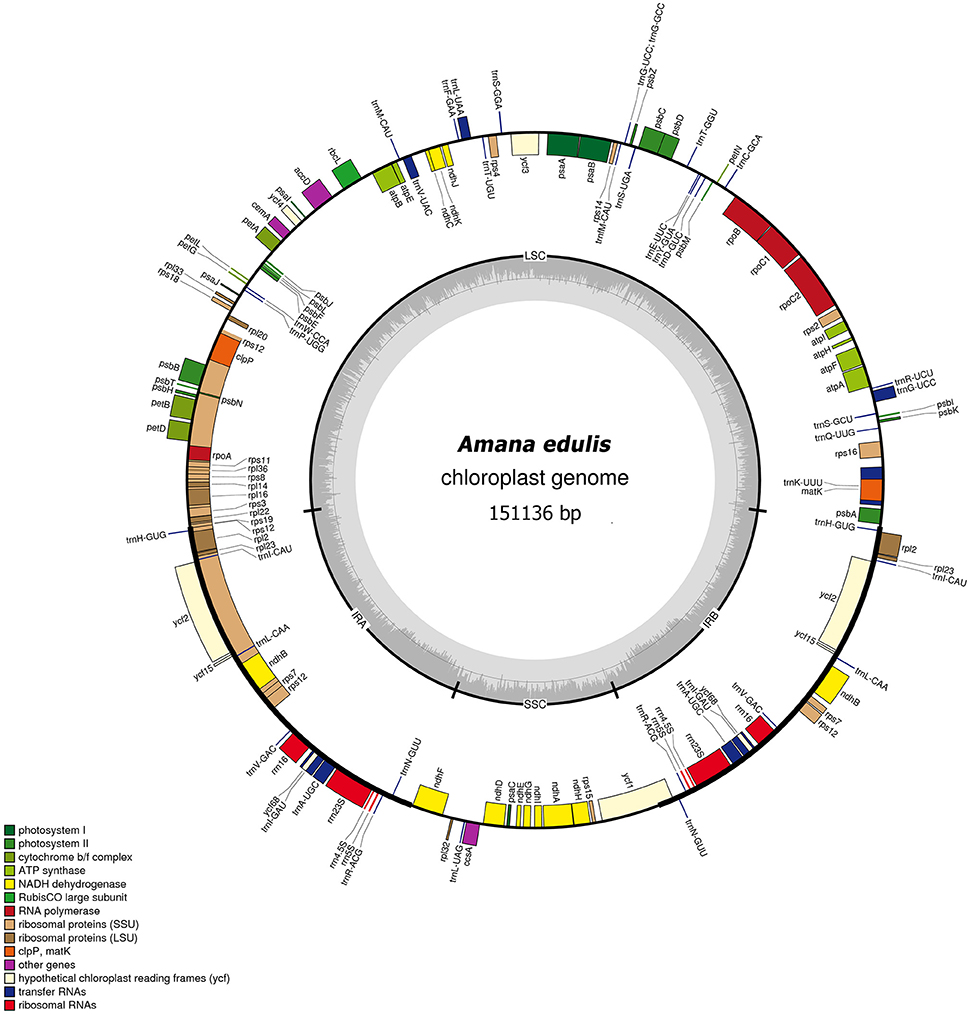
Figure 2. Gene map of the Amana edulis chloroplast genome. Genes shown on the outside of the circle are transcribed clockwise, and genes inside are transcribed counter-clockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner corresponds to the GC content, and the lighter gray to the AT content. The cp genomes of other five Amana species are slightly different with that of A. edulis in nucleotide composition, but do not vary in terms of gene content or order.
The six Amana chloroplast genomes contained the same 132 genes, of which 20 were duplicated in the IR regions and 112 were unique genes comprising four rRNA genes, 30 tRNA genes and 78 protein-coding genes (Table 2). Of the 112 distinct genes, 15 held a single intron (nine protein-coding genes and six tRNA genes) and three (ycf3, clpP, and rps12) possessed two introns. The gene infA was lost in all six Amana species. The gene rps12 was trans-spliced; the exon at the 5′ end was located in the LSC region, however the 3′ exon and intron were located in the IR regions. The regions ycf15, ycf68, and ycf1 were identified as pseudogenes because they contained several internal stop codons. Besides, the rps19 gene located in the IRa/LSC junction region lost their protein-coding ability because of incomplete gene duplication. The similar event was also observed in the ycf1 region at the IRb and SSC border (see below for detailed information).
