 Jayaraman Aravind1,2
Jayaraman Aravind1,2 Sharma Rinku1,3
Sharma Rinku1,3 Banduni Pooja1
Banduni Pooja1 Mittal Shikha1
Mittal Shikha1 Shiriga Kaliyugam1
Shiriga Kaliyugam1 Mallana Gowdra Mallikarjuna1
Mallana Gowdra Mallikarjuna1 Arun Kumar4
Arun Kumar4 Atmakuri Ramakrishna Rao5
Atmakuri Ramakrishna Rao5 Thirunavukkarasu Nepolean1*
Thirunavukkarasu Nepolean1*- 1Division of Genetics, Indian Agricultural Research Institute, New Delhi, India
- 2Division of Germplasm Conservation, National Bureau of Plant Genetic Resources, New Delhi, India
- 3Department of Life Sciences, Shiv Nadar University, Gautam Buddha Nagar, India
- 4National Phytotron Facility, Indian Agricultural Research Institute, New Delhi, India
- 5Centre for Agricultural Bioinformatics, Indian Agricultural Statistics Research Institute, New Delhi, India
MicroRNA-mediated gene regulation plays a crucial role in controlling drought tolerance. In the present investigation, 13 drought-associated miRNA families consisting of 65 members and regulating 42 unique target mRNAs were identified from drought-associated microarray expression data in maize and were subjected to structural and functional characterization. The largest number of members (14) was found in the zma-miR166 and zma-miR395 families, with several targets. However, zma-miR160, zma-miR390, zma-miR393, and zma-miR2275 each showed a single target. Twenty-three major drought-responsive cis-regulatory elements were found in the upstream regions of miRNAs. Many drought-related transcription factors, such as GAMYB, HD-Zip III, and NAC, were associated with the target mRNAs. Furthermore, two contrasting subtropical maize genotypes (tolerant: HKI-1532 and sensitive: V-372) were used to understand the miRNA-assisted regulation of target mRNA under drought stress. Approximately 35 and 31% of miRNAs were up-regulated in HKI-1532 and V-372, respectively. The up-regulation of target mRNAs was as high as 14.2% in HKI-1532 but was only 2.38% in V-372. The expression patterns of miRNA-target mRNA pairs were classified into four different types: Type I- up-regulation, Type II- down-regulation, Type III- neutral regulation, and Type IV- opposite regulation. HKI-1532 displayed 46 Type I, 13 Type II, and 23 Type III patterns, whereas V-372 had mostly Type IV interactions (151). A low level of negative regulations of miRNA associated with a higher level of mRNA activity in the tolerant genotype helped to maintain crucial biological functions such as ABA signaling, the auxin response pathway, the light-responsive pathway and endosperm expression under stress conditions, thereby leading to drought tolerance. Our study identified candidate miRNAs and mRNAs operating in important pathways under drought stress conditions, and these candidates will be useful in the development of drought-tolerant maize hybrids.
Introduction
Drought is one of the prevailing abiotic stresses that affect plant growth, development and grain yield (Ceccarelli and Grando, 1996). In particular, regions with insufficient water are more prone to drought due to uneven changes in weather conditions. Furthermore, shortages of water resources resulting from increasing human needs and growing climatic adversities exaggerate the effects of drought several fold (Rosegrant and Cline, 2003). Development of climate-resilient and drought-tolerant cultivars could help in sustaining food grain production in the present era of climate change. However, drought tolerance is a morphologically, physiologically, and genetically complex trait. Therefore, understanding the underlying molecular basis and regulation of drought tolerance could help in accelerating drought-tolerance breeding programs. Many reports have highlighted and proposed various genetic and molecular mechanisms of drought tolerance in different crops, including model plant systems (Ha et al., 2012; Shikha et al., 2017). Additionally, a functional understanding of stress responsive gene(s) and their regulation patterns can aid in devising new genetic tools (Langridge and Reynolds, 2015).
Post-transcriptional modification of RNAs is one of the major forms of regulation of gene expression and is primarily performed by microRNA (miRNA) molecules (Ding et al., 2009; Zhou et al., 2010). miRNAs belong to a non-coding family of RNAs and are known to play major roles in modulating gene expression to adjust plant metabolism to withstand stresses. The regulatory mechanism of miRNAs involves a change in self-concentration, targeting the mRNA quantity and modifying the mRNA expression via miRNA-protein complexes. These, in turn, change the ultimate expression of proteins upon exposure to stress (Ding et al., 2009; Wang B. et al., 2014). In plants, miRNA genes code for long pri-miRNA (primary miRNA) transcripts with imperfect stem-loop secondary structures, which are transcribed by RNA polymerase II (Lee et al., 2004; Xie et al., 2005). These transcripts are processed into ~70-nt pre-miRNAs and subsequently released as miRNA/miRNA duplexes by DCL-1 (dicer-like enzyme 1) in association with a dsRNA binding protein, HYL-1. Such duplexes are methylated by a dsRNA methylase, HEN1, and loaded into AGO1 (Kurihara and Watanabe, 2004; Vazquez et al., 2004; Yu et al., 2005). They are subsequently transported to the cytoplasm with the help of an exportin homolog, the HASTY protein (Bartel, 2004; Park et al., 2005), and cleaved into ~22-nt mature miRNAs. Mature miRNA strands are incorporated into a multiprotein complex, the RNA-induced silencing complex (RISC), where they guide the cleavage of complementary target mRNAs by AGO1, which possesses a PAZ domain and a catalytic PIWI domain (Vaucheret et al., 2004; Baumberger and Baulcombe, 2005).
The involvement of miRNAs in different abiotic stresses has been demonstrated in Arabidopsis. Overexpression of miR168, miR171 and miR396 under hypersalinity, mannitol, and cold stress was reported in Arabidopsis (Liu et al., 2008). Nevertheless, both the involvement of total miRNAs under drought stress and the systemic expression analysis of their drought-related mechanism are still in progress and necessitate further exploration.
Maize is an important crop in the world, contributing significantly to food and nutritional security. However, the production of maize is most vulnerable to various abiotic stresses, especially drought. To date, functional genomics approaches have revealed large amounts of information on target mRNA control through miRNAs for various traits in maize. The expression of a class III homeodomain-leucine zipper (HD-Zip III) protein that functions in asymmetrical leaf development and that of a floral meristem transcription factor, APETALA2, responsible for meristem identity were found to be targeted by miR166 and miR172, respectively (Juarez et al., 2004). Similarly, miR156 is reported to target the expression of tga1 (Teosinte glume architecture 1) (Chuck et al., 2007). Studies on differential expression of miRNAs have shed light on the regulatory roles of miRNAs in plant development (Kang et al., 2012; Liu et al., 2014) and stress responses in maize (Zhang et al., 2008; Ding et al., 2009; Wei et al., 2009; Zhai et al., 2013; Wu et al., 2014). Such investigations deciphering the regulatory control between miRNAs and target mRNAs will pave the way for a better understanding of the molecular mechanisms underlying drought stress responses. Differentially expressed genes (DEGs) have been identified in drought or low-moisture stress microarray studies (Yu, 2003; Yue et al., 2008; Hayano-Kanashiro et al., 2009; Li et al., 2009; Marino et al., 2009; Luo et al., 2010; Zheng et al., 2010; Lu et al., 2011; Hansen et al., 2013; Regulski et al., 2013). Such DEGs were found to be associated with drought-related miRNA targets.
Maize germplasm shows great genetic variability involving temperate, tropical, and subtropical groups, as well as dent, flint, semi flint and waxy types within germplasm groups. Efforts have been taken to characterize the drought-responsive miRNAs and target mRNAs in temperate maize germplasm (Li et al., 2013; Wang Y. G. et al., 2014). There have been no reports available on comprehensive characterization of drought-responsive miRNAs and target-mRNAs using subtropical maize germplasm. Therefore, in the present study, we identified putative regulatory miRNAs targeting drought-related mRNAs based on gene expression data from 12 drought/low-moisture stress experiments. The expression patterns of miRNA-target mRNA pairs were validated in the root and shoot tissues of two contrasting subtropical maize inbreds under drought stress. The functional annotation and the role of drought-related miRNA-target mRNA pairs were analyzed. Our study identified the differential interactions of miRNA-target mRNA pairs during drought stress in the tolerant genotype and explained their functional roles in drought tolerance.
Materials and Methods
Plant Growth and Experimentation
Seeds of two contrasting subtropical maize inbreds, HKI-1532 (drought-tolerant) and V-372 (drought-susceptible), were grown in the National Phytotron Facility, Indian Agricultural Research Institute, New Delhi. Potting was done in triplicate with sandy loam soil. The plants were maintained under controlled greenhouse conditions of 28/22°C (day/night) at a light intensity of 600 μmol m−2s−1 (16 h day/8 h night) with 50–55% relative humidity. Regular irrigation was provided for 15 days to the first set of plants (stress) after sowing and suspended for the next 5 days to induce severe drought stress (Kakumanu et al., 2012; Min et al., 2016; Nepolean et al., 2017). The second set of plants (control) was watered throughout the experiment. On the 21st day after sowing, leaf samples were collected for expression assays (Figure 1).

