 Wan Shi
Wan Shi Mingyang Quan
Mingyang Quan Qingzhang Du
Qingzhang Du Deqiang Zhang
Deqiang Zhang- 1Beijing Advanced Innovation Center for Tree Breeding by Molecular Design, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China
- 2National Engineering Laboratory for Tree Breeding, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China
- 3Key Laboratory of Genetics and Breeding in Forest Trees and Ornamental Plants, Ministry of Education, College of Biological Sciences and Technology, Beijing Forestry University, Beijing, China
Long non-coding RNAs (lncRNAs) are important regulatory factors for plant growth and development, but little is known about the allelic interactions of lncRNAs with mRNA in perennial plants. Here, we analyzed the interaction of the NERD (Needed for RDR2-independent DNA methylation) Populus tomentosa gene PtoNERD with its putative regulator, the lncRNA NERDL (NERD-related lncRNA), which partially overlaps with the promoter region of this gene. Expression analysis in eight tissues showed a positive correlation between NERDL and PtoNERD (r = 0.62), suggesting that the interaction of NERDL with its putative target might be involved in wood formation. We conducted association mapping in a natural population of P. tomentosa (435 unrelated individuals) to evaluate genetic variation and the interaction of the lncRNA NERDL with PtoNERD. Using additive and dominant models, we identified 30 SNPs (P < 0.01) associated with five tree growth and wood property traits. Each SNP explained 3.90–8.57% of phenotypic variance, suggesting that NERDL and its putative target play a common role in wood formation. Epistasis analysis uncovered nine SNP-SNP association pairs between NERDL and PtoNERD, with an information gain of -7.55 to 2.16%, reflecting the strong interactions between NERDL and its putative target. This analysis provides a powerful method for deciphering the genetic interactions of lncRNAs with mRNA and dissecting the complex genetic network of quantitative traits in trees.
Introduction
DNA methylation is a conserved epigenetic silencing mechanism that functions in many crucial plant growth and developmental processes, such as genomic imprinting and abiotic stress responses (Bird, 2002; Song et al., 2016). RdDM (RNA-directed DNA methylation), the most important and well-characterized DNA methylation pathway, is guided by 24-nt siRNAs, induces de novo cytosine methylation, and regulates gene expression (Le et al., 2014). RdDM occurs only in plants performing transcriptional gene silencing (TGS) involving the Pol IV–RDR2–DCL3 complex. Another siRNA-dependent DNA methylation pathway, the NERD (Needed for RDR2-independent DNA methylation)-dependent pathway (Matzke et al., 2015), involves RDR1/RDR6 and AGO2 and is guided by 21-nt siRNAs (Matzke and Mosher, 2014) to mediate gene silencing at the post-transcriptional level in Arabidopsis thaliana. The NERD pathway initiates de novo methylation of newly integrated transposons that initially undergo PTGS (post-transcriptional gene silencing), which may build a bridge between PTGS and TGS (Matzke and Mosher, 2014).
The NERD pathway requires Pol IV and Pol V, as well as several PTGS components, including NERD, RDR6, SGS3 (SUPPRESSOR OF GENE SILENCING 3), SDE3 (SILENCING DEFECTIVE 3), and SDE5 (Matzke and Mosher, 2014). NERD, a key protein in the NERD pathway, plays important roles in plant growth and development. In Arabidopsis, NERD is responsible for the accumulation of siRNA at NERD-targeted loci (Pontier et al., 2012). In rice, OsNERD1 affects pollen sterility via its effect on related transposons, as well as cold-related transposons (Chen, 2015). Natural allelic variation plays an important role in plant development and physiology, especially plant growth and morphology, the timing of germination and flowering, and primary metabolism (Alonso-Blanco et al., 2009). However, the specific genetic variants resulting in variations in growth and development in woody plants are only starting to be uncovered. NERD was identified in Populus trichocarpa through database searches (Pontier et al., 2012), but the effect of allelic variations within NERD on tree growth and wood formation, which could be utilized for genetic improvement of important economic traits in forest trees, remains unknown.
LncRNAs (long non-coding RNAs), which are longer than 200 nucleotides and have no apparent coding ability, are emerging as important participants in numerous biological processes (Quan et al., 2015). In animals, lncRNAs are involved at multiple levels in many biological regulatory processes, interacting with their target genes via various mechanisms (Ponting et al., 2009). In humans, lncRNAs in the promoter regions of protein-coding genes are thought to affect the expression of these genes, thus playing an important role in controlling cell growth (Hung et al., 2011). Several plant lncRNAs have been identified and their roles characterized, such as lncRNA XLOC_057324 in panicle development and fertility in rice (Zhang et al., 2014), lncRNAs involved in flowering in Arabidopsis (Heo and Sung, 2011) and drought responses in Populus (Shuai et al., 2014). The effects of ncRNAs and their target genes on plant growth and development have been explored. For example, ASCO-lncRNA, which hijacks nuclear regulators of alternative splicing machinery by regulating the expression levels of its target gene, contributes to lateral root formation in Arabidopsis (Bardou et al., 2014). However, few studies have focused on the roles of lncRNAs in perennial trees.
Stem growth and wood formation are important processes in the lifecycles of perennial plants such as forest trees. However, the long cycle of forest tree growth, the presence of highly lignified tissues, and the lack of suitable mutants, have limited the use of traditional transgenic analysis techniques in trees to explore specific interactions between ncRNAs and their target genes. SNP-based association mapping in conjunction with molecular marker-assisted selection (Thumma et al., 2005) and association studies can be used to decipher the interactions of ncRNAs with mRNA and their roles in tree growth and wood formation. For instance, a SNP-based association analysis shed light on the involvement of the interactions between the miRNA Pto-miR530a and its target gene Pto-KNAT1 in eight traits, which laid the foundation for exploring the multi-gene networks affecting tree growth and wood properties (Yang et al., 2015). However, the effects of the genetic interactions between lncRNAs and their targets on tree growth and wood formation remain largely unknown.
Here, we explored the genetic variation and interactions of the lncRNA NERDL and its putative target gene, PtoNERD, in a natural population of 435 unrelated individuals of the poplar Populus tomentosa, a major commercial tree species grown for pulp and timber production in northern China. Using SNP-based association mapping with additive and dominant models, we detected significant SNPs within NERD and PtoNERD associated with tree growth and wood properties, revealing the common role of NERDL and its potential target, PtoNERD, in wood formation. Expression profiling revealed a significant correlation between the expression of NERDL and PtoNERD, suggesting that they share a common pathway involved in wood formation. Epistasis analysis uncovered strong allelic genetic interactions between NERDL and PtoNERD. Our approach is useful for identifying genetic variation and deciphering the interactions between lncRNAs and mRNAs associated with multiple economic traits in forest trees.
Materials and Methods
Population Materials and Phenotypic Data
Plant Materials
The association population was composed of 435 unrelated individuals of P. tomentosa randomly selected from a collection of 1047 individuals covering the original distribution of P. tomentosa, as well as three climatic zones (southern, northwestern, and northeastern China) (Huang, 1992). A clonal arboretum of this collection was built in 1982 in Guan Xian County, Shandong Province, China (36°23′N, 115°47′E) using a randomized complete block design with three replications. Forty-four individuals were randomly selected from this association population to identify SNPs within NERDL and PtoNERD via direct sequencing.
Phenotypic Data
Phenotypic data for 10 growth and wood property traits were obtained from the 435 selected individuals in 2011 as previously described (Du et al., 2013b). Trees of this age are in a stable growth and developmental phase and can therefore be used to reflect the natural growth and developmental state of trees. Seven wood property traits and three tree growth traits were investigated: LC (lignin content, %), HC (holocellulose content, %), HEMC (hemicellulose content, %), AC (α-cellulose content, %), FL (fiber length, mm), FW (fiber width, μm), Angle (microfibril angle, °), H (tree height, m), D (tree diameter at breast, cm), and V (volume of wood, m3). The correlations of these phenotypic data in the association population were calculated as previously described (Du et al., 2013b).
Identification of the lncRNA NERDL and Its Putative Target, PtoNERD
Isolation and Sequence Analysis of PtoNERD
Cambium, developing xylem, and mature xylem tissues were obtained from 1-year-old P. tomentosa clone LM50 when the tree growth and development were in a relatively active state, allowing us to obtain important information about tree growth and wood formation. Total RNA was extracted from the tissues with a Plant RNeasy Kit (Qiagen) following the manufacturer’s instructions. DNA contamination was removed with three additional on-column DNase digestions during RNA purification with an RNase-Free DNase Set (Qiagen). The three RNA samples were used to construct RNA-seq libraries, and transcript sequencing was performed on an Illumina HiSeq 2500 by Shanghai Biotechnology Corporation (Shanghai, China). High-quality reads were mapped to the Populus reference genome (Tuskan et al., 2006) using TopHat v2.0.9 (Trapnell et al., 2012). The transcripts were assembled using Cufflinks v2.1.1 (Trapnell et al., 2012), and the expression levels were calculated and normalized by FPKM (fragments per kilobase of transcript per million fragments). The sequencing data are available in the NCBI Sequence Read Archive (SRA accession number SRP073689). Expression analysis of seven genes (RDR1, RDR6, NERD, SGS3, SDE3, SDE5, and AGO2) involved in the NERD pathway showed that PtoNERD was highly expressed in vascular tissue, suggesting that it might function in wood formation (Supplementary Figure S1). Therefore, PtoNERD (Accession number: KY344444–KY344487) was selected as a candidate gene for further analysis. RNA sequencing was performed to identify the mRNA sequence of PtoNERD, which was compared with available Arabidopsis or P. trichocarpa NERD sequences, showing that PtoNERD mRNA shares high sequence similarity with AtNERD AT2G16485 (45%) and Potri.009G1238000 (95%).
