 Panpan Yang
Panpan Yang Leifeng Xu
Leifeng Xu Hua Xu2
Hua Xu2- 1College of Landscape Architecture, Nanjing Forestry University, Nanjing, Jiangsu, China
- 2The Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, China
Aerial bulbils are an important propagative organ, playing an important role in population expansion. However, the detailed gene regulatory patterns and molecular mechanism underlying bulbil formation remain unclear. Triploid Lilium lancifolium, which develops many aerial bulbils on the leaf axils of middle-upper stem, is a useful species for investigating bulbil formation. To investigate the mechanism of bulbil formation in triploid L. lancifolium, we performed histological and transcriptomic analyses using samples of leaf axils located in the upper and lower stem of triploid L. lancifolium during bulbil formation. Histological results indicated that the bulbils of triploid L. lancifolium are derived from axillary meristems that initiate de novo from cells on the adaxial side of the petiole base. Transcriptomic analysis generated ~650 million high-quality reads and 11,871 differentially expressed genes (DEGs). Functional analysis showed that the DEGs were significantly enriched in starch and sucrose metabolism and plant hormone signal transduction. Starch synthesis and accumulation likely promoted the initiation of upper bulbils in triploid L. lancifolium. Hormone-associated pathways exhibited distinct patterns of change in each sample. Auxin likely promoted the initiation of bulbils and then inhibited further bulbil formation. High biosynthesis and low degradation of cytokinin might have led to bulbil formation in the upper leaf axil. The present study achieved a global transcriptomic analysis focused on gene expression changes and pathways' enrichment during upper bulbil formation in triploid L. lancifolium, laying a solid foundation for future molecular studies on bulbil formation.
Introduction
A bulbil, also known as a bulblet, consists of a small bud with a short stem (Bell and Bryan, 2008). The term also applies to aerial tubers that can be generated at the angle between a leaf and stem or in place of a flower (Ceplitis and Bengtsson, 2004). A bulbil can be released from the parent plant and grow independently into a new plant, making it an important reproductive organ (Callaghan et al., 1997). At higher elevations and in arid areas, some plants survive extreme conditions through bulbil reproduction (Steiner et al., 2012; Abraham-Juarez et al., 2015).
Bulbil formation is a rare natural phenomenon, but this character occurs in Lilium. The genus Lilium belongs to the family Liliaceae and is one of the most fascinating ornamental groups. Lilium species can be propagated via both sexual propagation through seeds and asexual propagation through underground bulb scales or aboveground bulbils (Bach and Sochacki, 2012). To maintain uniformity and genetic purity, most commercial cultivars are propagated by vegetative means (Maesato et al., 1991; Kumar et al., 2006). Scale-cutting propagation is an efficient method of asexual reproduction. However, scales must be manually separated from parent bulbs, and each scale can produce one or two bulblets, resulting in low propagation efficiency. As a special asexual reproductive organ, bulbils can be generated from each leaf axil (Figure S1B). In total, dozens or even hundreds of bulbils can be obtained per plant. Each mature bulbil eventually falls from the leaf axil to the ground and grows into a new plant as an individual (Suh and Roh, 2014). Consequently, bulbil propagation is more efficient and convenient than scale-cutting propagation. Elucidating the mechanisms that control bulbil formation can allow these mechanisms to be utilized to effectively improve the propagation coefficient of lilies.
In fact, not all species of the genus Lilium can form bulbils. Lilium includes ~115 species, but only four species (L. lancifolium, L. sulphureum, L. sargentiae, and L. bulbiferum) naturally form bulbils on the aboveground leaf axils of the stem (McRae, 1998; Liang and Tamura, 2000; Bach and Sochacki, 2012). Among bulbiferous species, L. lancifolium (Figure S1), the tiger lily, exhibits a number of special characters. This species is cultivated from cold zones to the subtropics as an ornamental plant, exhibiting strong adaptability (Liang and Tamura, 2000). L. lancifolium is widely cultivated in China for its edible bulbs and medicinal application (China Pharmacopoeia Committee, 2005; Yu et al., 2015), with a production value of approximately six billion Yuan per year. In addition, L. lancifolium is characterized by its complex ploidy. Most wild Lilium species are diploid (2n = 2x = 24). The only exception is L. lancifolium, which exhibits complex ploidy that includes both diploid and autotriploid (2n = 3x = 36), with triploids accounting for the majority of its distribution (Noda, 1986; Kim et al., 2006; Chung et al., 2015). The triploids are completely sterile, and L. lancifolium therefore cannot extend its population through seeds (Noda, 1986). However, triploid L. lancifolium produces large numbers of purple-black bulbils on the leaf axils along the stem, making bulbil propagation a significant asexual strategy for triploid forms of L. lancifolium (Suzuki and Yamagishi, 2016). An interesting phenomenon in this species is that bulbils are exclusively generated from the leaf axils in the middle-upper portion of the stem. Thus, it is interesting and meaningful to study the mechanisms of bulbil formation in triploid L. lancifolium.
Bulbil formation has been studied in several plant species, such as Dioscorea batatas, Allium sativum, Titanotrichum oldhamii, Lilium sulphureum, Pinellia ternata, and Agave tequilana. Histologic studies of bulbiferous species have shown that aerial bulbils are derived from axillary meristems (AMs). The process of axillary bulbil formation includes two steps: (1) meristematic cells in the leaf axil divide continuously to form bulbil primordia; and (2) the bulbil primordia grow and differentiate to form the bulbil structure (Zhang et al., 1994; Wang and Cronk, 2003; Li et al., 2012; Luo et al., 2014). It is widely recognized that plant hormones, particularly auxin (IAA), play important roles in the above processes. In Agave tequilana, removal of flower buds results in a reduction in IAA flux in pedicel vascular tissue, stimulating the development of new meristems and vegetative bulbils (Abraham-Juarez et al., 2015). Conversely, the application of exogenous IAA to cut pedicel tissue suppresses bulbil formation at the bracteoles, indicating an inhibitory effect of IAA on axillary bulbils. In addition, cytokinin (CK) stimulates the formation of axillary tubers, whereas gibberellin (GA) and strigolactone (SL) inhibit their formation in potato (Navarro et al., 2015).
At the molecular level, the Gesneriaceae FLO/LFY homolog (GFLO) is involved in the regulation of bulbil formation in Titanotrichum oldhamii, and its expression is down-regulated with bulbil development (Wang et al., 2004). In Agave tequilana, the expression of two class I KNOX genes (AtqKNOX1 and AtqKNOX2) occurs at bulbil initiation and increases as bulbils mature (Abraham-Juarez et al., 2010). The AtqMADS1, 2, 4, 6, and 7 genes all present the same pattern of reduced expression during bulbil development in Agave tequilana (Sandoval et al., 2012). Expression of the LONELY GUY (LOG1) gene confers the ability for axillary tomato meristems to undergo de novo formation of tuber-like organs (Eviatar-Ribak et al., 2013). Silencing of the SL biosynthetic StCCD8 gene in potato results in the formation of aerial tubers (Pasare et al., 2013). Although several genes related to bulbil formation have been isolated, the metabolic pathways and detailed regulatory patterns involved in the process of bulbil formation remain unclear.
RNA sequencing (RNA-Seq) is a recently developed, effective and powerful tool for analyzing the entire transcriptome composition and the gene expression patterns of various biological processes, particularly in non-model organisms for which genomic sequences are not available (Wang et al., 2009). To date, RNA-Seq technology has been successfully applied for the study of flower color (Xu et al., 2017), flowering (Liu et al., 2014), bulblet development (Li et al., 2014), growth characteristics (Zhu et al., 2016), and stress responses mechanisms (Wang et al., 2014a,b) of the lily. No detailed analysis has previously been performed on transcriptional changes during bulbil formation in Lilium.
To understand the process of bulbil formation in triploid L. lancifolium, we first performed a histological analysis of leaf axil samples from the upper and lower stem throughout the period of bulbil formation and evaluated developmental events. Using the Illumina HiSeq 4000 platform, we then performed de novo transcriptome sequencing of three types of leaf axil samples from the upper stem (axil, swelling axil and white dot structure axil) and one type of leaf axil sample from the lower stem (at the stage of swelling in the upper axil). This work provides comprehensive transcript information and gene expression data on bulbil initiation and formation, laying a solid foundation for future molecular studies on bulbil formation.
Materials and Methods
Plant Materials
The autotriploid species Lilium lancifolium was used as the material in this study. L. lancifolium bulbs of uniform size with 1-2-cm buds were grown in soil in a greenhouse at the Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, China, in early June of 2015. Samples of the upper and lower sectional stem nodes with ~0.4-mm petioles (Figure S2) were collected by hand-dissection with a double-edge blade every 5 days during the observation period, from the appearance of upper internodes (1 month after planting) to the appearance of white dot structures (early bulbils), producing four stages (S0, S1, S2, and S3) (Figures 1A–D), and these samples were used for histological analysis. The leaf axil zone samples (including leaf axil tissue, partial stem node and ~0.4-mm petiole) (Figure S2) from the upper stem at stage 1 (U1), stage 2 (U2), and stage 3 (U3) and from the lower stem at stage 2 (D2) were cut by hand with a disposable double-edge blade. All leaf axil samples were immediately frozen in liquid nitrogen and stored at −80 °C until further RNA-Seq and quantitative real-time PCR analyses. Three biological replicates for each sample were collected randomly from five individual plants, and each biological replicate contained five to seven leaf axils.
