Abstract
Transition from vegetative to floral buds is a critical physiological change during flower induction that determines fruit productivity. Small non-coding RNAs (sRNAs) including microRNAs (miRNAs) and small interfering RNAs (siRNAs) are pivotal regulators of plant growth and development. Although the key role of sRNAs in flowering regulation has been well-described in Arabidopsis and some other annual plants, their relevance to vegetative-to-floral transition (hereafter, referred to floral transition) in perennial woody trees remains under defined. Here, we performed Illumina sequencing of sRNA libraries prepared from vegetative and floral bud during flower induction of the apple trees. A large number of sRNAs exemplified by 33 previously annotated miRNAs and six novel members display significant differential expression (DE) patterns. Notably, most of these DE-miRNAs in floral transition displayed opposite expression changes in reported phase transition in apple trees. Bioinformatics analysis suggests most of the DE-miRNAs targeted transcripts involved in SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) gene regulation, stress responses, and auxin and gibberellin (GA) pathways, with further suggestion that there is an inherent link between physiological stress response and metabolism reprogramming during floral transition. We also observed significant changes in 24 nucleotide (nt) sRNAs that are hallmarks for RNA-dependent DNA methylation (RdDM) pathway, suggestive of the correlation between epigenetic modifications and the floral transition. The study not only provides new insight into our understanding of fundamental mechanism of poorly studied floral transition in apple and other woody plants, but also presents important sRNA resource for future in-depth research in the apple flowering physiology.
Introduction
Apple (Malus domestica Borkh.) is a major deciduous fruit tree crop in the world. Similar to other woody plants, apple tree has a long juvenile phase (Zhang et al., 2007). After the phase transition to the adult stage, apple trees undergo annual reproductive growth cycle. During the annual flower induction period, the developmental transition from vegetative to reproductive growth takes place in the floral bud (Mimida et al., 2009). In apple trees, flower induction occurs in the preceding summer of spring bloom (Abbott, 1970). This time window is typically in July the Northern Hemisphere (Kurokura et al., 2013), and approximately 2 weeks prior to floral differentiation, depending on species and the geographic and climate conditions (Kotoda et al., 1999; Kotoda and Wada, 2005). As flower induction determines fruit development and fruit productivity in the next year to a large extent (Williamson and Darnell, 1999; Link, 2000), elucidating the mechanism regulating floral transition is critical for both apple breeding and cultivation (Foster et al., 2003; Bangerth, 2009).
Many internal and external factors regulate floral transition. In annual model plant Arabidopsis, numerous interwoven genetic pathways exemplified by vernalization, photoperiod, senescence, and phytohromone signaling could regulate flowering timing (Moon et al., 2005). On the molecular level, the above pathways can end at promotion of the flowering process by repression of flowering repressor FLC (FLOWERING LOCUS C) and TFL1 (TERMINAL FLOWER 1), or by up-regulation of flowering activator FT (FLOWERING LOCUS T), SOC1 (SUPPERSSOR OF OVER EXPRESSION OF CONSTANS I), CO (COSTANS), AP1 (APETALA1), or LFY (LEAFY) (Teotia and Tang, 2015). Unlike annual plants that only flower one time during their life cycle, perennials live for many years and flower repeatedly (Albani et al., 2012). Moreover, for apple trees, the whole flowering process from floral bud initiation to blooming lasts for as long as 1 year. Thus, understanding the distinct mechanisms underline the floral transition in apple and other perennials is of high importance. Although some flowering-related genes have been cloned and analyzed in fruit crops, little is known about the sRNA's role in floral transition in apple and other fruit trees (Almada et al., 2009; Trankner et al., 2010; An et al., 2012; Lei et al., 2013; Porto et al., 2015; Wells et al., 2015; Ito et al., 2016).
miRNA plays pivotal roles in regulation of diverse biological processes including plant growth and development, flowering time, adaptation to the environment, and resistance to biotic and abiotic stress (Huijser and Schmid, 2011; Li et al., 2016). miRNAs are processed by Dicer-like machinery from primary miRNA precursors that contain a hairpin-like foldback, and then are incorporated into an Argonaute (AGO)-containing ribonucleoprotein complex to repress expression of target genes through cleavage of transcripts or translational repression in a sequence-specific manner (Zhang et al., 2015). In both annual model plant Arabidopsis and polycarpic perennial crops such as Cardamine flexuosa, miR156, and miR172 were first identified to regulate phase transition, the process that also represents the first time of floral transition during plant life circle. miR156 functions to extend juvenile phase and delay flowering, while miR172 leads to early flowering (Wu et al., 2009; Zhou C. M. et al., 2013). miR156 and miR172 could also regulate flowering time in response to vernalization through the opposite expression trends (Bergonzi et al., 2013). In Arabidopsis, miR156 targets a gene family of 11 SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) transcription factors; whereas miR172 regulates six members of the APETALA2 (AP2) transcription factors.
In apple, dozens of novel miRNAs or apple-specific miRNAs have been proposed through high-throughput sequencing (Xia et al., 2012; Ye et al., 2013; Xing et al., 2016). Meanwhile, more than 200 potential miRNA targets have been computationally predicted (Ye et al., 2013). Ectopic expression of apple miR156 h reduces expression levels of AtSPL9 and AtSPL15, and delays the flowering time in transgenic Arabidopsis (Sun et al., 2013). On the other hand, constitutive expression of apple miR172 leads to earlier flowering in transgenic Arabidopsis (Zhao et al., 2015). Notably, numerous miRNAs related to the abscisic acid (ABA) and gibberellins (GA) pathways, flowering gene expression and floral bud formation have been reported to respond to shoot bending that promotes apple flower induction (Xing et al., 2016). To date, the relevance of miRNAs to developmental transition from vegetative to floral buds remains poorly described in apple trees.
Different from miRNAs, another group of sRNAs, namely, 24 nucleotide (nt) small interfering RNA (siRNA), function in nucleus to repress target loci through epigenetic silencing. Briefly, in model plant Arabidopsis, siRNAs are derived from endogenous loci and repetitive sequences, and are loaded into AGO4 and/or its genetic paralogs like AGO3/6/9 to target sequence-complementary loci to eventually trigger chromatin methylation through a group of DNA and histone methyltransfereases (Borges and Martienssen, 2015; Du et al., 2015). However, correlation between the 24-nt siRNAs and floral transition in plants remains under defined.
In this study, we aimed to investigate the potential connection of miRNAs and siRNAs in apple vegetative-to-floral bud transition. We identified differentially expressed miRNAs and siRNAs between vegetative and floral buds through small RNA sequencing data analysis. Bioinformatics analysis of the sRNAs sheds new light on our understanding of floral transition in woody plants, and provide a new idea to design strategies to accelerate apple-breeding process.
Materials and methods
Plant material and growth condition
Sixteen-year-old Malus domestica “Golden Delicious”/M.26 trees grown at an experimental orchard of Cornell University, New York, USA (40° 43′ N, 74° 0′ W) were used in this study. Lateral buds from extension shoots and terminal buds from non-fruiting spurs were taken during flower induction process in late July of 2015, and designated as vegetative bud (VB) and floral buds (FB), respectively. There were three biological replicates per bud type. For each replicate, the VB and the FB were collected from the same tree. Each sample contained 20 buds. The collected samples were immediately frozen in liquid nitrogen and stored at −80°C.
sRNA extraction and library construction
For each sample, 100 mg of the ground powder was used for sRNA extraction as previously described (Wang C. et al., 2011). Six micro-grams (μg) of total RNA were spiked with 5′ 32P-labeled radioisotope-labeled 19–24 nt RNA oligos and then resolved in 15% urea-PAGE. sRNA library was constructed as previously described (Zhang et al., 2016). Briefly, sRNAs of 19–24 nt were gel-purified and ligated to a pair of adapters at the 5′ and 3′ ends by using T4 RNA ligase. sRNAs with adapters were transcribed into cDNA and amplified by Illumina sequencing-compatible primers. The final PCR products were sequenced at Texas A&M sequencing center. The sequences of 3′ and 5′ adapters, primers for amplification of the cDNA libraries are listed in Table S1.
