 Raghuprakash Kastoori Ramamurthy
Raghuprakash Kastoori Ramamurthy Brian M. Waters
Brian M. Waters- Department of Agronomy and Horticulture, University of Nebraska–Lincoln, Lincoln, NE, United States
Iron (Fe) deficiency in plants limits crop growth and productivity. Molecular mechanisms that plants use to sense and respond to Fe deficiency by coordinated expression of Fe-uptake genes are not fully understood. The C940-fe chlorotic melon (Cucumis melo) mutant known as fefe is unable to upregulate Fe-uptake genes, however, the FeFe gene had not been identified. In this study, we used two F2 mapping populations to map and identify the FeFe gene as bHLH38, a homolog of subgroup Ib bHLH genes from Arabidopsis thaliana that are involved in transcriptional regulation of Fe-uptake genes in partnership with the FIT gene. A Ty1-copia type retrotransposon insertion of 5.056 kb within bHLH38 is responsible for the defect in bHLH38 in fefe, based on sequencing and expression analysis. This retrotransposon insertion results in multiple non-functional transcripts expected to result in an altered and truncated protein sequence. Hairy root transformation of fefe plants using wild-type bHLH38 resulted in functional complementation of the chlorotic fefe phenotype. Using a yeast-2-hybrid assay, the transcription factor Fit interacted with the wild-type bHLH38 protein, but did not interact with the fefe bHLH38 protein, suggesting that heterodimer formation of Fit/bHLH38 to regulate Fe-uptake genes does not occur in fefe roots. The second subgroup Ib bHLH gene in the melon genome is not functionally redundant to bHLH38, in contrast to Arabidopsis where four subgroup Ib bHLH genes are functionally redundant. Whereas the Arabidopsis bHLH transcript levels are upregulated by Fe deficiency, melon bHLH38 was not regulated at the transcript level. Thus, the fefe mutant may provide a platform for studying bHLH38 genes and proteins from other plant species.
Introduction
Iron (Fe) is crucial for plant growth, development, and productivity (Briat et al., 2015). Iron is involved in chlorophyll synthesis and is a constituent of certain enzymes involved in metabolism (Kobayashi and Nishizawa, 2012; Bashir and Nishizawa, 2013). Iron deficiency is a major limiting factor for crop production, especially in alkaline soils, which occur on approximately 30% of the earth (Chen and Barak, 1982). Plant species can be classified into two categories based on their Fe uptake mechanisms (Marschner et al., 1986). Iron uptake in graminaceous species, known as Strategy II, is characterized by production of high-affinity Fe(III) binding compounds called phytosiderophores, which are secreted into the rhizosphere to form phytosiderophore-Fe(III) complexes. These complexes are taken up by the root cells through a specific plasma membrane transport system (Römheld and Marschner, 1986; Curie et al., 2001). Iron uptake in dicotyledonous and non-graminaceous monocots, known as Strategy I, is characterized by soil acidification by H+-ATPase proteins, reduction of Fe(III) to Fe(II) by ferric chelate reductase (FCR) proteins and subsequent uptake of Fe(II) by Fe transporter proteins (Kobayashi and Nishizawa, 2012). In Arabidopsis thaliana, H+-ATPase 2 (AHA2) encodes the proton extrusion component (Santi and Schmidt, 2009). The primary root surface FCR is encoded by Ferric Reduction Oxidase 2 (FRO2) (Robinson et al., 1999), and analogous genes in cucumber (Cucumis sativus L.) and melon (C. melo L.) are called FRO1 (Waters et al., 2007, 2014). Fe(II) transporters include IRT1 (Varotto et al., 2002; Vert et al., 2002) and Nramp1 (Curie et al., 2000; Castaings et al., 2016).
Strategy I Fe-uptake genes are regulated largely at the transcriptional level. AtFRO2, AtIRT1, AtNRAMP1 and various other genes are transcriptionally activated by the basic helix-loop-helix (bHLH) transcription factor AtFit1 (Colangelo and Guerinot, 2004; Yuan et al., 2005). The subgroup Ib genes of the bHLH superfamily, bHLH38, bHLH39, bHLH100 and bHLH101, are upregulated by Fe deficiency in Arabidopsis (Wang et al., 2007; Dinneny et al., 2008; Buckhout et al., 2009; Yang et al., 2010; Bauer and Blondet, 2011; Schuler et al., 2011; Stein and Waters, 2012; Waters et al., 2012; Andriankaja et al., 2014; Maurer et al., 2014). The AtFit1 protein interacts with these subgroup Ib bHLH proteins to regulate Fe-uptake genes as a heterodimer complex (Yuan et al., 2008; Wang et al., 2013). The bHLH38, bHLH39, bHLH100 and bHLH101 genes are functionally redundant in Arabidopsis, as single, double, or triple loss-of-function bHLH mutations do not have a chlorotic Fe deficiency phenotype under Fe sufficient conditions (Wang et al., 2007; Sivitz et al., 2012; Andriankaja et al., 2014; Maurer et al., 2014), except in one report (Wang et al., 2013).
The chlorotic melon C940-Fe (fefe) mutant originated spontaneously in the melon cultivar Edisto (Nugent and Bhella, 1988). The genetic basis for fefe was retained by outcrossing the original mutant plant to the cultivar Mainstream and self-pollinating chlorotic mutants until the F5 generation, resulting in the C940-fe germplasm (Nugent, 1994). The fefe mutant plants are incapable of inducing Strategy I Fe-uptake responses (Von Jolley et al., 1991; Welkie, 1996), and Fe-uptake gene expression (Waters et al., 2014; Hsieh and Waters, 2016), and cannot survive under normal culture conditions unless it is supplemented with Fe, similar to FIT mutants in Arabidopsis. 82 genes, including Fe-uptake genes and riboflavin synthesis genes, were not regulated by Fe-deficiency in fefe plants compared to their WT counterpart (Waters et al., 2014; Hsieh and Waters, 2016), suggesting that the fefe gene could be a transcription factor. Since the fefe lesion is not in the melon FIT gene, FeFe was predicted to act upstream of FIT or as a partner of Fit (Waters et al., 2014). The main objective of this work is to map and characterize the fefe gene. We used genetic, genomic, transcriptomic and molecular approaches to map and functionally characterize the fefe gene. The results of this research will provide increased understanding of Fe-homeostasis in Strategy I plant species.
Materials and Methods
Genetic Mapping
An F2 mapping population consisted of 269 individuals from a cross between “snake melon” (PI 435288) and C940-fe. The population was genotyped and scored, and the chlorosis trait associated with the fefe mutation was mapped to an 8 cM region of linkage group 8 (LG8) (Ramamurthy and Waters, 2015). To fine map the fefe mutation, a second, 288 individual F2 mapping population was developed from a cross between “pocket melon” (PI 536481) and C940-fe. This F2 mapping population was grown in hydroponics as described below prior to scoring leaf chlorosis in F2 plants as “0” (chlorotic) or “1” (normal). The normal and chlorotic fefe mutant F2 plants were distinguishable 1–2 weeks after planting in nutrient solution. DNA was isolated from a single young leaf from each plant (Kang et al., 1998), and quantity and purity of DNA was assessed using spectrophotometry at 260 and 280 nm. DNA was diluted to 25 ng/μl and stored at -20°C until genotyping.
A total of 112 SSR markers for LG8 from the consensus genetic map (Diaz et al., 2011) and markers for LG8 provided by Syngenta on the ICUGI website1 were tested, and in total, 27 markers were polymorphic and were used for genotyping the F2 mapping population (Table 1). PCR reactions were performed in a final volume of 10 μl with 1×Taq buffer [(16 mM (NH4)2SO4, 67 mM Tris–HCL (pH 8.8 at 25°C), 0.1% stabilizer], 2 mM MgCl2, 0.15 mM dNTP, 1 μM each primer, 0.2 U Taq DNA polymerase (Bioline USA Inc., Taunton, MA, United States), and 20 ng DNA. The cycling conditions were: an initial cycle at 94°C for 3 min, followed by 40 cycles at 94°C, 30 s, 55–58°C, 30 s and 72°C, 30 s, and a final extension step at 72°C, 5 min. PCR products were visualized with UV light after electrophoresis in 3% superfine resolution agarose (Amresco LLC, Solon, OH, United States) gels with 1× TBE (0.9 M Tris-borate, 0.002 M EDTA, pH 8.0), stained with ethidium bromide.

TABLE 1. Linkage mapping of the fefe gene using two different F2 mapping populations and a joint linkage map.
Phenotype scores for 288 pocket melon × C940-fe F2 individuals and their corresponding genotypes across 27 loci in population were entered into a spreadsheet as an input file for QTL analysis in R/qtl software (Broman et al., 2003). Genetic positions were deduced for markers that were not present in the consensus map (from Syngenta and Li et al., 2011). Data checking steps for genotyping were performed using standard R/qtl functions (Broman et al., 2003). To obtain a better resolution of the fefe locus, a joint linkage map consisting of 557 individuals in both snake melon X fefe and pocket melon X fefe populations was constructed using polymorphic markers (N = 35) on LG8. For joint map construction, genotypic (LG8) and phenotypic information from the two mapping populations was input for QTL analysis in R/qtl software (Broman et al., 2003). The genetic maps for individual and joint analysis were constructed using est.rf and est.map functions of R/qtl. Interval mapping was performed using the “scanone” function which detects a single QTL by performing genome wide scan with possible allowance for covariates, with a binary model to analyze the binary phenotype (i.e., fefe or FeFe).