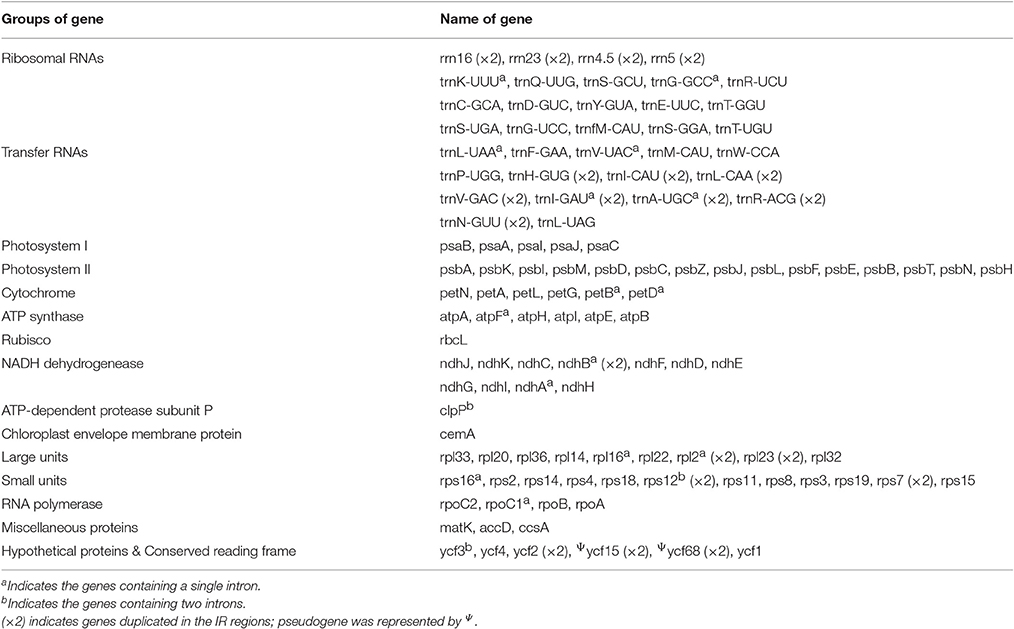
Table 2. Gene composition in six Amana chloroplast genomes.
The number of codons encoded by 78 protein-coding genes in the six Amana chloroplast genomes ranged from 24,349 to 24,380. Amana kuocangshanica was randomly selected as an example for detailed investigation, owing to the similar result of codon usage and RSCU values for these six species (Table S1). Among the 24,369 codons in A. kuocangshanica, 2,529 (10.38%) encoded leucine and 286 (1.17%) encode cysteine, which were the most and least frequent amino acids, respectively (Table S1, Figure S1). In synonymous codons, RSCU value increased with the number of the codons. Furthermore, the RSCU values of 30 codons were greater than 1, suggesting that they are biased codons in the A. kuocangshanica chloroplast protein-coding genes (Table S1, Figure S1).
Boundaries between IR and SC Regions
The IR/SC borders with full annotations for adjacent genes were compared among the six Amana chloroplast genomes (Figure 3). Except for A. anhuiensis and A. erythronioides, all the IRb regions expanded by 104–106 bp toward the rps19 gene with corresponding pseudogene fragment ψrps19 created at the IRa/LSC border. Long ψycf1 fragment with 1,121–1,154 bp was located at the IRb regions because the border between SSC and IRa extended into the ycf1 genes. In addition, the ndhF gene in A. wanzhensis overlapped with the IRa/SSC border by 66 bp. However, for the other five Amana species, the distance between ψycf1 and ndhF varied from 5 to 42 bp (Figure 3).
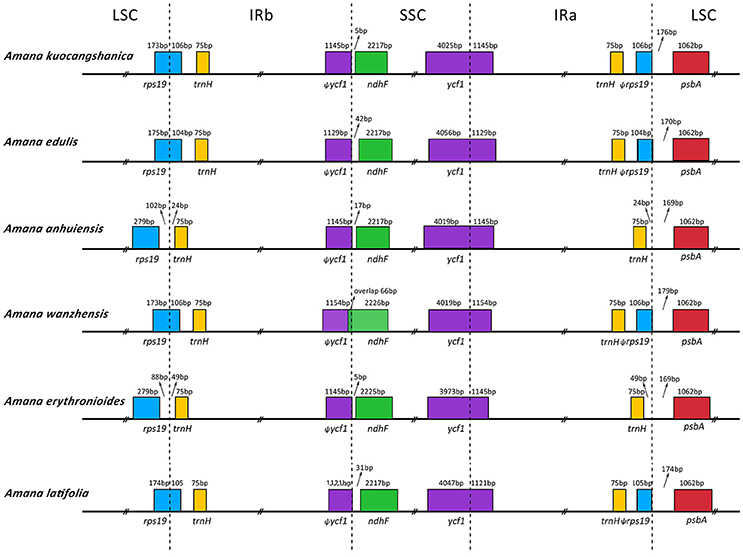
Figure 3. Comparison of the LSC, IR, and SSC junction positions among six Amana chloroplast genomes.
