 Bourlaye Fofana1*
Bourlaye Fofana1* Kaushik Ghose1†
Kaushik Ghose1† Ashok Somalraju1
Ashok Somalraju1 Jason McCallum1
Jason McCallum1 David Main1
David Main1 Michael K. Deyholos2
Michael K. Deyholos2 Gordon G. Rowland3
Gordon G. Rowland3 Sylvie Cloutier4
Sylvie Cloutier4- 1Charlottetown Research and Development Centre, Agriculture and Agri-Food Canada, Charlottetown, PE, Canada
- 2Department of Biology, University of British Columbia, Kelowna, BC, Canada
- 3Department of Plant Science, Crop Development Centre, University of Saskatchewan, Saskatoon, SK, Canada
- 4Ottawa Research and Development Centre, Agriculture and Agri-Food Canada, Ottawa, ON, Canada
Flax secoisolariciresinol (SECO) diglucoside (SDG) lignan is an emerging natural product purported to prevent chronic diseases in humans. SECO, the aglycone form of SDG, has shown higher intestinal cell absorption but it is not accumulated naturally in planta. Recently, we have identified and characterized a UDP-glucosyltransferase gene, UGT74S1, that glucosylates SECO into its monoglucoside (SMG) and SDG forms when expressed in yeast. However, whether this gene is unique in controlling SECO glucosylation into SDG in planta is unclear. Here, we report on the use of UGT74S1 in reverse and forward genetics to characterize an ethyl methane sulfonate (EMS) mutagenized flax population from cultivar CDC Bethune and consisting of 1996 M2 families. EMS mutagenesis generated 73 SNP variants causing 79 mutational events in the UGT74S1 exonic regions of 93 M2 families. The mutation frequency in the exonic regions was determined to be one per 28 Kb. Of these mutations, 13 homozygous missense mutations and two homozygous nonsense mutations were observed and all were transmitted into the M3 and M4 generations. Forward genetics screening of the population showed homozygous nonsense mutants completely lacking SDG biosynthesis while the production of SMG was observed only in a subset of the M4 lines. Heterozygous or homozygous M4 missense mutants displayed a wide range of SDG levels, some being greater than those of CDC Bethune. No additional deleterious mutations were detected in these mutant lines using a panel of 10 other genes potentially involved in the lignan biosynthesis. This study provides further evidence that UGT74S1 is unique in controlling SDG formation from SECO and this is the first report of non-transgenic flax germplasm with simultaneous knockout of SDG and presence of SMG in planta.
Introduction
Flax lines with high linolenic acid (Duguid et al., 2014), low linolenic acid (Green and Marshall, 1984; Rowland, 1991; Dribnenki et al., 1996), and low cyanogenic glucosides (Duguid, personal communications) are some of the major achievements in linseed genetics and breeding. Whereas, the first two attributes are associated with fatty acid content, the last one refers to flax seed content in linustatin and neolinustatin, two cyanogenic glucosides that are toxic to mammals (EFSA, 2007). The nutritional benefits of flaxseed linolenic acid in human and animal health are now well established, especially for the positive roles it plays in reducing incidence of cardiovascular diseases (Simopoulos, 2008). Flaxseed is also the richest source of lignans (Thompson et al., 2006; Pan et al., 2009; Touré and Xueming, 2010) for which a wide variety of purported health benefits have been reported (Webb and McCullough, 2005; Adolphe et al., 2010; Buck et al., 2011; Wang et al., 2015). In planta, flax lignans are usually found as secoisolariciresinol diglucosides (SDGs) ester-linked within an oligomeric matrix called the lignan macromolecule (Struijs et al., 2009; Kosińska et al., 2011). Its monomeric aglycone (SECO) and intermediate monoglucoside (SMG) forms do not naturally accumulate in planta.