Growing Condition of Plants Used for RNA-seq
Seeds were imbibed in germination paper soaked with 0.2 mM CaSO4 for 4 days, then were transferred to hydroponic containers. Seedlings were placed in sponge holders in lids of black plastic containers, four plants per 750 ml solution, with continuous aeration. The nutrient solution had the following composition: 0.8 mM KNO3, 0.4 mM Ca(NO3)2, 0.3 mM NH4H2PO4, 0.2 mM MgSO4, 25 μM CaCl2, 25 μM H3BO3, 2 μM MnCl2, 2 μM ZnSO4, 0.5 μM CuSO4, 0.5 μM Na2MoO4, 100 mM MES buffer (pH 5.5) and 10 μM Sprint 138 (Becker-Underwood, Ames, IA, United States). Plants were grown in a growth chamber with lighting provided by a mixture of incandescent and fluorescent sources at 250 μmol m-2 s-1 for a photoperiod of 16 h (on at 06:00 and off at 22:00). For the +/- Fe RNA-seq experiment, Edisto and fefe mutants were pretreated for 9 days on -Cu solution, and fefe mutants that had green leaves were used for treatments of 3 days duration in -Fe nutrient solution or 20 μM Sprint 138. The purpose for the -Cu pretreatment was to use only healthy fefe plants [since the fefe chlorotic phenotype can be rescued using -Cu treatment (Waters et al., 2014)].
RNA-seq and Differential Expression Analysis
Total RNA was extracted from roots using the Plant RNeasy kit (Qiagen, Hilden, Germany). RNA quality and concentration was determined by UV spectrophotometry. Sources of RNA samples were as described in the previous section. RNA-seq was performed at the University of Nebraska Medical Center Next Generation Sequencing Core Facility using an Illumina HiSeq 2000 instrument. Barcoded libraries were constructed from 3 μg of root total RNA, with three biological replicate libraries per treatment. Replicates were run in separate lanes, with a total of six samples from different treatments in each lane. The short reads are available as NCBI BioProject: PRJNA3718262. Because melon and cucumber genomes are orthologous and the cucumber genome sequence and annotation is complete (Huang et al., 2009; González et al., 2010), the cucumber transcriptome was used as a reference for read mapping. Trimming of primers and adapters was performed using Trimmomatic (V0.32), read mapping was performed using BOWTIE2 (Langmead and Salzberg, 2012) with –local-N1 option, conversion of mapped reads into sam format was performed using SAMtools (Li et al., 2009) and extraction of read counts from sam files was performed using perl scripts, as previously reported (Waters et al., 2014). For gene expression analysis, the data matrix was imported into R and analyzed using the Bioconductor package DESeq (Anders and Huber, 2010). The count data was normalized for library size and then transformed using variance stabilization. Poisson distributions of normalized counts for each transcript were compared for different conditions using a negative binomial test. Differential expression was called for genes with a false discovery rate moderated q-value < 0.05 (Benjamini and Hochberg, 1995), and also showed a 1.0 log fold-change in expression and >10 reads in at least one treatment. De novo reconstruction of the bHLH38 transcript was performed using Trinity software (V.r20131110) (Haas et al., 2013). IGVviewer 2.33 was used to view the reads that are mapped onto the reference sequence as applicable.
Reverse Transcription-PCR
One microgram of DNase treated RNA (RNase-free DNase I, New England Biolabs, Ipswich, MA, United States) from roots of -Fe and -Fe/-Cu treated Edisto and fefe and from eight normal and eight mutant snake melon X fefe F2 roots was used for cDNA synthesis, using the High Capacity cDNA Reverse Transcription kit (ABI, Foster City, CA, United States) with random hexamers at 2.5 μM final concentration. The cDNA templates were PCR amplified using primers spanning the insertion: fefe_mrkr_F-5′-AGAAACTGAGTAATCCGGCGA-3′ and fefe_mrkr_R-5′ TCGACTTGCAGAAATTATCGA-3′.
Edisto bHLH38 Cloning and Hairy Root Transformation
Edisto bHLH38 (MELO3C019065) full length genomic sequence (2.334 kb promoter +2.32 kb gene) was PCR amplified using the primers bHLH38_Promoter_F 5′-TCCCTTTGAACCAATGATGG-3′ and bHLH38_XbaI_R 5′-GCATGATCTAGAACACATTGATATATATGGTTAATAA-3′. Phusion High-fidelity DNA polymerase (Thermo Scientific) was used for PCR amplification of bHLH38 following manufacturer’s instructions. An Xba1 restriction site was present in the promoter region at position 379 bp of 2.334 kb, and an XbaI site was in the reverse primer, underlined above. After XbaI restriction (NEB Biolabs), the resulting bHLH38 Xba1 fragment was cloned into the pHairyRed (Lin et al., 2011) destination vector. The Edisto-bHLH38 genomic construct was transformed into the K599 strain of Agrobacterium rhizogenes (generously provided by Dr. Christopher Taylor, The Ohio State University) using freeze-thaw transformation (Wise et al., 2006). K599 containing pHairyRed-Edisto bHLH38 genomic fragment or K599 containing pHairyRed (empty vector) was grown in YEP plates with streptomycin and kanamycin selection. Agrobacteria suspension was prepared as previously described (Kereszt et al., 2007), and plug preparation and inoculation of Agrobacteria into rock wool plugs was performed as described (Chabaud et al., 2006), except we used hydroponic liquid media (described above) instead of half-strength MS. Stem sections with one or two axillary nodes from 1-month-old fefe plants were cut and inserted into the hole in the rock wool plug, and the plants were covered in a humid chamber for 4–5 days under ambient light. The humid chamber was opened for dehydration treatment for several hours until the leaves were not turgid, and the humid chamber was closed. Hairy roots developed 2–3 weeks after transformation. Transgenic roots were distinguished from non-transgenic roots, based on the presence of DsRed fluorescence. Z-series images were acquired on a Nikon A1+ CLSM mounted on a Nikon 90i compound microscope. Excitation of DsRed was at 561 nm and emission was detected at 575–625 nm. Image series were projected to form a single image. The transmitted light images were simultaneously acquired, but only a single image plane is presented. At least two biological experiments were performed to obtain the transgenic plants.
Ferric-Chelate Reductase Activity
Root ferric reductase assays were performed for 50 min on transgenic roots (positive for DsRed fluorescence) and non-transgenic roots of fefe plants that were grown on -Fe solution for 2 days, using 20 ml of an assay solution. The assay solution was composed of 0.1 mM ferrozine (3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-4′,4′-disulfonic acid sodium salt; Sigma–Aldrich), 0.1 mM Fe(III)-EDTA and 1 mM MES buffer (pH 5.5) (Fisher Scientific, Fair Lawn, NJ, United States). Change of assay solution from colorless to purple indicates ferric-chelate reductase activity.
Yeast 2-Hybrid Interactions
The MatchmakerTM GAL4 Two-Hybrid System 3 (Clontech) was used for the yeast 2-hybrid experiment. Coding sequences for Edisto FIT, Edisto bHLH38 and the fefe bHLH38 with its 14 bp insertion were amplified from cDNA and were cloned into pGEM-T vector and were later cloned as EcoRI fragment into pGADT7 and pGBKT7 yeast two-hybrid vectors. Drop assays were performed by growing AH109 transformants on synthetic dropout (SD) liquid media without leucine (L) and tryptophan (T) at 29°C until an OD600 of >0.5 is reached. Cultures were then diluted to an OD600 of 0.1 and diluted in a 10× dilution series. For each dilution, 5 μl of cell suspension was spotted on SD media without leucine, tryptophan, histidine (H) and adenine (A).
Results
FeFe Gene Mapping
The chlorotic phenotype (Figure 1) mapped to a single locus on LG8 using the snake melon X fefe F2 mapping population (Ramamurthy and Waters, 2015) (Figure 2A). Using Syngenta marker positions, the fefe locus was mapped to a 6 cM (2-LOD support) (Figure 2A) region in the snake melon X fefe population. The percentage of variation explained by LG8 locus was approximately 59% (Figure 2A and Table 1). Due to the low polymorphism rate of 12% in the snake melon X fefe population, we could not further narrow down the fefe genetic interval with the markers available. Therefore, we also mapped the chlorosis phenotype in a pocket melon X fefe F2 mapping population. Of 288 F2 plants, 196 had normal green leaves and 92 had chlorotic leaves. 27 markers on LG8 were polymorphic in the parents. The distribution of the parental genotypes in the F2 population was almost equal based on 27 polymorphic loci on LG8, with 20.7% pocket melon genotype, 30.1% C940-fe genotype and 42.2% heterozygous. The fefe locus was mapped to a 4 cM interval, with a LOD score of 62 (Figure 2B and Table 1), with 63% of the variation explained by the peak (Table 1). Since both populations in this study had a common parent, C940-fe (fefe), we used joint linkage analysis to refine the fefe genetic interval to a 1 cM peak on LG8 with a LOD score of 115 (Figure 2C and Supplementary Data S1A). The closest marker at the peak explained ∼61% of the variation (Figure 2C and Table 1).

