 Weiguo Li
Weiguo Li Lihui Zhang1
Lihui Zhang1 Yanwen Zhang
Yanwen Zhang- 1College of Life Sciences, Changchun Normal University, Changchun, China
- 2College of Resource and Environment, Henan Polytechnic University, Jiaozuo, China
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) is the most commonly used and powerful method for gene expression analysis due to its high sensitivity, specificity, and high throughput, and the accuracy of this approach depends on the stability of reference genes used for normalization. Taihangia rupestris Yu and Li (Rosaceae), an andromonoecious plant, produces both bisexual flowers and unisexual male flowers within the same individual. Using qRT-PCR technique, investigation of the gene expression profiling in staminate and perfect flowers would improve our understanding of the molecular mechanism in regulation of flower formation and sex differentiation in andromonoecious T. rupestris. To accurate normalize the gene expression level in Taihangia flower, 16 candidate reference genes, including 10 traditional housekeeping genes, and 6 newly stable genes, were selected based on transcriptome sequence data and previous studies. The expressions of these genes were assessed by qRT-PCR analysis in 51 samples, including 30 staminate and perfect flower samples across developmental stages and 21 different floral tissue samples from mature flowers. By using geNorm, NormFinder, BestKeeper, and comprehensive RefFinder algorithms, ADF3 combined with UFD1 were identified as the optimal reference genes for staminate flowers, while the combination of HIS3/ADF3 was the most accurate reference genes for perfect floral samples. For floral tissues, HIS3, UFD1, and TMP50 were the most suitable reference genes. Furthermore, two target genes, TruPI, and TruFBP24, involved in floral organ identity were selected to validate the most and least stable reference genes in staminate flowers, perfect flowers, and different floral tissues, indicating that the use of inappropriate reference genes for normalization will lead to the adverse results. The reference genes identified in this study will improve the accuracy of qRT-PCR quantification of target gene expression in andromonoecious T. rupestris flowers, and will facilitate the functional genomics studies on flower development and sex differentiation in the future.
Introduction
Quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR) has become the most common and powerful method for detecting and measuring transcripts abundance due to its high sensitivity, specificity, accuracy, and high throughput (Gachon et al., 2004). However, to obtain reliable and accurate results, qRT-PCR requires specific strategies to control for possible variability related to the serials of steps of the experimental procedure, such as discrepancy in initial sample amount, quantity of mRNA templates, and qRT-PCR amplification efficiency (Bustin, 2002; Huggett et al., 2005). Relative quantification is a widely accepted procedure to evaluate gene expression by normalization with one or more internal reference genes, which are required for stable expression regardless of experimental conditions (Bustin et al., 2005; Thellin et al., 2009).
Traditionally, the housekeeping genes, such as actin (ACT), elongation factor 1 alpha (EF-1α), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and ubiquitin (UBQ), involved in primary metabolism or other basic cellular processes are regarded as steadily expressed genes, (Thellin et al., 1999; Kozera and Rapacz, 2013), and thus these genes are commonly used for normalizing qPCR data without any validation. For non-model plant species, the reference genes are mainly identified by the search for orthologous sequences of common housekeeping genes reported in model plant species, due to limited genetic and sequence information (Gonzalezaguero et al., 2013). However, numerous studies have reported that the expression levels of traditional housekeeping genes vary considerably alterations in different biological sample and variable experimental condition (Hu et al., 2009; Zhang et al., 2017). Consequently, the use of improper traditional housekeeping genes for normalization in relative quantification of gene expression could result in bias of expression data and incorrect conclusion (Gutierrez et al., 2008).
With the development of high-throughput sequencing technology, a large number of transcriptome sequence data provide abundant information about gene expression profiling in many plant species. Benefitting from this technique, the growing plant transcriptome datasets have been widely used as new resource for identification of appropriate reference genes, especially in non-model plant (Zhuang et al., 2015; Ma et al., 2016). Furthermore, several statistical algorithms, such as geNorm (Vandesompele et al., 2002), NormFinder (Andersen et al., 2004), BestKeeper (Pfaffl et al., 2004), and RefFinder (Xie et al., 2012) have been developed to screen the most appropriate reference gene for qRT-PCR normalization in given biological samples. Therefore, next generation sequencing technology such as high-throughput RNA-seq provides new opportunities to explore appropriate reference gene candidates for non-model plant species (Demidenko et al., 2011).
Taihangia rupestris Yu and Li (Rosaceae), an ancient perennial herb belonging to the tribe Dryadeae, is distributed on cliff faces in the southern part of the Taihang Mountains of China (Yu and Li, 1980). T. rupestris produces both bisexual flowers and unisexual male flowers within the same individual, forming the andromonoecious sexual system (Yu and Li, 1983). The Taihangia staminate flowers are bisexual at initiation and become unisexual by arresting the pistil development in consequent developmental stages (Lu, 1996). By using qRT-PCR technique investigation of the gene expression profiling in staminate and perfect flowers would improve our understanding of the molecular mechanism in regulation of flower formation and sex differentiation in T. rupestris. In previous studies, a housekeeping gene Actin was used as the reference gene for normalizing qRT-PCR analysis in T. rupestris flowers (Du et al., 2008; Lu et al., 2010). However, little information is available concerning appropriate reference genes for qRT-PCR normalization, and no systematic selection of reference genes have not been performed in this plant species. Recently, we used de novo RNA sequencing to compare the transcriptome profiles of staminate and perfect flowers at early and late developmental stages (Li et al., 2017), and transcriptome sequences data available could be a potential source for identification of suitable reference genes in andromonoecious T. rupestris.
To accurate normalize the gene expression level in T. rupestris flower, 15 candidate reference genes, including nine traditional housekeeping genes and six novel genes, were selected based on the floral transcriptome datasets. In addition, the housekeeping genes Actin used as reference gene in previous studies was also included. The expression stability of these candidate genes were assessed by qRT-PCR analysis in 30 staminate and perfect flower samples across developmental stages and 21 different floral tissue samples from mature flowers. Moreover, two MADS-box genes TruPI and TruFBP24 involved in control of floral organ identity were used to validate the stable reference genes selected in this study. The aim of this work was to identify reference genes appropriate for transcript normalization in andromonoecious Taihangia flowers, and then validate their stability in staminate flowers, perfect flowers, and different floral tissues. This work will provide the basis for further research in exploring the molecular mechanism of flower development and sex differentiation in andromonoecuous plants.
Materials and Methods
Plant Material
The plant materials were transplanted from Zhuyufeng (35°27′ N, 113°22′E), Henan, China in April 2014, and were grown in the greenhouse of Henan Polytechnic University. During flowering phrase in early spring of 2016, the staminate and perfect flowers at different developmental stages were harvested from the same plant, respectively (Figure S1). Flowers were classified into five developmental stages: young flower buds (stage 1, < 0.5 cm), elongated buds (stage 2, 0.5–1 cm), pre-anthesis (stage 3), fully opened flowers (stage 4), and mature flowers (stage 5). Three independent samples of staminate and perfect flowers were taken from six different individuals at each of the developmental stage, respectively. In addition, floral tissues including sepals, petals, stamens, and carpals were collected from fully opened staminate and perfect flowers, respectively. All samples were immediately frozen in liquid nitrogen and stored at −80°C for RNA isolation.
