 Hoang Dang Khoa Do
Hoang Dang Khoa Do Joo-Hwan Kim
Joo-Hwan Kim- Plant Systematics Laboratory, Department of Biological Science, Gachon University, Seongnam, South Korea
Chloroplast genomes (cpDNA) are highly valuable resources for evolutionary studies of angiosperms, since they are highly conserved, are small in size, and play critical roles in plants. Slipped-strand mispairing (SSM) was assumed to be a mechanism for generating repeat units in cpDNA. However, research on the employment of different small repeated sequences through SSM events, which may induce the accumulation of distinct types of repeats within the same region in cpDNA, has not been documented. Here, we sequenced two chloroplast genomes from the endemic species Heloniopsis tubiflora (Korea) and Xerophyllum tenax (USA) to cover the gap between molecular data and explore “hot spots” for genomic events in Melanthiaceae. Comparative analysis of 23 complete cpDNA sequences revealed that there were different stages of deletion in the rps16 region across the Melanthiaceae. Based on the partial or complete loss of rps16 gene in cpDNA, we have firstly reported potential molecular markers for recognizing two sections (Veratrum and Fuscoveratrum) of Veratrum. Melathiaceae exhibits a significant change in the junction between large single copy and inverted repeat regions, ranging from trnH_GUG to a part of rps3. Our results show an accumulation of tandem repeats in the rpl23-ycf2 regions of cpDNAs. Small conserved sequences exist and flank tandem repeats in further observation of this region across most of the examined taxa of Liliales. Therefore, we propose three scenarios in which different small repeated sequences were used during SSM events to generate newly distinct types of repeats. Occasionally, prior to the SSM process, point mutation event and double strand break repair occurred and induced the formation of initial repeat units which are indispensable in the SSM process. SSM may have likely occurred more frequently for short repeats than for long repeat sequences in tribe Parideae (Melanthiaceae, Liliales). Collectively, these findings add new evidence of dynamic results from SSM in chloroplast genomes which can be useful for further evolutionary studies in angiosperms. Additionally, genomics events in cpDNA are potential resources for mining molecular markers in Liliales.
Introduction
Chloroplast genome sequences provide useful information for phylogenetic studies of higher level taxa, including families and orders (Zomlefer et al., 2001; Ji et al., 2006; Barrett et al., 2013; Kim and Kim, 2013; Kim et al., 2013, 2016a; Nguyen et al., 2013; Ruhfel et al., 2014). Structural changes such as small and large inversions, gene contents (duplication, triplication, and deletion), and pseudogenization have provided valuable resources for examining genome evolution among plants. Gene duplications have been reported in previous studies (Lee et al., 2007; Cai et al., 2008; Schmickl et al., 2009). Specifically, different copies of trnF_GAA were found in several genera of Brassicaceae (Schmickl et al., 2009). Additionally, repeated DNA sequences, which were assumed to have originated from different mechanisms such as gene conversion, unequal recombination, and slipped-strand mispairing (SSM), are main resources for genomic events of duplication, deletion, and rearrangement in chloroplast genomes (Levinson and Gutman, 1987; Cai et al., 2008; Huang et al., 2014; Sveinsson and Cronk, 2014).
Melanthiaceae is a family within the order Liliales that includes 16 genera divided into five tribes: Melanthieae (7 genera), Heloniadeae (3 genera), Parideae (3 genera), Chionographideae (2 genera), and Xerophylleae (1 genus) (Angiosperm Phylogeny Group, 2009, 2016; Govaerts, 2016; WCSP, 2016). Prior to its grouping within Liliales, these genera were classified into different orders of Dioscoreales and Melanthiales based on the morphological characteristics of their extrorse anthers and ovaries, and often with the presence of three styles (Rudall et al., 2000). The tribe Parideae, comprising Paris, Psedotrillium, and Trillium, was formerly treated as an independent family, Trilliaceae (Thorne, 1992; Takhtajan, 1997). However, based on molecular and morphological data, this tribe was later reclassified as monophyletic within Liliales (Chase et al., 2000; Angiosperm Phylogeny Group, 2009; Kim and Kim, 2013). Recently, Pellicer et al. (2014) identified the extreme variations in genome size and a significant reduction in the number of chromosomes in Parideae. The evolution of the chloroplast genome (cpDNA) has been investigated in Veratrum patulum (Do et al., 2013), Chionographis japonica (Bodin et al., 2013), Paris verticillata (Do et al., 2014), Trillium species (Kim et al., 2016b), and Paris sp. (Huang et al., 2016), which represent three tribes, Melanthieae, Chionographideae, and Parideae, respectively. Specifically, different numbers of trnI_CAU and repeat sequences in rpl23-ycf2 regions and inversion was detected in tribe Parideae (Do et al., 2014; Huang et al., 2016; Kim et al., 2016b). The rps16 gene was completely lost in C. japoncica and partially deleted in V. patulum (Bodin et al., 2013; Do et al., 2014). Collectively, these findings suggest that Melanthiaceae possess evidence of different genomic events in cpDNA. Nonetheless, these genomic events in Melanthiaceae have not been fully characterized because of the lack of cpDNA data.
In this study, we sequenced the complete chloroplast genomes of Heloniopsis tubiflora (GenBank Accession number KM078036) and Xerophyllum tenax (GenBank Accession number KM078035), representing the two unreported tribes of Heloniadeae and Xerophylleae, to cover the gap of cpDNA data within Melanthiaceae. Based on the complete cpDNA sequences, we characterized the differentiation, including gene loss, duplication, and fluctuation of IR-LSC boundary, among five tribes of Melanthiaceae. Then, we applied these features to create the first potential molecular marker for recognizing two sections of Veratrum. Additionally, we questioned the pattern and the mechanism of repeat's accumulation in rpl23-ycf2 regions. Therefore, we sequenced this region among representatives from other families in Liliales and conducted comparative analyses of sequence data to (1) investigate the pattern of repeat's accumulation within rpl23-ycf2 regions of examined species, and (2) propose hypothetical scenarios for the duplication process.
Materials and Methods
Sample Collection, DNA Extraction, Whole-Genome Sequencing, and Assembly
Fresh leaves of H. tubiflora were collected in Deogyusan National Park, South Korea. Voucher specimens were deposited in the Herbarium of Gachon University (GCU). Dried leaves of X. tenax were obtained from the Forestfarm Plant Nursery (Williams, Oregon, USA). The plant materials used in this study were collected through the KNRRC (Medicinal Plants Resources Bank NRF-2010-0005790), supported by the Korea Research Foundation (resources provided by the Ministry of Education, Science and Technology in 2014). Total DNA was extracted using a DNAEasy Plant Mini Kit (Qiagen, Seoul, South Korea). These DNA samples were sequenced using the 454 system for H. tubiflora and the Hiseq2000 system for X. tenax. After removal of reads with ambiguous “N” bases, the remaining reads were trimmed with no more than a 5% chance of error per base before being mapped to the reference chloroplast genome sequences of C. japonica (Bodin et al., 2013) and P. verticillata (Do et al., 2014), to isolate chloroplast genome sequences using Geneious (Biomatters Ltd., Auckland, New Zealand). Based on the tribal relationship in Melanthiaceae (Kim et al., 2016a), we mapped the reads of H. tubiflora and X. tenax to cpDNA sequences of C. japonica and P. verticillata, respectively. The assembled reads were then extracted and reanalyzed in Geneious using the De Novo Assembly tool with the option of “no gaps or mismatches per read.” The consensus sequences generated from De Novo Assembly were used as references to reassemble raw reads. These steps were repeated until the complete cpDNA sequences were identified. Occasionally, gaps were present among chloroplast contigs. These remaining gaps were closed using the Sanger method with newly designed primers based on homologous sequences between the reads and reference sequences. Additionally, borders between the LSC, small single copy (SSC), and IR regions, as well as ambiguous regions (i.e., insertion and deletion events in coding regions and low coverage regions) were confirmed by Sanger sequencing methods. A total of 1,093,684 and 8,719,277 reads of H. tubiflora and X. tenax were generated, respectively. The results showed that cpDNA of H. tubiflora consisted of 37,973 (3.47%) out of 1,093,684 reads with a coverage rate of 19.2 x. For X. tenax, 196,299 reads (2.25%) belonged to chloroplast genome sequences with a coverage rate of 112.6 × over the cpDNA. The complete cpDNA sequences of X. tenax and H. tubiflora were deposited into GenBank under accession numbers KM078035 and KM078036, respectively (Table 1).