FIGURE 1. Leaf color phenotypes of fefe (chlorotic) and wild-type Edisto plants grown under different conditions in this study. (A) Pocket melon X fefe F2 plants grown in hydroponics. Scoring for chlorosis phenotype is indicated by N (normal) or C (chlorotic); (B) Greenhouse grown plants cultivated in commercial potting mix. (C) Field grown fefe mutant plant; (D) Field grown wild-type Edisto plant.
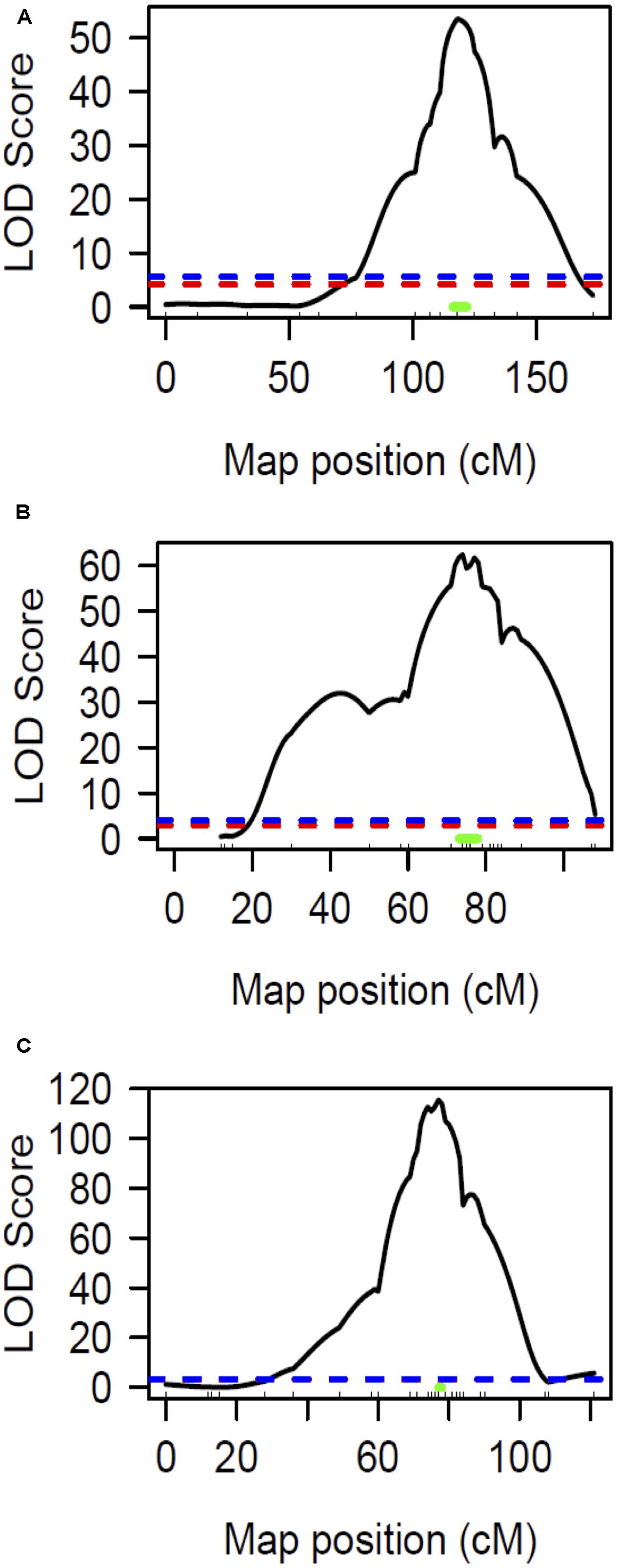
FIGURE 2. Linkage mapping of the fefe gene on chromosome 8. Ticks inside X-axis represent the position of markers. The blue and red dashed lines represent the permutation test specified LOD threshold at p-values of 0.05 and 0.01, respectively. The green bar represents Bayesian confidence interval associated with the fefe locus. (A) Interval mapping analysis using snake melon X fefe F2 (n = 269); (B) Interval mapping analysis using pocket melon X fefe F2 (n = 288); (C) Interval mapping analysis using joint linkage map (both populations) consisting of 557 F2 individuals.
FeFe Candidate Genes
Since the fefe gene was predicted to be a transcription factor (Waters et al., 2014), we explored possible candidates within the 1 cM confidence interval. The genetic interval spanning the fefe gene, between the markers DM0766 and DM0640, corresponded to two scaffolds (scaffold0068 and scaffold0036) of the melon genome draft (Garcia-Mas et al., 2012). There were 186 predicted genes (Supplementary Data S1B) in this interval. There were six genes annotated as transcription factors in the mapped interval, based on homology to Arabidopsis thaliana: AT3G56970.1 BHLH038; AT5G04150.1 BHLH101; AT3G57390.2 AGL18; AT5G62470.1 MYB96; AT3G57040.1 ARR9 (RESPONSE REGULATOR 9); and AT3G10760.1 myb family transcription factor. We ruled out AGL18, MYB96 and ARR9 as unlikely candidates for fefe based on their function (To et al., 2004; Verelst et al., 2007; Seo et al., 2011). The AT3G10760.1 myb family transcription factor is putatively involved in the fruit ripening process (Pillet et al., 2015). Two transcription factors in the interval, bHLH38 and bHLH101, are associated with regulation of Fe-uptake genes (Wang et al., 2007). To determine if these genes were polymorphic between fefe and WT Edisto, or were differentially regulated, we sequenced the cDNAs and observed the normalized RNA-seq read counts from two independent experiments. Sequencing the bHLH101 gene showed that it was not polymorphic between the mutant and WT plants. The expression of bHLH101 was extremely low in both WT and mutant plants, ranging from 0 to 3 total raw read counts (compared to average read counts of approximately 450) under Fe replete or Fe-deficient conditions, suggesting that bHLH101 can be considered not expressed. The bHLH38 transcript levels were much higher than bHLH101 both in WT and fefe, ranging from 965 to 3979 read counts under Fe replete or deficient conditions, respectively. Transcript abundance did not change significantly under Fe deficiency in Edisto and snake melon WT roots. Although the trend of Fe regulation appeared similar in WT and fefe, the fefe bHLH38 expression differed in the two RNA-seq experiments (Figure 3). In the first experiment, bHLH38 was not significantly up-regulated by Fe deficiency, but in the second experiment, bHLH38 abundance was about fourfold higher under Fe deficiency due mainly to an unusually low read count in the +Fe fefe sample. We also checked abundance of bHLH38 transcript by RT-PCR (data not shown) and confirmed that the transcript is not regulated by Fe deficiency.
