 Paolo Baldi
Paolo Baldi Nicola La Porta
Nicola La Porta- 1IASMA Research and Innovation Centre, Fondazione Edmund Mach, Trento, Italy
- 2MOUNTFOR Project Centre, European Forest Institute, Trento, Italy
In the never ending struggle against plant pathogenic bacteria, a major goal is the early identification and classification of infecting microorganisms. Xylella fastidiosa, a Gram-negative bacterium belonging to the family Xanthmonadaceae, is no exception as this pathogen showed a broad range of vectors and host plants, many of which may carry the pathogen for a long time without showing any symptom. Till the last years, most of the diseases caused by X. fastidiosa have been reported from North and South America, but recently a widespread infection of olive quick decline syndrome caused by this fastidious pathogen appeared in Apulia (south-eastern Italy), and several cases of X. fastidiosa infection have been reported in other European Countries. At least five different subspecies of X. fastidiosa have been reported and classified: fastidiosa, multiplex, pauca, sandyi, and tashke. A sixth subspecies (morus) has been recently proposed. Therefore, it is vital to develop fast and reliable methods that allow the pathogen detection during the very early stages of infection, in order to prevent further spreading of this dangerous bacterium. To this purpose, the classical immunological methods such as ELISA and immunofluorescence are not always sensitive enough. However, PCR-based methods exploiting specific primers for the amplification of target regions of genomic DNA have been developed and are becoming a powerful tool for the detection and identification of many species of bacteria. The aim of this review is to illustrate the application of the most commonly used PCR approaches to X. fastidiosa study, ranging from classical PCR, to several PCR-based detection methods: random amplified polymorphic DNA (RAPD), quantitative real-time PCR (qRT-PCR), nested-PCR (N-PCR), immunocapture PCR (IC-PCR), short sequence repeats (SSRs, also called VNTR), single nucleotide polymorphisms (SNPs) and multilocus sequence typing (MLST). Amplification and sequence analysis of specific targets is also mentioned. The fast progresses achieved during the last years in the DNA-based classification of this pathogen are described and discussed and specific primers designed for the different methods are listed, in order to provide a concise and useful tool to all the researchers working in the field.
Introduction
Xylella fastidiosa is a Gram-negative, slow growing and strictly aerobic bacterium in the family Xanthmonadaceae. It is a widely distributed plant pathogen as it can colonize the xylem of many different species, causing a variety of diseases such as Pierce's disease (PD) in grape (Vitis vinifera) or citrus variegated chlorosis (CVC) (Purcell, 2013). X. fastidiosa can move upstream and downstream along plant xylem, thanks to the presence of long type IV pili (Li et al., 2007). The bacteria actively multiply until, in the later stages of infection, they block the plant xylem by forming a biofilm. As a consequence, water stress and nutritional deficiencies can occur in the host plant, causing the appearance of disease symptoms (Hopkins, 1989). The first report about a disease caused by X. fastidiosa dates back to the end of the nineteenth century, when the so called “California vine disease” destroyed about 14,000 ha of grapes in the Los Angeles area (CA, USA). Newton Pierce (1856–1916), a bacteriologist, was assigned to study the epidemic and even though he was not able to identify the causal agent, he came to the conclusion that the disease was likely caused by a microscopic infectious agent (Pierce, 1892). The disease was named Pierce's disease in 1939, in a bulletin of the California Department of Agriculture (Gardner, 1974), but for a long time the etiological agent was thought to be a virus, until it was recognized as a bacterium in 1973 (Goheen et al., 1973; Hopkins and Mollenhauer, 1973). Pure cultures of the bacterium were isolated from grape in 1978 (Davis et al., 1978) and finally, in 1987, the causal agent of PD was properly classified and named Xylella fastidiosa (Wells et al., 1987).
Since the first report in grape, X. fastidiosa was isolated and identified from an increasingly large number of plant hosts, with or without symptoms, and recognized to be the causal agent of different diseases(Moller et al., 1974; Hearon et al., 1980; Chang et al., 1993; Grebus et al., 1996; McElrone et al., 1999; Hopkins and Purcell, 2002; Montero-Astua et al., 2008). In many cases also wild plant species were found to carry this pathogen, but often in a latent stage only (Freitag, 1951; Raju et al., 1983; Hopkins and Adlerz, 1988; Blake, 1993; Hill and Purcell, 1997; Li et al., 2001). The distribution range of X. fastidiosa is usually limited to tropical and subtropical areas, being its optimal growing temperature 26–28°C (Feil and Purcell, 2001). In some cases strains of X. fastidiosa have been found in much colder countries, such as Canada (Goodwin and Zhang, 1997), even if usually this pathogen does not occur in areas with low winter temperatures, such as New York and the Pacific Northwest of USA, or at high altitudes (Hopkins and Purcell, 2002). To date, most of the diseases caused by X. fastidiosa have been reported from North and South America. According to the EPPO Global Database (https://gd.eppo.int/taxon/XYLEFA/distribution), only few cases have been reported outside this area, like in Yugoslavia (Berisha et al., 1998), Switzerland (EPPO, 2015a), France (EPPO, 2015b; Marcelletti and Scortichini, 2016b; Denancé et al., 2017), Germany (EPPO, 2016), Iran (Amanifar et al., 2014), and Taiwan (Leu and Su, 1993). In Europe X. fastidiosa was first recorded in Puglia region (southern Italy, province of Lecce), where it was recognized to be the causal agent of a dangerous disease of olive trees, the so-called olive quick decline syndrome (OQDS) (Elbeaino et al., 2014; Loconsole et al., 2014).
In the beginning X. fastidiosa was regarded as an extended group of bacteria capable of infecting a wide range of host plants and it was only in the early nineties, with the introduction of DNA-based genotyping techniques, that the researchers started to divide the species into different genetic groups (Chen et al., 1992). To date, at least five different subspecies of X. fastidiosa have been reported and classified: X. fastidiosa subsp. fastidiosa, in one report called subsp. piercei and then corrected to fastidiosa according to the rules (Rule 13d) of the International Code of Nomenclature of Bacteria (Schaad et al., 2004); X. fastidiosa subsp. multiplex; X. fastidiosa subsp. pauca; X. fastidiosa subsp. Sandyi, and X. fastidiosa subsp. tashke (Schaad et al., 2004; Randall et al., 2009; Janse and Obradovic, 2010). The subsp. sandyi is associated with disease in oleander, Jacaranda spp., daylily and magnolia (Schuenzel et al., 2005; Hernandez-Martinez et al., 2007). Recently, a new subspecies, X. fastidiosa subsp. morus, has been proposed (Nunney et al., 2014c). Moreover, it cannot be ruled out that other subspecies still exist, as till now most of the studies about the genetic diversity of X. fastidiosa have been performed on cultivated crops of relevant economic importance, while little is known about the strains that colonize wild grasses, sedges and forest trees. Therefore, the development and application of molecular methods to the study of X. fastidiosa's genetic diversity can be of primary importance to fill such gap and extend our knowledge about the true diversity of this organism.
Many sap-feeding insects can function as vectors for the transmission of X. fastidiosa to host plants, especially sharpshooters and froghoppers or spittlebugs (Cicadellidae) (Janse and Obradovic, 2010; Bhowmick et al., 2016). After acquisition from the source plant, the bacterium is persistent in the vector (Severin, 1949) and can multiply in the foregut (Brlansky et al., 1983; Hill and Purcell, 1995). The process of acquisition and transmission of X. fastidiosa by the vector is very complex and can be dependent on many variables such as host plant, vector species and bacterium subspecies in interaction environmental variables, first at all climate. Nevertheless, one important factor has been proven to influence the efficiency of acquisition, that is X. fastidiosa population size (number of live cells per gram of plant tissue) (Hill and Purcell, 1997). In fact, the feeding apparatus of vectors is not an easy environment to be colonized, due to the fast flow of sap, that was estimated to reach an average speed of 8 cm/s (Purcell et al., 1979) and the possible turbulence caused by the fast contractions of the muscles allowing the insect to pump sap from the plant, that are contracted and relaxed approximately once every second (Dugravot et al., 2008). Therefore, it is likely that only few cells, out of the thousands acquired by the vector when feeding from an infected plant, actually succeed to colonize the vector's foregut. Another factor that can influence the efficiency of X. fastidiosa transmission, and particularly inoculation, is a long access period of the vector to the plant, as probably longer periods allow the insects to deliver a greater number of bacteria, having time to generate a large number of inoculation events (Almeida and Purcell, 2003).
The initial belief that X. fastidiosa was a generalist pathogen capable of infecting a very large range of host plants has gradually changed with the discovery and characterization of genetically different strains of X. fastidiosa, each capable of infecting distinct hosts. Also the infection characteristics can vary considerably in different species. In some hosts the bacteria multiply locally but cannot move and eventually the infection can regress spontaneously (Purcell and Saunders, 1999). In the past, reciprocal transmission tests have been conducted for X. fastidiosa strains. For example, the strains infecting grape cannot infect peach (Prunus persica) and peach strains cannot infect grape (Hopkins, 1989). Similarly, grape strains cannot infect oleander (Nerium oleander) and vice-versa (Purcell et al., 1999), while a CVC strain produced leaf scorch disease in coffee (Coffea arabica) (Li et al., 2001) and CVC and coffee strains can infect grape (Hopkins and Purcell, 2002). Natural isolates produced leaf scorch disease in American elm tree but failed in reciprocal transmission in sycamore (Sherald, 1993). However, in this puzzling host/pathogen interaction the host specificity of X. fastidiosa is probably a genetically-controlled character, even though in the genome of this bacterium no genes coding for effector proteins were found, nor it is present a type III secretion system (Van Sluys et al., 2003). Pathogenicity factors have been found in X. fastidiosa under the control of a cell-cell signaling system, similar but not equal to the one found in the sister genus Xanthomonas (Chatterjee et al., 2008). By altering such cell-cell signaling system it is possible to modify X. fastidiosa host specificity (Killiny and Almeida, 2011).
Hosts and Diseases
As already mentioned above, X. fastidiosa can infect a great number of plant species. A partial list of the main hosts is shown in Table 1 (EFSA, 2016). At present, according to the European Food Safety Authority (EFSA), the updated list of X. fastidiosa hosts consists of 359 plant species (including hybrids) from 75 different plant families (EFSA, 2016). Even if the infection process is always the same, the symptoms and the diseases caused by X. fastidiosa may vary among species. In addition to PD (Stevenson et al., 2005), CVC and ALS, the most known and dangerous diseases are phony peach disease (PPD) in peach and a number of leaf scorch diseases such as oleander leaf scorch (OLS), coffee leaf scorch (CLS) and plum (Prunus domestica) leaf scald (PLS).
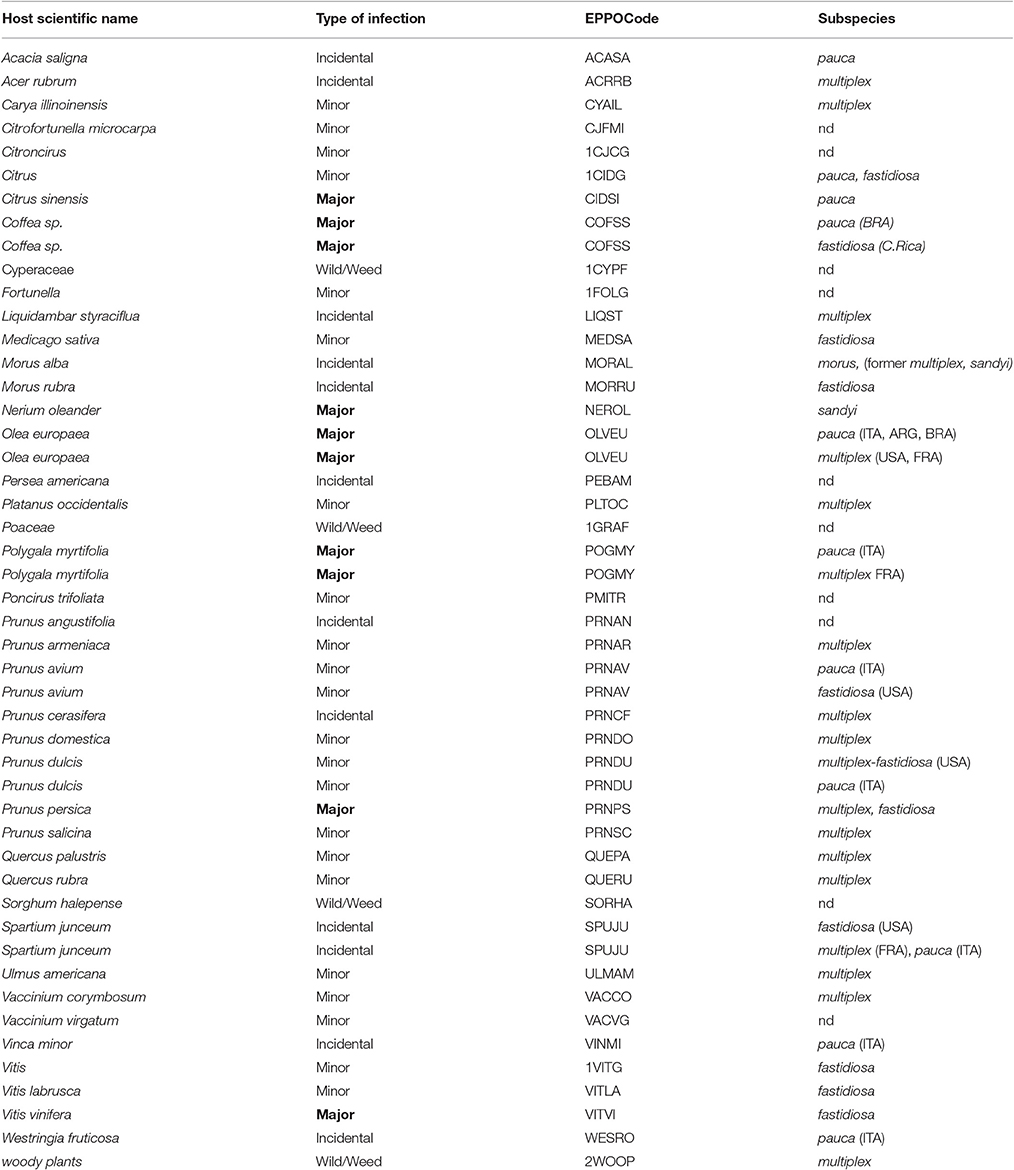
Table 1. Partial list of the main plant hosts of Xilella fastidiosa and their X. fastidiosa subspecies.
Grape (PD)
Symptoms of Pierce's disease may vary according to the characteristics of the infected cultivar as well as to the time of infection and seasonal factors (Goheen, 1988). Plants that were infected the previous growing season will generally show more severe symptoms when compared to those infected only in the current season. The first signs of the disease are a sudden drying and yellowing of the leaf margins (a red band can be present in red cultivars), due to the occlusion of leaf veins by bacterial infestation (Hopkins, 1981; Newman et al., 2003). Eventually, the affected (scorched) leaves will fall, usually from the distal part of the petiole, leaving the leaf stems attached to the cane (matchsticks) (Stevenson et al., 2005). Severely infected plants can be completely defoliated within late summer. During the first year of infection, the symptoms are limited to one or few shoots, but get worse over time. The seasonal concentration of bacteria in infected tissues is variable, being at a maximum in late spring and early summer. Usually, X. fastidiosa is not detectable in shoots from current season during the first 4 weeks of growth, but it's detectable in old wood (Hopkins, 1981). New wood will mature irregularly, producing patches of brown and green bark (the so called green islands), especially in the intermediate zone of a shoot, between the green tip and the browned basal part (Stevenson et al., 2005). The tips of canes may eventually die back and the new shoots will be shorter and stunted. Also fruit production will be progressively reduced, with most of the fruit clusters drying and wilting. In 1–5 years, depending on the susceptibility of the different genotypes, the infected plants usually die, even if differences may be due also to the climate and particularly to water stress (Thorne et al., 2006). Indeed, for many years most of the symptoms of PD were attributed to the limited water transport due to vessels occlusion (Hopkins, 1989) but recently it was shown that plants infected with X. fastidiosa show symptoms that are characteristic of PD and cannot be reproduced by water deficit (Thorne et al., 2006).
