 Ulrike Schumann
Ulrike Schumann Joanne Lee
Joanne Lee Kemal Kazan
Kemal Kazan Michael Ayliffe
Michael Ayliffe Ming-Bo Wang
Ming-Bo Wang- 1CSIRO Agriculture and Food, Canberra, ACT, Australia
- 2CSIRO Agriculture and Food, Queensland Bioscience Precinct, St. Lucia, QLD, Australia
Recent research has indicated that a subset of defense-related genes is downregulated in the Arabidopsis DNA demethylase triple mutant rdd (ros1 dml2 dml3) resulting in increased susceptibility to the fungal pathogen Fusarium oxysporum. In rdd plants these downregulated genes contain hypermethylated transposable element sequences (TE) in their promoters, suggesting that this methylation represses gene expression in the mutant and that these sequences are actively demethylated in wild-type plants to maintain gene expression. In this study, the tissue-specific and pathogen-inducible expression patterns of rdd-downregulated genes were investigated and the individual role of ROS1, DML2, and DML3 demethylases in these spatiotemporal regulation patterns was determined. Large differences in defense gene expression were observed between pathogen-infected and uninfected tissues and between root and shoot tissues in both WT and rdd plants, however, only subtle changes in promoter TE methylation patterns occurred. Therefore, while TE hypermethylation caused decreased gene expression in rdd plants it did not dramatically effect spatiotemporal gene regulation, suggesting that this latter regulation is largely methylation independent. Analysis of ros1-3, dml2-1, and dml3-1 single gene mutant lines showed that promoter TE hypermethylation and defense-related gene repression was predominantly, but not exclusively, due to loss of ROS1 activity. These data demonstrate that DNA demethylation of TE sequences, largely by ROS1, promotes defense-related gene expression but does not control spatiotemporal expression in Arabidopsis.
Summary: Ros1-mediated DNA demethylation of promoter transposable elements is essential for activation of defense-related gene expression in response to fungal infection in Arabidopsis thaliana.
Introduction
Epigenetic modifications, such as DNA cytosine methylation, have recently been implicated in plant defense against bacterial and fungal pathogens (Zhu et al., 2016). In plants, DNA methylation occurs in all cytosine contexts (CG, CHG, and CHH; H stands for A, C, or T) and involves both de novo DNA methylation and maintenance DNA methylation. De novo DNA methylation is mediated by RNA-directed DNA methylation (RdDM), a plant-specific RNA silencing pathway directed by 24-nt small interfering RNAs (siRNA) (Matzke and Mosher, 2014). Maintenance methylation of CG and CHG contexts occurs during DNA replication and is catalyzed by Methyltransferase 1 (MET1) and Chromomethylase 3 (CMT3), respectively. Maintenance of CHH methylation generally requires the continuous presence of 24-nt siRNAs and RdDM, although CHH methylation in long transposable element (TE) sequences in highly repetitive chromosomal regions can be maintained by CMT2 (Zemach et al., 2013). In plants, DNA methylation primarily targets TEs and repetitive sequences in the genome.
The DNA methylation level of specific subsets of genomic loci in plants is regulated by the DNA glycosylase family of DNA demethylases (Penterman et al., 2007; Lister et al., 2008; Stroud et al., 2013). DNA demethylases remove 5-methylcytosine and replace it with unmethylated cytosine through a base-excision-repair mechanism (Zhu, 2009). The Arabidopsis genome encodes four DNA demethylase genes; Demeter (dme), Repressor of silencing 1 (ros1)/Demeter-like 1 (dml1), dml2, and dml3. DME is required for the expression of specific imprinted maternal alleles during seed development (Bauer and Fischer, 2011), while ROS1 is required to maintain the activity of some transgenes and transposons (Zhu, 2009). ROS1 has also been implicated in plant developmental regulation and biotic and abiotic stress responses (Yu et al., 2013; Yamamuro et al., 2014; Bharti et al., 2015). The biological function of DML2 and DML3 remains unknown.
Recent studies using the Arabidopsis–Pseudomonas syringae pathosystem have shown that DNA demethylation plays an important role in pathogen defense. For example the DNA demethylase-deficient mutant ros1 shows increased susceptibility to P. syringae infection while de novo and maintenance DNA methylation mutants such as nrpe1, ddc (drm1 drm2 cmt3) and met1 display enhanced resistance to P. syringae (Lopez et al., 2011; Dowen et al., 2012; Yu et al., 2013). Using an Arabidopsis–Fusarium oxysporum pathosystem, we recently demonstrated that the triple DNA demethylase mutant rdd (ros1 dml2 dml3) is highly susceptible to this fungal pathogen when compared with wild-type (WT) plants (Le et al., 2014). Collectively these data demonstrate that DNA demethylation plays a role in both bacterial and fungal disease resistance.
How DNA demethylation facilitates plant defense remains unclear. In our previous study using whole Arabidopsis plant tissues, we found 279 genes to be downregulated in the rdd mutant compared with WT plants. A significant proportion of these genes have a role in stress responses and are induced upon Fusarium infection (Le et al., 2014). These data suggest that DNA demethylases play a role in plant defense by positively regulating the expression of stress response genes. Most of the rdd-downregulated stress response genes contain short TE sequences in their predicted promoter regions which had altered localized methylation changes at some CHH and CG sites in the rdd mutant (Le et al., 2014). Methylation of these sites therefore appears to play a regulatory role in the expression of these stress response genes.
Here, we extend our previous study by examining the role of DNA demethylases in regulating tissue-specific and F. oxysporum-inducible expression patterns of the rdd-downregulated stress response genes. F. oxysporum is a vascular pathogen that enters the plant mainly through the roots and colonizes the vascular system (Czymmek et al., 2007; Islam et al., 2012). Expression patterns of these rdd-regulated genes were compared in roots and shoots, with and without Fusarium infection, and the role of DNA methylation in regulating this spatiotemporal expression determined using Arabidopsis demethylase mutants. Furthermore, the role of the individual DNA demethylase genes, ros1, dml2, and dml3 in the regulation of these genes was investigated. Our findings indicate a role for DNA demethylases and promoter TEs in controlling the magnitude of defense gene expression but not spatiotemporal expression.
Materials and Methods
Plant Growth Conditions, Fusarium Infection, and Fungal Biomass Measurements
Plants used in this study included WT Arabidopsis Col-0, the triple demethylase mutant rdd (Col-0 background with ros1, dml2, dml3; SALK_045303, SALK_131712, SALK_056440, respectively) (Le et al., 2014) and the single demethylase mutants ros1-3, dml2-1, and dml3-2 (Penterman et al., 2007). Seeds were sterilized and sown on Murashige and Skoog (MS) agar (Murashige and Skoog, 1962) with 3% sucrose, incubated at 4°C for 2 days and then grown at 22°C with a 16 h light/8 h dark photoperiod. Seedlings were transferred to fresh media 1 week after germination.
Fusarium oxysporum f. sp. conglutinans strain 5176 (obtained from Dr. Roger Shivas, Queensland Department of Primary Industries and Fisheries, Australia) was grown in liquid potato dextrose broth (PDB) at 28°C for at least 2 days and filtered through miracloth. Eighteen day old Arabidopsis plants were Fusarium-infected by briefly dipping the roots into 1 × 106 spores/ml, draining excess solution and immediately transferring the plants to sucrose-free MS plates. Mock-infected plants were dipped in water to serve as a stress control. Disease scoring was performed at 8 days post infection (dpi) by counting the number of plants exhibiting the following symptoms: (0) no chlorotic leaves, (1) 1–3 leaves showing chlorosis, (2) 4–6 leaves showing chlorosis, (3) all leaves showing chlorosis, and (4) dead plant. The experiment was performed in duplicates using at least 15 plants per genotype and repeated independently.
Plant material was harvested by carefully lifting the plants from the plate and separating root and shoot tissue at the hypocotyl/epicotyl junction. Leaf sections were obtained by cutting individual leaves at the base and then separating the midrib and petiole from the leaf blade (Figure 1). Multiple individual plants were pooled for a single biological replicate as indicated. Quantitative measurement of fungal biomass in plant tissue was performed using the wheat germ agglutinin (WGA) chitin assay as described by Ayliffe et al. (2013). Briefly, tissue samples were autoclaved in 1 M KOH, gently neutralized in 50 mM Tris pH 7.0 and then macerated by sonication. A standard curve was created by spiking a known amount of macerated Fusarium mycelial tissue into macerated uninfected WT Arabidopsis tissue.