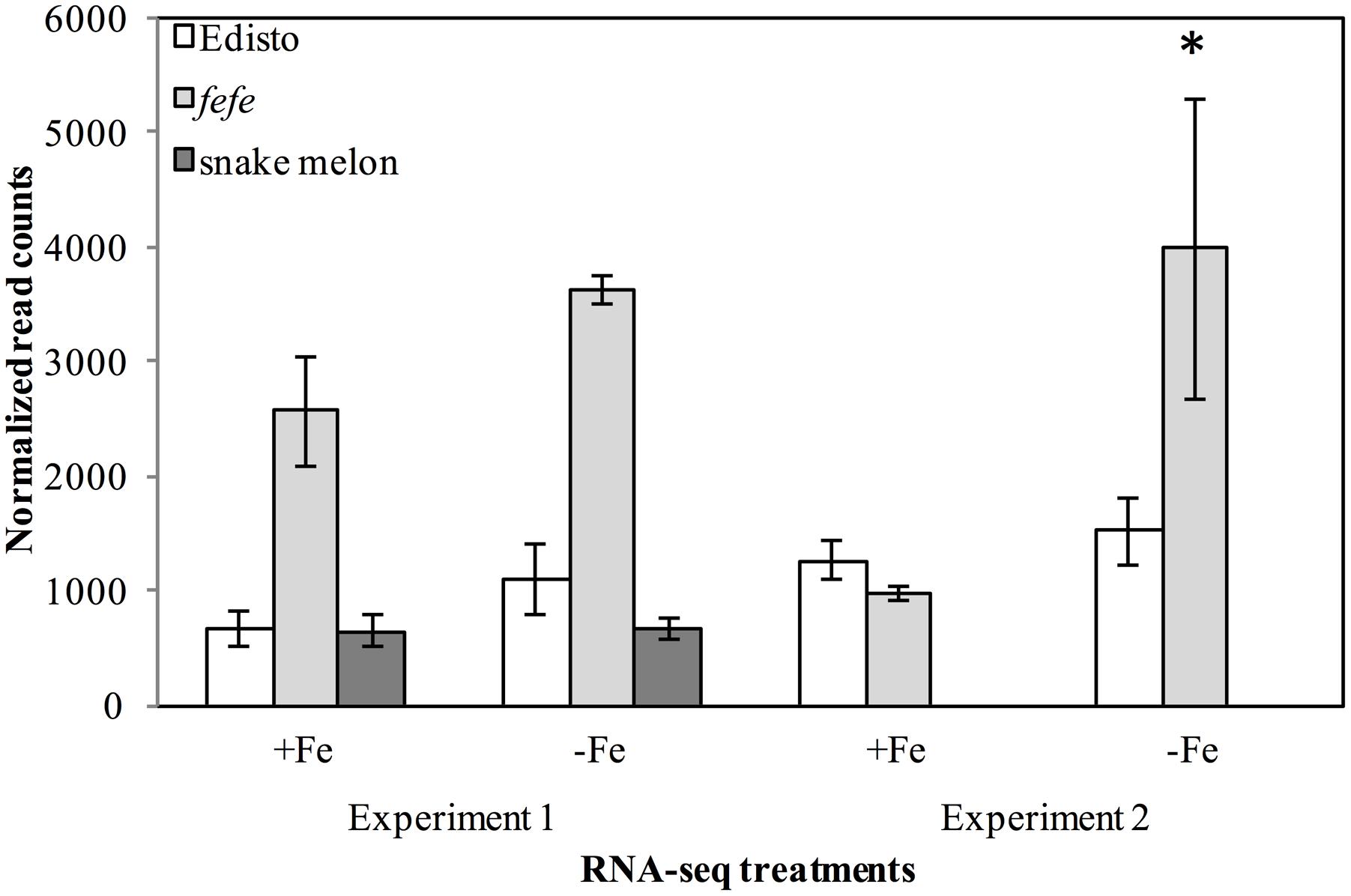
FIGURE 3. Gene expression (normalized read counts ± SD) of bHLH38 in wild-types Edisto and snake melon and the fefe mutant under Fe deficient and Fe sufficient control conditions in two independent RNA-seq experiments. Experiment 1 is from Waters et al. (2014); experiment 2 is from this study. ∗Indicates significant difference between Fe-deficient and control treatment means at FDR < 0.05.
To better understand the expression of the bHLH38 gene in fefe and WT plants, we visualized read mapping of fefe and Edisto bHLH38 against the reference cucumber CsbHLH38 transcript (Csa4M434480.1). The reads mapped uniformly to the reference CsbHLH38 transcript in Edisto (Figure 4A), but the fefe read counts were much higher at the beginning of the transcript and decreased abruptly at position 370 bp of the reference transcript (Figure 4B), for both Fe deficient and Fe sufficient roots, suggesting the presence of a transcript variant. We performed de novo assembly of the Edisto and fefe transcriptomes to reconstruct the bHLH38 transcripts. The assembled WT bHLH038 transcript was 943 bp, which includes predicted start and stop codons to include 309 deduced amino acids. The de novo assembly of the fefe mutant resulted in eight unique transcripts, which were longer than the WT transcript by 277–4789 bp. The extra length occurred beginning at 549 bp in the WT bHLH038 transcript, where we saw read mapping anomalies relative to the cucumber transcript. Both before 549 bp and after the insertion, from 550 bp onward, the fefe transcript sequence matched the WT transcript sequence. Using RT-PCR with primers spanning the insertion site to visualize cDNA, a single band was present in Edisto, whereas the fefe parent contained multiple bands, in agreement with the de novo assembly results, in -Fe, and -Fe/-Cu treatments (Figure 5A). Similar to Fe deficiency, Cu deficiency did not change the bHLH38 banding pattern. These results suggest that the fefe mutant was producing multiple insertion-containing bHLH38 transcripts, or one large transcript that had been differently or partially spliced. A cDNA laddering pattern was also observed in the snake melon X fefe F2 mapping population (Figure 5B), but only in individual plants with the chlorotic fefe phenotype. The fefe cDNA band that was closest in size to the Edisto bHLH38 cDNA was sequenced, to reveal the presence of a 14 bp insertion. Relative to the start codon, the fefe transcript had a reading frame shift followed by a premature in-frame stop codon, and would produce a different deduced amino acid sequence (Supplementary Data S2) that could negatively affect protein structure and function.
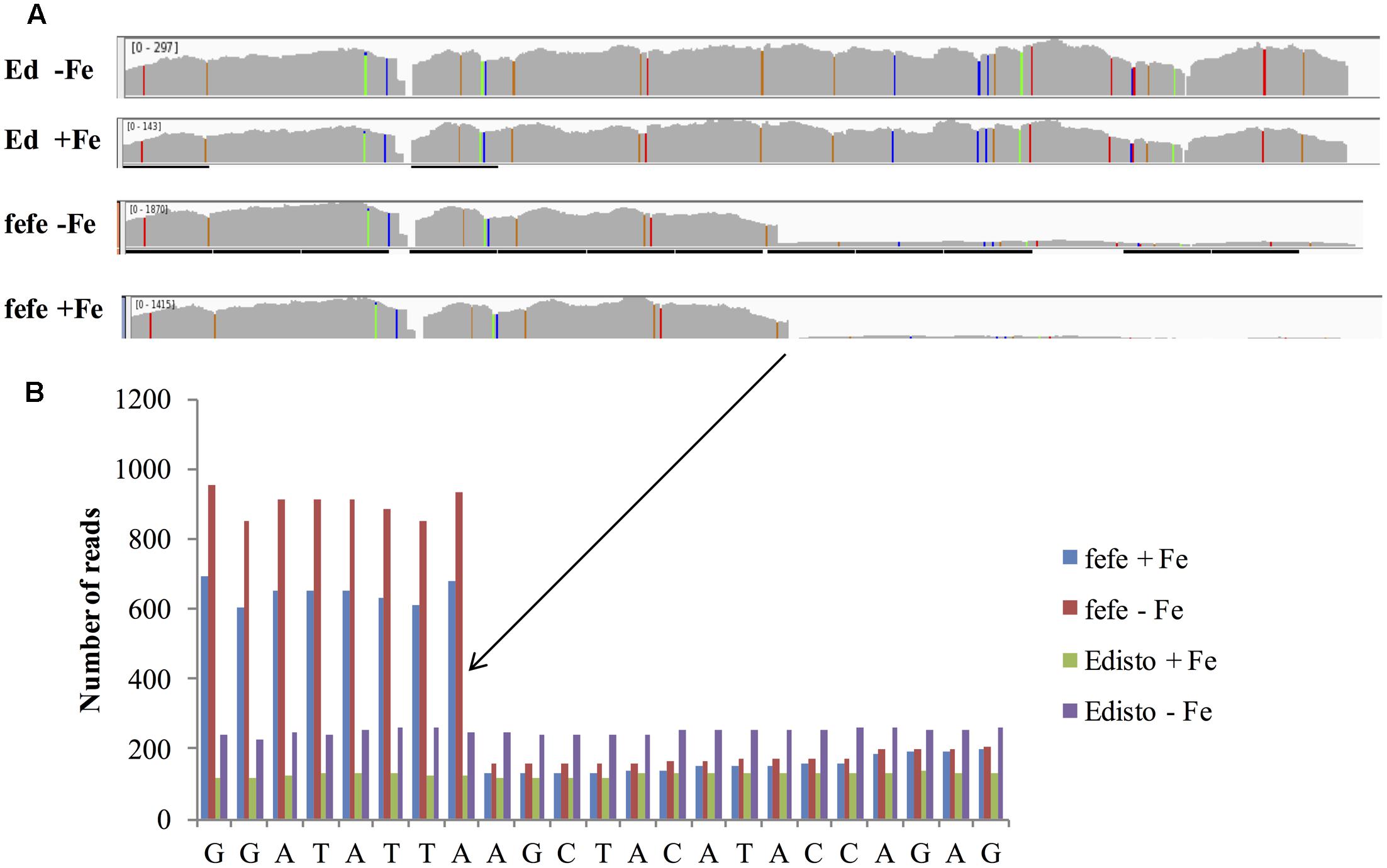
FIGURE 4. RNA-seq reads of melon bHLH38 mapped onto cucumber reference transcript Csa4M434480.1. (A) Read mapping of bHLH38 full length transcript in Edisto and fefe under Fe sufficient or deficient conditions; (B) Detail of sequence around site of the abrupt decrease in read counts in fefe bHLH38.
