 Jihye Jung
Jihye Jung Sang Chul Choi
Sang Chul Choi Sunghee Jung
Sunghee Jung Byung-Kwan Cho
Byung-Kwan Cho Gwang-Hwan Ahn4
Gwang-Hwan Ahn4 Stephen B. Ryu
Stephen B. Ryu- 1Department of Biological Sciences, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 2Plant Systems Engineering Research Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea
- 3Department of Biosystems and Bioengineering, KRIBB School of Biotechnology, Korea University of Science and Technology, Daejeon, South Korea
- 4Sweet Persimmon Research Institute, Gyeongsangnam-do Agricultural Research and Extension Services, Gimhae, South Korea
Persimmon (Diospyros kaki Thunb.), which is a climacteric fruit, softens in 3–5 weeks after harvest. However, little is known regarding the transcriptional changes that underlie persimmon ripening. In this study, high-throughput de novo RNA sequencing was performed to examine differential expression between freshly harvested (FH) and softened (ST) persimmon fruit peels. Using the Illumina HiSeq platform, we obtained 259,483,704 high quality reads and 94,856 transcripts. After the removal of redundant sequences, a total of 31,258 unigenes were predicted, 1,790 of which were differentially expressed between FH and ST persimmon (1,284 up-regulated and 506 down-regulated in ST compared with FH). The differentially expressed genes (DEGs) were further subjected to KEGG pathway analysis. Several pathways were found to be up-regulated in ST persimmon, including “amino sugar and nucleotide sugar metabolism.” Pathways down-regulated in ST persimmon included “photosynthesis” and “carbon fixation in photosynthetic organisms.” Expression patterns of genes in these pathways were further confirmed using quantitative real-time RT-PCR. Ethylene gas production during persimmon softening was monitored with gas chromatography and found to be correlated with the fruit softening. Transcription involved in ethylene biosynthesis, perception and signaling was up-regulated. On the whole, this study investigated the key genes involved in metabolic pathways of persimmon fruit softening, especially implicated in increased sugar metabolism, decreased photosynthetic capability, and increased ethylene production and other ethylene-related functions. This transcriptome analysis provides baseline information on the identity and modulation of genes involved in softening of persimmon fruits and can underpin the future development of technologies to delay softening in persimmon.
Introduction
Seven of the approximately 190 known species of persimmon (Diospyros) are grown commercially for fruit production, the most widely cultivated of which is Diospyros kaki. The D. kaki “Fuyu” sweet persimmon is a pollination-constant and non-astringent cultivar (Ito, 1971). After harvest, persimmon fruits often undergo rapid softening, darkening, and browning, resulting in short shelf life (Hobson, 1981). Persimmons are cultivated in sub-tropical and warm temperate regions, and their production is limited to certain areas in countries such as China, Korea, Japan, and Brazil (Mowat et al., 1995; FAOSTAT, 2013). The short shelf life is a barrier to successful exportation of persimmon fruit to other countries, and extending shelf life is economically desirable.
Persimmon produces ethylene during fruit ripening and is classified as a climacteric plant based on its pattern of respiration and ethylene production (Biale, 1981). Ethylene causes fruit softening by inducing the expression of cell wall degradation genes, resulting in a modified fruit texture (Mattoo and Suttle, 1991; Nakano et al., 2003; Nishiyama et al., 2007). Aminocyclopropane-1-carboxylic acid (ACC) synthase and ACC oxidase (ACO) are known key enzymes for synthesizing the ethylene and silencing of these genes inhibited ripening in tomato (Oeller and Min-Wong, 1991; Fluhr et al., 1996). Chemical treatment such as 1-methylcyclopropene (1-MCP), an inhibitor of ethylene perception, also delays fruit ripening (Mattoo and Suttle, 1991; Vilas-Boas and Kader, 2007). Therefore, ethylene perception in addition to ethylene production is a major cause of fruit softening.
Little is known regarding the genes associated with fruit softening in persimmon. Ethylene biosynthesizing genes, ACC synthase (DkACS1, -2, and -3) and ACC oxidase (DkACO1 and -2), were known in persimmon and the expression of these genes is temporally and spatially regulated during fruit softening (Nakano et al., 2003; Ortiz et al., 2006). Two persimmon homologs of Arabidopsis ethylene receptor genes, namely, DkERS1 and DkETR2, exhibited induced expression during softening (Pang et al., 2007). Expressed sequence tag (EST) analysis showed that cell wall degradation-related genes such as those encoding basic chitinase and endochitinase were also induced during later stages of persimmon fruit development (Nakagawa et al., 2008).
Transcriptional analysis of fruit ripening was previously examined in several species, including bayberry, tomato, and watermelon, and specific transcripts were linked to increases in fruit quality and reductions in softening (Guo et al., 2011; Feng et al., 2012; Lee et al., 2012). Softening-related expression profiles differed between fruit species (Brummell and Harpster, 2001). Previous transcriptional analysis of persimmon examined expression changes after ethanol treatment or under anoxic conditions (Luo et al., 2014; Min et al., 2014). However, to date, fruit softening in persimmon has only been examined at the individual gene level rather than with comprehensive transcriptional analysis (Hou et al., 2015).
De novo transcriptome assembly is a powerful technique for assessing transcription in the absence of a reference genome (Haas et al., 2013). In this study, de novo RNA sequencing (RNA-seq) using Illumina HiSeq 2000 technology was used to generate transcriptomes from freshly harvested (FH) and softened (ST) persimmon fruit peels. This investigation identified several genes involved in persimmon fruit softening and provides insights into the molecular events underlying the regulation of persimmon fruit softening.
Materials and Methods
Plant Material and Sample Preparation
Persimmon fruits were harvested and stored in constant conditions (22°C, 60% humidity). Persimmon fruit peels (n = 5) collected at 5 days after harvest (DAH) were used as the FH sample for RNA-seq. Peels were harvested for the ST sample when 80% of the stored fruits had softened. One and two (a total of three) FH and ST samples were collected at two different years, respectively. Persimmon peel samples were also taken at 1, 13, 25, and 34 DAH for gene expression analysis with quantitative real-time RT-PCR (qRT-PCR) to verify RNA-seq analysis results. All samples were frozen in liquid nitrogen immediately upon harvest.
RNA Isolation, cDNA Library Construction, and RNA-Seq
Total RNA was extracted from persimmon fruit peels using cetyl trimethyl ammonium bromide (CTAB) buffer [2.0% CTAB, 1.4 M NaCl, 20 mM EDTA, pH 8.0, 100 mM Tris-HCl, pH 8.0, 2.0% polyvinylpyrrolidone (PVP40), 1% β-mercaptoethanol] after sample grinding in liquid nitrogen. Total RNA was purified using an RNeasy mini prep kit (Qiagen) and treated with DNase I (Qiagen) to remove residual DNA. The cDNA libraries for RNA-seq were prepared using an Illumina TruSeq DNA/RNA library preparation kit according to the manufacturer's protocol. Briefly, RNA was fragmented to an average length of 200–300 bp by Mg2+-catalyzed hydrolysis and then reverse transcribed to cDNA with random priming.