Citrus (CVC)
X. fastidiosa can infect any type of citrus species and hybrids but the severity of symptoms may vary according to the genotype of the host. Sweet oranges are the most susceptible, while grapefruit, mandarins, lemons, limes and trifoliate orange are only moderately susceptible (Garcia et al., 2012; Gmitter et al., 2012; Casais et al., 2014; Fadel et al., 2014). In most cases CVC is not lethal but infected trees always display a reduced vigor and growth rate and a decreased productive lifespan. Symptoms develop faster in young trees, that are also more susceptible to new infections (Garcia et al., 2012) Symptoms can be easily confused with zinc deficiency, as plants show chlorotic spots on the upper surface of leaves, especially in the interveinal area (Beretta et al., 1997). Deficiencies of P and K have been reported in leaves of CVC affected trees, together with high concentrations of Fe, Mn, and Zn (Silva-Stenico et al., 2009). On the leaves, chlorotic lesions appear on the upper side, while on the lower leaf side gummy lesions may appear, due to the production by X. fastidiosa of fastidian gum, an exopolysaccharide that was proposed to be involved in the formation of biofilms that allow the attachment and survival of bacteria inside the xylem vessels (da Silva et al., 2001). While the leaf matures, the gummy lesions can enlarge and become necrotic. Fruits are also affected and this represents one of the major problems of CVC, especially from an economical point of view. Affected fruits remain smaller (thinning does not occur on infected branches), become hard and ripen earlier. The color is the same as healthy fruits but the juice content is reduced, while acidity is higher (Gonçalves et al., 2014). Bacteria can be found also in roots of infected plants (Hopkins et al., 1991).
Peach (PPD)
Symptoms of PPD are not immediately apparent on host plants until 1 year or more from the first infection. Infected plants usually show a reduced internode length of new growth, giving to the tree a rather bushy aspect (Hutchins, 1933). Leaves become flattened and dark-green. In spring, trees infected by X. fastidiosa flower and leaf earlier than normal and hold their foliage longer in the fall (Wells et al., 1981; Hopkins and Purcell, 2002). Unlike other diseases caused by X. fastidiosa, PPD is not lethal to the infected plants. Nevertheless, PPD may cause significant damage to orchards as one of the main consequences of the infection is loss of fruit production. If trees become infected prior to production age, they will never produce fruits, while plants infected after production age will exhibit reduced production and smaller and highly colored fruits that are not suitable for the market (Evert, 1985). After 2–4 years after first infection, diseased trees may stop producing fruit at all. In peach, X. fastidiosa was detected in roots xylem fluid both in symptomatic or asymptomatic trees (Aldrich et al., 1992).
Unlike other Prunus species such as almond and plum, peach does not show leaf scorch symptoms when infected by X. fastidiosa (Ledbetter and Rogers, 2009).
Other Hosts (Scorch Diseases)
Many plant species, when infected by X. fastidiosa, show very similar symptoms, especially on the leaves, and namely: early after infection a slight chlorosis appears, usually along the margins of leaves. In some species (e.g., coffee, olive), these symptoms may affect initially younger shoots, while in others (e.g., elm) may progress from older to younger leaves (Sherald, 1993) and this may confound the early diagnosis. Symptoms on leaf petioles of infected plants colonized by X. fastidiosa varied from coffee, plum, and sweet orange in clear decreasing order of severity (Alves et al., 2004). In all cases symptoms get worse with time, even if differences may exist depending on the genetic background of the host plant, timing of inoculation and overwinter survival of the pathogen (Cao et al., 2011). The disease gradually extend from one or few branches to the entire crown and the leaves may desiccate completely and eventually fall. One of the major problems caused by X. fastidiosa infections is the decreased fruit quality and yield in commercially important crops such as coffee and olive (Rocha et al., 2010; Della Coletta et al., 2016) Plants affected by scorch disease usually show weak and stunt growth, may be more susceptible to environmental stresses, such as water or heat stress, but do not always die. Instead, they can be removed to reduce the risk of new infections or because the weakened plant has become dangerous (e.g., ornamental trees). The problem of X. fastidiosa infection is getting particularly serious in the south-eastern part of Italy on olive trees as a strain of X. fastidiosa subsp. pauca was strongly associated to a severe burst of OQDS (Saponari et al., 2013). Only few years ago (2008–2010) the first cases where reported. Nowadays, according to the last surveys, an area of at least 10,000 ha in the province of Lecce (Salento) could be infected by X. fastidiosa (Martelli et al., 2016). Only recently two olive cultivars, Leccino and Favolosa FS-17 were selected, that appear to have some degree of tolerance to the disease (Giampetruzzi et al., 2016). The introduction in Puglia of X. fastidiosa could be due to infected plant material imported from Central America (Giampetruzzi et al., 2015a; Marcelletti and Scortichini, 2016b). Other recent reports describe the presence of X. fastidiosa in olive trees showing leaf scorch symptoms in Argentina (Haelterman et al., 2015) and Brazil (Della Coletta et al., 2016). The two South American strains resulted different from each other and from the Italian strain but were both classified as belonging to the subsp. pauca. Over the last few years, the presence of X. fastidiosa outside the American continent is becoming more and more common. In October 2015, the bacterium (X. fastidiosa subsp. multiplex) was discovered in France, initially on the island of Corsica, on Polygala myrtifolia plants (ornamentals) and later on the mainland (X. fastidiosa subsp. multiplex) (EPPO, 2015b), with almost 300 foci found and nearly 30 host plant species declared contaminated (Denancé et al., 2017). In order to prevent entry and spread of X. fastidiosa within the European Union territory, specific EU phytosanitary measures have been taken (PM 7/24 (2) 2016. Xylella fastidiosa. EPPO Bull, 46: 463–500. doi: 10.1111/epp.12327).
Detection and Identification
Detection and identification of X. fastidiosa is not an easy task. First of all, these bacteria are slow-growing (up to 2–3 weeks can be necessary to obtain colonies on agar media) and secondly many common culture media are not suitable to grow X. fastidiosa strains. Instead, selective media (PD2, PW, CS20) should be used (Schaad et al., 2001). Direct identification of bacteria is possible only using dark field or phase contrast microscopy, due to the dimensions of X. fastidiosa cells (0.2–0.4 μm radius and 0.9–3.5 μm length) (Wells et al., 1987). In addition, microscopy identification requires a quite specific professional training to be efficient. Immunological methods, such as enzyme-linked immunosorbent assay (ELISA) can be used (Chang et al., 1993; Leu et al., 1998). More recently, detection of X. fastidiosa by immunofluorescence technology has been described (Carbajal et al., 2004; Buzkan et al., 2005). Anyway, most of the antibody-based detection assay are effective only at the species level, while at our knowledge a single report is available describing the development of single chain variable fragment (scFv) antibodies specific for X. fastidiosa subsp. pauca (Yuan et al., 2015). Another detection method that since the nineties has become more and more popular relies on PCR amplification of bacterial DNA. Several protocols have been developed employing different techniques, all with the use of specific primers capable of recognizing target sequences on genomic DNA. The PCR-based detection methods are usually faster and cheaper than standard plating methods and more specific than immunological methods as it is often possible to design primers specific for the desired genus, species or sub-species. Despite all these advantages, some drawbacks are still present in the use of DNA amplification methods. First of all specific equipment is required and often the protocols must be optimized in order to work properly with the specific samples and conditions of a given laboratory. Then, even when all the technical requirement are met, there is still the possibility to detect false-positive or false-negative for several reasons. For example, the PCR-based methods all allow detection of bacterial DNA, that in some cases can still persist in the environment even when the bacterial cells are no longer viable (Willerslev and Cooper, 2005). Therefore these methods alone cannot be used to distinguish between viable and non-viable bacteria, with exception of few cases (Gedalanga and Olson, 2009). The genetic material for PCR amplification can be isolated with standard protocols, which may be optimized according to the user's needs, or using several commercial kits, many of which are suitable for robotized DNA extraction and allow the simultaneous processing of up 96 samples (Smit et al., 2001). In order to avoid the laborious and time-consuming step of DNA purification, several attempts were made using directly plant sap as a template, not always with reliable results (Minsavage et al., 1994; Banks et al., 1999).
The DNA-based methods are rapidly becoming the most widely used in all modern laboratories for the detection and identification of X. fastidiosa strains infecting a number of plant species and new protocols and primers are continuously being developed. Here follows a review of the most commonly used PCR approaches (summarized in Table 2).
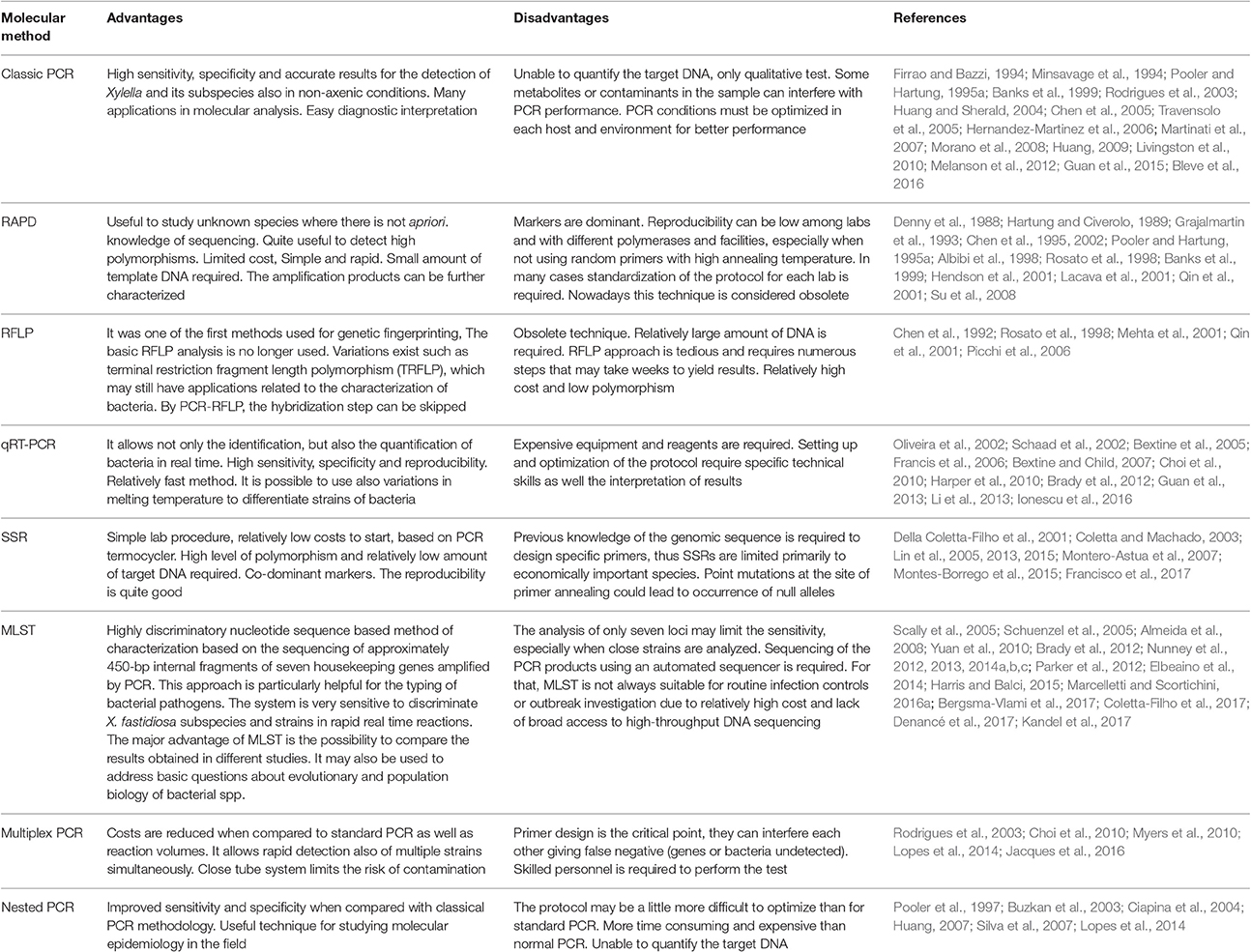
Table 2. Most commonly used PCR-based techniques for X. fastidiosa identification.
PCR Detection with Specific Primers
Since the first half of the nineties specific PCR primers have been used in order to identify X. fastidiosa from infected plants. In the first reports, the genomic region amplified could belong to a defined gene, such as 16S rDNA (Firrao and Bazzi, 1994), but also to selected fragments of bacterial DNA of unknown function (Minsavage et al., 1994). In Pooler and Hartung (1995a) developed a series of specific primers capable to detect X. fastidiosa in general and X. fastidiosa strains that cause CVC by cloning and sequencing randomly amplified polymorphic DNA (RAPD) products (Pooler and Hartung, 1995a). Since then a number of reports have been published describing the development of specific PCR primers for the identification of X. fastidiosa (Table 3) and if the 16S rDNA and the 16S-23S intergenic spacer region (ITS) are the most common targets (Rodrigues et al., 2003; Chen et al., 2005; Martinati et al., 2007), also several other genomic sequences, with or without a known function, have been used (Travensolo et al., 2005; Huang, 2009). The use of different genes, alone or in combinations, can be exploited in order to increase the level of sensitivity and specificity of the test. As a matter of fact, the early detection of pathogens before the development of any symptom by the plant host is crucial for the development of a correct defense strategy but when used for field analysis the detection test must be also as easy and fast as possible. Therefore, it is difficult to develop a single test that can be used in all conditions, but combining different approaches the researchers can find a balance among sensitivity, specificity and ease of use. As an example, the gene encoding the b-subunit polypeptide of the DNA gyrase (gyrB) was used in combination with the 16S rDNA in order to increase the specificity of the test, because gyrB is thought to evolve much faster than 16S rDNA (Yamamoto et al., 1999), therefore allowing a higher resolution when comparing closely related strains of bacteria (Rodrigues et al., 2003). For an initial genetic analysis of the population of X. fastidiosa in Texas, gyrB was used together with mopB, the latter coding for an outer membrane protein of the OmpA family with a fairly conserved sequence (Morano et al., 2008). When using a single primer pair to amplify a specific target, there is a low but non-zero possibility of false-positives due to primer recognition of non-target DNA with high sequence similarity to the target. On the contrary, false-negatives can be generated due to variations in bacterial genome, particularly to mutations in the recognition site of the primers (Scally et al., 2005). To avoid such problems, more complex approaches can be used. In a recent study, gyrB was combined with a set of housekeeping genes for the development of an array-PCR protocol that allowed to analyze a large number of samples avoiding the occurrence of false negatives due to the failure of PCR amplification of a single primer (Livingston et al., 2010).