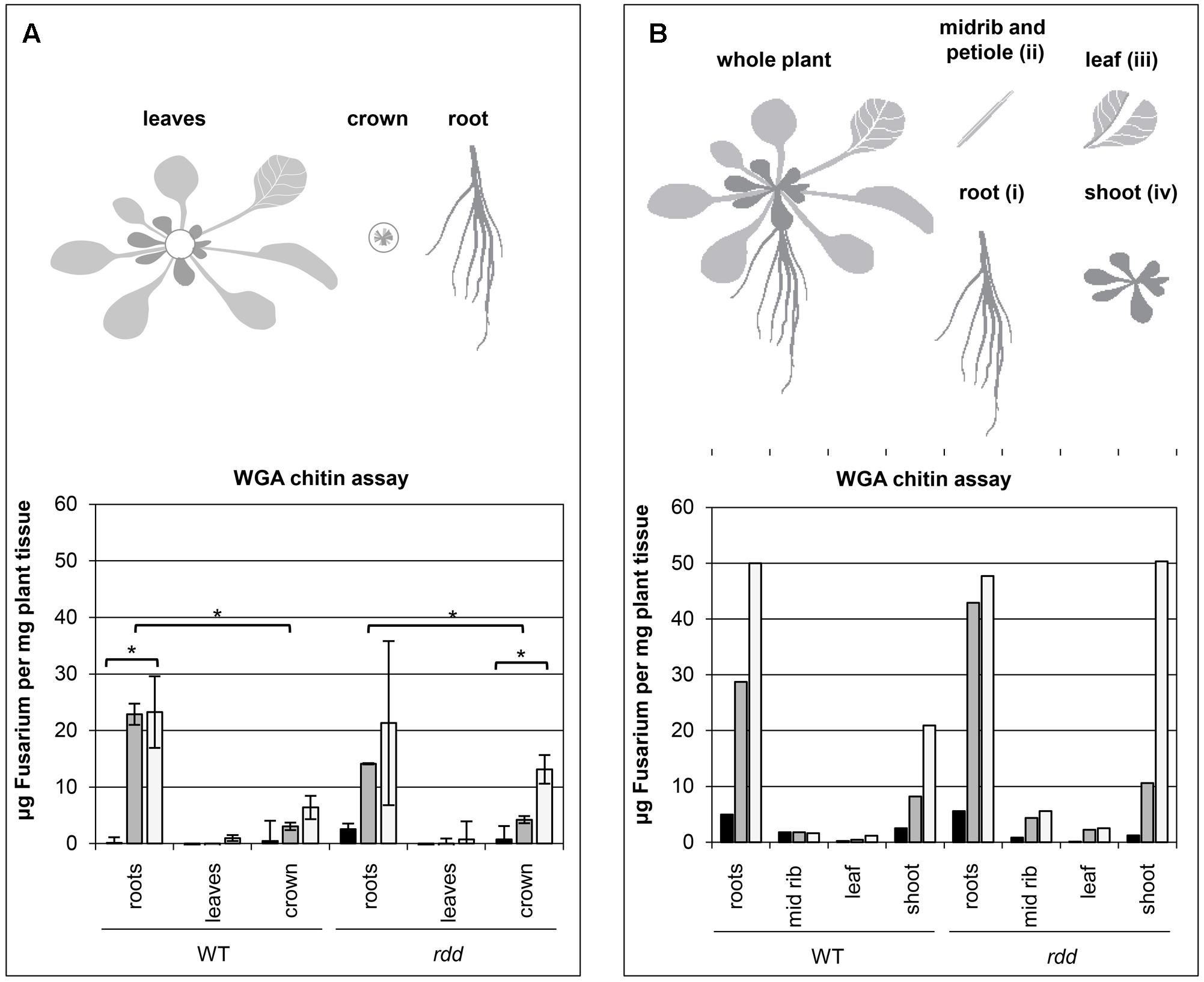
FIGURE 1. Temporal characteristics of fungal biomass accumulation in Arabidopsis WT and rdd mutant plants. (A) Top panel depicts a schematic detailing the plant subsections used for the WGA chitin assay. Bottom panel shows the accumulation of fungal biomass. The average of two biological replicates, nine plants per replicate, is shown with error bars indicating standard deviation. Asterisks indicate significance as determined by Student’s t-test p ≤ 0.05. The temporal characteristics of the Fusarium-infected sample are shown by black (1 dpi), gray (3 dpi) and white (6 dpi) bars; mock samples showed negligible fluorescence indicating low background and are omitted here. (B) Top panel depicts a schematic detailing the plant subsections used for the WGA chitin assay. Bottom panel shows the accumulation of fungal biomass determined in a single experiment using 12 individual plants per sample. The temporal characteristics of the Fusarium-infected sample are shown by black (1 dpi), gray (3 dpi) and white (6 dpi) bars; mock samples showed negligible fluorescence indicating low background and are omitted here.
Samples were incubated with a WGA-FITC conjugate antibody that specifically binds to chitin. Excess stain was removed by washing and the remaining fluorescence measured. Measurements were performed in technical quadruplicates. To test whether differential accumulation was significantly different between samples, Student’s t-tests were performed between indicated samples.
RNA Isolation and Quantitative RT-PCR (RT-qPCR)
RNA was either isolated using TRIzol® (Thermo Fischer Scientific) according to manufacturer’s instructions with inclusion of an additional second chloroform extraction, or by hot phenol extraction as described below. RNA samples were treated with RNase-free DNase (Promega) and reverse-transcribed using oligo-dT primers and Superscript III reverse transcriptase (Invitrogen) according to manufacturer’s instructions. RT-qPCR was performed in technical triplicates in the ABI 7900HT Real Time PCR System (Applied Biosystems) using Platinum Taq polymerase (Invitrogen) and SYBR green. Transcript abundance was measured using the standard curve method and normalized to Actin2 and FDH (primers are listed in Supplementary Table S1). Both normalisations gave similar results for all experiments so only the results against actin2 are shown. All measurements were performed in technical triplicates using biological duplicates. Student’s t-tests were performed to test whether differential expression was significant. Samples analyzed are indicated and significance ranges shown. Sample pairs that were not significantly different are not shown to simplify the figures.
DNA Isolation and Methylation Analysis by Bisulfite Sequencing
DNA (and RNA) was isolated by hot phenol extraction as previously described (Vries et al., 1989) with some modifications. Large RNA was precipitated with LiCl and DNA was precipitated from the supernatant using isopropanol. DNA was further purified by a second ethanol precipitation step. Ten μg of genomic DNA was bisulfite-treated as previously described (Wang et al., 2008). Regions of interest were amplified by nested PCR using the following cycling program: 12 min at 94°C followed by 10 cycles of 1 min at 94°C, 2:30 min at 50°C, 1:30 min at 72°C, and 30 cycles with 1 min at 94°C, 1:30 min at 55°C, 1:30 min at 72°C, with a final extension of 10 min at 72°C (primers were designed as described by Finn et al., 2011; Supplementary Table S1). Products were purified using the PCR clean-up system (Qiagen) and directly subjected to Sanger Sequencing using the BigDye Terminator system (Life Technologies) and the forward primer. Sequencing traces were uploaded into Mutation Surveyor (SoftGenetics, United States) and C to T conversion peak height ratios calculated. Analyses were performed in biological duplicates. To determine the efficiency of bisulfite conversion a region of the chloroplast encoded gene psaA was amplified from the bisulfite-treated DNA and sequenced. Conversion for all samples analyzed here was found to be efficient (examples of the traces of psaA are shown in Supplementary Figure S1). Significance of differential methylation was determined by unpaired, two-tailed Student’s t-test as indicated. Pairings with significant differential DNA methylation levels are indicated in the respective figures. p-values are not shown in figures for simplification but can be found in Extended Data Files 1 and 2.
Results
The Majority of Fungal Biomass Accumulates in Roots and Meristematic Crown Tissue during Fusarium Infection
Previously, we have shown that the expression of a subset of defense-related genes is downregulated at the whole plant level in the Arabidopsis triple DNA demethylase mutant rdd upon infection with F. oxysporum (Le et al., 2014). Gene expression changes upon pathogen infection can often be localized specifically to infection sites and therefore differ from that of whole plants where the majority of tissues may be uninfected. To further define the regulatory role of DNA demethylation in defense gene expression we first examined the spatial and temporal colonization of Arabidopsis tissue by F. oxysporum, which is a poorly characterized process (Czymmek et al., 2007; Islam et al., 2012).
A WGA chitin assay (Ayliffe et al., 2013) was performed on root, leaf (all leaves) and crown tissue of Fusarium infected WT and rdd plants to determine where the majority of the fungal biomass is located. Fungal biomass increased over a 6 day period and was located predominantly in root and crown tissue, with only limited amounts of fungal growth occurring in leaf tissue (Figure 1A). As leaf vein clearing is a major symptom of Fusarium infection (Pietro et al., 2003; Michielse and Rep, 2009) leaf petioles (section ii) were excised and compared with the remaining leaf tissue (section iii) as well as shoot tissue (section iv) in a single experiment, however, no obvious difference in Fusarium accumulation was apparent between these two tissues (Figure 1B). Leaf vein clearing is therefore not a direct consequence of Fusarium accumulation in this tissue. Interestingly, in the rdd mutant increased fungal biomass was observed in crown tissue at 6 dpi when compared with the equivalent WT tissue.