To evaluate the phylogenetic relationship of PtoNERD with similar genes in other species, the amino acid sequences of NERD from A. thaliana, P. trichocarpa, P. euphratica, Picea abies, Pinus taeda, Zea mays, and Oryza sativa ssp. japonica were identified by searching public databases at NCBI1 using BLAST, and sequence alignment and phylogenetic tree construction were performed with MEGA 5.0 as previously described (Tamura et al., 2011).
Identification of an lncRNA Targeting PtoNERD
RNA sequencing was performed on lncRNAs from three tissues. All lncRNA sequences identified by transcriptome sequencing were aligned against the Populus reference genome (SRA accession number SRP073689) (Tuskan et al., 2006). To identify the lncRNA potentially targeting PtoNERD, genome annotation and a genome browser were used to screen potential cis lncRNAs that are physically close to PtoNERD (within 10 kb) as described (Jia et al., 2010).
Tissue-Specific Expression Analysis of lncRNA and Its Putative Target, PtoNERD
Total RNA was extracted from fresh tissue samples from 1-year-old P. tomentosa clone LM50, including juvenile leaf, mature leaf, apical shoot meristem, phloem, cambium, developing xylem, mature xylem, and root tissue as described (Du et al., 2013a). The RNA was reverse-transcribed into cDNA using the SuperScript First-Strand Synthesis system with the supplied polythymine primers (Invitrogen, Life Technologies, New York, NY, United States) (Zhang et al., 2010a). All cDNA samples were used to investigate the tissue-specific expression of PtoNERD and NERDL through reverse-transcription quantitative PCR (RT-qPCR). The qPCR was performed on the 7500 Fast Real-Time PCR System with SYBR Premix EX Taq (TaKaRa, Dalian, China) with gene-specific primers (Supplementary Table S1) designed with Primer Express 3.0 (Applied Biosystems). The PCR conditions were as follows: 94°C for 5 min; 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s; and a final cycle at 70–95°C for melting curve analysis to confirm the specificity of the amplification. The qPCR was conducted as described by Du et al. (2013a). All reactions were conducted with three technical and three biological replicates. The results for the various tissues were standardized to the internal control (Actin, Accession number: EF145577). Data analysis was performed with Opticon Monitor Software 3.1 (MJ Research, Bio-Rad, Hercules, CA, United States).
SNP Discovery and Genotyping
Total genomic DNA was extracted from fresh young leaves with a Plant DNeasy Mini kit (Qiagen China, Shanghai) according to the manufacturer’s instructions. The primer sets used for amplification and sequencing were specifically designed based on the sequence of the lncRNA NERDL and the cDNA sequence of PtoNERD. The 16,119 bp genomic DNA sequence of PtoNERD, including the flanking region (2000 bp), was obtained by direct sequencing of the P. tomentosa (LM50 clone) using conserved primers and the BigDye Terminator Cycle Sequencing kit version 3.1 (Applied Biosystems, Beijing, China) on a Li-Cor 4300 genetic analyzer (Li-Cor Biosciences, Lincoln, NE, United States). The Prosite database2 was used to identify protein domains, families, and functional sites, and SMART3 was used to analyze the protein sequence. To identify SNPs, NERDL, and PtoNERD were sequenced and analyzed in 44 unrelated individuals from the association population with MEGA 4.0 and DnaSP v4.90.1 (Rozas et al., 2003). Insertions/deletions (InDels) were disregarded in the analysis. All 88 sequences have been deposited in the GenBank database (GenBank Accession No. KY344444-KY344487 for NERDL and KY344488-KY344531 for PtoNERD). Sequence alignment was performed with BioEdit, and manual editing was performed to remove primer sequences and to confirm sequence quality. All common SNPs (minor allele frequencies > 0.05) detected in NERDL and PtoNERD were genotyped in the association population using the Beckman Coulter sequencing system (Franklin Lakes, NJ, United States).
Nucleotide Diversity and Linkage Disequilibrium
Nucleotide Diversity
The summary statistics for SNP polymorphisms were obtained using DnaSP version 4.90.1 (Rozas et al., 2003). The average number of pairwise differences per site between the sequences, π (Nei, 1987), and the average number of segregating sites, 𝜃w (Watterson, 1975) were used to assess the nucleotide diversity. Diversity statistics were also calculated for non-coding, synonymous, and non-synonymous sites. Tajima’s D and Fu and Li’s D (Tajima, 1989; Fu and Li, 1993) statistics were obtained and the complete data set was tested using 10,000 simulations to determine whether a gene or genomic region is evolving randomly (neutral evolution) or whether the region is under selection (non-neutral evolution). The statistical significance of Tajima’s D statistics was determined using DnaSP v4.90.1.
Linkage Disequilibrium (LD)
Linkage disequilibrium between pairs of SNP markers within NERDL and PtoNERD were assessed using TASSEL V.5.0 (Bradbury et al., 2007) as the squared correlation of allele frequencies (r2), which can be affected by both recombination and differences in allele frequencies between sites (Hill and Robertson, 1968). To assess the level of LD within the sequenced regions of NERDL and PtoNERD, the decay of LD with featured physical distance (bp) between common SNP sites within both candidate genes was estimated by non-linear regression analysis with 105 permutations of the data (Remington et al., 2001), and singletons were excluded.
Detection of SNP-Based Phenotype–Genotype Associations
Single SNP-Based Associations
The phenotypic trait–SNP genotype associations between 159 common SNP markers within NERDL and PtoNERD and 10 traits were identified using a MLM (mixed linear model) with 105 permutations with the software package TASSEL v5.04 (Yu et al., 2006). The values of estimated membership probability (Q) and pairwise kinship (K) were used to evaluate the effects of population structure and relatedness among individuals for marker–trait associations. In this Q + K model, Q matrix was obtained based on the significant subpopulations (k = 3) (Du et al., 2012) using STRUCTURE version 2.3.45 (Pritchard et al., 2000), and K, the relative kinship matrix, was evaluated with SPAGeDi version 1.3 (Hardy and Vekemans, 2002) using the method proposed by Ritland (1996). The positive FDR method was used to correct for multiple testing using QVALUE software (Storey and Tibshirani, 2003). Additive effects and dominance effects were calculated with Tassel v5.0 to analyze the patterns of gene action.
Haplotype Analysis
Haplotype frequencies were calculated with SNP genotype data, and haplotype association tests were performed on a three-marker sliding window using haplotype trend regression software (Higuchi, 2012). The significance of each haplotype association was based on 104 permutation tests, and singleton alleles and haplotypes with frequencies less than 0.05 were excluded from further analysis.
Multi-SNP-Based Epistasis Analysis
To detect the epistatic effects among multiple factors, a multifactor dimensionality reduction (MDR) algorithm (Hahn et al., 2003) was developed. With this algorithm, it is possible to process high-dimensionality genetic data into a single dimension, allowing interactions to be investigated using a relatively small set of data. MDR 3.0.2, the Relief algorithm, was used to filter all unlinked SNPs (r2 < 0.1 or different genes), improving the reliability of probability approximation. The five most-significant SNPs were calculated for each trait. The genetic effects of significant SNP–SNP pairs were assessed by IG (information gain) analysis, which was calculated via an entropy-based measurement (Moore et al., 2006).
Transcript Analysis of SNP Genotypes
The overlapping significant SNPs were used to investigate the roles of different genotypes on the expression of NERDL and PtoNERD. For each SNP genotypic class, 10 individuals were selected and used to measure the expression of the lncRNA NERDL and PtoNERD via RT-qPCR. The differential expression across different genotypes was calculated with ANOVA. The effects of different genotypes of the shared locus on phenotypic variation were also investigated.
Results
Sequence Analysis of NERDL and Its Putative Target PtoNERD from P. tomentosa
To identify SNPs within PtoNERD, we cloned a cDNA from PtoNERD. This cDNA is 9527 bp long, including a 5163 bp open reading frame and 364 bp of 3′UTR. Based on the alignment of the full-length cDNA sequence to the genomic sequence, PtoNERD (16119 bp) has 10 introns and 11 exons flanked by 2000 bp of promoter region, 364 bp of 3′UTR, with a 2000 bp flanking region (Figure 1A); the length of each region is listed in Table 1. PtoNERD encodes a putative polypeptide of 1720 amino acids, with an estimated molecular mass of 188.4 kD and a pI of 4.82. Characterization of the amino acid sequence of PtoNERD revealed that it contains the essential domain PHD, SWIB, Plus-3, and GYF (Figure 1A). Phylogenetic analysis indicated that PtoNERD is an ortholog of AtNERD and PtrNERD (Figure 1B). Phylogenetic analysis using the neighbor-joining method showed that PtoNERD in P. tomentosa shares a close relationship with the homologous sequences in P. trichocarpa and P. euphratica, indicating that PtoNERD is highly conserved in Populus, whereas the conservation of PtoNERD in distantly related species is weak.