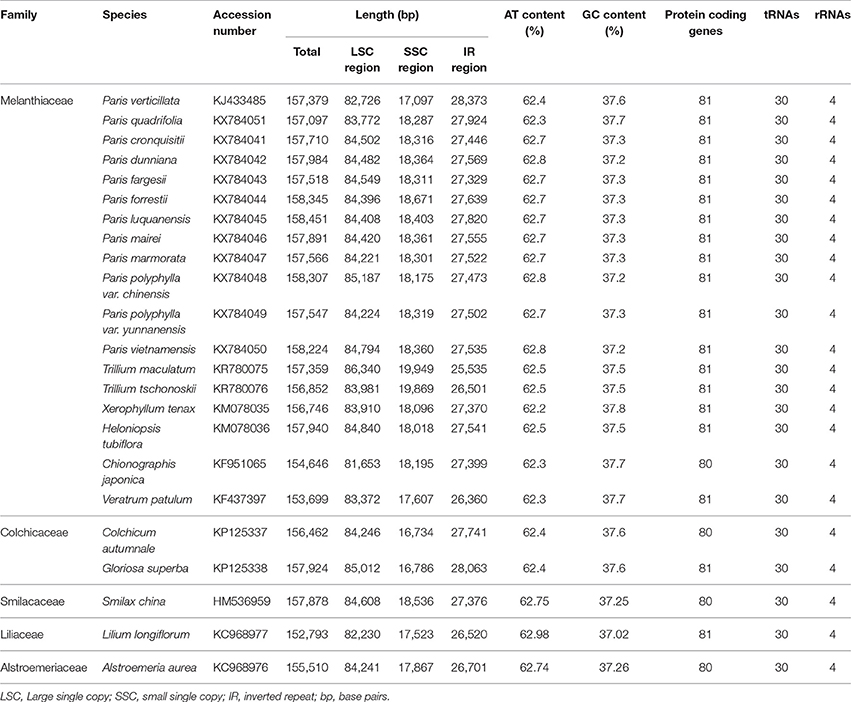
Table 1. Comparison of the features of cpDNAs from five families of Liliales.
Genome Annotation, Comparison, Visualization, and Characterization of Repeat Sequences
The complete cpDNA sequences of H. tubiflora and X. tenax were annotated using Geneious. All tRNA sequences were confirmed using the online web-based tool tRNAScan-SE (Schattner et al., 2005). The Mauve alignment, embedded in Geneious, was used to compare the 23 complete cpDNA sequences and to identify significant differentiation, including gene loss, duplication, and fluctuation of IR-LSC boundary with default settings (Darling et al., 2004). Genome maps were generated using OGDraw v1.2 (Lohse et al., 2013), followed by manual modifications. The maps of cpDNA sequences of X. tenax and H. tubiflora, which illustrate the genome structure and gene composition and order, were shown in Figure 1. The locations of repeat sequences were identified using Phobos (Mayer, 2006) with default settings.
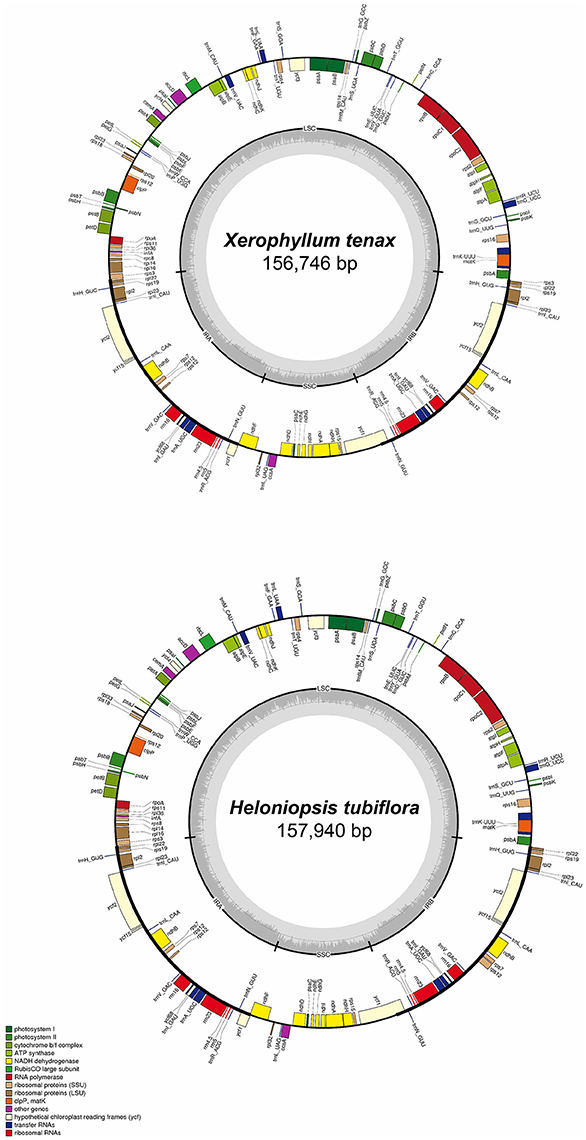
Figure 1. Map of Heloniopsis tubiflora and Xerophyllum tenax chloroplast genomes. Genes shown outside of the outer circle are transcribed counterclockwise, whereas those shown inside are transcribed clockwise. The thick lines in the small circles indicate the inverted repeat regions. The dark gray area in the inner circle indicates the CG content of the chloroplast genome. The colors represent different groups of genes in cpDNA. LSC, Large single copy; SSC, small single copy; IRA, inverted repeat region A; IRB, inverted repeat region B.
Characterization of rps16 Loss and Junction of IR-LSC Regions
After conducting alignment of complete cpDNA sequences using Geneious, we designed primer pairs for amplifying trnK_UUU-trnQ_UUG region, containing rps16 (data not shown). Because of different PCR results of verifying rps16 loss in tribe Melanthieae, primer pairs (rps16-F: 5′-GTCAATATGAATGTTGATAA-3′ and rps16-R: 5′-TTTTCTATTCCATACACATG-3′) were designed using Primer3 (Untergasser et al., 2012). The PCR profile consisted of denaturation at 94°C for 3 min followed by 35 cycles at 94°C for 1 min, 54°C for 1 min, and 72°C for 1 min, with a final extension at 72°C for 7 min. These newly designed primer pairs were applied for Veratrum species of two sections including Veratrum (V. oxysepalum—JK Hong 015; V. lobelianum—Chase 19618; V. grandiflorum—KUN0303565) and Fuscoveratrum (V. versicolor—GCU1411788; V. maackii—KWU03358; V. nigrum—GCU121205181). All PCR products were purified using the MEGAquick-spin Total Fragment DNA Purification Kit (iNtRON Biotechnology, Seoul, Korea) and sequenced using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions. These sequences were assembled and annotated in Geneious.