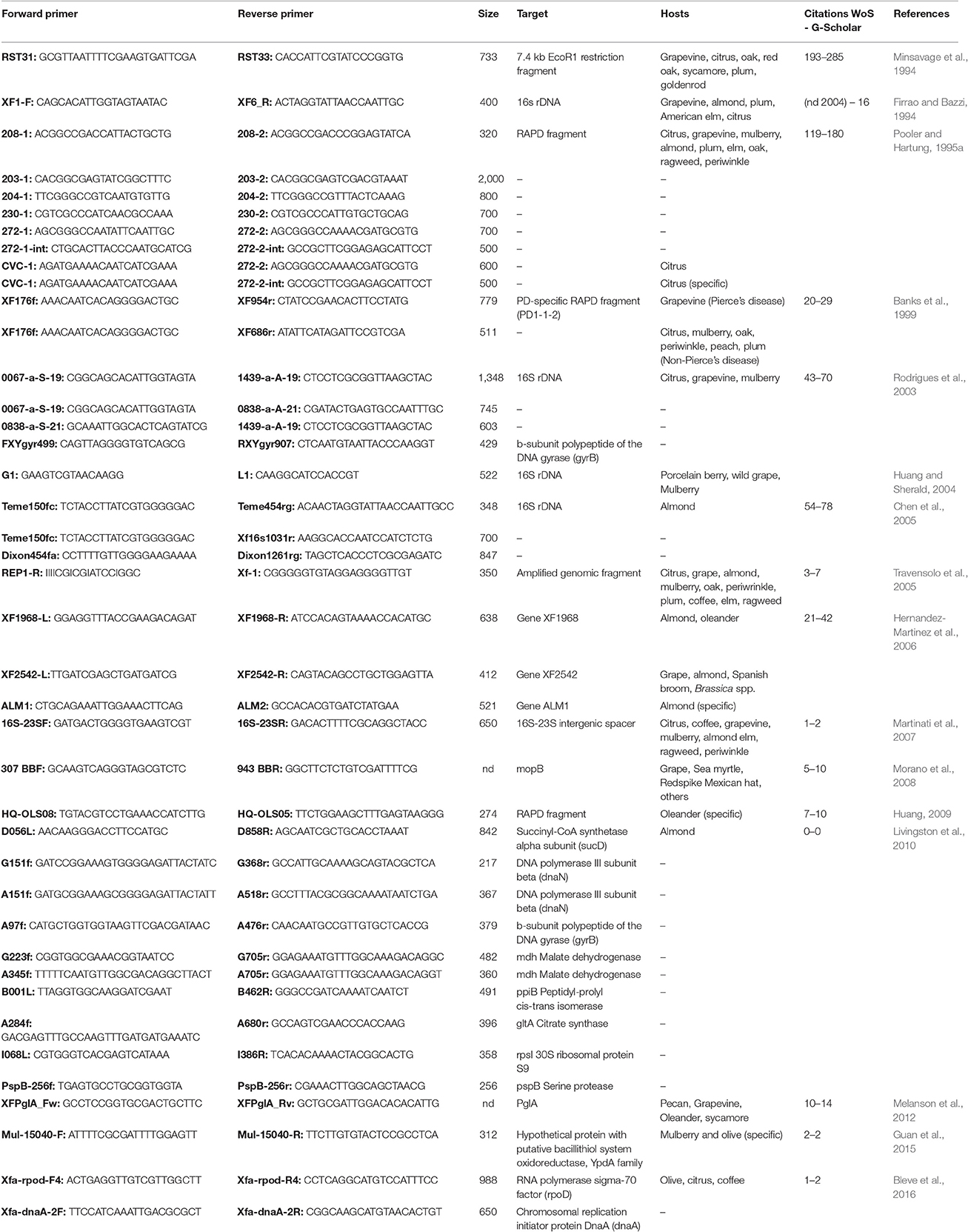
Table 3. List of specific primers designed for the PCR-based identification of X. fastidiosa.
Based on the target genomic region or selected bacterial gene, the specificity of the PCR primers used for the analysis can vary, from genus to sub-species and in some cases even to different strains belonging to the same sub-species, so it is crucial a correct primer choice in order to avoid misinterpretation of results. For X. fastidiosa analysis, a number of explicative examples can be found in literature. Starting from a pool of X. fastidiosa-specific RAPDs, Pooler and Hartung (Pooler and Hartung, 1995a) analyzed a group of 21 bacterial strains collected in different regions of USA and Brazil and developed a pair of primers capable to distinguish X. fastidiosa strains causing CVC from all the others. Similarly, Banks and colleagues designed two specific sets of primers from a PD strain collected in Florida: the first could be used to distinguish between X. fastidiosa and Xanthomonas campestris and the second to amplify a 511 bp fragment from 98 PD strains but not from CVC strains of X. fastidiosa (Banks et al., 1999). Therefore, both sets of primers can be exploited to identify X. fastidiosa but the second showed a greater specificity and can be used in more detailed studies. More recently a set of primers specific for OLS strains of X. fastidiosa was developed and tested successfully on cultured bacteria, infected plant samples and insect vectors capable of transmitting OLS (Huang, 2009). A particular case was reported when a set of primers specific for mulberry-infecting strains of X. fastidiosa was developed starting from the nucleotide sequence of a unique open reading frame identified only in mulberry-infecting strains among all the North and South American strains of X. fastidiosa sequenced at the date of that work (Guan et al., 2015). Such primers could distinguish between mulberry-infecting strains and those infecting other species, such as sycamore, elm, oak, plum, maple, and grape. Surprisingly, with the same set of primer a specific amplification could be obtained also from two isolates of X. fastidiosa belonging the recently sequenced CoDiRO strain infecting olive trees in Italy (Guan et al., 2015). This means that sometimes it can be difficult to identify unambiguously a given strain of X. fastidiosa by the use of a single approach. Even the use of advanced techniques such as multilocus sequence typing (described in more details in one of the next sections) is not always enough to distinguish among closely related strains, as for the characterization of a new X. fastidiosa strain (Salento-1) infecting olive trees in Italy (Bleve et al., 2016). In this case, the analysis of two additional genes, the polymerase sigma 70 factor (rpoD) and the chromosomal replication initiator protein DnaA (dnaA) was necessary to separate Salento-1 from closely related strains of X. fastidiosa subsp. pauca isolated from citrus and coffee in Brazil (Bleve et al., 2016).
A multiprimer combination with three different targets was developed in order to differentiate strains of X. fastidiosa infecting grape, almonds and oleander (Hernandez-Martinez et al., 2006). In this way, strains belonging to different subspecies (multiplex, fastidiosa, and sandyi) could be distinguished. Moreover, two different strains (ALSI and ALSII) belonging to subspecies multiplex and infecting almonds could be separated on the basis of amplification patterns. When the aim of the study is to characterize a new strain or a group of strains of X. fastidiosa rather than the fast and efficient identification of the pathogen under field conditions, the combination of multiple molecular techniques can greatly enhance the resolution power of the test. A good example is a study performed on pecan, where a multiprimer PCR assay was used in combination with other PCR-based techniques, such as amplification and sequence analysis of the 16S-23S ITS and pglA gene as well as enterobacterial repetitive intergenic consensus (ERIC)-PCR and repetitive extragenic palindromic (REP)-PCR. In this way, the authors were able to classify the X. fastidiosa strains infecting pecan as belonging to the subsp. multiplex (Melanson et al., 2012).
A different way to increase PCR sensitivity is the so-called nested-PCR (N-PCR) that involves the use of two different sets of primers specific for the same region in two successive runs. The second set of primers is designed to recognize a secondary target within the PCR product obtained with the first set. In Pooler et al. (1997) used N-PCR in combination with immunomagnetic separation to screen a population of 16 different species of leafhoppers, putative X. fastidiosa vectors, living on American elm (Ulmus americana L.) (Pooler et al., 1997). Two of these species regularly tested positive with this technique, with a sensitivity of as few as five bacteria per sample. Similarly, N-PCR was used in combination with immunocapture (IC) for detection of X. fastidiosa in grapevine tissue (Buzkan et al., 2003). When compared with standard non-IC-PCR, a 10,000-fold increase of sensitivity was obtained thanks to the IC procedure and a further 1,000-fold increase with N-PCR primers, achieving a maximum sensitivity of 2 cfu/ml of bacteria concentration in grape leaf extract. In another work, three different bacterial extraction and purification protocols were combined with N-PCR and use to identify alternative hosts of X. fastidiosa in the Washington D.C. area (McElrone et al., 1999). By the optimization of bacterial DNA extraction method using ionic exchange resin (Chelex 100) and N-PCR, it was possible to increase X. fastidiosa detection sensitivity from citrus plants and sharpshooter leafhoppers up to two bacteria per reaction (Ciapina et al., 2004). Therefore, once that primer pairs and PCR conditions have been optimized, N-PCR can be considered a very efficient way to increase detection sensitivity and could be used for the early detection of X. fastidiosa.
Real-Time PCR
A further improvement in PCR-based techniques of bacteria detection can be obtained by the use of quantitative real-time PCR (qRT-PCR), that allows not only the identification of the pathogen but also its quantification (Heid et al., 1996; Ionescu et al., 2016). Therefore, by qRT-PCR it is possible to study in more details the temporal and spatial distribution of X. fastidiosa in infected plants. The primers used for qRT-PCR are similar to those used for conventional PCR, while the amplification product is usually shorter that in normal PCR. Moreover, depending on the technique used (SYBR green or TaqMan), a fluorescent-labeled probe can be necessary (Heid et al., 1996; Wittwer et al., 1997) (see Supplementary Materials for a list of primers and probes used for X. fastidiosa detection). In one of the first reports published, qRT-PCR was used to quantify X. fastidiosa in naturally and artificially infected citrus (Oliveira et al., 2002). Temporal differences in bacterial cell number were detected, increasing with the age of the examined leaves. Spatial differences were also found, with no bacteria detected in the upper midrib section of young leaves. In the same work qRT-PCR was used to compare a resistant and a susceptible citrus cultivar. This is a good example of how qRT-PCR can be used to study the development of bacterial diseases in plants. By qRT-PCR it is possible for example to quantify bacteria in different plant organs at different time points after infection, correlate a given amount of bacteria to the appearance of the first symptoms and find differences between plants showing variable degrees of resistance. Nevertheless, qRT-PCR can be used also for field applications. A portable Smart Cycler for 1-h on-site diagnosis was used to detect X. fastidiosa in grape (Schaad et al., 2002). Using sap and samples of macerated chips of secondary xylem from trunks of grape trees in a direct qRT-PCR without extraction of DNA, the authors were able to positively detect X. fastidiosa in about 26% of the examined asymptomatic plants. The results were then confirmed by other techniques, such as direct isolation of bacteria. Even if these results suggest that qRT-PCR can be used effectively instead of standard PCR, it is always up to the researcher to choose the more suitable approach for each study.
As for conventional PCR methods, also for qRT-PCR the level of specificity can vary according to the genomic target and primer design. As an example, primers HL5 and HL6 (Table S1) were designed to amplify a unique region common to the sequenced genomes of four X. fastidiosa strains causing PD, ALS, OLS, and CVC. Such primers can be effectively used to distinguish between infections of X. fastidiosa and other plant pathogens and have been tested in several plant and insect species (Francis et al., 2006). In other works, primer sets were specifically developed in order to recognize oleander-infecting strains (Guan et al., 2013) or strains causing CVC (Li et al., 2013). In all, qRT-PCR showed the same advantages as conventional PCR, that are high sensitivity and specificity. Moreover, additional information about temporal and spatial distribution of the pathogen can be obtained. One of the drawbacks of this technique is that it can be more difficult to optimize the protocol, especially when using TaqMan probes. When using qRT-PCR, additional strategies can be used to distinguish among different genotypes. A protocol was developed, based on SYBR green qRT-PCT, for X. fastidiosa genotype differentiation using a single primer pair and exploiting differences in melting temperature (Tm) due to small differences of target sequence. Such protocol was tested on eight PD, six OLS, and six ALS strains that could be successfully placed into the respective strain group by the analysis of Tm curve (Bextine and Child, 2007). Similarly but using a set of fluorescent probes, Brady et al. developed a multilocus melt typing (MLMT) system to discriminate X. fastidiosa subspecies and strains in rapid real time reactions (Brady et al., 2012). These approaches are potentially very interesting, as theoretically it is possible to detect genetic variations determined by as few as a single base pair alteration. For practical uses, this high sensitivity could be a problem as small unspecific variations in Tm curve may lead to misinterpretation of the results. Therefore, a very reliable and reproducible protocol is necessary.
In order to improve sensitivity, speed and ease of detection, several approaches have been tested, such as novel DNA extraction protocols (Bextine et al., 2005; Harper et al., 2010; Yaseen et al., 2015), development of an agar absorption-based technique for elimination of PCR inhibitors (Fatmi et al., 2005) and use of multiplex PCR (Choi et al., 2010; Myers et al., 2010). A novel technique, the loop-mediated isothermal amplification (LAMP), was used as a promising alternative to PCR for X. fastidiosa detection as it is easy to use and provides a good reliability and specificity (Notomi et al., 2000). Moreover the LAMP reaction occurs at isothermal conditions, so it can be performed using a simple heat block and does not require any specific equipment. The results are displayed by colorimetric or fluorescent dyes, so the gel running phase of standard PCR is also skipped. The LAMP technique was successfully used to study 20 isolates of X. fastidiosa representing the four main subrgroups of the pathogen (Harper et al., 2010) and more recently applied to olive tree and other host plants and insect vectors (Yaseen et al., 2015). However, the main drawback of LAMP when compared with qRT-PCR is a lower sensitivity as the detection limit was about 500 copies of target template per reaction, while for qRT-PCR the limit was only 10 copies per reaction (Harper et al., 2010).
Genetic Variation
In order to characterize and differentiate strains and pathotypes of X. fastidiosa, several molecular techniques have been used. In the late eighties and early nineties, restriction fragment length polymorphism (RFLP) and randomly amplified polymorphic DNA (RAPD) analyses were largely diffused to study strain differentiations of pathogenic bacteria(Denny et al., 1988; Hartung and Civerolo, 1989; Grajalmartin et al., 1993; Cave et al., 1994; Albibi et al., 1998; da Costa et al., 2000; Ferreira et al., 2000; Mehta et al., 2000; Lacava et al., 2001; Su et al., 2008). In a study on X. fastidiosa, RFLP were used to characterize 24 strains of this pathogen from 8 different hosts (Chen et al., 1992). In the same period RAPD analysis was also used, for the ease of this technique that doesn't require any a priori knowledge of the DNA sequence (Chen et al., 1995; Pooler and Hartung, 1995b, #708). In some cases RFLP and RAPD were also used together, for example to study sweet orange plants affected by CVC (Rosato et al., 1998). In the following years, RFLP and RAPD were often used in combination with other techniques, such as REP-PCR, ERIC-PCR and contour-clamped homogeneous electric field (CHEF), in order to improve sensitivity and reliability of the test (Chu et al., 1986; Versalovic et al., 1991; de Bruijn, 1992; Mehta et al., 2001; Qin et al., 2001) The results obtained with the combined techniques always showed greater resolution than using any single method alone (Hendson et al., 2001). Simple techniques such as RAPDs, usually are not suitable to distinguish between closely related strains of X. Fastidiosa and nowadays have become quite obsolete. Nevertheless, as no complex equipment or sequencing steps are needed, RAPD analysis could still be useful for routinely analysis, where this technique usually showed good resolution. As an example, a comparison between RAPD and 16S rDNA sequence analysis was made to study three groups of X. fastidiosa strains from citrus, grape and mulberry. Phylogenetic trees were obtained separately with the two techniques. Three distinct groups, from a total of 21 strains collected in Florida, Nebraska and Brasil, were detected, causing PD, CVC and MLS, respectively, using RAPD analysis, while sequence analysis of 16S rDNA only distinguished two groups, causing PD and CVC, while the MLS group was included in the PD group (Chen et al., 2002).
Even though more advanced techniques have been developed when high resolution is needed, PCR-RFLP was used till recent years to study X. fastidiosa. Four open reading frames (ORFs) related to the restriction modification type I system, ordinarily named R–M have been characterized. The study was carried out on 43 different strains isolated from citrus, coffee, grapevine, periwinkle, almond and plum trees and allowed to define haplotypes for all the loci. By analysis of molecular variance (AMOVA) two distinct groups were obtained, the first comprising populations from citrus and coffee plants, the second including populations isolated from grapevine, periwinkle, mulberry and plum. Moreover gene transfer was detected between coffee and citrus strains coming from southern Brazil, probably due to geographic proximity (Picchi et al., 2006).
A more recent technique to study genetic variability among strains exploits the so called short sequence repeats (SSRs) that are located within the prokaryotic genome (Kremer et al., 1999). Such short repetitive regions, due to the potentially variable number of tandem repeats (VNTR), can be highly polymorphic among different strains of bacteria and therefore represent a valuable tool for molecular studies.
A set of 9 SSR markers were developed within the genome of X. fastidiosa and used for genotyping studies (Table 4), comparing the results with the RAPD method (Della Coletta et al., 2001). The genetic diversity estimated using SSR markers was considerably higher than using RAPDs, as the former specifically target hypervariable regions. Similar differences in variability were obtained when SSRs and RAPDs were used to characterize populations of X. fastidiosa isolated from citrus in two different studies, one of which comprising a total of 360 bacterial strains (Della Coletta-Filho and Machado, 2002; Coletta and Machado, 2003). A second set of 34 SSR loci was identified in a genome-wide search, specific primers were designed (Table 4) and used to evaluate genetic diversity among 43 isolates of X. fastidiosa collected from grape, almond, citrus and oleander (Lin et al., 2005). Again, SSR markers proved to be powerful tools to distinguish genetically similar isolates, as the average level of polymorphism found among the 34 SSRs was 11.3 alleles per locus. Numerous studies on X. fastidiosa have been reported using SSR markers for genotyping strains infecting different plant species, such as grape (Lin et al., 2013), sweet orange (Coletta et al., 2014), almond (Lin et al., 2015), and coffee (Francisco et al., 2017), sometimes in combination with other techniques such as RFLP (Montero-Astua et al., 2007) (Table 4). In all cases, by the analysis of multiple SSR loci, it was possible to study temporal and spatial differences between closely related populations of X. fastidiosa, as well as population structure.