The cDNA was then subjected to end repair, adaptor ligation, size selection, and PCR amplification. Each cDNA library was 101 bp paired-end sequenced using an Illumina HiSeq 2000 sequencer according to the manufacturer's protocols. The whole transcriptome sequence raw data were deposited in the NCBI database (https://www.ncbi.nlm.nih.gov/sra) with accession numbers SRR4345331 and SRR4345332 for FH and ST, respectively. The assembled sequence database was also deposited in the NCBI database with accession number GFUF01000000.
Sequence Preprocessing and De novo Assembly
Raw 101 bp paired-end reads were filtered by quality score (Q ≥ 20) using SolexaQA (Cox et al., 2010). Trimming resulted in reads with a mean length of 85.61 bp across all samples and a minimum length of 25 bp. De novo assembly of trimmed reads was carried out using the de Bruijn graph-based assemblers Velvet (v1.2.07) (Zerbino and Birney, 2008) and Oases (v0.2.08) (Schulz et al., 2012). To optimize data assembly, de novo assembly was performed using various hash lengths (k-mers) in the 51–75 range.
Validation of Unigene Assembly and Annotation
Unigene transcripts for persimmon were assembled from merged total reads from both the FH and ST mRNA samples. Translated transcripts were validated by comparison with sequences in the Phytozome database (http://www.phytozome.net) using BLASTx (E ≤ 1e−10) (Goodstein et al., 2012). Protein sequences with the highest sequence similarities were retrieved for further study.
Functional Annotation
Unigenes were functionally annotated using The Arabidopsis Information Resource (TAIR) database and subjected to KEGG (http://www.genome.jp/kegg/) pathway analysis (Kanehisa et al., 2004). In addition, translated transcripts were compared with the KOG (http://www.ncbi.nlm.nih.gov/COG/) protein database using BLASTx (E ≤ 1e−10) (Tatusov et al., 2003), and the best-matching results were selected to annotate the unigenes.
Short Read Mapping and Analysis of Differentially Expressed Genes (DEGs)
Reads for each sequence were mapped to the assembled unigenes using bowtie2 software with default parameters (Langmead and Salzberg, 2012). After counting the number of reads mapped to each transcript, expression values were normalized using DESeq method (Anders and Huber, 2010). DEGs that were differentially expressed between the FH and ST samples were determined from three replicates samples using the binomial test method with expression levels log2 > 1 or < −1, and false discovery rate (FDR) < 0.01. KEGG pathway analysis of DEGs was conducted with TAIR ID using DAVID (Huang et al., 2008) with a two-count option and p < 0.35.
Expression Analysis Using qRT-PCR
First strand cDNA for qRT-PCR was synthesized from total RNA (0.3 μg) using SuperScript III transcriptase (Invitrogen) according to the manufacturer's instructions. Expression analysis was performed using a CFX96 Real-time PCR detection system (Bio-Rad) with SYBR green fluorescent dye (Enzynomics). The thermal cycling program was 95°C for 5 min, 40 cycles of 95°C for 15 s, 55, 57, or 59°C for 30 s, and 72°C for 30 s. The D. kaki Actin gene (accession no. AB473616) served as a reference for expression normalization. PCR specificity was confirmed by electrophoresis gel, amplicon size and DNA sequencing of PCR products (Bustin et al., 2009). The ΔΔCt method was used to calculate gene expression (Livak and Schmittgen, 2001). For convenient comparisons, the expression levels were presented as relative values compared to those of the day 1 samples (expression level = 1). Gene-specific primer sequences are listed in Table S7.
Ethylene Quantification Analysis
Sequence of the genes were deposited in the NCBI database (https://www.ncbi.nlm.nih.gov/sra) with accession numbers in Table S8. The rate of ethylene production by whole persimmon fruits at 1, 13, 25, and 34 DAH was measured by enclosing two samples in 2 L airtight containers for 1 h at 22°C. Headspace gas (0.2 mL) was withdrawn and injected into a gas chromatograph (model GC-4CMPF, Shimadzu, Kyoto) equipped with a flame ionization detector and a 2,000 × 3 mm aluminum oxide column. Ethylene production was presented as nL·g FW−1·h−1, and five replicate measurements were performed.
Results
De novo RNA-Seq Analysis of FH or ST Persimmon
Persimmon fruits soften after harvesting as a result of metabolic processes induced by transcriptional changes. RNA-seq was used to examine differential gene expression between peel samples from FH and ST persimmon. In total, 289,161,674 raw reads and 29.1 Gbp sequence were obtained from all samples using the Illumina HiSeq 2000 platform (Table S1). To assess sequence data quality, raw reads were trimmed and sorted by length using the Dynamic Trim and Length Sort programs in the SolexaQA package, which is used (Cox et al., 2010). Of the raw reads, 89.74% had high-quality score thresholds of Q ≥ 20 and read length >25 bp. In total, 259,483,704 high-quality reads with 23,283,962,364 bases were obtained.
Processed sequence reads were assembled de novo into transcripts using Velvet and Oases based on the de Bruijn graph algorithm (Zerbino and Birney, 2008; Schulz et al., 2012). To optimize data assembly, de novo assembly was performed using various hash lengths (k-mers) in the 51–75 range. The assembled transcripts were combined using Velvet and Oases with k-mer = 61, resulting in 94,856 extended transcripts with lengths ≥200 bp. After removal of highly similar redundant transcripts, 31,258 unigene transcripts were defined and used for further analyses (Table 1). The unigenes comprised 30,524,524 bp in total and had an average length of 976 bp, a maximum length of 10,510 bp, and N50 of 1,695 bp (Table 1). The majority of the unigenes (27,046; 86.53%) were 0.2–2 kb in length (Figure S1).
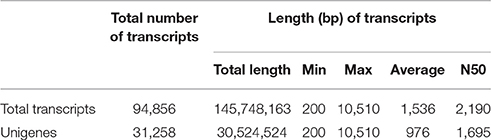
Table 1. Statistical summary of de novo assembly of D. kaki transcripts.