Total RNA Isolation and cDNA Synthesis
Total RNA was extracted from 100 mg homogenized flowers and floral tissues using an RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer's manuals. RNA quality was initially assessed by 1.5% agarose gel electrophoresis. After that, RNA was quantified with a UV-visible spectrophotometer (UV-2550, Japan), and were adjusted to the same concentration for each sample after measuring the RNA concentration. The RNA samples with A260/A280 ratios between 1.8 and 2.0 and A260/A230 ratios greater than 2.0 were used for subsequent analyses. To eliminate DNA contamination, 1 μg total RNA was digested using gDNA Eraser (Takara, Japan) at 42°C for 2 min in a 10 μl mixture. Then, the RNA sample was used to synthesis the first strand cDNA with the Perfect Real Time RT reagent Kit (Takara, Japan) in a 20 μl reaction mixture according to the manufacturer's protocol. The cDNA samples were diluted 1:10 with nuclease-free water prior to the qRT-PCR analysis.
Identification and Selection of Candidate Reference Genes
In our previous study, four cDNA libraries, including male floral bud, hermaphroditic floral bud, male flower, and hermaphroditic flower, were constructed and sequenced by using the Illumina RNA-Seq method. A total of 24,753 unigenes were annotated in the NCBI non-redundant protein database, and further used for mining candidate reference genes based on expression stability (Li et al., 2017). To estimate expression stability of each gene, the values of MV, SD, and CV, were calculated for each gene based on read count, according to the previous described methodology (Czechowski et al., 2005; Gonzalezaguero et al., 2013). The genes that had both a mean of read counts above 500 and a CV below 0.3 were considered to be stably expressed. To facilitate the identification of putative function, the stably expressed genes were further screened with E-value ≤ 10−50. Candidate reference genes were selected from the homologous of traditional housekeeping genes previous used for flower development according to gene Nr annotation (Mallona et al., 2010; Yeap et al., 2013; Li et al., 2016; Wang et al., 2016). Meanwhile, some novel genes with a minor variation in expression level were also considered. In addition, the housekeeping gene Actin (ACT2) used as reference gene in previous studies was included (Du et al., 2008).
PCR Primer Design and qRT-PCR Analysis
Primers were designed using the Primer Premier 5 software according to the following parameters: primer lengths 18–23 bp, GC content 40–60%, melting temperatures 55–60°C, and amplicon lengths 100–300 bp. In order to confirm the primer specificity, the specificities of all primer pairs were initially tested by standard RT-PCR, and amplification product for each gene was verified by electrophoresis on a 1.5% agarose gel followed by ethidium bromide staining.
The qRT-PCR reactions were performed with the MiniOpticon Real-Time Detection System (Bio-Rad) using the SsoFast EvaGreen Supermix RT-PCR kit (Bio-Rad Laboratories). The PCR reaction mixture (20 μl) contained 10 μl of EvaGreen Supermix, 2.0 μl of 1:10 diluted cDNA, 0.4 μl of each primer (10 mM), and 7.2 μl of water. The reactions were incubated under following cycling conditions: 2 min at 95°C, 40 cycles of 95°C for 15 s, a specific annealing temperature (Ta) for 15 s, and 72°C for 30 s, and finally 72°C for 2 min with a single melt cycle from 60 to 95°C in 5 s intervals. The specificity of primer pair was verified by the presence of a single peak in the melt curve analysis during qRT-PCR. Three independent biological replicates and three technical repetitions were performed for each of the quantitative PCR experiments.
The threshold cycle (Ct) was measured automatically, and was used to define the expression level of each reference gene. The correlation coefficients (R2) and the slope were calculated from standard curve based on 5-fold series dilution of the cDNA templates, and the corresponding qRT-PCR efficiencies (E) for each gene were determined from the given slope (Ginzinger, 2002).
Data Analysis
The Ct values were used to evaluate the stability of 16 candidate reference genes in staminate flowers, perfect flowers, floral tissues, and total floral samples, across developmental stages by using geNorm, NormFinder, and BestKeeper algorithms. Furthermore, the web-based tool RefFinder (http://150.216.56.64/referencegene.php), which integrates these methods, was used to evaluate and screen the appropriate reference genes for qRT-PCR normalization.
Validation of Reference Genes
To confirm the reliability of the candidate reference genes, the relative expression profiles of two MADS-box transcription factors TruPI and TruFBP24, involved in flower organ identity (Krizek and Meyerowitz, 1996; De Folter et al., 2006), were detected by using RT-qPCR analysis. The expressions of the target genes TruPI and TruFBP24 were normalized using with the most and the least stable reference genes, respectively, as suggested by RefFinder. The qRT-PCR amplification conditions were the same as described above. The relative expression of each gene was calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Results
Selection of Candidate Reference Genes and PCR Amplification
Based on transcriptome data from Taihangia staminate and perfect flowers, a total of 3,628 genes were identified as stable expressed genes with average read counts above 500 and CV < 0.3 (Supplementary Material Data Sheet 1). Based on Nr annotation, we identified nine traditional housekeeping genes, such as GAPDH, EF-1α, UBQ, and ACT, common used as reference genes for flower development (E-value ≤ 10−50). Meanwhile, we selected six novel reference genes including actin-depolymerizing factor 3 (ADF3), ubiquitin fusion degradation protein 1 (UFD1), iron-sulfur cluster assembly protein (ISP), thiosulfate/3-mercaptopyruvate sulfurtransferase (THS), transmembrane protein 50 (TMP50), and vacuolar protein sorting-associated protein 32 (VAP32), with relatively stable expression profiles among staminate and perfect flower across the developmental stages. In addition, the housekeeping gene Actin (ACT2) used as the reference gene in previous Taihangia studies was also included. The results showed that 16 candidate reference genes involved in many aspects of primary metabolism or other basic cellular processes such as cytoskeleton (ACT, ACT2, ADF3, and TUA), transport of ions (THS), transport in vacuoles (VAP32) or membranes (TMP50), glycolysis (GAPDH), energy metabolism (ISP), RNA processing and modification (SPF), protein translation (EF-1α), protein posttranslational modification (UBC, UFD1, and UBQ), cell signaling (PP2A), and chromatin structure, and dynamics (HIS3). Based on Taihangia transcriptome sequence data, we found varied transcriptional profiles for reference gene candidates. Average read count of each gene ranged from 703.25 (ACT) to 7,940.25 (GAPDH), while the CV value varied from 0.007(UFD1) to 0.203 (GAPDH).
The specificity of RT-PCR amplification for each reference gene was verified by 1.5% agarose gel electrophoresis and melting curve analysis (Figure S2). The results showed that all 16 primer pairs generated single fragments with expected size, ranging from 100 bp for ACT2 to 254 bp for ISP, indicating that no primer dimers or non-specific products amplified in RT-PCR reactions. The amplification specificity of each reference gene was further confirmed by the presence of single-peak melting curve in qRT-PCR analysis. For each primer pair, the amplification efficiencies of ranged from 90.4% for PP2A to 104.5% for TUA, while correlation coefficients (R2) ranged from 0.9901 (ACT2) to 0.9999 (ISP). The candidate reference genes, primer sequences, and characteristics of PCR amplifications were summarized in Table 1.