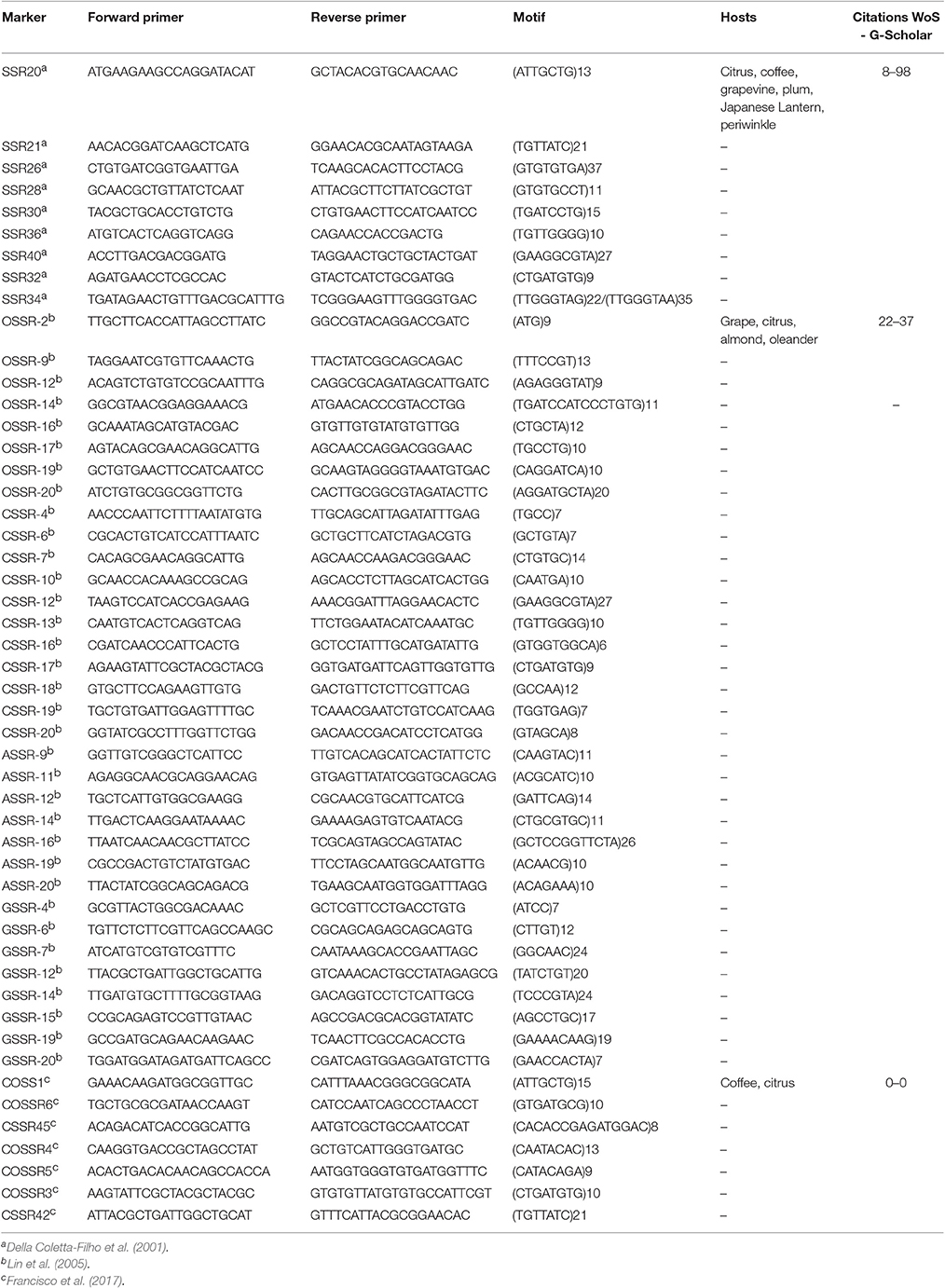
Table 4. List of SSR markers specific for X. fastidiosa.
A less common but useful technique to characterize X. fastidiosa from a molecular point of view, especially considering the increasing amount of genomic sequences available, is represented by single nucleotide polymorphisms (SNPs) (Stoneking, 2001). A genome-wide search for SNPs and insertion/deletions (INDELs) using genome sequence information from four X. fastidiosa strains was performed (Doddapaneni et al., 2006). A total of 12,754 SNPs and 14,449 INDELs in the 1528 common genes and 20,779 SNPs and 10,075 INDELs in the 194 non-coding sequences were found. SNP markers were developed from 16 distinct genomic regions of 24 strains of X. fastidiosa isolated from coffee and citrus and positively used to discriminate among genetically close genotypes (Wickert et al., 2007). Combined use of SNPs and other techniques allowed to assess genetic diversity of X. fastidiosa strains from citrus and coffee plants (Montes-Borrego et al., 2015) and olive trees (Mang et al., 2016).
Another molecular approach that can be useful to infer phylogenetic relationships in bacteria is the characterization of the rDNA genetic locus. Such locus is of extreme importance for all organisms and moreover it is conserved enough to allow a universal classification of evolutionary relationships among species (Cedergren et al., 1988; LeblondBourget et al., 1996). 16S rDNA sequence analysis was often used to study inter- and intraspecific phylogenetic relationships in X. fastidiosa. The 16S rDNA sequences from 16 strains of X. fastidiosa isolated from 9 different hosts were amplified by PCR, cloned and sequenced. The results indicated that the strains could be divided into three groups, one including PD and MLS strains, the second including PLS, PPD, OLS, ELS, and perwinkle wilt strains and the third CVC and CLS strains (Chen et al., 2000a). A 20-bp oligonucleotide from the same sequence was also identified to be highly characteristic of X. fastidiosa and different from other bacteria, including the closely related Xanthomonas genus. Therefore, 16S rDNA was proposed as a signature character for the identification of X. fastidiosa (Chen et al., 2000b). In all, 16S rDNA sequence analysis can be considered a fast and reliable method for the identification of bacteria at genus or species level. Nevertheless, when genetic distances decrease under the species level, the sequence differences found in the 16S rDNA aren't always enough to distinguish between closely related strains. A good way to partially overcome this problemis to analyze also the 16S-23S ITS.This intergenic region presents a higher variation in length and sequence than 16S rDNA and therefore can be used to increase the sensitivity of the analysis (Garcia-Martinez et al., 1999; Jeng et al., 2001). In X. fastidiosa, combined sequence analysis of 16S-23S ITS and 16S rDNA was performed in strains from grape, citrus, coffee, plum, and pear (Mehta and Rosato, 2001). The level of similarity was indeed higher for 16S rDNA (97.1–100%) than in 16S-23S ITS (79.8–100%). Phylogenetic relationships based on 16S-23S ITS sequence analysis among strains of X. fastidiosa isolated from a number of different hosts have been studied (Huang and Sherald, 2004; Martinati et al., 2005). Randall et al. (2009) have analyzed several strains of X. fastidiosa, collected from New Mexico, California and Arizona and infecting Chitalpa tashkentensis, a common ornamental landscape plant used throughout the southwestern USA. By sequence analysis of 16S rDNA and 16S-23S ITS, a differentiation of chitalpa strains from all the known X. fastidiosa subspecies was highlighted and therefore a new subspecies (subsp. tashke) was proposed. The differentiation of chitalpa strains from other known strains was further analyzed by several approaches (such as analysis of gyrB, SSRs and the virulence-associated protein VapD) that sometimes gave ambiguous results, such as for example the VapD analysis, showing that the chitalpa isolates from New Mexico were more similar to the CVC strain than to the Arizona isolates. Since the first report, the newly proposed subsp. tashke has not been reported anymore and remains poorly characterized. Moreover, it must be noted that the distinction between subspecies is not always clear (see below), depending on the method used for the characterization and the fast evolution of bacterial populations.
X. fastidiosa was the first plant bacterium to have its complete genome sequenced (Simpson et al., 2000). Since then, a lot of efforts have been made to elucidate the complete genome sequence of several X. fastidiosa strains infecting different plant hosts, such as grape (Van Sluys et al., 2003), almond (Chen et al., 2010), mulberry (Guan et al., 2014b), sycamore (Guan et al., 2014a), pear (Su et al., 2014), coffee (Giampetruzzi et al., 2015b), and olive (Giampetruzzi et al., 2015a). This huge amount of data can be used for extensive in silico analysis in order to identify similarities and differences among strains at a whole genome scale. Such approach allowed the researchers to study X. fastidiosa strains with unprecedented resolution. By whole genome approach, the complete set of unique genes present in each strain can be characterized, together with those that are common to all strains (Bhattacharyya et al., 2002; Barbosa et al., 2015). Sets of genes involved in important functions can be compared and studied (da Silva et al., 2007; Barbosa et al., 2015), SNPs can be identified and PCR primers can be designed for the identification of specific strains (Doddapaneni et al., 2006; Marcelletti and Scortichini, 2016b) and detailed phylogenetic analysis can be performed. As an example, by genome-wide comparison of 21 X. fastidiosa strains, Marcelletti and Scortichini (2016a) constructed a phylogenetic tree analyzing 820,088 nucleotides, ~30% of the entire X. fastidiosa genome. According to their results, three different, clearly defined subspecies of X: fastidiosa can be identified, while the two subsp. sandyi and morus are actually members of the subsp. fastidiosa (Marcelletti and Scortichini, 2016a). Although extremely powerful, the whole genome analysis is still a time-consuming and relatively expensive technique that cannot be considered an applicable method for a fast identification of pathogens, especially in field applications. Instead, it could be very useful to perform a whole genome study when a new site of infection is found, especially in those Countries where X. fastidiosa was never reported before, in order to efficiently classify the strain and its origin (Marcelletti and Scortichini, 2016b).
A relatively new method for bacteria identification is multilocus sequence typing (MLST). It was developed at the end of the nineties and the characterization of different strains is based on nucleotide sequence differences in a small number of housekeeping genes, typically seven (Maiden et al., 1998). Briefly, in MLST each allele of a given gene is assigned a number, so different strains of bacteria can be characterized by a series of numbers, representing one allele for each locus analyzed. The combination of the allele numbers at each locus determine the so called sequence type (ST) for each analyzed strain. The advantages of MLST are high resolution and reproducibility, fast analysis and easy interpretation of data that usually are made available in a public database (http://pubmlst.org/xfastidiosa/). Moreover MLST datasets can be used to estimate the relative contributions of recombination and point mutations in the formation of new alleles within a closely related group of strains that is defined as clonal complex (Enright and Spratt, 1998; Feil et al., 2000, 2001, 2004). MLST was applied to 25 strains of X. fastidiosa from five different host plants: grape, oleander, oak, almond and peach. An initial set of 10 sequences was used (Table 5) and all the bacterial strains were grouped into six clonal complexes corresponding to previously identified phylogenetic clades (Scally et al., 2005). The same approach was used to study the evolutionary relationships, geographic variation, and divergence times among 26 X. fastidiosa isolates from grape, oleander, almond oak, peach, plum and citrus (Schuenzel et al., 2005). MLST approach was compared to SSRs in a study on 26 CVC and 20 CLS strains (Almeida et al., 2008). Although both SSRs and MLST provided similar results, the authors concluded that SSRs are more useful to study X. fastidiosa at population level, while MLST may be more suitable for strain/subspecies studies. In the last years, MLST was largely employed in population studies of X. fastidiosa, for example to better understand the genetic diversity of subsp. fastidiosa and subsp. sandyi (Yuan et al., 2010) or to clarify the role of mutation and homologous recombination in bacterial evolution (Nunney et al., 2012). The introduction of two new taxa of X. fastidiosa into Central America was documented, together with their introgression into the native subspecies (Nunney et al., 2014b). Recently, MLST was used to better understand the epidemiology of leaf scorch disease in a urban environment, by the analysis of 101 samples isolated from 84 trees of 7 different species located in Washington D.C. (Harris and Balci, 2015). An important evolutionary issue studied by MLST is intersubspecific homologous recombination (IHR) and how it can affect X. fastidiosa host specialization and host shifts (Nunney et al., 2013, 2014a,c). As an example, by MLST analysis of 21 X. fastidiosa isolates from mulberry, it was determined that such strains represent a distinct group, different from all the others described in literature. Therefore a new subspecies was proposed (subsp. morus), originating by IHR from X. fastidiosa subsp. fastidiosa and X. fastidiosa subsp. multiplex (Nunney et al., 2014c). As mentioned above, in a study combining MLST and whole genome analysis, Marcelletti and Scortichini (2016a) proposed that the subsp. morus is included in the subsp. fastidiosa, thus contrasting with the results from Nunney et al. (2014c). It is therefore conceivable that, due to the continuous gene flow by IHR among different X. fastidiosa subspecies, in the future it will become more and more difficult to determine a clear distinction between subspecies. As a matter of fact, a new subspecies of X. fastidiosa was recently described in pears and grapevine in Taiwan (Su et al., 2012, 2013, 2016; Almeida and Nunney, 2015). However, additional genetic typing is needed to fully support the recognition of this new subspecies and to further study the isolates in Taiwan (Nunney et al., 2012; Retchless et al., 2014).
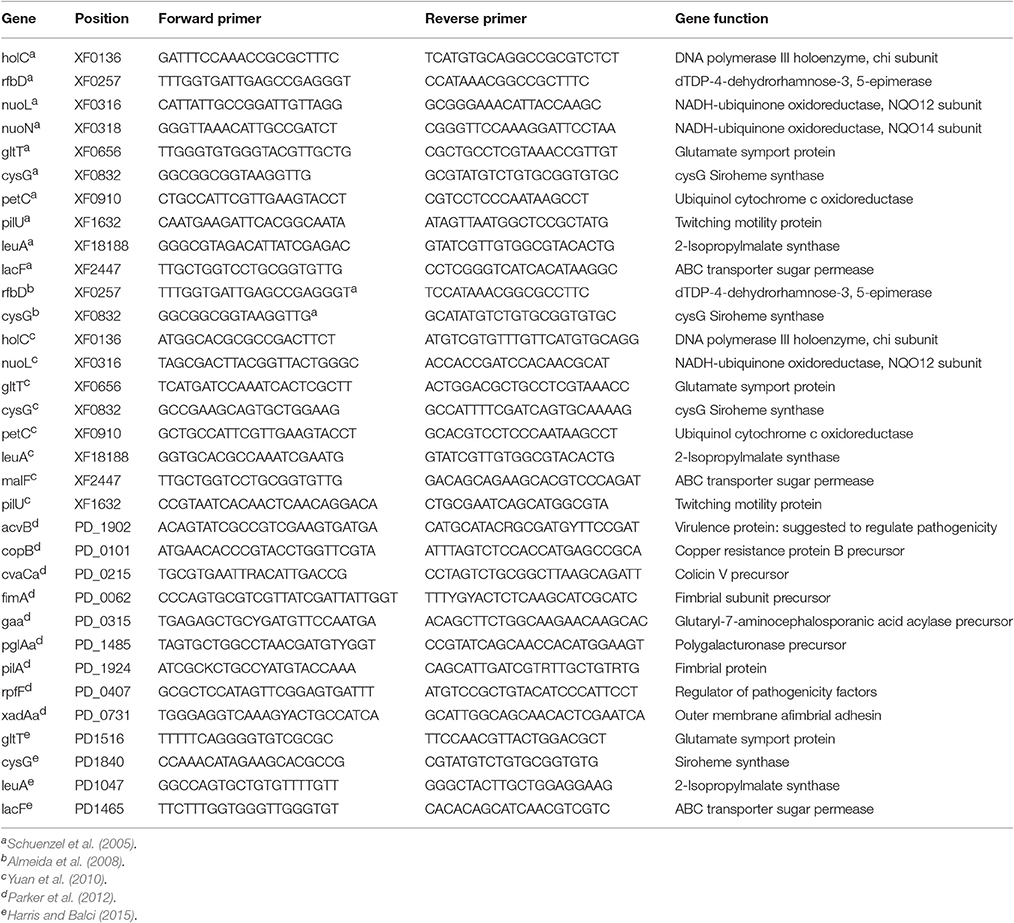
Table 5. Housekeeping genes and specific primers used for multilocus sequence typing (MLST) in X. fastidiosa.