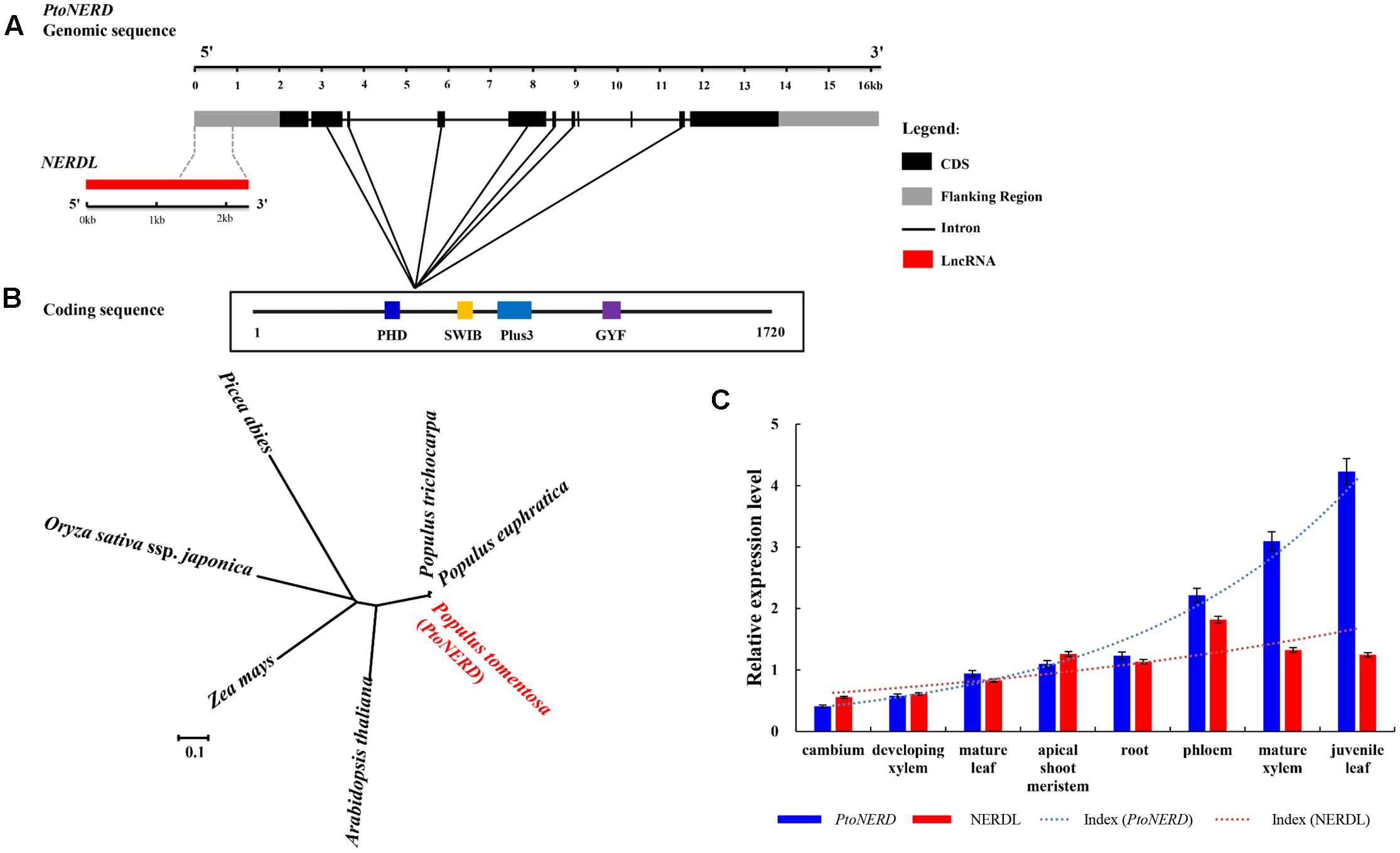
FIGURE 1. Gene structures of NERDL and PtoNERD, expression levels of NERDL and PtoNERD, and phylogenetic tree of PtoNERD. (A) Gene structures and SNP diversity of NERDL and PtoNERD. NERDL shares a 960 bp long overlapping region with PtoNERD. (B) Phylogenetic tree of homologs of Arabidopsis thaliana, P. trichocarpa, Populus euphratica, Picea abies, Pinus taeda, Zea mays, and Oryza sativa ssp. Japonica. (C) The relative expression levels of NERDL and PtoNERD in eight tissues measured by RT-qPCR with Actin as the internal control.
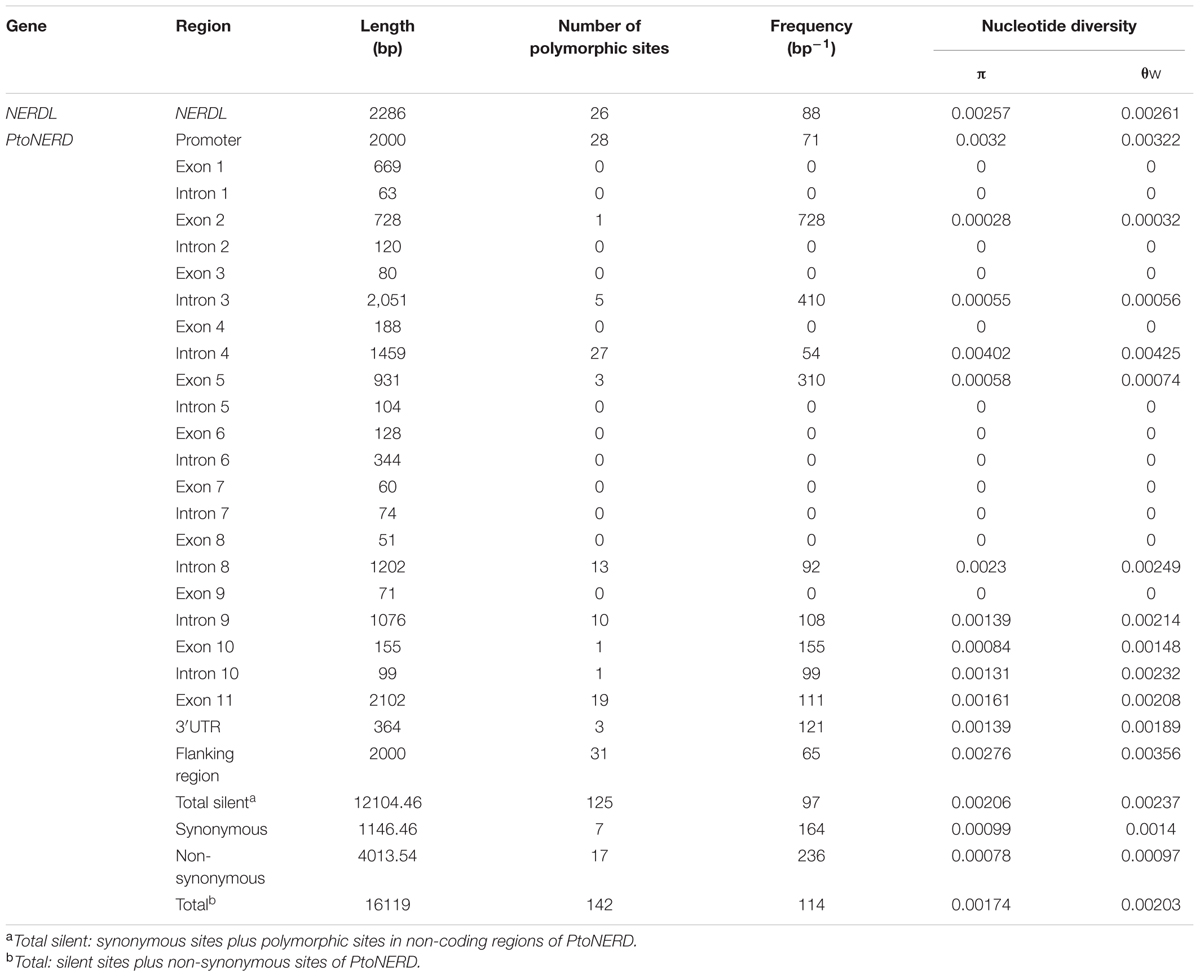
TABLE 1. Single nucleotide polymorphisms of NERDL and PtoNERD.
To further explore the roles of PtoNERD in plant development, using genome annotation and a genome browser, we identified NERDL, an lncRNA located in the antisense strand and overlapping with the promoter region of PtoNERD, which acts as a cis-regulator of PtoNERD. Sequencing analysis revealed the 2286-nt sequence of NERDL, which shares an overlapping region of 961 bp with PtoNERD (Figure 1A). Therefore, PtoNERD was regarded as the candidate cis target gene of the overlapping lncRNA NERDL (lncRNA related to PtoNERD). Several functional motifs are found in the overlapping region, such as light responsive element 3-AF1 binding site, Box I, and GT1-motif, as well as as-2-box involved in shoot-specific expression and light responsiveness.
NERDL and PtoNERD Expression Are Highly Correlated
We explored the tissue-specific expression patterns of the lncRNA NERDL and PtoNERD in eight different plant tissues via RT-qPCR (Figure 1C). NERDL was highly abundant in phloem (1.82 ± 0.09) (arbitrary units normalized to control), followed by mature xylem (1.33 ± 0.07), with the lowest abundance detected in cambium (0.56 ± 0.03), suggesting that NERDL plays a regulatory role in wood formation. PtoNERD mRNA was enriched in juvenile leaves (4.23 ± 0.20), followed by mature xylem (3.10 ± 0.14), with the lowest abundance in cambium (0.41 ± 0.19). Overall, PtoNERD was more highly expressed than NERDL. The Pearson’s product moment correlation was 0.62 (P < 0.05), indicating a strong correlation in transcript levels between NERDL and PtoNERD. The reads numbers of NERDL and PtoNERD obtained by RNA-seq showed a similar trend in cambium, developing xylem, and mature xylem tissue (Supplementary Figure S3).
Single Nucleotide Diversity and Linkage Disequilibrium in NERDL and Its Putative Target PtoNERD
To characterize the intraspecific polymorphisms for use in association mapping, we obtained the full-length genomic sequences of NERDL and PtoNERD from 44 unrelated individuals of P. tomentosa. In total, 26 SNPs were detected in NERDL at a density of 1/88, and 142 SNPs in PtoNERD were detected at a frequency of approximately one SNP every 114 bp. The nucleotide diversity (excluding InDels) was higher in NERDL (π = 0.00257 and 𝜃w = 0.00261) than in PtoNERD (π = 0.00174 and 𝜃w = 0.00203). As expected, the average nucleotide diversity in PtoNERD was higher in introns (π = 0.00096) than in exons (π = 0.00030), since under selection pressure, the coding region of a gene shows more conservation than the non-coding regions (Table 1). Within the coding region of PtoNERD, the average nucleotide diversity of synonymous polymorphisms (dS, π = 0.00099) was higher than that of non-synonymous polymorphisms (dN, π = 0.00078) at a ratio of dN/dS (0.788 < 1), indicating that non-synonymous sites in the exons are under purifying selection.