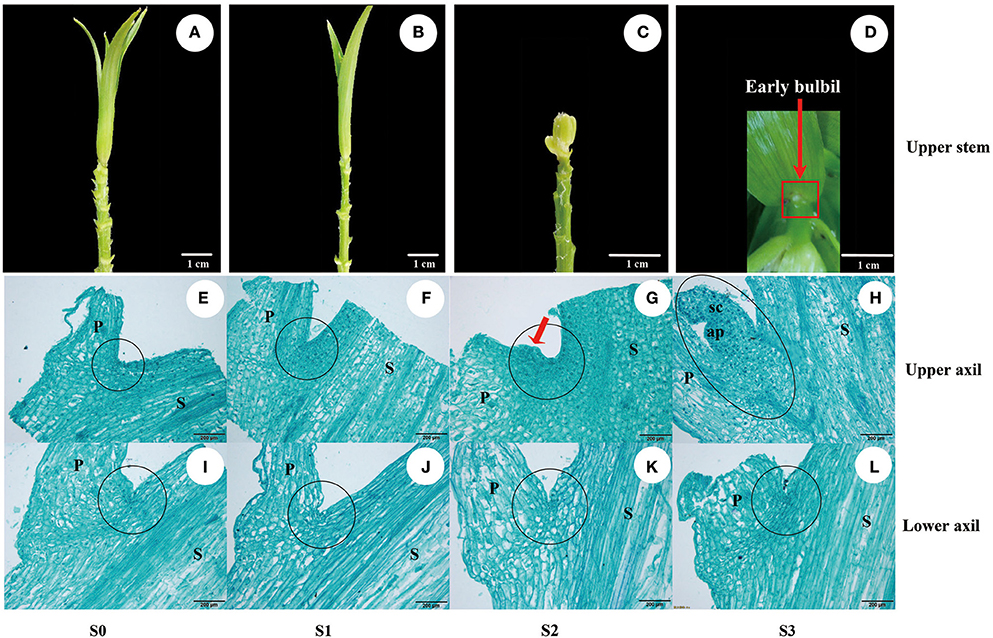
Figure 1. Bulbil formation in L. lancifolium. (A–D) Stages of bulbil formation in the upper leaf axil: (A,B) S0 and S1 stages exhibiting apparent upper internodes; (C) S2 stage exhibiting swelling; (D) S3 stage exhibiting a white dot structure (early bulbil). (E–L) Microscopy images of paraffin-embedded samples of leaf axils located on the upper (E–H) and lower stem (I–L) from four stages (S0–S3). Scale bar for samples images = 1 cm; Scale bar for microscopy images = 200 μm. P, Petiole; S, stem; sc, scale; ap, apical point; black circle, leaf axil.
Histological Analysis
Samples of the upper and lower sectional stem nodes with ~0.4-mm petioles (Figure S2) from each developmental stage were fixed in FAA fixation buffer (formaldehyde: glacial acetic acid: 70% ethanol, 1:1:18) for 48 h at 4°C. The fixed samples were dehydrated in a graded series of ethanol (70%, 85%, 95% and 100%), followed by a xylene/ethanol series (xylene: ethanol 1:2, 1:1, and 2:1 and 100% xylene). Xylene was gradually replaced with paraffin (melting point of 58°C–60°C) at 60°C for 3 days. Sections (10 μm thick) were obtained using a rotary microtome and were double-stained with 5% (w/v) Safranine T and 0.5% (w/v) Fast Green FCF.
RNA Extraction
Total RNA was extracted using the RNAprep Pure Plant Kit (Polysaccharide- & Polyphenolic-rich) (TIANGEN, Beijing, China) according to the manufacturer's instructions, and DNA contamination was removed with RNase-free DNase I. RNA degradation and contamination were monitored using 1% agarose gels. RNA purity and concentration were measured using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) and Qubit® RNA Assay Kit with a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA), respectively. RNA integrity (RIN) was assessed using the RNA Nano 6000 Assay Kit with the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA samples with RINs ≥ 8.0, A260/280 ranging from 1.8 to 2.1, and 25S:18S ranging from 1.7 to 2.0 were used for library preparation. Three biological replicates were used for RNA extraction and further RNA-Seq.
cDNA Library Construction and Sequencing
Twelve cDNA libraries (three replicates for each sample) were constructed using the NEBNext® Ultra™RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer's protocol. Briefly, mRNA was purified from 1.5 μg of total RNA using oligo (dT) magnetic beads, and then broken into short fragments of 300–500 bp by adding fragmentation buffer. First-strand cDNA was synthesized using random hexamer primer. Second-strand cDNA synthesis was subsequently performed using RNase H and DNA polymerase I. After adenylation of the 3′ ends of cDNA fragments, NEBNext adapter oligonucleotides were ligated to prepare for hybridization, and then the cDNA fragments were purified using AMPure XP system (Beckman Coulter, Beverly, USA) to select the fragments of preferentially 150–200 bp in length. Then 3 μL of USER Enzyme (NEB, USA) was used with size-selected and adaptor-ligated cDNA fragments. Then PCR was performed with Phusion High-Fidelity DNA polymerase, universal PCR primers and Index(X) Primer. PCR products were purified with the AMPure XP system, and library quality was assessed using the Agilent Bioanalyzer 2100 system. Finally, all cDNA libraries were sequenced on the Illumina HiSeq 4000 platform using the paired-end technology by Novogene Co. (Beijing, China), and 150-bp paired-end reads were generated.
De novo Transcriptome Assembly
Clean reads were obtained from the sequencing reads by removing reads containing adapters, reads with more than 10% unknown nucleotides, and low-quality reads. The Q20, Q30, GC content and sequence duplication levels were calculated from the clean data. Then, de novo assembly based on the clean data was performed using the Trinity program (Grabherr et al., 2011). Trinity includes three independent software modules: Inchworm, Chrysalis and Butterfly. Short reads with overlapping sequences are first assembled by Inchworm to form longer transcripts, known as contigs. Chrysalis then clusters the contigs into clusters and constructs complete de Bruijn graphs for each cluster. Finally, Butterfly reports the full-length transcripts. The final sequences of Trinity assembly were defined as unigenes.
Functional Annotation and Classification of Unigenes
All unigene sequences were aligned to Nt nucleotide databases using Blastn and were aligned by Blastx to protein databases such as the NCBI non-redundant (Nr) database (http://www.ncbi.nlm.nih.gov), the Swiss-Prot protein database (http://www.ebi.ac.uk/uniprot), the Pfam database (http://pfam.sanger.ac.uk), the KEGG database (http://www.genome.jp/kegg/) (Kanehisa and Goto, 2000), and the KOG/COG database (http://www.ncbi.nlm.nih.gov/COG). Each unigene was functionally annotated based on the protein sharing the highest sequence similarity with the given unigene. Gene Ontology (GO, http://www.geneontology.org/) annotation was performed using Blast2GO software (version 2.5) (Gotz et al., 2008) based on the results of Nr and Pfam annotation. Web Gene Ontology Annotation Plot (WEGO, http://wego.genomics.org.cn/cgi-bin/wego/index.pl) was then employed to produce GO functional classifications (Ye et al., 2006).
Analysis of Differential Gene Expression
The FPKM (fragments per kilobase per million base pairs) method was used to calculate the expression of unigenes. Then, differential expression analysis of two groups was performed using the DESeq R package (1.10.1). The resulting p-value was adjusted using the Benjamini and Hochberg's approach (Benjamini and Hochberg, 1995) for controlling the false discovery rate (FDR). The DEGs between two samples were determined based on an adjusted p-value (q-value) < 0.05 and |log2Foldchange| ≥ 1. GO enrichment analysis (corrected p < 0.05) of the DEGs was implemented with the GOseq R package-based Wallenius noncentral hypergeometric distribution (Young et al., 2010). KOBAS software was used to test the statistical enrichment of DEGs in KEGG pathways (Mao et al., 2005). KEGG pathways with a threshold q < 0.05 were considered significantly enriched in the DEGs.
Quantitative Real-Time PCR Validation and Expression Analysis
Sixteen DEGs were selected for RNA-Seq data validation and expression analysis using quantitative real-time PCR (qRT-PCR). For cDNA synthesis, 500 ng of total RNA (without DNA contamination) was reverse-transcribed using the TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TRAN, Beijing, China). Specific primers for qRT-PCR were designed with Primer 5.0 software, and the detailed information is listed in Table S1. All reactions were performed in 96-well plates using the CFX96 Real-Time System (Bio-Rad, USA). Each PCR reaction (20 μL) contained 2 μL of cDNA (1:3 dilution), 0.5 μL of each forward and reverse primer (10 μM), 10 μL of 2 × SYBR® Premix Ex Taq™ (Tli RNaseH Plus) (Takara) and 7 μL of ddH2O. Amplification reactions were performed under the following conditions: 95°C for 3 min, followed by 40 cycles of 95°C for 20 s, 60°C for 10 s, and 72°C for 20 s. A melting curve was generated for each PCR reaction to determine the amplification specificity and the presence of reaction contaminations. The melting curve was obtained by heating the amplification products from 60°C to 95°C in 5-s intervals. The primer efficiency was analyzed with CFX Manager™Software v3.1 (Bio-Rad). All qRT-PCR experiments were repeated with three biological and three technical replicates. The lilyActin (Gene Bank Accession Number: JX826390) gene (forward primer: 5′-GCATCACACCTTCTACAACG-3′; reverse primer: 5′-GAAGAGCATAACCCTCATAGA-3′) was used as the internal control for normalization, and the relative expression levels of the target genes were calculated using the 2−ΔΔCt method against the internal control (Livak and Schmittgen, 2001).