For identifying junction of IR/LSC regions, we designed primer pairs for rpl2-psbA and rpl2-rps3 regions (data not shown). The PCR products were sequenced and then annotated in Geneious. After getting annotation results, these sequences were aligned to identify the border among examined species.
Confirmation of Duplication Events in Liliales
To confirm the duplication patterns in Liliales, we sampled 68 taxa of 9 families within Liliales, except Corsiaceae which contains mycoheterotrophic species (Table 2). Total genomic DNA was extracted from dried leaves using a modified CTAB method based on Doyle and Doyle (1987). Primer pairs that amplify the entire rpl23-ycf2 IGS were applied with the same PCR profile from the description of Kim et al. (2016b). The further steps for PCR product's treatment were conducted identically as the description above.
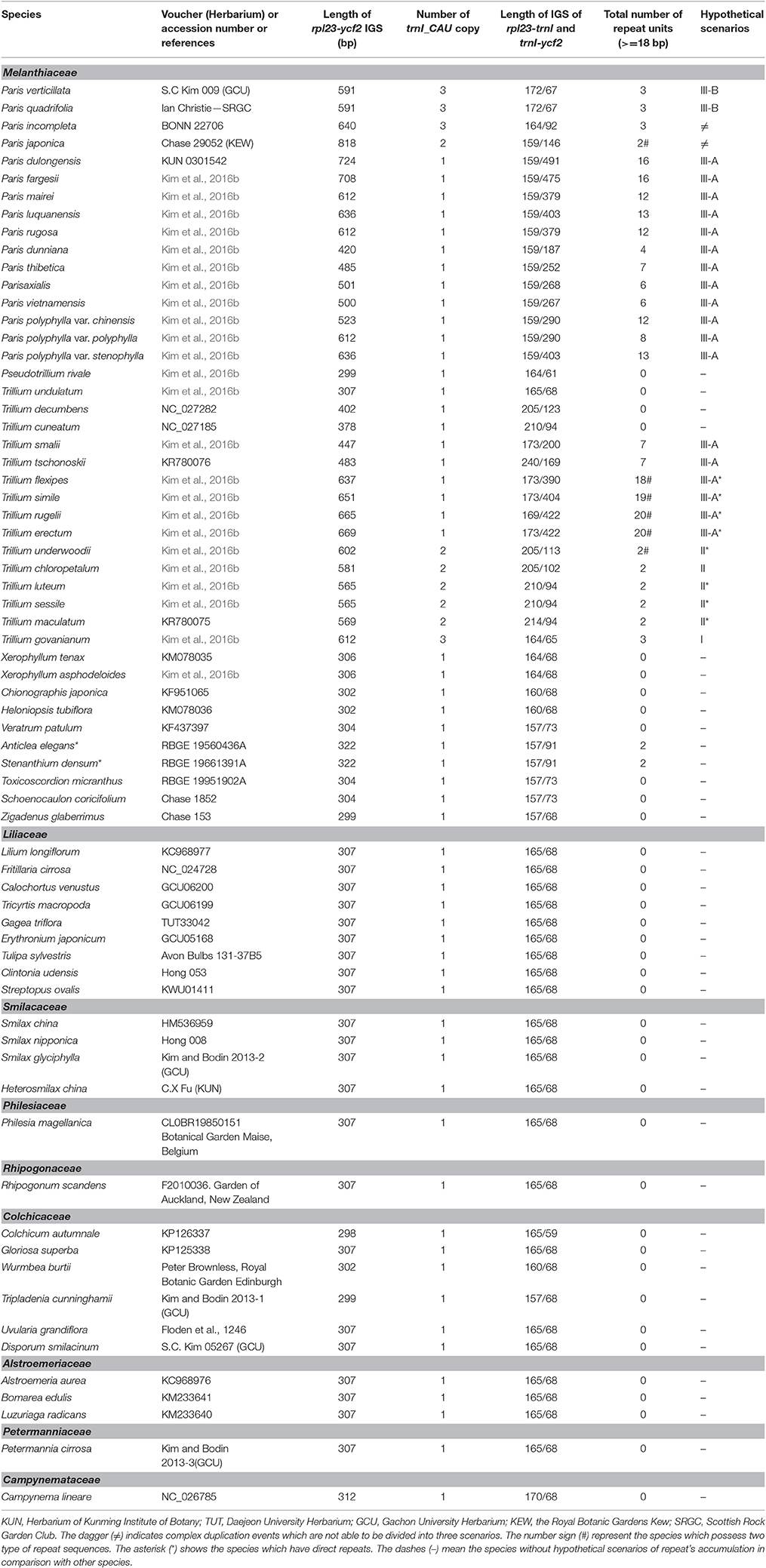
Table 2. List of the surveyed taxa with the length of rpl23-ycf2 intergenic space sequences, number of trnI_CAU copies, number of repeats and the hypothetical scenarios of repeat's accumulation.
Results
Features of cpDNA among Five Families of Liliales and Five Tribes of Melanthiaceae
The lengths of circular double-stranded DNA molecules differed among the families, ranging from 152,793 bp (Lilium longiflorum, Liliaceae) to 158,451 bp (Paris luquanensis, Melanthiaceae, Table 1). In Melanthiaceae, V. patulum (tribe Melanthieae) possesses the smallest cpDNA whereas the biggest cpDNA belongs to Paris species (tribe Parideae, Table 1). Although, the lengths varied, the AT and GC contents were relatively stable among the observed taxa (Table 1). Most of the examined species have 81 protein-coding genes, 30 tRNAs and 4 rRNAs in cpDNA sequences. However, there were 80 protein-coding genes in C. japonica, Colchicum autumnale, and Alstroemeria aurea because of the complete loss of rps16 in C. japonica and the deletion of whole region of ycf15 in C. autumnale and A. aurea. Comparative genomic analysis among five representative taxa of Melanthiaceae revealed that the deletion of rps16 was only found in tribes Chionographideae and Melanthieae. Further investigation on rps16 among genera of tribe Melanthieae revealed that the loss of rps16 was not common and only found in Veratrum, Toxicoscordion, and Schoenocaulon (Figure 2). Specifically, exon 1 of rps16 was deleted in Schoenocaulon whereas a part of exon 2 of this gene (47 bp) remained in Toxicoscordion. In contrast to complete loss of rps16 in C. japonica, only exon 2 of this gene was deleted in V. patulum, suggesting that there were different stages of this event in Veratrum genus which was divided into two sections: Veratrum and Fuscoveratrum. To track the loss of rps16, one primer pairs which covered the whole coding regions of rps16 was designed and applied for Veratrum species (Figure 3A). As expected, the PCR results revealed that there were two types of deletion of rps16 in Veratrum (Figure 3B). The first type was found in section Veratrum of which exon 2 of rps16 was lost. Remaining of exon 1 of rps16 resulted in a PCR product of ~1.5 kb in three examined taxa of section Veratrum (V. oxysepalum, V. lobelianum, and V. grandiflorum; Figures 3A,B). In contrast, the deletion of whole coding region of rps16 in section Fuscoveratrum caused a 400 bp-PCR product in V. versicolor, V. maackii, and V. nigrum (Figures 3A,B). In Liliaceae, Smilacaceae, Altroemeriaceae, and Colchicaceae, the intact coding sequence of rps16 was found (Table 1, Figure 2).