RNA blot analysis
sRNA blot assays were performed with aliquots of total RNAs used for sRNA-seq according to Zhang et al. (2016). Each lane contained 6 μg of total RNA. Blots were hybridized with 32P-radiolabeled oligo nucleotide probes complementary to related sRNAs. U6 served as loading controls. sRNA blots were detected after exposure to a phosphor plate and quantified using the Quantity One Version 4.6.9 according to the manufacturer's instructions. Probes for RNA blot were listed in Table S1.
Bioinformatic analysis
The sRNA reads from HiSeq 2000 were cleaned by passing the QC with the standard Illumina software first. After trimming adapters, sRNAs with lengths between 19- and 28-nt were selected and mapped using bowtie (version 1.1.2) to the Malus genomic sequences (Malus domestica Whole Genome v1.0 from http://www.rosaceae.org) with perfect genomic matches. The genomic and features of sRNAs were defined by the same version of genome annotation files. The previously annotated miRNAs were mapped to the reference genome M. domestica in miRase (release 21.0). sRNAs reads mapped to selected genomic features or miRNAs were count by BED Tools (v2.26) with 1 bp overlapping. All the loci with at least 1 readcount were retained, then edgeR (v3.3) were used for the differential expression (DE) analysis by normalization to total reads for each sample (Robinson et al., 2010). miRNAs with false discovery ration (FDR) < 0.05 were defined as DE-miRNAs.
Novel miRNA were first predicted by miRPlant V5 with cut off score over 0. Their secondary structures were predicted by PsRobot with the criteria of having large stem loop; mismatches in small RNA region between 0 and 7, and maximal precursor length of 200 nt (Wu et al., 2012; An et al., 2014). Then these candidates were examined individually according to the criteria from Meyers et al. (2008): (1) The miRNA and miRNA* are derived from opposite stem-arms such that they form a duplex with two nucleotide, 3′-end overhangs; (2) base-pairing between the miRNA and the other arm of the hairpin, which includes the miRNA*, is extensive such that there are typically four or fewer mismatched miRNA bases; (3) asymmetric bulges are minimal in size (one or two bases) and frequency (typically one or less), especially within the miRNA/miRNA* duplex. Only miRNAs that meet at least two criteria and showed consistent hairpin shaped secondary structures predicted by PsRobot (Wu et al., 2012), mfold (http://mfold.rna.albany.edu/?q= mfold), and miRPlant V5 were selected as final candidates.
miRNA targets were also predicted by PsRobot (Wu et al., 2012). Our criteria included: (1) Penalty score threshold = 2 (the penalty score of each candidate alignment is obtained by subtracting the actual alignment score from the ideal perfect global pairing score); (2) Maximal number of permitted mismatch = 1; (3) Position of mature miRNA sequence after which mismatch permitted is 17th. Gene ontology (GO) analysis (http://bioinfo.cau.edu.cn/agriGO/) was performed to classify the predicted target genes.
Results
Cloning of sRNAs from vegetative and floral bud in apple trees
To identify sRNAs potentially involved in apple flower induction, libraries of sRNAs prepared from vegetative and floral bud from M. domestic “Golden delicious” were constructed following the steps shown in Figure 1A. Two repeats for the vegetative bud (VB1 and VB2) and three repeats for the floral bud (FB1, FB2, and FB3) were finally obtained respectively. 5.2–9.1 million of reads were generated from each sample. Approximately 87–90% of the reads were perfectly mapped to the apple genome and included in further analysis (Table 1). In lines with previous reports, the reads of sRNAs were dominated by 21-nt and 24-nt long species, with the population of 24-nt sRNAs much larger than the 21-nt ones for both vegetative and floral bud (Figures 1B,C, Tables S2, S3). Related data have been deposited in Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE97777).
Figure 1
Table 1
| Sample | Raw reads | Mapped reads | Mapped percentage | Unmapped reads | Unmapped percentage |
|---|---|---|---|---|---|
| VB1 | 6018158 | 5339332 | 88.72 | 678826 | 11.28 |
| VB2 | 5268927 | 4718634 | 89.56 | 550293 | 10.44 |
| FB1 | 9078685 | 7910981 | 87.14 | 1167704 | 12.86 |
| FB2 | 7470033 | 6518364 | 87.26 | 951669 | 12.74 |
| FB3 | 7565678 | 6599438 | 87.23 | 966240 | 12.77 |
Statistics of raw and clean reads of small RNAs isolated from Malus domestica.
We then studied genomic features of the sRNAs. A substantial number of sRNAs (approximately 39–56%), were derived from repeat elements for all samples. Other species of RNAs detected included miRNAs (9.6–11.2%), small nucleolar RNA (snoRNAs) (0.1–0.2%), and small nuclear RNA (snRNAs) (0.2–0.3%). Notably, additional and in a large number, sRNAs were mapped to unknown regions for both the vegetative and floral buds (Figure 1D).
Differentially expressed miRNAs in floral transition of apple trees
The sRNAs in vegetative and floral bud libraries were queried using the known mature miRNAs of M. domestica in the miRBase 21.0 (http://www.mirbase.org/) database. The expression levels of these miRNAs were determined by normalizing their reads to the snoRNA reads, followed by comparison of their ratios in floral and vegetative bud (Log2FC-value). As such, 170 conserved miRNAs which belong to 34 families were finally identified (Table S4). Among these families, major malus miRNAs, including miRNA156 (9 members), miRNA171 (14), miRNA172 (14), miRNA167 (10), and miRNA395 (9), were detected (Figure 2A).
Figure 2
A number of 33 differentially expressed miRNAs (DE-miRNAs) (FDR < 0.05) between vegetative and floral bud were identified (Figure 2B). The expression of some DE-miRNAs was validated by RNA blot analysis (Figure 2C). Approximate 91% of the 33 DE-miRNAs showed enhanced expression in floral bud, whereas only levels of mdm-miR398b/c, mdm-miR408a, and mdm-miR159c enriched in vegetative bud. The flowering regulators miR156 expressed stronger in the vegetative buds, while miR172 was abundant in the floral buds (Table S4). Notably, mdm-miR398b/c, instead of miR156 or miR172, represents the most significant difference among all the DE-miRNAs (Figure 2B, Table S4).
miRNAs in floral transition of apple trees
Next we predicted the potential targets for DE-miRNAs and the potential gene networks by using PsRobot (Wu et al., 2012; http://omicslab.genetics.ac.cn) as described in Methods. Under those scenarios, bioinformatics analysis revealed numerous targets for all miRNAs (Table 2). Notably, GO ontology analysis revealed that the potential targets of DE-miRNAs were mainly related to SPL gene regulation, stress response, and hormone pathways, providing three important clues to underline the mechanism of floral transition (Table 2, Table S5).