Moreover, new variants of X. fastidiosa infecting coffee were isolated and characterized in South America (Jacques et al., 2016; Bergsma-Vlami et al., 2017). In any case, the genetic diversity of X. fastidiosa is probably broad and still poorly characterized as suggested by a very recent work, studying genetic diversity of subsp. pauca and multiplex in several locations and plant hosts in South America (Coletta-Filho et al., 2017). By MLST analysis, the authors could identify 7 new ST for subsp. pauca and 2 new ST for subsp. multiplex. Also in this work, IHR between the two subspecies was observed, suggesting that in X. fastidiosa gene flow can occur in short time, as the subsp. multiplex is thought to be introduced in South America from North America and it was first reported only in the seventies (French, 1978). Despite all the recent works suggest that MLST is a very effective technique that can be used to successfully study X. fastidiosa genetic diversity, especially at subspecies level, one possible limitation is due to the fact that the housekeeping genes used for the analysis are usually relatively conserved among strains, limiting the resolution power when studying closely related genotypes. One possible way to overcome this problem is by multilocus sequence analysis (MLSA) of genes influenced by environmental factors, termed environmentally mediated genes. Such genes are supposed to be subject to positive selection pressure and therefore to have a greater sequence variability than conserved housekeeping genes (Hoffmann and Willi, 2008). Multilocus sequence analysis of environmentally mediated genes (MLSA-E) was applied to 54 X. fastidiosa strains isolated from different species (Parker et al., 2012). Nine genes that resulted from previous studies to have a role in the infection process and to be environmentally mediated were selected, amplified by specific primers (Table 5) and sequenced. Phylogenetic results indicated that MLSA-E can increase differentiation of X. fastidiosa isolates when compared with MLST and identify novel variations within the same subspecies. In another work, MLST was combined with the analysis of 6 genes involved in different biochemical functions in order to increase the genetic information and therefore the resolution power of the experiment (Elbeaino et al., 2014). Such approach was used for characterizing X. fastidiosa isolated from olive trees affected by OQDS in Italy and the results showed that olive strain is distinct from the four previously defined species fastidiosa, multiplex, sandyi and pauca, showing the highest level of similarity with the latter. In conclusion, at present MLST is probably the best PCR-based approach to identify and classify X. fastidiosa, as testified by the wide use of this technique in analyzing the new strains identified in the recent outbreaks of this pathogen in Europe (Elbeaino et al., 2014; Marcelletti and Scortichini, 2016a; Denancé et al., 2017). The diffusion of MLST will be probably even wider in the next years as the drawbacks of the method, that are mainly due to costs and availability of an automated DNA sequencer, will be overcome very quickly by the fast evolution and cost reduction that this kind of technology is undergoing in the last years. Nevertheless, for the same reasons, it is conceivable that in the close future whole genome approaches will gradually become the classification method of choice, due to the superior level of sensitivity and amount of information that can be provided by such techniques.
Conclusion
X. fastidiosa has recently emerged as a potential threat to European agriculture after its outbreak in southern Italy were it was found associated to OQDS (Saponari et al., 2013; Cariddi et al., 2014; Elbeaino et al., 2014; Martelli et al., 2016). Among the most dangerous features of this pathogen are: (i) the host range is very wide, as X. fastidiosa is capable to infect more than 300 plant species; (ii) the initial stages of infection are often symptomless, so there's a high risk of contaminating new areas by importation of diseased plants; (iii) bacteria can be irregularly distributed in host tissues making the pathogen difficult to be detected; (iv) many insects of the Cicadellidae family are capable of transmitting X. fastidiosa, including some European species such as Cicadella viridis and Philaenus spumarius (Saponari et al., 2014). Therefore, it's become mandatory to strengthen the surveillance, looking for infected plant material, in order to limit the spread of this pathogen. Moreover, the geographic expansion of the olive epidemic area in Italy should encourage new studies to identify possible vectors and hosts of X. fastidiosa in this newly colonized environment. DNA-based molecular approaches have proven to be a valuable tool for detection and characterization of plant pathogens and in the last years they are gradually substituting the traditional serological and biochemical methods. Even though some limitations still exist, the fast development of new and more sensitive technologies, a concomitant reduction of costs and a greater ease of use, together with the increasing amount of Xylella's genomic sequences available (Simpson et al., 2000; Bhattacharyya et al., 2002; Van Sluys et al., 2003; Chen et al., 2010; Barbosa et al., 2015; Giampetruzzi et al., 2015a) will allow a further diffusion of the DNA-based methods for studying the ecology, epidemiology and taxonomy of X. fastidiosa. New researches on topics such as bacteria distribution, identification and classification of new strains, host specificity and transmission vectors will be of primary importance in order to efficiently fight this fastidious pathogen.
Author Contributions
Both authors have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was carried out by the Grant P1611034I provided by the Reseacrh and Innovation Centre, Fondazione Edmund Mach.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00944/full#supplementary-material
References
Albibi, R., Chen, J., Lamikanra, O., Banks, D., Jarret, R. L., and Smith, B. J. (1998). RAPD fingerprinting Xylella fastidiosa Pierce's disease strains isolated from a vineyard in north Florida. FEMS Microbiol. Lett. 165, 347–352. doi: 10.1111/j.1574-6968.1998.tb13168.x
Aldrich, J. H., Gould, A. B., and Martin, F. G. (1992). Distribution of Xylella fastidiosa within roots of peach. Plant Dis. 76, 885–888. doi: 10.1094/PD-76-0885
Almeida, R. P. P., Nascimento, F. E., Chau, J., Prado, S. S., Tsai, C. W., Lopes, S. A., et al. (2008). Genetic structure and biology of Xylella fastidiosa strains causing disease in citrus and coffee in Brazil. Appl. Environ. Microbiol. 74, 3690–3701. doi: 10.1128/AEM.02388-07
Almeida, R. P. P., and Nunney, L. (2015). How do plant diseases caused by Xylella fastidiosa emerge? Plant Dis. 99, 1457–1467. doi: 10.1094/PDIS-02-15-0159-FE
Almeida, R. P. P., and Purcell, A. H. (2003). Transmission of Xylella fastidiosa to grapevines by Homalodisca coagulata (Hemiptera: Cicadellidae). J. Econ. Entomol. 96, 264–271. doi: 10.1093/jee/96.2.264
Alves, E., Marucci, C. R., Lopes, J. R. S., and Leite, B. (2004). Leaf symptoms on plum, coffee and citrus and the relationship with the extent of xylem vessels colonized by Xylella fastidiosa. J. Phytopathol. 152, 291–297. doi: 10.1111/j.1439-0434.2004.00843.x
Amanifar, N., Taghavi, M., Izadpanah, K., and Babaei, G. (2014). Isolation and pathogenicity of Xylella fastidiosa from grapevine and almond in Iran. Phytopathol. Mediterr. 53, 318–327. doi: 10.14601/Phytopathol_Mediterr-12647
Banks, D., Albibi, R., Chen, J. C., Lamikanra, O., Jarret, R. L., and Smith, B. J. (1999). Specific detection of Xylella fastidiosa Pierce's disease strains. Curr. Microbiol. 39, 85–88. doi: 10.1007/s002849900423
Barbosa, D., Alencar, V. C., Santos, D. S., de Freitas Oliveira, A. C., de Souza, A. A., Coletta-Filho, H. D., et al. (2015). Comparative genomic analysis of coffee-infecting Xylella fastidiosa strains isolated from Brazil. Microbiology 161, 1018–1033. doi: 10.1099/mic.0.000068
Beretta, M. J. G., Barthe, G. A., Ceccardi, T. L., Lee, R. F., and Derrick, K. S. (1997). A survey for strains of Xylella fastidiosa in citrus affected by citrus variegated chlorosis and citrus blight in Brazil. Plant Dis. 81, 1196–1198. doi: 10.1094/PDIS.1997.81.10.1196
Bergsma-Vlami, M., Bilt, J. L. J., Tjou-Tam-Sin, N. N. A., Helderman, C. M., Gorkink-Smits, P. P. M. A., Landman, N. M., et al. (2017). Assessment of the genetic diversity of Xylella fastidiosa in imported ornamental Coffea arabica plants. Plant Pathol. doi: 10.1111/ppa.12696
Berisha, B., Chen, Y. D., Zhang, G. Y., Xu, B. Y., and Chen, T. A. (1998). Isolation of Peirce's disease bacteria from grapevines in Europe. Eur. J. Plant Pathol. 104, 427–433. doi: 10.1023/A:1008655621235
Bextine, B., Blua, M., Harshman, D., and Miller, T. A. (2005). A SYBR green-based real-time polymerase chain reaction protocol and novel DNA extraction technique to detect Xylella fastidiosa in Homalodisca coagulata. J. Econ. Entomol. 98, 667–672. doi: 10.1603/0022-0493-98.3.667
Bextine, B., and Child, B. (2007). Xylella fastidiosa genotype differentiation by SYBR Green-based QRT-PCR. FEMS Microbiol. Lett. 276, 48–54. doi: 10.1111/j.1574-6968.2007.00910.x
Bhattacharyya, A., Stilwagen, S., Ivanova, N., D'Souza, M., Bernal, A., Lykidis, A., et al. (2002). Whole-genome comparative analysis of three phytopathogenic Xylella fastidiosa strains. Proc. Natl. Acad. Sci. U.S.A. 99, 12403–12408. doi: 10.1073/pnas.132393999
Bhowmick, T. S., Das, M., Heinz, K. M., Krauter, P. C., and Gonzalez, C. F. (2016). Transmission of phage by glassy-winged sharpshooters, a vector of Xylella fastidiosa. Bacteriophage 6:e1218411. doi: 10.1080/21597081.2016.1218411
Blake, J. H. (1993). Distribution of Xylella fastidiosa in Oak, Maple and Sycamore in South-Carolina. Plant Dis. 77, 1262–1262. doi: 10.1094/PD-77-1262B
Bleve, G., Marchi, G., Ranaldi, F., Gallo, A., Cimaglia, F., Logrieco, A. F., et al. (2016). Molecular characteristics of a strain (Salento-1) of Xylella fastidiosa isolated in Apulia (Italy) from an olive plant with the quick decline syndrome. Phytopathol. Mediterr. 55, 139–146. doi: 10.14601/Phytopathol_Mediterr-17867
Brady, J. A., Faske, J. B., Ator, R. A., Castaneda-Gill, J. M., and Mitchell, F. L. (2012). Probe-based real-time PCR method for multilocus melt typing of Xylella fastidiosa strains. J. Microbiol. Methods 89, 12–17. doi: 10.1016/j.mimet.2012.02.002
Brlansky, R. H., Timmer, L. W., French, W. J., and McCoy, R. E. (1983). Colonization of the sharpshooter vectors Oncometopia-nigricans and Homalodisca-coagulata by xylem-limited bacteria. Phytopathology 73, 530–535. doi: 10.1094/Phyto-73-530
Buzkan, N., Kocsis, L., and Walker, M. A. (2005). Detection of Xylella fastidiosa from resistant and susceptible grapevine by tissue sectioning and membrane entrapment immunofluorescence. Microbiol. Res. 160, 225–231. doi: 10.1016/j.micres.2004.05.006
Buzkan, N., Krivanek, A. F., Eskalen, A., and Walker, M. A. (2003). Improvements in sample preparation and polymerase chain reaction techniques for detection of Xylella fastidiosa in grapevine tissue. Am. J. Enol. Vitic. 54, 307–312.
Cao, T. S., Connell, J. H., Wilhelm, M., and Kirkpatrick, B. C. (2011). Influence of inoculation date on the colonization of Xylella fastidiosa and the persistence of almond leaf scorch disease among almond cultivars. Plant Dis. 95, 158–165. doi: 10.1094/PDIS-05-10-0327
Carbajal, D., Morano, K. A., and Morano, L. D. (2004). Indirect immunofluorescence microscopy for direct detection of Xylella fastidiosa in xylem sap. Curr. Microbiol. 49, 372–375. doi: 10.1007/s00284-004-4369-5
Cariddi, C., Saponari, M., Boscia, D., De Stradis, A., Loconsole, G., Nigro, F., et al. (2014). Isolation of a Xylella fastidiosa strain infecting olive and oleander in Apulia, Italy. J. Plant Pathol. 96, 425–429. doi: 10.4454/JPP.V96I2.024
Casais, V. O., Patrocínio, E. D., Oliveira, S. A. S. D., Schnadelbach, A. S., Barbosa, C. D. J., and Barbosa, L. V. (2014). Genetic diversity of Xylella fastidiosa in citrus producing regions in the state of Bahia, Brazil. Pesquisa Agropecuária Brasileira 49, 26–33. doi: 10.1590/S0100-204X2014000100004
Cave, H., Bingen, E., Elion, J., and Denamur, E. (1994). Differentiation of Escherichia coli strains using randomly amplified polymorphic DNA analysis. Res. Microbiol. 145, 141–150. doi: 10.1016/0923-2508(94)90007-8
Cedergren, R., Gray, M. W., Abel, Y., and Sankoff, D. (1988). The evolutionary relationships among known life forms. J. Mol. Evol. 28, 98–112. doi: 10.1007/BF02143501
Chang, C. J., Garnier, M., Zreik, L., Rossetti, V., and Bove, J. M. (1993). Culture and serological detection of the xylem-limited bacterium causing citrus variegated chlorosis and its identification as a strain of Xylella fastidiosa. Curr. Microbiol. 27, 137–142. doi: 10.1007/BF01576010
Chatterjee, S., Almeida, R. P. P., and Lindow, S. (2008). Living in two worlds: the plant and insect lifestyles of Xylella fastidiosa. Ann. Rev. Phytopathol. 46, 243–271. doi: 10.1146/annurev.phyto.45.062806.094342
Chen, J. C., Banks, D., Jarret, R. L., Chang, C. J., and Smith, B. J. (2000b). Use of 16S rDNA sequences as signature characters to identify Xylella fastidiosa. Curr. Microbiol. 40, 29–33. doi: 10.1007/s002849910006
Chen, J., Chang, C. J., Jarret, R. L., and Gawel, N. (1992). Genetic variations among Xylella fastidiosa strains. Phytopathology 82, 973–977. doi: 10.1094/Phyto-82-973
Chen, J. C., Hartung, J. S., Chang, C. J., and Vidaver, A. K. (2002). An evolutionary perspective of Pierce's disease of grapevine, citrus variegated chlorosis, and mulberry leaf scorch diseases. Curr. Microbiol. 45, 423–428. doi: 10.1007/s00284-002-3785-7
Chen, J., Groves, R., Civerolo, E. L., Viveros, A., Freeman, A., and Zheng, Y. (2005). Two Xylella fastidiosa genotypes associated with almond leaf scorch disease on the same location in California. Phytopathology 95, 708–714. doi: 10.1094/PHYTO-95-0708
Chen, J., Jarret, R. L., Qin, X., Hartung, J. S., Banks, D., Chang, C. J., et al. (2000a). 16S rDNA sequence analysis of Xylella fastidiosa strains. Syst. Appl. Microbiol. 23, 349–354. doi: 10.1016/S0723-2020(00)80064-8
Chen, J., Lamikanra, O., Chang, C. J., and Hopkins, D. L. (1995). Randomly amplified polymorphic DNA analysis of Xylella fastidiosa Pierce's disease and oak leaf scorch pathotypes. Appl. Environ. Microbiol. 61, 1688–1690.