Soluble Sugar and Starch Content Measurement
Soluble sugar and starch were measured using an anthrone colorimetric method. Briefly, samples of leaf axils from the upper and lower stem at different stages were ground into powder using liquid nitrogen. Then, 0.2 g of powder was weighed in a 10-mL centrifuge tube, to which 6 mL of distilled water was added. The mixed homogenate was then incubated in boiling water for 30 min. Then, the extracted solution was centrifuged at 5,000 rpm for 10 min. The supernatant was collected, and the extraction was repeated three times (5,000 rpm for 10 min each). All supernatants were collected into 25-mL volumetric flasks for the determination of soluble sugar. The residual was re-dissolved in 20 mL of distilled water and incubated in boiling water for 15 min. Then, 2 mL of cold 9.2 mol/L perchloric acid was added, and the mixture was again incubated in boiling water for 15 min. The solution was finally filtered in a 50-mL volumetric flask, and the filtrate was used for the determination of starch. The soluble sugar and starch contents were measured by their absorption values at 630 nm and 485 nm, respectively, and calibration curves were constructed with standard solutions of sucrose and soluble starch, respectively. Three biological replicates for each sample and three technical replicates for each biological replicate were performed.
Determination of IAA Concentration
Samples of leaf axils from the upper and lower stem at different stages were ground into powder using liquid nitrogen. Then, 0.2 g of powder was weighed in a 10-mL centrifuge tube and extracted overnight at 4°C in cold 80% (v/v) methanol containing 1 mM butylated hydroxytoluene. The extracts were collected after centrifugation at 10,000 × g at 4°C for 20 min and were then passed through a C18 Sep-Pak cartridge (Waters). The efflux was dried with nitrogen (N2), and the IAA content was determined by means of enzyme-linked immunosorbent assay (ELISA) (He, 1993; Yang et al., 2001). Three biological replicates for each sample and three technical replicates for each biological replicate were performed.
Statistical Analysis
The data for soluble sugar, starch and endogenous IAA contents and the relative expression analyses of genes were presented as the means with standard errors from three biological replicates. All statistical analyses were performed using SAS 9.1 (SAS Institute, Cary, NC, USA), and statistical significance was defined at p ≤ 0.05. Non-overlapping letters (a-d) indicated significant differences between the comparisons, based on the ANOVA analysis and Duncan's multiple range test.
Results
Morphological and Histological Description of Bulbil Formation in Triploid L. lancifolium
To describe the process of bulbil formation in triploid L. lancifolium, the upper and lower sectional stem nodes with ~0.4-mm petioles (Figure S2) were collected at each developmental stage (S0–S3). The process of bulbil formation was observed on leaf axils of the upper stem (Figures 1A–D), and a defined early bulbil structure (white dot structure) was discovered in the upper axil at stage S3 (Figure 1D: red arrow). Based on morphological and histological observations, no morphological changes in the upper leaf axil were detectable at stage S0 or S1 (Figures 1E,F). At stage S2, the adaxial side of the petiole base in the upper stem showed obvious bulging (Figure 1G: red arrow). Cells in the swelling region exhibited large nuclei and dense protoplasm. They were small and serried, whereas the relative size of the cells was larger outside of this region (Figure 1G). At stage S3, actively dividing cells differentiated to form the bulbil structure (Figure 1H) on the upper leaf axil. In contrast, there were no obvious structural changes in the leaf axils of the lower stem (Figures 1I–L). Visual analyses of sections suggested that the bulbils originated from several layers of parenchyma cells in the leaf axil at stage S2. In addition, upper axil swelling was found to be a key stage for initiating bulbil formation.
Transcriptome Sequencing and Assembly
To obtain a comprehensive overview of the triploid L. lancifolium transcriptome during bulbil initiation and formation, leaf axils on the upper stem at stages S1, S2 and S3 (U1, U2, and U3) and leaf axils showing no bulbil formation on the lower stem at stage S2 (D2) were obtained for an RNA-Seq analysis with three biological replicates. Sequencing of cDNA libraries from the twelve samples resulted in 46–70 million raw paired-end reads with greater than 6.5 Gb per sample (Table 1). All raw reads were deposited in the NCBI Sequence Reads Archive (SRA) under the accession number SRP103184. Given the lack of reference genome sequences for lilies, a de novo assembly strategy was used to construct transcripts. After ambiguous nucleotides, low-quality sequences and contaminated sequences were removed, ~45–67 million clean reads were obtained for assembly from each library. For all the sequence data, the Q20 and Q30 percentages were greater than 94 and 86%, respectively (Table 1). A total of 598,060,858 clean reads obtained from 649,714,466 raw reads (92.05%) were used in the assembly, and 389,636 transcripts (contigs) were generated, with a mean length of 674 bp and an N50 of 1105 bp (Tables S2, S3, Figure S3). All contigs were assembled into 293,858 non-redundant unigenes with an average length of 549 bp and an N50 of 752 bp (Tables S2, S3, Figure S3). All unigenes were longer than 200 bp. In total, 211,581 of the unigenes (72%) were 200–500 bp in length, and 11,337 (3.90%) were longer than 2 kb (Tables S3).
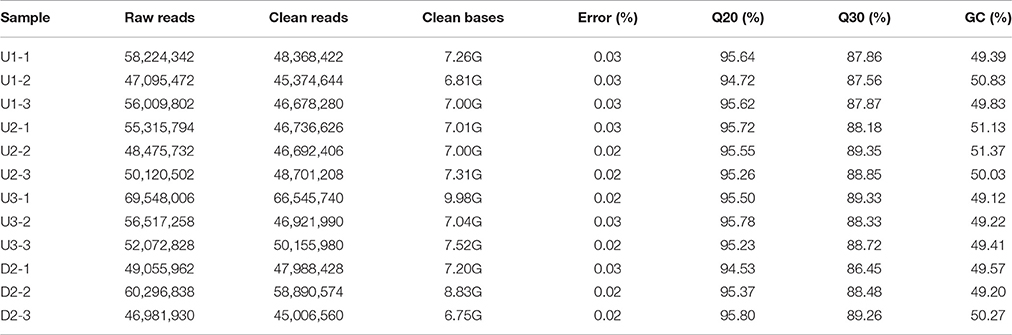
Table 1. Summary of RNA-Seq data from leaf axils in the upper (U) and lower (D) stems of L. lancifolium.
To assess the quality of the assembled transcripts, the clean reads were mapped back to the assembly. The results showed that 75% of the clean reads could be mapped back to the assembled transcripts, and more than 68.4% of the paired reads of each library could be mapped (Table S4). These data revealed that the quality of transcriptome sequencing and assembly was sufficiently high for further analysis.
Functional Annotation and Classification of Unigenes
The 293,858 unigenes were annotated via alignment with seven public databases (Table 2). The results showed that 79,950 (27.20%), 53,597 (18.23%), 63,967 (21.76%), 64,874 (22.07%), 67,672 (23.02%), and 36,214 (12.32%) unigenes were matched in the Nr, Nt, Swiss-Prot, Pfam, Gene Ontology (GO) and Clusters of Orthologous Groups of proteins (KOG/COG) databases, respectively. In total, 34,491 unigenes (11.73%) were annotated in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and 112,185 unigenes (38.17%) were successfully annotated in at least one of the seven databases, among which 15,569 unigenes (5.29%) were simultaneously annotated in all databases.
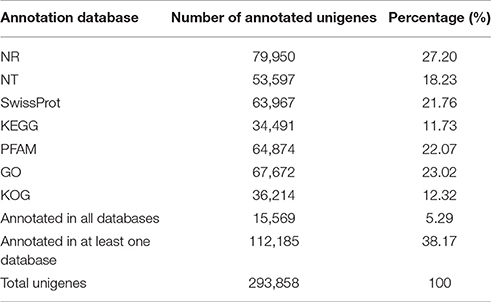
Table 2. Annotation of assembled unigenes against seven public databases.
GO assignment was performed to classify the functions of the predicted unigenes. The 67,672 unigenes annotated in the GO database were categorized into 56 functional groups and divided into three main ontologies: biological process (BP), cellular component (CC), and molecular function (MF) (Figure S4). Regarding the MF category, genes involved in “binding” (51.82%) and “catalytic activity” (42.87%) accounted for the largest proportion. Within the CC category, the “cell” (31.14%), “cell part” (31.13%), “organelle” (20.33%) and “macromolecular complex” (19.73%) categories were highly represented. Moreover, “cellular processes” (55.00%) and “metabolic processes” (52.51%) were dominant in the BP category (Table S5, Figure S4).