Figure 1. Phenotypic response of tolerant (HKI-1532) and sensitive (V-372) genotypes under drought stress.
Prediction of miRNA-target mRNA Interactions
The entire set of maize mature miRNA sequences available (321) was downloaded from miRbase v19 (Griffiths-Jones et al., 2008; Kozomara and Griffiths-Jones, 2011). Four popular miRNA target prediction tools—RNAHybrid (Rehmsmeier et al., 2004), TargetFinder (Allen et al., 2005; http://carringtonlab.org/resources/targetfinder), TAPIR (FASTA search engine) (Bonnet et al., 2010), and psRNATarget (Dai and Zhao, 2011)—were used to identify targets for the miRNAs from among the 76,617 Zea mays B73 RegGen v2 mRNAs downloaded from NCBI (National Center for Biotechnology Information) Genome database identified by the GNOMON v2 gene prediction tool (Souvorov et al., 2010; ftp://ftp.ncbi.nlm.nih.gov/genomes/PLANTS/Zea_mays/Gnomon_v2/). Consensus predictions of miRNA-target mRNA interactions detected among the four tools were used for further analysis.
Identification of Putative Drought-Related miRNAs
DEGs identified in 12 microarray-based gene expression studies in maize under drought/low moisture stress and/or recovery irrigation conditions (Table S1) were selected for comparison with the target mRNAs identified in the consensus predictions of miRNA-target mRNA interactions. For the experiments where DEG lists were not available, the raw data from NCBI GEO (Gene Expression Omnibus) (Barrett et al., 2005) were reanalyzed to generate a list. The regulatory miRNAs of the DEG sequences that were also predicted to be miRNA targets were identified as putative drought-related miRNAs. For both the mature miRNA sequences and the target mRNA sequences with the respective target sites masked, multiple sequence alignment was done using the Clustal Omega program (Sievers et al., 2011). A distance matrix was constructed based on pairwise distances using an identity matrix with the “dist.alignment” function of the R package seqinR (Charif et al., 2008) and used to performing hierarchical clustering by complete linkage. The clusterings of miRNAs and their respective targets were compared visually by plotting a tanglegram using “cophyloplot” function of the R package ape (Paradis et al., 2004). Furthermore, the functional annotation of the corresponding target mRNAs was carried out by Blast2GO (Conesa et al., 2005).
Analysis of cis-Regulatory Elements (CREs) in miRNA Promoters in Maize
DNA sequences upstream of the starts of the precursor miRNA structures were identified and extracted using NCBI tools (https://www.ncbi.nlm.nih.gov). The transcription start sites (TSS) were predicted from the upstream regions of pri-miRNA sequences using softberry TSSP (Shahmuradov et al., 2005; http://www.softberry.com/berry.phtml?topic = tss). The promoter sequence, i.e., the 2-kb region upstream of the TSS, was retrieved using Perl Script. For families having >1 promoter, the sequence between the two promoters was considered for the identification of CREs using the PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) database.
RNA Isolation and qRT-PCR Assay
Total RNA was isolated from seedling leaf tissue using the Ultra Clean Plant RNA isolation kit (MO BIO Laboratories, USA). Primers specific to the target mRNA sequences were designed using BatchPrimer3 (You et al., 2008) and optimized to avoid primary and amplicon secondary structures using Beacon DesignerTM Free Edition (http://www.premierbiosoft.com/qpcr/) and IDT OligoAnalyzer (http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer/), respectively (Table S2). Sequence specificity of the primers was assured by comparing their sequences with maize gene sequences using NCBI nucleotide BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). qRT-PCR for mature miRNAs was performed using a modified version of stem-loop qRT-PCR (Chen et al., 2005). The stem-loop primers, miRNA forward primers and universal reverse primers were designed according to Kramer (2011) (Table S3). The primer for the reference transcript U6snRNA was designed as previously described (Turner et al., 2013).
For target mRNAs, the first-strand cDNA synthesis was carried out with the RNA ProtoScript First Strand cDNA Synthesis Kit (New England Biolabs, Ipswich, Massachusetts) using 50 ng total RNA and 1 μM Oligo d(T)23 VN primer. For miRNAs, reverse transcription reactions were carried out with 50 ng total RNA and 1 μM stem-loop RT primer. The reactions were incubated in a thermocycler for 30 min at 16°C, 30 min at 42°C and 5 min at 85°C and then held at 4°C.
For both mature miRNAs and their mRNA targets, real-time quantitative PCR with SYBR Green I was performed on an Agilent Technologies Mx3005P QPCR (Agilent Technologies, Santa Clara, California, United States) instrument. Briefly, each 25-μl PCR mixture contained approximately 100 ng cDNA, 9 ml Hotstart IT SYBR Green qPCR Master Mix (Affymetrix), ROX (Affymetrix) and 250 nM of each primer. The reactions were mixed gently and incubated at 94°C for 2 min (pre-heating), followed by 40 cycles of 94°C for 15 s (denaturation), 60°C for 30 s (primer annealing), and 72°C for 30 s (primer extension). Analysis was conducted on three biological replicates and two technical replicates for each treatment and genotype combination. U6 snRNA and 18S rRNA were used as internal controls for miRNA and mRNA, respectively. The ΔΔCt method was used to determine the differences in expression levels among samples (Livak and Schmittgen, 2001).
Results
Identification of Putative Drought-Related miRNAs and Target mRNAs
Among the DEGs identified in 12 microarray-based gene expression studies under drought/low moisture stress and/or recovery irrigation conditions, 42 differentially expressed mRNA sequences were predicted to be putatively regulated by 65 miRNAs belonging to 13 families in 183 miRNA-target mRNA interactions (Table 1). These were considered for further expression analysis. The predicted interactions involved the same miRNAs targeting multiple mRNAs, as well as the same mRNA being targeted by multiple miRNAs. For most of the interactions, miRNAs with similar sequences targeted mRNA sequences that clustered together (Figure 2). The miRNA families with the largest numbers of members included zma-miR166, zma-miR395 and zma-miR156, with 14, 14, and 12 members, respectively. On the other hand, some miRNA families, such as zma-miR160, zma-miR399, zma-miR529, and zma-miR2275, had only one member each (Figure 3). The target distributions of miRNA families themselves were investigated, which showed that zma-miR166, zma-miR396, zma-miR529, zma-miR164, and zma-miR169 had the most targets, with 6, 6, 5, and 5 target mRNAs, respectively, while zma-miR160, zma-miR390, zma-miR393, and zma-miR2275 had the fewest target mRNAs, with a single target each (Figure 3).