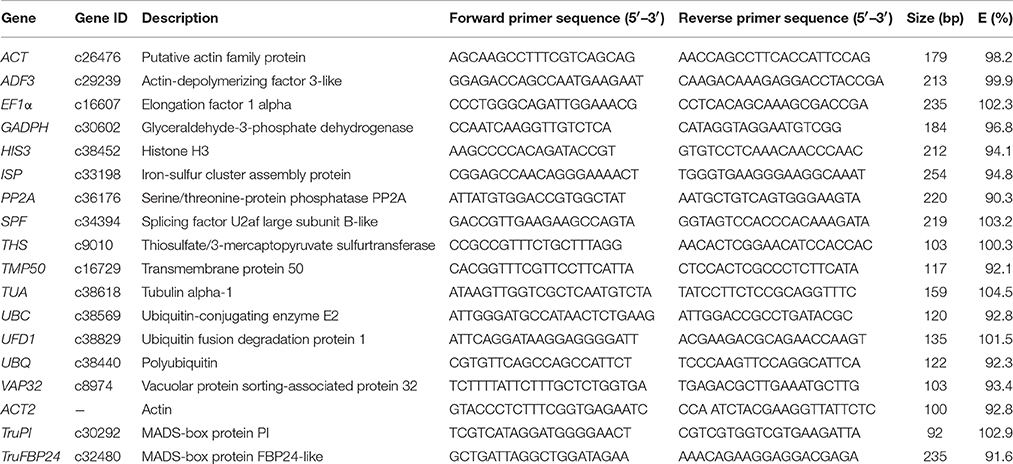
Table 1. Candidate reference genes, primer sequences, and characteristics of PCR amplifications in Taihangia rupestris.
Expression Stability of the Candidate Reference Genes
To detect overall expression profiles of the 16 reference genes in Taihangia flowers, the 30 floral samples, including staminate and perfect flowers at different developmental stages, and 21 floral tissue samples (sepal, petal, stamen, and carpel) were used for the RT-qPCR analyses. The results showed that the mean Ct values of the 16 reference genes varied from 19.11 ± 0.85 (HIS3) (mean ± SD) to 23.96 ± 0.77 (SPF) for highest and lowest expression levels, respectively, in all samples (Figure 1). In addition, standard deviation (SD) of Ct values, which represented as expression stability, ranged from 0.63 to 1.45. The genes with higher SD of Ct values indicated more variable expression compared to these with lower SD. UFD1 showed the smallest variation in gene expression with the lowest SD (22.43 ± 0.63), while GAPDH with the most variable levels of expression (22.22 ± 1.41).
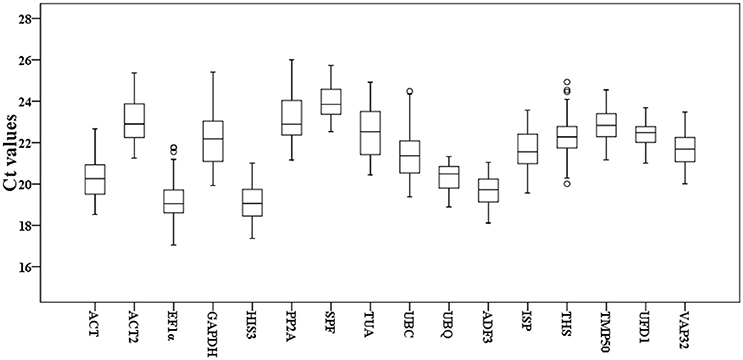
Figure 1. Cycle threshold (Ct) values of the 16 candidate reference genes for Taihangia flowers. Box-plot graph of Ct values show the median values as line across the box. The outside box is determined by the 25th and 75th percentiles. The whiskers indicate the maximum and minimum values.
To further select the most appropriate reference gene for qRT-PCR analysis in investigation of flower development and sex differentiation within andromonoecious Taihangia, we divided these floral samples into four groups: staminate flower, perfect flower, floral tissues, and total samples, and four statistical approaches (geNorm, NormFinder, BestKeeper, and RefFinder) were used to analyze the stability of each reference gene from different floral groups.
Genorm Analysis
The geNorm program was employed to assess the expression stability (M) for each reference gene based on the ratio of average pair-wise variation within all gene candidates. According to geNorm algorithm, the reference gene with M value below 1.5 is considered to be stably expressed, and lower M value represents the more stable expression (Vandesompele et al., 2002). In this study, all of the tested candidate genes showed an M value < 1.5, indicating that the 16 selected genes should be considered relatively stable. ADF3 and HIS3 were the most stable reference genes for both staminate and perfect flowers, while UBQ and SPF were identified as the most stable in sepal, petal, stamen, and carpel samples. For all samples tested, HIS3 and UFD1 were recommended as the most stable genes. In contrast, TUA in staminate flowers and GAPDH in perfect flowers, floral tissues, and total samples with the highest M value were identified as the least stable reference genes, respectively (Figure 2).
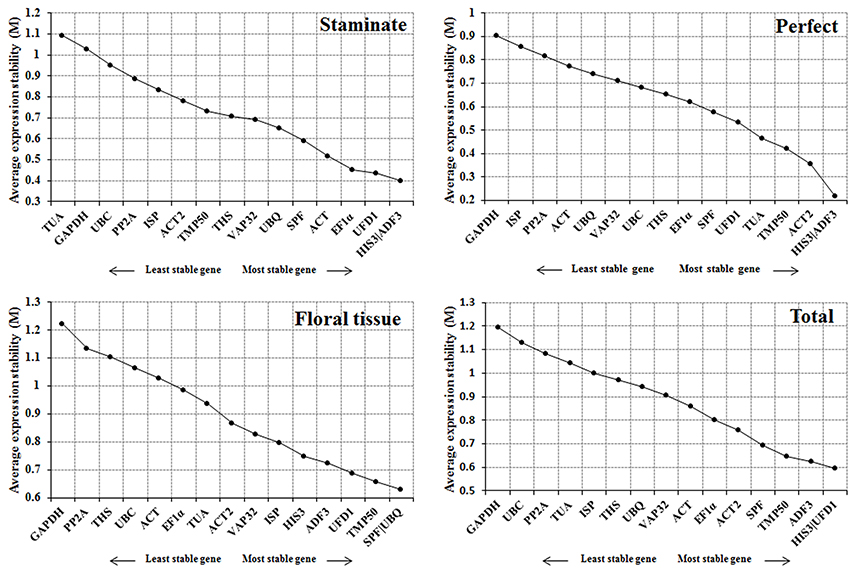
Figure 2. Expression stability and ranking order of the 16 reference genes for Taihangia flowers predicted by the geNorm software. The genes with M ≤ 1.5 are considered significant with stable expression.
The optimal number of the reference genes was also determined based on the pairwise variation between sequential ranked genes (Vn/Vn+1) with the cut-off value of 0.15 in geNorm program. If the value of Vn/n+1 was below 0.15, it is not necessary to append additional genes for accurate normalization. The V2/V3 values were below 0.15 in both staminate and perfect flowers, suggesting that adding an extra gene to obtain accurate results was not necessary for normalization. For the floral tissues and total samples, V2/3 value was 0.219 and 0.185 showed that additional reference genes may be required (Figure 3).