To perform LD analysis, 24 and 135 common SNPs (minor allele frequency > 0.05) from NERDL and PtoNERD, respectively, were successfully genotyped across 435 individuals in the association population. Four common SNPs were located in the overlapping region and were shared by NERDL and PtoNERD (SNP21–24 in NERDL and SNP1–4 in PtoNERD). A plot of the r2 combined with physical distance between SNPs within NERDL and the PtoNERD genes is shown in Supplementary Figure S2. In this population, the rate of LD declined rapidly, and the r2 values decreased to 0.1 within 1200 and 8000 bp in NERDL (Supplementary Figure S2A) and PtoNERD (Supplementary Figure S2B), respectively, indicating that LD does not extend across the entire gene region.
Genetic Effect of Allelic Variation in NERDL and Its Target Gene Revealed by Association Studies
Single SNP-Based Association
To thoroughly investigate the effect of NERDL and its putative target PtoNERD on growth and wood formation in P. tomentosa, we performed an association study among 159 common SNPs from NERDL and PtoNERD and 10 quantitative traits using MLM. In all, we detected 35 significant associations (P < 0.01, Q < 0.10) between 30 unique SNPs and 5 quantitative traits (Supplementary Table S2), with the phenotype variance (r2) explained by each association ranging from 3.90 to 8.57%. Four SNPs in NERDL are associated with three growth and wood property traits in P. tomentosa (D, AC, and HEC), with an average r2 of 5.78%. For PtoNERD, 26 significant SNPs from the promoter, exon, intron, and 3′UTR regions are associated with five traits (D, AC, HEC, FW, and LC), with r2 ranging from 3.90 to 8.57%. Of the seven SNPs in the coding region, three non-synonymous SNPs (SNP33, SNP49, and SNP75) were detected that are significantly associated with FW (r2, 4.13–7.76%). For example, SNP33, a missense mutation in exon 1, results in an encoded amino acid change from Thr to Ile, which is significantly associated with FW.
Among the identified associations, the most-significant SNP–trait associations affected wood property traits, including 14 for FW and 9 for wood chemical composition (AC, HEC, and LC); of these associations, 11.54% involve SNPs within NERDL. In addition, three traits (D, AC, and HEC) formed 1–14 associations with SNPs from both NERDL and PtoNERD simultaneously, with various contributions to phenotypic variation, suggesting that NERDL and its target gene PtoNERD play common roles in phenotypic variation (Supplementary Table S2). These finding, combined with the high correlation between NERDL and PtoNERD, suggest that NERDL and its target may play a common role in wood formation. For example, two SNPs in NERDL and seven in PtoNERD were associated with D, with r2 ranging from 4.21 to 6.10%. In addition, SNP9 from NERDL and four SNPs (SNP9, SNP11, SNP12, and SNP105) in PtoNERD were associated with more than one trait. Interestingly, an overlapping significant SNP locus, L23/N3, was detected in NERDL (L23, short for SNP23) and its putative target gene PtoNERD (N3, short for SNP3) that associated with D (r2 = 4.70%). In general, the single SNP-based associations of lncRNA NERDL and its potential target suggest that they play a role in tree growth and wood properties.
To further examine the allelic roles of NERD and PtoNERD in tree growth and wood formation, we investigated the additive and dominant effects of the 35 SNP–trait associations, with an average r2 of 5.48% (Figure 2A). The additive effect of these associations varied from 0.17 to 14.50% (Figure 2A and Table 2). In particular, SNP12 in PtoNERD, associated with AC, had the largest additive effect (14.50%); this additive effect was lower when associated with FW (2.80%) (Supplementary Table S2). The dominant values of these associations ranged from -10.34 to 12.32%, with 68.57% possessing positive dominant effects and the trait FW having the most associations. SNP12 in PtoNERD associated with AC and had the largest positive dominant effect, but it had a negative dominant effect when associated with FW. SNP 8 in NERDL had the largest negative dominant effect (Supplementary Table S2).
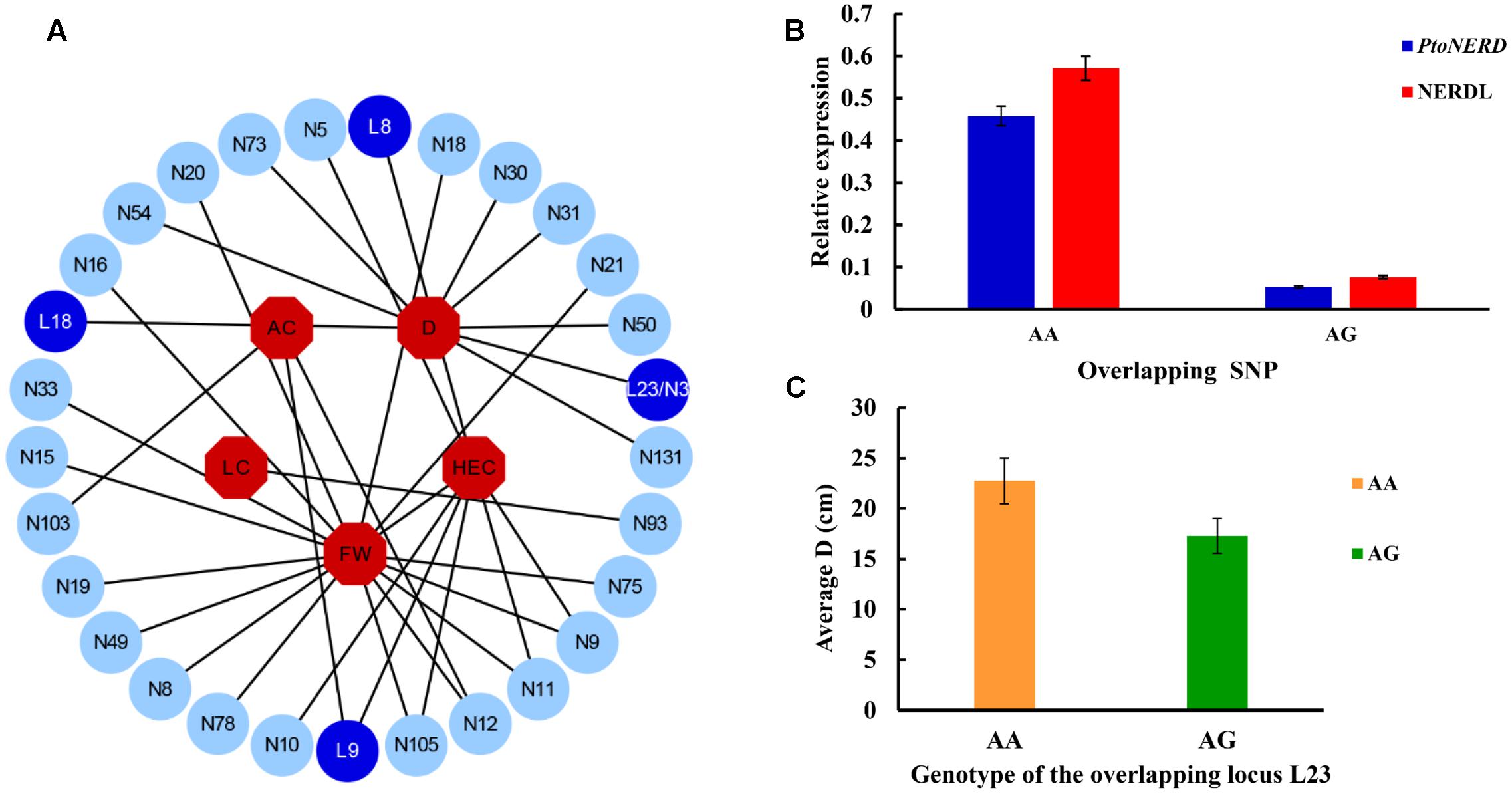
FIGURE 2. SNP-trait associations under additive and dominant effects in the association population of P. tomentosa, expression levels of genotypic classes for the overlapping SNP loci, and variations in average values of D depending on the genotype of L23/N3. (A) The associations with additive and dominant effect between 30 SNPs from NERDL and its target and five traits. Dark blue and light blue circles represent SNPs from NERDL (L) and PtoNERD (N), respectively, and red octagons indicate the five associated traits. (B) Expression levels of genotypic classes for the overlapping SNP loci. Error bars indicate standard deviations. (C) Average D depends on the genotype (AA or AG) of L23/N3.
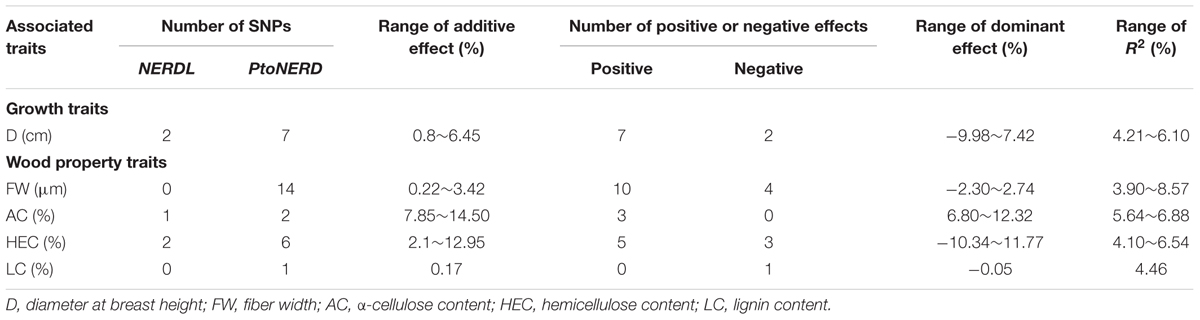
TABLE 2. Summary of additive and dominant effects of all significant SNPs associated with each trait in the association population of Populus tomentosa.