To further evaluate the functions of all assembled unigenes, we performed a search against the KOG/COG database. Based on sequence homology, 36,214 unigenes were assigned to a KOG functional classification, divided into 26 specific categories. Among these categories, the “general functional prediction only” (16.22%) cluster represented the largest category, followed by “posttranslational modification, protein turnover, chaperones” (13.26%), “translation, ribosomal structure and biogenesis” (9.81%), “signal transduction mechanisms” (9.54%), “energy production and conversion” (6.01%) and “intracellular trafficking, secretion, and vesicular transport” (5.98%). The “cell motility” (0.10%) and “unnamed protein” (0.01%) clusters were the smallest categories (Table S6).
KEGG pathways were also searched to allow a biological interpretation of the functions of the assembled unigenes. According to the KEGG results, 34,491 unigenes were mapped to predicted metabolic pathways (Figure S5, Table S7), and the majority of unigenes were classified into pathways related to translation (11.32%), carbohydrate metabolism (8.36%), folding, sorting and degradation (8.12%). All the pathways were divided into five categories, with “metabolism” (42.90%) representing the largest category (Figure S5D class). Within “metabolism,” carbohydrate metabolism was the most represented category; within “carbohydrate metabolism,” starch and sucrose metabolism was most dominant (Table S7).
Analysis of Differentially Expressed Genes (DEGs)
Verification of RNA-Seq data via qRT-PCR
To validate the differential expression by RNA-Seq, sixteen unigenes were selected from the identified DEGs for qRT-PCR analysis, including six randomly selected genes (WOX4_c109232_g2, AS1_c104426_g1, LOX1.1_c128051_g2, FTSZ1_c111017_g1, HOX21_c106431_g1, and TPS1_c100758_g1), six genes related to starch biosynthesis (SUS2_c111432_g1, UGP2_c123358_g1, AGPS1_c121621_g2, SSS3_ c122034_g2, WAXY_c123636_g2, and SBE1_ c97206_g1) and four genes involved in auxin signaling (YUC_c111323_g1, YUC10_c128166_g2, IAA17_c115342_g1, and PIN1B_c108190_g1). The result showed that most of the expression patterns revealed by qRT-PCR were in good agreement with the RNA-Seq results, indicating the reliability of the RNA-Seq data (Figures 2, 4C, 6D).
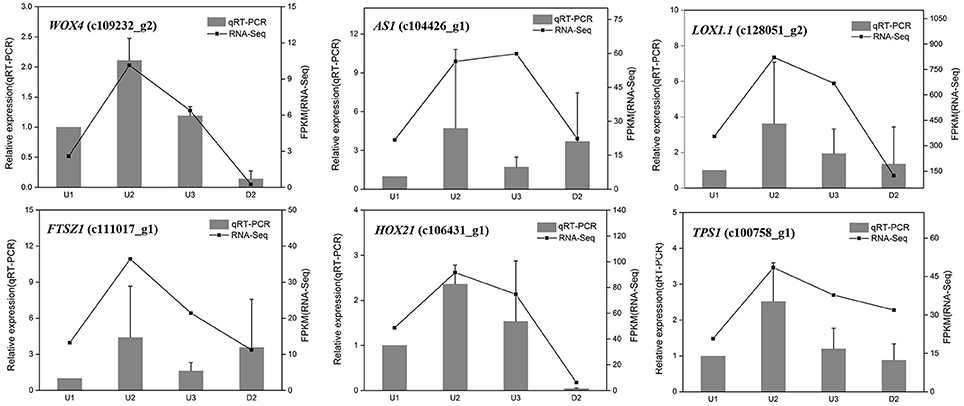
Figure 2. qRT-PCR validation of differential gene expression in U1, U2, U3, and D2. The left y-axis indicates relative gene expression levels. The right y-axis indicates gene expression levels calculated based on mean FPKM value of each sample.
Global Analysis of DEGs
We sought to identify genes exhibiting significant changes in expression levels during bulbil formation in triploid L. lancifolium. In total, 11,871 DEGs were identified by comparing the four libraries in pairs (Table S8). In total, 4,061, 4,820, and 5,045 of the DEGs were differentially expressed between U2 vs. U1, U3 vs. U1, and U2 vs. D2, respectively (Figure 3A). The smallest difference existed between U3 and U2, with only 257 up-regulated and 516 down-regulated unigenes identified (Figure 3B). 2,176, 157, 3,302, and 3,857 DEGs were specific for U2 vs. U1, U3 vs. U2, U3 vs. U1 and U2 vs. D2, respectively (Figure 3A). Moreover, the principal component analysis (PCA) of the RNA-Seq data was performed based on the FRPM values of three replicates for each sample, and U2 and U3 groups were clustered in the same quadrant (Figure S6). The results suggested that U3 is developmentally close to U2, and significant differential gene expression was observed in the initial stage (U2) of bulbil formation. In addition, more genes were up-regulated than down-regulated in U2 vs. U1, U3 vs. U1 and U2 vs. D2 (Figure 3B), indicating that the initiation process was likely caused by the expression of up-regulated genes.
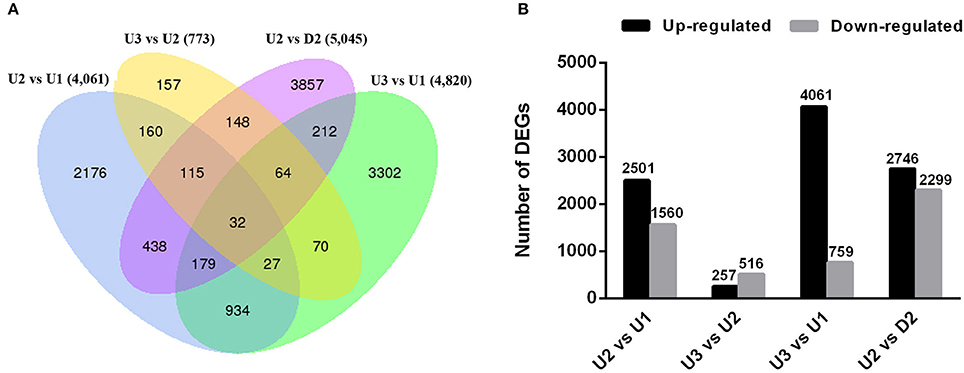
Figure 3. Overview of numbers of DEGs in pairwise comparisons. (A) Venn diagram of DEGs among the four comparisons performed (U2 vs. U1, U3 vs. U2, U2 vs. D2 and U3 vs. U1). (B) Histogram showing the number of DEGs that were up- or down-regulated between different libraries. The black columns indicate up-regulated DEGs, and the gray columns represent down-regulated DEGs in the four pairwise comparisons.
GO enrichment analysis for the DEGs was performed to classify the functions of the DEGs in pairwise of upper leaf axils at different stages during bulbil formation (U1, U2, and U3) and in pairwise of U2 and D2. The DEGs between U2 and U1, U3 and U2, U3 and U1, and U2 and D2 were assigned to 34, 10, 104, and 108 GO terms based on “biological process (BP),” “cellular component (CC),” and “molecular function” (Table S9). In all comparisons, DEGs were mainly enriched for “biological process (BP)” and were significantly enriched (corrected p < 0.05) for “metabolic process” in “biological process (BP).” To further explore the detailed metabolic pathways, all DEGs were mapped to terms in the KEGG database, and a KEGG pathway enrichment analysis was subsequently performed. In total, 4,061 DEGs (U2 vs. U1) were assigned to 115 pathways; 773 DEGs (U3 vs. U2) to 71 pathways; 4,820 DEGs (U3 vs. U1) to 113 pathways; and 5,045 DEGs (U2 vs. D2) to 110 pathways (Table S10). Pathway enrichment analysis revealed that the initiation and formation of bulbils involved the integration of several pathways. A q < 0.05 was used as the thresholds to determine significant enrichment. The annotated changes between U2 and U1 were mainly involved in starch and sucrose metabolism, amino sugar and nucleotide sugar metabolism, and phenylpropanoid biosynthesis (Figure S7A). The annotated changes between U3 and U2 were mainly involved in photosynthesis, protein processing in the endoplasmic reticulum, amino sugar and nucleotide sugar metabolism (Figure S7B). The annotated changes between U2 and D2 were mainly involved in phenylpropanoid biosynthesis, starch and sucrose metabolism, and plant hormone signal transduction (Figure S7C).
DEGs Involved in Starch and Sucrose Metabolism
In total, 43, 8, 26, and 56 DEGs between U2 vs. U1, U3 vs. U2, U3 vs. U1, and U2 vs. D2, respectively, were enriched in starch and sucrose metabolism pathways (Ko00500) (Table S10). Most of the DEGs were up-regulated during the process of bulbil initiation and formation in the upper leaf axil in L. lancifolium. Among starch and sucrose metabolism genes, genes encoding starch synthases, such as sucrose synthase (SUS) (c129100_g1, c128869_g1, c111432_g1, c126221_g1, c111800_g1 and c126221_g2), HK (c115235_g3), PGM (c115630_g1), UGP (c123358_g1), AGPS (c121621_g2), SSS (c4952_g1 and c122034_g2), GBSS (c123636_g2), and SBE (c97206_g1 and c108260_g1), exhibited high expression levels during upper bulbil initiation (up-regulated in U2 vs. U1), which declined slightly in U3 (Figure 4B, Table S11). Additionally, the above DEGs were significantly up-regulated in U2 compared with D2. These results suggested that starch biosynthesis was likely predominant in bulbil initiation. Validation of SUS2 (c111432_g1), UGP2 (c123358_g1), AGPS1 (c121621_g2), SSS3 (c122034_g2), WAXY (c123636_g2), and SBE1 (c97206_g1) via qRT-PCR further confirmed our observations (Figure 4C). In addition, we analyzed changes in soluble sugar content and starch content during bulbil formation in triploid L. lancifolium using an anthrone colorimetric method. During the process of bulbil formation in the upper portion of stem, the starch content increased slowly (from 0.94 to 1.04%) from stage S0 to S2, followed by a rapid increase to 1.65% at S3 (Figure 5B). In contrast, the soluble sugar content dropped from S0 to S2 and then increased in S3 (Figure 5A). During this process, the starch content of lower axils was less than that of the upper axils and exhibited minimal change (Figure 5B).