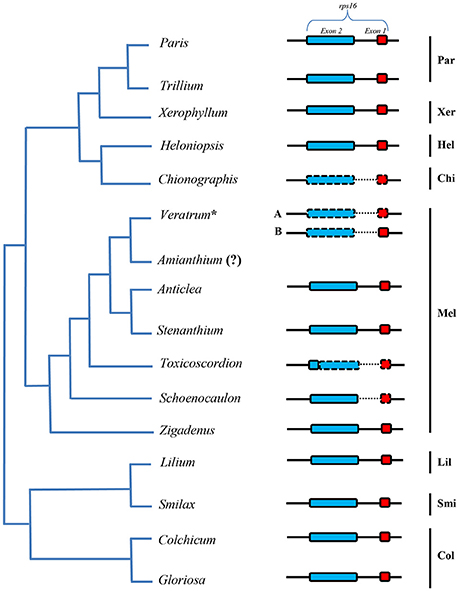
Figure 2. Comparison of rps16 among species of Melanthiaceae and other families. The simplified phylogenetic tree was based on Kim et al. (2016a). The dashed line box indicates the lost sequences and the dotted line showed the significantly changed sequences in comparison with others. Par, Parideae; Xer, Xerophylleae; Hel, Heloniadeae; Chi, Chionographideae; Mel, Melanthieae; Lil, Liliaceae; Smi, Smilacaceae; Col, Colchicaceae. The question mark (?) means missing data. The asterisk shows the genus which has two types of rps16 loss (A) completely lost and (B) partially lost.
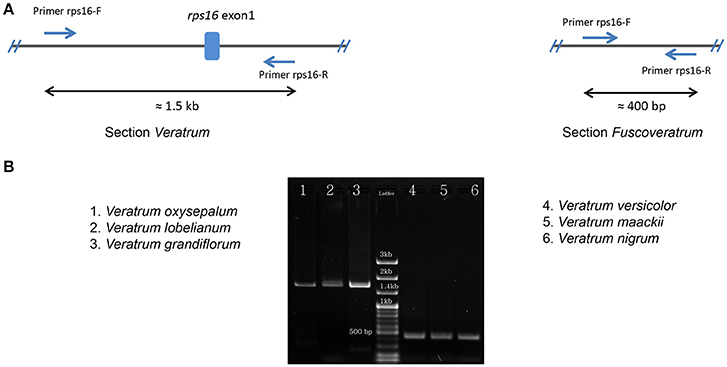
Figure 3. Confirmation of rps16 gene loss in Veratrum species. (A) The design of primers (Forward primer: rps16-F; Reverse primer: rps16-R) which cover the whole coding sequences of rps16 based on the cpDNA sequence of V. patulum (Accession number KF437397) and their positions in two types of rps16 gene loss. Expected lengths are ~1.5 kb in section Veratrum and 400 bp in section Fuscoveratrum. (B) Results of PCR among species of Veratrum. The section Veratrum includes V. oxysepalum, V. Lobelianum, and V. grandiflorum. The section Fuscoveratrum includes V. versicolor, V. maackii, and V. nigrum.
Sequences flanking the LSC/IR junction were compared between other taxa of the Liliales (Figure 4). The IR/LSC borders varied among the taxa. Specifically, the IR/LSC borders located in coding region of rps19 in Liliaceae and Colchicaceae. Meanwhile, it expanded to a part of rpl22 in Smilacaceae (Figure 4). In Melanthiaceae, it occurred in the trnH_GUG/rps19 intergenic spacer in Veratrum and Toxicoscordion. However, in other taxa, it expanded into a part of rps19 (350 bp in Trillium), into the rps19/rpl22 intergenic spacer (IGS; Anticlea and Stenanthium), into the rpl22/rps3 IGS (Heloniopsis), and into a section of rps3 (6 bp in Xerophyllum and Paris; 83 bp in Chionographis; 65 bp in Schoenocaulon; 161 bp in Zigadenus). Compared to other tribes, Melanthieae possessed a wide range of IR/LSC junctions (Figure 4).
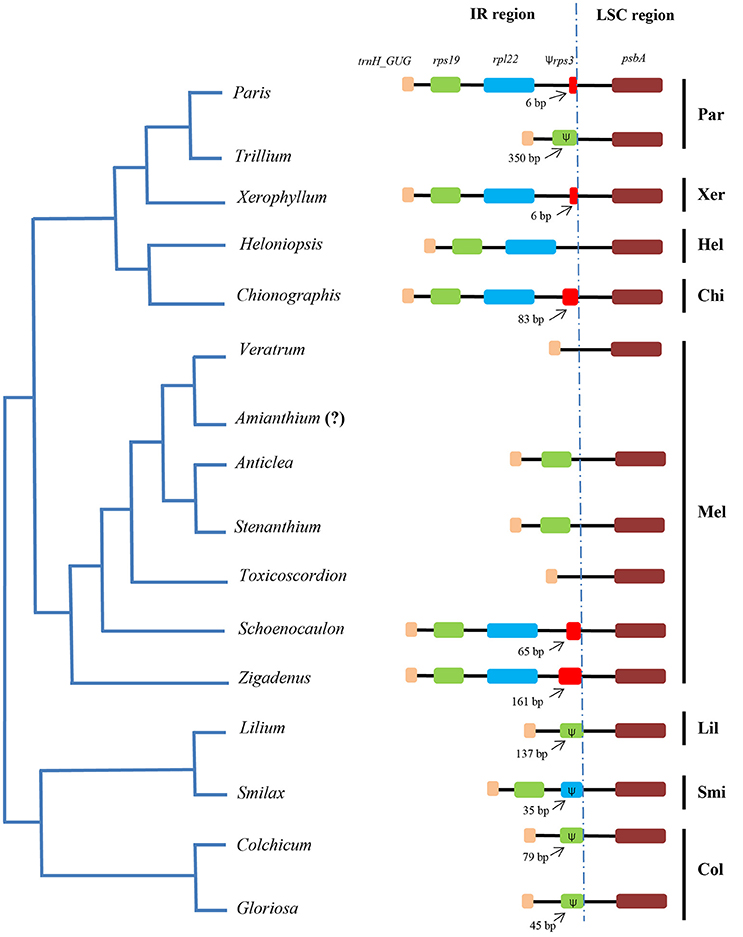
Figure 4. Comparison of borders between IR and LSC regions among species of Melanthiaceae and other families. The simplified phylogenetic tree was based on Kim et al. (2016a). The dotted line indicates the junction of IR-LSC regions. The (Ψ) represents pseudogenes. Par, Parideae; Xer, Xerophylleae; Hel, Heloniadeae; Chi, Chionographideae; Mel, Melanthieae; Lil, Liliaceae; Smi, Smilacaceae; Col, Colchicaceae. The question mark (?) means missing data.