To further explore whether the allelic variation in the overlapping SNP L23/N3 influences the expression of lncRNA and its putative target, we used RT-qPCR to measure the transcript levels of NERDL and PtoNERD. The presence of different genotypes of L23/N3 led to significant differences in the transcript levels of NERDL and PtoNERD (Figure 2B). The highest PtoNERD mRNA levels were detected in the AA genotype (0.46 ± 0.02), followed by the AG genotype (0.05 ± 0.002). For NERDL, the expression levels were higher for the AA genotype (0.57 ± 0.02) than for the AG genotype (0.08 ± 0.004), indicating that the same locus can influence the expression of different transcripts. In addition, the average D depended on the genotype of L23/N3, with an average D of 22.74 cm in this association population for the AA genotype and 17.28 cm for AG (Figure 2C).
Haplotype-Based Associations
We detected 15 common haplotypes (frequency ≥ 0.05) within NERDL (3) and PtoNERD (14) associated with four traits (D, FW, AC, and HEC), explaining 1.06–13.16% of the phenotypic variance (Table 3). Two haplotypes (G-A-C and T-G-A from NERDL_SNP22-24/PtoNERD_SNP2-4) associated with D were shared by NERDL and PtoNERD since they share an overlapping region. Each of these four traits is associated with one to nine haplotypes. For example, FW is associated with nine haplotypes from PtoNERD (r2, 1.06–2.85%) (Table 3). In addition, one haplotype (T-A-G-A) from PtoNERD_SNP6-9 is associated with both HEC and FW, with different r2 (2.08 and 2.72%, respectively), implying the pleiotropy effects and conspicuousness of the gene. In total, the results of haplotype association analysis were validated by single-SNP-based association analysis and explained more phenotypic variation. In addition, the association of L23/N3 with D supports the notion that the two overlapping haplotypes (G-A-C and T-G-A) from NERDL and PtoNERD are associated with the same trait, D, indicating that different SNPs located in different regions might have a common effect on the same trait (Figures 3A,B).
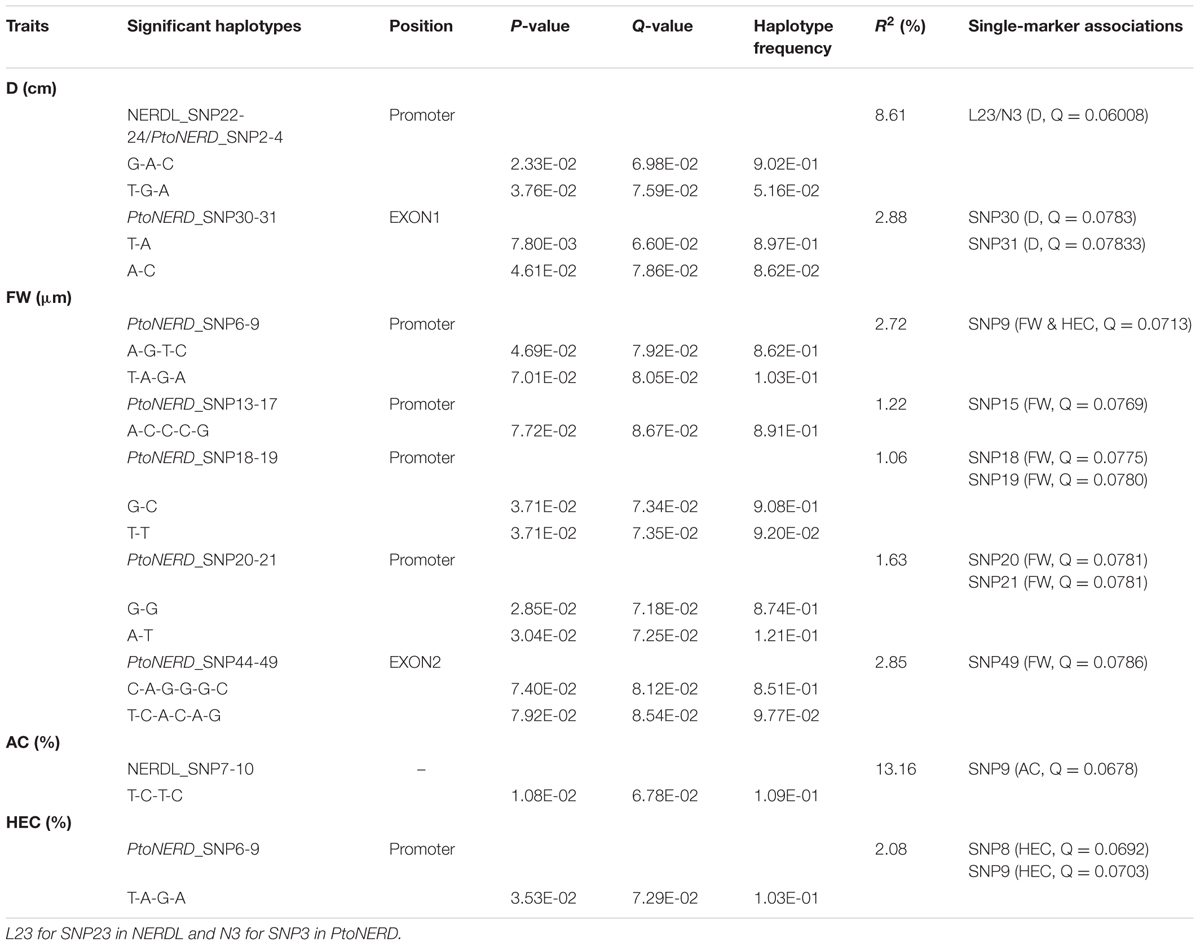
TABLE 3. Significant haplotypes from NERDL and its putative target PtoNERD associated with growth and wood properties traits in the association population of Populus tomentosa.
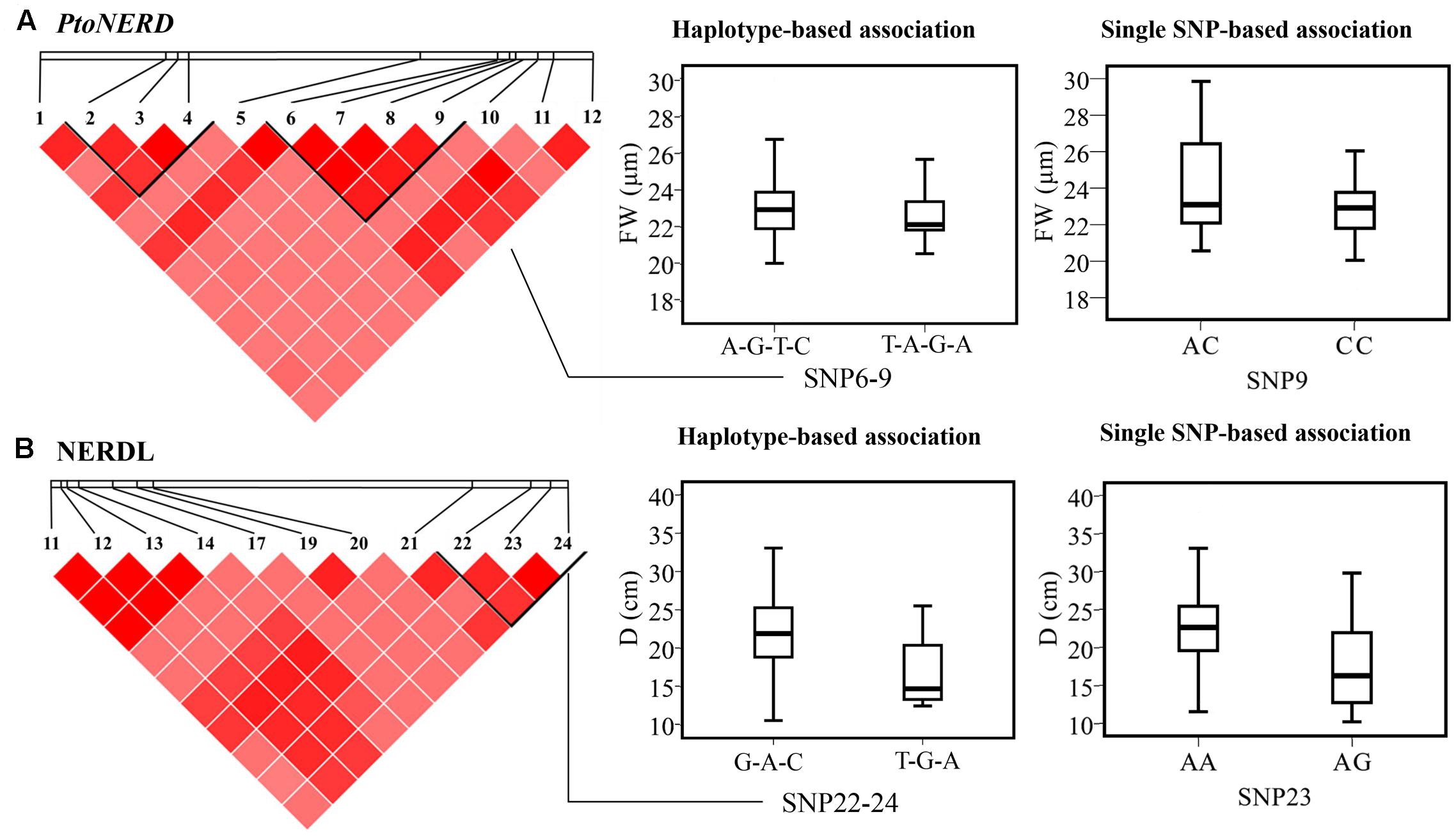
FIGURE 3. Haplotype-based epistasis associations. (A,B) The genotypic effect for significant haplotypes of NERDL_SNP22-24 and PtoNERD_SNP6-9 with that of the single loci of NERDL_SNP23 and PtoNERD_SNP9 based on single SNP-based associations.