Lignan metabolism in mammals has been the focus of many detailed studies (During et al., 2012; Udani et al., 2013; Setchell et al., 2014; Mukker et al., 2015). After consumption, the lignan macromolecule complex is hydrolyzed, SDG is deglucosylated into SECO and absorbed in the gut (Clavel et al., 2007). Non-absorbed SECO (50–72% of ingested SECO) is subsequently metabolized into the enterolignans, enterolactone (ENL), and enterodiol (END) mainly in the colon by the intestinal microflora (Clavel et al., 2006, 2007; Landete et al., 2015). Enterolignans have been reported to elicit modest estrogen-like activity by binding estrogen receptors (ERs), ERα or ERβ (Satake et al., 2015). Nonetheless, low concentrations of intact lignans have been detected in the sera of mammals fed lignan-rich diets, suggesting that non-metabolized lignans are taken up by the mammalian digestive system and manifest ER-independent activities in vivo and in vitro (During et al., 2012; Satake et al., 2015). As a β-glucoside however, SDG metabolism may vary between individuals due to differences in their gut microflora (Rowland et al., 2000; Clavel et al., 2005; Landete et al., 2015), thereby affecting its bioavailability in some individuals (Possemiers et al., 2007) similar to what has already been shown with isoflavone diazein, which is metabolized only by one third of humans into its bioactive S-equol form (Setchell and Cole, 2006; Landete et al., 2015). Recently, During et al. (2012) reported a linear increase in the uptake of lignan aglycone forms, including pinolariciresinol (PINO), SECO, and ENL, by human intestinal Caco-2 cells by simple diffusion or low affinity transporter. Less than 0.1% SDG absorption was observed, compared to 2% for SECO and PINO, and 7% for ENL, evidencing the effect of glucosylation on absorption and bioavailability. However, no SMG of flax lignan has been available so far for such testing. Due to its lower molecular weight of 524.6 g/mol and the lack of a second bulky and polar glucoside moiety, it is logical to conceive that SMG uptake by Caco-2 cells through diffusion would exceed that of SDG which has a molecular weight of 686.7 g/mol. Currently, SECO and SMG are only obtained after acid hydrolysis of SDG (Li et al., 2008), a process that is not environmentally friendly. Because SDG deglucosylation is a pre-requisite to any absorption or conversion into END and ENL in vivo, production of seeds with altered SDG glucosylation in vivo is of interest for designing a highly bioavailable functional flaxseed food. It would be advantageous to create flax lines with reduced lignan glucosylation in the forms of SECO or SMG in planta.
Glycosylation is a biological process that leads to chemical complexity and diversity of plant natural products (Osmani et al., 2009; Wang and Hou, 2009). This synthesis modification is catalyzed by enzymes of the glycosyltransferases (GT) superfamily in which family 1 GT, refers to uridine glycosyl transferases (UGTs) (Caputi et al., 2012) that are characterized by a PSPG box consisting of a 44 amino acid consensus signature motif (Gachon et al., 2005). UGTs transfer UDP-activated sugar moieties to specific acceptor molecules (Witte et al., 2009) and their glycosylation activities on acceptors can modulate their pharmacological activity (Osmani et al., 2009). Recently, UGT74S1 (JX011632), a family 1 GT, was identified in flax and characterized to sequentially glucosylate SECO into SMG and SDG (Ghose et al., 2014; Figure 1). UGT74S1 has also been reported to be a single-copy gene and the key player in controlling SDG formation in flax (Fofana et al., 2017). Furthermore, Trp355 and His352 were found to be critical for the UGT74S1 glucosylation activity toward SECO whereas Gln337 and Ser357 appeared essential for SMG conversion into SDG in vitro (Ghose et al., 2015). Until now however, no study has assessed the direct effect of UGT74S1 mutations in planta. Moreover, despite growing interests in flax lignans, breeding for SDG per se has so far received little attention (Pavelek et al., 2015), with most studies being limited to observed variations among flax varieties and accessions within collections (Saastamoinen et al., 2013). So far, no flax lines with altered lignan glucosylation have been reported.