Comparative Genomic Analysis and Divergence Hotspot Regions
We analyzed the comprehensive sequence divergence of the six Amana cp genomes using the mVISTA software with the annotation of A. kuocangshanica as a reference. A genome-wide alignment revealed globally high sequence similarity (>90% identity) among them (Figure S2). Inverted repeat regions show a lower level of sequence divergence than LSC and SSC regions. In addition, 120 regions were eventually extracted to calculate the nucleotide variability, and the Pi value ranged from 0.02% (rrn23s) to 1.66% (rps15–ycf1). A total of nine regions (rps15–ycf1, accD–psaI, petA–psbJ, rpl32–trnL, atpH–atpI, petD–rpoA, trnS–trnG, psbM–trnD, and ycf4–cemA) with a nucleotide diversity >0.9% were recognized as hotspot regions that could be developed as molecular markers for future phylogenetic analysis and plant identification studies (Figure 4, Table S2).
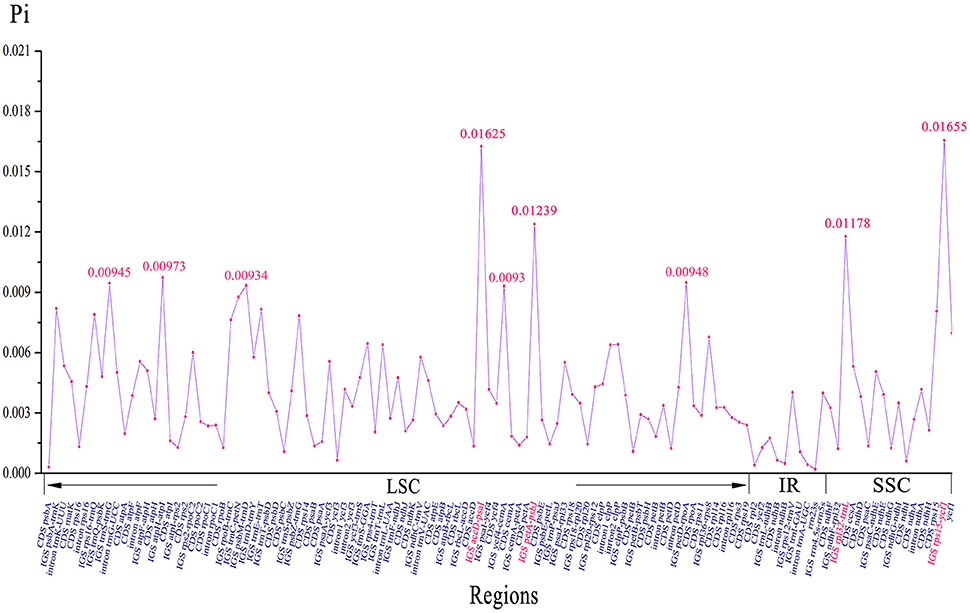
Figure 4. Nucleotide variability (Pi) values of six Amana chloroplast genomes.
Repeat Analysis and SSR Polymorphisms
A total of 371 repeats including 161 forward, 195 palindromic and 15 reverse repeats were identified in the six Amana cp genomes using the REPUTER software. Amana kuocangshanica possessed the greatest total number of repeats (69), while A. anhuiensis contained the fewest (54) (Figure S3A). For each Amana species, the majority of repeats (62.3% in A. kuocangshanica – 76.2% in A. edulis) ranged in size between 30 and 40 bp (Figure S3B). Repeats located in homologous regions with the same lengths were identified as shared repeats. Under this criterion, 38 repeats were shared by all Amana species and seven repeats were shared by five of the Amana species (all except A. edulis). Amana edulis showed the most distinct repeats (19), whereas A. anhuiensis had the least number (2) (Figure S3C, Table S3).