Functional Annotation of Assembled Persimmon Transcripts
The 31,258 assembled unigenes were searched against the GenBank non-redundant database using BLASTx (E ≤ 1e−10) (http://www.ncbi.nlm.nih.gov/). Annotated proteins were identified in the NR database with hits against 16,806 (53.76%) of the unigenes (Table 2). The reliability of the assembled transcripts was assessed by BLASTx comparison of the 31,258 unigenes against the plant-specific Phytozome database (http://www.phytozome.net). Phytozome provides a view of the evolutionary history of each plant gene at the level of sequence, gene structure, gene family, and genome organization, and provides sequence and functional annotation information for each gene (Goodstein et al., 2012). Of the 31,258 unigenes, 18,424 (58.94%) transcripts shared sequence similarity with genes from 41 plant species in Phytozome (E ≤ 1e−10; Table S2). The plant species with the highest numbers of BLASTx hits were Vitis vinifera (3,720 transcripts; 20.19%), Theobroma cacao (2,037 transcripts; 11.06%), Manihot esculenta (1,324 transcripts; 7.19%), Prunus persica (1,229 transcripts; 6.67%), Populus trichocarpa (1,122 transcripts; 6.09%), and Solanum tuberosum (1,053 transcripts; 5.72%) (Figure S2). These results showed that the assembled D. kaki transcripts shared similarity with transcripts from several reference species that were not evolutionarily close to D. kaki.
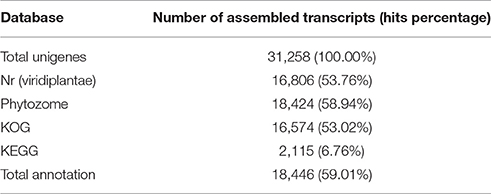
Table 2. Annotation summary of the assembled transcripts.
Clusters of orthologous groups for eukaryotic genome (KOG; http://www.ncbi.nlm.nih.gov/COG/) analysis can be used for functional annotation of transcriptomes and newly sequenced genomes (Tatusov et al., 2003). D. kaki unigenes were matched to sequences in the KOG database using BLASTx (E ≤ 1e−10). Of the 31,258 unigenes, 16,574 (53.02%) were assigned to 26 KOG categories (Figure 1; Table S3). Of these 26 categories, the cluster for “general function prediction only” was the largest group (3,518; 21.23%), followed by “signal transduction mechanisms” (1,929; 11.64%), and “nuclear structure” (1,686; 10.17%).
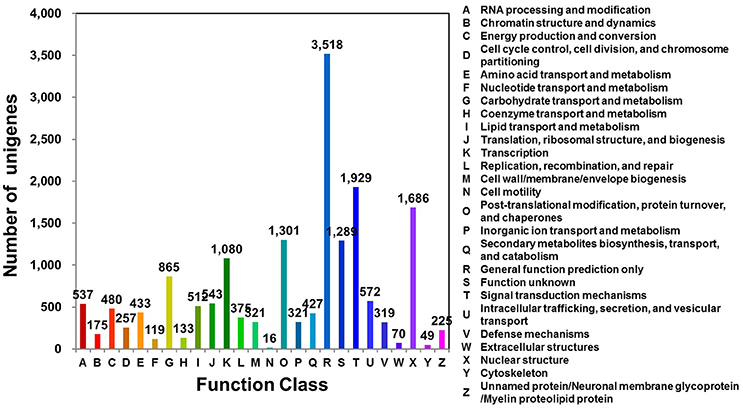
Figure 1. KOG functional classification of D. kaki. The unigenes of persimmon fruit peels were aligned to the KOG database to predict and classify possible functions. A total of 16,574 unigenes were assigned to functional orthologs.
KEGG, an alternative functional gene annotation system, examines molecular interaction networks. KEGG pathway analysis was performed with the 31,258 unigenes using the KEGG Automatic Annotation Server with TAIR as a reference (Kanehisa et al., 2004). KEGG mapped 2,115 unigenes (6.76%) to 17 predicted pathways (Figure 2). Of the five categories identified, the largest was “Metabolism” (2,115 unigenes), which contained “carbohydrate metabolism” (640 unigenes), “amino acid metabolism” (423 unigenes), “energy metabolism” (255 unigenes), “lipid metabolism” (235 unigenes), and other sub-categories (Figure 2; Table S4). The category “Genetic Information Pathways” also contained a large number of unigenes (867), namely, “translation” (206 unigenes), “transcription” (186 unigenes), “replication and repair” (208 unigenes), and “folding, sorting, and degradation” (267 unigenes) (Figure 2; Table S4). The major categories of “Cellular Processes,” “Environmental Information Processing,” and “Organismal Systems” contained 97, 38, and 38 unigenes, respectively (Figure 2; Table S4).
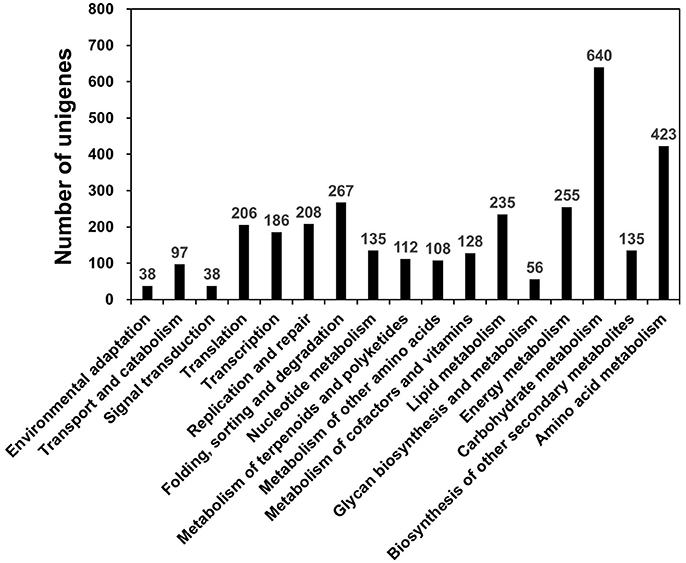
Figure 2. KEGG pathway mapping for D. kaki. The unigenes of persimmon fruit peels were functionally classified according to metabolic function.
Biochemical Pathways of DEGs in FH and ST Persimmon
Unigenes with >2-fold expression differences between ST and FH persimmon were defined as DEGs. DEGs were considered significant at FDR adjusted p < 0.01 (Figure 3A; Table S5). Of the 1,790 DEGs identified, 1,284 unigenes exhibited elevated expression and 506 unigenes exhibited lower expression in ST compared with FH persimmon (Figure 3B; Table S5).
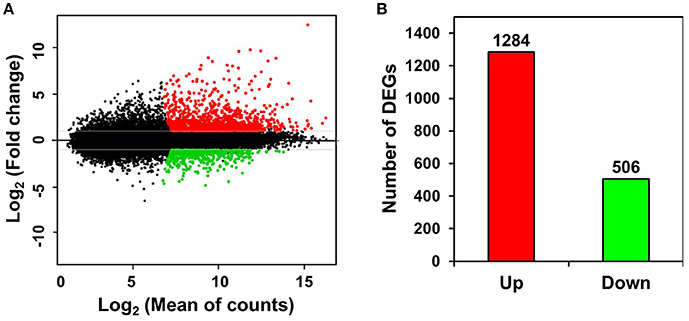
Figure 3. Differential gene expression between freshly harvested (FH) and softened (ST) persimmon. (A) MA plot of DEGs between FH and ST. Genes up- and down-regulated in ST are shown in red and green, respectively. (B) Numbers of up- and down-regulated genes in ST compared with FH persimmon.