Chen, J., Xie, G., Han, S., Chertkov, O., Sims, D., and Civerolo, E. L. (2010). Whole genome sequences of two Xylella fastidiosa strains (M12 and M23) causing almond leaf scorch disease in California. J. Bacteriol. 192, 4534–4534. doi: 10.1128/JB.00651-10
Choi, H. K., Goes da Silva, F., Lim, H. J., Iandolino, A., Seo, Y. S., Lee, S. W., et al. (2010). Diagnosis of Pierce's disease using biomarkers specific to Xylella fastidiosa rRNA and Vitis vinifera gene expression. Phytopathology 100, 1089–1099. doi: 10.1094/PHYTO-01-10-0014
Chu, G., Vollrath, D., and Davis, R. W. (1986). Separation of large DNA-molecules by contour-clamped homogeneous electric-fields. Science 234, 1582–1585. doi: 10.1126/science.3538420
Ciapina, L. P., Carareto Alves, L. M. C., and Lemos, E. G. M. (2004). A nested-PCR assay for detection of Xylella fastidiosa in citrus plants and sharpshooter leafhoppers. J. Appl. Microbiol. 96, 546–551. doi: 10.1111/j.1365-2672.2004.02176.x
Coletta, H. D., Francisco, C. S., and Almeida, R. P. P. (2014). Temporal and spatial scaling of the genetic structure of a vector-borne plant pathogen. Phytopathology 104, 120–125. doi: 10.1094/PHYTO-06-13-0154-R
Coletta, H. D., and Machado, M. A. (2003). Geographical genetic structure of Xylella fastidiosa from citrus in São Paulo State, Brazil. Phytopathology 93, 28–34. doi: 10.1094/PHYTO.2003.93.1.28
Coletta-Filho, H. D., Francisco, C. S., Lopes, J. R., Muller, C., and Almeida, R. P. (2017). Homologous recombination and Xylella fastidiosa host-pathogen associations in South America. Phytopathology 107, 305–312. doi: 10.1094/PHYTO-09-16-0321-R
da Costa, P. I., Franco, C. F., Miranda, V. S., Teixeira, D. C., and Hartung, J. S. (2000). Strains of Xylella fastidiosa rapidly distinguished by arbitrarily primed-PCR. Curr. Microbiol. 40, 279–282. doi: 10.1007/s002849910055
da Silva, F. R., Vettore, A. L., Kemper, E. L., Leite, A., and Arruda, P. (2001). Fastidian gum: the Xylella fastidiosa exopolysaccharide possibly involved in bacterial pathogenicity. FEMS Microbiol. Lett. 203, 165–171. doi: 10.1111/j.1574-6968.2001.tb10836.x
da Silva, V. S., Shida, C. S., Rodrigues, F. B., Ribeiro, D. C., de Souza, A. A., Coletta-Filho, H. D., et al. (2007). Comparative genomic characterization of citrus-associated Xylella fastidiosa strains. BMC Genomics 8:474. doi: 10.1186/1471-2164-8-474
Davis, M. J., Purcell, A. H., and Thomson, S. V. (1978). Pierce's disease of grapevines - Isolation of causal bacterium. Science 199, 75–77. doi: 10.1126/science.199.4324.75
de Bruijn, F. J. (1992). Use of repetitive (repetitive extragenic palindromic and enterobacterial repetitive intergeneric consensus) sequences and the polymerase chain reaction to fingerprint the genomes of Rhizobium meliloti isolates and other soil bacteria. Appl. Environ. Microbiol. 58, 2180–2187.
Della Coletta-Filho, H., and Machado, M. A. (2002). Evaluation of the genetic structure of Xylella fastidiosa populations from different Citrus sinensis varieties. Appl. Environ. Microbiol. 68, 3731–3736. doi: 10.1128/AEM.68.8.3731-3736.2002
Della Coletta, H., Francisco, C. S., Lopes, J. R. S., De Oliveira, A. F., and Da Silva, L. F. D. (2016). First report of olive leaf scorch in Brazil, associated with Xylella fastidiosa subsp pauca. Phytopathol. Mediterr. 55, 130–135. doi: 10.14601/Phytopathol_Mediterr-17259
Della Coletta, H., Takita, M. A., de Souza, A. A., Aguilar-Vildoso, C. I., and Machado, M. A. (2001). Differentiation of strains of Xylella fastidiosa by a variable number of tandem repeat analysis. Appl. Environ. Microbiol. 67, 4091–4095. doi: 10.1128/AEM.67.9.4091-4095.2001
Della Coletta-Filho, H., Takita, M. A., de Souza, A. A., Aguilar-Vildoso, C. I., and Machado, M. A. (2001). Differentiation of strains of Xylella fastidiosa by a variable number of tandem repeat analysis. Appl. Env. Microbiol. 6, 4091–4095. doi: 10.1128/AEM.67.9.4091-4095.2001
Denancé, N., Legendre, B., Briand, M., Olivier, V., de Boisseson, C., Poliakoff, F., et al. (2017). Several subspecies and sequence types are associated with the emergence of Xylella fastidiosa in natural settings in France. Plant Pathol. doi: 10.1111/ppa.12695
Denny, T. P., Gilmour, M. N., and Selander, R. K. (1988). Genetic diversity and relationships of 2 pathovars of Pseudomonas-syringae. J. Gen. Microbiol. 134, 1949–1960. doi: 10.1099/00221287-134-7-1949
Doddapaneni, H., Yao, J., Lin, H., Walker, M. A., and Civerolo, E. L. (2006). Analysis of the genome-wide variations among multiple strains of the plant pathogenic bacterium Xylella fastidiosa. BMC Genomics 7:225. doi: 10.1186/1471-2164-7-225
Dugravot, S., Backus, E. A., Reardon, B. J., and Miller, T. A. (2008). Correlations of cibarial muscle activities of Homalodisca spp. sharpshooters (Hemiptera: Cicadellidae) with EPG ingestion waveform and excretion. J. Insect Physiol. 54, 1467–1478. doi: 10.1016/j.jinsphys.2008.05.008
EFSA (2016). Scientific report on the update of a database of host plants of Xylella fastidiosa: 20 November 2015. EFSA J. 14, 4378–4418. doi: 10.2903/j.efsa.2016.4378
Elbeaino, T., Valentini, F., Abou Kubaa, R., Moubarak, P., Yaseen, T., and Digiaro, M. (2014). Multilocus sequence typing of Xylella fastidiosa isolated from olive affected by “olive quick decline syndrome” in Italy. Phytopathol. Mediterr. 53, 533–542. doi: 10.14601/Phytopathol_Mediterr-15000
Enright, M. C., and Spratt, B. G. (1998). A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144, 3049–3060. doi: 10.1099/00221287-144-11-3049
EPPO (2015a). EPPO Reporting Service 2015/181 Xylella fastidiosa Detected in Coffea spp. Plants Imported into Switzerland. Available online at: https://gd.eppo.int/reporting/article-5128 (Accessed May 28, 2017).
EPPO (2015b). EPPO Global Database. First Report of Xylella fastidiosa in France. EPPO Reporting Service no. 08-2015. Available online at: https://gd.eppo.int/reporting/article-4942 (Accessed May 28, 2017).
EPPO (2016). Normes OEPP EPPO Standards - PM 7/24 (2) Xylella fastidiosa. EPPO Bull. 46, 463–500. doi: 10.1111/epp.12327
Evert, D. R. (1985). Influence of phony disease of peach on the relationship between fruit weight and firmness near harvest. Hortscience 20, 87–88.
Fadel, A. L., Stuchi, E. S., de Carvalho, S. A., Federici, M. T., and Della Coletta, H. (2014). Navelina ISA 315: a cultivar resistant to citrus variegated chlorosis. Crop Protect. 64, 115–121. doi: 10.1016/j.cropro.2014.06.014
Fatmi, M., Damsteegt, V. D., and Schaad, N. W. (2005). A combined agar-absorption and BIO-PCR assay for rapid, sensitive detection of Xylella fastidiosa in grape and citrus. Plant Pathol. 54, 1–7. doi: 10.1111/j.1365-3059.2005.01114.x
Feil, E. J., Enright, M. C., and Spratt, B. G. (2000). Estimating the relative contributions of mutation and recombination to clonal diversification: a comparison between Neisseria meningitidis and Streptococcus pneumoniae. Res. Microbiol. 151, 465–469. doi: 10.1016/S0923-2508(00)00168-6
Feil, E. J., Holmes, E. C., Bessen, D. E., Chan, M. S., Day, N. P. J., Enright, M. C., et al. (2001). Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. U.S.A. 98, 182–187. doi: 10.1073/pnas.98.1.182
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Feil, H., and Purcell, A. H. (2001). Temperature-dependent growth and survival of Xylella fastidiosa in vitro and in potted grapevines. Plant Dis. 85, 1230–1234. doi: 10.1094/PDIS.2001.85.12.1230
Ferreira, H., Gonçalves, E. R., Rodrigues Neto, J., and Rosato, Y. B. (2000). Primers specific for Xylella fastidiosa based on RAPD differential fragments. Summa Phytopathol. 26, 15–20.
Firrao, G., and Bazzi, C. (1994). Specific identification of Xylella fastidiosa using the polymerase chain reaction. Phytopathol. Mediterr. 33, 90–92.
Francis, M., Lin, H., Cabrera-La Rosa, J., Doddapaneni, H., and Civerolo, E. L. (2006). Genome-based PCR primers for specific and sensitive detection and quantification of Xylella fastidiosa. Eur. J. Plant Pathol. 115, 203–213. doi: 10.1007/s10658-006-9009-4
Francisco, C. S., Ceresini, P. C., Almeida, R. P. P., and Coletta-Filho, H. D. (2017). Spatial genetic structure of coffee-associated Xylella fastidiosa populations indicates that cross infection does not occur with sympatric citrus orchards. Phytopathology. 107, 395–402. doi: 10.1094/phyto-08-16-0300-r
Freitag, J. H. (1951). Host range of the Pierce's disease virus of Grapes as determined by insect transmission. Phytopathology 41, 920–934.
French, W. J. K. (1978). Occurrence of plum leaf scald in Brazil and Paraguay. Plant Dis. Rep. 62, 1035–1038.
Garcia, A. L., Torres, S. C. Z., Heredia, M., and Lopes, S. A. (2012). Citrus Responses to Xylella fastidiosa Infection. Plant Dis. 96, 1245–1249. doi: 10.1094/PDIS-10-11-0868-RE
Garcia-Martinez, J., Acinas, S. G., Anton, A. I., and Rodriguez-Valera, F. (1999). Use of the 16S-23S ribosomal genes spacer region in studies of prokaryotic diversity. J. Microbiol. Methods 36, 55–64. doi: 10.1016/S0167-7012(99)00011-1
Gardner, M. W. (1974). Pierce's Disease of Grapevine: The Anaheim Disease and the California Vine Disease. Berkeley, CA: University of California Press.
Gedalanga, P. B., and Olson, B. H. (2009). Development of a quantitative PCR method to differentiate between viable and nonviable bacteria in environmental water samples. Appl. Microbiol. Biotechnol. 82, 587–596. doi: 10.1007/s00253-008-1846-y
Giampetruzzi, A., Chiumenti, M., Saponari, M., Donvito, G., Italiano, A., Loconsole, G., et al. (2015a). Draft genome sequence of the Xylella fastidiosa CoDiRO strain. Genome Announc. 3, e01538–e01514. doi: 10.1128/genomea.01538-14
Giampetruzzi, A., Loconsole, G., Boscia, D., Calzolari, A., Chiumenti, M., Martelli, G. P., et al. (2015b). Draft genome sequence of CO33, a coffee-infecting isolate of Xylella fastidiosa. Genome Announc. 3:e01472–15. doi: 10.1128/genomeA.01472-15
Giampetruzzi, A., Morelli, M., Saponari, M., Loconsole, G., Chiumenti, M., Boscia, D., et al. (2016). Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp pauca. BMC Genomics 17:475. doi: 10.1186/s12864-016-2833-9
Gmitter, F. G., Chen, C., Machado, M. A., de Souza, A. A., Ollitrault, P., Froehlicher, Y., et al. (2012). Citrus genomics. Tree Genet. Genomes 8, 611–626. doi: 10.1007/s11295-012-0499-2
Goheen, A. C. H. (1988). “Pierce's disease,” in Compendium of Grape Diseases, eds R. C. Pearson and A. C. Goheen (St Paul, MN: Am. Phytopatol. Soc.), 44–45.
Goheen, A. C., Nyland, G., and Lowe, S. K. (1973). Association of a rickettsialike organism with Pierce's disease of grapevines and alfalfa dwarf and heat therapy of the disease in grapevines. Phytopathology 63, 341–345. doi: 10.1094/Phyto-63-341
Gonçalves, F. P., Stuchi, E. S., Lourenço, S. A., Kriss, A. B., Gottwald, T. R., and Amorim, L. (2014). The effect of irrigation on development of citrus variegated chlorosis symptoms. Crop Protect. 57, 8–14. doi: 10.1016/j.cropro.2013.11.016
Goodwin, P. H., and Zhang, S. (1997). Distribution of Xylella fastidiosa in southern Ontario as determined by the polymerase chain reaction. Can. J. Plant Pathol. 19, 13–18. doi: 10.1080/07060669709500564
Grajalmartin, M. J., Simon, C. J., and Muehlbauer, F. J. (1993). Use of random amplified polymorphic DNA (RAPD) to characterize Race 2 of Fusarium-oxysporum F sp pisi. Phytopathology 83, 612–614. doi: 10.1094/Phyto-83-612
Grebus, M. E., Henry, J. M., Hartin, J. E., and Wilen, C. A. (1996). Bacterial leaf scorch of oleander: a new disease in southern California. Phytopathology 86(11 Suppl.), S110–S110.