Expression of Defense-Related Genes Is Tissue-Specific, Highly Responsive to Fusarium Infection and Compromised in rdd
Having established the preferential growth patterns of Fusarium in Arabidopsis tissues shown above we investigated if defense-related genes containing promoter TE sequences are expressed in a tissue-specific manner upon infection. We selected six defense-related genes which we previously identified to be strongly downregulated in the rdd mutant compared to WT and which showed induction upon Fusarium infection (Le et al., 2014) (Table 1, genes 1–6). We analysed their expression in leaf (iii), midrib and petiole (ii), and the remaining shoot tissue (iv) (Figure 1B). These genes showed either similar levels of expression (At5G24210, At2G15040, At1G58602, and At4G09420) or displayed a distinct expression pattern among the three aerial tissue types (At5G39110, At5G38550) (Supplementary Figure S2). To simplify the analyses, all aerial tissues (shoot tissue) were combined and compared with root tissue in subsequent experiments. It is noteworthy that significant gene induction occurred in shoot tissues despite the reduced fungal colonization when compared with root tissue.
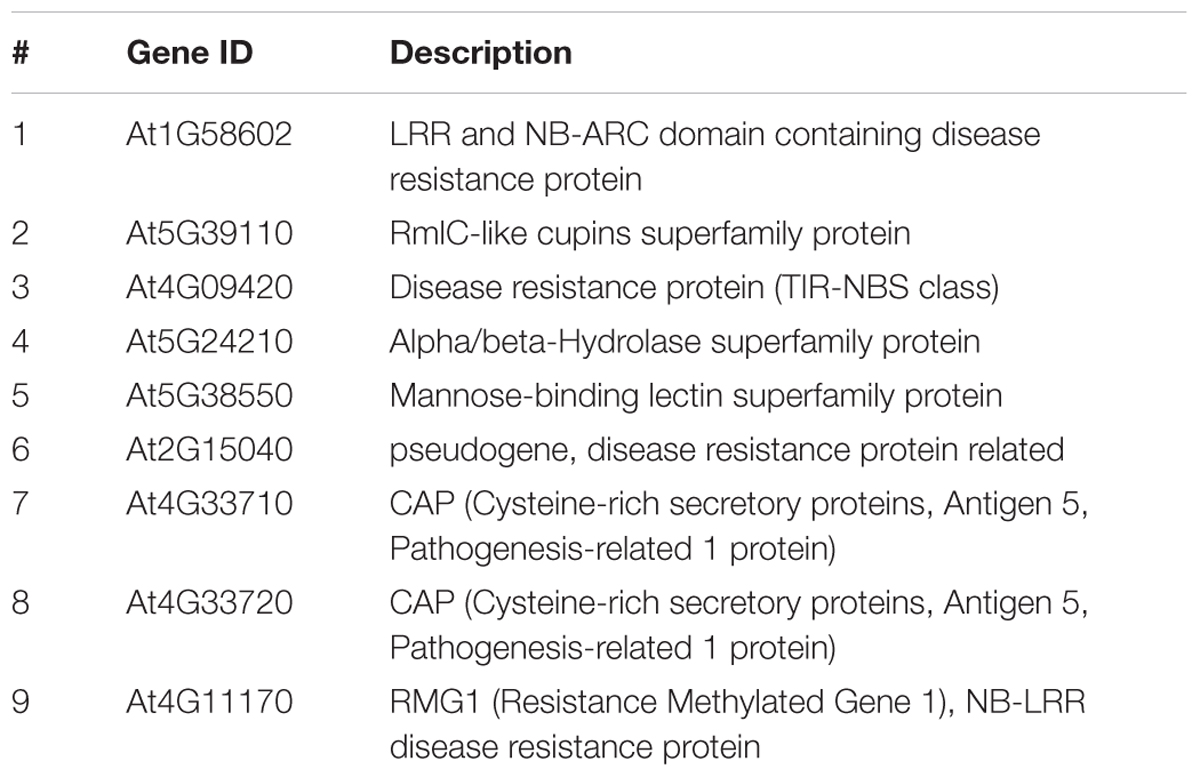
TABLE 1. List of all Arabidopsis genes analyzed in this study for their root/shoot expression and DNA promoter methylation levels.
The expression profile of nine defense-related genes containing promoter TE sequences was analyzed in shoot and root tissues of WT and rdd, including all six Fusarium-inducible genes mentioned above and three additional defense-related genes (Table 1), the two neighboring genes AT4G33710 and AT4G33720 (both CAP superfamily proteins, PR1-related), and AT4G11170 (RMG1, NB-LRR disease resistance protein), which was shown to be induced by the bacterial elicitor flg22 (Yu et al., 2013). As shown in Figure 2, these genes in general showed differential expression between shoots and roots, and displayed four types of Fusarium-responsive expression patterns; (i) expression detectable under mock conditions only in roots, but induced in both roots and shoots upon Fusarium infection (At5G39110, At5G38550, and At4G33720), (ii) expression detectable under mock conditions only in shoots, but induced in roots and shoots following Fusarium infection (At1G58602, At2G15040, and At5G24210), (iii) expression detectable in both shoots and roots but only following Fusarium infection (At4G11170, At4G33710), and (iv) expression detectable in both shoots and roots under mock conditions and induced by Fusarium infection in both tissues (At4G09420) (Supplementary Table S2). The level of induction was gene-specific with Fusarium-induced expression levels in the rdd mutant usually being lower than in WT plants. Consistent with previous reports using the ros1 DNA demethylase single mutant (Yu et al., 2013), At4G11170 showed higher expression in the rdd mutant following infection. Furthermore, we observed tissue-specificity at the level of gene induction following infection for almost all genes (Supplementary Table S2).
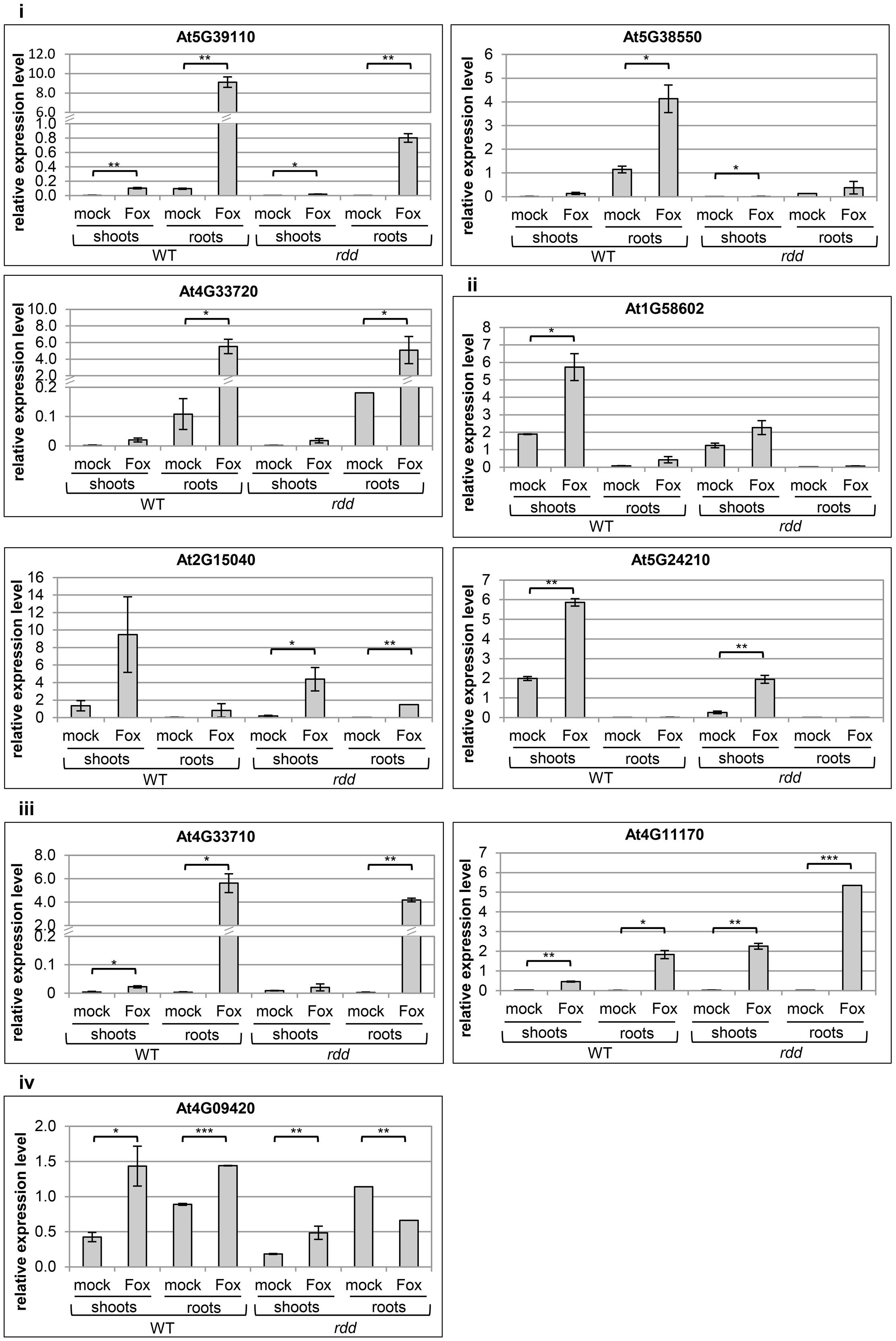
FIGURE 2. Expression levels of defense-related genes in WT and rdd root and shoot samples. Plants were infected with Fusarium (Fox) or mock treated and biological duplicate samples were harvested at 3 dpi. Roots and shoots of five individual plants per replicate were separated at the epicotyl/hypocotyl junction and RNA isolated for expression analysis. Root expression is induced for gene At5G24210 following Fusarium infection, but this induction is not significant and at such low levels that it was not feasible to portray this in the figure. Expression levels are relative to actin2 and error bars indicate standard deviation. Significance levels used for the Student’s t-test analysis were: ∗p ≤ 0.05; ∗∗p ≤ 0.005; ∗∗∗p ≤ 0.0005; non-significant pairings are omitted to simplify the figure.