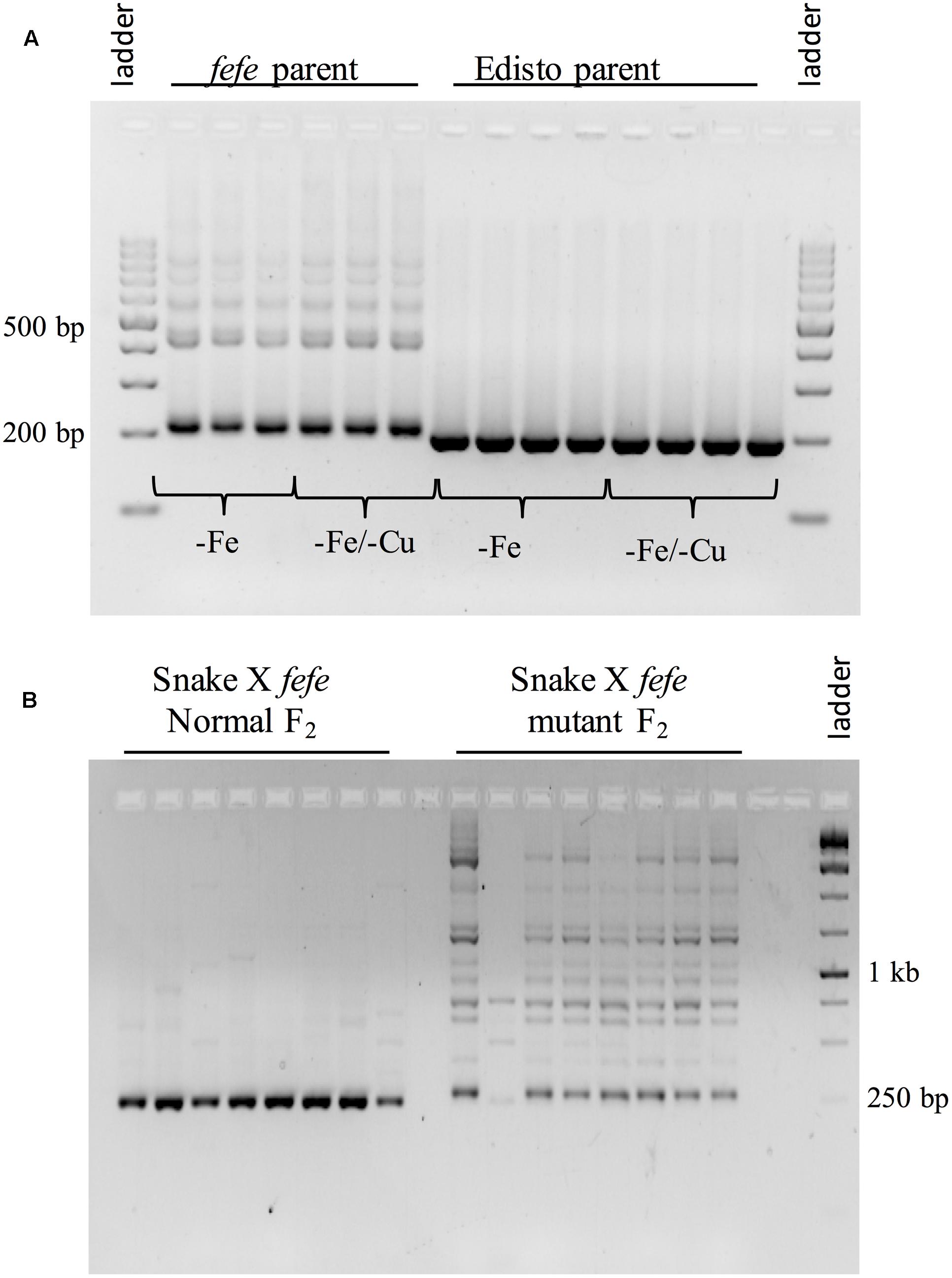
FIGURE 5. Reverse-transcriptase PCR of bHLH38 using primers that span the retrotransposon insertion site. (A) Transcripts from roots of multiple individual plants of fefe or Edisto grown under Fe deficiency or simultaneous Fe and Cu deficiency; (B) Transcripts from roots of multiple individual plants of the snake melon X fefe F2 mapping population scored as normal leaf color (left) or chlorotic, indicating fefe mutant (right).
To further investigate the source of the transcript insertions in fefe bHLH38, we amplified genomic bHLH38 in normal and fefe plants by PCR and sequenced the products. All WT lines produced an amplicon of 1.019 kb, while fefe produced a 6.076 kb fragment (Figure 6). The fefe bHLH38 contained a 5.057 kb insertion relative to WT Edisto (Supplementary Data S3). The sequence of the insertion had identical 278 bp sequences at both extremes of the insertion (Supplementary Data S3 and Figure S1). A BLAST search of the fefe genomic bHLH38 sequence against the melon reference genome had hits to six melon genomic scaffolds, unassembled sequences and scaffold 36. Length of the hits for fefe bHLH38 using BLAST search against the melon genome ranged between 272 and 1368 bp, and the total scores from BLAST search ranged between 608 and 2510. An NCBI conserved domain search identified the insertion as a long terminal repeat (LTR) Ty1-copia type retrotransposon. The fefe bHLH38 contained helix-loop-helix, polypurine tract, RNAseH1-RT-Ty1, Reverse transcriptase, Integrase, gag-polypeptide and primer binding site of LTR-copia type domains (Supplementary Figure S1). There were also large genomic bands in WTs Mainstream and pocket melon, of a similar but not identical size as the band in fefe (Figure 6). From the banding pattern and BLAST search, it appears that the retrotransposon in fefe-bHLH38 could be present in other loci in the melon genome, potentially as an intact sequence. A global BLAST search for sequences similar to fefe bHLH38 in the NCBI nucleotide database indicated that the fefe bHLH38 retrotransposon was specific to melon.
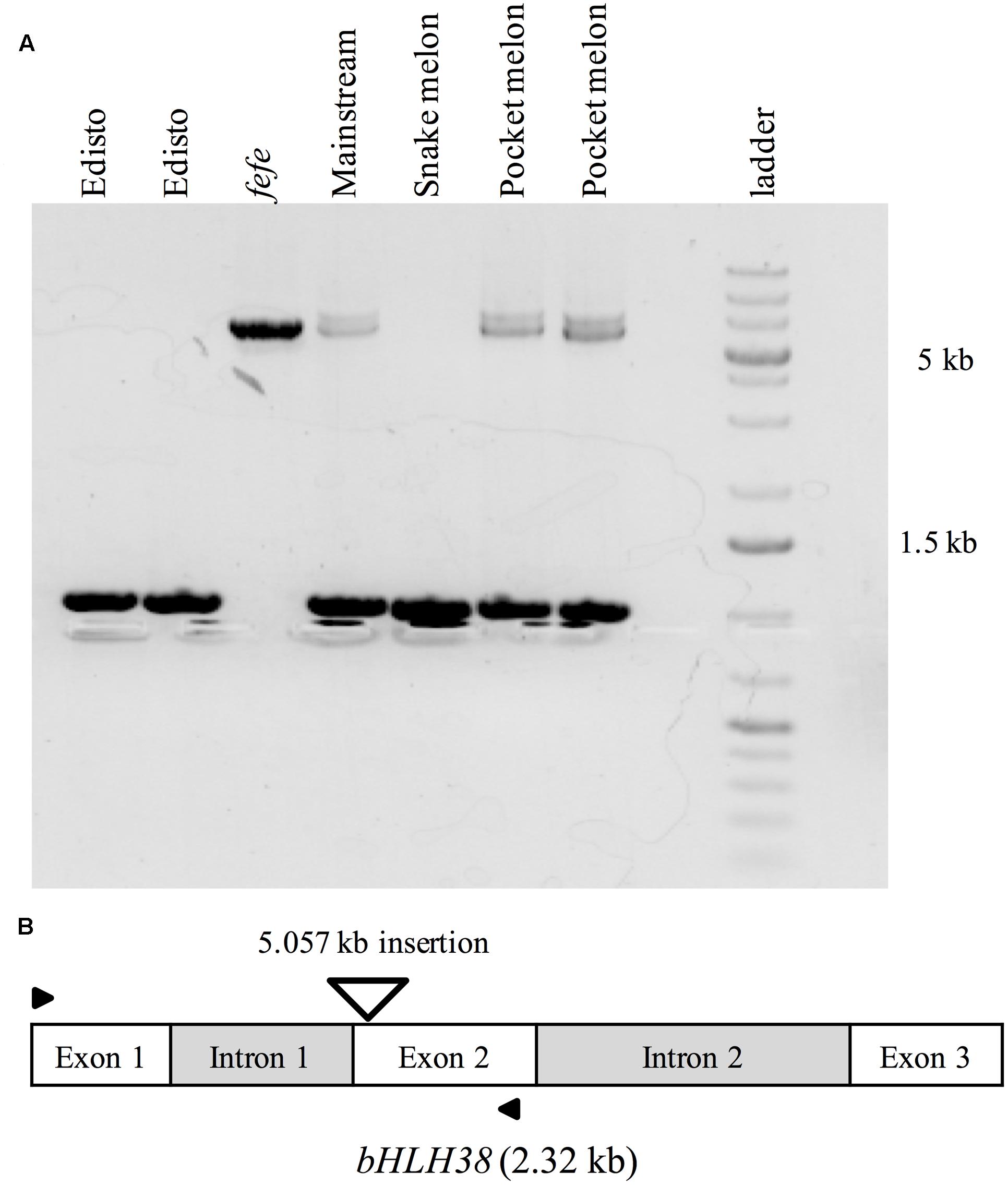
FIGURE 6. PCR amplification of genomic bHLH38 from wild-type varieties and the fefe mutant. (A) From left to right, two individual plants of the wild-type Edisto, fefe mutant, wild-type Mainstream, wild-type snake melon, and two individual plants of the wild-type pocket melon; (B) Diagram depicting the melon bHLH38 gene structure and insertion site of the 5.057 kb retrotransposon in fefe bHLH38 (triangle). Arrows indicate the location of primers used for amplification of genomic bHLH38 in (A).