Table 2
| Name | Target protein | Protein ID | GO terms | GO ID | Function in plants |
|---|---|---|---|---|---|
| FLORAL TRANSITION AND PHASE TRANSITION RELATED | |||||
| miR156 | Squamosa promoter-binding protein-like (SPL; Sun et al., 2013)** | MDP0000155354 MDP0000158607 MDP0000146640 MDP0000297978 MDP0000263766 | DNA binding | GO:0003677 | Vegetative-to-floral phase transition Stress response Anthocyanin pathway |
| miR172 | AP2 (Zhao et al., 2015)** | MDP0000281079 MDP0000130802 MDP0000137561 MDP0000204900 MDP0000163645 MDP0000200319 | Transcription factor | GO:0003700 | Vegetative-to-floral phase transition Stress response |
| AUXIN PATHWAY | |||||
| miR393 | RNA polymerase III subunit RPC82 family protein* | MDP0000249678 | DNA-directed RNA polymerase | GO:0003899 | Auxin pathway |
| F-box/RNI-like superfamily protein (TIR1)* | MDP0000498419 | DNA binding | GO:0003677 | ||
| DNA-directed RNA polymerase | MDP0000313025 | Protein binding | GO:0005515 | ||
| miR167 | Auxin response factor 6 (ARF6)* | MDP0000550049 | Protein dimerization activity DNA binding | GO:0046983 GO:0003677 | Auxin pathway Light inducible |
| miR390 | Auxin response factor (At: ARF2/3/4) | MDP0000568581 MDP0000475144 MDP0000284274 MDP0000284274 MDP0000379904 | Protein tyrosine kinase activity Protein binding | GO:0004713 GO:0005515 | Auxin pathway |
| miR164 | NAM/ATAF/CUC (NAC) domain containing protein1 (NAC1)* | MDP0000911724 MDP0000121265 | DNA binding | GO:0003677 | Multiple development stage Auxin pathway |
| STRESS RESPONSE | |||||
| miR398 | DC1 domain-containing protein* | MDP0000152817 | Protein-disulfide reductase Nucleotide binding | GO:0047134 GO:0000166 | Oxidative stress Nutritional stress |
| GroES-like zinc-binding dehydrogenase family protein* | MDP0000193167 | ||||
| Ctr copper transporter family* | MDP0000530255 | Oxidoreductase activity | GO:0016491 | Photosynthesis | |
| miR408 | Plantacyanin (ARPN)* | MDP0000124552 MDP0000150953 | Copper ion binding | GO:0005507 | Oxidative stress Nutritional stress Anthocyanin pathway |
| miR399 | Inorganic phosphate transmembrane transporter [At: PHOSPHATE2 (PHO2)] | MDP0000166425 | Transmembrane transport | GO:0055085 | Nutritional stress |
| miR482 | Plant invertase/pectinmethylesterase inhibitor superfamily* | MDP0000296741 | Cell wall Pectinesterase | GO:0005618 GO:0030599 | Defense response Conserved in trees |
| Protein serine/threonine kinase | MDP0000305229 | Protein binding | GO:0005515 | ||
| miR2118 | Unknown (At: TIP-NBS-LRR) | MDP0000184039 | Protein serine/threonine kinase | Stress response phasiRNA biogenesis | |
| miR159 | Transcription factor | MDP0000233948 MDP0000574158 | Transcription factor Nucleus | GO:0003700 GO:0005634 | Pollen tube growth Abiotic stress Defining plant morphology |
| MYB domain protein 65 (MYB65)* | MDP0000321057 | ||||
| miR1511 | Ca2+ binding | MDP0000283945 | Peptidyl-prolyl cis-trans isomerase activity | GO:0003755 | Abiotic tress response |
| Isomerase | MDP0000293965 | ||||
| miR319 | TEOSINTEBRANCHED1/CYCLOIDEA/ PRO-LIFERATING CELL FACTOR transcription factor4 (EE35,TCP4)* | MDP0000916623 | Regulation of development | GO:0045962 | Cell proliferation Abiotic stress response |
| miR171 | DNA binding [At: Scarecrow-like proteins (SCL)] | MDP0000288614 | DNA binding | GO:0006298 GO:0005524 GO:0030983 | Plant development Light inducible |
| miR396 | Transcription regulator [At; Growth-regulating factor (GRF)] | MDP0000194223 | Transcription activator activity | GO:0016563 | Cell proliferation Secondary metabolism |
| TREE SPECIFIC | |||||
| miR3627 | Integral to membrane cellular component | MDP0000941000 | Unknown | Unanootated | Conserved in trees |
| AT hook motif DNA-bindingprotein* | MDP0000237744 | ||||
| miR477 | Unknown | MDP0000232264 | Unknown | Unannotated | Conserved in trees |
| Transcription factor | MDP0000431628 | ||||
| miR7124 | WD40/YVTN repeat-like domain | MDP0000162030 MDP0000296050 | Transmembrane transport | GO:0055085 | Only found in apple |
| P-loop containing nucleosidetriphosphate hydrolases superfamily protein* | MDP0000319328 | Microtubule-based movement | GO:0007018 | ||
| OTHERS | |||||
| miR162 | Leucine zipper EF-hand containing transmembrane protein 1 (LETM1-LIKE protein)* (At:DCL1) | MDP0000187512 | Developmental growth | GO:0048589 | miRNA processing |
| miR5225 | RNA-directed RNA polymerase | MDP0000303619 | RNA-directed RNA polymerase oxidation reduction | GO:0003968 GO:0055114 | Unknown |
| miR535 | Transferase activity (At: SPLs) | MDP0000177623 | Biosynthetic process | GO:0009058 | Plant development |
| MDP0000173587 | Protein binding | GO:0005515 | |||
GO analysis of potential targets of differentially expressed known miRNAs during floral transition of Malus domestica.
Several DE-miRNAs were shown to correlate with SPL genes, such as the vegetative bud-enriched miR156, miR159, miR398, and miR408. They were all reported to either target SPLs or involve in the SPL regulation (Wu et al., 2009; Yamasaki et al., 2009; Zhang et al., 2014). Besides, miR159 involves in GA pathway during flower development (Reyes and Chua, 2007).
Many DE-miRNAs in our results were reported to be stress responsive, such as the floral bud-enriched ones including miR159, miR171, miR319, miR396, miR399, miR482, miR1511, and miR2118 (Table 2). They were reported to involve in various abiotic (e.g., copper, drought, or ABA) and biotic stress resistance (e.g., fungal pathogen; Abdel-Ghany and Pilon, 2008; Jia et al., 2009; Pantaleo et al., 2010; Zhai et al., 2011; Zhou M. et al., 2013; Ma Z. et al., 2014).
Auxin is a phytohromone that play a critical role in plant growth and development including the flowering process. Of the DE-miRNAs detected, miR164 targets NAC (NAM, ATAF, CUC) genes, miR167 targets AUXIN RESPONSIVE FACTOR6/8 (ARF6/8), miR390 targets trans-acting small interfering RNA3 (TAS3) transcripts to produce ta-siRNAs, which in turn regulates plant development by repressing ARF2/3/4, and miR393 targets TIR1 genes. All the above miRNAs were up-regulated in floral bud, indicating the potential changes on auxin response during the floral transition.
In addition, some of the DE-miRNAs have been reported to regulate floral development in some other plants. For instance, miR396, which was expressed higher in floral bud, was reported to target GROWTH REGULATING FACTORS (GRFs) and to regulate cell proliferation and function in floral organ specification in Populus (Yang C. Y. et al., 2015). Last but not least, some DE-miRNAs, exemplified by miR1522, have been also recovered but their functions appear to be elusive at this stage. Some others such as miR3627, miR477, and miR7124 were specific to trees or apple according to miRBase 21.0 but their roles remain to be explored yet.
Re-annotation of recently reported new miRNAs from apple tree
Recent effort of sRNA-seq in the different materials from apple has recovered 349 novel miRNAs (Ma C. et al., 2014; Xing et al., 2014; Kaja et al., 2015). We mined the public database and compared them with ours. Among them, 172 novel miRNAs were predicted to involve in phase transition in apple trees (Xing et al., 2014). To our surprise, only 17 novel miRNAs could be detected in our materials (Table S6). Among them, 10 showed opposite patterns whereas the rest displayed consistent changes between our results and previous data (Figure S1A). Based on FDR-values, only four novel miRNAs showed significant changes in our data (Table S6). Specifically, three novel miRNAs displayed stronger expression in floral bud (which are designated as Xing-novel-mir334/276/4 here), whereas one novel miRNA had stronger expression in vegetative bud (designated as Xing-novel-mir262).