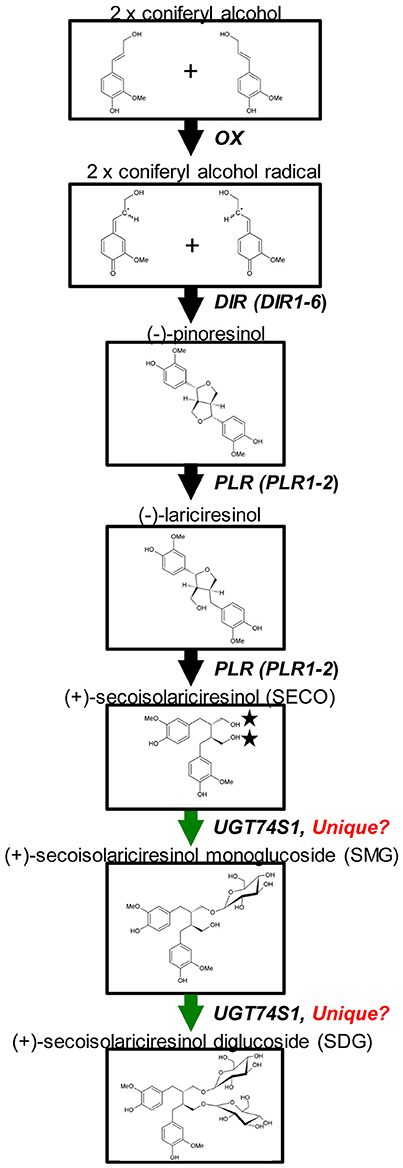
Figure 1. Current knowledge of flax lignan biosynthetic pathways. Genes with known functions are shown (adapted from Kim et al., 2009) with the permission of Dr. Honoo Satake and PCP editorial office and from Ghose et al. (2014) with the permission of BMC Plant Biology editorial office.
Flaxseed is a health-functional crop and the commercial production of genetically modified flax remains an impediment to conquering such markets (Fofana et al., 2010). Mutation breeding is a well-established method in flax improvement and has led to the development of cultivars with reduced linolenic acid (Green and Marshall, 1984; Rowland et al., 1995; Ntiamoah and Rowland, 1997). Targeting Induced Local Lesions in Genomes (TILLING) is a reverse genetic tool used in functional genomics and crop improvement (McCallum et al., 2000; Sikora et al., 2011). Traditional mutagenesis, including radiation or chemical mutagenesis, is followed by the detection of mutations in the causal gene(s) responsible for the phenotype(s) of interest (McCallum et al., 2000; Slade and Knauf, 2005; Uchida et al., 2011; Chantreau et al., 2013). Recently, for example, Chantreau et al. (2013) developed a flax EMS mutagenized population and characterized the roles of coumarate-3-hydroxylase (C3H) and cinnamyl alcohol dehydrogenase (CAD) in fiber biosynthesis in flax stems by TILLING via forward and reverse genetics. The chemical mutagen, ethyl methane sulfonate (EMS), generates a wide range of mutant alleles, primarily point mutations (Comai and Henikoff, 2006) and, current advances in next generation sequencing have greatly improved the efficiency of induced-mutation detection (Galindo-González et al., 2015). With the release of the flax genome sequence (Wang et al., 2012), the DNA sequence of almost all flax genes is known. However, the gene functions and their link to phenotypic traits remain largely incomplete or completely unknown.
The present study was undertaken to (1) characterize an EMS mutagenized flaxseed population through the identification of lines carrying mutations in UGT74S1 and displaying altered lignan glucosylation phenotypes, and (2) determine the extent of the role played by UGT74S1 in SDG formation through SECO glucosylation. Using a next generation sequencing platform for the amplicon re-sequencing of the UGT74S1 gene in an EMS mutagenized flax population, we identified stable mutant lines with altered SDG lignan profile and demonstrated that the loss-of-function of SECO glucosylation into SDG was attributable solely to UGT74S1.
Materials and Methods
Plant Materials
A total of 1996 M2 EMS mutagenized flax families were obtained from Dr. Gordon G. Rowland (Crop Development Centre, University of Saskatchewan, Saskatoon, SK) who multiplied them before distribution. These M2 families were originally developed by Dr. Michael K. Deyholos' laboratory (University of Alberta, Edmonton, AB) as part of the Genome Canada's Total Utilization Flax GENomics (TUFGEN) project. To generate these lines, seeds of flax (Linum usitatissimum L.) cultivar CDC Bethune (Rowland et al., 2002) were treated with 0.5% (v/v) EMS for 4 h, planted and grown to seed set. M2 seeds from M1 plants were harvested separately as previously described (Galindo-González et al., 2015). Five seeds from each of the 1996 M2 families were hand sown in 30 cm rows in 2011 at the Harrington Research Farm of Agriculture and Agri-Food Canada (Charlottetown, PEI) along with 10 rows of untreated CDC Bethune. Individual EMS plants within a row are referred to as lines and, all plants within a row constitute a family. Leaves of one line from each M2 family and 20 CDC Bethune plants were collected, frozen in liquid nitrogen and stored at −80°C for DNA extraction and reverse genetic analysis. All M2 plants were destroyed before flowering to avoid any potential cross pollination and seed production in the field. Re-sampling from the original M2 seed stock was also performed, whenever necessary, by planting in greenhouse to generate subsequent mutant generations.