Complementation of the fefe Phenotype Using Hairy Root Transformation
Since the FeFe gene is necessary for normal plant growth and Fe uptake only in roots (Waters et al., 2014), we tested whether the chlorotic fefe phenotype could be complemented with a normal copy of bHLH38. We transformed fefe plants with Agrobacterium rhizogenes to generate hairy roots containing the WT bHLH38 gene (Figure 7). The chlorotic phenotype of the fefe plants was rescued in the pHairyRed-Edisto-bHLH38 treated plants, but not in mock or empty vector treated plants (Figure 7A). The rescued fefe plant roots were positive for the dsRed reporter gene (Figures 7B–G), and were able to initiate ferric reductase activity under Fe deficiency (Figure 7H), suggesting that normal root Fe uptake responses were recovered in the transgenic fefe plant roots expressing Edisto-bHLH38.

FIGURE 7. Complementation of the chlorotic fefe phenotype by hairy root transformation. (A) Plant mock-transformed with Agrobacterium rhizogenes K599, transformed with empty pHairyRed vector, and transformed with pHairyRed-Edisto-bHLH038. (B–D) DsRed expression in hairy roots of fefe plants viewed by confocal microscopy, (B) mock transformation, (C) pHairyRed and (D) pHairyRed-Edisto-bHLH038. Bright field view of same roots in (B–D); (E) mock transformation, (F) pHairyRed and (G) pHairyRed-Edisto-bHLH038 roots; (H) Ferric chelate reductase activity of rescued fefe plant roots transformed with pHairyRed-Edisto-bHLH38 compared to non-transgenic roots.
Yeast Two-hybrid Assays for Protein–Protein Interactions
To determine whether the bHLH38 protein interacts with the Fit protein, as it does in Arabidopsis (Yuan et al., 2008), we performed a yeast two-hybrid experiment. Yeast cells transformed with different combinations of bait and prey plasmids were tested for auxotrophic growth. The melon Fit protein tested positive for interacting with Edisto bHLH38, but did not interact with fefe bHLH38 (Figure 8). The Edisto bHLH38 protein was capable of forming a homodimer, although yeast growth was less robust than in other combinations (Supplementary Figure S2A). The fefe bHLH38 did not form homodimers (Supplementary Figure S2B). Yeast growth suggested that there was some degree of interaction between Edisto bHLH38 and fefe bHLH38. This test could not be used to determine whether melon Fit forms a homodimer, since Fit was capable of auto-activation, as indicated by growth of yeast transformed with pAD+pBD-FIT (Figure 8).

FIGURE 8. Yeast 2-hybrid assay of bHLH38 and Fit in Edisto (ed) and fefe plated on SD media without Leu, Trp, His, and Ade in a serial dilution series. pAD-FIT+pBD ed-bHLH38 tests the interaction of FIT with Edisto-bHLH38, pAD-FIT+pBD fefe-bHLH38 tests the interaction of FIT with fefe-bHLH38, pAD-FIT+pBD and pAD+pBD-FIT are controls to test if FIT is capable of auto-activation, pAD-FIT+pBD-FIT is a test to check homodimer formation of FIT. pAD-T7-T+pBD-T7-53 is a positive control for the assay.
Discussion
In this paper, we seek new understanding of how Strategy I plants respond to Fe-deficiency stress by mapping the fefe gene that controls Fe uptake in melon. We mapped the fefe gene to bHLH38, which contains a 5.056 kb Ty1-copia type retrotransposon insertion. Multiple length transcripts were observed in fefe-bHLH38 (Figures 5A,B), apparently due to the full retrotransposon being incorporated into the transcript and partially spliced out to varying degrees. The altered RNA-seq read mapping we observed, with about three times higher reads before the retrotransposon insertion site in fefe-bHLH38 (Figures 3, 4), may arise from promoters within LTR regions of LTR retrotransposons (Kumar and Bennetzen, 1999). Coincidentally, the loss of Fe homeostasis in the tomato fer mutant (Ling et al., 2002) is due to an insertion of a copia-type retrotransposon, called Rider, in the first exon of the FER gene (Cheng et al., 2009). Rider replicates by reverse transcribing an aberrant and novel cDNA that can include nearby genes, and this novel cDNA is then integrated into a new location in the genome (Lisch, 2012). We saw evidence for a pseudogene in some varieties of melon (Mainstream and pocket melon) that was amplified by PCR primers located in the exons of the bHLH38 gene (Figure 6), suggesting that the retrotransposon in fefe exists in other loci in the melon genome. To our knowledge this retrotransposon has not been described in detail, since BLAST searches of the reference melon genome only hit unassembled scaffolds, however, it could be an important feature in melon evolution and diversity (Kumar and Bennetzen, 1999).
Regulation of Fe-uptake genes, such as FRO1, Nramp1, and IRT1, was abolished in the fefe mutant (Waters et al., 2014), similar to the Fit mutant of Arabidopsis (Colangelo and Guerinot, 2004) and the fer mutant of tomato (Ling et al., 2002), however, the lesion in the fefe mutant is not in the FIT gene (Waters et al., 2014). The formation of a heterodimer between the Fit protein and subgroup Ib bHLH proteins is a hallmark of transcriptional regulation of Fe-uptake genes (Yuan et al., 2008; Du et al., 2015). The fefe-bHLH38 transcript that was closest in size to the normal bHLH38 transcript would be translated to a protein that has an altered sequence after the first 120 aa, and terminates at 144 aa instead of 249 aa, due to the 14-bp insertion (Supplementary Data S2). Using the yeast 2-hybrid technique, the WT-bHLH38 interacted with the Fit protein, however, the fefe-bHLH38 protein did not form a heterodimer with the Fit protein (Figure 8). We propose that this lack of Fit-bHLH38 heterodimer formation is the cause of abolished upregulation of Fe-uptake gene expression in fefe. The severity of the fefe phenotype under Fe sufficient conditions from mutation of a single bHLH gene is surprising considering that in Arabidopsis, single, double, or even triple bHLH mutants (with bHLH38 or bHLH39 remaining and the other three subgroup Ib bHLH genes knocked out) had no Fe deficiency phenotype under Fe sufficient conditions (Wang et al., 2007; Sivitz et al., 2012; Andriankaja et al., 2014; Maurer et al., 2014), suggesting that one of the four bHLH proteins is adequate for Fe-uptake gene regulation. The tomato genome has three subgroup Ib bHLH genes (Sun et al., 2015), and the soybean genome has two subgroup Ib bHLH genes. A 12 bp deletion in one of the soybean bHLH genes was suggested to cause increased sensitivity to alkalinity-induced Fe deficiency chlorosis (Peiffer et al., 2012). But, like the Arabidopsis bHLH mutants, the soybean lines with this deletion are not chlorotic under Fe sufficient conditions (O’Rourke et al., 2007). A knockout line for the soybean bHLH genes has not been reported, and quadruple Arabidopsis bHLH lines have not been generated. So far, fefe is the only subgroup Ib bHLH mutant with an Fe uptake phenotype as severe as the fit or fer mutants. While the melon genome has a second subgroup Ib bHLH gene, bHLH101 was not polymorphic between WT and the fefe mutant, its transcript abundance was strikingly low, which together with genetic results suggests that melon bHLH101 is not functionally redundant with melon bHLH38.
Another key difference between melon bHLH38 and subgroup Ib bHLH genes in other plant species is their transcriptional regulation in roots by Fe status. In Arabidopsis and tomato, subgroup Ib bHLH genes are upregulated upon Fe deficiency in roots (Wang et al., 2007; Dinneny et al., 2008; Buckhout et al., 2009; Yang et al., 2010; Bauer and Blondet, 2011; Schuler et al., 2011; Stein and Waters, 2012; Waters et al., 2012; Andriankaja et al., 2014; Maurer et al., 2014; Sun et al., 2015). However, bHLH38 was not upregulated in Fe deficient melon roots (Figure 3; Waters et al., 2014; Hsieh and Waters, 2016). Copper deficiency also did not change melon bHLH38 expression, or its transcript pattern in fefe (Figure 5A), suggesting that the rescue of the chlorotic phenotype of fefe under simultaneous Fe and Cu deficiency (Waters et al., 2014) was not due to a change in bHLH38 expression, splicing, or protein function. However, since the bHLH38 protein is crucial to Fe homeostasis, its regulation may be entirely at the post-transcriptional or post-translational level. Arabidopsis FIT is regulated at both the transcriptional and post-transcriptional levels (Meiser et al., 2011; Sivitz et al., 2011).