Each Amana species contained 69 (A. latifolia) to 76 (A. wanzhensis) SSRs, and more than half were composed of A or T bases (Figure S4A, Table S4). In the total 438 SSR regions, the proportion of the repeats situated in the intergenic spacer (IGS) regions led to 58.68%, while the regions located in the coding DNA sequence (CDS), CDS introns, tRNA introns and ψycf1 accounted for 15.53, 14.38, 4.34, and 7.08%, respectively (Figure S4B, Table S4). In addition, 29 SSRs (excluding mononucleotide SSRs) were identified as polymorphic SSRs between Amana species (Table S5), which could be useful for further population studies. Three criteria for identification were followed: (1) SSRs possessed the same repeat units (2) the number of repeat units is different and (3) SSRs located in the homologous regions.
Phylogenetic Analyses
In the present study, two datasets (whole chloroplast genome sequences and shared protein-coding genes) from the six Amana species, together with Erythronium sibiricum, Tulipa altaica, Lloydia tibetica and two outgroups, were used to conduct various phylogenetic analyses. Both ML and BI methods, based on different datasets and partitioning strategies, produced highly congruent topologies (Figure 5). The phylogenetic trees based on complete genome sequences, which had full support at every nodes [ML bootstrap (BS) = 100%, Bayesian posterior probabilities (PP) = 1], are shown here. Amana, Erythronium, and Tulipa were fully supported as a monophyletic group, in which the Amana species were resolved as a monophyletic clade that was sister to E. sibiricum. For the six Amana species, A. edulis was sister to a clade of the other remaining species with (A. erythronioides + A. kuocangshanica) and A. latifolia sharing a common ancestor and sister to (A. anhuiensis + A. wanzhensis) (Figure 5).
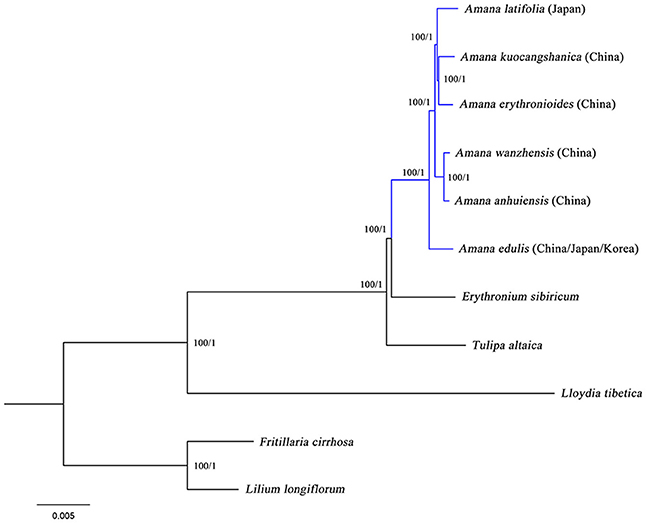
Figure 5. Phylogenetic relationships among Amana, Erythronium, Tulipa and within Amana inferred from maximum likelihood (ML) and Bayesian inference (BI) based on complete genome sequences (162,505 bp). Numbers above the lines represent ML bootstrap values and BI posterior probabilities. A phylogenetic tree resulting from analysis of 74 protein-coding genes (78,815 bp) was fully congruent with this topology.
Discussion
Comparative Genomics
Our results revealed that the overall gene content and arrangement within the six Amana cp genomes are largely similar. This is expected considering the morphological similarities and presumed recent age of divergence among them. At the same time, however, we have been able to document that the plastomes of these species do, indeed, vary, even if their differences are small. The IR/LSC boundaries in the Amana chloroplast genomes (except those of A. erythronioides and A. anhuiensis) expand into the rps19 gene. This is congruent with a typical monocot cp genome structure (Yang et al., 2013). Among other taxa in the family Liliaceae, IR expansion into rps19 has been observed in Lilium (Kim and Kim, 2013), Fritillaria (Li et al., 2014), and Cardiocrinum (Lu et al., 2016). This suggests that the expansion of the IR/LSC junctions into rps19 may be an ancestral symplesiomorphy of the family Liliaceae, and thus provides no relevant phylogenetic information for addressing intrafamilial questions.
Millen et al. (2001) suggested that infA, which codes for translation initiation factor 1, has been entirely lost or has become a pseudogene approximately 24 separate times in 309 angiosperms. According to their results, the parallel loss of infA from the chloroplast genome occurred in both Tricyrtis (Liliaceae) and Smilax (Smilacaceae), which are members of Liliales. Kim and Kim (2013) revealed that this event also occurred in Alstroemeria (Alstroemeriaceae), which is closer to the basal Liliales than Smilax. Besides, they found that infA existed but seemed to have lost its function in the Lilium (Liliaceae) cp genome, because it had AAT instead of ATG in the start codon position and includes two premature stop codons. The pseudogenization of infA was also found in Fritillaria (Li et al., 2014) and Cardiocrinum (Lu et al., 2016), close relatives of Lilium. However, our study indicates the loss of infA in Amana. Overall, it shows that infA may have been lost in the most recent common ancestor (MRCA) of Liliales, and the pseudogenization of infA seems to be a synapomorphy of the (Lilium + Fritillaria) + Cardiocrinum clade. Further study is needed to improve our understanding of infA gene evolution in Liliaceae.