By sharp contrast, we were unable to detect the remaining tentative novel miRNAs reported (Xing et al., 2014). These sRNAs either had no hits in our database or numbers of the reads were extremely low, thus we considered these sRNAs might be siRNAs, rather bona fide miRNAs. Similarly, we failed to detect all novel miRNAs reported by Kaja et al. (2015) and Ma C. et al. (2014) in our materials. As we could not access the sequence, structure information, and reads mapping of precursors for the tentative miRNAs, we could not evaluate the viability of the reported novel miRNA candidates.
Then we did target prediction and GO analysis of the 17 novel miRNAs overlapped between our sRNA-seq and the one reported by Xing et al. (2014). For the novel miRNAs with a consistent pattern between our studies, their targets were mainly related to the cell component, catalytic, metabolic process (Figure S1B). While for the opposite part, the targets were related to stimulus response, signaling process and biological regulation, which might represents some of the differences between buds and leaves (Figure S1C, Table S7).
Six differentially expressed novel miRNAs in floral transition of apple trees
We next aimed at identifying novel miRNAs that are potentially engaged in floral transition in apple tree. To this end, we mined the remaining unannotated reads that could be mapped to the M. domestica genomic exon antisense strand, intron and intergenic regions and conducted bioinformatics predict for potential novel miRNAs. Based on the standards in Methods, our initial screening recovered 425 candidates (Figure S2).
Next, we pursued more stringent criteria according to Meyers et al. (2008) as described in Methods. Only candidates having at least two characteristics of the above could be annotated as miRNAs. Under these filters, we narrow down the candidate lists and finally recovered 6 novel miRNA candidates with high confidence. According to mfold, we included 20–25 nt upstream or downstream of miRNA/miRNA* sequence and predicted pri-miRNA secondary structures. We also mapped all sRNAs that are derived from the pri-miRNAs and found that the distribution patterns of the novel miRNA/miRNA*s were characteristic of the previously established ones in model plants (Figure 3A). Thus, these newly identified sRNAs are most likely bona fide miRNAs in the apple tree.
Figure 3
Among these 6 novel miRNAs, novel mdm-miR3279, and mdm-miR3803 were proved to be the same one as their sequences are identical. As such, we re-annotated them mdm-miR3279a and miR3279b. Only novel mdm-miR3279a/b, miR2819, miR940, and miR4672 were significantly different expressed miRNAs according to FDR < 0.05 (Figure 3B, Table S6). Phylogeny tree analysis showed the floral bud-enriched novel mdm-miR3279a/b/2819 are close to miR319c and miR482d, which were also expressed higher in floral bud (Figures 2A, 3C). On the other hand, the vegetative bud-enriched novel mdm-miR940/4672 showed high similarity with floral bud-enriched miR1511 and miR319c.
We then performed target prediction by PsRobot (Wu et al., 2012). As shown in Table 3, miR3279a/b target WD40 and MYB domain transcription factors that are correlated with anthocyanin biosynthesis and metabolic process; miR2819 may also target a transcription factor that is involved in metabolic process; miR2398 and miR940 repress the transcripts encoding factors that are engaged in protein-protein interaction; whereas miR4672 and miR331 repress genes that encode zinc binding protein.
Table 3
| Name | Target protein | Protein ID | GO terms | GO ID | Pairing with target sequence (Upper line indicates miRNA, lower line indicates target) |
|---|---|---|---|---|---|
| novel mdm-miR2398 | Protein binding | MDP0000306873 | Protein binding | GO:0005515 |  |
| Calcium-binding protein | MDP0000191848 | GO:0008219 GO:0016021 | |||
| novel mdm-miR2819 | Initiate or regulate RNA polymerase II | MDP0000288574 | CaFBohydrate metabolic process | GO:0005975 | 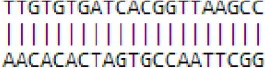 |
| Hydrolase | MDP0000447149 | Polygalacturonase | GO:0004650 | ||
| Transcription factor | MDP0000280817 | Binding | GO:0005488 | ||
| novel mdm-miR3279/3803 | Nucleus cellular componentwith WD40 domain | MDP0000251730 | Protein binding | GO:0005515 | 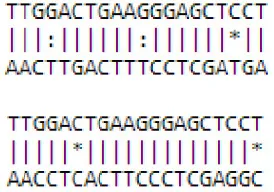 |
| Nucleus cellular componentwith MYB domain | MDP0000147309 | Catalytic activity Metabolic processs DNA binding | GO:0003824 GO:0005515 GO:0008152 GO:0003677 | ||
| novel mdm-miR331 | Zinc ion binding | MDP0000286463 | ATP-dependent peptidase activity Proteolysis Zinc ion binding | GO:0004176 GO:0006508 GO:0008270 | 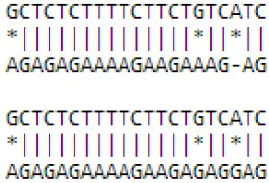 |
| Iron ion binding | MDP0000308925 | Protein binding | GO:0005515 | ||
| novel mdm-miR4672 | Iron ion binding | MDP0000161181 | Iron ion binding Oxidoreductase activity | GO:0005506 GO:0016491 |  |
| novel mdm-miR940 | Protein binding | MDP0000281236 | Protein domain specific binding | GO:0019904 | 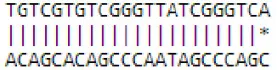 |
GO analysis of potential targets of novel miRNAs during floral transition from Malus domestica.
24-nt siRNAs in floral transition of apple trees
Increasing evidence has shown that chromatin methylation is closely related to flowering regulation in plants. siRNAs of 24 nt length are signatures of the RdDM pathway and play critical role in epigenetic silencing in plants (Matzke et al., 2015).
We investigated the 24-nt sRNA population in vegetative and floral bud. 261, 702 differentially expressed (FDR < 0.05) 24-nt siRNAs were found; and among them, 2,469 were predominant in vegetative bud and 259, 233 were predominant in floral bud, which count for 9.4 and 90.6%, respectively (Figure 4A). This result suggests that DNA methylation status might be altered during the phase change in apple.
Figure 4
Among the DE-24nt-siRNAs, more than 60% were distributed over transposable element (TE) regions (Figure 4B), suggesting that a substantial amount of TEs are de-regulated in the floral buds compared to the vegetative buds. Then the top 40 differentially expressed 24 nt siRNAs (DE-siRNAs) screened by FDR < 0.05 were selected for further analysis (Table 4, Table S8).