We confirmed that the fefe defect in root Fe-uptake is due to loss of function of bHLH38 by complementation of the fefe chlorotic phenotype with WT-bHLH38 (Figure 7). The mapping and identification of the fefe mutation as bHLH38 has given new insight into regulation of Fe homeostasis in Strategy I plants. The fefe mutant may prove to be a valuable platform for studying bHLH genes and proteins from other plant species, since it can be complemented by hairy root transformation. Further characterization of bHLH38 protein regulation in melon is a needed future direction to help provide understanding of Fe-uptake control mechanisms.
Author Contributions
RR and BW planned and designed experiments. RR conducted experiments and performed statistical and bioinformatics analysis. RR and BW wrote the manuscript. All authors read and approved the whole manuscript.
Funding
This work was supported by funding from United States Department of Agriculture-National Institute for Food and Agriculture (USDA-NIFA) competitive grant (2014-67013-21658) and the National Science Foundation (NSF IOS-1257568).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Keenan Amundsen for RNA-seq analysis, Dr. Puneet Paul for help with yeast 2-hybrid assays, and Morrison Microscopy Core Research Facility (Dr. Christian Elowsky) for help with dissection and confocal microscopy. Authors would also like to thank Tony Delaney, Grace Troupe, Laura Armbrust and Dr. Ricardo Stein for technical support and Dr. Mary Guttieri for helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01003/full#supplementary-material
Footnotes
- ^ http://www.icugi.org/cgi-bin/cmap/map_set_info?species_acc=CM
- ^ https://trace.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA371826&go=go
- ^ http://www.broadinstitute.org/igv
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Andriankaja, M. E., Danisman, S., Mignolet-Spruyt, L. F., Claeys, H., Kochanke, I., Vermeersch, M., et al. (2014). Transcriptional coordination between leaf cell differentiation and chloroplast development established by TCP20 and the subgroup Ib bHLH transcription factors. Plant Mol. Biol. 85, 233–245. doi: 10.1007/s11103-014-0180-2
Bashir, K., and Nishizawa, N. K. (2013). “Iron proteins, plant iron transporters,” in Encyclopedia of Metalloproteins, eds R. H. Kretsinger, V. N. Uversky, and E. A. Permyakov (New York, NY: Springer), 1015–1023. doi: 10.1007/978-1-4614-1533-6_356
Bauer, P., and Blondet, E. (2011). Transcriptome analysis of ein3 eil1 mutants in response to iron deficiency. Plant Signal. Behav. 6, 1669–1671. doi: 10.4161/psb.6.11.17847
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. doi: 10.2307/2346101
Briat, J. F., Dubos, C., and Gaymard, F. (2015). Iron nutrition, biomass production, and plant product quality. Trends Plant Sci. 20, 33–40. doi: 10.1016/j.tplants.2014.07.005
Broman, K. W., Wu, H., Sen, S., and Churchill, G. A. (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890.
Buckhout, T. J., Yang, T. J., and Schmidt, W. (2009). Early iron-deficiency-induced transcriptional changes in Arabidopsis roots as revealed by microarray analyses. BMC Genomics 10:147. doi: 10.1186/1471-2164-10-147
Castaings, L., Caquot, A., Loubet, S., and Curie, C. (2016). The high-affinity metal transporters NRAMP1 and IRT1 team up to take up iron under sufficient metal provision. Sci. Rep. 6:37222. doi: 10.1038/srep37222
Chabaud, M., Boisson-Dernier, A., Zhang, J., Taylor, C. G., Yu, O., and Barker, D. G. (2006). “Agrobacterium rhizogenes-mediated root transformation,” in The Medicago Truncatula Handbook, Version November, eds U. Mathesius, E. P. Journer, and L. W. Sumne (Ardmore, OK: The Samuel Roberts Noble Foundation).
Chen, Y., and Barak, P. (1982). Iron nutrition of plants in calcareous soils. Adv. Agron. 35, 217–240.
Cheng, X., Zhang, D., Cheng, Z., Keller, B., and Ling, H.-Q. (2009). A new family of Ty1-copia-like retrotransposons originated in the tomato genome by a recent horizontal transfer event. Genetics 181, 1183–1193. doi: 10.1534/genetics.108.099150
Colangelo, E. P., and Guerinot, M. L. (2004). The essential basic helix-loop-helix protein FIT1 is required for the iron deficiency response. Plant Cell 16, 3400–3412. doi: 10.1105/tpc.104.024315
Curie, C., Alonso, J. M., Le Jean, M., Ecker, J. R., and Briat, J. F. (2000). Involvement of NRAMP1 from Arabidopsis thaliana in iron transport. Biochem. J. 347, 749–755.
Curie, C., Panaviene, Z., Loulergue, C., Dellaporta, S. L., Briat, J. F., and Walker, E. L. (2001). Maize yellow stripe1 encodes a membrane protein directly involved in Fe(III) uptake. Nature 409, 346–349. doi: 10.1038/35053080
Diaz, A., Fergany, M., Formisano, G., Ziarsolo, P., Blanca, J., Fei, Z., et al. (2011). A consensus linkage map for molecular markers and quantitative trait loci associated with economically important traits in melon (Cucumis melo L.). BMC Plant Biol. 11:111. doi: 10.1186/1471-2229-11-111
Dinneny, J. R., Long, T. A., Wang, J. Y., Jung, J. W., Mace, D., Pointer, S., et al. (2008). Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 320, 942–945. doi: 10.1126/science.1153795
Du, J., Huang, Z., Wang, B., Sun, H., Chen, C., Ling, H.-Q., et al. (2015). SlbHLH068 interacts with FER to regulate the iron-deficiency response in tomato. Ann. Bot. 116, 23–34. doi: 10.1093/aob/mcv058
Garcia-Mas, J., Benjak, A., Sanseverino, W., Bourgeois, M., Mir, G., González, V. M., et al. (2012). The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. U.S.A. 109, 11872–11877. doi: 10.1073/pnas.1205415109
González, V. M., Rodríguez-Moreno, L., Centeno, E., Benjak, A., Garcia-Mas, J., Puigdomènech, P., et al. (2010). Genome-wide BAC-end sequencing of Cucumis melo using two BAC libraries. BMC Genomics 11:618. doi: 10.1186/1471-2164-11-618
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hsieh, E.-J., and Waters, B. M. (2016). Alkaline stress and iron deficiency regulate iron uptake and riboflavin synthesis gene expression differently in root and leaf tissue: implications for iron deficiency chlorosis. J. Exp. Bot. 67, 5671–5685.
Huang, S., Li, R., Zhang, Z., Li, L., Gu, X., Fan, W., et al. (2009). The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281. doi: 10.1038/ng.475
Kang, H., Cho, Y., Yoon, U., and Eun, M. (1998). A rapid DNA extraction method for RFLP and PCR analysis from a single dry seed. Plant Mol. Biol. Rep. 16, 1–9. doi: 10.1023/a:1007418606098
Kereszt, A., Li, D., Indrasumunar, A., Nguyen, C. D., Nontachaiyapoom, S., Kinkema, M., et al. (2007). Agrobacterium rhizogenes-mediated transformation of soybean to study root biology. Nat. Protoc. 2, 948–952. doi: 10.1038/nprot.2007.141
Kobayashi, T., and Nishizawa, N. K. (2012). Iron uptake, translocation, and regulation in higher plants. Annu. Rev. Plant Biol. 63, 131–152. doi: 10.1146/annurev-arplant-042811-105522
Kumar, A., and Bennetzen, J. L. (1999). Plant retrotransposons. Annu. Rev. Genet. 33, 479–532. doi: 10.1146/annurev.genet.33.1.479
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, D., Cuevas, H. E., Yang, L., Li, Y., Garcia-Mas, J., Zalapa, J., et al. (2011). Syntenic relationships between cucumber (Cucumis sativus L.) and melon (C. melo L.) chromosomes as revealed by comparative genetic mapping. BMC Genomics 12:396. doi: 10.1186/1471-2164-12-396
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin, M.-H., Gresshoff, P. M., Indrasumunar, A., and Ferguson, B. J. (2011). pHairyRed: a novel binary vector containing the DsRed2 reporter gene for visual selection of transgenic hairy roots. Mol. Plant 4, 537–545. doi: 10.1093/mp/ssq084
Ling, H.-Q., Bauer, P., Bereczky, Z., Keller, B., and Ganal, M. (2002). The tomato fer gene encoding a bHLH protein controls iron-uptake responses in roots. Proc. Natl. Acad. Sci. U.S.A. 99, 13938–13943. doi: 10.1073/pnas.212448699
Lisch, D. (2012). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Marschner, H., Romheld, V., and Kissel, M. (1986). Different strategies in higher plants in mobilization and uptake of iron. J. Plant Nutr. 9, 695–713. doi: 10.1080/01904168609363475
Maurer, F., Naranjo Arcos, M. A., and Bauer, P. (2014). Responses of a triple mutant defective in three iron deficiency-induced Basic Helix-Loop-Helix genes of the subgroup Ib(2) to iron deficiency and salicylic acid. PLoS ONE 9:e99234. doi: 10.1371/journal.pone.0099234
Meiser, J., Lingam, S., and Bauer, P. (2011). Posttranslational regulation of the iron deficiency basic helix-loop-helix transcription factor FIT is affected by iron and nitric oxide. Plant Physiol. 157, 2154–2166. doi: 10.1104/pp.111.183285
Nugent, P. E., and Bhella, H. S. (1988). A new chlorotic mutant of muskmelon. HortScience 23, 379–381.