KEGG pathway analysis of DEGs was used to identify the major biochemical pathways modulated during persimmon softening. Carbohydrate and amino acid metabolic pathways were up-regulated in ST persimmon, as were secondary metabolite biosynthetic pathways (Table S6). Previously, starch breakdown and sucrose synthesis were observed during the rapid stage of fruit ripening and softening (Cordenunsi and Lajolo, 1995). These results support that enhancement of sugar metabolism pathways in persimmon is related to fruit softening. Differential expression of the three identified genes in the amino sugar and nucleotide sugar metabolism category during softening were verified using qRT-PCR (Figure 5). After harvest, samples were taken at days 1 and 13 (prior to softening onset at 16 DAH), and 25 and 34 (Figure 4). Consistent with the RNA-seq analysis, all the examined genes (DkUGD1, DkChiA3, and DkB-Chi1) were up-regulated at 25 and 34 DAH compared with 1 and 13 DAH (Figure 5A).
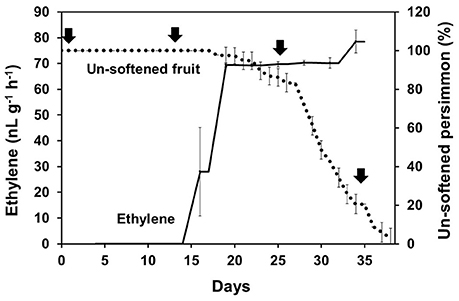
Figure 4. Persimmon softening and production of ethylene gas. Persimmon softening is shown in terms of unsoftened persimmon (%) with a black dotted line. Ethylene abundance is indicated with a solid black line. Arrows indicate sampling points for qRT-PCR. Error bars represent standard deviation (SD).
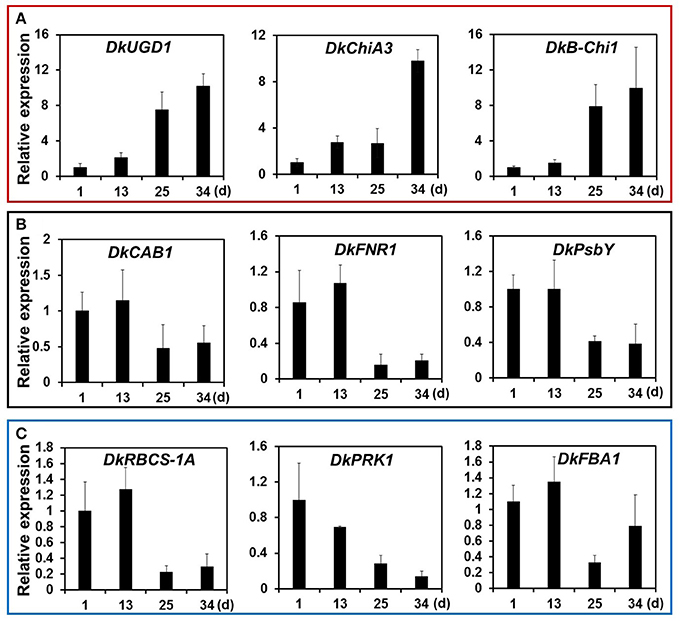
Figure 5. Quantitative real-time PCR analysis of representative DEGs of KEGG pathways modulated during persimmon fruit softening. (A) “Sugar metabolism,” (B) “Photosynthesis,” and (C) “Carbon fixation.” Transcription is shown relative to day 1 (expression level = 1) with the DkActin gene as an internal reference. Each qRT-PCR analysis was repeated three times. Error bars represent standard deviation (SD).
Photosynthetic pathways were down-regulated in ST persimmon, as were pathways involved in carbon fixation in photosynthetic organisms, carotenoid biosynthesis, and glycolysis/gluconeogenesis (Table 3; Table S6). Genes identified in the photosynthesis and carbon fixation pathways were examined using qRT-PCR. Four of the six photosynthetic genes and all five carbon fixation genes exhibited lower expression at 25 and 34 DAH than at 1 and 13 DAH (Figures 5B,C; Figures S3 and S4). Minor discrepancies between the RNA-seq and qRT-PCR results were observed for two genes, DkPsaG and DkPsaD-1(Figure S3). This discrepancy may be explained by the low expression levels and minimal differential expression observed for DkPsaG and DkPsaD-1 (Table 3). Similar discrepancies between RNA-seq and qRT-PCR results were previously observed in other studies (Wang R. et al., 2017).
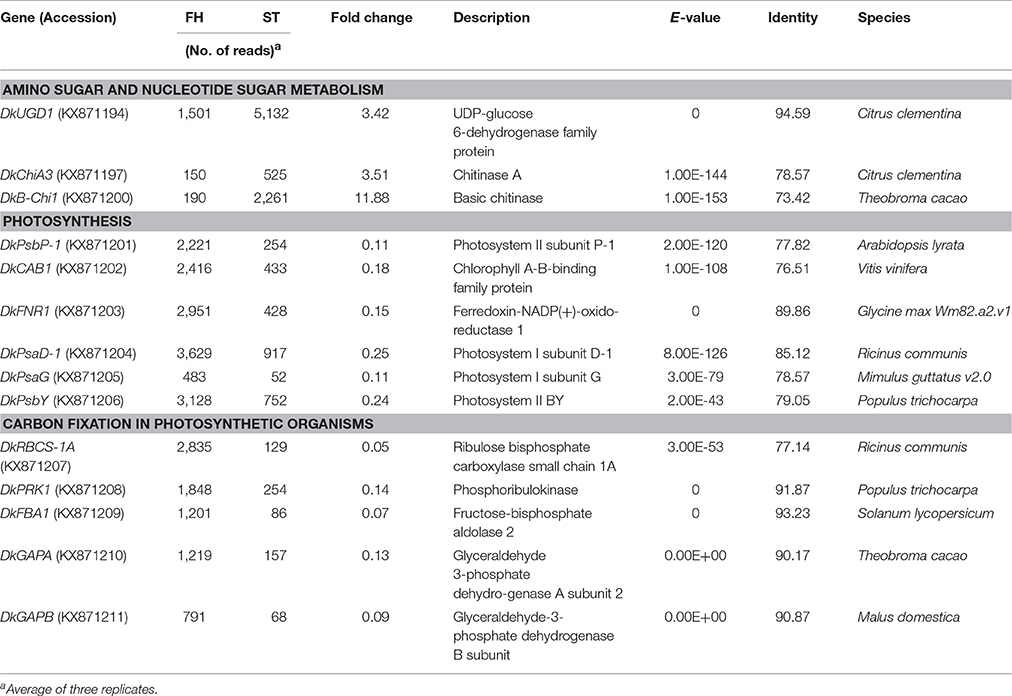
Table 3. Summary of DEGs annotated using KEGG.