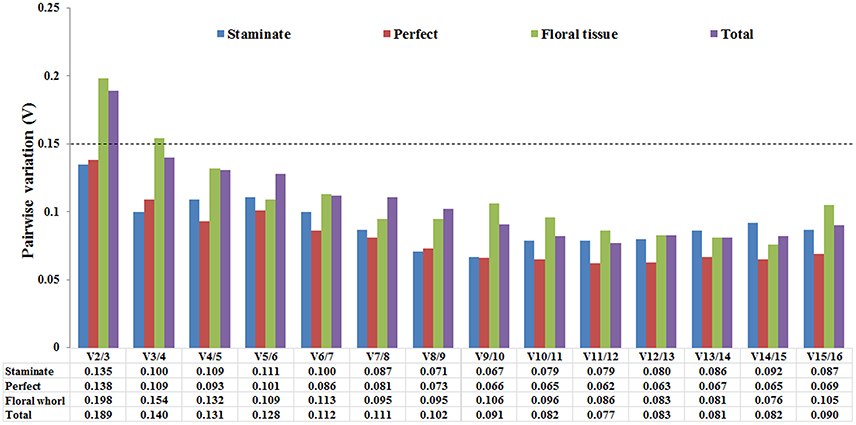
Figure 3. Pairwise variation (V) of 16 candidate reference genes calculated by GeNorm software. The optimal number of the reference genes required for accurate normalization was determined by pairwise variation (Vn/Vn+1). The dotted line indicates the recommended threshold value of 0.15 below which the inclusion of an additional reference gene is not necessary.
Normfinder Analysis
As a mathematical model-based algorithm, NormFinder is widely applied in determination of the stability value of reference genes, based on inter- and intra-group variance estimation in given serials of specific samples (Andersen et al., 2004). According to the stability value for each reference gene, ranking order was arranged from smallest to largest, with the lower stability value indicating the higher stability. In staminate flowers, UFD1 was ranked first for expression stability, whereas TUA with the largest stability value was the least stably expressed. For perfect flowers and floral tissues samples, HIS3 was identified as the most suitable reference gene, while GAPDH was the most unstably expressed. When total samples were taken into considered, UFD1 was ranked first as the most stable expressed gene, followed by HIS3 and ADF3 (Table 2).
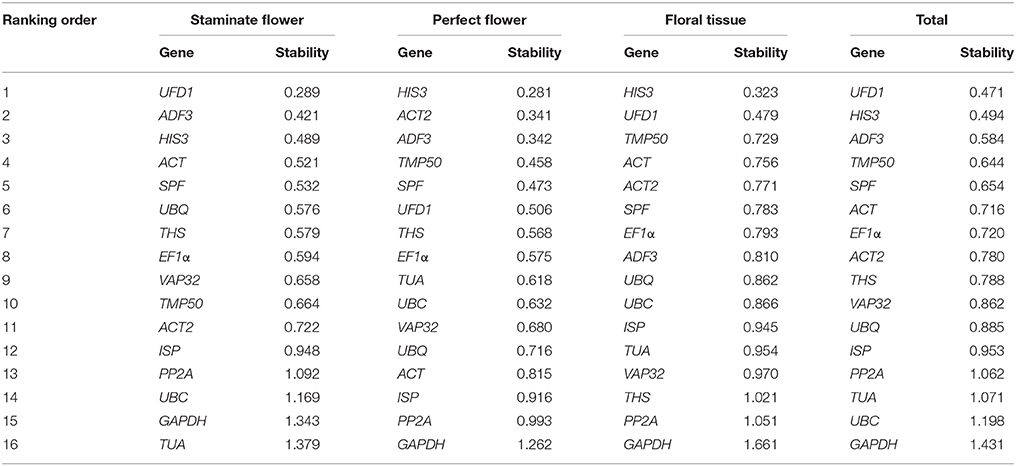
Table 2. Expression stability analysis of reference genes assayed by NormFinder software.
BestKeeper Analysis
BestKeeper program evaluates the stability of reference genes by means of the standard deviation (SD) and coefficient of variance (CV) of the Ct values, with the lowest SD value indicating the most stable reference gene (Pfaffl et al., 2004). The genes with a SD [± CP] value below 1.0 are considered to be stably expressed and used for the gene expression normalization. According to these criteria, UFD1 was recommended as the most stable gene in staminate flowers, floral tissues, and all samples, while SPF was the most stable in perfect flowers. On the contrary, TUA in staminate flower, PP2A in perfect flower, and GAPDH in floral tissues and total samples with the highest SD value displayed the least stable expression as determined by Bestkeeper software, respectively (Table 3).
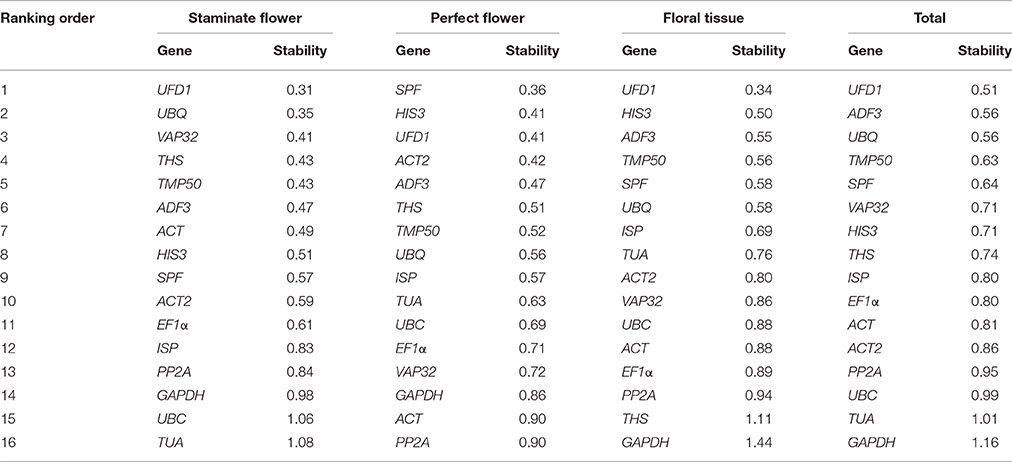
Table 3. Expression stability analysis of reference genes assayed by Bestkeeper software.
RefFinder Analysis
RefFinder was applied to calculate the geometric mean of weights for the comprehensive ranking order recommended by ΔCt, geNorm, NormFinder, and BestKeeper (Xie et al., 2012). Based on RefFinder analysis, UFD1 and ADF3 were the optimal reference genes for staminate flowers, while HIS3 and ADF3 were the most stable in perfect floral samples. For floral tissues samples tested, HIS3 and UFD1 were recommended as the suitable reference genes. When total samples were taken into considered, two novel genes (UFD1 and ADF3) and one traditional housekeeping gene (HIS3) were identified as the most stable expressed genes. Conversely, several traditional housekeeping genes showed unstable expression profiles in T. rupestris flowers. GAPDH was the least stable in floral tissues, perfect flowers, and total samples, while TUA was unstable for staminate flowers. The rankings of the four algorithms were integrated by RefFinder and the results are shown in Table 4.