Table 4
| Repeat | Chromosome | Start | End | TE element | Target protein ID | Target |
|---|---|---|---|---|---|---|
| INTRAGENIC REGION (INTRON) | ||||||
| repeat_1085094 | Unanchored | 51760264 | 51760412 | LTR trim terminal | MDP0000664744 | Heat shock protein HSP20 |
| repeat_1117604 | Chr2 | 10581879 | 10582001 | Unclassified | MDP0000314081 | Rhodanese-like protein |
| repeat_1028150 | Chr3 | 32067196 | 32067416 | LTR copia termial | MDP0000247441 | Pectinesterase; DNA binding |
| repeat_1265460 | Chr8 | 4276443 | 4276475 | Unclassified tandem | MDP0000490697 | Phosphatase |
| repeat_1265461 | Chr8 | 4291702 | 4291734 | Unclassified tandem | MDP0000488182 | Unknown |
| repeat_853832 | Chr9 | 30625201 | 30625309 | TIR spring inverted | MDP0000438624 | Unknown |
| MDP0000266324 | WD40-like protein | |||||
| repeat_853530 | Chr14 | 27263462 | 27263596 | TIR spring inverted | MDP0000241035 | Zinc ion binding |
| MDP0000312371 | ATP binding; nucleic binding | |||||
| INTRAGENIC REGION (EXTRON) | ||||||
| repeat_1270144 | Chr2 | 4978879 | 4979011 | Unclassified tandem | MDP0000197534 | DNA binding |
| repeat_1039882 | Chr4 | 22346821 | 22346934 | Unclassified | MDP0000692845 | Peptidase |
| repeat_1289450 | Chr7 | 14280602 | 14280669 | Ribosomal unclassified | MDP0000268430 | Nucleus protein |
| repeat_1265459 | Chr8 | 4285173 | 4285205 | Unclassified tandem | MDP0000265846 | Zinc ion binding |
| repeat_1267023 | Chr9 | 30630665 | 30630773 | Unclassified tandem | MDP0000321586 | Isoprenoid synthase |
| repeat_1039981 | Chr11 | 908937 | 909060 | Unclassified | MDP0000849784 | Unknown |
| repeat_1117835 | Chr15 | 20360355 | 20360459 | Unclassified | MDP0000304869 | Unknown |
| repeat_1267817 | Chr17 | 311713 | 311849 | Unclassified tandem | MDP0000083486 | ATase |
| repeat_853499 | Chr17 | 5016279 | 5016409 | TIR spring inverted | MDP0000286141 | Glucosyltransferase |
| INTERGENIC REGION | ||||||
| repeat_1269060 | Unanchored | 6189646 | 6189801 | Unclassified tandem | MDP0000135966 | Unknown |
| MDP0000734661 | Leucine-rich repeat | |||||
| repeat_1270145 | Chr2 | 4978251 | 4978383 | Unclassified tandem | MDP0000197534 | DNA binding |
| repeat_1039411 | Chr4 | 1228773 | 1228888 | Unclassified | MDP0000124365 | Unknown |
| repeat_1269362 | Chr4 | 1278056 | 1278216 | Unclassified tandem | MDP0000872787 | Unknown |
| repeat_1265373 | Chr5 | 10388169 | 10388199 | Unclassified tandem | MDP0000319295 | protein binding |
| repeat_1267275 | Chr7 | 1274745 | 1274848 | Unclassified tandem | MDP0000183959 | Galactose-like |
| repeat_1260363 | Chr8 | 365188 | 365314 | Unclassified | MDP0000376261 | Unknown |
| repeat_1040200 | Chr9 | 12254661 | 12254789 | Unclassified | MDP0000311464 | Oxidoreducta |
| repeat_873607 | Chr10 | 24553191 | 24553235 | LTR trim terminal | MDP0000289531 | Calcium ion binding |
| repeat_1119810 | Chr12 | 1189324 | 1189408 | Unclassified | MDP0000257983 | Unknown |
| repeat_1062664 | Chr14 | 16793739 | 16794145 | LTR gypsy terminal | MDP0000286350 | Endopeptidase |
| repeat_1040003 | Chr15 | 11077070 | 11077182 | Unclassified | MDP0000197911 | Protein kinase |
| repeat_1039784 | Unanchored | 9053589 | 9053706 | Unclassified | MDP0000228072 | Unknown |
| repeat_1265811 | Unanchored | 92494467 | 92494514 | Unclassified tandem | MDP00001634 | Actin-binding FH2 |
| repeat_1265458 | Unanchored | 102659034 | 102659066 | Unclassified tandem | MDP0000156554 | Iron ion binding |
| repeat_1266082 | Chr3 | 8503155 | 8503210 | Unclassified tandem | MDP0000165108 | Ribosome |
| MDP0000152862 | Unknown | |||||
| repeat_853972 | Chr3 | 8672342 | 8672447 | TIR spring inverted | MDP0000252384 | Unknown |
| repeat_854466 | Chr8 | 4291702 | 4291734 | Unclassified tandem | MDP0000209821 | Iron ion binding |
| repeat_1270178 | Chr10 | 12548160 | 12548315 | Unclassified tandem | MDP0000649828 | Calcium ion binding |
| repeat_855585 | Chr12 | 29732429 | 29732584 | TIR spring inverted | MDP0000214500 | Diacylglycerol acyltransferase |
| repeat_1265663 | Chr15 | 25979552 | 25979599 | Unclassified tandem | MDP0000228862 | Actin-binding FH2 |
| repeat_1039352 | Chr15 | 28620140 | 28620275 | Unclassified tandem | MDP0000088811 | Protein binding |
| repeat_856374 | Chr16 | 7712559 | 7712712 | TIR spring inverted | MDP0000307719 | Glutamate_decaFBoxylase |
| repeat_854465 | Chr17 | 311713 | 311849 | Unclassified tandem | MDP0000203334 | Protein binding |
Potential targets and TE elements of top 40 differentially expressed siRNAs during the floral transition of Malus domestica.
IGV analysis showed that except five siRNAs unanchored, the distribution of other top 40 DE-siRNAs almost covers all the 17 chromosomes of apple genome (Table 4). Among them, 28 DE-siRNAs were located at the intergenic regions and 17 DE-siRNAs located at the intragenic regions. As examples, repeat 1028150 was distributed at intron of MDP0000247441, and repeat 1062664 was distributed at intergenic region of MDP0000286350 (Figure 4C). These results suggested that siRNAs might regulate the expression of the loci flanking the siRNA producing regions during the developmental transition. GO analysis of potential targeted loci revealed that the genes affected by the top 40 DE-siRNAs mainly encode ion binding proteins, enzymes related to metabolic, catalytic, and cell processes, electron carrier and cellular parts (Figure 4D, Table 4, Table S9). These results indicated that these 24 nt-siRNAs might be involved in apple floral transition by targeting TEs that are distributed around the genes related to cellular signal transduction, cell growth, and metabolic process.
Discussion
Correlation of miRNA-mediated floral transition to SPL gene regulation, stress response, and auxin and GA pathways
In this study, we identified numerous known miRNAs that show differential expression patterns in floral transition during flower induction in apple trees. The numbers of DE miRNA members were comparable with the ones reported with peach vegetative buds, pear floral buds, apple floral bud (“Fuji”), and apple leaves (Barakat et al., 2012; Wu et al., 2014; Xing et al., 2014, 2016). These results suggest the conserved distribution of miRNAs among species.
In DE-miRNAs analysis, only miR398, miR408, miR159, and miR156 expressed stronger in vegetative bud. Among them, miR398, miR408, and miR156 are all related with the repression of SPL genes, a key factor in juvenile growth and a positive regulator of flowering process (Wu et al., 2009; Zhou C. M. et al., 2013). miR156 has been widely reported to targeted SPLs. Moreover, in Citrus trees, fewer fruit load in an OFF year (light yield) increases floral bud numbers and the expression of SPLs in floral bud during flower induction the following year, demonstrating the positive role of SPLs in flowering (Shalom et al., 2012). miR398 represented the miRNA with the most significant changes among all the DE-miRNAs. As reported, SPL7 could activate miR398 expression by binding to its core elements in the promoter (Yamasaki et al., 2009). This result is also consistent with the finding of a previous study where the expression of miR398 was significantly decreased in the floral buds in response to shoot bending, a technique used to promote flowering in apple (Xing et al., 2016). miR408 functions in copper homeostasis regulation together with miR398 in Arabidopsis and Populus (Lu et al., 2005; Abdel-Ghany and Pilon, 2008), and it also involves in the HY5-SPL7 network that mediates the coordinated response to light and copper (Yamasaki et al., 2009; Zhang et al., 2014). Besides, miR159 is regulated by GA pathway during flower development (Reyes and Chua, 2007). High expression of miR159 could reduce LFY activity and results in delayed flowering (Achard et al., 2004). Together, the above evidence indicate that in apple trees, miRNAs might control the vegetative growth through the regulation of SPLs and GA pathway (Figure 4E).