These results highlight the advantage of analyzing specific tissues over whole plant samples given the tissue specificity of Fusarium-responsive defense-related gene expression coupled with the preferential pathogen accumulation in specific tissues. For example, using whole plant tissues we previously detected a 10-fold upregulation of gene At5G39110 upon Fusarium infection at 5 dpi (Le et al., 2014), whereas in the present study a 100-fold induction by Fusarium infection was detected when analyzing root tissue alone (Figure 2). Gene expression changes in specific tissue types are therefore not accurately determined when using whole plant tissue for analyses.
Promoters of Defense-Related Genes Are Hypermethylated in the rdd Mutant
To investigate the molecular basis for the differential expression of these genes in rdd and WT plants the DNA methylation level of the promoter region of all the above genes, except At4G11170 and At4G33710, was examined (Table 1, genes 1–6 and 8). In particular, short TE sequences in promoter regions were examined as they are often targeted by RdDM (Figure 3, pink bars) and we have previously shown differential cytosine methylation of these sequences in rdd plants (Le et al., 2014). DNA was isolated from the same samples used for gene expression analysis and cytosine methylation analysis undertaken for genomic regions of interest (Figure 3, green bars). Methylation levels were determined by sequencing PCR amplified products derived from bisulfite treated DNA (for primer sequences see Supplementary Table S1).
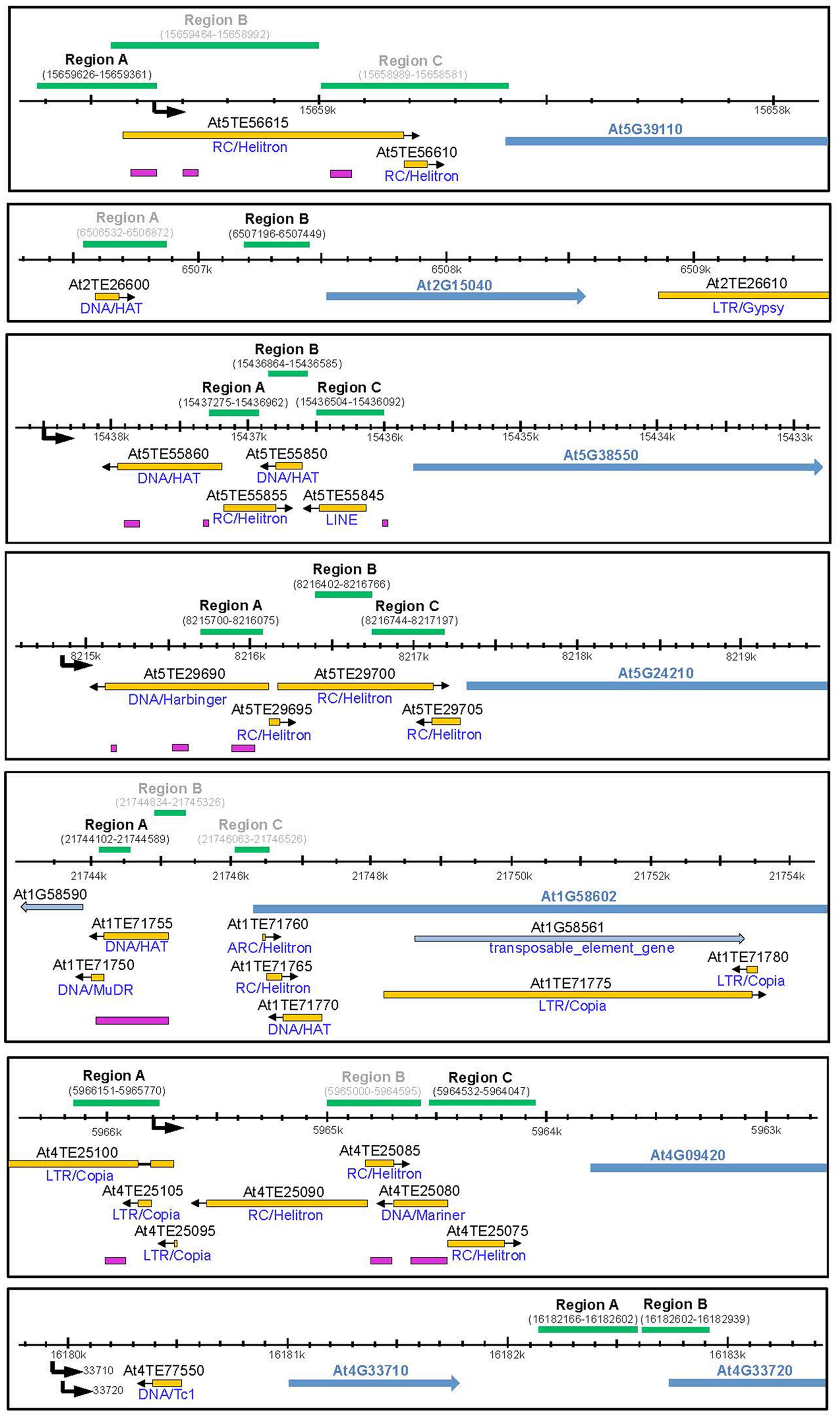
FIGURE 3. Schematic diagram showing promoter regions of defense-related genes subjected to bisulfite sequencing. Schematics were created according to TAIR10 with the gene model and name indicated by blue arrows. All genes of interest are displayed in 5′–3′ orientation. Neighboring genes models and TE genes are indicted by light blue arrows. Natural transposons, as annotated by TAIR, are shown by yellow bars with the orientation indicated by a black arrow and the TE family indicated below each bar. Regions targeted by small RNAs are indicated by pink bars (ASRP; http://asrp.danforthcenter.org/). Promoter start sites were predicted using the starPRO database (Baev et al., 2010) and are indicated by a black arrows. Genomic regions (coordinates in brackets) subjected to bisulfite sequencing are indicated above by green bars. Regions for which data could be obtained are indicated in black, regions that did not yield a PCR product are indicated in gray.
Increased promoter methylation levels in regions near (∼500–1000 bp) the putative gene transcription start sites (TSSs) were consistently observed in the rdd mutant when compared with WT for most genes analyzed. Increased DNA methylation in the mutant was largely irrespective of tissue type or treatment with only minor localized differences occurring between different tissues and infected/uninfected tissues (for full dataset see Extended Data File 1). Hypermethylation in rdd was particularly obvious for genes At5G38550, At4G09420 and At5G39110, and to a lesser extent for gene At2G15040 (Figure 4, black boxes). Specifically for region B of gene At5G38550 and region C of gene At4G09420, cytosine methylation levels in all three sequence contexts were low in the WT and comparatively higher in rdd. Increased CG methylation was also observed in region A of the At5G39110 gene in rdd plants. Similarly, some CHH and CHG sites in region B of gene At2G15040 also showed hypermethylation in rdd. In contrast, for two regions of gene At5G24210 (region A – highly methylated, region C – lowly methylated) and the highly methylated region A of gene At1G58602, no clear differences in methylation pattern were observed between mutant and WT plants (Figure 4). However, analysis of whole genome bisulfite sequencing data (Stroud et al., 2013) showed hypermethylation in rdd plants of region B of gene At5G24210 (between the two regions analyzed here) and region B of gene At1G58602 (downstream of region analyzed here) (Supplementary Figure S3). No DNA methylation was detected for gene At4G33720 (Figure 4).

FIGURE 4. DNA methylation levels of defense-related gene promoters. Plants were infected with Fusarium or mock treated and samples harvested at 3 dpi. Methylation percentage was calculated using Mutation Surveyor and the mean methylation level for each site is represented as a heatmap. Each square represents a single cytosine and lines above graphs show location of TE elements. Boxes indicate regions where the majority of cytosines showed significant differential methylation (p ≤ 0.05) between rdd and WT. The TSS for each gene is located to the right of the sequences shown here (see Figure 3). Extended regions of non-differential methylation have been removed for easier visualization. Fusarium infection data were largely similar and are not shown for easier visualization. For the complete dataset including Student’s t-test p-values see Extended Data File 1.