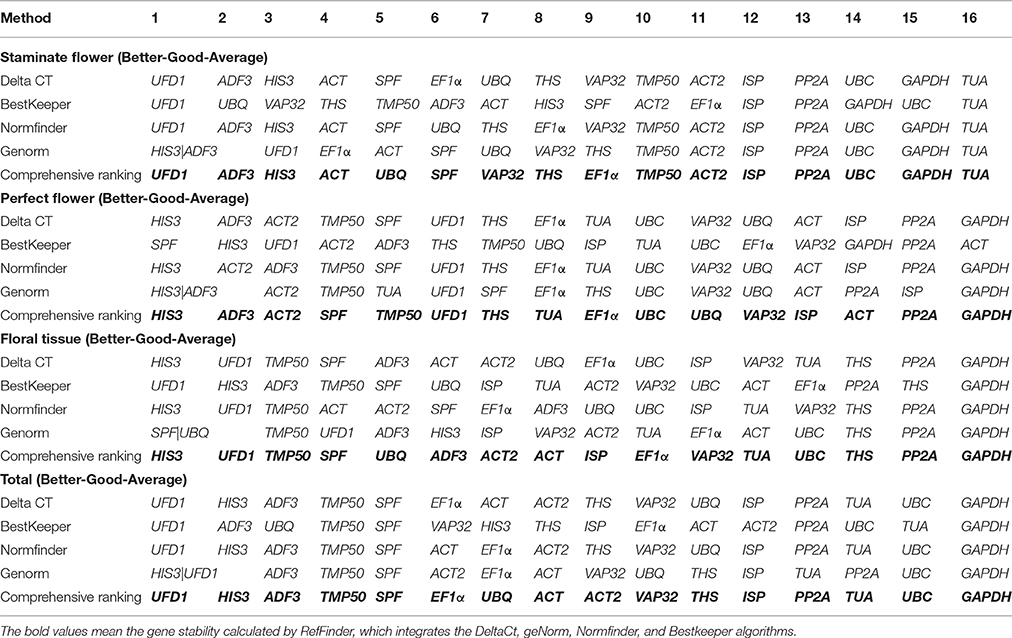
Table 4. Expression stability ranking of the 16 candidate reference genes as calculated by RefFinder.
Validation of the Reference Genes
In order to examine reliability of reference genes for normalization, the relative expression patterns of two MADS-box genes TruPI and TruFBP24, which are required to specify flower organ identity, were evaluated in floral tissues, staminate and perfect flowers using qRT-PCR analysis (Krizek and Meyerowitz, 1996; De Folter et al., 2006). According to geNorm pairwise variation (Vn/Vn+1) in staminate flowers, perfect flowers, and floral tissues, we selected the most stable reference genes (UFD1, ADF3, and UFD1/ADF3 for staminate flowers, HIS3, ADF3, and HIS3/ADF3 for perfect flowers, and HIS3/UFD1, HIS3/TMP50, and HIS3/UFD1/TMP50 for floral tissues, respectively) to normalize TruPI gene expression in staminate and perfect flowers across developmental stages. Meanwhile, the expression levels of TruPI were also investigated in different floral tissues of staminate and perfect flowers such as sepal, petal, stamen and carpel. Moreover, we evaluated target genes expression following normalization with the least stable reference gene (TUA for staminate and GAPDH for perfect flowers and floral tissues, respectively) for a comparative analysis. When the stable reference gene(s) were used for normalization, TruPI transcripts increased at early developmental stage with maximum at stage 2, and then steadily decreased with developmental stage in both staminate and perfect flowers (Figure 4). By using different stable reference genes, either single or the combination, the expression patterns of TruPI were similar with minor differences at stage 2. Based on geNorm pairwise variation, the combination of UFD1/ADF3 was recommended to be the optimum pairs of reference gene for staminate flowers, and HIS3/ADF3 was the suitable pairs of reference gene for accurate normalization in perfect flowers. However, the relative expression levels of target gene TruPI were underestimated in Taihangia flowers at developmental stage 2 and 3, when normalized using the most unstable reference genes. Normalization by TUA in staminate and GAPDH in perfect flowers showed decrease trends in the relative expression levels of TruPI at early developmental stages, indicating that the adverse effect of using an inappropriate reference gene. For floral tissues of staminate and perfect flowers, TruPI was expressed in all of the floral organs, including sepals, petals, stamens, and carpels, but at different levels (Figure 5). When using the combination of HIS3/UFD1, HIS3/TMP50, and HIS3/UFD1/TMP50 for normalization, the qRT-PCR analyses showed that TruPI was strongly expressed petals and stamens, and weakly in sepals and carpels. However, different expression profiles of TruPI were observed in floral tissues, especially in petals and stamens, by using GAPDH for normalization. Despite the higher expression levels of TruPI also detected in petals and stamens, the overestimation of relative expression was observed.
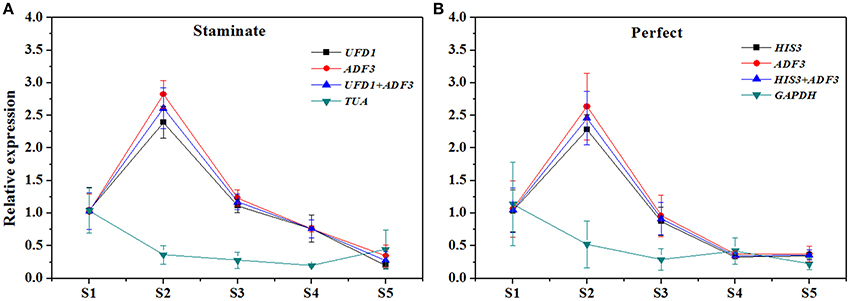
Figure 4. Relative quantification of TruPI gene expression in staminate and perfect flowers across developmental stages by using different reference genes for normalization. (A) Normalization was performed using single and combined reference genes for staminate flower. The most stable reference gene(s): UFD1, ADF3, and UFD1/ADF3, and the most unstable one: TUA. (B) Normalization was performed using single and combined reference genes for perfect flower. The most stable reference gene(s): HIS3, ADF3, and HIS3/ADF3, and the most unstable one: GAPDH.
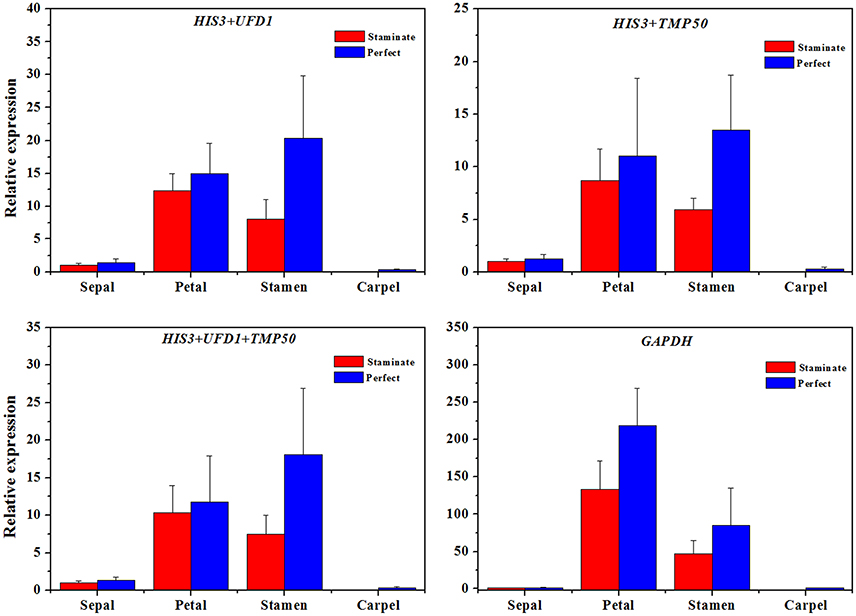
Figure 5. The expression level of the TruPI gene in floral tissues by using different reference genes for normalization. Combinations of HIS3/UFD1, HIS3/TMP50, HIS3/UFD1/TMP50 were used for normalization as the most stable reference genes, GAPDH was used as worst stable reference gene.