Reverse Genetics of UGT74S1
Genomic DNA Extraction and Targeted UGT74S1 Gene Amplification
Leaf samples of each 1996 M2 lines and 20 CDC Bethune plants were arranged in individual wells of 21 × 96-well plates containing sterile tungsten beads (Sigma Aldrich, CA, USA), freeze-dried and genomic DNA was extracted using the Qiagen DNeasy Plant Mini Kit (Qiagen, Mississauga, ON) following the manufacturer's recommendations. DNA was quantified using Picogreen (Life technologies, CA, USA) and the quality was verified by agarose gel electrophoresis.
For the amplification and re-sequencing, the genomic DNA region of UGT74S1 (Supplementary Figures 1A,B) corresponding to locus Lus10017825.g (scaffold35, position 7,827–9,972) of the flax draft genome (Phytozme.jgi.doe.gov) was targeted using universal and standard tagging approaches. The first exon of the gene was amplified and tagged using the universal tagging approach, where the gene specific primers flax1_F1b (GAAGGTGACCAAGTTCATGCACCCCACTTCTCCAATTCTC) and flax1_R1 (AATGCGTTACAAGCACATCTCGATTATCATTGCTCGAAACGTG) contained a 5′ tail identical to primers TRS_F (GAAGGTGACCAAGTTCATGC) and TRS_R (AATGCGTTACAAGCACATCTC), respectively. From the TRS_F and TRS_R primers, 96 versions carrying an additional 8-base barcode at the 5′-end were used (Supplementary Table 1). The second exon was amplified and tagged by a more conventional approach using only two primers per PCR reaction. For each gene-specific primer, 96 versions of UGT74S1 gene-specific primer (flax1_F2b GTCAATCGAGCCAACTCTCG) carrying an additional 8-base barcode at the 5′ end (Supplementary Table 2) were used in combination with gene-specific primer flax1_R2b (ACAAAACCTATCTTCCCAAGTCA). The 20 μl PCR reactions were set up using the 5 × MyTaq Mastermix (Bioline, Berlin, Germany). The standard tagging reactions contained 5 pM of each primer, while the universal tagging reactions contained 2 pM of each gene-specific primer and 20 pM of each TRS primer. Cycling conditions consisted of 2 min at 96°C for pre-denaturation followed by 35 cycles of 15 s at 96°C, 30 s at 55°C, and 2 min at 72°C.
Library Construction and Pyrosequencing
After PCR amplifications, amplicons from each exonic region in each 96-well plate were pooled, purified using MinElute gel purification kit (Qiagen) to remove non-specific amplification products, size-selected and quantified. The two gel-purified amplicon pools derived from each individual exonic DNA plate were mixed in equimolar amounts to create 21 pools, which were further split into a 10 and an 11 pool sets. DNA from each of the 11 or 10 pool sets were ligated to 11 or 10 different 454 multiplex identifier (MID) adaptors and two Roche/454-compatible sequencing libraries were constructed and sequenced by pyrosequencing at LGC Genomics GmbH (Berlin, Germany). All sequencing reactions were based upon FLX-Titanium chemistry (Roche/454 Life Sciences, Branford, CT, USA; www.454.com) and all methods were performed according to manufacturer's protocols. The resulting sequences were processed using the standard Roche software for base calling, adaptor and quality trimming (Genome Sequencer FLX System Software manual version 2.3).
Bioinformatics of Sequence Reads and EMS-Induced SNP Detection in M2 Plants
The sequencing reads were deconvoluted using an in-house-developed tag sorting and clipping program, allowing no more than a single nucleotide mismatch. The barcodes were sorted and trimmed, and individual FASTQ files were generated for each sample.