Epistatic Modeling Demonstrates the Strong Genetic Interactions of lncRNA NERDL with Its Target
We uncovered strong interactions of different genes with phenotypic variation by investigating the epistatic effects between the lncRNA NERDL and its target. In total, we detected 90 significant SNP pairwise associations (P < 0.01, Q < 0.1) representing 32 SNPs from NERDL (2 SNPs) and PtoNERD (30 SNPs) (Supplementary Table S3), including nine SNP–SNP pairs representing lncRNA–mRNA interactions (Table 4). The effects of these nine lncRNA–mRNA interactions ranged from -7.55 to 2.16%, and nearly half showed negative IGs, indicating the redundant information for the same traits.
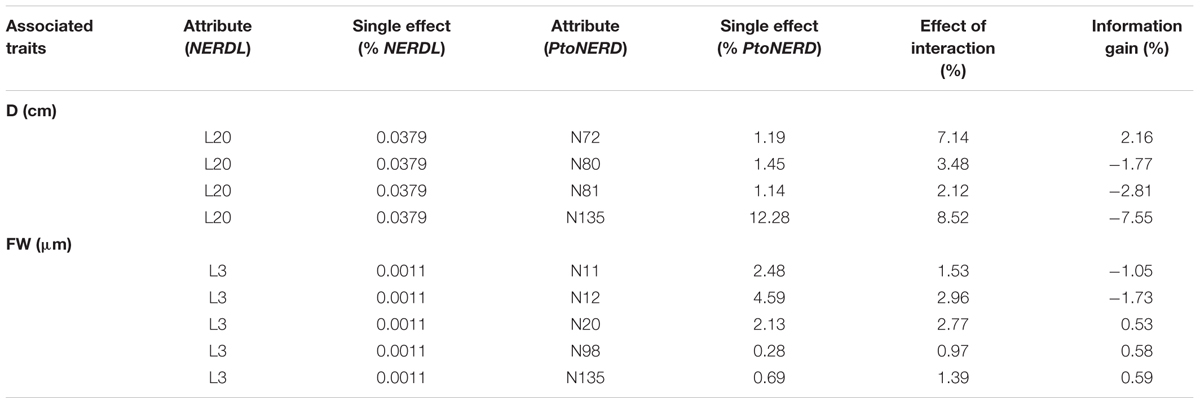
TABLE 4. The SNP pairs and their main effects detected from NERDL and PtoNERD under the epistasis model in the association population of Populus tomentosa.
Of these associated SNPs detected in the epistasis model, 23.08% of unique SNPs also showed significant associations under the additive and dominant models (Tables 2, 4). The epistasis network consisting of SNPs from NERDL and its putative target offered strong genetic evidence for the lncRNA–mRNA interactions and their potential function in wood formation and tree growth (Figures 4D,E). For example, L3 and N11 showed an epistatic interaction for FW, and the epistasis value for the combination of the AA genotype of L3 and the AC genotype of N11 was higher than those of the other combinations. In addition, the epistasis values of the AA-AC combination (2.829%) in SNP pair L3-N11 were significantly higher than the values for single genotypes (-0.201% for AA and 2.229% for AC) (Table 4, Supplementary Table S3, and Figures 4D,E). L3 formed five SNP pairs with SNPs from the target gene, showing different pairwise effects (0.97–2.96%) on FW, indicating the pleiotropy of L3 and the important roles of these interactions in tree growth and development.
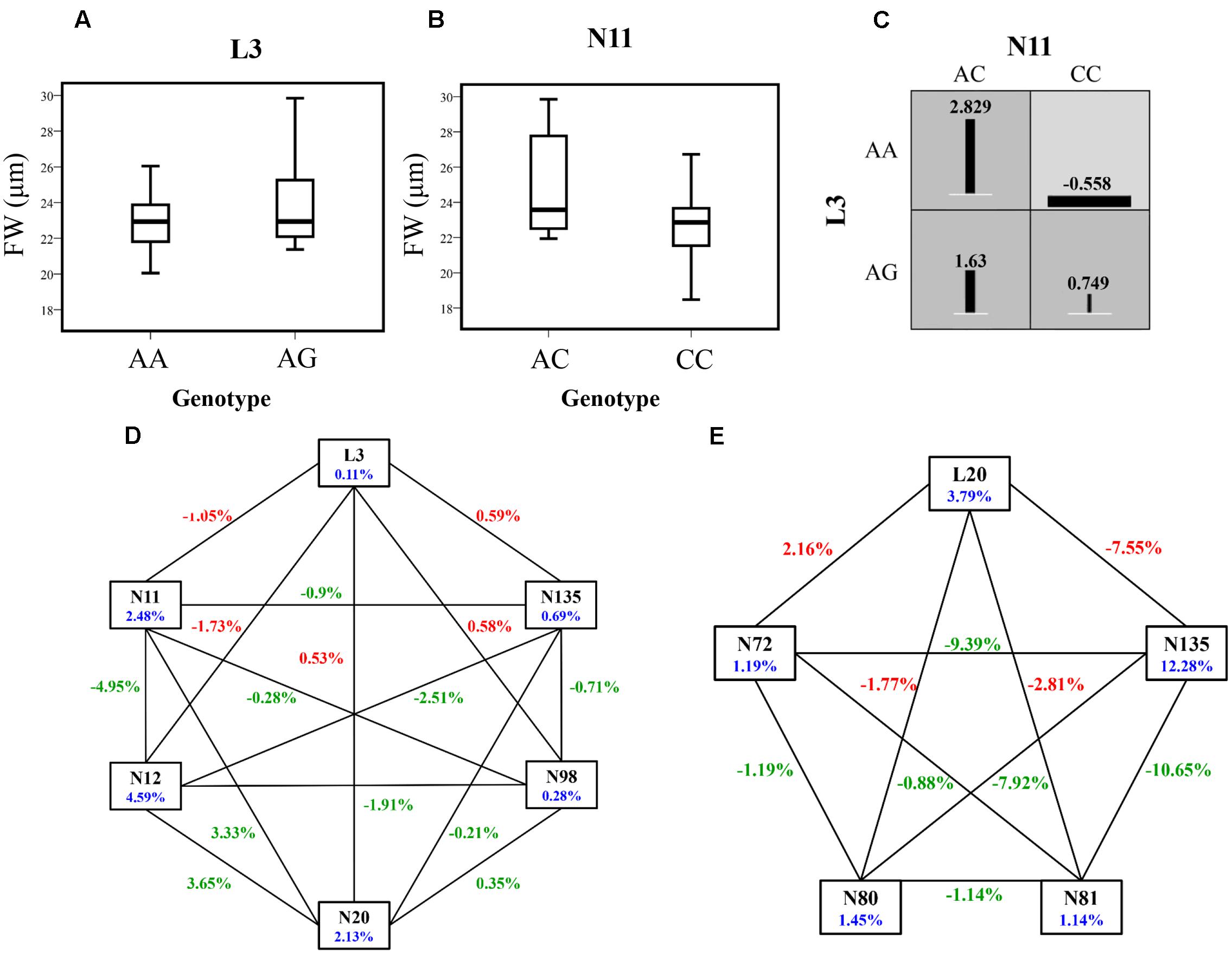
FIGURE 4. The epistatic network within the SNPs from NERDL and its target gene PtoNERD, and the phenotypic variations of pairwise genotypic combinations and single-locus genotypes. (A,B) Box plots showing the single-locus phenotypic variation of different genotypes of SNPs. (C) Square boxes show the pairwise phenotypic variation in different genotypic combinations of SNP pair L3-N11. (D,E) Interaction graph for FW and D among SNPs in NERDL and PtoNERD. Blue numbers represent the single-marker effect, red numbers indicate the pairwise epistatic effect between SNPs from NERDL and PtoNERD, respectively, and green numbers indicate the pairwise epistatic effect between SNPs from PtoNERD.
Discussion
Characterization of the lncRNA NERDL and Its Putative Target Gene PtoNERD
Unlike microRNAs, whose roles in post-transcriptional gene regulation are well-characterized (Bartel, 2009), the biological functions of lncRNAs are still largely unknown, although studies have increasingly focusing on the functional mechanisms of lncRNAs. LncRNAs function in a variety of process in plants, such as flowering, RdDM, and responses to biotic and abiotic stress (Quan et al., 2015). LncRNAs function through various mechanisms, including mutual interactions with RNAs and proteins (Guhaniyogi and Brewer, 2001). Base pairing-dependent lncRNA–mRNA interactions are a common mode of action for such interactions. In this study, we found that the lncRNA NERDL partly overlaps with the promoter region of PtoNERD and shares a 961 bp fragment containing four common SNPs and two haplotypes (Figure 1A and Tables 2, 3), suggesting a potential correlation between this lncRNA and PtoNERD. The overlap of lncRNAs with the promoter regions of protein-coding genes can affect the expression of these genes (Hung et al., 2011). Transcript expression analysis revealed a Pearson Correlation Coefficient of 0.62 (P < 0.05), providing evidence for a strong correlation between NERDL and its putative target gene, PtoNERD (Figure 1C). LncRNAs are usually shorter and are expressed at lower levels than protein-coding transcripts in P. tomentosa (Chen et al., 2015) and in mammals (Pauli et al., 2012). Here, RT-qPCR analysis and RNA-seq showed that the expression level of PtoNERD was higher than that of NERDL and that they both exhibit tissue-specific expression in P. tomentosa (Figure 1C and Supplementary Figure S3). NERDL was expressed at the highest level in phloem, suggesting that it may play a regulatory role in secondary wood formation. Furthermore, NERDL and PtoNERD were highly expressed in xylem (Figure 1C), suggesting that they play important roles in wood formation.