To further explore the stability of reference genes for normalization qRT-PCR between staminate and perfect flowers, the expression levels of TruFBP24 was investigated for all floral samples across five developmental stages. Based on geNorm pairwise variation analysis in total samples (V3/V4 value below 0.15), we selected single and combinations of two or three reference genes to normalize TruFBP24 gene expression suggested by RefFinder. As shown in Figure 6, when using the combinations of UFD1/ADF3, UFD1/HIS3, ADF3/HIS3, and ADF3/HIS3/UFD1, as reference genes for normalization, similar expression profiles of TruFBP24 were detected in both staminate and perfect flowers at each developmental stage. When the least stable gene GAPDH was used for normalization, TruFBP24 was 7 times higher expression in perfect flower than that in staminate flower at stage 1. Moreover, relative gene expression of TruFBP24 (20.07 ± 4.12) at stage 5 was remarkably lower than those obtained by normalizing with the suitable reference genes, indicating the low expression stability of GAPDH as reference gene for normalization qRT-PCR in all floral samples. Overall, our analysis suggested that the target gene's expression profiles are strongly affected by the choice of the reference genes.
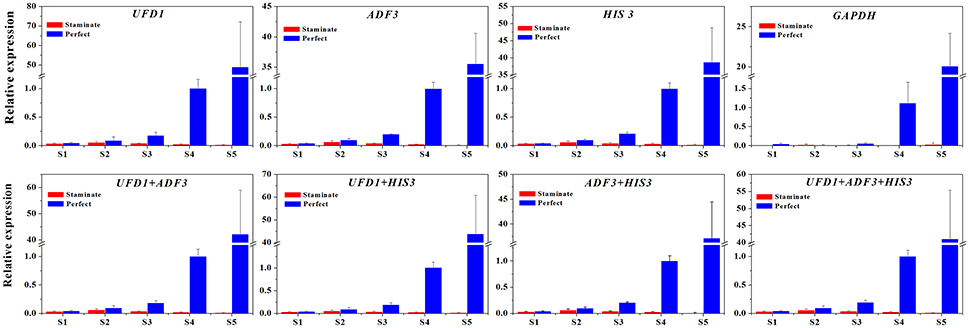
Figure 6. The expression level of the TruFBP24 gene in staminate and perfect flowers across five developmental stages by using different reference genes for normalization. The single or combinations of UFD1, ADF3, and HIS3, were used for normalization as the most stable reference genes, GAPDH was used as worst stable reference gene.
Discussion
Andromonoecious plant species, which produces both bisexual flowers and unisexual male flowers within the same individual, provides an excellent model system for studying establishment and development of bi- and unisexual flowers under a uniform genetic background. Using qRT-PCR technique, investigation of gene expression profiles in staminate and perfect flowers not only better understands the underlying molecular mechanism in regulation of flower formation, but also provides insights into the complex regulatory networks in flower sex differentiation. To obtain reliable and accurate quantification results in qRT-PCR analysis, it is fundamental to select and validate of appropriate reference genes for normalizing qRT-PCR data. In this study, ten traditional housekeeping genes and six newly candidate reference genes were selected for evaluation of expression stability based on T. rupestris transcriptome data and previous studies (Du et al., 2008; Li et al., 2017). To the best of our knowledge, this is the first report on the identification and validation of suitable reference genes for normalizing qRT-PCR analysis in andromonoecious plant species.
Ideally, an accurate reference gene should be expressed stably regardless of varied organ, tissue type or developmental stages. By using geNorm and NormFinder, and BestKeeper software, the 16 reference gene candidates showed various performances in expression stability. However, the ranked orders generated by three algorithms were not completely identical. The results of geNorm and NormFinder were similar in some cases, but they showed quite differences from the results obtained from Bestkeeper. For example, SPF was ranked first by BestKeeper in perfect flowers, while it was ranked at a medium position by geNorm and NormFinder. The results were similar to many previous studies, and this apparent inconsistency was probably due to the different principles in the three statistical algorithms (Xiao et al., 2015; Qi et al., 2016). Considered as an integrative statistical program, RefFinder has been widely applied to evaluate the overall stability of reference gene expression and determine appropriate reference genes for diverse plant species (Kim et al., 2015; Qi et al., 2016). In order to obtain an integral assessment of the most suitable reference gene for Taihangia, a comprehensive tool RefFinder was used to determine the final overall ranking by a comparison of different algorithms (Xie et al., 2012). Based on comprehensive RefFinder analysis, ADF3, HIS3, and UFD1 were identified as the most appropriate reference genes for normalization in Taihangia floral tissues and flowers. As a whole, the integrated results obtained from different programs, could lead to better accuracy for each gene in this study, suggesting that more than two algorithms should be used for stability evaluation of reference gene.
Increasing evidences indicated that the expressions of classical housekeeping genes do not always express stably not only across developmental stages but also in variable biological samples (Li et al., 2016; Niu et al., 2017). In this study, the expression stability of commonly used housekeeping genes such as EF-1α, GAPDH, ACT, UBQ, TUA, and HIS3, displayed remarkably different expressed patterns in Taihangia floral tissues and flowers. Of these selected housekeeping genes, HIS3 was identified as one of the most stable reference genes for staminate flowers, perfect flowers, and different floral tissues. As a major component of chromatin, HIS3 is thought to be crucial for maintenance of a stable chromatin structure (Oliver et al., 2013). In T. rupestris, they may remain expressed constitutively, and showed minimal changes in mRNA transcription in staminate and perfect flowers across developmental stages. For majority of common used housekeeping genes, including ACT, EF-1α, and UBQ, showed relatively stable expression with middle ranking orders calculated by RefFinder analysis. For example, the ACT gene, used as reference gene in previous study, is not the best choice for normalization in T. rupestris. A possible explanation was that that reference genes are implicated in multiple cell processes. Although TUA and GAPDH were also identified as stable reference genes for flower development in other plant species (Li et al., 2016; Karuppaiya et al., 2017), their expressions were the least stable in Taihangia flowers. In addition, the transcriptional profiles of target genes showed a strong bias via qRT-PCR validation, when these genes were used as reference gene for normalization gene expression in staminate and perfect flowers of T. rupestris. Similar to our analysis, a number of reports have demonstrated that the expressions of several traditional housekeeping genes, such as GAPDH and TUA, varied across flower developmental stage and were unsuitable for normalizing qRT-PCR data as internal control gene (Jin et al., 2013; Yuan et al., 2014). These results reported here supported that the traditional reference transcripts may not always show constitutive expression as has often been assumed, and thus it is prerequisite for verification of their expression stability before qRT-PCR normalization in specific biological samples.
Compare to traditional reference genes, the newly discovered reference genes could perform better in qRT-PCR normalization for non-model plants (Narsai et al., 2010; Gonzalezaguero et al., 2013). In this study, UFD1 showed the most stable expression profile in staminate flowers, while ADF3 was identified as the second stable gene in both staminate and perfect flowers. ADF are small actin-binding proteins, which involved in regulation of actin dynamics, play an essential role in plant growth and development (Kandasamy et al., 2007). In a previous study, ADF was identified as a reference gene candidate for grapevine flower development based on transcriptome data because of stable expression during anthesis (Gonzalezaguero et al., 2013). In rubber tree, ADF was also identified as a candidate reference gene because its transcription abundance maintained relatively stable duration of latex flow (Chao et al., 2016). For andromonoecious T. rupestris, the stability of ADF3 in staminate and perfect flowers probably attributed to its function for basic cellular processes, such as cell proliferation and regulation of actin dynamics in cells (Ruzicka et al., 2007). In plants, UFD1 is an essential ubiquitin recognition component in the ubiquitin-mediated degradation pathway (Wei et al., 2009). Given that possible basic function in cell processes, UFD1 should be suitable as reference gene for qRT-PCR normalization in T. rupestris, although it was not reported as reference gene in plant species. It was note worth that ADF3, HIS3, and UFD1 showed high stability among staminate and perfect flowers when total samples were taken into consideration, implying that those genes could provide accurate RT-qPCR analysis in comparison gene expression between uni- and bisexual flowers within andromonoecious system.