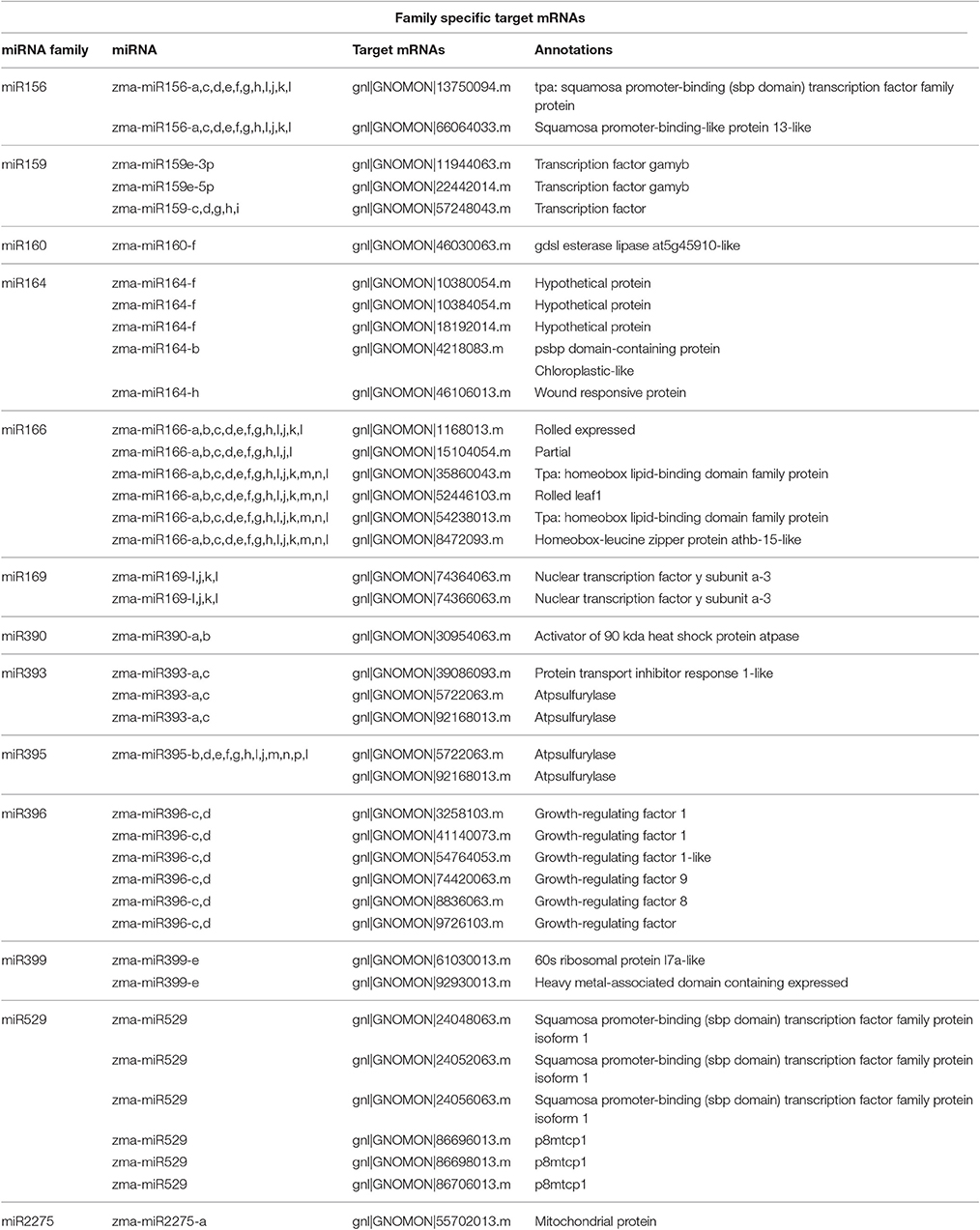
Table 1. List of 13 drought-related miRNA families and their respective targets with annotations.
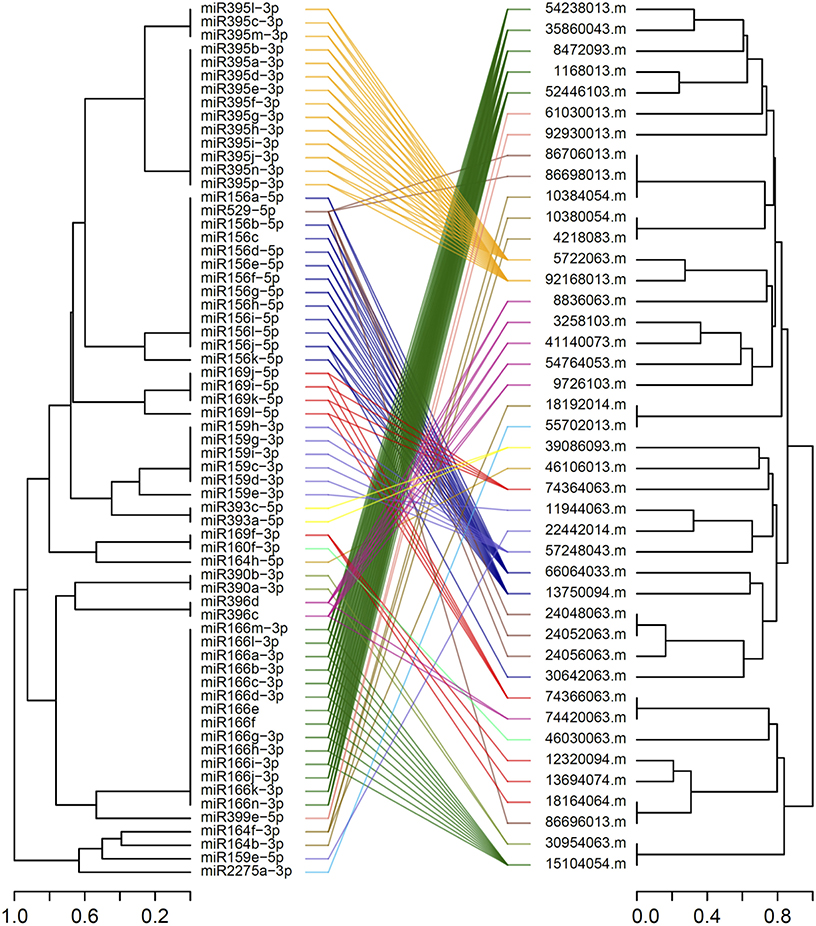
Figure 2. Tanglegram of predicted mature miRNA and target mRNA sequences (with the target site masked) related to drought/low-moisture stress. Lines colored according to the miRNA family, connect the miRNA sequences to their respective target mRNAs.
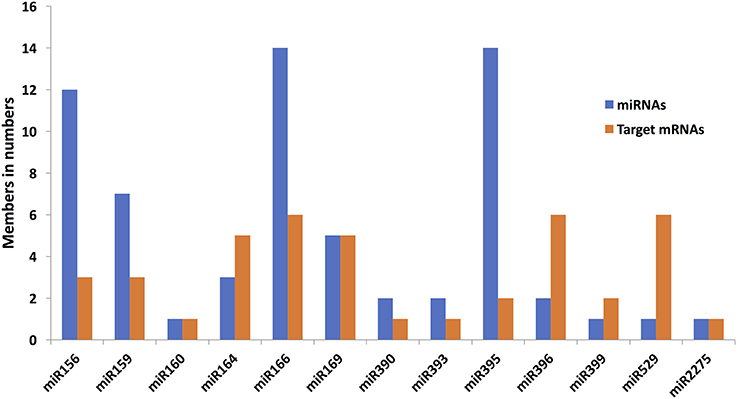
Figure 3. Distribution of 13-drought related miRNA families members and respective target mRNAs. Blue bar represents the members count in each miRNA family and orange bar represents the total number of target mRNAs in each miRNA family.
Prediction of miRNA-Target mRNA Interactions
The four miRNA target prediction tools detected different but overlapping sets of miRNA-target mRNA pairs from among the 321 miRNAs and 76617 mRNAs of maize. Among these dissimilar sets, 594 consensus predictions of miRNA-target mRNA interactions could be identified. Compared to single prediction tool-based identification of miRNA-target mRNA interactions, we employed four prediction tools to choose the robust miRNA-target mRNA interactions. The total numbers of interactions supported by RNAHybrid, TargetFinder, TAPIR FASTA and psRNATarget were 31,262, 4,522, 3,406, and 3,072, respectively, including both unique and commonly identified interactions from each tool. The numbers of unique interactions identified by RNAHybrid, TargetFinder, TAPIR FASTA and psRNATarget were 29869, 1264, 222, and 1293, respectively (Figure 4). The consensus predictions of miRNA-target mRNAs interactions involved 156 miRNAs belonging to 25 families, and 150 unique target mRNAs.
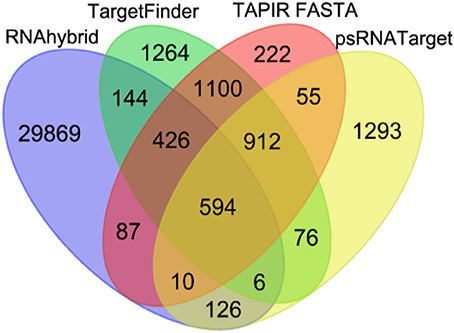
Figure 4. Venn diagram illustrating the number of common and differently identified miRNA-target mRNA interactions through four different source tools (RNAhybrid, TargetFinder, TAPIR FASTA, and psRNATarget). The color of each leaf represents the total and overlapped number of miRNA-target mRNA interactions obtained from each source tool.
GO Enrichment Analysis of miRNA Target Genes
To gain insights into the functional roles of miRNAs, annotation of their predicted target mRNAs was carried out. GO terms were identified under three different functional categories: biological (57.14%), cellular (23.80%) and molecular (30.95%). In the biological process category, major functions included anatomical structure morphogenesis, multicellular organismal development and post-embryonic development. In the molecular process category, major functions included DNA binding, hydrolase activity and nucleotide binding (Figure 5). Furthermore, most of the target genes were confined to the nucleus.