We undertook further analysis of promoter methylation for over 100 of the most significantly rdd-downregulated genes using the Stroud et al. (2013) bisulfite sequencing data which revealed a positive, quantitative correlation between gene repression and the presence of hypermethylation within 1kb of gene TSS’s. Ten (53%) out of 19 highly downregulated rdd genes (logFC < -3.0; Le et al., 2014) had hypermethylated promoter regions close to gene TSS’s, compared with 39% (21/54 genes, logFC -3.0 to -2.0) and 26% (7/27, logFC > -2.0) of genes that were less severely repressed in the rdd background (Figure 5). Together these results suggest that DNA demethylases specifically target TE sequences in promoter regions of defense-related genes for active demethylation and this demethylation process is essential to maintain gene expression levels.
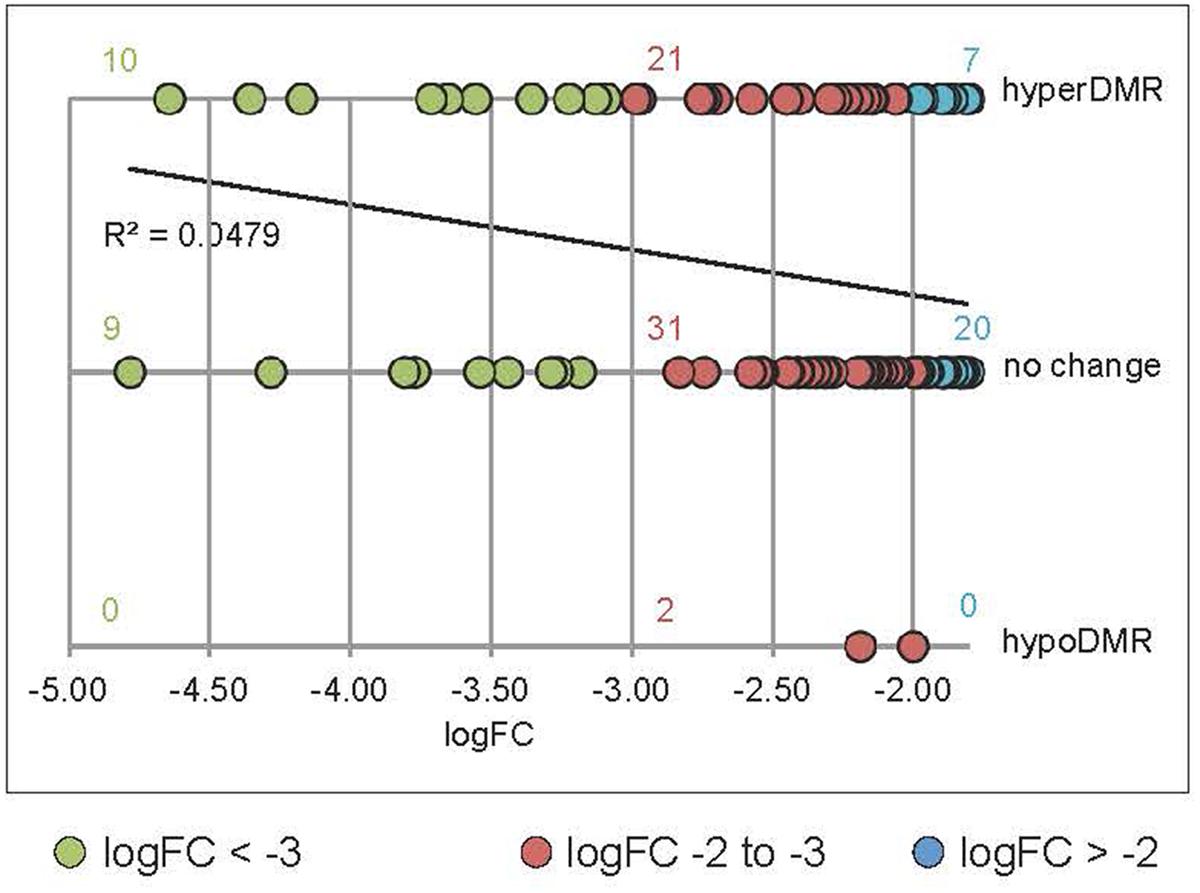
FIGURE 5. Positive correlation between the level of downregulation in rdd and hypermethylation within 1 kb of the genes TSS’s. Data were obtained by interrogating a publicly available database (Stroud et al., 2013) for rdd and WT DNA methylation levels. rdd-downregulated protein coding genes (Le et al., 2014) were analyzed (downregulation logFC between –4.78 and –1.8). Transposable element (TE) genes were eliminated from this analysis. For multiple splice variants, only the most downregulated variant was included. The number of gene promoters with hyper-, hypomethylation (hyperDMR, hypoDMR) and no change in rdd compared to WT were plotted against the level of downregulation (logFC). Linear regression of all data shows a positive correlation between the level of downregulation and hyperDMR.
Root and Shoot Tissues Show Low-Level Differential Methylation Patterns of Defense-Related Gene Promoters
While WT and rdd plants show clear differential methylation in promoter regions, only low levels (10–15% change) of localized methylation differences were observed for the equivalent sequences in root and shoot tissues of the same genotype (Supplementary Table S3). Methylation differences between these tissues were most apparent in the CHH context in gene At5G24210 where both rdd and WT plants showed increased CHH methylation in shoots compared with roots, while in genes At4G09420 and At1G58602 this increased shoot CHH methylation was only observed in the rdd genotype (Figure 6, boxes). Differential methylation was particularly localized for genes At1G58602 and At4G09420, affecting only few cytosine residues. Fusarium infection increased CHH methylation in gene At5G24210 and to a lesser extent in At1G58602 in WT and rdd roots, but not shoots (Figure 6, brackets).

FIGURE 6. Tissue specific differences in CHH methylation in defense-related gene promoters occur in a distinct localized manner. Data are identical to Figure 4 but selected regions of CHH methylation are shown for all datasets. The order in which the data are represented varies to Figure 4 to highlight the differences. Boxes highlight regions where the majority of cytosines show significant hypermethylation (p ≤ 0.05) in shoots compared to roots. Brackets highlight regions where Fusarium infection increased CHH methylation in roots compared to shoots (p ≤ 0.1). Student’s t-test p-values can be found in the Extended Data File 1.
Although promoter methylation is generally considered to repress gene expression, four of the genes analyzed showed a positive correlation between hypermethylation and increased expression in a specific tissue (Supplementary Table S3). The context of this hypermethylation was primarily CHH and not CG, which is consistent with a previous observation that repressed gene expression in rdd plants tended to be associated with CG hypermethylation but not CHH hypomethylation (Le et al., 2014). Interestingly, three of these four genes showed an increase in CHH methylation in response to Fusarium infection in tissues in which expression is induced upon infection. These results suggest that CHH methylation, indicative of RdDM, may play a regulatory role in the expression of defense-related genes during stress response, as previously speculated (Le et al., 2014).
ROS1 Is Essential for the Regulation of rdd-Downregulated Defense-Related Genes
ros1, dml2, and dml3 were previously shown to have some functional redundancy in the response of Arabidopsis to Fusarium infection (Le et al., 2014). To dissect the contribution of each demethylase gene to DNA methylation and gene expression changes in the rdd mutant and in response to Fusarium infection, single demethylase mutants (Penterman et al., 2007) were analyzed. The rdd triple mutant showed the strongest symptoms to Fusarium infection amongst all the genotypes assayed, confirming that some functional redundancy exists amongst the three demethylases (Supplementary Figure S4). dml3-1 showed a similar phenotype to WT Col-0, whereas dml2-1 showed more leaf yellowing than WT plants, with ros1-3 displaying the strongest disease symptoms amongst the single mutants. This suggests that ROS1 is the dominant DNA demethylase in the regulation of rdd-controlled defense-related genes. Although the rdd mutant used in our analysis carried different mutations to the single mutants analyzed here (for details see Materials and Methods), the equivalent rdd triple mutant comprising these three single mutants showed comparable symptoms to the rdd mutant analyzed here.
Defense-related gene expression and promoter DNA methylation levels were also analyzed in root and shoot tissue of single mutants and compared with WT and rdd plants. Suppressed gene expression under mock conditions and reduced gene induction upon Fusarium infection, characteristic of the rdd mutant, was largely replicated in the ros1-3 mutant (Figure 7, red boxes). In contrast, dml2-1 and dml3-1 mutants showed expression profiles similar to WT. Repression of basal and pathogen-induced gene expression in ros1-3 occurred in both root and shoot tissue for At3G33710, At5G38550, At5G39110, and At5G24210 while suppression of At3G33720 and At1G58602 occurred in root tissue only (Figure 7, red boxes). These results further indicated that ROS1 is the dominant DNA demethylase required for positive regulation of these defense-related genes.