Sister Relationship of Amana and Erythronium
Phylogenetic analyses of the complete chloroplast genome sequences and separate analyses restricted only to 74 common plastid protein-coding genes both produced a well-resolved phylogenetic tree (Figure 5). The close relationship and relatively short branches among Amana, Erythronium, and Tulipa was confirmed, which is congruent with previous studies (Hayashi and Kawano, 2000; Allen et al., 2003; Rønsted et al., 2005; Zarrei et al., 2009; Clennett et al., 2012; Kim et al., 2013; Petersen et al., 2013). However, our phylogenetic trees unambiguously revealed a sister relationship between Amana and Erythronium (BS = 100%, PP = 1), clearly supporting the separation of Amana from Tulipa, as others have argued (Rønsted et al., 2005; Zarrei et al., 2009; Clennett et al., 2012; Kim et al., 2013). Although a closer relationship between Amana and Tulipa, or of Erythronium and Tulipa, has been suggested by some former studies, most of them used only a single locus and/or found only weak to medium support for their topologies (rbcL: Hayashi and Kawano, 2000; matK: Allen et al., 2003; ITS: Zarrei et al., 2009). In at least one case (Christenhusz et al., 2013), although a multi-locus phylogenetic analysis of these three genera showed Erythronium sister to Tulipa rather than to Amana, no external outgroup taxa from the sister clade were used to orient the tree, and so that the relative branching order of Amana, Erythronium, and Tulipa was actually undetermined in that study. The chloroplast genome gives the most strongly supported indication of relationships among the three genera, but the possible concordance between plastid gene trees and species trees remains tentative, given that the chloroplast genome sequences of ~150 kbp still essentially represent a single-locus (linkage group) phylogeny (Ruhsam et al., 2015).
Phylogenetic Relationships within Amana
Within Amana, the rare species may be recently evolved ecotypes of the widespread A. edulis, quickly adapting lineage. As ecotypes, these rare species are expected to show no genetic distinction at neutral loci, and may not merit species recognition lineage (Oberle and Schaal, 2011). Nevertheless, the six Amana species exhibit sequence divergences in plastid genomes (Figure S2), ruling out the possibility of being ecotypes. Furthermore, our phylogenomic analyses as expected recovered Amana as a monophyletic genus (BS = 100%, PP = 1), and strongly supported its division into two clades: a widespread species (A. edulis) and a clade of five rare species (BS = 100%, PP = 1; Figure 5). The result indicates that although they are allopatrically distributed across East China/South Japan, the five rare species share a more recent common ancestor with each other than they do with A. edulis. Therefore, it is unlikely that any of these rare species originated from the widespread species through local geographical and ecological isolation by progenitor–derivative speciation (Crawford, 2010). In fact, the two sister lineages exhibit different eco-geographies: while A. edulis is widespread in lowland evergreen broad-leaved or temperate deciduous forests of East/North China, Japan, and the Korean peninsula, the rare species are endemic to the montane warm-temperate-deciduous (WTD) forest in East China/South Japan. In line with evidence from palaeomodeling of East Asian forest biomes (Harrison et al., 2001) and recent phylogeographic studies (reviewed in Qiu et al., 2011), the exceptionally high diversification rate in the “rare-species” clade is mainly driven by long-term allopatric population isolation (viz. vicariance) in which climate-induced eco-geographic isolation through (a)biotic displacement of WTD forested habitats at different spatial–temporal scales and over glacial and interglacial periods is the primary vicariance factor (see also Qiu et al., 2009). Overall, our phylogenomic analyses based on chloroplast genomes have provided the first successful attempt to clarify intrageneric relationships within Amana. However, based on distributional considerations, hybridization is still expected to occur between the widespread A. edulis and rare species within their zone of sympatry. Although these cp genome data have generated a fully resolved phylogeny of the genus Amana (Figure 5), it is not possible to use such data to classify hybridization events because cpDNA is generally uniparentally inherited (Birky, 1995). In the future, multi-locus phylogenies, phylogeography and palaeo-climatic niche modeling are required to explore the time scales and demographies of species divergences as well as hybridization in this genus.