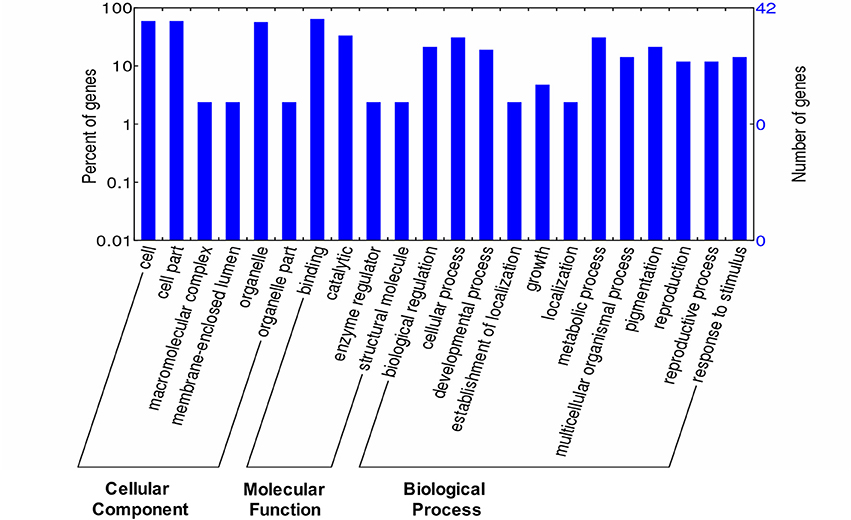
Figure 5. Overview of GO annotations of target mRNAs. The data represents the three major functional categories- biological, molecular, and cellular functions.
Analysis of cis-Regulatory Elements
The promoter regions of 65 drought-related miRNAs were examined to detect the occurrence of conserved cis-regulatory elements. A total of 61 conserved CRE motifs were identified from the miRNA promoters. Among these, 41 were major cis-regulatory elements that were observed in >10 members, including the drought-related cis-elements ABRE (ABA-responsive element) and MBS (MYB-binding site) (Table 2). In miR169n of rice, the ABRE cis-acting element resided in the promoter region, implying an ABA-mediated response to stress (Zhao et al., 2007). An ABRE cis-element was also found in sorghum ABA-responsive genes (Buchanan et al., 2005). Similarly, in Arabidopsis, MBS was found in the upstream regions of all the miRNA genes in the rice shoot apexes. Of the 65 miRNA members, miR164f showed the highest number of CREs (41), whereas miR156d had the lowest number (8).
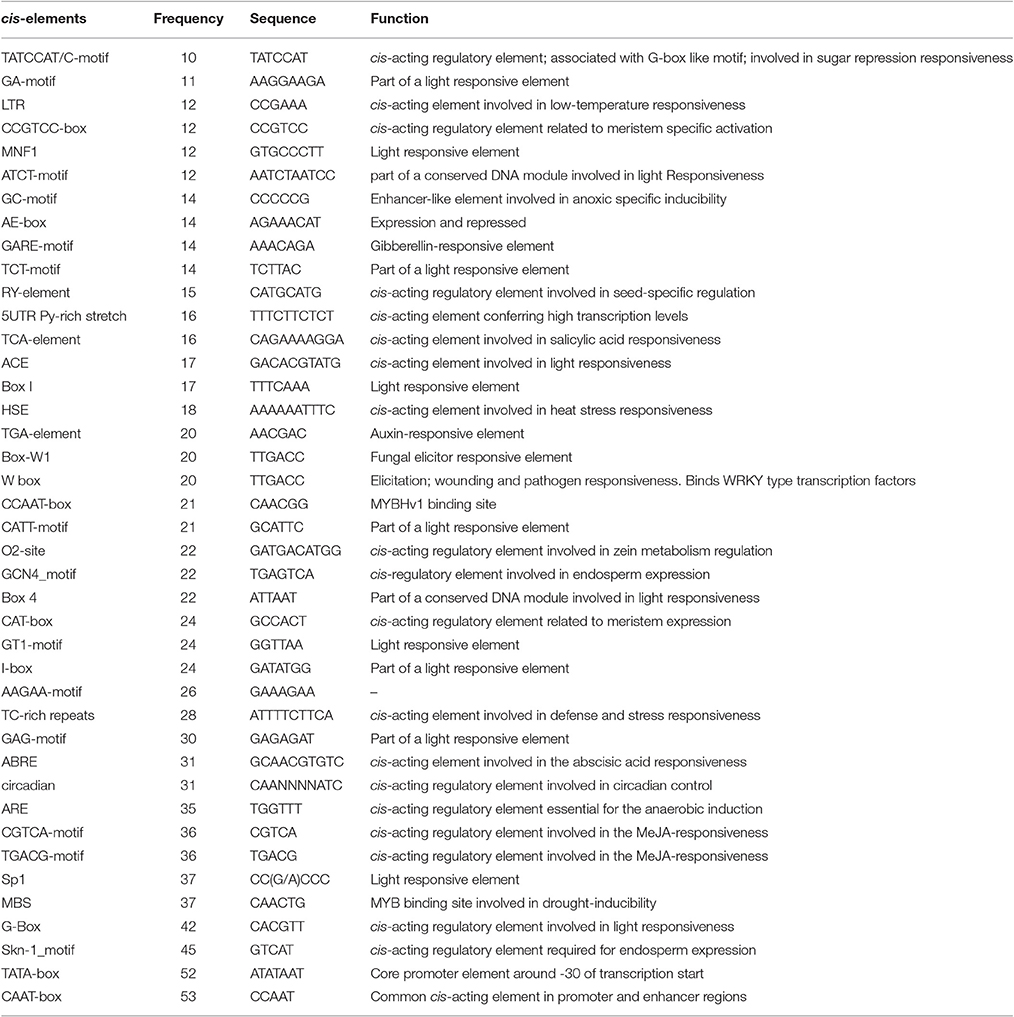
Table 2. Putative cis-regulatory elements identified in the upstream region of drought responsive miRNA in maize.
Expression Patterns of miRNAs and Their Target mRNA(s)
To analyze the responses of contrasting genotypes to drought, transcriptional profiling of the identified putative drought-responsive miRNAs and their target-mRNAs was done at the seedling stage, revealing their interactions with each other. The significantly up-regulated miRNAs common to both genotypes (HKI-1532 and V-372) included zma-miR164h, zma-miR169l, zma-miR396c, zma-miR396d, and zma-miR399e. In tolerant genotype HKI-1532, 16 miRNAs belonging to the zma-miR159, zma-miR160, zma-miR164, zma-miR166, zma-miR169, zma-miR390, zma-miR395, zma-miR396, and zma-miR399 families were significantly up-regulated. However, zma-miR156 and zma-miR159 were significantly down-regulated. Conversely, in V-372, members of 7 families—zma-miR164, zma-miR169, zma-miR393, zma-miR396, zma-miR399, zma-miR529, and zma-miR2275—were significantly up-regulated; and zma-miR156, zma-miR159, zma-miR166 and zma-miR395 families were significantly down-regulated. These results indicate disparate patterns of regulation in the tolerant and susceptible genotypes (Figure 6).
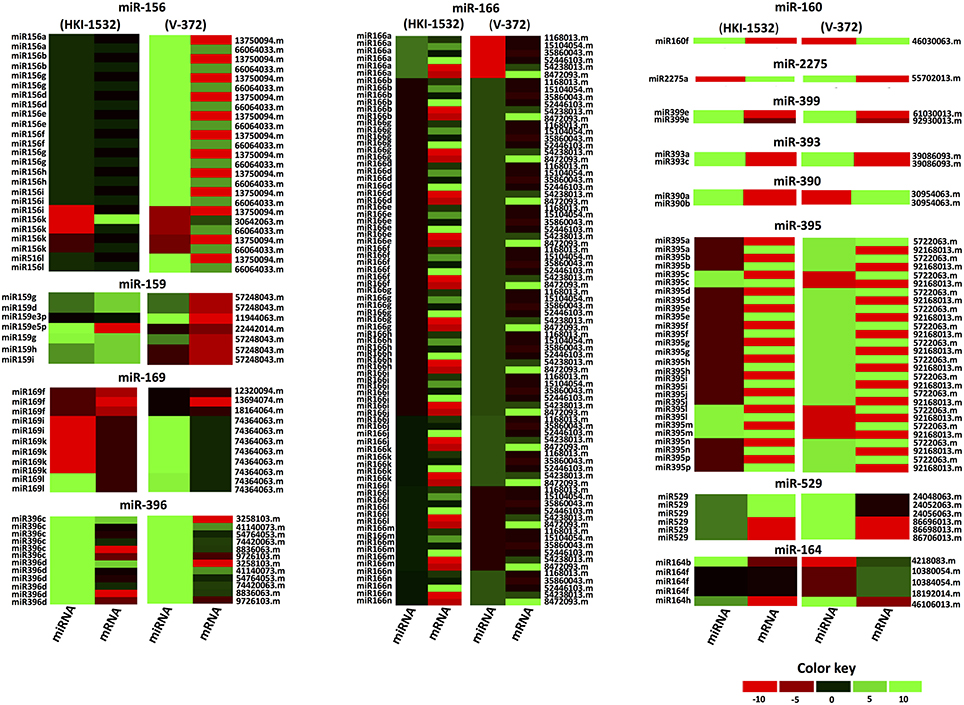
Figure 6. Heat map for altered expression of miRNAs and their respective targets within each family under drought stress. Different color codings have been assigned for specific expression pattern and comparative expression is made in maize genotypes (Left: HKI-1532 and Right: V-372).