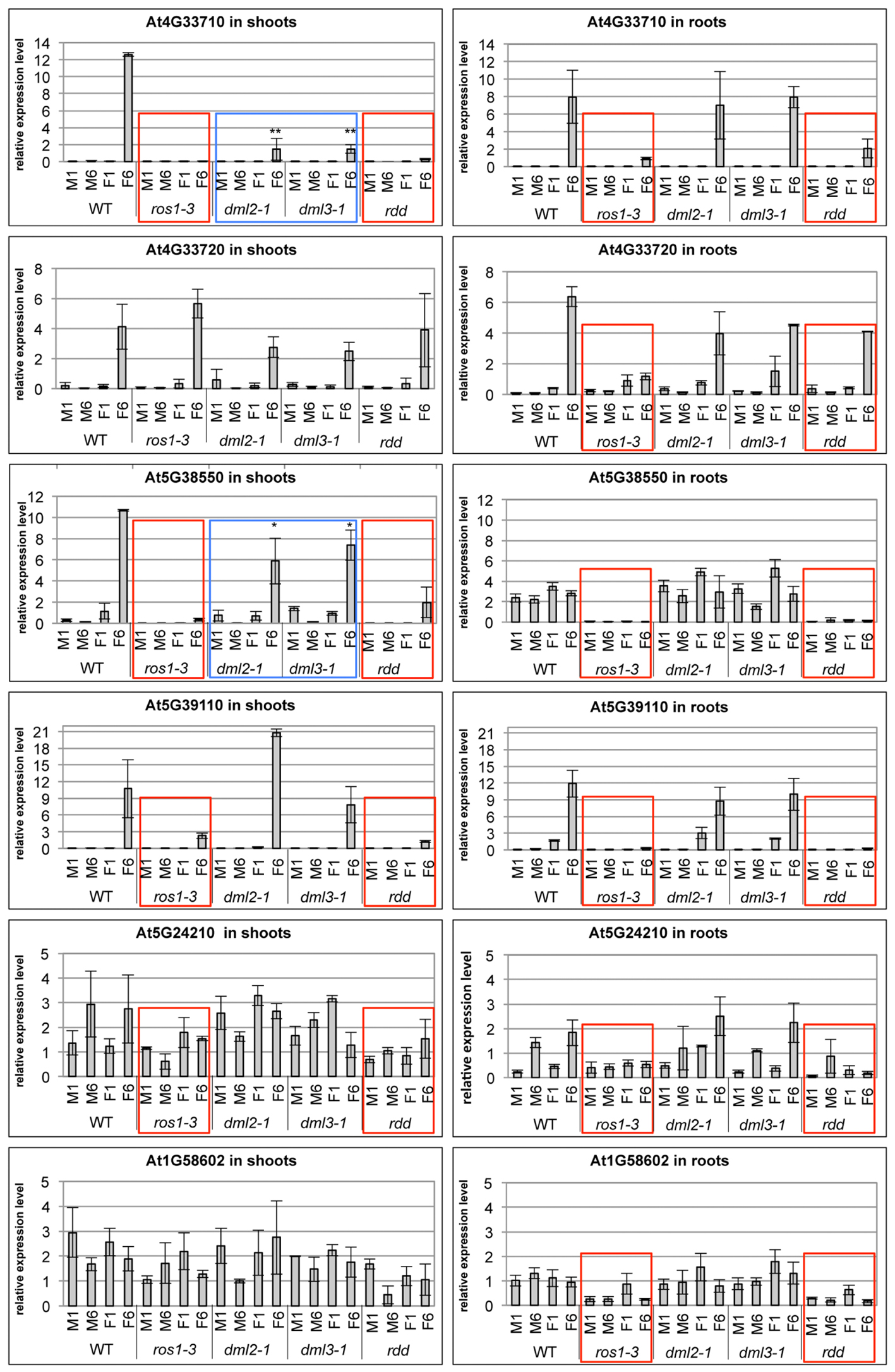
FIGURE 7. The ros1-3 mutant shows a similar gene expression profile of defense-related genes as the rdd mutant. Plants were Fusarium infected (F) or mock treated (M) and the expression level in shoots (left panels) and roots (right panels) examined at 1 and 6 dpi. Expression levels of defense-related genes are shown relative to FDH; comparable results were obtained using actin2 as reference. Bars show the average of three independent biological replicates with error bars indicating standard deviation. Expression profiles that are similar between rdd and ros1-3 are indicated with red boxes, while expression profiles intermediate between WT and rdd in the dml2-1 and dml3-1 mutants are indicated by blue boxes. ∗p ≤ 0.1 and ∗∗p ≤ 0.05, according to Student’s t-test.
However, ros1-3 does not entirely phenocopy rdd, as downregulation of basal expression of At5G24210 (both shoots and roots) and At1G58602 (shoots only) was less compromised in this mutant compared to rdd. In addition, At4G33710 and At5G38550 showed reduced levels of Fusarium-induced gene induction in shoots of dml2-1 and dml3-1 compared to WT (Figure 7, blue boxes). These data suggest that DML2 and DML3 also contribute to the rdd-mediated downregulation of some genes.
To verify that the repressed gene expression in ros1-3 and to a lesser extent dml2-1 and dml3-1 is due to localized DNA hypermethylation in promoter regions of these genes, bisulfite sequencing was undertaken on four genes (At4G09420, At5G38550, At5G39110, and At5G24210) in both root and shoot tissue (for full dataset see Extended Data File 2). Regions that showed increased methylation in the rdd mutant also showed significant hypermethylation in ros1-3 (Figure 8, red boxes). This increased methylation was not observed in dml2-1 and dml3-1, which had similar methylation levels to WT Col-0. At5G24210 was an exception and showed no clear difference in methylation levels among the five genotypes (WT, rdd, ros1-3, dml2-1, and dml3-1) except in CG methylation in region C (Figure 8, blue box).

FIGURE 8. The ros1-3 mutant shows hypermethylation of TE sequences in defense-related gene promoters, similar to rdd. DNA methylation in single demethylase mutants ros1-3, dml2-1, dml3-2 as well as the triple mutant rdd and WT was analyzed at 3 dpi. Methylation level was determined using Mutant Surveyor and the average of two biological replicates is shown as a heatmap. Each square represents a single cytosine in the sequenced region. Cytosines with significant hypermethylation (p ≤ 0.1) in ros1-3 (and rdd) compared to WT are indicated by red boxes. Regions where rdd and ros1-3 show significant differential methylation (p ≤ 0.1) are highlighted by blue boxes. Shoot data were largely similar and are not shown for easier visualization. Extended regions of non-differential methylation have been removed for easier visualization. The full dataset including the t-test p-values can be found in Extended Data File 2.
Interestingly, for gene At5G38550, the level of methylation in region B was lower in ros1-3 than in rdd. Similarly, the level of CG methylation in region C of At5G24210 was also lower in ros1-3 than in rdd (Figure 8, blue boxes). These results suggest that while ROS1 is the dominant player among the three DNA demethylases, it does not account for all DNA demethylase activities, consistent with the existence of functional redundancy as suggested by the disease phenotypes. The methylation data again supports a direct role for localized differential DNA methylation resulting in repressed gene expression in ros1-3 and rdd. The contribution of DML2 and DML3 to methylation could not be confirmed by bisulfite sequencing as only subtle differences were observed between these two mutants and WT Col-0 for the four genes analyzed.
Similar to the observation with WT and rdd (Figure 6, brackets), localized differences in methylation were also observed between roots and shoots and between Fusarium-infected and mock-treated ros1-3 plant tissues (Supplementary Figure S5). However, these methylation differences were not striking and unlikely to account for the tissue-specific or Fusarium-responsive expression patterns of the respective genes in ros1-3.
Discussion
DNA Demethylase-Regulated Fusarium-Response Genes Show Tissue-Specific Expression Patterns
In our previous study whole plant tissue of WT Col-0 and the rdd mutant was used to analyze gene expression and DNA methylation differences between each genotype and between F. oxysporum-infected and mock-treated plants (Le et al., 2014). We observed that the repressed expression of a subset of defense-related genes in rdd was associated with altered DNA methylation of short TE sequences in the gene promoters, which suggests that these genes are regulated by DNA demethylases at these TE sequences. We also observed that these rdd-downregulated genes tend to show increased expression upon Fusarium infection, however, this increased expression is not associated with a clear change in DNA methylation of these short TE sequences. This suggested that Fusarium-responsive gene expression might be tissue-specific and changes in DNA methylation might occur in a tissue-specific manner and escape detection in whole plant tissue analysis.
In this study we first examined the spatiotemporal distribution of F. oxysporum in Col-0 and rdd plants. The majority of fungal biomass was restricted to root tissue at 1–3 dpi and remained largely confined to the roots even at 6 dpi. In the aerial part of the plants, fungal biomass was detected at a significant level only in the crown tissue at the later stage of infection. Very little fungal growth was observed in tissue surrounding leaf veins or expanded leaves, even after leaf yellowing symptoms were visible. In addition, the increased fungal disease symptoms in rdd were not associated with a large increase in fungal biomass. These results suggest that the increased symptoms in rdd leaf tissues are not caused by direct fungal colonization, but rather are a secondary response to infection.