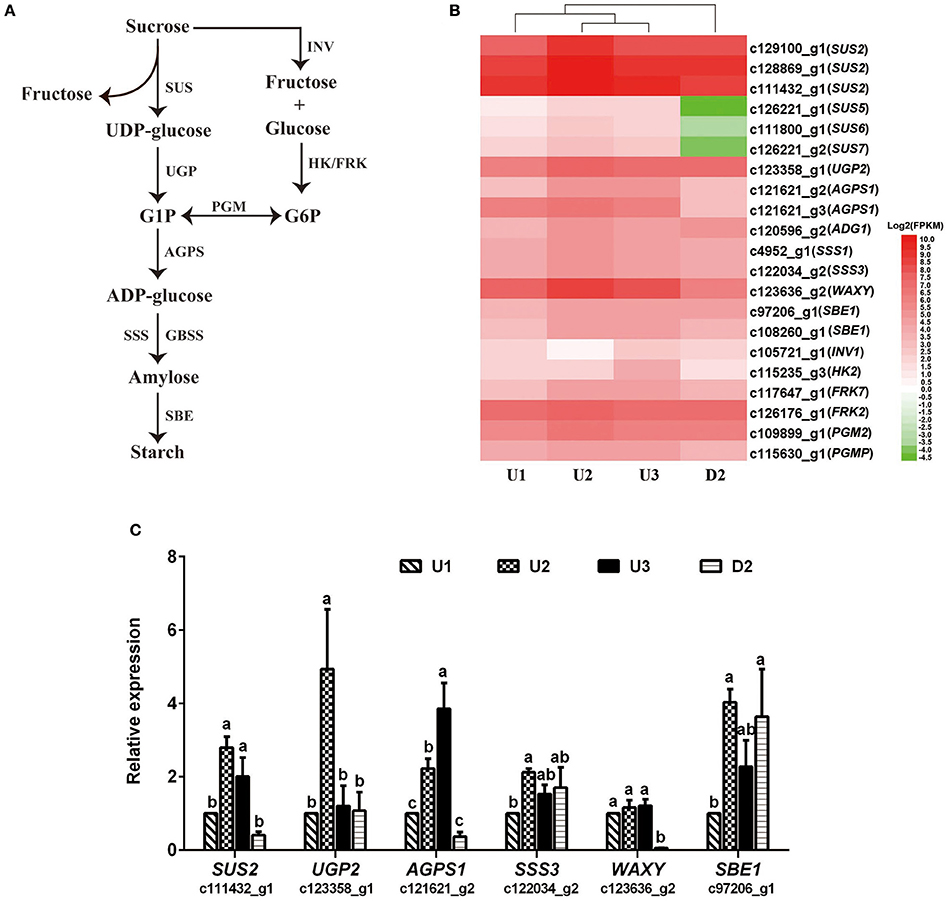
Figure 4. Expression patterns of DEGs assigned to starch biosynthesis and qRT-PCR analysis. (A) The starch biosynthesis pathway shown in this figure was modified from Yang et al. (2015). (B) Heatmap of the expressed genes. The FPKM values of the unigenes were log2 transformed, and a heatmap was generated using HemI 1.0. Red and green indicate up- and down-regulated transcripts, respectively. SUS, Sucrose synthase; UGP, UTP-glucose-1-phosphate uridylyltransferase; AGPS, glucose-1-phosphate adenylyltransferase; SSS, soluble starch synthase; GBSS, granule-bound starch synthase; SBE, starch branching enzyme; INV, beta-fructofuranosidase; HK, hexokinase; FRK, fructokinase; PGM, phosphoglucomutase. (C) qRT-PCR analysis of the expression profiles of six genes involved in starch biosynthesis. The x-axis represents different samples, and the y-axis shows relative gene expression levels. Columns and error bars indicate means and standard deviations, respectively. Non-overlapping letters (a-c) indicate significant differences (p < 0.05) between the comparisons of different samples.
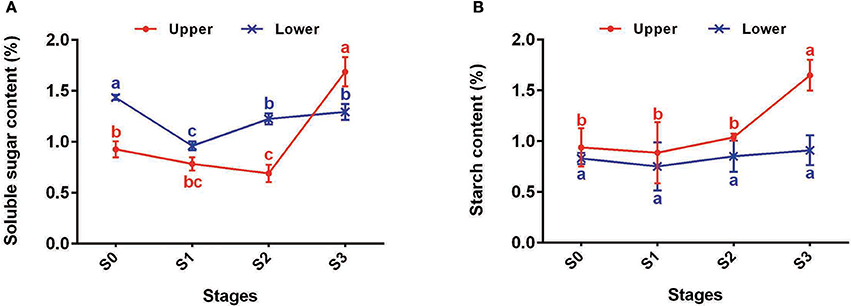
Figure 5. Changes of starch and sucrose contents of upper (red) and lower (blue) axils at different stages during bulbil formation. (A) Soluble sugar contents of upper and lower axils at different stages during bulbil formation. (B) Starch contents of upper and lower axils at different stages during bulbil formation. The x-axis represents different stages, and the y-axis shows content. Lines and error bars indicate means and standard deviations, respectively. Non-overlapping letters indicate significant differences (p < 0.05) for the different stages.
DEGs Involved in Plant Hormone Signal Transduction
The KEGG enrichment analysis of our transcriptome revealed that a fairly large number of DEGs were assigned to “plant hormone signal transduction” (ko04075) pathways, including auxin (IAA), cytokinin (CK), ethylene, gibberellin (GA), abscisic acid (ABA), brassinosteroid (BR), jasmonic acid (JA) and strigolactone (SL) signaling (Figure 6B, Table S12). IAA and CK signaling were enriched in all pairwise comparisons of the four libraries, with the number of genes related to IAA accounting for the highest percentage (Figure 6A).
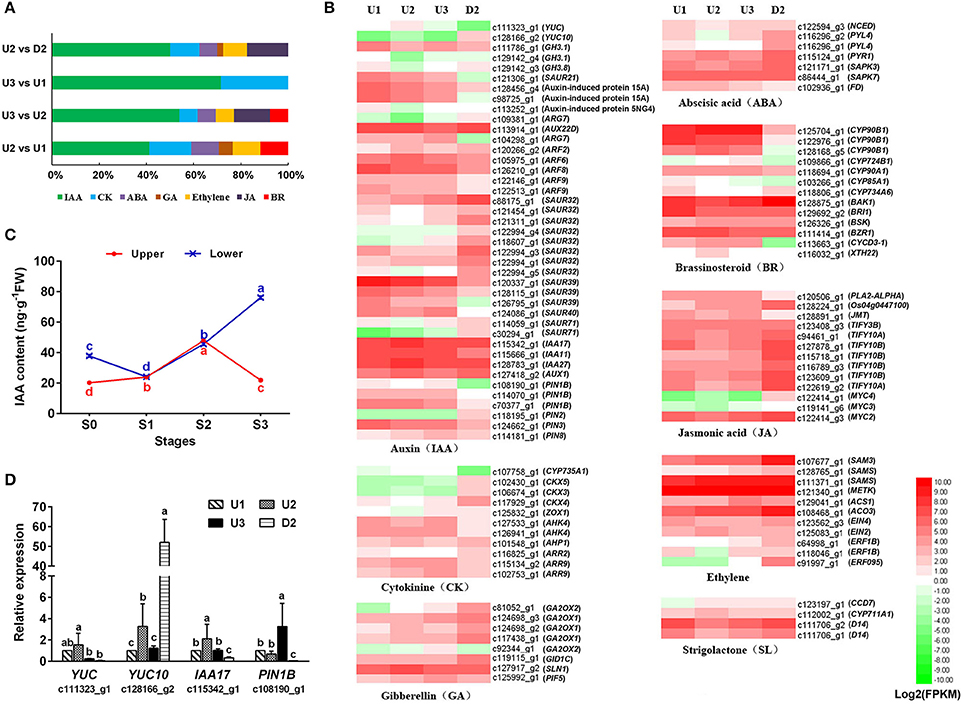
Figure 6. Regulation patterns of plant hormones during the process of bulbil formation. (A) Distribution of DEGs related to plant hormone signal transduction compared pairwise among the four libraries. (B) Heatmap of the unigenes assigned to hormone synthesis and signal transduction pathways in the four transcriptomes: U1, U2, U3, and D2. The FPKM values of the unigenes were log2 transformed, and a heatmap was generated using HemI 1.0. Red and green indicate up- and down-regulated transcripts, respectively. (C) IAA contents of the upper (red) and lower (blue) axils at different stages during bulbil formation. The representation of the x-axis, y-axis, significance tests and error bars are as described in Figure 5. (D) qRT-PCR analysis of the expression profiles of four genes related to auxin signailing. The representation of the x-axis, y-axis, significance tests and error bars are as described in Figure 4C. Non-overlapping letters (a–d) in (C,D) indicate significant differences (p < 0.05) for between the comparisons of different samples.