Furthermore, expression analysis of target mRNAs revealed six up-regulated, 18 down-regulated and 18 neutrally expressed mRNAs in HKI-1532. V-372, however, had one up-regulated, 10 down-regulated and 26 neutrally expressed mRNAs. The miRNA-target mRNA interactions were classified into four different groups: Type I, with miRNA-mRNA up-regulations; Type II, with miRNA-mRNA down-regulations; Type III, with neutral miRNA-mRNA expressions; and Type IV, with opposite interactions. The tolerant genotype, HKI-1532, had 46 Type I, 13 Type II, 23 Type III, and 101 Type IV combinations. The susceptible genotype, V-372, had eight Type I, four Type II, 20 Type III, and 151 Type IV combinations (Figure 6).
Discussion
Several investigations have revealed the role of miRNAs in modulating gene expression under abiotic stresses—cold (Lv et al., 2010), salt (Ding et al., 2009), aluminum tolerance (Kong et al., 2014), waterlogging (Zhai et al., 2013), and drought (Li et al., 2013; Wang Y. G. et al., 2014). Among the several abiotic stresses, drought is the most prominent stress in the sub-tropical maize production systems. A number of plant parameters, such as growth, yield, membrane integrity, pigment composition, osmotic relations and photosynthesis, are commonly affected by drought stress (Praba et al., 2009). The change in miRNA expression pattern under drought is an indication of stress responses in plants. Such findings provide an insight into drought tolerance mechanisms and can potentially aid in designing drought-tolerant cultivars (Zhao et al., 2007; Chen et al., 2012). The role of miRNAs in drought tolerance has been previously explored in maize using temperate germplasm. Wang Y. G. et al. (2014) identified 301 drought-responsive miRNAs in temperate maize germplasm and Li et al. (2013) detected differentially expressed 68 miRNAs falling under 29 miRNA families in a maize inbred R09. In the present investigation, 42 differentially expressed mRNA sequences were predicted to be putatively regulated by 65 miRNAs belonging to 13 families. Furthermore, a combination of 183 miRNA-target mRNA interactions were identified and validated in a contrasting pair of subtropical maize germplasm. Four prediction tools were employed to reveal high-confidence predictions of miRNA-target mRNA interactions due to enhanced accuracy and site recognition efficacy as compared to previous single tool-based prediction (Ding et al., 2012; Li et al., 2013; Wang Y. G. et al., 2014). All 13 of the miRNA families were found to be conserved across taxonomic groups related to drought, ABA or oxidative stress response (Kim et al., 2003; Ding et al., 2013). These results suggested that specific microarray expression profiling data could be effectively used for identifying regulatory miRNAs.
miRNA genes originated from the preexisting genes through duplication events (Nozawa et al., 2012). miRNAs that are evolutionarily conserved are usually encoded by miRNA gene families. This, coupled with the strong similarity requirements of plant miRNA - target site interactions, leads to overlapping functions of miRNAs belonging to the same families, buffering against loss of individual members (Jones-Rhoades et al., 2006). It was found that the families such as miR2118, miR395, and miR159 were highly conserved in maize and rice (Xu et al., 2017). In a few interactions, similar miRNA sequences belonging to the same families were found to target dissimilar target mRNA sequences, such as mature miRNAs from the 3′ arm of miRNA family 166 and those from the 5′ arm of miRNA family 169. In Arabidopsis, miR395 has been found to regulate an ATP sulfurylase and an unrelated sulfate transporter (Allen et al., 2005), and miR159 has been reported to target both MYB101 and MYB120 and two unrelated genes,OPT1 and ACS8 (Schwab et al., 2005). In maize, miR395 showed differential expression in response to salt stress, and was found to regulate ATP sulfurylase, L-Isoaspartyl methyltransferase, and Beta-D-xylosidase genes (Ding et al., 2009).
The promoter analysis of differentially expressed miRNAs indicated that they were predominantly involved in ABA signaling, the auxin response pathway, light-responsive pathways and endosperm expression. Among the 13 drought-related miRNA families, some families had common targets, i.e., zma-miR160, zma-miR390, and zma-miR393 were mostly related to the ARF (auxin response factor) transcription factor, which plays an active role in ABA and auxin mediated signaling under drought conditions (Ding et al., 2013). Similarly, miR164 targets the NAC transcription factor and CUC (cup shaped cotyledon) genes in Arabidopsis (Rhoades et al., 2002); these genes are responsible for root and shoot development. Furthermore, zma-miR164 showed higher expression in crown roots but not in seminal roots of maize, suggesting that it could play a crucial role in development of crown roots (Kong et al., 2014).
Members of the miR166 family generally participate in regulation of their target, HD-Zip III (homeodomain-leucine zipper III), which is engaged in lateral root development, initiation of axillary leaf meristems and leaf polarity (Boualem et al., 2008) (Figure 7). In maize, it was reported that the leaf polarity was controlled by a subset of miR166 family members (Nogueira et al., 2009). The regulatory mechanism of miR390 is somewhat different, as it prompts the production of tasiRNA (TAS-3 derived small interfering RNA), which targets the ARFs (ARF2, ARF3, and ARF4) that function in lateral root emergence and organ polarity (Nogueira et al., 2009; Meng et al., 2010). miR399 is involved in regulation of a phosphate transporter that regulates the uptake of phosphate and its translocation (Pant et al., 2008). Our in-silico analysis identified various target mRNAs, including SPL (SQUAMOSA promoter-binding-like proteins), GAMYB (a gibberellin- and abscisic acid-related MYB), ARF (auxin response factor), AST (a sulfate transporter), and GRFs (growth regulating factors), among others. The majority of the identified targets are also conserved across other plant species, including model systems such as Arabidopsis (Adai et al., 2005) and rice (Wang et al., 2004; Luo et al., 2006).
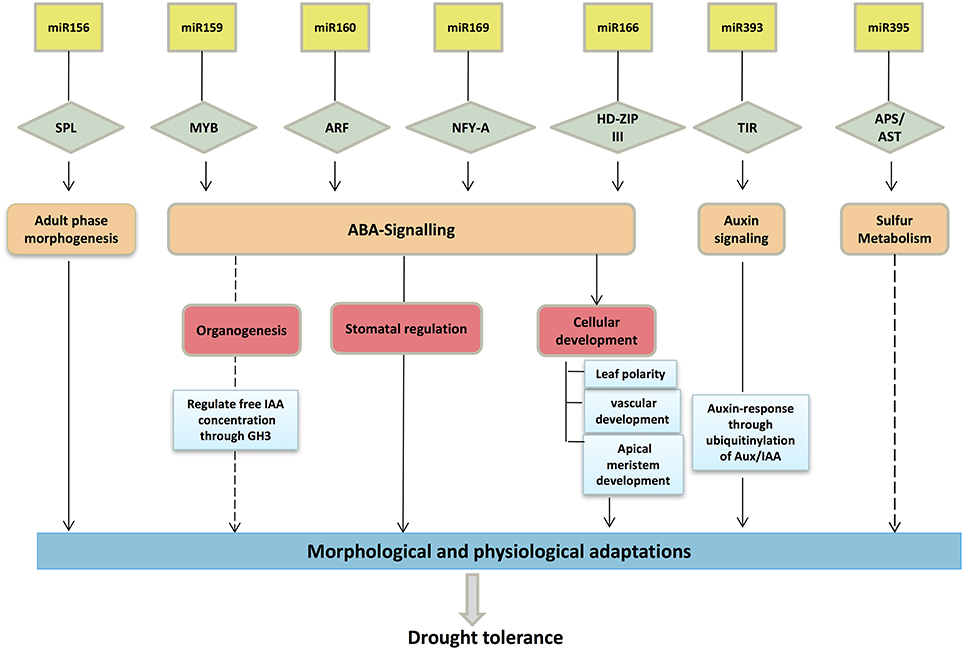
Figure 7. Schematic representation of important drought-related miRNAs and their targets involved in drought tolerance. The diagram depicts the possible role of targets and pathways in assigning in drought tolerance.