Accumulation of Repeat Sequences in Tribe Parideae
Further investigation of repeat sequences showed that the IGS between rpl23 and ycf2 containing trnI_CAU was extremely variable in length, ranging from 299 to 818 bp among Paris and Trillium, while a more stable length was detected in other species (Table 2). In Paris, the trnI-ycf2 IGS was ranged from 67 to 491 bp. As is the case in Paris, Trillium has varying lengths of the trnI–ycf2 IGS (from 68 to 422 bp). Notably, nearly all species in other Liliales families have equal lengths of the trnI-ycf2 IGS (68 bp). The length variation in the rpl23-ycf2 IGS of tribe Parideae can be attributed to the presence of tandem repeat sequences (Table 2, Supplementary Data S1). The results of repeat analysis in rpl23-ycf2 IGS regions among Liliales species showed that accumulation of repeat occurred only in tribes Parideae and Melanthieae of Melanthiaceae. Additionally, the number of repeat units was different among examined species (Table 2). In Paris species, this region contained a ranging copy number from 2 to 16 whereas the number of copy varied from 2 to 20 in Trillium taxa (Table 2, Supplementary Data S1). Also, the length of these repeats was different in both genera, ranging from 24 to 155 bp in Paris and from 18 to 209 bp in Trillium (Supplementary Data S1). Additionally, we observed upstream and downstream of repeats because of the important role of initial repeats in SSM mechanism. As a result, we found two groups of small conserved repeated sequences in most of the surveyed taxa (Figure 5A, Supplementary Data S1). In the first group, there were two 7 bp—direct repeats which were located upstream and within the coding sequence of trnI_CAU. In the rest group, there was a cluster of direct repeats including R1 (5′-CAAATTCCAAT-3′), R1a (5′-CCAATTCCAAT-3′), and R1b (5′-ATTCCA-3′).
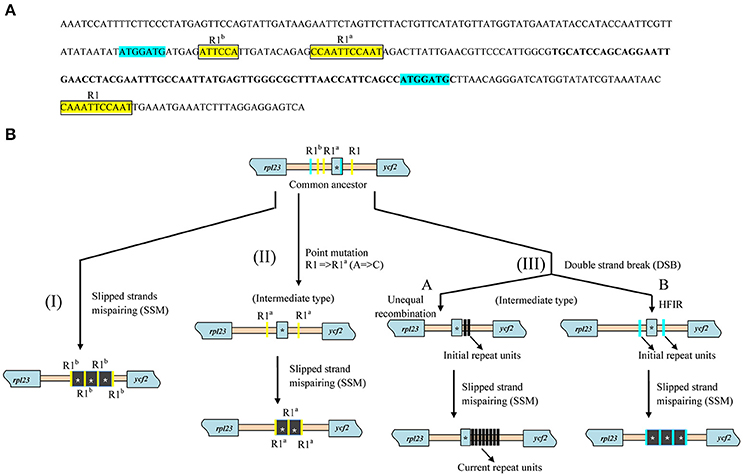
Figure 5. Conserved repeated sequences in rpl23-ycf2 IGS among Liliales species and hypothetical pathways of repeat's accumulation. (A) The representative sequence of rpl23-ycf2 IGS extracted from Campynema lineare (Accession number NC026785) and positions and two groups of conserved repeats in Liliales. Blue shaded and squared yellow shaded sequences represent two groups of repeats. The letters of R1, R1a, and R1b above sequence indicate variety of repeats. The bold letters indicate the coding region of trnI_CAU in rpl23-ycf2 IGS sequence. (B) Hypothetical scenarios for formation of repeats. The blue and yellow bars represent repeat units. Black bars indicate new generated repeats. HIFR stands for homology facilitated illegitimate recombination. Asterisks showed the trnI_CAU sequence which can be included in repeat units.
Discussion
Comparative Characteristics of cpDNA among Melanthiaceae Species and Its Implication
The cpDNA structures of representative species of Melanthiaceae consist of typical double-stranded DNA molecules and are highly conserved, as reported in previous angiosperm cpDNA studies (Palmer, 1991; Yang et al., 2010; Liu et al., 2012; Huang et al., 2013, 2014, 2016; Kim and Kim, 2013; Luo et al., 2014; Nguyen et al., 2015). In this study, length variations were identified among the Melanthiaceae taxa (Table 1). The longer sequences of cpDNA were found in Paris, Trillium, Xerophyllum, and Heloniopsis species which possessed either repeat units in rpl23-ycf2 regions or expansion of IR/LSC border. Although, C. japonica has the expansion of IR/LSC junction to rps3 (83 bp), the loss of rps16 caused a shorten length of its cpDNA. Therefore, it is suggested that the length variations within Melanthiaceae could have been led by the deletion and duplication of genes, as well as the expansion of IR regions. The comparative analysis among five families of Liliales revealed a notable variety of length and different losses of genes in cpDNA (Table 1). However, further studies, which cover all 10 families, should be conducted to investigate the overall trends of genomic events in Liliales.
The rps16 gene, encoding ribosomal protein S16, is commonly detected in the plant chloroplast genomes. However, the loss of this gene was also recorded in different taxa including Connarus, Epifagus, Pinus, Viola, Fagus, legume species, and etc. (Downie and Palmer, 1992; Doyle et al., 1995). For understanding this loss, it was proposed that the rps16 gene was transferred to the nucleus and its protein product was able to target both chloroplast and mitochondria in the case of Medicago truncatula and Populus alba (Ueda et al., 2008). Additionally, deletion of rps16 was found in a moss species of Physcomitrella patens subsp. patens (Sugiura et al., 2003). These results suggest that the transfer event of rps16 occurred independently at the early divergence of plants. It was lost in C. japonica and partially or completely deleted in V. patulum, S. densum, and T. micranthus among the Melanthiaceae; therefore, ribosomal protein S16 was predicted to be untranscribed and untranslated from cpDNA. However, this deficiency could be compensated from nuclear rps16 products as described in a previous study (Ueda et al., 2008). In contrast to the deletion of exon 2 and complete loss of rps16 in Chionographis and Veratrum, the deletion of exon 1 and remains of a piece of exon 2 were recorded in Schoenocaulon and Toxicoscordion, respectively. Additionally, 22 out of 26 species of Schoenocaulon are endemic from the Southern of United States of America to Peru (Zomlefer and Judd, 2008). Therefore, this genomic feature might contribute to investigating the evolution of cpDNA in this genus. Further studies which cover all species of Schoenocaulon and Toxicoscordion should be conducted to clarify the overview of this feature in the tribe Melanthieae. Furthermore, two types of rps16 deletions were found in two sections of Veratrum which were distinguished by characteristics of leaf, style, and sheath of stem base (Chen and Takahashi, 2000; Zomlefer et al., 2003; Figure 3). Previously, genomic events in chloroplast genome sequences were specifically detected in some species and could be molecular markers. For example, the inversion of the trnV_UAC-atpB region was only detected in species of Trillium subgenus Phyllantherum of Melanthiaceae and the loss of ycf15 was observed in tribe Colchiceae of Colchicaceae (Nguyen et al., 2015; Kim et al., 2016b). In this study, based on the finding of partial or complete loss of rps16, we provide the first potential molecular maker for recognizing two sections among Veratrum (Figure 3). From these results, it is likely that genomic events in chloroplast genomes are effective for making molecular markers and reflect the phylogeny among Liliales taxa.