Identification of Ethylene-Related DEGs in Persimmon
Ethylene, which is produced by persimmon fruits during maturation, induces expression of softening-related genes (Tucker and Brady, 1987; Sitrit and Bennett, 1998). As ethylene is a major effector of fruit softening, several previous studies have examined the effects of reducing ethylene impacts on persimmon by treatment with ethylene inhibiting chemicals such as 1-MCP (Nakano et al., 2003; Vilas-Boas and Kader, 2007). However, only a few genes involved in ethylene production or in the response to ethylene during softening in persimmon were discovered (Nakano et al., 2003; Pang et al., 2007; Min et al., 2014). In this study, ethylene gas production during softening was quantitated using gas chromatography. Ethylene gas was first detected 16 DAH and corresponded to the onset of fruit softening (Figure 4). DEG analysis revealed six transcripts that were up-regulated in ST compared with FH and encoded predicted ethylene-related genes (Table 4; Table S5). These genes included ethylene response factor, ethylene receptor, and EIN3-binding F-BOX protein. Two of the transcripts, namely, DkETR2 and DkERS1, were identified as D. kaki transcripts during BLASTx annotation, indicating their previous identification in D. kaki (Pang et al., 2007). The remaining four transcripts (DkERF25, DkEBF1, DkERF24, and DkETR3) were newly identified in D. kaki (Table 4). Up-regulation of the ethylene-related DEGs during softening was verified at 1, 13, 25, and 34 DAH using qRT-PCR analysis (Figure 6). Consistent with RNA-seq data, expression of all six genes was elevated at 25 and 34 DAH compared with 1 and 13 DAH (Figure 6). Interestingly, the transcripts involved in ethylene biosynthesis were not detected in the DEG analysis (Table S5). Only DkACO1 (gene ID: 1SL019260t0001), but not the others, was identified in the RNA-seq assembly database (Table S2). Nevertheless, since the ethylene biosynthesis genes, ACC synthase (DkACS1, -2, and -3) and ACC oxidase (DkACO1 and -2), have been previously reported (Nakano et al., 2003; Ortiz et al., 2006), we determined the expression patterns of the genes by qRT-PCR analysis (Figure 7). All five genes showed significant increase at days 25 and 34 after harvest, while no distinct modulation of expression until 13 days (Figure 7), consistent with ethylene production pattern (Figure 4).
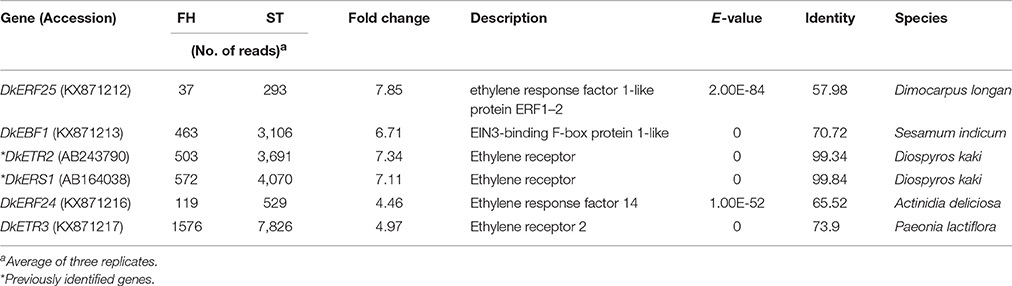
Table 4. Transcripts encoding predicted ethylene-related genes.
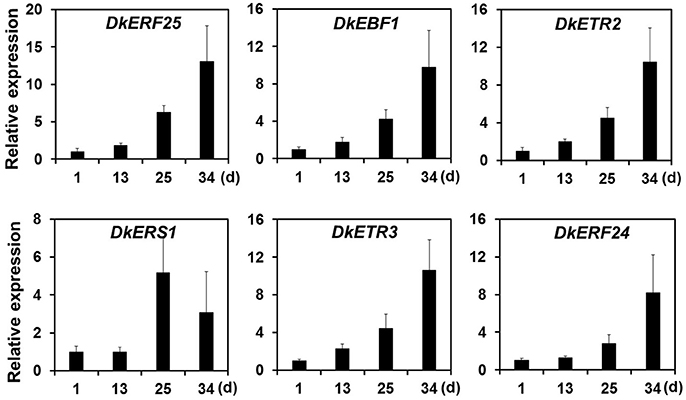
Figure 6. Quantitative real-time PCR analysis of DEGs involved in ethylene-perception and signaling during persimmon fruit softening. Transcription is shown relative to day 1 (expression level = 1) with the DkActin gene as an internal reference. Each qRT-PCR analysis was repeated three times. Error bars represent standard deviation (SD).
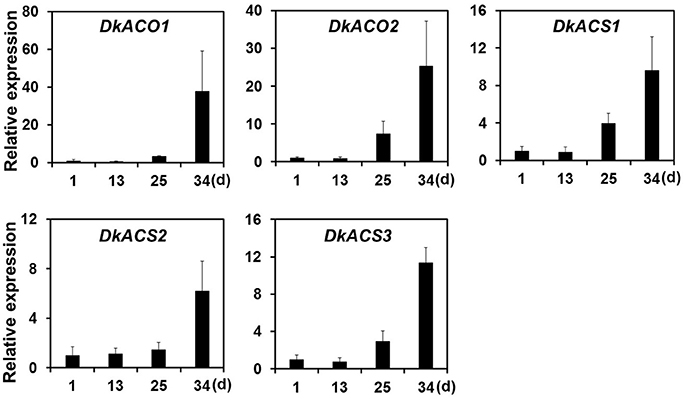
Figure 7. Quantitative real-time PCR analysis of ethylene-biosynthesizing genes during persimmon fruit softening. Transcription is shown relative to day 1 (expression level = 1) with the DkActin gene as an internal reference. Each qRT-PCR analysis was repeated three times. Error bars represent standard deviation (SD).
Discussion
The ability to delay fruit ripening is important in the food industry to prevent fruit decay and prolong storage time. Persimmon production is limited to a small number of geographical locations, and persimmon softening hampers successful fruit export (FAOSTAT, 2013). Persimmon is hexaploid (2n = 6x = 90) and does not have a sequenced genome, both of which hamper the study of genes related to fruit softening. Although NCBI contains data from previous studies for 14,482 ESTs, 1,238 nucleotides, and 755 protein sequences of D. kaki (as of January 2017), little is known regarding the biochemical and physiological pathways of relevance to persimmon softening. De novo assembly is an established technique for developing genome-level resources for non-model organisms, including plant species (Vera et al., 2008; Meyer et al., 2009; Der et al., 2011). In this study, de novo assembly of RNA-seq data was used to identify and compare transcripts from FH and ST persimmon peel samples. Genes related to fruit softening were identified using differential expression analysis.