To confirm the reference identity of the wild type target sequence, reads from the 20 wild type CDC Bethune plants were aligned to the genomic reference sequence of UGT74S1 (Lus10017825.g) using Bowtie2 v2.1.0 (Langmead and Salzberg, 2012) with default parameters in “local” alignment mode. The alignments were coordinate-sorted and saved in a standard Binary Sequence Alignment/Map (BAM) format. Subsequently, GATK v1.6-11 (http://www.broadinstitute.org/gatk/) was used to call SNPs by running the GATK walkers on the alignment files. Using GATK, IndelRealigner and UnifiedGenotyper were run to realign reads around indels called and to generate raw SNP calls, respectively. To identify and retain high quality SNPs, QualFilter (QUAL ≥ 30.0), QDFilter (QD ≥ 5.0, Quality-by-Depth), DPFilter (DP ≥ 10, sample read depth at locus), StrandFilter (FS ≤ 60.0, Phred-scaled p-value using Fisher's Exact Test to detect strand bias), and ABHetFilter (ABHet ≤ 0.9, testing allele balance for heterozygous) were applied using Variant Filtration. The final results were stored in raw and filtered standard variant call format (VCF) files. The VCF files from GATK were further subjected to homopolymer filtering to reduce false-positive calls in homopolymer-dense regions and, this was followed by a final manual inspection of the SNP calls to generate consensus sequences.
To detect EMS-induced SNP in M2 plants, reads from all 1996 M2 EMS lines were aligned using Bowtie2 v2.1.0 as described earlier, and raw and filtered SNPs were called. SNPs causing codon changes were identified using SnpEff v3.2 (http://snpeff.sourceforge.net/). Exon positions described for UGT74S1 (Ghose et al., 2014) were used to set up a SnpEff database for automatic annotation of VCF files (http://snpeff.sourceforge.net/SnpEff_manual.html#output) and an allele table was constructed for all samples.
A consensus sequence was generated using sequences derived from all samples. Final VCF files from the 1996 M2 lines were used to produce one putative genomic sequence FASTA file per sample. Regions not covered by the PCR amplicons were soft-masked using sequence changed into lowercase, likewise for regions with a read coverage below 10× (for bases with Phred score ≥20). Heterozygous SNP calls were annotated with standard ambiguous nucleotide sequence code (http://www.bioinformatics.org/sms2/iupac.html). The genomic sequences were subsequently used to generate putative coding sequence (CDS) FASTA files and haplotype sequences were generated. Predicted peptide sequences were obtained from the CDS using a standard DNA codon table.
The mutation rate was determined from UGT74S1 sequence for all the EMS lines sequenced, as performed routinely in EMS TILLING population. The mutation rate in the exonic region was calculated as followed: [(exon 1 + exon 2) × total number of original EMS lines sequenced]/total number of variants observed. Mutational effects for each of the missense and nonsense mutations were predicted using the SuSPect method, a standalone web server (Yates et al., 2014; http://www.sbg.bio.ic.ac.uk/phyre2/html/help.cgi?id=help/investigator). The wild type UGT74S1 protein sequence was uploaded and a heat map was generated for each amino acid position in the protein against a substitution by any of the 20 possible amino acids with color-coded effect level.
Forward Genetics Screening for Altered SDG Lignan Phenotypes in M2 Families Carrying UGT74S1 EMS-Induced Mutations
A total of 93 M2 families were identified to carry at least one mutation in UGT74S1, of which 18 carried sense mutations and were not considered for further analysis. Among the remaining 75 M2 families carrying EMS-induced missense and nonsense mutations, six had few or no seeds available and could also not be further tested. Because seeds were not collected from the M2 plants grown in the field that were used in reverse genetics, a new set of original M2 seeds were resampled from the remaining 69 M2 families, grown in a greenhouse and subjected to further analyses.
In a first step, forward genetics was performed using a subsample of original M2 seeds from each of the 69 M2 families in which missense or nonsense mutations were detected for assessing the effect of mutations on SDG production. Total lignan was extracted from 50 mg of seeds per family and hydrolyzed by alkaline hydrolysis as described by Ghose et al. (2014). The wild type CDC Bethune was used as control. Non-hydrolyzed lignan polymer and hydrolyzed lignan were purified from families 1230 and 2340 which had nonsense mutations and separated by UPLC to assess the potential effect of mutations on the lignan macromolecule and its constituent phenolic acid glucoside profile. SDG was hydrolyzed from all 69 M2 families, purified, separated by UPLC, confirmed by mass spectrometry and quantitated as previously reported (Ghose et al., 2014). Three independent extractions were performed and analyzed separately. SDG level from each M2 family was compared to that of CDC Bethune.