Guan, W., Shao, J., Davis, R. E., Zhao, T., and Huang, Q. (2014a). Genome sequence of a Xylella fastidiosa strain causing sycamore leaf scorch disease in Virginia. Genome Announc. 2:e00773–14. doi: 10.1128/genomeA.00773-14
Guan, W., Shao, J., Elbeaino, T., Davis, R. E., Zhao, T. C., and Huang, Q. (2015). Specific detection and identification of American Mulberry-infecting and Italian olive-associated strains of Xylella fastidiosa by polymerase chain reaction. PLoS ONE 10:e0129330. doi: 10.1371/journal.pone.0129330
Guan, W., Shao, J., Singh, R., Davis, R. E., Zhao, T. C., and Huang, Q. (2013). A TaqMan-based real time PCR assay for specific detection and quantification of Xylella fastidiosa strains causing bacterial leaf scorch in oleander. J. Microbiol. Methods 92, 108–112. doi: 10.1016/j.mimet.2012.11.008
Guan, W., Shao, J., Zhao, T., and Huang, Q. (2014b). Genome sequence of a Xylella fastidiosa strain causing mulberry leaf scorch disease in Maryland. Genome Announc. 2:e00916–13. doi: 10.1128/genomeA.00916-13
Haelterman, R. M., Tolocka, P. A., Roca, M. E., Guzman, F. A., Fernandez, F. D., and Otero, M. L. (2015). First presumptive diagnosis of Xylella fastidiosa causing olive scorch in Argentina. J. Plant Pathol. 97:393. doi: 10.4454/JPP.V97I2.023
Harper, S. J., Ward, L. I., and Clover, G. R. G. (2010). Development of LAMP and real-time PCR methods for the rapid detection of Xylella fastidiosa for quarantine and field applications. Phytopathology 100, 1282–1288. doi: 10.1094/PHYTO-06-10-0168
Harris, J. L., and Balci, Y. (2015). Population structure of the bacterial pathogen Xylella fastidiosa among street trees in Washington DC. PLoS ONE 10:e0121297. doi: 10.1371/journal.pone.0121297
Hartung, J. S., and Civerolo, E. L. (1989). Restriction fragent length polymorphisms distinguish Xanthomonas-campestris strains isolated from Florida citrus nurseries from Xc pv-citri. Phytopathology 79, 793–799. doi: 10.1094/Phyto-79-793
Hearon, S. S., Sherald, J. L., and Kostka, S. J. (1980). Association of xylem-limited bacteria with elm, sycamore and oak leaf scorch. Can. J. Bot. 58, 1986–1993. doi: 10.1139/b80-228
Heid, C. A., Stevens, J., Livak, K. J., and Williams, P. M. (1996). Real time quantitative PCR. Genome Res. 6, 986–994. doi: 10.1101/gr.6.10.986
Hendson, M., Purcell, A. H., Chen, D. Q., Smart, C., Guilhabert, M., and Kirkpatrick, B. (2001). Genetic diversity of Pierce's disease strains and other pathotypes of Xylella fastidiosa. Appl. Environ. Microbiol. 67, 895–903. doi: 10.1128/AEM.67.2.895-903.2001
Hernandez-Martinez, R., Costa, H. S., Dumenyo, C. K., and Cooksey, D. A. (2006). Differentiation of strains of Xylella fastidiosa infecting grape, almonds, and oleander using a multiprimer PCR assay. Plant Dis. 90, 1382–1388. doi: 10.1094/PD-90-1382
Hernandez-Martinez, R., de la Cerda, K. A., Costa, H. S., Cooksey, D. A., and Wong, F. P. (2007). Phylogenetic relationships of Xylella fastidiosa strains isolated from landscape ornamentals in southern California. Phytopathology 97, 857–864. doi: 10.1094/PHYTO-97-7-0857
Hill, B. L., and Purcell, A. H. (1995). Acquisition and retention of Xylella fastidiosa by an efficient vector, Graphocephala atropunctata. Phytopathology 85, 209–212. doi: 10.1094/Phyto-85-209
Hill, B. L., and Purcell, A. H. (1997). Populations of Xylella fastidiosa in plants required for transmission by an efficient vector. Phytopathology 87, 1197–1201. doi: 10.1094/PHYTO.1997.87.12.1197
Hoffmann, A. A., and Willi, Y. (2008). Detecting genetic responses to environmental change. Nat. Rev. Genet. 9, 421–432. doi: 10.1038/nrg2339
Hopkins, D. L. (1981). Seasonal concentration of the Pierce's disease bacterium in grapevine stems, petioles, and leaf veins. Phytopathology 71, 415–418. doi: 10.1094/Phyto-71-415
Hopkins, D. L. (1989). Xylella fastidiosa. Xylem-limited bacterial pathogen of plants. Annu. Rev. Phytopathol. 27, 271–290. doi: 10.1146/annurev.py.27.090189.001415
Hopkins, D. L., and Adlerz, W. C. (1988). Natural hosts of Xylella fastidiosa in Florida. Plant Dis. 72, 429–431. doi: 10.1094/PD-72-0429
Hopkins, D. L., Bistline, F. W., Russo, L. W., and Thompson, C. M. (1991). Seasonal fluctuation in the occurrence of Xylella fastidiosa in root and stem extracts from citru with blight. Plant Dis. 75, 145–147. doi: 10.1094/PD-75-0145
Hopkins, D. L., and Mollenhauer, H. H. (1973). Rickettsia-like bacterium associated with Pierce's disease of grapes. Science 179, 298–300. doi: 10.1126/science.179.4070.298
Hopkins, D. L., and Purcell, A. H. (2002). Xylella fastidiosa: cause of Pierce's disease of grapevine and other emergent diseases. Plant Dis. 86, 1056–1066. doi: 10.1094/PDIS.2002.86.10.1056
Huang, Q. (2007). Natural occurrence of Xylella fastidiosa in a commercial nursery in Maryland. Can. J. Plant Pathol. 29, 299–303. doi: 10.1080/07060660709507473
Huang, Q. (2009). Specific detection and identification of Xylella fastidiosa strains causing oleander leaf scorch using polymerase chain reaction. Curr. Microbiol. 58, 393–398. doi: 10.1007/s00284-008-9324-4
Huang, Q., and Sherald, J. L. (2004). Isolation and phylogenetic analysis of Xylella fastidiosa from its invasive alternative host, porcelain berry. Curr. Microbiol. 48, 73–76. doi: 10.1007/s00284-003-4109-2
Hutchins, L. M. (1933). Identification and control of the phony disease of the peach. U.S. Dep. Agric. Agric. Bull. 78:55.
Ionescu, M., Yokota, K., Antonova, E., Garcia, A., Beaulieu, E., Hayes, T., et al. (2016). Promiscuous diffusible signal factor production and responsiveness of the Xylella fastidiosa Rpf system. MBio 7, e01054–16. doi: 10.1128/mBio.01054-16
Jacques, M. A., Denance, N., Legendre, B., Morel, E., Briand, M., Mississipi, S., et al. (2016). New coffee plant-infecting Xylella fastidiosa variants derived via homologous recombination. Appl. Environ. Microbiol. 82, 1556–1568. doi: 10.1128/AEM.03299-15
Janse, J. D., and Obradovic, A. (2010). Xylella fastidiosa. Its biology, diagnosis, control and risks. J. Plant Pathol. 92, S35–S48.
Jeng, R. S., Svircev, A. M., Myers, A. L., Beliaeva, L., Hunter, D. M., and Hubbes, M. (2001). The use of 16S and 16S-23S rDNA to easily detect and differentiate common Gram-negative orchard epiphytes. J. Microbiol. Methods 44, 69–77. doi: 10.1016/S0167-7012(00)00230-X
Kandel, P. P., Almeida, R. P., Cobine, P. A., and De La Fuente, L. (2017). Natural competence rates are variable among Xylella fastidiosa strains and homologous recombination occurs in vitro between subspecies fastidiosa and multiplex. Mol. Plant-Microbe Interact. doi: 10.1094/MPMI-02-17-0053-R
Killiny, N., and Almeida, R. P. P. (2011). Gene regulation mediates host specificity of a bacterial pathogen. Environ. Microbiol. Rep. 3, 791–797. doi: 10.1111/j.1758-2229.2011.00288.x
Kremer, K., van Soolingen, D., Frothingham, R., Haas, W. H., Hermans, P. W. M., Martin, C., et al. (1999). Comparison of methods based on different molecular epidemiological markers for typing of Mycobacterium tuberculosis complex strains: interlaboratory study of discriminatory power and reproducibility. J. Clin. Microbiol. 37, 2607–2618.
Lacava, P. T., Araujo, W. L., Maccheroni, W., and Azevedo, J. L. (2001). RAPD profile and antibiotic susceptibility of Xylella fastidiosa, causal agent of citrus variegated chlorosis. Lett. Appl. Microbiol. 33, 302–306. doi: 10.1046/j.1472-765X.2001.01000.x
LeblondBourget, N., Philippe, H., Mangin, I., and Decaris, B. (1996). 16S rRNA and 16S to 23S internal transcribed spacer sequence analyses reveal inter- and intraspecific Bifidobacterium phylogeny. Int. J. Syst. Bacteriol. 46, 102–111. doi: 10.1099/00207713-46-1-102
Ledbetter, C. A., and Rogers, E. E. (2009). Differential susceptibility of prunus germplasm (Subgenus Amygdalus) to a California isolate of Xylella fastidiosa. HortScience 44, 1928–1931.
Leu, H. H., Leu, L. S., and Lin, C. P. (1998). Development and application of monoclonal antibodies against Xylella fastidiosa, the causal bacterium of pear leaf scorch. J. Phytopathol. 146, 31–37. doi: 10.1111/j.1439-0434.1998.tb04747.x
Leu, L. S., and Su, C. C. (1993). Isolation, cultivation, and pathogenicity of Xylella fastidiosa, the causal bacterium of pear leaf scald in Taiwan. Plant Dis. 77, 642–646. doi: 10.1094/PD-77-0642
Li, W. B., Pria, W. D., Teixeira, C., Miranda, V. S., Ayres, A. J., Franco, C. F., et al. (2001). Coffee leaf scorch caused by a strain of Xylella fastidiosa from citrus. Plant Dis. 85, 501–505. doi: 10.1094/PDIS.2001.85.5.501
Li, W. B., Teixeira, D. C., Hartung, J. S., Huang, Q., Duan, Y. P., Zhou, L. J., et al. (2013). Development and systematic validation of qPCR assays for rapid and reliable differentiation of Xylella fastidiosa strains causing citrus variegated chlorosis. J. Microbiol. Methods 92, 79–89. doi: 10.1016/j.mimet.2012.10.008
Li, Y., Hao, G., Galvani, C. D., Meng, Y., De La Fuente, L., Hoch, H. C., et al. (2007). Type I and type IV pili of Xylella fastidiosa affect twitching motility, biofilm formation and cell-cell aggregation. Microbiology 153(Pt 3), 719–726. doi: 10.1099/mic.0.2006/002311-0
Lin, H., Civerolo, E. L., Hu, R., Barros, S., Francis, M., and Walker, M. A. (2005). Multilocus simple sequence repeat markers for differentiating strains and evaluating genetic diversity of Xylella fastidiosa. Appl. Environ. Microbiol. 71, 4888–4892. doi: 10.1128/AEM.71.8.4888-4892.2005
Lin, H., Islam, M. S., Cabrera-La Rosa, J. C., Civerolo, E. L., and Groves, R. L. (2015). Population structure of Xylella fastidiosa associated with almond leaf scorch disease in the San Joaquin Valley of California. Phytopathology 105, 825–832. doi: 10.1094/PHYTO-09-14-0254-R
Lin, H., Islam, M. S., Morano, L., Groves, R., Bextine, B., Civerolo, E., et al. (2013). Genetic variation of Xylella fastidiosa associated with grapevines in two major viticultural regions in the United States California and Texas. J. Plant Pathol. 95, 329–337. doi: 10.4454/JPP.V95I2.028
Livingston, S., Chen, J. C., and Civerolo, E. L. (2010). Seasonal behavior of Xylella fastidiosa causing almond leafscorch disease under field conditions and improved detection of the bacteria by means of array-PCR. J. Phytopathol. 158, 40–45. doi: 10.1111/j.1439-0434.2009.01577.x
Loconsole, G., Potere, O., Boscia, D., Altamura, G., Djelouah, K., Elbeaino, T., et al. (2014). Detection of Xylella fastidiosa in olive trees by molecular and serological methods. J. Plant Pathol. 96, 7–14. doi: 10.4454/JPP.V96I1.041
Lopes, J. R., Landa, B. B., and Fereres, A. (2014). Short communication. A survey of potential insect vectors of the plant pathogenic bacterium Xylella fastidiosa in three regions of Spain. Span. J. Agric. Res. 12, 795–800. doi: 10.5424/sjar/2014123-5613
Maiden, M. C. J., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Mang, S. M., Frisullo, S., Elshafie, H. S., and Camele, I. (2016). Diversity evaluation of Xylella fastidiosa from infected olive trees in Apulia (Southern Italy). Plant Pathol. J. 32, 102–111. doi: 10.5423/PPJ.OA.08.2015.0153
Marcelletti, S., and Scortichini, M. (2016a). Genome-wide comparison and taxonomic relatedness of multiple Xylella fastidiosa strains reveal the occurrence of three subspecies and a new Xylella species. Arch. Microbiol. 198, 803–812. doi: 10.1007/s00203-016-1245-1
Marcelletti, S., and Scortichini, M. (2016b). Xylella fastidiosa CoDiRO strain associated with the olive quick decline syndrome in southern Italy belongs to a clonal complex of the subspecies pauca that evolved in Central America. Microbiology 162, 2087–2098. doi: 10.1099/mic.0.000388
Martelli, G. P., Boscia, D., Porcelli, F., and Saponari, M. (2016). The olive quick decline syndrome in south-east Italy: a threatening phytosanitary emergency. Eur. J. Plant Pathol. 144, 235–243. doi: 10.1007/s10658-015-0784-7
Martinati, J. C., Pacheco, F. T. H., de Miranda, V. F. O., and Tsai, S. M. (2005). Phylogenetic relationships of Xylella fastidiosa strains based on 16S-23S rDNA sequences. Curr. Microbiol. 50, 190–195. doi: 10.1007/s00284-004-4405-5
Martinati, J. C., Pacheco, F. T. H., de Miranda, V. F. O., and Tsai, S. M. (2007). 16S-23S RDNA: polymorphisms and their use for detection and identification of Xylella fastidiosa strains. Braz. J. Microbiol. 38, 159–165. doi: 10.1590/S1517-83822007000100033
McElrone, A. J., Sherald, J. L., and Pooler, M. R. (1999). Identification of alternative hosts of Xylella fastidiosa in the Washington, D.C., area using nested polymerase chain reaction (PCR). J. Arboricult. 25, 258–263.
Mehta, A., Leite, R. P., and Rosato, Y. B. (2000). Polymorphism of Xylella fastidiosa strains by RAPD-PCR and SDS-PAGE of proteins. Fitopatol. Bras. 25, 651–656.
Mehta, A., Leite, R. P., and Rosato, Y. B. (2001). Assessment of the genetic diversity of Xylella fastidiosa isolated from citrus in Brazil by PCR-RFLP of the 16S rDNA and 16S-23S intergenic spacer and rep-PCR fingerprinting. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 79, 53–59. doi: 10.1023/A:1010219811555
Mehta, A., and Rosato, Y. B. (2001). Phylogenetic relationships of Xylella fastidiosa strains from different hosts, based on 16S rDNA and 16S-23S intergenic spacer sequences. Int. J. Syst. Evol. Microbiol. 51, 311–318. doi: 10.1099/00207713-51-2-311
Melanson, R. A., Sanderlin, R. S., McTaggart, A. R., and Ham, J. H. (2012). A systematic study reveals that Xylella fastidiosa strains from pecan are part of X. fastidiosa subsp multiplex. Plant Dis. 96, 1123–1134. doi: 10.1094/PDIS-09-11-0730-RE
Minsavage, G. V., Thompson, C. M., Hopkins, D. L., Leite, R., and Stall, R. E. (1994). Development of a polymerase chain-reaction protocol for detection of Xylella fstidiosa in plant tissue. Phytopathology 84, 456–461. doi: 10.1094/Phyto-84-456
Moller, W. J., Sanborn, R. R., Mircetich, S. M., Williams, H. E., and Beutel, J. A. (1974). A newly recognized and serious leaf scorch disease of almond. Plant Dis. Report. 58, 99–101.
Montero-Astua, M., Chacon-Diaz, C., Aguilar, E., Mario Rodriguez, C., Garita, L., Villalobos, W., et al. (2008). Isolation and molecular characterization of Xylella fastidiosa from coffee plants in costa rica. J. Microbiol. 46, 482–490. doi: 10.1007/s12275-008-0072-8
Montero-Astua, M., Hartung, J. S., Aguilar, E., Chacon, C., Li, W., Albertazzi, F. J., et al. (2007). Genetic diversity of Xylella fastidiosa strains from Costa Rica, Sao Paulo, Brazil, and United States. Phytopathology 97, 1338–1347. doi: 10.1094/PHYTO-97-10-1338
Montes-Borrego, M., Lopes, J. R. S., Jimenez-Diaz, R. M., and Landa, B. B. (2015). Combined use of a new SNP-based assay and multilocus SSR markers to assess genetic diversity of Xylella fastidiosa subsp pauca infecting citrus and coffee plants. Int. Microbiol. 18, 13–24. doi: 10.2436/20.1501.01.230
Morano, L. D., Bextine, B. R., Garcia, D. A., Maddox, S. V., Gunawan, S., Vitovsky, N. J., et al. (2008). Initial genetic analysis of Xylella fastidiosa in Texas. Curr. Microbiol. 56, 346–351. doi: 10.1007/s00284-007-9088-2
Myers, B. A., Brady, J. A., Mitchell, F. L., and Rathburn, H. B. (2010). Genotyping Xylella fastidiosa strains using multiplex PCR. Phytopathology 100:S88.