The interactions of each miRNA with a specific target are of prime importance, as these interactions can help to discern the variation in drought-induced gene expression. For example, the miR156 family regulates the expression of SPL, leading to plant developmental phase transitions (Wang et al., 2009; Chen et al., 2010) (Figure 6). Altered expression of this family during drought is evidence of its functional novelty in drought stress. Li et al. (2013) reported the up-regulation of miR156 at the early stage of drought stress in the maize seedlings. The regulation of SPL TFs through miR156 was also reported during somatic embryogenesis of maize (Chávez-Hernández et al., 2015). Additionally, Liu et al. (2014) reported that miR156 controlled several SPL genes during the juvenile-to-adult phase transition in maize. Over-expression of miR156 encoding the maize Cg1 gene showed to prevent the flowering, and improved the digestibility and starch content in switchgrass (Chuck et al., 2011). Similarly, the expression of GRF, which plays important role in leaf growth by transforming cell proliferation, is regulated by miR396c (Kim et al., 2003).
It was interesting to note that the expression patterns of some miRNAs were genotype-specific under drought stress. In this experiment, the Type IV (opposite) pattern identified between miRNA and target mRNAs suggested negative regulation under drought stress. The up-regulation of miR396 in HKI-1532 suppressed the expression of its target, GRF1, whereas the up-regulation of miR396 in V-372 led to neutral expression of its target. This suggests that suppressed GRF expression under drought can provide tolerance by precluding leaf growth and function under stress (Figure 7). Our hypothesis is supported by the fact that GRF8 reduces stomatal density in Arabidopsis (Liu et al., 2009). However, expression of miR396c and miR396d, targeting GRFs, was the same in both genotypes. It may be deduced that the small up-regulation of miRNAs in HKI-1532 contributes to drought tolerance. Similar results were obtained for miR396 in Arabidopsis (Liu et al., 2008) and tobacco (Frazier et al., 2011).
In the same way, Type I (up-regulated) miRNA-mRNA interaction was observed for miR169l, which targets transcription factor NF-YA3 (nuclear transcription factor subunit A-3) in HKI1532. However, the same miRNA exhibited a Type IV interaction in V-372. Our hypothesis is supported by findings in Arabidopsis, where the role of transcription factor NF-YA in drought tolerance is well documented (Liu et al., 2008). Similarly, Type IV interaction was also observed for the miR159 family, targeting the transcription factor GAMYB that is involved in flowering development and flowering time. Furthermore, overexpression of the miR159 family during stress aids in germination under stress conditions. In Arabidopsis, overexpression of MYB transcription factors (MYB33 and MYB101) provides tolerance to drought by adapting ABA hypersensitivity (Reyes and Chua, 2007). Regulation of MYB transcription factors by the member of miR159 was also reported in temperate maize (Li et al., 2013) and was found to play a crucial role in stress tolerance (Wang Y. G. et al., 2014).
The miR393 family that targets the TIR1 (transport inhibitor response 1) enzyme was up-regulated in V-372. This enzyme directly participates in the ubiquitinylation of inhibitors of the auxin response pathway (Dharmasiri and Estelle, 2002). miR160, representing a Type IV interaction in HKI-1532, plays a role in ABA-auxin interaction during drought (Liu et al., 2007). Similarly, the Type IV interaction between up-regulated miR395 up-regulated and its down-regulated targets ATP sulfurylase and HD-Zip transcription factor (Buchner et al., 2004) were involved in seedling growth and seed germination in HKI-1532. This negative link indicates the role of miR395 in conferring drought tolerance. Our results are supported by the findings of studies conducted in Arabidopsis under drought stress, where overexpression of miR395 decreased seed growth and germination upon drought exposure (Kim et al., 2010). Similar to observations for miR156 family, Type II (down-regulated) interactions were found for SPL genes in both genotypes. SPL genes have been found to be involved in deferred blooming and the adult phase transition in Arabidopsis (Park et al., 2010). This can be correlated with the similar studies conducted in rice and maize (Wei et al., 2009; Zhou et al., 2010).
Conclusions
Drought-specific miRNAs and their target mRNAs were identified from existing drought-associated microarray expression profiling data, and their expression was assayed in two contrasting subtropical maize genotypes, HKI-1532 and V-372. The promoters of all drought-related miRNAs were analyzed and confirmed the presence of important drought-responsive CREs. Eleven miRNAs belonging to nine families in HKI-1532 and seven miRNAs from three families in V-372 were differentially regulated in both genotypes. Among different miRNA-mRNA interactions, suppression of biologically important genes by miRNAs in V-372 suggests their weak performance under drought stress. It was noticed that the fact that some crucial genes being unaffected by regulatory miRNAs in tolerant genotype HKI1532 could have led to drought tolerance. Our experiment provided a clear understanding of miRNA regulation in drought response. To our knowledge, this is the first report on the role of miRNAs in regulation of drought tolerance in subtropical maize inbreds. Many of the identified candidate miRNAs and mRNAs from the present investigation could be used as potential candidates for development of drought-tolerant maize hybrids for the subtropical production system.
Author Contributions
JA and TN: conceived and designed the experiments; JA, SK, AK, BP, and MGM: performed the experiments; SR, MS, and ARR: analyzed the data; All authors contributed to manuscript preparation. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the ICAR Network Projects on Transgenics in Crop Plants (Maize Functional Genomics Component: 21-22) and the Computational Biology and Agricultural Bioinformatics (Agril.Edn.14 (44)/2014-A&P) for funding the study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00941/full#supplementary-material
References
Adai, A., Johnson, C., Mlotshwa, S., Archer-Evans, S., Manocha, V., Vance, V., et al. (2005). Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res. 15, 78–91. doi: 10.1101/gr.2908205
Allen, E., Xie, Z., Gustafson, A. M., and Carrington, J. C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221. doi: 10.1016/j.cell.2005.04.004
Barrett, T., Suzek, T. O., Troup, D. B., Wilhite, S. E., Ngau, W.-C., Ledoux, P., et al. (2005). NCBI GEO: mining millions of expression profiles-database and tools. Nucleic Acids Res. 33, D562–D566. doi: 10.1093/nar/gki022
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Baumberger, N., and Baulcombe, D. C. (2005). Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. U.S.A. 102, 11928–11933. doi: 10.1073/pnas.0505461102
Bonnet, E., He, Y., Billiau, K., and van de Peer, Y. (2010). TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26, 1566–1568. doi: 10.1093/bioinformatics/btq233
Boualem, A., Laporte, P., Jovanovic, M., Laffont, C., Plet, J., Combier, J. P., et al. (2008). MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J. 54, 876–887. doi: 10.1111/j.1365-313X.2008.03448.x
Buchanan, C. D., Lim, S., Salzman, R. A., Kagiampakis, I., Morishige, D. T., Weers, B. D., et al. (2005). Sorghum bicolor's transcriptome response to dehydration, high salinity and ABA. Plant Mol. Biol. 58, 699–720. doi: 10.1007/s11103-005-7876-2
Buchner, P., Takahashi, H., and Hawkesford, M. J. (2004). Plant sulphate transporters: co-ordination of uptake, intracellular and long-distance transport. J. Exp. Bot. 1765–1773. doi: 10.1093/jxb/erh206
Ceccarelli, S., and Grando, S. (1996). Drought as a challenge for the plant breeder. Plant Growth Regul. 20, 149–155. doi: 10.1007/BF00024011
Charif, D., Humblot, L., Lobry, J. R., Necsulea, A., Palmeira, L., and Penel, S. (2008). SeqinR 2.0-1: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrievel and Analysis. 268