Ripening is a major determinant of fruit quality. Traits such as fruit color, texture, flavor, and aroma are affected by ripening and influence final fruit quality (Miszczak et al., 1995; Brummell et al., 2004; Cherian et al., 2014). During ripening, changes in gene expression or protein regulation lead to physiological and biochemical effects such as ethylene biosynthesis, pigmentation synthesis, chlorophyll degradation, cell wall degradation, and organic acid accumulation (Brady, 1987; Cherian et al., 2014).
KEGG pathway analysis showed that the up-regulated pathways during persimmon softening include “amino sugar and nucleotide sugar metabolism.” In this pathway, transcription of basic chitinase, chitinase A, and UDP-glucose 6-dehydrogenase was up-regulated during softening. Expression of these genes was also found in previous studies. For example, EST analysis identified basic chitinase and endochitinase gene expression at a late stage of fruit development in persimmon (Nakagawa et al., 2008). Expression of the MaECHI1 gene encoding banana endochitinase also increased markedly during banana ripening and after exogenous ethylene treatment (Liu et al., 2012). Functionally, chitinase hydrolyzes colloidal chitin into chito-oligomers, including chitotriose and chitobiose, and its monomer N-acetylglucosamine (Guthrie et al., 2005). The major function of endochitinase is thought to be hydrolysis of chitin and formation of a structural polysaccharide, resulting in modification of cell wall morphogenesis (Sahai and Manocha, 1993; Arakane and Muthukrishnan, 2009). Chitinase and endochitinase activities would therefore have the physical effect of loosening cell walls. UDP-glucose 6-dehydrogenase was identified as a member of the sucrose metabolism pathway (Table 3). UDP-glucose 6-dehydrogenase is a pectin-degrading enzyme (Wei et al., 2015). Fruit firmness is closely related to soluble pectin content, and up-regulation of UDP-glucose 6-dehydrogenase expression may thus lead to fruit softening (Fischer and Bennett, 1991). Expression of UDP-glucose 6-dehydrogenase was found to be increased in melon fruits at a late stage of maturation (Guo et al., 2017). Additional KEGG pathways, including those related to sucrose metabolism (“starch and sucrose metabolism” and “galactose metabolism”) and amino acid metabolism (“alanine, aspartate, and glutamate metabolism,” “glycine, serine, and threonine metabolism,” and “tryptophan metabolism”), were shown to be up-regulated in the fruit peels of ST persimmon (Table S6). The up-regulation of sugar metabolism was reported to be consistent with the increased sugar content seen in persimmon and other fruits, such as apple and tomato, during ripening (Lee et al., 2007; Gautier et al., 2008). Biosynthetic pathways for secondary metabolites such as terpenoids, steroids, carotenoids, and alkaloids were also significantly up-regulated in fruit peels of ST persimmon (Table S6), indicating that fruit softening is a complex process accompanied by the dynamic degradation and generation of different metabolites.
KEGG analysis also showed that genes related to “photosynthesis” and “carbon fixation in photosynthetic organisms” decreased in expression as persimmons matured. Decreased photosynthesis during ripening was reported for other fruits such as apple, tomato, and strawberry (Paul and Driscoll, 1997; Lee et al., 2007; Wang Q. H. et al., 2017). During the transition from green to red fruits (chloroplast/chromoplast differentiation), mRNA levels for photosynthesis-specific polypeptides decreased in tomato (Piechulla et al., 1985). Tomato fruits undergo a physiological transition during the differentiation of photosynthetically active chloroplasts into non-photosynthetic chromoplasts, and it was suggested that this transition was coupled to changes in gene expression (Lytovchenko et al., 2011). RNA-seq analysis of strawberry also showed decreased expression of genes in the “photosynthesis” and “carbon fixation” pathways during ripening. Expression of the genes encoding the components of photosystems I and II decreased during persimmon softening (Figure 5B; Table 3). Similarly, chlorophyll content of photosystems I and II decreased significantly during rice leaf senescence (Tang et al., 2005). Reduced photosynthetic activity may lead to reduction of carbon fixation, but expression of genes involved in carbon fixation has not been well-characterized with respect to fruit ripening. This study revealed the identities of genes involved in carbon fixation, which was down-regulated during persimmon fruit softening (Figure 5C and Figure S4; Table 3).
After harvest, persimmon produces ethylene gas, which may stimulate fruit softening in climacteric fruits. Persimmons undergo stress following harvest due to loss of water and nutrients provided by the parent tree (Nakano et al., 2003). In persimmon, water loss induces expression of several ethylene biosynthesis genes, including ACC synthase (DkACS1, -2, and -3) and ACC oxidase (DkACO1 and -2). Expression of these genes is temporally and spatially regulated during fruit softening (Nakano et al., 2003). Consistently, our study also detected up-regulation of all the five ethylene biosynthesis genes by qRT-PCR analysis (Figure 7). However, these genes were undetected in assembled database except DkACO1 (persimmon1SL019260t0001), possibly, due to low expression levels at different sampling times (Table S2). DkACO1 was then cut off in our DEG analysis because of poor consistency among replicates. In contrast, ethylene response factor, ethylene receptor, and EIN3-binding F-BOX protein were consistently detected. Previous study had shown that ethylene biosynthesis were induced mainly in the calyx of persimmon and subsequently diffused to other fruit tissues (Nakano et al., 2003). Our results support that the peel of persimmon fruit is a site of ethylene perception as well as a site of ethylene biosynthesis.
Twelve ethylene response factors (DkERF11-22) were previously identified from persimmon (Min et al., 2014). In this study, DkERF21 and DkERF22 (gene IDs: 1SL019317t0001 and 1SL020456t0001) were up-regulated during softening (Table S2). The ethylene receptor genes DkERS1, DkETR1, and DkETR2 were identified in persimmon as homologs of Arabidopsis genes (Pang et al., 2007). Expression of DkERS1 and DkETR2 correlated with ethylene quantities during fruit softening, and expression was enhanced after ethylene treatment (Pang et al., 2007). The present study also found that DkERS1 and DkETR2 were up-regulated during persimmon fruit softening (Figure 6). In addition, four new ethylene-related transcripts (ERF25, EBF1, ETR3, and ERF24) that were up-regulated during persimmon softening were identified in this study. These genes may be good targets for future studies of persimmon softening.
This study investigated the transcriptional events occurring during persimmon fruit softening by identifying genes implicated in increased sugar metabolism, decreased photosynthetic capability, and increased ethylene production and ethylene-related functions. This transcriptome analysis provides baseline information on the identity and modulation of genes involved in softening of persimmon fruits.