Author Contributions
PL, YQ, and CF conceived the ideas; PL, RL, TO, and MC contributed to the sampling; MC performed the experiment; RL and WX analyzed the data. The manuscript was written by PL, RL, YQ, and KC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No. 31500184), the International Cooperation and Exchange of the National Natural Science Foundation of China (Grant Nos. 31511140095, 31561143015), the National Science and Technology Basic Project of China (Grant No. 2015FY110200), the Special Project for National Industry of TCM (201407002), and Student Research and Innovation Program (Xinmiao Talent Program) of Zhejiang Province (2016R401261). We thank Mrs. Sarah Friedrich for her kind help to improve the figures.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00451/full#supplementary-material
Figure S1. Codon usage (gray bar) and relative synonymous codon usage (RSCU) value (red dot) of six Amana chloroplast genomes.
Figure S2. Sequence identity plots between five Amana chloroplast genomes, with A. kuocangshanica as a reference. Annotated genes are displayed along the top. The vertical scale represents the percent identity between 50 and 100%. Genome regions are color coded as exon, intron, and conserved non-coding sequences (CNS).
Figure S3. Repeat analyses in six Amana chloroplast genomes. (A) Frequency of repeat types. (B) Frequency of repeats by length. (C) Summary of the shared repeats among species (ED, A. edulis; LA, A. latifolia; ER, A. erythronioides; AN, A. anhuiensis; KU, A. kuocangshanica; WA, A. wanzhensis).
Figure S4. Simple sequence repeats (SSRs) in six Amana chloroplast genomes. (A) Numbers of SSRs by length. (B) Distribution of SSR loci in the cp genomes. IGS, intergenic spacer region.
Table S1. Codon usage and relative synonymous codon usage (RSCU) value of six Amana chloroplast genomes.
Table S2. Nucleotide variability (Pi) values and total number of mutation (Eta) in Amana.
Table S3. Analyses of repeat sequences in six Amana chloroplast genomes.
Table S4. Simple sequence repeat (SSR) distribution in six Amana chloroplast genomes.
Table S5. Simple sequence repeat (SSR) polymorphism in six Amana chloroplast genomes.
References
Allen, G. A., Soltis, D. E., and Soltis, P. S. (2003). Phylogeny and biogeography of Erythronium (Liliaceae) inferred from chloroplast matK and nuclear rDNA ITS sequences. Syst. Bot. 28, 512–523. doi: 10.1043/02-18.1
Barrett, C. F., Davis, J. I., Leebens-Mack, J., Conran, J. G., and Stevenson, D. W. (2013). Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29, 65–87. doi: 10.1111/j.1096-0031.2012.00418.x
Birky, C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. U.S.A. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Birky, C. W. Jr., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527.
Burke, S. V., Grennan, C. P., and Duvall, M. R. (2012). Plastome sequences of two New World bamboos—Arundinaria gigantea and Cryptochloa strictiflora (Poaceae)—extend phylogenomic understanding of Bambusoideae. Am. J. Bot. 99, 1951–1961. doi: 10.3732/ajb.1200365
Carbonell-Caballero, J., Alonso, R., Ibañez, V., Terol, J., Talon, M., and Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus. Mol. Biol. Evol. 32, 2015–2035. doi: 10.1093/molbev/msv082
Chen, X. Q., and Mordak, H. V. (2000). “Tulipa,” in Flora of China, Vol. 24, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press/St. Louis; Botanical Garden Press), 123–126.
Chinese Herbalism Editorial Board (1999). Chinese Materia Medica. Shanghai: Shanghai Science and Technology Press.