During the process of bulbil formation in upper leaf axils of triploid L. lancifolium, the IAA content increased from S0 to S2, peaked at S2 (47.95 ng/g FW), and then decreased significantly in S3 (21.95 ng/g FW). In contrast, the IAA content of the lower leaf axils increased continuously from S1 to S3 and was 3.5-fold greater than that in the upper part at S3 (Figure 6C). Forty-one DEGs related to IAA biosynthesis and signal transduction were identified (Table S12). In the IAA biosynthesis pathway, two YUCCA genes (c111323_g1 and c128166_g2) were up-regulated in U2 and then down-regulated in U3 during bulbil formation. In the IAA signaling pathway, the AUX1 (c127418_g2) gene encoding the auxin transporter protein 1 and genes encoding the auxin-responsive protein (IAA17_c115342_g1, IAA11_c115666_g1, and IAA27_c128783_g1) also exhibited increased expression in U2 and reduced expression in U3. GH3 genes (GH3.1_c111786_g1, GH3.1_c129142_g4, and GH3.8_c129142_g3) encoding indole-3-acetic acid-amido synthetase, which conjugates IAA to amino acids and then reduces IAA concentrations, were down-regulated in U2 vs. U1 and up-regulated in U3 vs. U2. In addition, an auxin efflux carrier component (PIN1B_ c108190_g1) exhibited the same regulatory pattern as GH3 genes (Figure 6B). Validation of YUC (c111323_g1), YUC10 (c128166_g2), IAA17 (c115342_g1), and PIN1B (c108190_g1) via qRT-PCR further confirmed our observations (Figure 6D). These results indicated that IAA accumulation in a short time likely promoted bulbil initiation and subsequently inhibited further axillary bulbil formation.
CYP735A1 (c107758_g1), which catalyzes the biosynthesis of trans-zeatin, was up-regulated during upper bulbil formation and was obviously up-regulated in U2 vs. D2 (Figure 6B, Table S12). Genes (CKX5_c102430_g1, CKX3_c106674_g1, and CKX4_c117929_g1) related to the degradation of CK exhibited increased expression in D2 compared with U1, U2, and U3. In the CK signal transduction pathway, the expression of AHK4 (c127533_g1 and c126941_g1) and AHP1 (c101548_g1) was increased in U1, U2, and U3 compared with D2, and the expression level of AHP was the highest in U2. These results indicated that the CK signaling pathway likely played a role in promoting bulbil formation in L. lancifolium. In addition, BR signaling exhibited a promoting function. CYCD3-1 (c113663_g1), which functions downstream of BR signaling, was up-regulated during bulbil initiation and formation but showed low expression in D2 (Figure 6B, Table S12).
In the GA biosynthesis pathway, four GA2OX (c81052_g1, c124698_g3, c124698_g2, and c117438_g1) genes were slightly up-regulated during bulbil formation, but their expression levels in U1, U2, and U3 were reduced compared with D2. In the GA signaling pathway, GID1C (c119115_g1) was down-regulated in U2. GID1C levels were increased in D2 compared with U1, U2 and U3. The SLN1 gene (c127917_g2) encoding the DELLA protein, a repressor of the GA signaling pathway, exhibited a regulatory pattern opposite that of GID1C (Table S12). The expression of D14 (c111706_g2 and c111706_g1) in the SL signaling pathway was the lowest in U2; furthermore, SL potentially inhibits bulbil initiation (Figure 6B, Table S12).
Discussion
To understand the process of bulbil formation in triploid L. lancifolium and to obtain complete transcriptome information during bulbil formation, we first analyzed the anatomical structures of the upper leaf axils, which generate bulbils, and the lower leaf axils, which cannot form bulbils, throughout the period of bulbil formation. Bulbil formation in triploid L. lancifolium is a notable illustration of axillary organogenesis, by which axillary cells undergo dedifferentiation to form AM cells on the adaxial side of the petiole base and then differentiate into a bulbil structure (Figures 1E–H) (Grbic and Bleecker, 2000; Wang and Cronk, 2003; Li et al., 2012). During this process, AM of triploid L. lancifolium initiated de novo from cells in the leaf axil after internodes of stem elongation and leaf unfolding; the same process occurs in Arabidopsis (Long and Barton, 2000; Leyser, 2003). In addition, upper axil swelling was observed on the adaxial side of the petiole base at stage S2, which provided a reference for the transcriptome sampling.
Based on the results of histological analysis, three types of samples from leaf axils located on the upper stem (U1, axil; U2, swelling axil; and U3, white dot structure axil) and one from leaf axils located on the lower stem at the stage of swelling in the upper axil (D2) were obtained for RNA-Seq analysis. Unlike other studies on bulbil formation, our analysis was focused not only on bulbil initiation in the leaf axil but also on the reason that bulbils exclusively form on the upper part of the stem in L. lancifolium. Approximately 46–70 million raw paired-end reads, with greater than 6.5 Gb per sample, were obtained (Table 1). The results of DEG analysis revealed that bulbil initiation associated with swelling on the adaxial side of the petiole base was particularly important in the process of bulbil formation (Figure 3, Figure S6). Starch and sucrose metabolism and plant hormone signal transduction may play important roles in bulbil formation in triploid L. lancifolium (Figures 4, 6).
Starch Synthesis Plays a Crucial Role in Bulbil Initiation in L. lancifolium
Based on the KEGG enrichment analysis, numerous DEGs were found to be enriched in starch and sucrose metabolism pathways, and most of these DEGs were involved in the process of starch synthesis (Table S11). The synthesis of starch in triploid L. lancifolium was positively related to the initiation of bulbils, according to the results of RNA-Seq analysis, qRT-PCR verification and starch and soluble sugar determination (Figures 4, 5); this finding is consistent with the swelling of storage organs in potato and Tulipa edulis (Geigenberger et al., 1994; Abelenda et al., 2011; Yang et al., 2015; Miao et al., 2016b) and the formation of bulblets from scales in L. davidii var. unicolor (Li et al., 2014).
In the process of starch synthesis, sucrose serves as the basic metabolic substance and is converted to fructose and UDP-glucose by SUS, generating the preconditions for starch synthesis (Wang et al., 1993). SUS is an exclusive and necessary enzyme for the entry of sucrose into the pathway of sucrose-starch metabolism (Sung et al., 1989). In our study, four of the six SUS genes (SUS2_c111432_g1, SUS5_c126221_g1, SUS6_c111800_g1, SUS7_c126221_g2) exhibited the highest expression in U2 and the lowest expression in D2 (Figure 4B, Table S11). SUS is expressed at high levels in sink tissues, such as potato tubers (Zrenner et al., 1995). During stolon formation in Tulipa edulis, the activity of SUS peaks in the initial period, and the contents of soluble sugars and starch are significantly enhanced in the middle period, when the stolons emerge (Miao et al., 2016a). Thus, the up-regulation of SUS may play a key role in bulbil initiation.
In addition, the AGPS (AGPS1_c121621_g2, AGPS1_c121621_g3 and ADG1_c120596_g2), SSS (SSS1_c4952_g1 and SSS3_c122034_g2), GBSS (WAXY_c123636_g2) and SBE1 (c97206_g1 and c108260_g1) genes exhibited the same up-regulated expression pattern during bulbil initiation (Figures 4B, C). AGPS encodes AGPase, which catalyzes the first committed step of starch synthesis in the plastids, converting glucose 1-phosphate (G1P) to ADP-glucose. SSS, GBSS and SBE are key enzymes in starch synthesis (Tetlow et al., 2004). All of these results indicated that starch synthesis likely promotes bulbil initiation in the early stage of bulbil formation and support the view that starch synthesis can satisfy the demands for reserve substances and energy in active cells, including those related to cell division and differentiation as well as starch accumulation in preparation for the initiation of new plant organs (Miao et al., 2016a).
Plant Hormone Signaling Exhibits Complex Regulation during Bulbil Formation
Our transcriptome analysis identified numerous genes involved in the plant hormone response to bulbil formation (Table S12), and different hormone-associated pathways exhibited distinct regulation patterns (Figure 6B).