To understand the forces of evolutionary change and to evaluate the precision and power of association mapping, thorough knowledge of the levels of nucleotide diversity and the extent of LD in natural populations is required (Ingvarsson, 2008; Zhang et al., 2010b). In general, we found that the levels of average nucleotide diversity were lower in coding regions than in the non-coding regions (Table 1), implying that the coding regions are conserved relative to the other regions under natural pressure, with a dN/dS ratio (0.7879) less than 1 for PtoNERD (Neale and Kremer, 2011). Indeed, synonymous mutations occurring during evolution are more likely to be fixed than neutral ones owing to purifying selection (Zhang et al., 2010b). Also, the nucleotide diversity of NERDL (π = 0.00257) is slightly higher than that of PtoNERD (π = 0.00174), further confirming the conservative nature of the protein-coding gene. SNP diversity in genes has primarily been explored in exons, introns, and UTRs, while less attention has been paid to promoter regions and lncRNAs. It would be quite useful to analyze the levels of nucleotide diversity within the promoters of numerous genes compared with other gene regions. In the current study, we found that the promoter region possessed a significantly higher frequency of SNPs than the other regions in PtoNERD (π = 0.0032; Table 1). This finding suggests that this region may be an unstable regulatory region that plays important roles during the evolutionary process (Weickert et al., 2012).
The Association of SNPs in NERDL and PtoNERD with Tree Growth and Wood Property Traits
We identified 4 significant SNPs in NERDL and 26 in PtoNERD associated with D, FW, AC, HEC, and/or LC, and 15 haplotype-based associations that were strongly supported by single-marker associations (Table 3), leading to the suggestion that NERDL and its potential target might play a role in tree growth and wood formation. We identified 35 associations with both additive and dominant effects associated with five traits (D, FW, AC, HEC, and LC), representing 30 significant SNPs from NERDL and PtoNERD (Figure 2A and Table 2), suggesting that the lncRNA–mRNA pair might have a common effect on certain traits. Eight SNPs from NERDL and PtoNERD were associated with HEC, with additive and dominant effects (r2, 4.10–6.54%), an association considered to indicate generalized pleiotropy (Mackay et al., 2009). Potential pleiotropy within trait categories and within different phenotypic traits has been reported in P. trichocarpa (Porth et al., 2014). Together, these findings suggest that NERDL and its putative target PtoNERD might affect HEC through a shared pathway. A similar trend was found for D, which is associated with nine SNPs from NERDL and PtoNERD (r2, 4.21–6.10%). Considering the associations based on additive and dominant effects, our findings suggest that NERDL and PtoNERD co-regulate HEC and D and that the role of NERDL might be similar to that of its target, PtoNERD. Furthermore, under the additive and dominant models, several SNPs are associated with multiple traits but with different contributions, such as SNP9 in NERDL, which is associated with two traits, with r2 varying from 11.28 to 13.08%, revealing the pleiotropy of specific SNPs, as observed in a previous study (Yang et al., 2015).
Three non-synonymous markers (SNP33, SNP49, and SNP75), one synonymous marker (SNP105), and 10 non-coding markers in promoter and introns were associated with FW simultaneously (Supplementary Table S2), confirming the necessity of using an integrated approach to characterize the genetic basis of wood traits. In addition, the existence of seven markers in exons suggests that these differences in PtoNERD might affect phenotypic variation by causing amino acid changes or influencing gene expression levels, whereas the five SNPs in introns might affect the regulatory roles of introns or exon splicing, thereby leading to phenotypic variation. One common SNP (L23/N3) overlapping in NERDL and PtoNERD was significantly associated with D (r2 = 4.7%). Notably, genotypes of the overlapping SNP L23/N3 led to significant differences in the transcript levels of NERDL and PtoNERD (Figure 2B). The transcript levels of the AA and AG genotypes showed similar trends in both NERDL and its putative target PtoNERD, indicating that this overlapping locus can influence the expression of different transcripts. Overall, the average D was higher in the AA genotype (22.74 cm) than in the AG genotype (17.28 cm) (Figure 2C), indicating that variations in genotype may lead to changes in phenotype. Moreover, two shared common haplotypes from NERDL_SNP22-24/PtoNERD_SNP2-4 associated with D surround L23/N3. The locus might serve as a functional marker involved in the regulation of D, which should be explored in the future.
The Interactions of the lncRNA NERDL and Its Potential Targets Have Epistatic Effects on Wood Formation
Epistasis analysis is a crucial method for deciphering the effects of genetic variation and interactions (Mackay, 2014). Epistasis analysis can address the limitations of single-locus analysis and is a convenient method for further investigating the interactions of lncRNAs and mRNA and their effects on quantitative traits (Quan et al., 2016). Complex gene interactions have a stronger effect on disease susceptibility than single genes (Chou et al., 2011). Here, we characterized nine significant lncRNA–mRNA associations involved in two traits (D and FW) and 11 unique SNPs from NERDL and PtoNERD (Figures 4D,E), reflecting the possible genetic interaction of the lncRNA NERDL with PtoNERD. Furthermore, 55.56% of these SNP–SNP pairs had negative IG values, implying that redundant information is carried in the pairs for each trait and that their similar functional modes contribute to phenotypic variation. In associations with positive IG values, the contribution of pairwise interactions to phenotypic variation is more than the sum of the contributions of two SNP individuals. The genotypic combinations with the highest and lowest IG values for FW were found for SNP pair L3-N11 (Figures 4A–C), where the value for the genotypic combinations was significantly higher than that of the single-locus genotype. The identification of positive and negative IGs contributes to our understanding of the close interactions of NERDL with its target.
Of the SNPs showing lncRNA–mRNA interactions identified under the epistasis model, L3 in NERDL formed five SNP pairs with N11, N12, N20, N98, and N135 in PtoNERD under the epistasis model associated with FW with diverse IGs, revealing the complexity of the interactions between NERDL and PtoNERD (Figure 2B). L20 also showed similar associations with four SNPs associating with D from PtoNERD (N72, N80, N81, and N135) (Figure 2C). These observations further demonstrate the important functions of L3 and L20 in NERDL and the importance of the SNP pairs under the epistasis model for phenotypic variation, and they help confirm the notion that the interaction of NERDL with PtoNERD contributes to wood formation. In addition, 3 SNPs (N11, N12, and N20) out of 11 also showed significant effects in the additive and dominant models, while the remaining epistatic effects involved pairs of SNPs that showed deficiency for both additive and dominant effects. These findings are consistent with the suggestion by Xu and Jia (2007) that epistatic variance affects genetic variance. Moreover, whether or not two SNP loci have additive and dominant effects does not influence whether they have epistatic effects. Interestingly, we detected an intragenic SNP pair, N4-N131, in which N4 had no effect on HEMC, whereas it did have pairwise effects (4.03%) on HEMC in combination with N131 (Supplementary Table S3). Epistasis analysis indicated that the lncRNA NERDL might interact with PtoNERD via similar pathways and that their complex interactions play a role in regulating tree growth and determining wood properties.
Our RT-qPCR analysis uncovered a positive correlation in expression of NERDL and its putative target (r = 0.62; Figure 1C). This finding is consistent with the results of association studies of additive and dominance effects, strongly suggesting that NERDL and its potential target affect phenotypic variation in a similar manner. NERDL and PtoNERD were more highly expressed in secondary tissues (xylem) than in primary tissues (cambium), indicating that they may be involved in secondary wood formation. The high abundance of the two transcripts in phloem and xylem and the 35 associations with growth and wood formation traits suggest that NERDL and PtoNERD together perform a regulatory function in secondary xylem formation in trees. The overlapping SNP L23/N3 also had significant effects on the transcript levels of NERDL and PtoNERD (Figure 2B). Analysis of genotype data indicated that the 15 haplotypes explained 1.06–13.16% of phenotypic variation, which was confirmed by the finding that significant SNPs are located in blocks of haplotypes associated with the same traits.
Conclusion
In the present study, we identified PtoNERD, as an important player in the non-canonical RdDM pathway, as well as the lncRNA NERDL, which is located in the antisense strand and overlaps with the promoter region of PtoNERD. We used association (SNP-based and haplotype-based) studies and expression profiling to examine the common roles of NERDL and its putative target PtoNERD in tree growth and wood formation. Epistasis modeling helped reveal interactions between NERDL and PtoNERD for the traits D and FW, suggesting that the genetic interactions of NERDL and PtoNERD affect wood formation. Taken together, these results show that our method was successfully used to help decipher the allelic variation and genetic interactions of lncRNA and its target in trees through population genetics studies. The phenotype-related SNPs detected in this study could be applied to marker-assisted breeding in the near future.
Data Archiving Statement
Sequence data for NERDL and PtoNERD in this article have been deposited in the GenBank Data Library (accession numbers KX344444-KX344531), and the transcriptome sequencing data are available in the SRA database (accession number SRP073689).
Author Contributions
DZ conceived and designed experiment; WS and MQ performed the experiments and data analysis; WS wrote the manuscript; MQ and QD provided valuable advice on the manuscript; QD and DZ revised the manuscript; DZ obtained funding and is responsible for this article. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Project of Shandong Province Agriculture Improved Variety Project (No. 2012213), and the National Natural Science Foundation of China (No. 31670333).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01035/full#supplementary-material
FIGURE S1 | The expression levels of seven genes involved in the NERD pathway in three tissues revealed by RNA-seq. The expression level of each gene was generated from the FPKM value and used to visualize the expression patterns of the genes according to the RNA-seq data.
FIGURE S2 | Linkage disequilibrium within NERDL (A) and PtoNERD (B). Pairwise correlations between SNPs were plotted against the physical distance between them (in base pairs). The curves indicate the non-linear regressions of r2 onto the physical distance (in base pairs).
FIGURE S3 | The expression levels of NERDL and PtoNERD in three vascular tissues revealed by RNA-seq. The expression level of each gene was generated from the FPKM value and used to visualize the expression patterns of the genes according to the RNA-seq data.