Christenhusz, M. J. M., Govaerts, R., David, J. C., Hall, T., Borland, K., Roberts, P. S., et al. (2013). Tiptoe through the tulips-cultural history, molecular phylogenetics and classification of Tulipa (Liliaceae). Bot. J. Linn. Soc. 172, 280–328. doi: 10.1111/boj.12061
Clennett, J. C. B., Chase, M. W., Forest, F., Maurin, O., and Wilkin, P. (2012). Phylogenetic systematics of Erythronium (Liliaceae): morphological and molecular analyses. Bot. J. Linn. Soc. 170, 504–528. doi: 10.1111/j.1095-8339.2012.01302.x
Crawford, D. J. (2010). Progenitor–derivative species pairs and plant speciation. Taxon 59, 1413–1423. doi: 10.2307/20774038
Doorduin, L., Gravendeel, B., Lammers, Y., Ariyurek, Y., Chin-A-Woeng, T., and Vrieling, K. (2011). The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 18, 93–105. doi: 10.1093/dnares/dsr002
Eijk, J. V., Raamsdonk, L. V., Eikelboom, W., and Bino, R. J. (1991). Interspecific crosses between Tulipa gesneriana cultivars and wild Tulipa species: a survey. Sexual Plant Reprod. 4, 1–5. doi: 10.1007/BF00194563
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Han, B. X., Zhang, K., and Huang, L. Q. (2014). Amana wanzhensis (Liliaceae), a new species from Anhui, China. Phytotaxa 177, 118–124. doi: 10.11646/phytotaxa.177.2.3
Harrison, S. P., Yu, G., Takahara, H., and Prentice, I. C. (2001). Palaeovegetation (Communications arising): diversity of temperate plants in east Asia. Nature 413, 129–130. doi: 10.1038/35093166
Hayashi, K., and Kawano, H. (2000). Molecular systematics of Lilium and allied genera (Liliaceae): phylogenetic relationships among Lilium and related genera based on the rbcL and matK gene sequence data. Plant Spec. Biol. 15, 73–93. doi: 10.1046/j.1442-1984.2000.00025.x
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., dePamphilis, C. W., Leebens-Mack, J., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U.S.A. 104:19369–19374. doi: 10.1073/pnas.0709121104
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J. S., Hong, J. K., Chase, M. W., Fay, M. F., and Kim, J. H. (2013). Familial relationships of the monocot order Liliales based on a molecular phylogenetic analysis using four plastid loci: matK, rbcL, atpB and atpF-H. Bot. J. Linn. Soc. 172, 5–21. doi: 10.1111/boj.12039
Kim, J. S., and Kim, J. H. (2013). Comparative genome analysis and phylogenetic relationship of order Liliales insight from the complete plastid genome sequences of two Lilies (Lilium longiflorum and Alstroemeria aurea). PLoS ONE 8:e68180. doi: 10.1371/journal.pone.0068180
Kurtz, S., and Schleiermacher, C. (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15, 426–427. doi: 10.1093/bioinformatics/15.5.426
Li, Q., Li, Y., Song, J., Xu, H., Xu, J., Zhu, Y., et al. (2014). High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy. New Phytol. 204, 1041–1049. doi: 10.1111/nph.12966
Li, R., Ma, P. F., Wen, J., and Yi, T. S. (2013). Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 8:e78568. doi: 10.1371/journal.pone.0078568
Liang, S. Y. (1995). Chorology of Liliaceae (s. str.) and its bearing on the Chinese flora. Acta Phytotaxon. Sin. 33, 27–51.
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lu, R. S., Li, P., and Qiu, Y. X. (2016). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7:2054. doi: 10.3389/fpls.2016.02054
Ma, H. L., Zhu, Z. B., Zhang, X. M., Miao, Y. Y., and Guo, Q. S. (2014). Species identification of the medicinal plant Tulipa edulis (Liliaceae) by DNA barcode marker. Biochem. Syst. Ecol. 55, 362–368. doi: 10.1016/j.bse.2014.03.038
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H., and Li, D. Z. (2014). Chloroplast phylogenomic analysis resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Mao, Z. M. (1980). “Tulipa,” in Flora Reipublicae Popularis Sinicae, Vol. 14, eds F. Z. Wang and J. Tang (Beijing: Science Press), 87–93.
Millen, R. S., Olmstead, R. G., Adams, K. L., Palmer, J. D., Lao, N. T., Heggie, L., et al. (2001). Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13, 645–658. doi: 10.1105/tpc.13.3.645
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Gateway Computing Environments Workshop (GCE), (New Orleans, LA: IEEE), 1–8.