GO analysis showed floral bud-enriched DE-miRNAs mainly related with stress response and auxin signaling. The link between stress response and floral transition in plants has been demonstrated by previous studies. In Arabidopsis, constitutive expression of ABI5, which could positively response to ABA signaling, causes delay in flowering by up-regulating FLC expression (Wang et al., 2013). While drought stress at the beginning of the flowering results in early arrest of flowering by regulating genes including DREB, MYB, VEGETATIVEY, and CO1, etc. (Su et al., 2013). Here, the floral bud-enriched miR482 and miR2118 are involved in plant fungal pathogen resistance (Zhai et al., 2011); miR171 play roles in ABA and GA pathways during flower development (Ma Z. et al., 2014). miR319 participates in abiotic stress response and regulate cell proliferation (Zhou M. et al., 2013). miR1511 and miR2118 could also respond to drought stress (Pantaleo et al., 2010; Wu et al., 2015).
miR164, miR167, miR390, and miR393 were found to involve in auxin signaling. In Arabidopsis, miR167-targeted ARF6/8 are activators of auxin-responsive genes and could accelerate flowering timing (Nagpal et al., 2005); while the miR390-TAS3-repressed ARF2/3/4 are known to be repressors of auxin-responsive genes and flowering (Fahlgren et al., 2006). The miR393-targeted TIR1 is also known to repress flowering in Arabidopsis (Chen et al., 2011). Thus, since all these four miRNAs expressed at higher levels in the floral buds, it is unable to conclude the final auxin changes during apple floral transition. Since apple is a woody perennial plant, the above conflict might indicate a more complex network of auxin regulations during its floral transition.
Together, we propose that apple floral transition might cause physiological stress responses in floral bud, and as such, the stress responsive miRNAs increase. The miRNAs involved in auxin pathways might be further induced by the stress responsive miRNAs or directly induced by physiological changes, and consequently stimulating downstream cascade of physiological changes through the floral transition (Figure 4E).
Correlation of miRNA-mediated floral transition to phase transition
In perennial plants, there is one-time of phase transition (from juvenile to reproductive) and many times of floral transition (from vegetative to reproductive) in their life cycle. The phase transition also represents the first time of floral transition. In many previous studies in Arabidopsis and other plants, it has shown that the function of miR156 and miR172 in phase transition also include the repression/promotion of flowering process, respectively. In our results, miR156 was expressed at a higher level in the vegetative buds, suggesting that miR156 could also control vegetative growth. However, the difference of miR156 expression between the vegetative and the floral buds was not as significant as the ones during phase transition in apple and other woody plants (Wang J. W. et al., 2011; Xing et al., 2014). Thus, miR156 might differentially regulate juvenile phase and vegetative phase by its abundance: higher abundance might be important for juvenile phase, while lower abundance might be important for vegetative growth.
Then, we compared our data with previous sRNA sequencing data of apple phase transition. In addition to miR156 and miR172, numerous DE-miRNAs were also identified to involve in floral transition in apple trees. Among them, miR164, miR171, miR482, and miR5225 and several reported novel miRNAs enriched in floral bud and also expressed stronger in adult materials in the previous studies (Xia et al., 2012; Xing et al., 2014), indicating their conserved roles in apple flowering process.
Interestingly, the majority of DE-miRNAs identified in our study (miR162, miR167, miR390, miR393, miR396, miR398, miR408, miR535, miR1511, miR2118, miR3627, and miR7124) showed opposite expression patterns compared to the ones in the previous study on phase transition in apple trees (Xing et al., 2014). Moreover, many previously reported novel miRNAs also displayed opposite patterns in our study (Figure S1). Together, these results suggested that although there are some common mechanisms shared by phase transition and floral transition, many miRNAs might function in these two different processes by changing their abundance or showing opposite expression patterns. If so, how the opposite expression patterns of the miRNAs contribute to the regulation of phase transition or floral transition would be exciting topics in the apple developmental studies.
Correlation between floral transition and siRNA guided DNA methylation
Floral transition in perennial tree is an integrate response of many factors including both environmental stimuli and plant phsiological changes. Accumulating evidence indicates that epigenetic mechanisms, including DNA methylation, play essential roles in the whole process of flower development, including bud stage. DNA methylation could involve in bud dormancy in azalea and floral bud differentiation in Castanea sativa (Santamaria et al., 2009; Meijon et al., 2010). siRNAs (24 nt) involve in epigenetic silencing by guiding DNA methylation. We have identified numerous DE-24 nt-siRNAs during the developmental transition from vegetative to floral bud. More than 90% of the DE 24 nt-siRNAs showed stronger expression in a whole genome pattern in the floral buds, suggesting an increased level of DNA methylation during floral transition (Figure 4A, Table 4). Our results were reminiscent of the previous observations in other plants. In Arabidopsis, increased DNA methylation levels have been observed during the transition from floral meristem to early flower stage (Yang H. et al., 2015). In radiate pine, the basal portion of the growing needles (<1 cm) of vegetative trees displayed 42% less of DNA methylation level than the same portions of trees that finish floral transition (Fraga et al., 2002). In Castanea sativa, vegetative tree shoots have the lowest level of DNA methylation (13.7%), while mature-tree shoots have a DNA methylation level of 15.0% (Hasbun et al., 2005). Such results were also demonstrated in the vegetative shoots and flower shoots of the chestnut tree (Hasbun et al., 2007). Thus, it appears that the floral transition is positively correlated with DNA methylation. Together, all these results suggested the siRNA might function in floral transition by increasing DNA methylation level. The detailed regulation and coordination of siRNA-mediated RdDM during floral transition need to be further investigated in the near future.
In conclusion, in this study we found that the classic model of miR156-miR172 in flowering appears to apply to floral transition of apple trees. Moreover, many other miRNAs, annotated or newly discovered, also appear to play critical roles during this process and showed different regulation with the ones in apple phase transition in previous study. These miRNAs may function in floral transition mainly through SPL genes, the response to stress stimuli and GA and auxin signaling. Last but not least, 24 nt siRNA mediated RdDM appears to be engaged in apple floral transition. Taken together, this study provides useful information and resource for future study on the roles of small non-coding RNAs in the floral transition in apple trees or other perennial plants.
Statements
Author contributions
XG, LC, XZ, and TL designed research; XG and ZZ performed research; LC provided samples; ZM, ZZ, and XG performed bioinformatic analysis; XG, LC, and XZ wrote the paper.
Funding
This work was supported by NSF CAREER (MCB-1253369) to XZ.
Acknowledgments
XG was supported by Chinese Scholarship Council.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00873/full#supplementary-material
References
1
AbbottD. L. (1970). The role of budscales in the morphogenesis and dormancy of the apple fruit bud, in Physiology of Tree Crops, eds LuckwillL. C.CuttingC. V. (London: Academic Press), 65–80.