The differential accumulation of fungal biomass between shoot and root tissues prompted us to investigate the expression pattern of the rdd-downregulated, Fusarium-responsive, defense-related genes in these two tissue types in both rdd and WT Col-0 plants. Tissue-specific or tissue-preferential expression was observed for eight of the nine genes analyzed, while all nine genes were induced by Fusarium infection. Interestingly, only At5G38550 was identified to be induced in roots by Fusarium in a previous transcriptome-wide study (Lyons et al., 2015). These results demonstrate that some defense-related genes are highly regulated in a tissue-specific manner, thereby highlighting the necessity for separating tissues in gene expression studies.
ROS1 Is the Major Demethylase Mediating Active Demethylation of Promoter TE Sequences
The majority of rdd-downregulated defense-related genes analyzed accumulated increased DNA methylation in TE sequences located in promoter regions close to gene TSS in the rdd mutant. Expression of these defense-related genes in rdd were still inducible by Fusarium infection, but the level of induced expression did not reach that of WT plants, consistent with the increased DNA methylation of promoter TE sequences repressing gene expression under both mock and Fusarium infection conditions. These observations indicate that DNA demethylases positively regulate defense-related gene expression under both these conditions.
To understand the role of each individual DNA demethylase present in the rdd triple mutant, we analyzed single demethylase mutant lines ros1-3, dml2-1 and dml3-1 for Fusarium susceptibility, defense-related gene expression and promoter methylation. Dml3-1 plants showed a similar response to Fusarium infection as WT, while dml2-1 plants were only slightly more susceptible than WT plants. However, ros1-3 plants were highly susceptible although the symptoms were less severe than those observed for rdd. These data suggest that ROS1 is the dominant DNA demethylase that regulates genes involved in Fusarium resistance in Arabidopsis. Consistent with this hypothesis, gene expression analysis showed that expression patterns of all six defense-related genes analyzed in ros1-3 was similar to the expression profiles observed in rdd. Interestingly, both dml2-1 and dml3-1 mutants did not always show a WT-like gene expression profile; for three of the six genes the expression level in these two mutants was intermediate between WT and rdd/ros1-3. This suggests that DML2 and DML3 do contribute to the regulation of some genes, possibly in a tissue-specific manner.
Similar to Fusarium infection and gene expression analysis, DNA methylation analysis showed that loss of ROS1 results in hypermethylation of promoter TE sequences, whereas DNA methylation profiles of dml2-1 and dml3-1 were virtually identical to those of the WT Col-0 plants. This further confirms that ROS1 is the dominant demethylase required to positively regulate these defense-related genes by actively demethylating promoter TE sequences. However, for one gene (At5G38550, region B) the level of methylation in ros1-3 was not as high as in rdd, and some regions showed tissue-specific differential methylation in ros1-3. These results further suggest that DML2 and DML3 participate in the demethylation of some genes, and that they may play a role in fine-tuning methylation or in tissue-specific methylation. This notion is also supported by the observation that some genes show intermediate expression patterns in shoot tissue in both dml2-1 and dml3-1.
Tissue-Specific and Fusarium-Inducible Gene Expression Patterns Are Not Directly Regulated by Differential DNA Methylation
Unlike the clear differential DNA methylation occurring between rdd/ros1-3 and Col-0, we only observed small, localized methylation differences in promoter TE sequences when root and shoot tissues were compared within a genotype, despite distinct expression patterns existing between these tissues. These differences in methylation level occurred mainly in the CHH context, particularly in the rdd mutant, and usually involved a few cytosine residues in close proximity. This hyper-CHH methylation in a specific tissue type tended to be associated with increased gene expression. For instance, shoots showed higher levels of CHH methylation than the roots for genes At5G24210 and At4G09420 and these two genes were expressed specifically in shoots. However, the lack of widespread methylation differences between roots and shoots suggests that tissue-specific expression patterns are not determined by DNA methylation in promoter TE regions. Our observation is consistent with a previous report that showed no significant global methylation differences between roots and shoots in the Arabidopsis Col-0 ecotype (Widman et al., 2014). This study identified 2424 genes as differentially expressed between the two tissues. Although these genes showed some enrichment of differential methylation between tissues, only one in every 173 cytosines was actually differentially methylated (Widman et al., 2014).
Similar to the small methylation difference between root and shoot tissues, no widespread methylation changes were observed between Fusarium-infected and mock-treated plants in promoter TE sequences of the defense-related genes analyzed. Only one gene (At5G58602) was found to have increased CHH methylation in response to Fusarium in both tissues in rdd plants. These data suggest that the Fusarium-inducible expression patterns of these genes are independent of DNA methylation in the promoter TE sequences.
Possible Mechanisms for the Regulation of DNA Methylation-Controlled Defense-Related Genes
Our results show that the repressed expression of defense-related genes in rdd and ros1-3 plants is largely explained by differential DNA methylation of short promoter TE sequences repressing gene expression. However, spatiotemporal regulation of these same genes in shoot and root tissues, and upon Fusarium infection, appears independent of DNA methylation. TE insertions in the mammalian genome are suggested to contribute cis-acting elements to adjacent genes, thereby conferring altered gene regulation (Bourque et al., 2008; Makarevitch et al., 2015). Similarly, in maize stress-response genes are enriched for TE insertions near genes and these TE elements may provide enhancers for stress-induced gene expression (Makarevitch et al., 2015). However, a genome-wide methylation analysis of maize plants after abiotic stresses indicated that stress-responsive gene expression was not usually associated with DNA methylation changes (Eichten and Springer, 2015).
In light of these studies and our own results, it is conceivable that the Fusarium-responsive and tissue-specific expression patterns of these defense-related genes could be due to cis-elements encoded by adjacent TEs. This is consistent with TEs being inducible by environmental stresses and therefore containing stress-responsive elements (Capy et al., 2000; Casacuberta and Gonzalez, 2013). In support of this hypothesis, we searched the upstream region (250–1500 bp) of rdd-downregulated genes for transcription factor (TF)-binding site sequence using the AthaMAP database (Steffens et al., 2004). Enrichment for bZIP_DOF and MYB class TF-binding sites was observed, particularly in the differentially methylated TE regions present within these gene promoters (Supplementary Table S4). For instance, 38 of the most downregulated protein-coding genes in the rdd mutant (Le et al., 2014) had bZIP_DOF and MYB-type TF-binding sites in 45–50% of the promoters, which was twice the frequency when compared with a randomly selected set of genes (Supplementary Table S4). In addition, some of these detected putative cis-elements have known roles in stress-inducible or tissue-specific gene expression (Villain et al., 1996; Zhou, 1999; Yanagisawa, 2004; Luo et al., 2010; Noguero et al., 2013; Behringer and Schwechheimer, 2015; Gupta et al., 2015) (Supplementary Table S5).
However, TEs are the main target of RdDM, and consequently, while these TE sequences are required to confer tissue-specific or pathogen-inducible expression patterns, they are also subject to DNA methylation affecting the transcriptional activity of the adjacent genes. This is consistent with DNA hypermethylation of these regions in rdd repressing gene expression (Le et al., 2014; and this study). DNA demethylases are therefore required to minimize the DNA methylation level, thus allowing accessibility of Fusarium-inducible TFs and concomitant induced gene expression. Further genome-wide investigations are required to substantiate this model, but the combination of TE insertions, RdDM and DNA demethylases may play a major role in the evolution of plant transcriptional responses to phytopathogens.
Author Contributions
Conceived and designed the study: US, M-BW, MA, KK; performed experiments: US, JL; analyzed the data: US, JL; provided reagents: MA; wrote the manuscript: US, M-BW, KK, MA.
Funding
US was supported by an CSIRO Office of Chief Executive (OCE) Post-doctoral Fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Robert Fischer for kind provision of the single demethylase mutants ros1-3, dml 2-1, dml3-1, the respective triple mutant rdd and the segregating WT, as wells as Dr. Jian-Kang Zhu for providing the rdd triple demethylase mutant used throughout this manuscript. Furthermore, we thank Ms. Melanie Solivieres for help with performing the WGA chitin assay and staff at SoftGenetics (United States) for assistance with Mutation Surveyor. We also thank Neil Smith for technical advice and Liz Dennis for helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01449/full#supplementary-material
References
Ayliffe, M., Periyannan, S. K., Feechan, A., Dry, I., Schumann, U., Wang, M. B., et al. (2013). A simple method for comparing fungal biomass in infected plant tissues. Mol. Plant Microbe Interact. 26, 658–667. doi: 10.1094/MPMI-12-12-0291-R
Baev, V., Naydenov, M., Apostolova, E., Ivanova, D., Doncheva, S., Minkov, I., et al. (2010). Identification of RNA-dependent DNA-methylation regulated promoters in Arabidopsis. Plant Physiol. Biochem. 48, 393–400. doi: 10.1016/j.plaphy.2010.03.013
Bauer, M. J., and Fischer, R. L. (2011). Genome demethylation and imprinting in the endosperm. Curr. Opin. Plant Biol. 14, 162–167. doi: 10.1016/j.pbi.2011.02.006
Behringer, C., and Schwechheimer, C. (2015). B-GATA transcription factors - insights into their structure, regulation, and role in plant development. Front Plant Sci 6:90. doi: 10.3389/fpls.2015.00090
Bharti, P., Mahajan, M., Vishwakarma, A. K., Bhardwaj, J., and Yadav, S. K. (2015). AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J. Exp. Bot. 66, 5959–5969. doi: 10.1093/jxb/erv304
Bourque, G., Leong, B., Vega, V. B., Chen, X., Lee, Y. L., Srinivasan, K. G., et al. (2008). Evolution of the mammalian transcription factor binding repertoire via transposable elements. Genome Res. 18, 1752–1762. doi: 10.1101/gr.080663.108
Capy, P., Gasperi, G., Biemont, C., and Bazin, C. (2000). Stress and transposable elements: co-evolution or useful parasites? Heredity 85(Pt 2), 101–106.