Footnotes
- ^ http://www.ncbi.nlm.nih.gov
- ^ http://prosite.expasy.org/prosite.html
- ^ http://smart.embl-heidelberg.de//
- ^ http://www.maizegenetics.net/
- ^ http://pritch.bsd.uchicago.edu/structure.html
References
Alonso-Blanco, C., Aarts, M. G., Bentsink, L., Keurentjes, J. J., Reymond, M., Vreugdenhil, D., et al. (2009). What has natural variation taught us about plant development, physiology, and adaptation? Plant Cell 21, 1877–1896. doi: 10.1105/tpc.109.068114
Bardou, F., Ariel, F., Simpson, C. G., Romero-Barrios, N., Laporte, P., Balzergue, S., et al. (2014). Long noncoding RNA modulates alternative splicing regulators in Arabidopsis. Dev. Cell 30, 166–176. doi: 10.1016/j.devcel.2014.06.017
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Chen, J., Quan, M., and Zhang, D. (2015). Genome-wide identification of novel long non-coding RNAs in Populus tomentosa tension wood, opposite wood and normal wood xylem by RNA-seq. Planta 241, 125–143. doi: 10.1007/s00425-014-2168-1
Chen, X. (2015). The function of a OsNERD1 gene in NERD pathway of Oryza sativa. Hubei Agric. Sci. 54, 992–998.
Chou, H. H., Chiu, H. C., Delaney, N. F., Segrè, D., and Marx, C. J. (2011). Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science 332, 1190–1192. doi: 10.1126/science.1203799
Du, Q., Pan, W., Tian, J., Li, B., and Zhang, D. (2013a). The UDP-glucuronate decarboxylase gene family in Populus: structure, expression, and association genetics. PLoS ONE 8:e60880. doi: 10.1371/journal.pone.0060880
Du, Q., Pan, W., Xu, B., Li, B., and Zhang, D. (2013b). Polymorphic simple sequence repeat (SSR) loci within cellulose synthase (PtoCesA) genes are associated with growth and wood properties in Populus tomentosa. New Phytol. 197, 763–776. doi: 10.1111/nph.12072
Du, Q., Wang, B., Wei, Z., Zhang, D., and Li, B. (2012). Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 103, 853–862. doi: 10.1093/jhered/ess061
Fu, Y.-X., and Li, W.-H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709.
Guhaniyogi, J., and Brewer, G. (2001). Regulation of mRNA stability in mammalian cells. Gene 265, 11–23. doi: 10.1016/S0378-1119(01)00350-X
Hahn, L. W., Ritchie, M. D., and Moore, J. H. (2003). Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics 19, 376–382. doi: 10.1093/bioinformatics/btf869
Hardy, O. J., and Vekemans, X. (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2, 618–620. doi: 10.1046/j.1471-8286.2002.00305.x
Heo, J. B., and Sung, S. (2011). Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 331, 76–79. doi: 10.1126/science.1197349
Higuchi, T. (2012). Biochemistry and Molecular Biology of Wood. Berlin: Springer Science & Business Media.
Hill, W., and Robertson, A. (1968). Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38, 226–231. doi: 10.1007/BF01245622
Huang, Z. (1992). The study on the climatic regionalization of the distributional region of Populus tomentosa. J. Beijing For. Univ. 14, 26–32.
Hung, T., Wang, Y. L., Lin, M. F., Koegel, A. K., Kotake, Y., Grant, G. D., et al. (2011). Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 43, 621–629. doi: 10.1038/ng.848
Ingvarsson, P. K. (2008). Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 180, 329–340. doi: 10.1534/genetics.108.090431
Jia, H., Osak, M., Bogu, G. K., Stanton, L. W., Johnson, R., and Lipovich, L. (2010). Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16, 1478–1487. doi: 10.1261/rna.1951310
Le, T.-N., Schumann, U., Smith, N. A., Tiwari, S., Au, P., Zhu, Q.-H., et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome. Biol. 15, 458. doi: 10.1186/s13059-014-0458-3
Mackay, T. F. (2014). Epistasis and quantitative traits: using model organisms to study gene-gene interactions. Nat. Rev. Genet. 15, 22–33. doi: 10.1038/nrg3627
Mackay, T. F., Stone, E. A., and Ayroles, J. F. (2009). The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 10, 565–577. doi: 10.1038/nrg2612
Matzke, M. A., Kanno, T., and Matzke, A. J. (2015). RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant. Biol. 66, 243–267. doi: 10.1146/annurev-arplant-043014-114633
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Moore, J. H., Gilbert, J. C., Tsai, C. T., Chiang, F. T., Holden, T., Barney, N., et al. (2006). A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J. Theor. Biol. 241, 252–261. doi: 10.1016/j.jtbi.2005.11.036
Neale, D. B., and Kremer, A. (2011). Forest tree genomics: growing resources and applications. Nat. Rev. Genet. 12, 111–122. doi: 10.1038/nrg2931
Pauli, A., Valen, E., Lin, M. F., Garber, M., Vastenhouw, N. L., Levin, J. Z., et al. (2012). Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome. Res. 22, 577–591. doi: 10.1101/gr.133009.111
Pontier, D., Picart, C., Roudier, F., Garcia, D., Lahmy, S., Azevedo, J., et al. (2012). NERD, a plant-specific GW protein, defines an additional RNAi-dependent chromatin-based pathway in Arabidopsis. Mol. Cell. 48, 121–132. doi: 10.1016/j.molcel.2012.07.027
Ponting, C. P., Oliver, P. L., and Reik, W. (2009). Evolution and functions of long noncoding RNAs. Cell 136, 629–641. doi: 10.1016/j.cell.2009.02.006
Porth, I., Klapste, J., McKown, A. D., La Mantia, J., Hamelin, R. C., Skyba, O., et al. (2014). Extensive functional pleiotropy of REVOLUTA substantiated through forward genetics. Plant Physiol. 164, 548–554. doi: 10.1104/pp.113.228783
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Quan, M., Chen, J., and Zhang, D. (2015). Exploring the secrets of long noncoding RNAs. Int. J. Mol. Sci. 16, 5467–5496. doi: 10.3390/ijms16035467
Quan, M., Wang, Q., Phangthavong, S., Yang, X., Song, Y., Du, Q., et al. (2016). Association studies in Populus tomentosa reveal the genetic interactions of Pto-MIR156c and its targets in wood formation. Front. Plant Sci. 7:1159. doi: 10.3389/fpls.2016.01159
Remington, D. L., Thornsberry, J. M., Matsuoka, Y., Wilson, L. M., Whitt, S. R., Doebley, J., et al. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 98, 11479–11484. doi: 10.1073/pnas.201394398
Ritland, K. (1996). Estimators for pairwise relatedness and individual inbreeding coefficients. Genet. Res. 67, 175–185. doi: 10.1017/S0016672300033620
Rozas, J., Sánchez-DelBarrio, J. C., Messeguer, X., and Rozas, R. (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19, 2496–2497. doi: 10.1093/bioinformatics/btg359
Shuai, P., Liang, D., Tang, S., Zhang, Z., Ye, C. Y., Su, Y., et al. (2014). Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 65, 4975–4983. doi: 10.1093/jxb/eru256
Song, Y., Ci, D., Tian, M., and Zhang, D. (2016). Stable methylation of a non-coding RNA gene regulates gene expression in response to abiotic stress in Populus simonii. J. Exp. Bot. 67, 1477–1492. doi: 10.1093/jxb/erv543
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445. doi: 10.1073/pnas.1530509100
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thumma, B. R., Nolan, M. F., Evans, R., and Moran, G. F. (2005). Polymorphisms in cinnamoyl CoA reductase (CCR) are associated with variation in microfibril angle in Eucalyptus spp. Genetics 171, 1257–1265. doi: 10.1534/genetics.105.042028
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Watterson, G. (1975). On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276. doi: 10.1016/0040-5809(75)90020-9
Weickert, C., Tiwari, Y., Schofield, P., Mowry, B., and Fullerton, J. (2012). Schizophrenia-associated HapICE haplotype is associated with increased NRG1 type III expression and high nucleotide diversity. Transl. Psychiatry. 2, e104. doi: 10.1038/tp.2012.25
Xu, S., and Jia, Z. (2007). Genomewide analysis of epistatic effects for quantitative traits in barley. Genetics 175, 1955–1963. doi: 10.1534/genetics.106.066571
Yang, X., Du, Q., Chen, J., Wang, B., and Zhang, D. (2015). Association mapping in Populus reveals the interaction between Pto-miR530a and its target Pto-KNAT1. Planta 242, 77–95. doi: 10.1007/s00425-015-2287-3
Yu, J., Pressoir, G., Briggs, W. H., Bi, I. V., Yamasaki, M., Doebley, J. F., et al. (2006). A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38, 203–208. doi: 10.1038/ng1702
Zhang, D., Du, Q., Xu, B., Zhang, Z., and Li, B. (2010a). The actin multigene family in Populus: organization, expression and phylogenetic analysis. Mol. Genet. Genomics 284, 105–119. doi: 10.1007/s00438-010-0552-5
Zhang, D., Yang, X., Zhang, Z., and Li, B. (2010b). Expression and nucleotide diversity of the poplar COBL gene. Tree Genet. Genomes 6, 331–344. doi: 10.1007/s11295-009-0252-7
Keywords: association genetics, Nerdl, lncRna–mRna interactions, epistasis, wood formation
Citation: Shi W, Quan M, Du Q and Zhang D (2017) The Interactions between the Long Non-coding RNA NERDL and Its Target Gene Affect Wood Formation in Populus tomentosa. Front. Plant Sci. 8:1035. doi: 10.3389/fpls.2017.01035
Received: 12 February 2017; Accepted: 30 May 2017;
Published: 15 June 2017.
Edited by:
Junhua Peng, Center for Life Science and Technology of China National Seed Group Co. Ltd., ChinaReviewed by:
Xiaoli Jin, Zhejiang University, ChinaQing Liu, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright © 2017 Shi, Quan, Du and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deqiang Zhang, ZGVxaWFuZ1poYW5nQGJqZnUuZWR1LmNu