2
Abdel-GhanyS. E.PilonM. (2008). MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem.283, 15932–15945. 10.1074/jbc.M801406200
3
AchardP.HerrA.BaulcombeD. C.HarberdN. P. (2004). Modulation of floral development by a gibberellin-regulated microRNA. Development131, 3357–3365. 10.1242/dev.01206
4
AlbaniM. C.CastaingsL.WötzelS.MateosJ. L.WunderJ.WangR.et al. (2012). PEP1 of Arabis alpina is encoded by two overlapping genes that contribute to natural genetic variation in perennial flowering. PLoS Genet.8:e1003130. 10.1371/journal.pgen.1003130
5
AlmadaR.CabreraN.CasarettoJ. A.Ruiz-LaraS.VillanuevaE. G. (2009). VvCO and VvCOL1, two CONSTANS homologous genes, are regulated during flower induction and dormancy in grapevine buds. Plant Cell Rep.28, 1193–1203. 10.1007/s00299-009-0720-4
6
AnJ.LaiJ.SajjanharA.LehmanM. L.NelsonC. C. (2014). miRPlant: an integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform.15:275. 10.1186/1471-2105-15-275
7
AnL.LeiH.ShenX.LiT. (2012). Identification and characterization of PpLFL, a homolog of floricaula/leafy, in peach (Prunus persica). Plant Mol. Biol. Rep.30, 1488–1495. 10.1007/s11105-012-0459-x
8
BangerthK. F. (2009). Flower induction in mature, perennial angiosperm fruit trees: similarities and discrepancies with annual/biennial plants and the involvement of plant hormones. Sci. Hortic.122, 153–163. 10.1016/j.scienta.2009.06.014
9
BarakatA.SriramA.ParkJ.ZhebentyayevaT.MainD.AbbottA. (2012). Genome wide identification of chilling responsive microRNAs in Prunuspersica. BMC Genomics13:481. 10.1186/1471-2164-13-481
10
BergonziS.AlbaniM. C.van ThemaatE. V. L.NordströmK. J.WangR.SchneebergerK.et al. (2013). Mechanisms of age-dependent response to winter temperature in perennial flowering of Arabisalpina. Science340, 1094–1097. 10.1126/science.1234116
11
BorgesF.MartienssenR. (2015). The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol.16, 727–741. 10.1038/nrm4085
12
ChenZ. H.BaoM. L.SunY. Z.YangY. J.XuX. H.WangJ. H.et al. (2011). Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol.77, 619–629. 10.1007/s11103-011-9838-1
13
DuJ.JohnsonL.JacobsenS.PatelD. (2015). DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol.16, 519–532. 10.1038/nrm4043
14
FahlgrenN.MontgomeryT. A.HowellM. D.AllenE.DvorakS. K.AlexanderA. L.et al. (2006). Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr. Biol.16, 939–944. 10.1016/j.cub.2006.03.065
15
FosterT.JohnstonR.SeleznyovaA. (2003). A morphological and quantitative characterization of early floral development in apple (Malus domestica Borkh.). Ann. Bot.92, 199–206. 10.1093/aob/mcg120
16
FragaM.CanalM.RodriguezR. (2002). Phase-change related epigenetic and physiological changes in Pinus radiata D. Don. Planta215, 672–678. 10.1007/s00425-002-0795-4
17
HasbunR.ValledorL.BerdascoM.SantamariaE.CanalM. J.RodriguezR.et al. (2005). In vitro proliferation and genome DNA methylation in adult chestnuts. Acta Hortic.693:333. 10.17660/ActaHortic.2005.693.42
18
HasbunR.ValledorL.SantamariaE.CanalM. J.RodriguezR.BerdascoM. (2007). Dynamics of DNA methylation in chestnut trees development. Acta Hortic.760:563. 10.17660/ActaHortic.2007.760.80
19
HuijserP.SchmidM. (2011). The control of developmental phase transitions in plants. Development138, 4117–4129. 10.1242/dev.063511
20
ItoA.SaitoT.SakamotoD.SugiuraT.BaiS.MoriguchiT. (2016). Physiological differences between bud breaking and flowering after dormancy completion revealed by DAM and FT/TFL1 expression in Japanese pear (Pyrus pyrifolia). Tree Physiol.36, 109–120. 10.1093/treephys/tpv115
21
JiaX.WangW. X.RenL.ChenQ. J.MenduV.WillcutB.et al. (2009). Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol. Biol.71, 51–59. 10.1007/s11103-009-9508-8
22
KajaE.SzcześniakM. W.JensenP. J.AxtellM. J.McNellisT.MakałowskaI. (2015). Identification of apple miRNAs and their potential role in fire blight resistance. Tree Genet. Genomes11, 1–11. 10.1007/s11295-014-0812-3
23
KotodaN.WadaM. (2005). MdTFL1, a TFL1-like gene of apple, retards the transition from the vegetative to reproductive phase in transgenic Arabidopsis. Plant Sci.168, 95–104. 10.1016/j.plantsci.2004.07.024
24
KotodaN.WadaM.KomoriS.KidouS. I.AbeK.MasudaT.et al. (1999). Expression pattern of homologues of floral bud identity genes LFY and AP1 during flower development in apple. J. Am. Soc. Hortic. Sci.125, 398–403.
25
KurokuraT.MimidaN.BatteyN. H.HytönenT. (2013). The regulation of seasonal flowering in the Rosaceae. J. Exp. Bot.64, 4131–4141. 10.1093/jxb/ert233
26
LeiH. J.YuanH. Z.LiuY.GuoX. W.LiaoX.LiuL. L.et al. (2013). Identification and characterization of FaSOC1, a homolog of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 from strawberry. Gene531, 158–167. 10.1016/j.gene.2013.09.036
27
LiS.Castillo-GonzalezC.YuB.ZhangX. (2016). The functions of plant small RNAs in development and in stress responses. Plant J.90, 654–670. 10.1111/tpj.13444
28
LinkH. (2000). Significance of flower and fruit thinning on fruit quality. Plant Growth Reg.31, 17–26. 10.1023/A:1006334110068
29
LuS.SunY. H.ShiR.ClarkC.LiL.ChiangV. L. (2005). Novel and mechanical stress–responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell17, 2186–2203. 10.1105/tpc.105.033456
30
MaC.LuY.BaiS.ZhangW.DuanX.MengD.et al. (2014). Cloning and characterization of miRNAs and their targets, including a novel miRNA-targeted NBS–LRR protein class gene in apple (Golden Delicious). Mol. Plant7, 218–230. 10.1093/mp/sst101
31
MaZ.HuX.CaiW.HuangW.ZhouX.LuoQ.et al. (2014). Arabidopsis miR171-targeted scarecrow-like proteins bind to GT cis-elements and mediate gibberellin-regulated chlorophyll biosynthesis under light conditions. PLoS Genet.10:e1004519. 10.1371/journal.pgen.1004519
32
MatzkeM. A.KannoT.MatzkeA. J. (2015). RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol.66, 243–267. 10.1146/annurev-arplant-043014-114633
33
MeijonM.FeitoI.ValledorL.RodriguezR.CanalM. J. (2010). Dynamics of DNA methylation and Histone H4 acetylation during floral bud differentiation in azalea. BMC Plant Boil.10:10. 10.1186/1471-2229-10-10
34
MeyersB. C.AxtellM. J.BartelB.BartelD. P.BaulcombeD.BowmanJ. L.et al. (2008). Criteria for annotation of plant MicroRNAs. Plant Cell20, 3186–3190. 10.1105/tpc.108.064311
35
MimidaN.KotodaN.UedaT.IgarashiM.HatsuyamaY.IwanamiH.etal. (2009). Four TFL1/CEN-like genes on distinct linkage groups show different expression patterns to regulate vegetative and reproductive development in apple (Malus domestica Borkh.). Plant Cell Physiol.50, 394–412. 10.1093/pcp/pcp001
36
MoonJ.LeeH.KimM.LeeI. (2005). Analysis of flowering pathway integrators in Arabidopsis. Plant Cell Physiol. 46, 292–299. 10.1093/pcp/pci024
37
NagpalP.EllisC. M.WeberH.PloenseS. E.BarkawiL. S.GuilfoyleT. J.et al. (2005). Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development132, 4107–4118. 10.1242/dev.01955
38
PantaleoV.SzittyaG.MoxonS.MiozziL.MoultonV.DalmayT.et al. (2010). Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J.62, 960–976. 10.1111/j.0960-7412.2010.04208.x
39