Casacuberta, E., and Gonzalez, J. (2013). The impact of transposable elements in environmental adaptation. Mol. Ecol. 22, 1503–1517. doi: 10.1111/mec.12170
Czymmek, K. J., Fogg, M., Powell, D. H., Sweigard, J., Park, S. Y., and Kang, S. (2007). In vivo time-lapse documentation using confocal and multi-photon microscopy reveals the mechanisms of invasion into the Arabidopsis root vascular system by Fusarium oxysporum. Fungal Genet Biol. 44, 1011–1023.
Dowen, R. H., Pelizzola, M., Schmitz, R. J., Lister, R., Dowen, J. M., Nery, J. R., et al. (2012). Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. U.S.A. 109, E2183–E2191. doi: 10.1073/pnas.1209329109
Eichten, S. R., and Springer, N. M. (2015). Minimal evidence for consistent changes in maize DNA methylation patterns following environmental stress. Front. Plant Sci. 6:308. doi: 10.3389/fpls.2015.00308
Finn, T. E., Wang, L., Smolilo, D., Smith, N. A., White, R., Chaudhury, A., et al. (2011). Transgene expression and transgene-induced silencing in diploid and autotetraploid Arabidopsis. Genetics 187, 409–423. doi: 10.1534/genetics.110.124370
Gupta, S., Malviya, N., Kushwaha, H., Nasim, J., Bisht, N. C., Singh, V. K., et al. (2015). Insights into structural and functional diversity of Dof (DNA binding with one finger) transcription factor. Planta 241, 549–562. doi: 10.1007/s00425-014-2239-3
Islam, M. N., Nizam, S., and Verma, P. K. (2012). A highly efficient Agrobacterium mediated transformation system for chickpea wilt pathogen Fusarium oxysporum f. sp. ciceri using DsRed-Express to follow root colonisation. Microbiol. Res. 167, 332–338. doi: 10.1016/j.micres.2012.02.001
Le, T. N., Schumann, U., Smith, N. A., Tiwari, S., Au, P. C., Zhu, Q. H., et al. (2014). DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 15, 458. doi: 10.1186/s13059-014-0458-3
Lister, R., O’Malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536. doi: 10.1016/j.cell.2008.03.029
Lopez, A., Ramirez, V., Garcia-Andrade, J., Flors, V., and Vera, P. (2011). The RNA silencing enzyme RNA polymerase v is required for plant immunity. PLoS Genet. 7:e1002434. doi: 10.1371/journal.pgen.1002434
Luo, X. M., Lin, W. H., Zhu, S., Zhu, J. Y., Sun, Y., Fan, X. Y., et al. (2010). Integration of light- and brassinosteroid-signaling pathways by a GATA transcription factor in Arabidopsis. Dev. Cell 19, 872–883. doi: 10.1016/j.devcel.2010.10.023
Lyons, R., Stiller, J., Powell, J., Rusu, A., Manners, J. M., and Kazan, K. (2015). Fusarium oxysporum triggers tissue-specific transcriptional reprogramming in Arabidopsis thaliana. PLoS ONE 10:e0121902. doi: 10.1371/journal.pone.0121902
Makarevitch, I., Waters, A. J., West, P. T., Stitzer, M., Hirsch, C. N., Ross-Ibarra, J., et al. (2015). Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet. 11:e1004915. doi: 10.1371/journal.pgen.1004915
Matzke, M. A., and Mosher, R. A. (2014). RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408. doi: 10.1038/nrg3683
Michielse, C. B., and Rep, M. (2009). Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol. 10, 311–324. doi: 10.1111/j.1364-3703.2009.00538.x
Murashige, T., and Skoog, F. (1962). A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant. 15, 473–497.
Noguero, M., Atif, R. M., Ochatt, S., and Thompson, R. D. (2013). The role of the DNA-binding one zinc finger (DOF) transcription factor family in plants. Plant Sci. 209, 32–45. doi: 10.1016/j.plantsci.2013.03.016
Penterman, J., Zilberman, D., Huh, J. H., Ballinger, T., Henikoff, S., and Fischer, R. L. (2007). DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. U.S.A. 104, 6752–6757.
Pietro, A. D., Madrid, M. P., Caracuel, Z., Delgado-Jarana, J., and Roncero, M. I. (2003). Fusarium oxysporum: exploring the molecular arsenal of a vascular wilt fungus. Mol. Plant Pathol. 4, 315–325. doi: 10.1046/j.1364-3703.2003.00180.x
Steffens, N. O., Galuschka, C., Schindler, M., Bulow, L., and Hehl, R. (2004). AthaMap: an online resource for in silico transcription factor binding sites in the Arabidopsis thaliana genome. Nucleic Acids Res. 32, D368–D372.
Stroud, H., Greenberg, M. V., Feng, S., Bernatavichute, Y. V., and Jacobsen, S. E. (2013). Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152, 352–364. doi: 10.1016/j.cell.2012.10.054
Villain, P., Mache, R., and Zhou, D. X. (1996). The mechanism of GT element-mediated cell type-specific transcriptional control. J. Biol. Chem. 271, 32593–32598.
Vries, S., Hoge, H., and Bisseling, T. (1989). “Isolation of total and polysomal RNA from plant tissues,”,” in Plant Molecular Biology Manual, eds, eds S. B. Gelvin, R. A. Schilperoort, and D. P. S. Verma (Dordrecht): Springer:), 323–335.
Wang, M. B., Helliwell, C. A., Wu, L. M., Waterhouse, P. M., Peacock, W. J., and Dennis, E. S. (2008). Hairpin RNAs derived from RNA polymerase II and polymerase III promoter-directed transgenes are processed differently in plants. RNA 14, 903–913. doi: 10.1261/rna.760908
Widman, N., Feng, S., Jacobsen, S. E., and Pellegrini, M. (2014). Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 9, 236–242. doi: 10.4161/epi.26869
Yamamuro, C., Miki, D., Zheng, Z., Ma, J., Wang, J., Yang, Z., et al. (2014). Overproduction of stomatal lineage cells in Arabidopsis mutants defective in active DNA demethylation. Nat Commun. 5, 4062. doi: 10.1038/ncomms5062
Yanagisawa, S. (2004). Dof domain proteins: plant-specific transcription factors associated with diverse phenomena unique to plants. Plant Cell Physiol. 45, 386–391.
Yu, A., Lepere, G., Jay, F., Wang, J., Bapaume, L., Wang, Y., et al. (2013). Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 110, 2389–2394. doi: 10.1073/pnas.1211757110
Zemach, A., Kim, M. Y., Hsieh, P. H., Coleman-Derr, D., Eshed-Williams, L., Thao, K., et al. (2013). The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 153, 193–205. doi: 10.1016/j.cell.2013.02.033
Zhou, D. X. (1999). Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci. 4, 210–214.
Zhu, J. K. (2009). Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 43, 143–166. doi: 10.1146/annurev-genet-102108-134205
Keywords: DNA-demethylation, ROS1, Demeter-like, Fusarium, Arabidopsis thaliana
Citation: Schumann U, Lee J, Kazan K, Ayliffe M and Wang M-B (2017) DNA-Demethylase Regulated Genes Show Methylation-Independent Spatiotemporal Expression Patterns. Front. Plant Sci. 8:1449. doi: 10.3389/fpls.2017.01449
Received: 03 May 2017; Accepted: 04 August 2017;
Published: 28 August 2017.
Edited by:
Keqiang Wu, National Taiwan University, TaiwanCopyright © 2017 Schumann, Lee, Kazan, Ayliffe and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming-Bo Wang, bWluZy1iby53YW5nQGNzaXJvLmF1 Ulrike Schumann, dWxyaWtlLnNjaHVtYW5uQGFudS5lZHUuYXU=
†Present address: Ulrike Schumann, EMBL–Australia Collaborating Group, Department of Genome Sciences, The John Curtin School of Medical Research, The Australian National University, Canberra, ACT, Australia; Joanne Lee, Plant Health Australia, Deakin, ACT, Australia