O’Rourke, J. A., Charlson, D. V., Gonzalez, D. O., Vodkin, L. O., Graham, M. A., Cianzio, S. R., et al. (2007). Microarray analysis of iron deficiency chlorosis in near-isogenic soybean lines. BMC Genomics 8:476. doi: 10.1186/1471-2164-8-476
Peiffer, G. A., King, K. E., Severin, A. J., May, G. D., Cianzio, S. R., Lin, S. F., et al. (2012). Identification of candidate genes underlying an iron efficiency quantitative trait locus in soybean. Plant Physiol. 158, 1745–1754. doi: 10.1104/pp.111.189860
Pillet, J., Yu, H.-W., Chambers, A. H., Whitaker, V. M., and Folta, K. M. (2015). Identification of candidate flavonoid pathway genes using transcriptome correlation network analysis in ripe strawberry (Fragaria × ananassa) fruits. J. Exp. Bot. 66, 4455–4467. doi: 10.1093/jxb/erv205
Ramamurthy, R. K., and Waters, B. M. (2015). Identification of fruit quality and morphology QTLs in melon (Cucumis melo) using a population derived from flexuosus and cantalupensis botanical groups. Euphytica 204, 163–177. doi: 10.1007/s10681-015-1361-z
Robinson, N. J., Procter, C. M., Connolly, E. L., and Guerinot, M. L. (1999). A ferric-chelate reductase for iron uptake from soils. Nature 397, 694–697.
Römheld, V., and Marschner, H. (1986). Evidence for a specific uptake system for iron phytosiderophores in roots of grasses. Plant Physiol. 80, 175–180.
Santi, S., and Schmidt, W. (2009). Dissecting iron deficiency-induced proton extrusion in Arabidopsis roots. New Phytol. 183, 1072–1084. doi: 10.1111/j.1469-8137.2009.02908.x
Schuler, M., Keller, A., Backes, C., Philippar, K., Lenhof, H.-P., and Bauer, P. (2011). Transcriptome analysis by GeneTrail revealed regulation of functional categories in response to alterations of iron homeostasis in Arabidopsis thaliana. BMC Plant Biol. 11:87. doi: 10.1186/1471-2229-11-87
Seo, P. J., Lee, S. B., Suh, M. C., Park, M.-J., Go, Y. S., and Park, C.-M. (2011). The MYB96 transcription factor regulates cuticular wax biosynthesis under drought conditions in Arabidopsis. Plant Cell 23, 1138–1152. doi: 10.1105/tpc.111.083485
Sivitz, A., Grinvalds, C., Barberon, M., Curie, C., and Vert, G. (2011). Proteasome-mediated turnover of the transcriptional activator FIT is required for plant iron-deficiency responses. Plant J. 66, 1044–1052. doi: 10.1111/j.1365-313X.2011.04565.x
Sivitz, A. B., Hermand, V., Curie, C., and Vert, G. (2012). Arabidopsis bHLH100 and bHLH101 control iron homeostasis via a FIT-independent pathway. PLoS ONE 7:e44843. doi: 10.1371/journal.pone.0044843
Stein, R. J., and Waters, B. M. (2012). Use of natural variation reveals core genes in the transcriptome of iron-deficient Arabidopsis thaliana roots. J. Exp. Bot. 63, 1039–1055. doi: 10.1093/jxb/err343
Sun, H., Fan, H.-J., and Ling, H.-Q. (2015). Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genomics 16:9. doi: 10.1186/s12864-014-1209-2
To, J. P. C., Haberer, G., Ferreira, F. J., Deruère, J., Mason, M. G., Schaller, G. E., et al. (2004). Type-A Arabidopsis response regulators are partially redundant negative regulators of cytokinin signaling. Plant Cell 16, 658–671. doi: 10.1105/tpc.018978
Varotto, C., Maiwald, D., Pesaresi, P., Jahns, P., Salamini, F., and Leister, D. (2002). The metal ion transporter IRT1 is necessary for iron homeostasis and efficient photosynthesis in Arabidopsis thaliana. Plant J. 31, 589–599.
Verelst, W., Twell, D., de Folter, S., Immink, R., Saedler, H., and Münster, T. (2007). MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol. 8:R249. doi: 10.1186/gb-2007-8-11-r249
Vert, G., Grotz, N., Dédaldéchamp, F., Gaymard, F., Guerinot, M. L., Briat, J.-F., et al. (2002). IRT1, an Arabidopsis transporter essential for iron uptake from the soil and for plant growth. Plant Cell 14, 1223–1233. doi: 10.1105/TPC.001388
Von Jolley, D., Brown, J. C., and Nugent, P. E. (1991). A genetically related response to iron deficiency stress in muskmelon. Plant Soil 130, 87–92. doi: 10.1007/BF00011860
Wang, H.-Y., Klatte, M., Jakoby, M., Bäumlein, H., Weisshaar, B., and Bauer, P. (2007). Iron deficiency-mediated stress regulation of four subgroup Ib BHLH genes in Arabidopsis thaliana. Planta 226, 897–908. doi: 10.1007/s00425-007-0535-x
Wang, N., Cui, Y., Liu, Y., Fan, H., Du, J., Huang, Z., et al. (2013). Requirement and functional redundancy of Ib subgroup bHLH proteins for iron deficiency responses and uptake in Arabidopsis thaliana. Mol. Plant 6, 503–513. doi: 10.1093/mp/sss089
Waters, B. M., Lucena, C., Romera, F. J., Jester, G. G., Wynn, A. N., Rojas, C. L., et al. (2007). Ethylene involvement in the regulation of the H+-ATPase CsHA1 gene and of the new isolated ferric reductase CsFRO1 and iron transporter CsIRT1 genes in cucumber plants. Plant Physiol. Biochem. 45, 293–301. doi: 10.1016/j.plaphy.2007.03.011
Waters, B. M., McInturf, S. A., and Amundsen, K. (2014). Transcriptomic and physiological characterization of the fefe mutant of melon (Cucumis melo) reveals new aspects of iron-copper crosstalk. New Phytol. 203, 1128–1145. doi: 10.1111/nph.12911
Waters, B. M., McInturf, S. A., and Stein, R. J. (2012). Rosette iron deficiency transcript and microRNA profiling reveals links between copper and iron homeostasis in Arabidopsis thaliana. J. Exp. Bot. 63, 5903–5918. doi: 10.1093/jxb/ers239
Welkie, G. W. (1996). Iron-deficiency stress responses of a chlorosis-susceptible and a chlorosis-resistant cultivar of muskmelon as related to root riboflavin excretion. J. Plant Nutr. 19, 1157–1169. doi: 10.1080/01904169609365187
Wise, A. A., Liu, Z., and Binns, A. N. (2006). “Three methods for the introduction of foreign DNA into Agrobacterium,” in Agrobacterium Protocols, ed. K. Wang (New York, NY: Humana Press), 43–54. doi: 10.1385/1-59745-130-4:43
Yang, T. J. W., Lin, W.-D., and Schmidt, W. (2010). Transcriptional profiling of the Arabidopsis iron deficiency response reveals conserved transition metal homeostasis networks. Plant Physiol. 152, 2130–2141. doi: 10.1104/pp.109.152728
Yuan, Y., Wu, H., Wang, N., Li, J., Zhao, W., Du, J., et al. (2008). FIT interacts with AtbHLH38 and AtbHLH39 in regulating iron uptake gene expression for iron homeostasis in Arabidopsis. Cell Res. 18, 385–397. doi: 10.1038/cr.2008.26
Keywords: iron uptake and metabolism, bHLH transcription factor, mutant proteins, Cucumis melo, gene expression regulation
Citation: Ramamurthy RK and Waters BM (2017) Mapping and Characterization of the fefe Gene That Controls Iron Uptake in Melon (Cucumis melo L.). Front. Plant Sci. 8:1003. doi: 10.3389/fpls.2017.01003
Received: 08 March 2017; Accepted: 26 May 2017;
Published: 14 June 2017.
Edited by:
Sebastien Thomine, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Hannetz Roschzttardtz, Pontificia Universidad Católica de Chile, ChileGang Liang, Xishuangbanna Tropical Botanical Garden (CAS), China
Hong-Qing Ling, Institute of Genetics and Developmental Biology (CAS), China
Copyright © 2017 Ramamurthy and Waters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian M. Waters, YndhdGVyczJAdW5sLmVkdQ==