Moore, M. J., Bell, C. D., Soltis, P. S., and Soltis, D. E. (2007). Using plastid genome scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Oberle, B., and Schaal, B. A. (2011). Responses to historical climate change identify contemporary threats to diversity in Dodecatheon. Proc. Natl. Acad. Sci. U.S.A. 108, 5655–5660. doi: 10.1073/pnas.1012302108
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Petersen, G., Seberg, O., and Davis, J. I. (2013). Phylogeny of the Liliales (Monocotyledons) with special emphasis on data partition congruence and RNA editing. Cladistics 29, 274–295. doi: 10.1111/j.1096-0031.2012.00427.x
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Qiu, Y. X., Fu, C. X., and Comes, H. P. (2011). Plant molecular phylogeography in China and adjacent regions: tracing the genetic imprints of Quaternary climate and environmental change in the world's most diverse temperate flora. Mol. Phylogenet. Evol. 59, 225–244. doi: 10.1016/j.ympev.2011.01.012
Qiu, Y. X., Sun, Y., Zhang, X. P., Lee, J., Fu, C. X., and Comes, H. P. (2009). Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol. 183, 480–495. doi: 10.1111/j.1469-8137.2009.02876.x
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rønsted, N., Law, S., Thornton, H., Fay, M. F., and Chase, M. W. (2005). Molecular phylogenetic evidence for the monophyly of Fritillaria and Lilium (Liliaceae; Liliales) and the infrageneric classification of Fritillaria. Mol. Phylogenet. Evol. 35, 509–527. doi: 10.1016/j.ympev.2004.12.023
Ruhsam, M., Rai, H. S., Mathews, S., Ross, T. G., Graham, S. W., Raubeson, L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Res. 15, 1067–1078. doi: 10.1111/1755-0998.12375
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Sharp, P. M., and Li, W. H. (1987). The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295.
Shaw, J., Shafer, H. L., Leonard, O. R., Kovach, M. J., Schorr, M., and Morris, A. B. (2014). Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. Am. J. Bot. 101, 1987–2004. doi: 10.3732/ajb.1400398
Shen, X. S. (2001). A new species of Tulipa (Liliaceae) from China. Acta Bot. Yunnan. 23, 39–40. [In Chinese with English Summary].
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tamura, M. N. (1998). “Liliaceae” in The Families and Genera of Vascular Plants. III. Flowering Plants-Monocotyledons, Lilianae (except Orchidaceae), ed K. Kubitzki (Berlin: Springer Press), 343–353.
Tan, D. Y., Li, X. R., and Hong, D. Y. (2007). Amana kuocangshanica (Liliaceae), a new species from south-east China. Bot. J. Linn. Soc. 154, 435–442. doi: 10.1111/j.1095-8339.2007.00660.x
Tan, D. Y., Li, X. R., and Hong, D. Y. (2008). Neotypification and additional description of Amana anhuiensis (X.S.Shen) D.Y.Tan & D.Y.Hong (Liliaceae) from Anhui, China. Acta Bot. Boreal. Occident. Sin. 28, 393–395. [In Chinese with English Summary].
Tan, D. Y., Zhang, Z., Li, X. R., and Hong, D. Y. (2005). Restoration of the genus Amana Honda (Liliaceae) on the basis of cladistic analysis of morphological characters. Acta Phytotaxon. Sin. 43, 262–270. [In Chinese with English Summary].
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Van Creij, M. G. M., Kerckhoffs, D. M. F. J., and Van Tuyl, J. M. (1997). Interspecific crosses in the genus Tulipa L.: identification of pre-fertilization barriers. Sexual Plant Reprod. 10, 116–123. doi: 10.1007/s004970050077
van Rossum, M. W., Alberda, M., and van der Plas, L. H. (1998). Tulipaline and tuliposide in cultured explants of tulip bulb scales. Phytochemistry 49, 723–729. doi: 10.1016/S0031-9422(98)00199-X
van Tunen, A. J., Eikelboom, W., and Angenent, G. C. (1993). Floral organogenesis in Tulipa. Flower. Newsl. 16, 33–38.
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Zarrei, M., Wilkin, P., Fay, M. F., Ingrouille, M. J., Zarre, S., and Chase, M. W. (2009). Molecular systematics of Gagea and Lloydia (Liliaceae; Liliales): implications of analyses of nuclear ribosomal and plastid DNA sequences for infrageneric classification. Ann. Bot. 104, 125–142. doi: 10.1093/aob/mcp103
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Keywords: Amana, Tulipa, Erythronium, Liliaceae, chloroplast genome, genomic structure, phylogenomics
Citation: Li P, Lu R-S, Xu W-Q, Ohi-Toma T, Cai M-Q, Qiu Y-X, Cameron KM and Fu C-X (2017) Comparative Genomics and Phylogenomics of East Asian Tulips (Amana, Liliaceae). Front. Plant Sci. 8:451. doi: 10.3389/fpls.2017.00451
Received: 08 January 2017; Accepted: 15 March 2017;
Published: 04 April 2017.
Edited by:
Fulvio Cruciani, Sapienza University of Rome, ItalyCopyright © 2017 Li, Lu, Xu, Ohi-Toma, Cai, Qiu, Cameron and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying-Xiong Qiu, cXl4aGVyb0B6anUuZWR1LmNu
†These authors have contributed equally to this work.