Author Contributions
SR conceived, designed, supervised the research, and made final revision of the manuscript. JJ collected samples, generated data, and wrote a draft of the manuscript. SC assisted data analysis. SJ confirmed some data by repeating the qRT-PCR assay. BC revised the manucript. GA were involved in the sample collection. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The investigations reported here were supported by grants from the Next-Generation BioGreen 21 Program (SSAC) (PJ011091032015) and Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ01130803), Korea Rural Development Administration, and in part from the KRIBB Creative Project Program.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01556/full#supplementary-material
References
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. doi: 10.1186/gb-2010-11-10-r106
Arakane, Y., and Muthukrishnan, S. (2009). Insect chitinase and chitinase-like proteins. Cell. Mol. Life Sci. 67, 201–216. doi: 10.1007/s00018-009-0161-9
Biale, J. B. (1981). “Respiration and ripening in fruits retrospect and prospect,” in Recent Advances in the Biochemistry of Fruits and Vegetables, eds J Friend, and M. J. C. Rhodes (London: Academic Press), 1–39.
Brady, C. J. (1987). Fruit Ripening. Annu. Rev. Plant Physiol. 38, 155–178. doi: 10.1146/annurev.pp.38.060187.001103
Brummell, D. A., and Harpster, M. H. (2001). Cell wall metabolism in fruit softening and quality and its manipulation in transgenic plants. Plant Mol. Biol. 47, 311–340. doi: 10.1023/A:1010656104304
Brummell, D. A., Dal Cin, V., Crisosto, C. H., and Labavitch, J. M. (2004). Cell wall metabolism during maturation, ripening and senescence of peach fruit. J. Exp. Bot. 55, 2029–2039. doi: 10.1093/jxb/erh227
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Cherian, S., Figueroa, C. R., and Nair, H. (2014). ‘Movers and Shakers’ in the regulation of fruit ripening: a cross-dissection of climacteric vs. non-climacteric fruit. J. Exp. Bot. 65, 4705–4722. doi: 10.1093/jxb/eru280
Cordenunsi, B. R., and Lajolo, F. M. (1995). Starch breakdown during banana ripening: Sucrose synthase and sucrose phosphate synthase. J. Agric. Food Chem. 43, 347–351. doi: 10.1021/jf00050a016
Cox, M. P., Peterson, D. A., and Biggs, P. J. (2010). SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11:485. doi: 10.1186/1471-2105-11-485
Der, J. P., Barker, M. S., Wickett, N. J., and Wolf, P. G. (2011). De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics 12:99. doi: 10.1186/1471-2164-12-99
FAOSTAT. (2013). FAOSTAT Database. Available online at: http://faostat.fao.org/site
Feng, C., Chen, M., Xu, C. J., Bai, L., Yin, X. R., Li, X., et al. (2012). Transcriptomic analysis of Chinese bayberry (Myrica rubra) fruit development and ripening using RNA-Seq. BMC Genomics 13:19. doi: 10.1186/1471-2164-13-19
Fischer, R. L., and Bennett, A. B. (1991). Role of cell wall hydrolases in fruit ripening. Annu. Rev. Plant biol. 42, 675–703. doi: 10.1146/annurev.pp.42.060191.003331
Fluhr, R., Mattoo, A. K., and Dilley, D. R. (1996). Ethylene-biosynthesis and perception. Crit. Rev. Plant Sci. 15, 479–523.
Gautier, H., Diakou-Verdin, V., Bénard, C., Reich, M., Buret, M., Bourgaud, F., et al. (2008). How does tomato quality (sugar, acid, and nutritional quality) vary with ripening stage, temperature, and irradiance? J. Agric. Food. Chem. 56, 1241–1250. doi: 10.1021/jf072196t
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, 1178–1186. doi: 10.1093/nar/gkr944
Guo, S., Liu, J., Zheng, Y., Huang, M., Zhang, H., Gong, G., et al. (2011). Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genomics 12:454. doi: 10.1186/1471-2164-12-454
Guo, X., Xu, J., Cui, X., Chen, H., and Qi, H. (2017). iTRAQ-based protein profiling and fruit quality changes at different development stages of oriental melon. BMC Plant Biol. 17:28. doi: 10.1186/s12870-017-0977-7
Guthrie, J. L., Khalif, S., and Castle, A. J. (2005). An improved method for detection and quantification of chitinase activities. Can. J. Microbiol. 51, 491–495. doi: 10.1139/w05-020
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat. Protoc. 8, 10. doi: 10.1038/nprot.2013.084
Hobson, G. E. (1981). “Enzymes and texture changes during ripening,” in Recent Advances in the Biochemistry of Fruit and Vegetables, eds J. Friend, and M. J. C. Rhodes (London: Academic Press), 123–132.
Hou, Y., Meng, K., Han, Y., Ban, Q., Wang, B., Suo, J., et al. (2015). The persimmon 9-lipoxygenase gene DkLOX3 plays positive roles in both promoting senescence and enhancing tolerance to abiotic stress. Front. Plant Sci. 6:1073. doi: 10.3389/fpls.2015.01073
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Ito, S. (1971). “The persimmon” in The Biochemistry of Fruits and their Products, Vol. 2, ed A. C. Hulme (London: Academic Press), 281–301.
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, 277–280. doi: 10.1093/nar/gkh063
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, J. M., Joung, J. G., McQuinn, R., Chung, M. Y., Fei, Z., Tieman, D., et al. (2012). Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 70, 191–204. doi: 10.1111/j.1365-313X.2011.04863.x
Lee, Y. P., Yu, G. H., Seo, Y. S., Han, S. E., Choi, Y. O., Kim, D., et al. (2007). Microarray analysis of apple gene expression engaged in early fruit development. Plant Cell Rep. 26, 917–926. doi: 10.1007/s00299-007-0308-9
Liu, J. H., Zhang, J., Xu, B. Y., Zhang, J. B., Jia, C. H., Wang, J. S., et al. (2012). Expression analysis of banana MaECHI1 during fruit ripening with different treatments. Afr. J. Biotechnol. 11, 12951–12957. doi: 10.5897/AJB12.763
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, C., Zhang, Q., and Luo, Z. (2014). Genome-wide transcriptome analysis of Chinese pollination-constant nonastringent persimmon fruit treated with ethanol. BMC Genomics 15:112. doi: 10.1186/1471-2164-15-112
Lytovchenko, A., Eickmeier, I., Pons, C., Osorio, S., Szecowka, M., Lehmberg, K., et al. (2011). Tomato fruit photosynthesis is seemingly unimportant in primary metabolism and ripening but plays a considerable role in seed development. Plant Physiol. 157, 1650–1663. doi: 10.1104/pp.111.186874
Meyer, E., Aglyamova, G. V., Wang, S., Buchanan-Carter, J., Abrego, D., Colbourne, J. K., et al. (2009). Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 10:219. doi: 10.1186/1471-2164-10-219
Min, T., Fang, F., Ge, H., Shi, Y. N., Luo, Z. R., Yao, Y. C., et al. (2014). Two novel anoxia-induced ethylene response factors that interact with promoters of deastringency-related genes from persimmon. PLoS ONE 9:e97043. doi: 10.1371/journal.pone.0097043
Miszczak, A., Forney, C. F., and Prange, R. K. (1995). Development of aroma volatiles and color during postharvest ripening of ‘Kent’ strawberries. J. Amer. Soc. Hort. Sci. 120, 650–655.