PortoD. D.BruneauM.PeriniP.AnzanelloR.RenouJ. P.dos SantosH. P.et al. (2015). Transcription profiling of the chilling requirement for bud break in apples: a putative role for FLC-like genes. J. Exp. Bot.66, 2659–2672. 10.1093/jxb/erv061
40
ReyesJ. L.ChuaN. H. (2007). ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J.49, 592–606. 10.1111/j.1365-313X.2006.02980.x
41
RobinsonM. D.McCarthyD. J.SmythG. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics26, 139–140. 10.1093/bioinformatics/btp616
42
SantamariaM.HasbunR.ValeraM.MeijonM.ValledorL.RodríguezJ. L.et al. (2009). Acetylated H4 histone and genomic DNA methylation patterns during bud set and bud burst in Castanea sativa. J. Plant Physiol.166, 1360–1369. 10.1016/j.jplph.2009.02.014
43
ShalomL.SamuelsS.ZurN.ShlizermanL.ZemachH.WeissbergM.et al. (2012). Alternate bearing in citrus: changes in the expression of flowering control genes and in global gene expression in on-versus off-crop trees. PLoS ONE7:e46930. 10.1371/journal.pone.0046930
44
SuZ.MaX.GuoH.SukiranN. L.GuoB.AssmannS. M.et al. (2013). Flower development under drought stress: morphological and transcriptomic analyses reveal acute responses and long-term acclimation in Arabidopsis. Plant Cell25, 3785–3807. 10.1105/tpc.113.115428
45
SunC.ZhaoQ.LiuD. D.YouC. X.HaoY. J. (2013). Ectopic expression of the apple Md-miRNA156h gene regulates flower and fruit development in Arabidopsis. Plant Cell Tiss. Org.112, 343–351. 10.1007/s11240-012-0241-7
46
TeotiaS.TangG. (2015). To bloom or not to bloom: role of microRNAs in plant flowering. Mol. Plant8, 359–377. 10.1016/j.molp.2014.12.018
47
TranknerC.LehmannS.HoenickaH.HankeM. V.FladungM.LenhardtD.et al. (2010). Over-expression of an FT-homologous gene of apple induces early flowering in annual and perennial plants. Planta232, 1309–1324. 10.1007/s00425-010-1254-2
48
WangC.WangX.KibetN. K.SongC.ZhangC.LiX.et al. (2011). Deep sequencing of grapevine flower and berry short RNA library for discovery of novel microRNAs and validation of precise sequences of grapevine microRNAs deposited in miRBase. Physiol. Pantarum143, 64–81. 10.1111/j.1399-3054.2011.01481.x
49
WangJ. W.ParkM. Y.WangL. J.KooY.ChenX. Y.WeigelD.et al. (2011). miRNA control of vegetative phase change in trees. PLoS Genet.7:e1002012. 10.1371/journal.pgen.1002012
50
WangY.LiL.YeT.LuY.ChenX.WuY. (2013). The inhibitory effect of ABA on floral transition is mediated by ABI5 in Arabidopsis. J. Exp. Bot.64, 675–684. 10.1093/jxb/ers361
51
WellsC. E.VendraminE.TarodoS. J.VerdeI.BielenbergD. G. (2015). A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Boil.15:41. 10.1186/s12870-015-0436-2
52
WilliamsonG.DarnellR. L. (1999). Flower bud density af fects vegetative and fruit development in field-grown southern highbush blueberry. Hort. Science34, 607–610.
53
WuB. F.LiW. F.XuH. Y.QiL. W.HanS. Y. (2015). Role of cin-miR2118 in drought stress responses in Caragana intermedia and tobacco. Gene574, 34–40. 10.1016/j.gene.2015.07.072
54
WuG.ParkM. Y.ConwayS. R.WangJ. W.WeigelD.PoethigR. S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell138, 750–759. 10.1016/j.cell.2009.06.031
55
WuH. J.MaY. K.ChenT.WangM.WangX. J. (2012). PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res.40, 22–28. 10.1093/nar/gks554
56
WuJ.WangD.LiuY.WangL.QiaoX.ZhangS. (2014). Identification of miRNAs involved in pear fruit development and quality. BMC Genomics15:953. 10.1186/1471-2164-15-953
57
XiaR.ZhuH.AnY. Q.BeersE. P.LiuZ. (2012). Apple miRNAs and tasiRNAs with novel regulatory networks. Genome Biol.13, 47–64. 10.1186/gb-2012-13-6-r47
58
XingL.ZhangD.LiY.ZhaoC.ZhangS.ShenY.et al. (2014). Genome-wide identification of vegetative phase transition-associated microRNAs and target predictions using degradome sequencing in Malus hupehensis. BMC Genomics15:1125. 10.1186/1471-2164-15-1125
59
XingL.ZhangD.ZhaoC.LiY.MaJ.AnN.et al. (2016). Shoot bending promotes flower bud formation by miRNA-mediated regulation in apple (Malus domestica Borkh.). Plant Biotech. J.14, 749–770. 10.1111/pbi.12425
60
YamasakiH.HayashiM.FukazawaM.KobayashiY.ShikanaiT. (2009). SQUAMOSA promoter binding protein–like7 is a central regulator for copper homeostasis in Arabidopsis. Plant Cell21, 347–361. 10.1105/tpc.108.060137
61
YangC. Y.HuangY. H.LinC. P.LinY. Y.HsuH. C.WangC. N.et al. (2015). MiR396-targeted SHORT VEGETATIVE PHASE is required to repress flowering and is related to the development of abnormal flower symptoms by the PHYL1 effector. Plant Physiol.168, 1702–1716. 10.1104/pp.15.00307
62
YangH.ChangF.YouC.CuiJ.ZhuG.WangL.et al. (2015). Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J.81, 268–281. 10.1111/tpj.12726
63
YeK.ChenY.HuX.GuoJ. (2013). Computational identification of microRNAs and their targets in apple. Genes Genom.35, 377–385. 10.1007/s13258-013-0070-z
64
ZhaiJ.JeongD. H.De PaoliE.ParkS.RosenB. D.LiY.et al. (2011). MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev.25, 2540–2553. 10.1101/gad.177527.111
65
ZhangH.ZhaoX.LiJ.CaiH.DengX. W.LiL. (2014). MicroRNA408 is critical for the HY5-SPL7 gene network that mediates the coordinated response to light and copper. Plant Cell26, 4933–4953. 10.1105/tpc.114.127340
66
ZhangS.LiuY.YuB. (2015). New insights into pri-miRNA processing and accumulation in plants. Wiley Interdiscip. Rev. RNA6, 533–545. 10.1002/wrna.1292
67
ZhangX. Z.ZhaoY. B.LiC. M.ChenD. M.WangG. P.ChangR. F.et al. (2007). Potential polyphenol markers of phase change in apple (Malus domestica). J. Plant Physiol.164, 574–580. 10.1016/j.jplph.2006.03.011
68
ZhangZ.LiuX.GuoX.WangX.ZhangX. (2016). Arabidopsis AGO3 predominantly recruits 24-nt small RNAs to regulate epigenetic silencing. Nat. Plants2:16049. 10.1038/nplants.2016.49
69
ZhaoQ.SunC.LiuD. D.HaoY. J.YouC. X. (2015). Ectopic expression of the apple Md-miR172e gene alters flowering time and floral organ identity in Arabidopsis. Plant Cell Tiss. Org.123, 535–546. 10.1007/s11240-015-0857-5
70
ZhouC. M.ZhangT. Q.WangX.YuS.LianH.TangH.et al. (2013). Molecular basis of age-dependent vernalization in Cardamine flexuosa. Science340, 1097–1100. 10.1126/science.1234340
71
ZhouM.LiD.LiZ.HuQ.YangC.ZhuL.et al. (2013). Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol.161, 1375–1391. 10.1104/pp.112.208702
Summary
Keywords
apple, vegetative and floral buds, floral transition, sRNA, miRNA, RdDM
Citation
Guo X, Ma Z, Zhang Z, Cheng L, Zhang X and Li T (2017) Small RNA-Sequencing Links Physiological Changes and RdDM Process to Vegetative-to-Floral Transition in Apple. Front. Plant Sci. 8:873. doi: 10.3389/fpls.2017.00873
Received
12 March 2017
Accepted
10 May 2017
Published
29 May 2017
Volume
8 - 2017
Edited by
Claudio Bonghi, University of Padua, Italy
Reviewed by
Bin Yu, University of Nebraska Lincoln, United States; Li-Song Chen, Fujian Agriculture and Forestry University, China; Guo-Qing Song, Michigan State University, United States
Updates
Copyright
© 2017 Guo, Ma, Zhang, Cheng, Zhang and Li.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuren Zhang xiuren.zhang@tamu.edu
This article was submitted to Crop Science and Horticulture, a section of the journal Frontiers in Plant Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.