Auxin may promoted bulbil initiation and then inhibited the further formation of axil bulbils according to our results. We observed that YUCCA genes (c111323_g1 and c128166_g2) were up-regulated in U2 vs. U1 and then down-regulated in U3 (Figures 6B, D). These genes play a key role in the synthesis of IAA; thus, their up-regulated expression is positively related to IAA accumulation (Zhao, 2010). At the same time, GH3 genes (c111786_g1, c129142_g4, and c129142_g3) and an IAA efflux carrier component (PIN1B_ c108190_g1), which reduce IAA concentrations, were down-regulated in U2 vs. U1 and then up-regulated in U3. These results are consistent with the notion that IAA accumulation induces new primordia in maize and potato and inhibits the outgrowth of axillary organs in Arabidopsis (Greb et al., 2003; Reinhardt et al., 2003; Roumeliotis et al., 2012). However, these results are inconsistent with what occurs in Agave tequilana and tomato, which exhibit an inhibitory effect of IAA on the development of new meristems and vegetative bulbils (Wang et al., 2014; Abraham-Juarez et al., 2015). This discrepancy may be caused by the different locations of IAA within plants and differences in IAA concentrations. IAA is largely produced in young expanding leaves at the shoot apex and is transported basipetally down the stem (Ljung et al., 2001; Durbak et al., 2012). Consequently, the concentration of IAA at the shoot apex is higher than in the middle of the stem, and the concentration at the base of stem is the lowest. L. lancifolium bulbils first arise at a certain position in the mid-upper leaf axil, instead of at the shoot apex, and the lower stem cannot form bulbils during its life cycle. These findings indicate that bulbil initiation in L. lancifolium is related to the concentration of IAA, which can induce bulbil formation after surpassing a certain threshold. However, the exact threshold range is unclear and will be investigated in further studies.
CKs play critical roles in the entire process of bulbil formation. In our study, CYP735A1 (c107758_g1), which is involved in CK biosynthesis, was obviously up-regulated during bulbil formation in the upper leaf axil. In addition, the expression of CKX (CKX 5_c102430_g1, CKX3_ c106674_g1, CKX4_c117929_g1) exhibited minimal changes in the upper leaf axil during bulbil formation and was highest in D2 (Figure 6B, Table S12). High CK biosynthesis and low degradation occurred in the upper stem might promote bulbil formation in the upper leaf axil. This finding is closely related to the roles of CKs in regulating cell proliferation and tissue differentiation in many plants (Miller et al., 1955; Hwang et al., 2012). Similar results were reported by Guivarc'h et al. (2002). In addition, a homolog of CYCD3-1 (c113663_g1), which is induced by CK and plays a key role in integrating cell division and lateral organ development (Riou-Khamlichi et al., 1999), was up-regulated during bulbil formation, and its expression level was lowest in D2. This result further verifies the important role of the promotion of CK in cell division in bulbil formation.
Based on the KEGG analysis, we also identified GA-, SL-, ABA-, ethylene-, and JA-related genes, such as GID1, D14, SNRK2, PYL4, ERF, JAZ, and MYC. These genes all showed the same expression pattern (Figure 6B): they were down-regulated in U2 vs. U1 and presented the highest expression in D2, indicating an inhibitory effect on bulbil initiation.
Overall, bulbil formation in the upper portion of the stem of triploid L. lancifolium is a complex process. Bulbil initiation is particularly important for bulbil formation and might be controlled by genes involved in hormone signal transduction and starch biosynthesis. Further studies on the functions of these candidate genes might be helpful for understanding bulbil formation in lily.
Author Contributions
JM conceived and designed the study. PY performed most of the experiments, with assistance from LX, HX, GH, YF, YC, YT, and SY. The manuscript was written by PY.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31672196, 31272205), the National Key Technology Support Program (Grant No. 2012BAD01B0701), and the Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was conducted at the Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Ministry of Agriculture, China.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01508/full#supplementary-material
References
Abelenda, J. A., Navarro, C., and Prat, S. (2011). From the model to the crop: genes controlling tuber formation in potato. Curr. Opin. Biotechnol. 22, 287–292. doi: 10.1016/j.copbio.2010.11.013
Abraham-Juarez, M. J., Hernandez-Cardenas, R., Santoyo-Villa, J. N., O'Connor, D., Sluis, A., Hake, S., et al. (2015). Functionally different PIN proteins control auxin flux during bulbil development in Agave tequilana. J. Exp. Bot. 66, 3893–3905. doi: 10.1093/jxb/erv191
Abraham-Juarez, M. J., Martinez-Hernandez, A., Leyva-Gonzalez, M. A., Herrera-Estrella, L., and Simpson, J. (2010). Class I KNOX genes are associated with organogenesis during bulbil formation in Agave tequilana. J. Exp. Bot. 61, 4055–4067. doi: 10.1093/jxb/erq215
Bach, A., and Sochacki, D. (2012). “Propagation of ornamental geophytes: physiology and management,” in Ornamental Geophytes: From Basic Science to Sustainable Production, eds R. Kamenetsky and H. Okubo (Boca Raton, FL: CRC Press), 261–286.
Bell, A. D., and Bryan, A. (2008). Plant Form: An Illustrated Guide to Flowering Plant Morphology. Portland, OR: Timber Press.
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300.
Callaghan, T., Jonasson, S., and Brooker, R. (1997). “Arctic clonal plants and global change,” in The Ecology and Evolution of Clonal Plants, eds H. de Kroon and J. M. van Groenendael (Leiden: Backhuys Publishers), 381–403.
Ceplitis, A., and Bengtsson, B. O. (2004). Genetic variation, disequilibrium and natural selection on reproductive traits in Allium vineale. J. Evol. Biol. 17, 302–311. doi: 10.1046/j.1420-9101.2003.00678.x
Chung, M. Y., López-Pujol, J., Chung, J. M., Kim, K.-J., Park, S. J., and Chung, M. G. (2015). Polyploidy in Lilium lancifolium: evidence of autotriploidy and no niche divergence between diploid and triploid cytotypes in their native ranges. Flora 213, 57–68. doi: 10.1016/j.flora.2015.04.002
Durbak, A., Yao, H., and McSteen, P. (2012). Hormone signaling in plant development. Curr. Opin. Biotechnol. 15, 92–96. doi: 10.1016/j.pbi.2011.12.004
Eviatar-Ribak, T., Shalit-Kaneh, A., Chappell-Maor, L., Amsellem, Z., Eshed, Y., and Lifschitz, E. (2013). A cytokinin-activating enzyme promotes tuber formation in tomato. Curr. Biol. 23, 1057–1064. doi: 10.1016/j.cub.2013.04.061
Geigenberger, P., Merlo, L., Reimholz, R., and Stitt, M. (1994). When growing potato tubers are detached from their mother plant there is a rapid inhibition of starch synthesis, involving inhibition of ADP-glucose pyrophosphorylase. Planta 193, 486–493. doi: 10.1007/BF02411552
Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Grbic, V., and Bleecker, A. B. (2000). Axillary meristem development in Arabidopsis thaliana. Plant J. 21, 215–223. doi: 10.1046/j.1365-313x.2000.00670.x
Greb, T., Clarenz, O., Schafer, E., Muller, D., Herrero, R., Schmitz, G., et al. (2003). Molecular analysis of the LATERAL SUPPRESSOR gene in Arabidopsis reveals a conserved control mechanism for axillary meristem formation. Genes Dev. 17, 1175–1187. doi: 10.1101/gad.260703
Guivarc'h, A., Rembur, J., Goetz, M., Roitsch, T., Noin, M., Schmülling, T., et al. (2002). Local expression of the ipt gene in transgenic tobacco (Nicotiana tabacum L. cv. SR1) axillary buds establishes a role for cytokinins in tuberization and sink formation. J. Exp. Bot. 53, 621–629. doi: 10.1093/jexbot/53.369.621
He, Z. P. (1993). A Laboratory Guide to Chemical Control Technology on Field Crop. Beijing: Agricultural University Press.
Hwang, I., Sheen, J., and Müller, B. (2012). Cytokinin signaling networks. Annu. Rev. Plant Biol. 63, 353–380. doi: 10.1146/annurev-arplant-042811-105503
Kanehisa, M., and Goto, S. (2000). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim, J. H., Kyung, H. Y., Choi, Y. S., Lee, J. K., Hiramatsu, M., and Okubo, H. (2006). Geographic distribution and habitat differentiation in diploid and triploid Lilium lancifolium of South Korea. J. Fac. Agric. Kyushu Univ. 51, 239–243.
Kumar, S., Kanwar, J. K., and Sharma, D. R. (2006). In vitro propagation of Lilium. Adv. Hortic. Sci. 20, 181–188.
Leyser, O. (2003). Regulation of shoot branching by auxin. Trends Plant Sci. 8, 541–545. doi: 10.1016/j.tplants.2003.09.008
Li, T., Li, S. Q., and Luo, R. (2012). Morphological observation and anatomical study on bulbil development of Lilium sulphureum. Acta Bot. Boreali-Occident. Sin. 32, 85–89. doi: 10.3969/j.issn.1000-4025.2012.01.014
Li, X. Y., Wang, C. X., Cheng, J. Y., Zhang, J., Teixeira da Silva, J. A., Liu, X. Y., et al. (2014). Transcriptome analysis of carbohydrate metabolism during bulblet formation and development in Lilium davidii var. unicolor. BMC Plant Biol. 14:358. doi: 10.1186/s12870-014-0358-4
Liang, S. Y., and Tamura, M. N. (2000). “Lillium L., nomocharis franchet,” in Flora of China, eds Z. Y. Wu and P. H. Raven (Beijing: Science Press), 135–159.