In general, IR expansion affects length variation in cpDNA. For example, the expansion of the IR region (36,501 bp) into psbB in Mahonia bealei cpDNA led to an increased total genome length (164,792 bp; Ma et al., 2013). In Melanthiaceae, the IR/LSC junctions were also variable (Figure 4). This variability affected the total length of the cpDNA region. For instance, the IR/LSC junction expansion from trnH_GUG into rps3 resulted in an increased length of the IR region from 26,360 bp in V. patulum to 28,373 bp in P. verticillata (Table 1). Wang et al. (2008) suggested that the IR/LSC junctions in the Liliales taxa contained the trnH_GUG—psbA cluster, but variable patterns of junction existed in the order Liliales (Figure 4). Within the same family of monocots, IR/LSC junctions contained similarities; for example, the boundaries located in the rps19 and rpl22 genes of the Arecaceae and Orchidaceae, respectively (Huang et al., 2013; Luo et al., 2014). A similar trend was observed in dicots species of the Araliaceae, in which a common IR/LSC boundary was detected in the rps19 gene (Li et al., 2013). In contrast, the border of IR/LSC varied among Melanthiaceae in which the IR region was expanded from trnH_GUG into a part of rps3 (Figure 4). Significantly, there were three different borders in tribe Melanthieae (Figure 4). The unique expansion into 161 bp of rps3 might be a potentially molecular marker for monotypic species—Z. glaberrimus (Figure 4).
Hypothetical Scenarios for Dynamic SSM Events in cpDNA
Previously, the DSB mechanism induced recombination in Chlomydomonas reinhardtii cpDNA (Dürrenberger et al., 1996). Kwon et al. (2010) reported the DSB repair pathways from both microhomology and no homology in Arabidopsis. Additionally, cpDNA sequences typically contain two inverted repeat regions which can be reversely used as a template for repairing the break of DNA through recombination. Tandem repeats ranging from 6 to 33 bp in IR region was discovered in Oenothera species (Onagraceae, Myrtales) (Blasko et al., 1988; Nimzyk et al., 1993; Sears et al., 1996), and tandem repeats comprising a 29-bp sequence have been found in the rps8-rpl14 IGS of the LSC region of Oenothera (Wolfson et al., 1991). The copy correction of IR regions after imprecise alignment, replication slippage, and recombination have also been proposed as a mechanism for the accumulation of tandem repeats in Oenothrea cpDNA (Blasko et al., 1988; Wolfson et al., 1991; Sears et al., 1996). Recently, Massouh et al. (2016) surveyed and found spontaneous mutants in chloroplast genomes of Oenothera which were mostly caused by the replication slippage events. SSM was believed to be a major factor for DNA evolution (Levinson and Gutman, 1987). Although, results of SSM were previously reported in cpDNA of angiosperms, there have not been records of utilization of different small conserved repeats in the same region of cpDNA for generating newly repeated sequences. In this study, due to the presence of the conserved regions which flanked tandem repeats, we proposed three different patterns for generating the repeated sequences among Parideae taxa (Figure 5A). In the first scenario (I), the R1b sequence was utilized through SSM mechanism to form three tandem repeats of 164 bp in Trillium govanianum which includes the whole trnI_CAU sequence (Supplementary Data S1). Within the second pathway (II), prior to the process of SSM, a point mutation which changed the adenine base to cytosine base to create a perfect direct repeat between R1 and R1a occurred. The present of direct repeat (R1a) induced SSM process which resulted in formation of two repeats in Trillium taxa. Generally, in the SSM mechanism, initial repeats play an important role. Therefore, in the third case (III), we proposed the formation of initial repeats through the double-strand break (DSB) repair mechanism (Figure 5B). Specifically, two repair mechanisms may be involved in this case due to the difference in repeat contents among species. First, in the III-A subcase, unequal recombination occurred downstream of trnI_CAU and induced the formation of initial repeats which were employed in SSM process. Meanwhile, in the second subcase (III-B), repairing mechanism of DSB through homology facilitated illegitimate recombination (HFIR) occurred based on direct repeat sequences of 7 bp (5′-ATGGATG-3′) to create a longer 16 bp- repeat unit (5′-ATGGATGCTTAACAGG-3′) which was assumed to be an initial repeat unit for SSM event. Because of the different initial repeat units, SSM events occurred and resulted in newly distinct types of repeat sequences in both Paris and Trillium species (Figure 5B, Table 2, Supplementary Data S1). Albeit the sequence data supported our hypothetical scenarios, there was not essential evidence of in vivo experiment in this study. However, GuhaMajumdar et al. (2008) previously attempted to trace replication slippage in vivo and successfully confirmed this event from results of deletion and duplication in C. reinhardtii and Escherichia coli. Although, this study employed only one type of short tandem repeat, it fundamentally supported the reliability of three hypothetical scenarios in our study. Further studies, which use more types of small sequence repeats in the same region, should be conducted to provide substantial evidence for our hypothesis.
Recently, Kim et al. (2016b) used the number of trnI_CAU to classify the type of duplication events across Parideae. This classification was incongruent with infrageneric circumscription of Paris members, but not for Trillium species. In contrast, in terms of the origin of repeat sequences, there were no relationships between the classification within tribe Parideae and mechanisms of repeat's accumulation. For instance, in Paris, the formation of repeats could be explained by the (III) scenario, except P. incompleta whose repeats have likely arisen from the (I) pathway followed by point mutation events and P. japonica which reflected complex duplication processes (Table 2, Supplementary Data S1). In Trillium, all three pathways can be found. For example, the (I) scenario was recorded only in T. govanianum. Meanwhile, the (II) and (III-A) pathways could be found in subgenus Phylantherum and subgenus Trillium, respectively. Moreover, repeats were not found in rpl23-ycf2 IGS of T. undulatum (Subgenus Trillium), or in T. decumbens and T. cuneatum (subgenus Phylantherum). These findings suggested that SSM events occurred independently across the tribe Parideae. Additionally, the number of <24 bp—repeat units was more abundant than those of over 24 bp in length, suggesting that that SSM occurred more frequently for short repeats than for long repeat sequences in the tribe Parideae (Melanthiaceae, Liliales). In Anticlea elegans and S. densum, two repeats (19 bp) were found (Table 2), suggesting that the accumulation of repeats may also occur in tribe Melanthieae, which is composed of 7 genera and 78 species of Melanthiceae.
Although, sequence data provided evidence for different scenarios of SSM and its independence, there was not enough evidence regarding the alternation of initial repeats during the SSM process in Parideae. Notably, small conserved units were found in most of the examined taxa; however, within Liliales, the repeats in the rpl23-ycf2 IGS were mainly present in the tribe Parideae of Melanthiaceae. Therefore, variation within this region may be due to a unique genomic event in Parideae. Previous studies have found diverse genome sizes among Melanthiaceae (Pellicer et al., 2014). In contrast to the trend of reduced genome size in other tribes, Parideae exhibit significant increases in chromosome size and possess the largest nuclear genome in Melanthiaceae. This trend can also been seen in the patterns of repeats between Parideae and other tribes. It is likely that the causes of chromosome changes in Parideae might be related to the accumulation of repeats within this tribe. More studies should be conducted to shed light on the significance of these two unique features of the genomes of Parideae. Additionally, accumulation of repeat sequences was also found in rpl23-ycf2 IGS of monocots such as Acorus calamus (Accession number NC_007407), Sagittaria lichuanensis (Accession number NC_029815), Anomochloa marantoidea (Accession number NC_014062), Eustrephus latifolius (Accession number KM_233639), Curcuma roscoeana (Accession number KF_601574), and Musa acuminata subp malaccensis (Accession number HF677508; Data not shown), suggesting that the rpl23-ycf2 IGS may be one of the “hot spots” for genomic events in angiosperm species.