It is widely acceptable that the normalization with combination of multiple reference genes could provide more accurate relative expression quantification than that with a single gene in qRT-PCR analysis (Reid et al., 2006; Expositorodriguez et al., 2008). Based on validation of target gene expression, UFD1 combined with ADF3 for staminate flowers, the combination of ADF3/HIS3 for perfect flowers, HIS3/UFD1/TMP50 for floral tissues, and UFD1/HIS3 for total samples were recommended as appropriate reference genes for qRT-PCR normalization. As the three stable reference genes ADF3, HIS3, and UFD1 belong to the different functional classes, they should be used together as suggested by Vandesompele et al. (2002). Therefore, our results supported that the more accurate quantification of gene expression could be obtain when multiple reference genes were applied in qRT-PCR analysis. Interestingly, the combinations, including one traditional housekeeping gene and one new reference gene, were identified as the most stable reference genes for majority of group tested, suggesting that the selection of internal reference genes with traditional house-keeping gene combined novel reference gene may be a good strategy for normalizing qRT-PCR on T. rupestris flowers.
In summary, 16 reference gene candidates were selected based on our transcriptome sequence data and previous reports, and their expression stability were assessed in staminate and perfect flowers within andromonoecious plant T. rupestris. ADF3 combined with UFD1 were identified as the optimal reference genes for staminate flowers, while combination of HIS3/ADF3 was recommended to be the best one in perfect flowers, respectively. For floral tissues, combination of HIS3/UFD1/TMP50 was the most suitable reference genes for normalization. The stable reference genes reported in this study will be helpful to improve the accuracy of qRT-PCR analysis in andromonoecious T. rupestris, and will facilitate the functional genomics studies on flower development and sex differentiation in the future.
Author Contributions
WL and YWZ conceived and designed the work. LZ, YDZ, and GW performed experiments and analyzed data. WL wrote the paper, DS partially revised the manuscript. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Dr. Sen Liu and Ms Shangtong Jiang for assistance in sample collection and experiment. This work was supported by National Natural Science Foundation of China (No. 31370434 and 31170354), and Scientific Innovation Fund from Henan Polytechnic University (B2008-25 and T2013-2).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00729/full#supplementary-material
References
Andersen, C. L., Jensen, J. L., and Orntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-pcr data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Bustin, S. A. (2002). Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. 29, 23–39. doi: 10.1677/jme.0.0290023
Bustin, S. A., Benes, V., Nolan, T., and Pfaffl, M. W. (2005). Quantitative real-time RT-PCR - a perspective. J. Mol. Endocrinol. 34, 597–601. doi: 10.1677/jme.1.01755
Chao, J., Yang, S., Chen, Y., and Tian, W. (2016). Evaluation of reference genes for quantitative real-time PCR analysis of the gene expression in laticifers on the basis of latex flow in rubber tree (Hevea brasiliensis Muell. Arg.). Front. Plant Sci. 7:1149. doi: 10.3389/fpls.2016.01149
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K., and Scheible, W. (2005). Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139, 5–17. doi: 10.1104/pp.105.063743
De Folter, S., Shchennikova, A. V., Franken, J., Busscher, M., Baskar, R., Grossniklaus, U., et al. (2006). A Bsister MADS-box gene involved in ovule and seed development in petunia and Arabidopsis. Plant J. 47, 934–946. doi: 10.1111/j.1365-313X.2006.02846.x
Demidenko, N. V., Logacheva, M. D., and Penin, A. A. (2011). Selection and validation of reference genes for quantitative real-time PCR in buckwheat (Fagopyrum esculentum) based on transcriptome sequence data. PLoS ONE 6:e19434. doi: 10.1371/journal.pone.0019434
Du, X., Xiao, Q., Zhao, R., Wu, F., Xu, Q., Chong, K., et al. (2008). TrMADS3, a new MADS-box gene, from a perennial species Taihangia rupestris (Rosaceae) is upregulated by cold and experiences seasonal fluctuation in expression level. Dev. Genes Evol. 218, 281–292. doi: 10.1007/s00427-008-0218-z
Expositorodriguez, M., Borges, A. A., Borgesperez, A., and Perez, J. A. (2008). Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 8, 131–131. doi: 10.1186/1471-2229-8-131
Gachon, C. M. M., Mingam, A., and Charrier, B. (2004). real-time PCR: what relevance to plant studies? J. Exp. Bot. 55, 1445–1454. doi: 10.1093/jxb/erh181
Ginzinger, D. G. (2002). Gene quantification using real-time quantitative PCR. Exp. Hematol. 30, 503–512. doi: 10.1016/S0301-472X(02)00806-8
Gonzalezaguero, M., Garciarojas, M., Genova, A. D., Correa, J. J., Maass, A., Orellana, A., et al. (2013). Identification of two putative reference genes from grapevine suitable for gene expression analysis in berry and related tissues derived from RNA-Seq data. BMC Genomics 14:878. doi: 10.1186/1471-2164-14-878
Gutierrez, L., Mauriat, M., Guenin, S., Pelloux, J., Lefebvre, J., Louvet, R., et al. (2008). The lack of a systematic validation of reference genes: a serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 6, 609–618. doi: 10.1111/j.1467-7652.2008.00346.x
Hu, R., Fan, C., Li, H., Zhang, Q., and Fu, Y. (2009). Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol. Biol. 10:93. doi: 10.1186/1471-2199-10-93
Huggett, J., Dheda, K., Bustin, S. A., and Zumla, A. (2005). Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 6, 279–284. doi: 10.1038/sj.gene.6364190
Jin, X., Fu, J., Dai, S., Sun, Y., and Hong, Y. (2013). Reference gene selection for qPCR analysis in cineraria developing flowers. Sci. Hortic. 153, 64–70. doi: 10.1016/j.scienta.2013.01.023
Kandasamy, M. K., Burgosrivera, B., Mckinney, E. C., Ruzicka, D. R., and Meagher, R. B. (2007). Class-specific interaction of profilin and ADF isovariants with actin in the regulation of plant development. Plant Cell 19, 3111–3126. doi: 10.1105/tpc.107.052621
Karuppaiya, P., Yan, X.-X., Liao, W., Wu, J., Chen, F., and Tang, L. (2017). Identification and validation of superior reference gene for gene expression normalization via RT-qPCR in staminate and pistillate flowers of Jatropha curcas – a biodiesel plant. PLoS ONE 12:e0172460. doi: 10.1371/journal.pone.0172460
Kim, H., Saha, P., Farcuh, M., Li, B., Sadka, A., and Blumwald, E. (2015). RNA-Seq analysis of spatiotemporal gene expression patterns during fruit development revealed reference genes for transcript normalization in plums. Plant Mol. Biol. Report. 33, 1634–1649. doi: 10.1007/s11105-015-0860-3
Kozera, B., and Rapacz, M. (2013). Reference genes in real-time PCR. J. Appl. Genet. 4, 391–406. doi: 10.1007/s13353-013-0173-x
Krizek, B. A., and Meyerowitz, E. M. (1996). The Arabidopsis homeotic genes APETALA3 and PISTILLATA are sufficient to provide the B class organ identity function. Development 122, 11–22.