Mowat, A. D., Collins, R. J., and George, A. P. (1995). Cultivation of persimmon (Diospyros kaki L.) under tropical conditions. Acta Hortic. 409, 141–150. doi: 10.17660/ActaHortic.1995.409.17
Nakagawa, T., Nakatsuka, A., Yano, K., Yasugahira, S., Nakamura, R., Sun, N., et al. (2008). Expressed sequence tags from persimmon at different developmental stages. Plant Cell Rep. 27, 931–938. doi: 10.1007/s00299-008-0518-9
Nakano, R., Ogura, E., Kubo, Y., and Inaba, A. (2003). Ethylene biosynthesis in detached young persimmon fruit is initiated in calyx and modulated by water loss from the fruit. Plant Physiol. 131, 276–286. doi: 10.1104/pp.010462
Nishiyama, K., Guis, M., Rose, J. K., Kubo, Y., Bennett, K. A., Wangjin, L., et al. (2007). Ethylene regulation of fruit softening and cell wall disassembly in Charentais melon. J. Exp. Bot. 58, 1281–1290. doi: 10.1093/jxb/erl283
Oeller, P. W., and Min-Wong, L. (1991). Reversible inhibition of tomato fruit senescence by antisense RNA. Science 254, 437. doi: 10.1126/science.1925603
Ortiz, G., Sugaya, S., Sekozawa, Y., Ito, H., Wada, K., and Gemma, H. (2006). Expression of 1-aminocyclopropane-1-carboxylate synthase and 1-aminocyclopropane-1-carboxylate oxidase genes during ripening in ‘Rendaiji’ persimmon fruit. J. Jpn. Soc. Hort. Sci. 75, 178–184. doi: 10.2503/jjshs.75.178
Pang, J. H., Ma, B., Sun, H. J., Ortiz, G. I., Imanishi, S., Sugaya, S., et al. (2007). Identification and characterization of ethylene receptor homologs expressed during fruit development and ripening in persimmon (Diospyros kaki Thumb.). Postharvest Biol. Technol. 44, 195–203. doi: 10.1016/j.postharvbio.2006.12.017
Paul, M. J., and Driscoll, S. P. (1997). Sugar repression of photosynthesis: the role of carbohydrates in signalling nitrogen deficiency through source: sink imbalance. Plant Cell Environ. 20, 110–116. doi: 10.1046/j.1365-3040.1997.d01-17.x
Piechulla, B., Imlay, K. R. C., and Gruissem, W. (1985). Plastid gene expression during fruit ripening in tomato. Plant Mol. Biol. 5, 373–384. doi: 10.1007/BF00037558
Sahai, A. S., and Manocha, M. S. (1993). Chitinases of fungi and plants: their involvement in morphogenesis and host-parasite interaction. FEMS Microbiol. Rev. 11, 317–338. doi: 10.1111/j.1574-6976.1993.tb00004.x
Schulz, M. H., Zerbino, D. R., Vingron, M., and Birney, E. (2012). Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092. doi: 10.1093/bioinformatics/bts094
Sitrit, Y., and Bennett, A. B. (1998). Regulation of tomato fruit polygalacturonase mRNA accumulation by ethylene: A re-examination. Plant Physiol. 116, 1145–1150. doi: 10.1104/pp.116.3.1145
Tang, Y., Wen, X., and Lu, C. (2005). Differential changes in degradation of chlorophyll–protein complexes of photosystem I and photosystem II during flag leaf senescence of rice. Plant Physiol. Biochem. 43, 193–201. doi: 10.1016/j.plaphy.2004.12.009
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Tucker, G. A., and Brady, C. J. (1987). Silver ions interrupt tomato fruit ripening. J. Plant Physiol. 127, 165–169. doi: 10.1016/S0176-1617(87)80051-2
Vera, J. C., Wheat, C. W., Fescemyer, H. W., Frilander, M. J., Crawford, D. L., Hanski, I., et al. (2008). Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol. 17, 1636–1647. doi: 10.1111/j.1365-294X.2008.03666.x
Vilas-Boas, E. V. D. B., and Kader, A. A. (2007). Effect of 1-methylcyclopropene (1-MCP) on softening of fresh-cut kiwifruit, mango and persimmon slices. Postharvest Biol. Technol. 43, 238–244. doi: 10.1016/j.postharvbio.2006.09.010
Wang, Q. H., Zhao, C., Zhang, M., Li, Y. Z., Shen, Y. Y., and Guo, J. X. (2017). Transcriptome analysis around the onset of strawberry fruit ripening uncovers an important role of oxidative phosphorylation in ripening. Sci. Rep. 7:41477. doi: 10.1038/srep41477.
Wang, R., Xu, S., Wang, N., Xia, B., Jiang, Y., and Wang, R. (2017). Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea. Front. Plant Sci. 7:1971. doi: 10.3389/fpls.2016.01971
Wei, J., Qi, X., Cheng, Y., and Guan, J. (2015). Difference in activity and gene expression of pectin-degrading enzymes during softening process in two cultivars of Chinese pear fruit. Sci. Hortic. 197, 434–440. doi: 10.1016/j.scienta.2015.10.002
Keywords: Diospyros kaki, persimmon softening, de novo RNA sequencing, ethylene, transcriptional events
Citation: Jung J, Choi SC, Jung S, Cho B-K, Ahn G-H and Ryu SB (2017) A Transcriptome Approach Toward Understanding Fruit Softening in Persimmon. Front. Plant Sci. 8:1556. doi: 10.3389/fpls.2017.01556
Received: 17 February 2017; Accepted: 25 August 2017;
Published: 12 September 2017.
Edited by:
Prem Bhalla, University of Melbourne, AustraliaReviewed by:
Ryo Fujimoto, Kobe University, JapanAutar Krishen Mattoo, United States Department of Agriculture, United States
Copyright © 2017 Jung, Choi, Jung, Cho, Ahn and Ryu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen B. Ryu, c2JyeXVAa3JpYmIucmUua3I=
†Present Address: Sang Chul Choi, Department of Crop Science, College of Agriculture and Life Sciences, Chungnam National University, Daejeon, South Korea