Conclusions
In conclusion, comparative analysis of cpDNA in Melanthiaceae revealed that genomic events including pseudogenization, duplication, and deletion in the chloroplast genome are precise sources for mining molecular marker in plants. Specifically, gene loss events of rps16 were potentially valuable molecular data for identifying two sections of the Veratrum species. Melanthiaceae also exhibits a significant change in junctions between LSC and IR regions. Additionally, we provided the first evidence of different employments of small repeat sequences for SSM in chloroplast genomes of monocots species. Though the origin of these differences remains unclear, these data highlight the dynamic molecular evolution in chloroplast genomes. With the increasing number of complete organelle genomes, these patterns could be detected in other species and be useful references for tracing genomic evolution among plants.
Author Contributions
HDKD carried out the genomic experiment and drafted the manuscript. HDKD and J-HK participated in the design of the study and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) Grant Fund (MEST 2010-0029131) and Scientific Research of Korea National Arboretum (KNA) Grant Fund (KNA 1-2-13, 14-2).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Sang-Chul Kim of Gachon University (Korea), Anett Krämer of the Botanische Gärten der Universität Bonn (Germany), Peter Brownless of Royal Botanic Garden Edinburgh (United Kingdom), Botanical Garden Maise (Belgium), Garden of Auckland (New Zealand), and Ian Christie from the Scottish Rock Garden Club for collecting and providing the plant materials for this study. Also, we would like to thank Dr. Jung Sung Kim (Gachon University, Korea), Dr. Michael A. Vincent (Miami University, Ohio, USA) and reviewers for helpful suggestions for preparing and improving this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00693/full#supplementary-material
References
Angiosperm Phylogeny Group (2009). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 161, 105–121. doi: 10.1111/j.1095-8339.2009.00996.x
Angiosperm Phylogeny Group (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Barrett, C. F., Davis, J. I., Leebens-Mack, J., Conran, J. G., and Stevenson, D. W. (2013). Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29, 65–87. doi: 10.1111/j.1096-0031.2012.00418.x
Blasko, K., Kaplan, S. A., Higgins, K. G., Wolfson, R., and Sears, B. B. (1988). Variation in copy number of a 24-base pair tandem repeat in the chloroplast DNA of Oenothera hookeri strain Johansen. Curr. Genet. 14, 287–292. doi: 10.1007/BF00376749
Bodin, S. S., Kim, J. S., and Kim, J. H. (2013). Complete chloroplast genome of Chionographis japonica (Willd.) Maxim. (Melanthiaceae): comparative genomics and evaluation of universal primers for Liliales. Plant Mol. Biol. Rep. 31, 1407–1421. doi: 10.1007/s11105-013-0616-x
Cai, Z., Guisinger, M., Kim, H. G., Ruck, E., Blazier, J. C., McMurtry, V., et al. (2008). Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J. Mol. Evol. 67, 696–704. doi: 10.1007/s00239-008-9180-7
Chase, M. W., Soltis, D. E., Soltis, P. S., Rudall, P. J., Fay, M. F., Hahn, W. H., et al. (2000). “Higher-level systematics of the monocotyledons: an assessment of current knowledge and a new classification,” in Monocots: Systematics and Evolution, eds K. L. Wilson and D. A. Morrison (Melbourne, VIC: CSIRO Publishing), 3–16.
Chen, X., and Takahashi, H. (2000). “Veratrum,” in Flora of China, Vol. 24, eds Z. Y. Wu and P. H. Raven (Beijing; St. Louis, MO: Science Press; Missouri Botanical Garden Press), 82–85.
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Do, H. D. K., Kim, J. S., and Kim, J. H. (2013). Comparative genomics of four Liliales families inferred from the complete chloroplast genome sequence of Veratrum patulum O. Loes. (Melanthiaceae). Gene 530, 229–235. doi: 10.1016/j.gene.2013.07.100
Do, H. D. K., Kim, J. S., and Kim, J. H. (2014). A trnI_CAU triplication event in the complete chloroplast genome of Paris verticillata M.Bieb. (Melanthiaceae, Liliales). Genome Biol. Evol. 6, 1699–1706. doi: 10.1093/gbe/evu138
Downie, S. R., and Palmer, J. D. (1992). “Use of chloroplast DNA rearrangements in reconstructing plant phylogeny,” in Molecular Systematics of Plants, eds E. S. Soltis, D. E. Soltis, and J. J. Doyle (New York, NY: Chapman and Hall), 14–35.
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Doyle, J. J., Doyle, J. L., and Palmer, J. D. (1995). Multiple independent losses of two genes and one intron from legume chloroplast genomes. Syst. Bot. 20, 272–294. doi: 10.2307/2419496
Dürrenberger, F., Thompson, A. J., Herrin, D. L., and Rochaix, J. D. (1996). Double strand break-induced recombination in Chlamydomonas reinhardtii chloroplasts. Nucleic Acids Res. 24, 3323–3331. doi: 10.1093/nar/24.17.3323
Govaerts, R. (2016). World Checklist of Melanthiaceae. Facilitated by the Royal Botanic Gardens, Kew. 2012. Available online at: http://apps.kew.org/wcsp/ (Accessed December 24, 2016).
GuhaMajumdar, M., Dawson-Baglien, E., and Sears, B. B. (2008). Creation of a chloroplast microsatellites reporter for detection of replication Slippage in Chlamydomonas reinhardtii. Eukaryot. Cell 7, 639–646. doi: 10.1128/EC.00447-07
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huang, Y., Li, X., Yang, Z., Yang, C., Yang, J., and Ji, Y. (2016). Analysis of complete chloroplast genome sequences improves phylogenetic resolution in Paris (Melanthiaceae). Front. Plant Sci. 7:1797. doi: 10.3389/fpls.2016.01797
Huang, Y. Y., Matzke, A. J., and Matzke, M. (2013). Complete sequence and comparative analysis of the chloroplast genome of coconut palm (Cocosnucifera). PLoS ONE 8:e74736. doi: 10.1371/journal.pone.0074736
Ji, Y., Fritsch, P. W., Li, H., Xiao, T. J., and Zhou, Z. K. (2006). Phylogeny and classification of Paris (Melanthiaceae) inferred from DNA sequence data. Ann. Bot. 98, 245–256. doi: 10.1093/aob/mcl095
Kim, J. S., Hong, J. K., Chase, M. W., Fay, M. F., and Kim, J. H. (2013). Familial relationships of the monocot order Liliales based on a molecular phylogenetic analysis using four plastid loci: matK, rbcL, atpB and atpF-H. Bot. J. Linn. Soc. 172, 5–21. doi: 10.1111/boj.12039
Kim, J. S., and Kim, J. H. (2013). Comparative genome analysis and phylogenetic relationship of order liliales insight from the complete plastid genome sequences of two lilies (Lilium longiflorum and Alstroemeria aurea). PLoS ONE 8:e68180. doi: 10.1371/journal.pone.0068180
Kim, S. C., Kim, J. S., Chase, M. W., and Kim, J. H. (2016a). Molecular phylogenetic relationships of Melanthiaceae (Liliales) based on plastid DNA sequences. Bot. J. Linn. Soc. 181, 567–584. doi: 10.1111/boj.12405
Kim, S. C., Kim, J. S., and Kim, J. H. (2016b). Insight into infrageneric circumscription through complete chloroplast genome sequences of two Trillium species. AoB Plants 8:plw015. doi: 10.1093/aobpla/plw015
Kwon, T., Huq, E., and Herrin, D. L. (2010). Microhomology-mediated and nonhomologous repair of a double-strand break in the chloroplast genome of Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 107, 13954–13959. doi: 10.1073/pnas.1004326107
Lee, H. L., Jansen, R. K., Chumley, T. W., and Kim, K. J. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 24, 1161–1180. doi: 10.1093/molbev/msm036
Levinson, G., and Gutman, G. A. (1987). Slipped-strand mispairing: a major Mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221.