Li, J., Han, J., Hu, Y., and Yang, J. (2016). Selection of reference genes for quantitative real-time PCR during flower development in tree peony (Paeonia suffruticosa Andr.). Front. Plant Sci. 7:516. doi: 10.3389/fpls.2016.00516
Li, W., Zhang, L., Ding, Z., Wang, G., Zhang, Y., Gong, H., et al. (2017). De novo sequencing and comparative transcriptome analysis of the male and hermaphroditic flowers provide insights into the regulation of flower formation in andromonoecious Taihangia rupestris. BMC Plant Biol. 17:54. doi: 10.1186/s12870-017-0990-x
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, S., Fan, Y., Liu, L., Liu, S., Zhang, W., and Meng, Z. (2010). Ectopic expression of TrPI, a Taihangia rupestris (Rosaceae) PI ortholog, causes modifications of vegetative architecture in Arabidopsis. J. Plant Physiol. 167, 1613–1621. doi: 10.1016/j.jplph.2010.06.028
Lu, W. (1996). Development of sexual organs in Taihangia rupestris different temperature requirements for both sexual organ development in a bisexual flower. Acta Bot. Sin. 38, 174–179.
Ma, R., Xu, S., Zhao, Y., Xia, B., and Wang, R. (2016). Selection and validation of appropriate reference genes for quantitative real-time PCR analysis of gene expression in Lycoris aurea. Front. Plant Sci. 7:536. doi: 10.3389/fpls.2016.00536
Mallona, I., Lischewski, S., Weiss, J., Hause, B., and Egeacortines, M. (2010). Validation of reference genes for quantitative real-time PCR during leaf and flower development in Petunia hybrida. BMC Plant Biol. 10:4. doi: 10.1186/1471-2229-10-4
Narsai, R., Ivanova, A., Ng, S., and Whelan, J. (2010). Defining reference genes in Oryza sativa using organ, development, biotic and abiotic transcriptome datasets. BMC Plant Biol. 10:56. doi: 10.1186/1471-2229-10-56
Niu, K., Shi, Y., and Ma, H. (2017). Selection of candidate reference genes for gene expression analysis in Kentucky Bluegrass (Poa pratensis L.) under abiotic stress. Front. Plant Sci. 8:193. doi: 10.3389/fpls.2017.00193
Oliver, C., Pradillo, M., Corredor, E., and Cunado, N. (2013). The dynamics of histone H3 modifications is species-specific in plant meiosis. Planta 238, 23–33. doi: 10.1007/s00425-013-1885-1
Pfaffl, M. W., Tichopad, A., Prgomet, C., and Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515. doi: 10.1023/B:BILE.0000019559.84305.47
Qi, S., Yang, L., Wen, X., Hong, Y., Song, X., Zhang, M., et al. (2016). Reference gene selection for RT-qPCR analysis of flower development in Chrysanthemum morifolium and Chrysanthemum lavandulifolium. Front. Plant Sci. 7:287. doi: 10.3389/fpls.2016.00287
Reid, K. E., Olsson, N., Schlosser, J., Peng, F. Y., and Lund, S. T. (2006). An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 6:27. doi: 10.1186/1471-2229-6-27
Ruzicka, D. R., Kandasamy, M. K., Mckinney, E. C., Burgosrivera, B., and Meagher, R. B. (2007). The ancient subclasses of Arabidopsis actin depolymerizing factor genes exhibit novel and differential expression. Plant J. 52, 460–472. doi: 10.1111/j.1365-313X.2007.03257.x
Thellin, O., Elmoualij, B., Heinen, E., and Zorzi, W. (2009). A decade of improvements in quantification of gene expression and internal standard selection. Biotechnol. Adv. 27, 323–333. doi: 10.1016/j.biotechadv.2009.01.010
Thellin, O., Zorzi, W., Lakaye, B., De Borman, B., Coumans, B., Hennen, G., et al. (1999). Housekeeping genes as internal standards: use and limits. J. Biotechnol. 75, 291–295. doi: 10.1016/S0168-1656(99)00163-7
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., and Speleman, F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 1–12. doi: 10.1186/gb-2002-3-7-research0034
Wang, C., Cui, H.-M., Huang, T.-H., Liu, T.-K., Hou, X.-L., and Li, Y. (2016). Identification and validation of reference genes for RT-qPCR analysis in non-heading Chinese Cabbage flowers. Front. Plant Sci. 7:811. doi: 10.3389/fpls.2016.00811
Wei, L., Tao, Y., Jia, H., Zhang, L., Xu, P., Wang, Y., et al. (2009). Highly conserved UFD1 proteins among Eukaryotes exhibit considerable C-Terminus diversity in different taxa. Plant Mol. Biol. Report. 27, 439–447. doi: 10.1007/s11105-009-0099-y
Xiao, X., Ma, J., Wang, J., Wu, X., Li, P., and Yao, Y. (2015). Validation of suitable reference genes for gene expression analysis in the halophyte Salicornia europaea by real-time quantitative PCR. Front. Plant Sci. 5:788. doi: 10.3389/fpls.2014.00788
Xie, F., Xiao, P., Chen, D., Xu, L., and Zhang, B. (2012). miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 80, 75–84. doi: 10.1007/s11103-012-9885-2
Yeap, W., Loo, J. M., Wong, Y. C., and Kulaveerasingam, H. (2013). Evaluation of suitable reference genes for qRT-PCR gene expression normalization in reproductive, vegetative tissues and during fruit development in oil palm. Plant Cell Tissue Organ Cult. 116, 55–66. doi: 10.1007/s11240-013-0382-3
Yu, T., and Li, C. (1980). Taihangia- a new genus of Rosaceae from China. Acta Phytotaxon Sin. 18, 469–472.
Yu, T., and Li, C. (1983). The systematic position of genus Taihangia in Rosaceae. Acta Phytotaxon Sin. 21, 229–235.
Yuan, X., Jiang, S., Wang, M., Ma, J., Zhang, X., and Cui, B. (2014). Evaluation of internal control for gene expression in phalaenopsis by quantitative real-time PCR. Appl. Biochem. Biotechnol. 173, 1431–1445. doi: 10.1007/s12010-014-0951-x
Zhang, Y., Han, X., Chen, S., Zheng, L., He, X., Liu, M., et al. (2017). Selection of suitable reference genes for quantitative real-time PCR gene expression analysis in Salix matsudana under different abiotic stresses. Sci. Rep. 7:40290. doi: 10.1038/srep40290
Keywords: reference genes, quantitative real-time PCR, Taihangia rupestris, flower development, staminate flower, perfect flower
Citation: Li W, Zhang L, Zhang Y, Wang G, Song D and Zhang Y (2017) Selection and Validation of Appropriate Reference Genes for Quantitative Real-Time PCR Normalization in Staminate and Perfect Flowers of Andromonoecious Taihangia rupestris. Front. Plant Sci. 8:729. doi: 10.3389/fpls.2017.00729
Received: 12 March 2017; Accepted: 19 April 2017;
Published: 19 May 2017.
Edited by:
Stefan De Folter, National Polytechnic Institute (Cinvestav-IPN), MexicoReviewed by:
Vagner Benedito, West Virginia University, USAFernando Andres Lalaguna, Genetic Improvement and Adaptation of Mediterranean and Tropical Plants (INRA), France
Copyright © 2017 Li, Zhang, Zhang, Wang, Song and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiguo Li, d2dsaTA3MDhAMTYzLmNvbQ==
Yanwen Zhang, eWFud2VuemhhbmcwMjA5QDE2My5jb20=