Newman, K. L., Almeida, R. P., Purcell, A. H., and Lindow, S. E. (2003). Use of a green fluorescent strain for analysis of Xylella fastidiosa colonization of Vitis vinifera. Appl. Environ. Microbiol. 69, 7319–7327. doi: 10.1128/AEM.69.12.7319-7327.2003
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., et al. (2000). Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28:E63. doi: 10.1093/nar/28.12.e63
Nunney, L., Hopkins, D. L., Morano, L. D., Russell, S. E., and Stouthamer, R. (2014a). Intersubspecific recombination in Xylella fastidiosa strains native to the United States: infection of novel hosts associated with an unsuccessful invasion. Appl. Environ. Microbiol. 80, 1159–1169. doi: 10.1128/AEM.02920-13
Nunney, L., Ortiz, B., Russell, S. A., Ruiz Sanchez, R., and Stouthamer, R. (2014b). The complex biogeography of the plant pathogen Xylella fastidiosa: genetic evidence of introductions and Subspecific introgression in Central America. PLoS ONE 9:e112463. doi: 10.1371/journal.pone.0112463
Nunney, L., Schuenzel, E. L., Scally, M., Bromley, R. E., and Stouthamer, R. (2014c). Large-scale intersubspecific recombination in the plant-pathogenic bacterium Xylella fastidiosa is associated with the host shift to mulberry. Appl. Environ. Microbiol. 80, 3025–3033. doi: 10.1128/AEM.04112-13
Nunney, L., Vickerman, D. B., Bromley, R. E., Russell, S. A., Hartman, J. R., Morano, L. D., et al. (2013). Recent evolutionary radiation and host plant specialization in the Xylella fastidiosa subspecies native to the United States. Appl. Environ. Microbiol. 79, 2189–2200. doi: 10.1128/AEM.03208-12
Nunney, L., Yuan, X., Bromley, R. E., and Stouthamer, R. (2012). Detecting genetic introgression: high levels of intersubspecific recombination found in Xylella fastidiosa in Brazil. Appl. Environ. Microbiol. 78, 4702–4714. doi: 10.1128/AEM.01126-12
Oliveira, A. C., Vallim, M. A., Semighini, C. P., Araujo, W. L., Goldman, G. H., and Machado, M. A. (2002). Quantification of Xylella fastidiosa from citrus trees by real-time polymerase chain reaction assay. Phytopathology 92, 1048–1054. doi: 10.1094/PHYTO.2002.92.10.1048
Parker, J. K., Havird, J. C., and De La Fuente, L. (2012). Differentiation of Xylella fastidiosa strains via multilocus sequence analysis of environmentally mediated genes (MLSA-E). Appl. Environ. Microbiol. 78, 1385–1396. doi: 10.1128/AEM.06679-11
Picchi, S. C., Vilas-Boas, L. A., Ceresini, P. U., Lemos, E. G. D., and Lemos, M. V. F. (2006). Strain variability in the DNA immigration control region (ICR) of Xylella fastidiosa. Res. Microbiol. 157, 254–262. doi: 10.1016/j.resmic.2005.07.001
Pooler, M. R., and Hartung, J. S. (1995a). Specific PCR detection and identification of Xylella fastidiosa strains causing citru variegated chlorosis. Curr. Microbiol. 31, 377–381. doi: 10.1007/BF00294703
Pooler, M. R., and Hartung, J. S. (1995b). Genetic relationships among strains of Xylella fastidiosa from RAPD-PCR data. Curr. Microbiol. 31, 134–137. doi: 10.1007/BF00294290
Pooler, M. R., Myung, I. S., Bentz, J., Sherald, J., and Hartung, J. S. (1997). Detection of Xylella fastidiosa in potential insect vectors by immunomagnetic separation and nested polymerase chain reaction. Lett. Appl. Microbiol. 25, 123–126. doi: 10.1046/j.1472-765X.1997.00188.x
Purcell, A. (2013). Paradigms: examples from the bacterium Xylella fastidiosa. Ann. Rev. Phytopathol. 51, 339–356. doi: 10.1146/annurev-phyto-082712-102325
Purcell, A. H., Finlay, A. H., and McLean, D. L. (1979). Pierce's disease bacterium. Mechanism of transmission by leafhopper vectors. Science 206, 839–841. doi: 10.1126/science.206.4420.839
Purcell, A. H., and Saunders, S. R. (1999). Fate of Pierce's disease strains of Xylella fastidiosa in common riparian plants in California. Plant Dis. 83, 825–830. doi: 10.1094/PDIS.1999.83.9.825
Purcell, A. H., Saunders, S. R., Hendson, M., Grebus, M. E., and Henry, M. J. (1999). Causal role of Xylella fastidiosa in oleander leaf scorch disease. Phytopathology 89, 53–58. doi: 10.1094/PHYTO.1999.89.1.53
Qin, X. T., Miranda, V. S., Machado, M. A., Lemos, E. G. M., and Hartung, J. S. (2001). An evaluation of the genetic diversity of Xylella fastidiosa isolated from diseased citrus and coffee in Sao Paulo, Brazil. Phytopathology 91, 599–605. doi: 10.1094/PHYTO.2001.91.6.599
Raju, B. C., Goheen, A. C., and Frazier, N. W. (1983). Occurrence of Pierce's disease bacteria in plants and vectorsin California. Phytopathology 73, 1309–1313. doi: 10.1094/Phyto-73-1309
Randall, J. J., Goldberg, N. P., Kemp, J. D., Radionenko, M., French, J. M., Olsen, M. W., et al. (2009). Genetic analysis of a novel Xylella fastidiosa subspecies found in the southwestern United States. Appl. Environ. Microbiol. 75, 5631–5638. doi: 10.1128/AEM.00609-09
Retchless, A. C., Labroussaa, F., Shapiro, L., Stenger, D. C., Lindow, S. E., and Almeida, R. P. (2014). “Genomic insights into Xylella fastidiosa interactions with plant and insect hosts,” in Genomics of Plant-Associated Bacteria, eds D. C. Gross, A. Lichens-Park, and C. Kole (Berlin; Heidelberg: Springer), 177–202.
Rocha, J. G., Zambolim, L., Zambolim, E. M., do Vale, F. X. R., Junior, W. C. J., and Bergamin, A. (2010). Quantification of yield loss due to coffee leaf scorch. Crop Protect. 29, 1100–1104. doi: 10.1016/j.cropro.2010.04.011
Rodrigues, J. L. M., Silva-Stenico, M. E., Gomes, J. E., Lopes, J. R. S., and Tsai, S. M. (2003). Detection and diversity assessment of Xylella fastidiosa in field-collected plant and insect samples by using 16S rRNA and gyrB sequences. Appl. Environ. Microbiol. 69, 4249–4255. doi: 10.1128/AEM.69.7.4249-4255.2003
Rosato, Y., Rodrigues Neto, J., Miranda, V., Carlos, E., and Manfio, G. (1998). Diversity of a Xylella fastidiosa Population Isolated from Citrus sinensis Affected by Citrus Variegated Chlorosis in Brazil. Syst. Appl. Microbiol. 21, 593–598. doi: 10.1016/S0723-2020(98)80072-6
Saponari, M., Boscia, D., Nigro, F., and Martelli, G. P. (2013). Identification of DNA sequences related to Xylella fastidiosa in oleander, almond and olive trees exhibiting leaf scorch symptoms in Apulia (Southern Italy). J. Plant Pathol. 95:668. doi: 10.4454/JPP.V95I3.035
Saponari, M., Loconsole, G., Cornara, D., Yokomi, R. K., De Stradis, A., Boscia, D., et al. (2014). Infectivity and Transmission of Xylella fastidiosa by Philaenus spumarius (Hemiptera: Aphrophoridae) in Apulia, Italy. J. Econ. Entomol. 107, 1316–1319. doi: 10.1603/EC14142
Scally, M., Schuenzel, E. L., Stouthamer, R., and Nunney, L. (2005). Multilocus sequence type system for the plant pathogen Xylella fastidiosa and relative contributions of recombination and point mutation to clonal diversity. Appl. Environ. Microbiol. 71, 8491–8499. doi: 10.1128/AEM.71.12.8491-8499.2005
Schaad, N. W., Jones, J. B., and Chun, W. (eds.). (2001). Laboratory Guide for the Identification of Plant Pathogenic Bacteria, 3rd Edn. St. Paul, MN: American Phytopathological Society Press.
Schaad, N. W., Opgenorth, D., and Gaush, P. (2002). Real-time polymerase chain reaction for one-hour on-site diagnosis of Pierce's disease of grape in early season asymptomatic vines. Phytopathology 92, 721–728. doi: 10.1094/PHYTO.2002.92.7.721
Schaad, N. W., Postnikova, E., Lacy, G., Fatmi, M. B., and Chang, C. J. (2004). Xylella fastidiosa subspecies: X-fastidiosa subsp piercei, subsp nov., X-fastidiosa subsp multiplex subsp. nov., and X-fastidiosa subsp pauca subsp nov. Syst. Appl. Microbiol. 27, 290–300. doi: 10.1078/0723-2020-00263
Schuenzel, E. L., Scally, M., Stouthamer, R., and Nunney, L. (2005). A multigene phylogenetic study of clonal diversity and divergence in North American strains of the plant pathogen Xylella fastidiosa. Appl. Environ. Microbiol. 71, 3832–3839. doi: 10.1128/AEM.71.7.3832-3839.2005
Severin, H. (1949). Transmission of the virus of Pierce's disease of grapevines by leafhoppers. Hilgardia 19, 190–206. doi: 10.3733/hilg.v19n06p190
Sherald, J. L. (1993). Pathogenicity of Xylella fastidiosa in american elm and failure of reciprocal transmission between strains from elm and sycamore. Plant Dis. 77, 190–193. doi: 10.1094/PD-77-0190
Silva, M. R., Meneguim, A. M., Paião, F. G., Meneguim, L., Canteri, M. G., and Leite, R. P. Jr. (2007). Natural infectivity of Xylella fastidiosa Wells et al. in sharpshooters (Hemiptera: Cicadellidae) from coffee plantations of Parana, Brazil. Neotrop. Entomol. 36, 274–281. doi: 10.1590/S1519-566X2007000200015
Silva-Stenico, M. E., Pacheco, F. T. H., Pereira, E. R., Rodrigues, J. L. M., Souza, A. N., Etchegaray, A., et al. (2009). Nutritional deficiency in citrus with symptoms of citrus variegated chlorosis disease. Braz. J. Biol. 69, 859–864. doi: 10.1590/S1519-69842009000400013
Simpson, A. J. G., Reinach, F. C., Arruda, P., Abreu, F. A., Acencio, M., Alvarenga, R., et al. (2000). The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406, 151–157. doi: 10.1038/35018003
Smit, M. L., Giesendorf, B. A. J., Vet, J. A. M., Trijbels, F. J. M., and Blom, H. J. (2001). Semiautomated DNA mutation analysis using a robotic workstation and molecular beacons. Clin. Chem. 47, 739–744.
Stevenson, J. F., Matthews, M. A., and Rost, T. L. (2005). The developmental anatomy of Pierce's disease symptoms in grapevines: green islands and matchsticks. Plant Dis. 89, 543–548. doi: 10.1094/PD-89-0543
Stoneking, M. (2001). Single nucleotide polymorphisms - From the evolutionary past. Nature 409, 821–822. doi: 10.1038/35057279
Su, C. C., Chang, C. J., Yang, W. J., Hsu, S. T., Tzeng, K. C., Jan, F. J., et al. (2012). Specific characters of 16S rRNA gene and 16S-23S rRNA internal transcribed spacer sequences of Xylella fastidiosa pear leaf scorch strains. Eur. J. Plant Pathol. 132, 203–216. doi: 10.1007/s10658-011-9863-6
Su, C. C., Chang, C. J., Chang, C. M., Shih, H. T., Tzeng, K. C., Jan, F. J., et al. (2013). Pierce's disease of grapevines in Taiwan: isolation, cultivation, and pathogenicity of Xylella fastidiosa. J. Phytopathol. 161, 389–396. doi: 10.1111/jph.12075
Su, C. C., Deng, W. L., Jan, F. J., Chang, C. J., Huang, H., and Chen, J. (2014). Draft genome sequence of Xylella fastidiosa pear leaf scorch strain in Taiwan. Genome Announc. 2:e00166–14. doi: 10.1128/genomeA.00166-14
Su, C. C., Deng, W. L., Jan, F. J., Chang, C. J., Huang, H., Shih, H. T., et al. (2016). Xylella taiwanensis sp. nov., causing pear leaf scorch disease. Int. J. Syst. Evol. Microbiol. 66, 4766–4771. doi: 10.1099/ijsem.0.001426
Su, C. C., Yang, W. J., Feng, C. Y., Hsu, S. T., and Tzeng, K. C. (2008). The application of DNA fingerprintings amplified by arbitrary primers in differentiating pear leaf scorch bacterium from other Xylella fastidiosa strains. Plant Pathol. Bull. 17, 261–269.
Thorne, E. T., Stevenson, J. F., Rost, T. L., Labavitch, J. M., and Matthews, M. A. (2006). Pierce's disease symptoms: comparison with symptoms of water deficit and the impact of water deficits. Am. J. Enol. Vitic. 57, 1–11.
Travensolo, R. F., Ciapina, L. P., and Lemos, E. C. M. (2005). Development of a scar marker for pierce's disease strains of Xylella fastidiosa. Fitopatol. Bras. 30, 115–120. doi: 10.1590/S0100-41582005000200002
Van Sluys, M. A., de Oliveira, M. C., Monteiro-Vitorello, C. B., Miyaki, C. Y., Furlan, L. R., Camargo, L. E. A., et al. (2003). Comparative analyses of the complete genome sequences of Pierce's disease and citrus variegated chlorosis strains of Xylella fastidiosa. J. Bacteriol. 185, 1018–1026. doi: 10.1128/JB.185.3.1018-1026.2003
Versalovic, J., Koeuth, T., and Lupski, J. R. (1991). Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 19, 6823–6831. doi: 10.1093/nar/19.24.6823
Wells, J. M., Raju, B. C., Hung, H. Y., Weisburg, W. G., Mandelcopaul, L., and Brenner, D. J. (1987). Xylella fastidiosa gen-nov. sp-nov. - Gram negative, xylem-limited, fastidious plant bacteria related to Hanthomonas spp. Int. J. Syst. Bacteriol. 37, 136–143. doi: 10.1099/00207713-37-2-136
Wells, J. M. R., Thompson, J. M., and Lowe, S. K. (1981). Ethiology of phony peach and plum leaf scald diseases. Phytopathology 71, 1156–1161. doi: 10.1094/Phyto-71-1156
Wickert, E., Machado, M. A., and Lemos, E. G. M. (2007). Evaluation of the genetic diversity of Xylella fastidiosa strains from citrus and coffee hosts by single-nucleotide polymorphism markers. Phytopathology 97, 1543–1549. doi: 10.1094/PHYTO-97-12-1543
Willerslev, E., and Cooper, A. (2005). Ancient DNA. Proc. R. Soc. B Biol. Sci. 272, 3–16. doi: 10.1098/rspb.2004.2813
Wittwer, C. T., Herrmann, M. G., Moss, A. A., and Rasmussen, R. P. (1997). Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques 22:130.
Yamamoto, S., Bouvet, P. J. M., and Harayama, S. (1999). Phylogenetic structures of the genus Acinetobacter based on gyrB sequences: comparison with the grouping by DNA-DNA hybridization. Int. J. Syst. Bacteriol. 49, 87–95. doi: 10.1099/00207713-49-1-87
Yaseen, T., Drago, S., Valentini, F., Elbeaino, T., Stampone, G., Digiaro, M., et al. (2015). On-site detection of Xylella fastidiosa in host plants and in “spy insects” using the real-time loop-mediated isothermal amplification method. Phytopathol. Mediterr. 54, 488–496. doi: 10.14601/Phytopathol_Mediterr-15250
Yuan, Q., Jordan, R., Brlansky, R. H., Istomina, O., and Hartung, J. (2015). Development of single chain variable fragment (scFv) antibodies against Xylella fastidiosa subsp pauca by phage display. J. Microbiol. Methods 117, 148–154. doi: 10.1016/j.mimet.2015.07.020
Keywords: grape pierce's disease, citrus variegated chlorosis, OQDS, CoDiRO, Xylella diagnosis, asymptomatic, leaf scorch disease, quarantine organism
Citation: Baldi P and La Porta N (2017) Xylella fastidiosa: Host Range and Advance in Molecular Identification Techniques. Front. Plant Sci. 8:944. doi: 10.3389/fpls.2017.00944
Received: 21 March 2017; Accepted: 22 May 2017;
Published: 08 June 2017.
Edited by:
Jesús Mercado-Blanco, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Leonardo De La Fuente, Auburn University, United StatesJoao Lucio Azevedo, University of São Paulo, Brazil
Copyright © 2017 Baldi and La Porta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Baldi, cGFvbG8uYmFsZGlAZm1hY2guaXQ=