Chávez-Hernández, E. C., Alejandri-Ramírez, N. D., Juárez-González, V. T., and Dinkova, T. D. (2015). Maize miRNA and target regulation in response to hormone depletion and light exposure during somatic embryogenesis. Front. Plant Sci. 6:555. doi: 10.3389/fpls.2015.00555
Chen, C., Ridzon, D. A., Broomer, A. J., Zhou, Z., Lee, D. H., Nguyen, J. T., et al. (2005). Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33:e179. doi: 10.1093/nar/gni178
Chen, H., Li, Z., and Xiong, L. (2012). A plant microRNA regulates the adaptation of roots to drought stress. FEBS Lett. 586, 1742–1747. doi: 10.1016/j.febslet.2012.05.013
Chen, X., Zhang, Z., Liu, D., Zhang, K., Li, A., and Mao, L. (2010). SQUAMOSA promoter-binding protein-like transcription factors: star players for plant growth and development. J. Integr. Plant Biol. 52, 946–951. doi: 10.1111/j.1744-7909.2010.00987.x
Chuck, G., Cigan, A. M., Saeteurn, K., and Hake, S. (2007). The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 39, 544–549. doi: 10.1038/ng2001
Chuck, G. S., Tobias, C., Sun, L., Kraemer, F., Li, C., Dibble, D., et al. (2011). Overexpression of the maize Corngrass1 microRNA prevents flowering, improves digestibility, and increases starch content of switchgrass. Proc. Natl. Acad. Sci. U.S.A. 108, 17550–17555. doi: 10.1073/pnas.1113971108
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dai, X., and Zhao, P. X. (2011). PsRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, 155–159. doi: 10.1093/nar/gkr319
Dharmasiri, S., and Estelle, M. (2002). The role of regulated protein degradation in auxin response. Plant Mol. Biol. 49, 401–409. doi: 10.1023/A:1015203013208
Ding, D., Zhang, L., Wang, H., Liu, Z., Zhang, Z., and Zheng, Y. (2009). Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot. 103, 29–38. doi: 10.1093/aob/mcn205
Ding, J., Zhou, S., and Guan, J. (2012). Finding MicroRNA targets in plants: current status and perspectives. Genomics Proteomics Bioinformatics 10, 264–275. doi: 10.1016/j.gpb.2012.09.003
Ding, Y., Tao, Y., and Zhu, C. (2013). Emerging roles of microRNAs in the mediation of drought stress response in plants. J. Exp. Bot. 64, 3077–3086. doi: 10.1093/jxb/ert164
Frazier, T. P., Sun, G., Burklew, C. E., and Zhang, B. (2011). Salt and drought stresses induce the aberrant expression of microRNA genes in tobacco. Mol. Biotechnol. 49, 159–165. doi: 10.1007/s12033-011-9387-5
Griffiths-Jones, S., Saini, H. K., Van Dongen, S., and Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, 154–158. doi: 10.1093/nar/gkm952
Ha, S., Vankova, R., Yamaguchi-Shinozaki, K., Shinozaki, K., and Tran, L. S. P. (2012). Cytokinins: metabolism and function in plant adaptation to environmental stresses. Trends Plant Sci. 17, 172–179. doi: 10.1016/j.tplants.2011.12.005
Hansen, S., Clay, S. A., Clay, D. E., Carlson, C. G., Reicks, G., Jarachi, Y., et al. (2013). Landscape features impact on soil available water, corn biomass, and gene expression during the late vegetative stage. Plant Genome 6, 1–9. doi: 10.3835/plantgenome2012.11.0029
Hayano-Kanashiro, C., Calderón-Vásquez, C., Ibarra-Laclette, E., Herrera-Estrella, L., and Simpson, J. (2009). Analysis of gene expression and physiological responses in three Mexican maize landraces under drought stress and recovery irrigation. PLoS ONE 4:e7531. doi: 10.1371/journal.pone.0007531
Jones-Rhoades, M. W., Bartel, D. P., and Bartel, B. (2006). MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 57, 19–53. doi: 10.1146/annurev.arplant.57.032905.105218
Juarez, M. T., Kui, J. S., Thomas, J., Heller, B. A., and Timmermans, M. C. P. (2004). microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428, 84–88. doi: 10.1038/nature02363
Kakumanu, A., Ambavaram, M. M. R., Klumas, C., Krishnan, A., Batlang, U., Myers, E., et al. (2012). Effects of drought on gene expression in maize reproductive and leaf meristem tissue revealed by RNA-Seq. Plant Physiol. 160, 846–867. doi: 10.1104/pp.112.200444
Kang, M., Zhao, Q., Zhu, D., and Yu, J. (2012). Characterization of microRNAs expression during maize seed development. BMC Genomics 13:360. doi: 10.1186/1471-2164-13-360
Kim, J. H., Choi, D., and Kende, H. (2003). The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant J. 36, 94–104. doi: 10.1046/j.1365-313X.2003.01862.x
Kim, J. Y., Lee, H. J., Jung, H. J., Maruyama, K., Suzuki, N., and Kang, H. (2010). Overexpression of microRNA395c or 395e affects differently the seed germination of Arabidopsis thaliana under stress conditions. Planta 232, 1447–1454. doi: 10.1007/s00425-010-1267-x
Kong, X., Zhang, M., Xu, X., Li, X., Li, C., and Ding, Z. (2014). System analysis of microRNAs in the development and aluminium stress responses of the maize root system. Plant Biotechnol. J. 12, 1108–1121. doi: 10.1111/pbi.1221
Kozomara, A., and Griffiths-Jones, S. (2011). MiRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, 152–157. doi: 10.1093/nar/gkq1027
Kramer, M. F. (2011). Stem-loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. Chapter 15:Unit 15.10. doi: 10.1002/0471142727.mb1510s95
Kurihara, Y., and Watanabe, Y. (2004). Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758. doi: 10.1073/pnas.0403115101
Langridge, P., and Reynolds, M. P. (2015). Genomic tools to assist breeding for drought tolerance. Curr. Opin. Biotechnol. 32, 130–135. doi: 10.1016/j.copbio.2014.11.027
Lee, Y., Kim, M., Han, J., Yeom, K.-H., Lee, S., Baek, S. H., et al. (2004). MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 23, 4051–4060. doi: 10.1038/sj.emboj.7600385
Li, J. S., Fu, F. L., Ming, A. N., Zhou, S. F., She, Y. H., and Li, W. C. (2013). Differential expression of microRNAs in response to drought stress in maize. J. Integr. Agricult. 12, 1414–1422. doi: 10.1016/S2095-3119(13)60311-1
Li, Y., Sun, C., Huang, Z., Pan, J., Wang, L., and Fan, X. (2009). Mechanisms of progressive water deficit tolerance and growth recovery of chinese maize foundation genotypes huangzao 4 and chang 7-2, which are proposed on the basis of comparison of physiological and transcriptomic responses. Plant Cell Physiol. 50, 2092–2111. doi: 10.1093/pcp/pcp145
Liu, D., Song, Y., Chen, Z., and Yu, D. (2009). Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 136, 223–236. doi: 10.1111/j.1399-3054.2009.01229.x
Liu, H.-H., Tian, X., Li, Y.-J., Wu, C.-A., and Zheng, C.-C. (2008). Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14, 836–843. doi: 10.1261/rna.895308
Liu, H., Qin, C., Chen, Z., Zuo, T., Yang, X., Zhou, H., et al. (2014). Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 15:25. doi: 10.1186/1471-2164-15-25
Liu, P. P., Montgomery, T. A., Fahlgren, N., Kasschau, K. D., Nonogaki, H., and Carrington, J. C. (2007). Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J. 52, 133–146. doi: 10.1111/j.1365-313X.2007.03218.x
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H. F., Dong, H. T., Sun, C. B., Qing, D. J., Li, N., Wu, Z. K., et al. (2011). The panorama of physiological responses and gene expression of whole plant of maize inbred line YQ7-96 at the three-leaf stage under water deficit and re-watering. Theor. Appl. Genet. 123, 943–958. doi: 10.1007/s00122-011-1638-0
Luo, M., Liu, J., Lee, R. D., Scully, B. T., and Guo, B. (2010). Monitoring the Expression of Maize Genes in Developing Kernels under Drought Stress using Oligo-microarray. J. Integr. Plant Biol. 52, 1059–1074. doi: 10.1111/j.1744-7909.2010.01000.x
Luo, Y. C., Zhou, H., Li, Y., Chen, J. Y., Yang, J. H., Chen, Y. Q., et al. (2006). Rice embryogenic calli express a unique set of microRNAs, suggesting regulatory roles of microRNAs in plant post-embryogenic development. FEBS Lett. 580, 5111–5116. doi: 10.1016/j.febslet.2006.08.046
Lv, D. K., Bai, X., Li, Y., Ding, X. D., Ge, Y., Cai, H., et al. (2010). Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene 459, 39–47. doi: 10.1016/j.gene.2010.03.011
Marino, R., Ponnaiah, M., Krajewski, P., Frova, C., Gianfranceschi, L., Pè, M. E., et al. (2009). Addressing drought tolerance in maize by transcriptional profiling and mapping. Mol. Genet. Genomics 281, 163–179. doi: 10.1007/s00438-008-0401-y