Liu, X. H., Huang, J., Wang, J. M., and Lu, Y. M. (2014). RNA-seq analysis reveals genetic bases of the flowering process in oriental hybrid lily cv. Sorbonne. Russ. J. Plant Physiol. 61, 880–892. doi: 10.1134/S1021443714060132
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔct. CT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ljung, K., Bhalerao, R. P., and Sandberg, G. (2001). Sites and homeostatic control of auxin biosynthesis in Arabidopsis during vegetative growth. Plant J. 28, 465–474. doi: 10.1046/j.1365-313X.2001.01173.x
Long, J., and Barton, M. K. (2000). Initiation of axillary and floral meristems in Arabidopsis. Dev. Biol. 218, 341–353. doi: 10.1006/dbio.1999.9572
Luo, R., Du, Y. S., Sun, Y. Y., and Cao, Z. J. (2014). Morphological observation and anatomical study on bulbil development of Pinellia ternata. Acta Bot. Boreali-Occident. Sin. 34, 1776–1781. doi: 10.7606/j.issn.1000-4025.2014.09.1776
Maesato, K., Sarma, K., Fukui, H., and Hara, T. (1991). In vitro bulblet induction from shoot apices of Lilium japonicum. Hortscience 26, 211.
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Miao, Y., Guo, Q., Zhu, Z., Yang, X., Wang, C., Sun, Y., et al. (2016a). Dynamic changes in starch metabolism and endogenous hormones during stolon formation in Tulipa edulis. J. Am. Soc. Hortic. Sci. 141, 211–221.
Miao, Y., Zhu, Z., Guo, Q., Yang, X., Liu, L., Sun, Y., et al. (2016b). Dynamic changes in carbohydrate metabolism and endogenous hormones during Tulipa edulis stolon development into a new bulb. J. Plant Biol. 59, 121–132. doi: 10.1007/s12374-016-0456-y
Miller, C. O., Skoog, F., Okumura, F. S., Von Saltza, M. H., and Strong, F. M. (1955). Isolation, structure and synthesis of kinetin, a substance promoting cell division1,2. J. Am. Chem. Soc. 78, 1375–1380. doi: 10.1021/ja01588a032
Navarro, C., Cruz-Oró, E., and Prat, S. (2015). Conserved function of flowering locus T (FT) homologues as signals for storage organ differentiation. Curr. Opin. Plant Biol. 23, 45–53. doi: 10.1016/j.pbi.2014.10.008
Noda, S. (1986). Cytogenetic behavior, chromosomal differentiations, and geographic distribution in Lilium lancifolium (Liliaceae). Plant Species Biol. 1, 69–78. doi: 10.1111/j.1442-1984.1986.tb00016.x
Pasare, S. A., Ducreux, L. J. M., Morris, W. L., Campbell, R., Sharma, S. K., Roumeliotis, E., et al. (2013). The role of the potato (Solanum tuberosum) CCD8 gene in stolon and tuber development. New Phytol. 198, 1108–1120. doi: 10.1111/nph.12217
Reinhardt, D., Pesce, E.-R., Stieger, P., Mandel, T., Baltensperger, K., Bennett, M., et al. (2003). Regulation of phyllotaxis by polar auxin transport. Nature 426, 255–260. doi: 10.1038/nature02081
Riou-Khamlichi, C., Huntley, R., Jacqmard, A., and Murray, J. A. (1999). Cytokinin activation of Arabidopsis cell division through a D-type cyclin. Science 283, 1541–1544. doi: 10.1126/science.283.5407.1541
Roumeliotis, E., Kloosterman, B., Oortwijn, M., Kohlen, W., Bouwmeester, H. J., Visser, R. G. F., et al. (2012). The effects of auxin and strigolactones on tuber initiation and stolon architecture in potato. J. Exp. Bot. 63, 4539–4547. doi: 10.1093/jxb/ers132
Sandoval, S. D. C., Abraham-Juárez, M. J., and Simpson, J. (2012). Agave tequilana MADS genes show novel expression patterns in meristems, developing bulbils and floral organs. Sex. Plant Reprod. 25, 11–26. doi: 10.1007/s00497-011-0176-x
Steiner, B. L., Armbruster, G. F. J., Scheepens, J. F., and Stocklin, J. (2012). Distribution of bulbil- and seed-producing plants of Poa alpina (Poaceae) and their growth and reproduction in common gardens suggest adaptation to different elevations. Am. J. Bot. 99, 2035–2044. doi: 10.3732/ajb.1200213
Suh, J. K., and Roh, M. S. (2014). New technique for cut flower production from bulbils of the Asiatic hybrid lily (Lilium×elegans Thunb). Sci. Hortic. 165, 374–383. doi: 10.1016/j.scienta.2013.11.012
Sung, S. J. S., Xu, D. P., and Black, C. C. (1989). Identification of actively filling sucrose sinks. Plant Physiol. 89, 1117–1121. doi: 10.1104/pp.89.4.1117
Suzuki, T., and Yamagishi, M. (2016). Aneuploids without bulbils segregated in F1 hybrids derived from triploid Lilium lancifolium and diploid L. leichtlinii crosses. Horticul. J. 85, 224–231. doi: 10.2503/hortj.MI-089
Tetlow, I. J., Morell, M. K., and Emes, M. J. (2004). Recent developments in understanding the regulation of starch metabolism in higher plants. J. Exp. Bot. 55, 2131–2145. doi: 10.1093/jxb/erh248
Wang, C. N., and Cronk, Q. C. B. (2003). Meristem fate and bulbil formation in Titanotrichum (Gesneriaceae). Am. J. Bot. 90, 1696–1707. doi: 10.3732/ajb.90.12.1696
Wang, C.-N., Möller, M., and Cronk, Q. C. B. (2004). Altered expression of GFLO, the Gesneriaceae homologue of Floricaula/Leafy, is associated with the transition to bulbil formation in Titanotrichum oldhamii. Dev. Genes Evol. 214, 122–127. doi: 10.1007/s00427-004-0388-2
Wang, F., Sanz, A., Brenner, M. L., and Smith, A. (1993). Sucrose synthase, starch accumulation, and tomato fruit sink strength. Plant Physiol. 101, 321–327. doi: 10.1104/pp.101.1.321
Wang, J., Wang, Q., Yang, Y., Liu, X. H., Gu, J. H., Li, W. Q., et al. (2014a). De novo assembly and characterization of stress transcriptome and regulatory networks under temperature, salt and hormone stresses in Lilium lancifolium. Mol. Biol. Rep. 41, 8231–8245. doi: 10.1007/s11033-014-3725-1
Wang, J., Yang, Y., Liu, X., Huang, J., Wang, Q., Gu, J., et al. (2014b). Transcriptome profiling of the cold response and signaling pathways in Lilium lancifolium. BMC Genomics 15:203. doi: 10.1186/1471-2164-15-203
Wang, Q., Kohlen, W., Rossmann, S., Vernoux, T., and Theres, K. (2014). Auxin depletion from the leaf axil conditions competence for axillary meristem formation in Arabidopsis and Tomato. Plant Cell 26, 2068–2079. doi: 10.1105/tpc.114.123059
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genetics 10, 57–63. doi: 10.1038/nrg2484
Xu, L., Yang, P., Feng, Y., Xu, H., Cao, Y., Tang, Y., et al. (2017). Spatiotemporal transcriptome analysis provides insights into bicolor tepal development in Lilium “Tiny Padhye.” Front. Plant Sci. 8:398. doi: 10.3389/fpls.2017.00398
Yang, M., Zhu, L., Pan, C., Xu, L., Liu, Y., Ke, W., et al. (2015). Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 5:13059. doi: 10.1038/srep13059
Yang, Y. M., Xu, C. N., Wang, B. M., and Jia, J. Z. (2001). Effects of plant growth regulators on secondary wall thickening of cotton fibres. Plant Growth Regul. 35, 233–237. doi: 10.1023/A:1014442015872
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Yu, X., Zhang, J., Shao, S., Yu, H., Xiong, F., and Wang, Z. (2015). Morphological and physicochemical properties of bulb and bulbil starches from Lilium lancifolium. Starch Stärke 67, 448–458. doi: 10.1002/star.201400209
Zhang, Y., Yu, Y., and Chen, B. (1994). Anatomical study on the generation of the buibil of the Chinese yam. Acta Agric. Univer. Pekinensis 20, 413–418
Zhao, Y. (2010). Auxin biosynthesis and its role in plant development. Ann. Rev. Plant biol. 61, 49–64. doi: 10.1146/annurev-arplant-042809-112308
Zhu, X., Chai, M., Li, Y., Sun, M., Zhang, J., Sun, G., et al. (2016). Global transcriptome profiling analysis of inhibitory effects of paclobutrazol on leaf growth in lily (Lilium Longiflorum-Asiatic Hybrid). Front. Plant Sci. 7:491. doi: 10.3389/fpls.2016.00491
Keywords: Lilium lancifolium, bulbil formation, histology, transcriptome, starch synthesis, plant hormones
Citation: Yang P, Xu L, Xu H, Tang Y, He G, Cao Y, Feng Y, Yuan S and Ming J (2017) Histological and Transcriptomic Analysis during Bulbil Formation in Lilium lancifolium. Front. Plant Sci. 8:1508. doi: 10.3389/fpls.2017.01508
Received: 09 April 2017; Accepted: 15 August 2017;
Published: 30 August 2017.
Edited by:
Stefan de Folter, Center for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV), MexicoReviewed by:
Annette Becker, Justus Liebig Universität Gießen, GermanyMiguel Angel Flores-Vergara, North Carolina State University, United States
Copyright © 2017 Yang, Xu, Xu, Tang, He, Cao, Feng, Yuan and Ming. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Ming, bWluZ2p1bkBjYWFzLmNu