Li, R., Ma, P. F., Wen, J., and Yi, T. S. (2013). Complete sequencing of five araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 8:e78568. doi: 10.1371/journal.pone.0078568
Liu, J., Qi, Z., Zhao, Y. P., Fu, C. X., and Xiang, Q. Y. (2012). Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales – Influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 64, 545–562. doi: 10.1016/j.ympev.2012.05.010
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Luo, J., Hou, B. W., Niu, Z. T., Liu, W., Xue, Q. Y., and Ding, X. Y. (2014). Comparative chloroplast genomes of photosynthetic orchids: insights into evolution of the orchidaceae and development of molecular markers for phylogenetic applications. PLoS ONE 9:e99016. doi: 10.1371/journal.pone.0099016
Ma, J., Yang, B., Zhu, W., Sun, L., Tian, J., and Wang, X. (2013). The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 528, 120–131. doi: 10.1016/j.gene.2013.07.037
Massouh, A., Schubert, J., Yaneva-Roder, L., Ulbrricht-Jones, E. S., Zupok, A., Johnson, M. T. J., et al. (2016). Spontaneous chloroplast mutants mostly occur by replication slippage and show a biased pattern in the plastome of oenothera. Plant Cell 28, 911–929. doi: 10.1105/tpc.15.00879
Mayer, C. (2006). Phobos 3.3.11, 2006-2010, Available online at: http://www.rub.de/spezzoo/cm/cm_phobos.htm
Nguyen, P. A., Kim, J. S., and Kim, J. H. (2015). The complete chloroplast genome of colchicine plants (Colchicum autumnale L. and Gloriosa superba L.) and its application for identifying the genus. J. Planta 242, 223. doi: 10.1007/s00425-015-2303-7
Nguyen, T. P., Kim, J. S., and Kim, J. H. (2013). Molecular phylogenetic relationships and implications for the circumscription of Colchicaceae (Liliales). Bot. J. Linn. Soc. 172, 255–269. doi: 10.1111/boj.12037
Nimzyk, R., Schondorf, T., and Hachtel, W. (1993). In-frame length mutations associated with short tandem repeats are located in unassigned open reading frames of Oenothera. Curr. Genet. 23, 265–270. doi: 10.1007/BF00351505
Palmer, J. D. (1991). “Plastid chromosomes: structure and evolution,” in Cell Culture and Somatic Genetics of Plant Vol. 7A, Molecular Biology of Plastids, eds L. Bogorad and I. K. Vasil (San Diego, CA: Academic Press), 5–53.
Pellicer, J., Kelly, L. J., Leitch, I. J., Zomlefer, W. B., and Fay, M. F. (2014). A universe of dwarfs and giants: genome size and chromosome evolution in the monocot family Melanthiaceae. New Phytol. 201, 1484–1497. doi: 10.1111/nph.12617
Rudall, P. J., Stobart, K. L., Hong, W. P., Conran, J. G., Furness, C. A., Kite, G. C., et al. (2000). “Consider the lilies: systematics of liliales,” in Monocots: Systematics and Evolution, eds K. Wilson and D. A. Morrison (Melbourne, VIC: CSIRO Publishing), 347–359.
Ruhfel, B. R., Gitzendanner, M. A., Soltis, P. S., Soltis, D. E., and Burleigh, J. G. (2014). From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 14:23. doi: 10.1186/1471-2148-14-23
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, 686–689. doi: 10.1093/nar/gki366
Schmickl, R., Kiefer, C., Dobes, C., and Koch, M. A. (2009). Evolution of trnF (GAA) pseudogenes in cruciferous plants. Plant Syst. Evol. 282, 229–240. doi: 10.1007/s00606-008-0030-2
Sears, B. B., Stoike, L. L., and Chiu, W. L. (1996). Proliferation of direct repeats near the Oenothera chloroplast DNA origin of Replication. Mol. Biol. Evol. 13, 850–863. doi: 10.1093/oxfordjournals.molbev.a025645
Sugiura, C., Kobayashi, Y., Aoki, S., Sugita, C., and Sugita, M. (2003). Complete chloroplast DNA sequence of the moss Physcomitrella patens: evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucleic Acids Res. 31, 5324–5331. doi: 10.1093/nar/gkg726
Sveinsson, S., and Cronk, Q. (2014). Evolutionary origin of highly repetitive plastid genomes within the clover genus (Trifolium). BMC Evol. Biol. 14:228. doi: 10.1186/s12862-014-0228-6
Takhtajan, A. (1997). Diversity and Classification of Lowering Plants. New York, NY: Columbia University of Press.
Thorne, R. F. (1992). Classification and geography of the flowering plants. Bot. Rev. 58, 225–348. doi: 10.1007/BF02858611
Ueda, M., Nishikawa, T., Fujimoto, M., Takanashi, H., Arimura, S., Tsutsumi, N., et al. (2008). Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Mol. Biol. Evol. 25, 1566–1575. doi: 10.1093/molbev/msn102
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3 - new capabilities and interfaces. Nucleic Acids Res. 40, e115. doi: 10.1093/nar/gks596
Wang, R. J., Cheng, C. L., Chang, C. C., Wu, C. L., Su, T. M., and Chaw, S. M. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
WCSP (2016). World Checklist of Selected Plant Families. Facilitated by the Royal Botanic Gardens, Kew. Available online at: http://apps.kew.org/wcsp/ (Accessed December 21, 2016).
Wolfson, R., Higgins, K. G., and Sears, B. B. (1991). Evidence for replication slippage in the evolution of Oenothera chloroplast DNA. Mol. Biol. Evol. 8, 709–720.
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 5:e12762. doi: 10.1371/journal.pone.0012762
Zomlefer, W. B., and Judd, W. S. (2008). Two new species of schoenocaulon (Liliales: Melanthiaceae) from mexico supported by ITS sequence data. Syst. Bot. 33, 117–124. doi: 10.1600/036364408783887474
Zomlefer, W. B., Whitten, W. M., Williams, N. H., and Judd, W. S. (2003). An overview of Veratrum s.l. (Liliales: Melanthiaceae) and an infrageneric phylogeny based on ITS sequence data. Syst. Bot. 28, 250–269. doi: 10.1043/0363-6445-28.2.250
Keywords: Xerophyllum tenax, Heloniopsis tubiflora, chloroplast genome, tandem repeats, slipped-strand mispairing, Parideae, Melanthiaceae, Liliales
Citation: Do HDK and Kim J-H (2017) A Dynamic Tandem Repeat in Monocotyledons Inferred from a Comparative Analysis of Chloroplast Genomes in Melanthiaceae. Front. Plant Sci. 8:693. doi: 10.3389/fpls.2017.00693
Received: 18 January 2017; Accepted: 18 April 2017;
Published: 22 May 2017.
Edited by:
Badri Padhukasahasram, Illumina (United States), USAReviewed by:
Tina T. Hu, Princeton University, USAKi-Hong Jung, Kyung Hee University, South Korea
Jingkui Tian, Zhejiang University, China
Copyright © 2017 Do and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo-Hwan Kim, a2ltamgyMDA5QGdhY2hvbi5hYy5rcg==