Meng, Y., Ma, X., Chen, D., Wu, P., and Chen, M. (2010). MicroRNA-mediated signaling involved in plant root development. Biochem. Biophys. Res. Commun. 393, 345–349. doi: 10.1016/j.bbrc.2010.01.129
Min, H., Chen, C., Wei, S., Shang, X., Sun, M., Xia, R., et al. (2016). Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 7:1080. doi: 10.3389/fpls.2016.01080
Nepolean, T., Sharma, R., Singh, N., Shiriga, K., Mohan, S., Mittal, S., et al. (2017). Genome-wide expression and functional interactions of genes under drought stress in maize. Int J. Genomics 2017:2568706. doi: 10.1155/2017/2568706
Nogueira, F. T., Chitwood, D. H., Madi, S., Ohtsu, K., Schnable, P. S., Scanlon, M. J., et al. (2009). Regulation of small RNA accumulation in the maize shoot apex. PLoS Genet. 5:e1000320. doi: 10.1371/journal.pgen.1000320
Nozawa, M., Miura, S., and Nei, M. (2012). Origins and evolution of microRNA genes in plant species. Genome Biol. Evol. 4, 230–239. doi: 10.1093/gbe/evs002
Pant, B. D., Buhtz, A., Kehr, J., and Scheible, W. R. (2008). MicroRNA399 is a long-distance signal for the regulation of plant phosphate homeostasis. Plant J. 53, 731–738. doi: 10.1111/j.1365-313X.2007.03363.x
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Park, M. Y., Wu, G., Gonzalez-Sulser, A., Vaucheret, H., and Poethig, R. S. (2005). Nuclear processing and export of microRNAs in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 102, 3691–3696. doi: 10.1073/pnas.0405570102
Park, W., Scheffler, B. E., Bauer, P. J., and Campbell, B. T. (2010). Identification of the family of aquaporin genes and their expression in upland cotton (Gossypium hirsutum L.). BMC Plant Biol. 10:142. doi: 10.1186/1471-2229-10-142
Praba, M. L., Cairns, J. E., Babu, R. C., and Lafitte, H. R. (2009). Identification of physiological traits underlying cultivar differences in drought tolerance in rice and wheat. J. Agron. Crop Sci. 195, 30–46. doi: 10.1111/j.1439-037X.2008.00341.x
Regulski, M., Lu, Z., Kendall, J., Donoghue, M. T. A., Reinders, J., Llaca, V., et al. (2013). The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 23, 1651–1662. doi: 10.1101/gr.153510.112
Rehmsmeier, M., Steffen, P., Höchsmann, M., Giegerich, R., and Ho, M. (2004). Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–1517. doi: 10.1261/rna.5248604
Reyes, J. L., and Chua, N. H. (2007). ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 49, 592–606. doi: 10.1111/j.1365-313X.2006.02980.x
Rhoades, M. W., Reinhart, B. J., Lim, L. P., Burge, C. B., Bartel, B., and Bartel, D. P. (2002). Prediction of plant microRNA targets. Cell 110, 513–520. doi: 10.1016/S0092-8674(02)00863-2
Rosegrant, M. W., and Cline, S. A. (2003). Global food security: challenges and policies. Science 302, 1917–1919. doi: 10.1126/science.1092958
Schwab, R., Palatnik, J. F., Riester, M., Schommer, C., Schmid, M., and Weigel, D. (2005). Specific effects of microRNAs on the plant transcriptome. Dev. Cell 8, 517–527. doi: 10.1016/j.devcel.2005.01.018
Shahmuradov, I. A., Solovyev, V. V., and Gammerman, A. J. (2005). Plant promoter prediction with confidence estimation. Nucleic Acids Res. 33, 1069–1076. doi: 10.1093/nar/gki247
Shikha, M., Kanika, A., Rao, A. R., Mallikarjuna, M. G., Gupta, H. S., and Nepolean, T. (2017). Genomic selection for drought tolerance using genome-wide SNPs in maize. Front. Plant Sci. 8:550. doi: 10.3389/fpls.2017.00550
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Souvorov, A., Kapustin, Y., Kiryutin, B., Chetvernin, V., Tatusova, T., and Lipman, D. (2010). Gnomon–NCBI Eukaryotic Gene Prediction Tool. Bethesda, MD: National Center for Biotechnology Information.
Turner, M., Adhikari, S., and Subramanian, S. (2013). Optimizing stem-loop qPCR assays through multiplexed cDNA synthesis of U6 and miRNAs. Plant Signal. Behav. 8:e24918. doi: 10.4161/psb.24918
Vaucheret, H., Vazquez, F., Crété, P., and Bartel, D. P. (2004). The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 18, 1187–1197. doi: 10.1101/gad.1201404
Vazquez, F., Gasciolli, V., Crété, P., and Vaucheret, H. (2004). The nuclear dsRNA binding protein HYL1 is required for microRNA accumulation and plant development, but not posttranscriptional transgene silencing. Curr. Biol. 14, 346–351. doi: 10.1016/S0960-9822(04)00047-8
Wang, B., Sun, Y. F., Song, N., Wei, J. P., Wang, X. J., Feng, H., et al. (2014). MicroRNAs involving in cold, wounding and salt stresses in Triticum aestivum L. Plant Physiol. Biochem. 80, 90–96. doi: 10.1016/j.plaphy.2014.03.020
Wang, J. F., Zhou, H., Chen, Y. Q., Luo, Q. J., and Qu, L. H. (2004). Identification of 20 microRNAs from Oryza sativa. Nucleic Acids Res. 32, 1688–1695. doi: 10.1093/nar/gkh332
Wang, J. W., Czech, B., and Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, Y. G., An, M., Zhou, S. F., She, Y. H., Li, W. C., and Fu, F. L. (2014). Expression profile of maize microRNAs corresponding to their target genes under drought stress. Biochem. Genet. 52, 474–493. doi: 10.1007/s10535-016-0590-x
Wei, L., Zhang, D., Xiang, F., and Zhang, Z. (2009). Differentially expressed miRNAs potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int. J. Plant Sci. 170, 979–989. doi: 10.1086/605122
Wu, F., Shu, J., and Jin, W. (2014). Identification and validation of miRNAs associated with the resistance of maize (Zea mays L.) to Exserohilum turcicum. PLoS ONE 9:e87251. doi: 10.1371/journal.pone.0087251
Xie, Z., Allen, E., Fahlgren, N., Calamar, A., Givan, S. A., and Carrington, J. C. (2005). Expression of Arabidopsis MIRNA genes. Plant Physiol. 138, 2145–2154. doi: 10.1104/pp.105.062943
Xu, J., Li, Y., Wang, Y., Liu, X., and Zhu, X. G. (2017). Altered expression profiles of microRNA families during de-etiolation of maize and rice leaves. BMC Res. Notes 10:108. doi: 10.1186/s13104-016-2367-x
You, F. M., Huo, N., Gu, Y. Q., Luo, M.-C., Ma, Y., Hane, D., et al. (2008). BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 9:253. doi: 10.1186/1471-2105-9-253
Yu, B., Yang, Z. Y., Li, J. J., Minakhina, S., Yang, M. C., Padgett, R. W., et al. (2005). Methylation as a crucial step in plant microRNA biogenesis. Science 307, 932–935. doi: 10.1126/science.1107130
Yu, L.-X. (2003). Comparative transcriptional profiling of placenta and endosperm in developing maize kernels in response to water deficit. Plant Physiol. 131, 568–582. doi: 10.1104/pp.014365
Yue, G., Zhuang, Y., Li, Z., Sun, L., and Zhang, J. (2008). Differential gene expression analysis of maize leaf at heading stage in response to water-deficit stress. Biosci. Rep. 28, 125–134. doi: 10.1042/BSR20070023
Zhai, L., Liu, Z., Zou, X., Jiang, Y., Qiu, F., Zheng, Y., et al. (2013). Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol Plant. 147, 181–193. doi: 10.1111/j.13993054.2012.01653.x
Zhang, Z., Wei, L., Zou, X., Tao, Y., Liu, Z., and Zheng, Y. (2008). Submergence-responsive microRNAs are potentially involved in the regulation of morphological and metabolic adaptations in maize root cells. Ann. Bot. 102, 509–519. doi: 10.1093/aob/mcn129
Zhao, B., Liang, R., Ge, L., Li, W., Xiao, H., Lin, H., et al. (2007). Identification of drought-induced microRNAs in rice. Biochem. Biophys. Res. Commun. 354, 585–590. doi: 10.1016/j.bbrc.2007.01.022
Zheng, J., Fu, J., Gou, M., Huai, J., Liu, Y., Jian, M., et al. (2010). Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 72, 407–421. doi: 10.1007/s11103-009-9579-6
Keywords: drought, gene expression, maize, miRNA, mRNA, post-transcriptional changes
Citation: Aravind J, Rinku S, Pooja B, Shikha M, Kaliyugam S, Mallikarjuna MG, Kumar A, Rao AR and Nepolean T (2017) Identification, Characterization, and Functional Validation of Drought-responsive MicroRNAs in Subtropical Maize Inbreds. Front. Plant Sci. 8:941. doi: 10.3389/fpls.2017.00941
Received: 20 February 2017; Accepted: 19 May 2017;
Published: 02 June 2017.
Edited by:
Purificación Lisón, Universitat Politècnica de València, SpainReviewed by:
Hao Li, Northwest A&F University, ChinaAnil Kumar Singh, ICAR-Indian Institute of Agricultural Biotechnology, India
Copyright © 2017 Aravind, Rinku, Pooja, Shikha, Kaliyugam, Mallikarjuna, Kumar, Rao and Nepolean. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thirunavukkarasu Nepolean, dG5lcG9sZWFuQHlhaG9vLmNvbQ==; dG5lcG9sZWFuQGdtYWlsLmNvbQ==