Validation of EMS-Induced SNPs in M2 Plants
After initial SDG lignan determination in the 69 M2 families, we sought to validate the mutations detected by amplicon resequencing in the M2 seeds. Five M2 seeds from each of the 69 M2 families were re-sampled, planted and grown as individual lines in the greenhouse along with CDC Bethune. Each germinated plant was assigned a number and grown to maturity. Hereafter, individual mutant lines are designated by the family number from which they were derived, followed by a number and, if needed, a letter identifier. M3 seeds and leaf tissues were collected separately from each individual M2 plant (or line). At this point, we retained plants (lines) of 28 M2 families with nonsense or missense mutations and that produced enough M3 seeds for further testing (Supplementary Table 3). Genomic DNA was extracted using the Mag-Bind Plant DNA plus Kit (Omega Bio-tek, Norcross, GA, USA) following manufacturer recommendations. SNP re-validation was performed by genotyping 1 or 2 plants (or lines) per family using Kompetitive allele specific PCR (KASP, LGC Genomics, Beverly, MA) assays. A total of 20 KASP assay, were designed to target 16 missense and four nonsense mutations (Supplementary Table 4). Each reaction was carried out using 11–22 technical replicates of each DNA sample and two no template controls to ensure proper clustering as recommended by the supplier. All reactions were performed on a CFX 96 Real Time PCR system (BioRad, Mississauga, ON, Canada) in a total volume of 10 μL containing 5 ng of DNA, 5 μL of 2X KASP master mix, and 0.14 μL of KASP-specific assay mix. The first step of the touchdown PCR included an initial denaturation at 94°C for 15 min, followed by 10 cycles consisting of a denaturation at 94°C for 20 s, a touchdown annealing step starting at 61°C for 60 s and achieving a final annealing at 55°C with 0.6°C decreases per cycle. The second step consisted of 26 cycles, each including a denaturation at 94°C for 20 s and a combined annealing and extension step at 55°C for 60 s. Completed KASP reactions were read at 37°C for 1 min. Two to four additional KASP recycling programs were performed for each assay to improve sample grouping, obtain tighter data point clusters and increase confidence in genotype assignment. A single recycling program consists of three cycles with denaturation at 94°C for 20 s followed by an annealing/extension step at 57°C for 60 s.
Stability of EMS-Induced Mutations in M3 Plants
To determine the stability and heritability of the EMS-induced mutations confirmed in the M2 flax lines by KASP genotyping, 3–45 viable M3 seeds per M2 plants were planted and grown to maturity in single pots in a greenhouse and from which M4 seeds were harvested. Leaf tissue was collected from each M3 plant, genomic DNA extracted, and KASP genotyping was performed as described above.
Forward Genetics Screening for Altered SDG Lignan Phenotypes in M4 Plants Carrying UGT74S1 EMS-Induced Mutations
After re-validation of the EMS-induced mutations in M2 and M3 plants, the stability of changes in the SDG lignan profiles were assessed in the M4 seeds produced by single seed descent of M3 plants derived from eight families (Table 1) as previously described (Ghose et al., 2014).
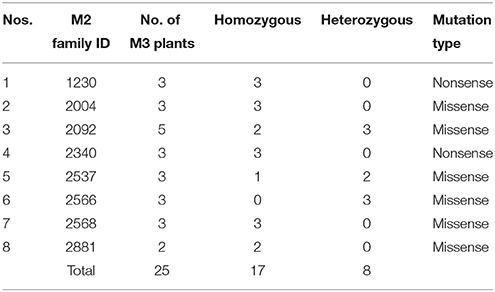
Table 1. M2 families and single seed descent M3 plants with UGTS74S1 mutations from which M4 seeds were collected and used for phenotypic characterization of SDG lignan content.
Confirmation or Detection of EMS-Induced Mutations in UGT74S1 and Other SDG Biosynthetic Genes Using Targeted Gene Panel Ampliseq Sequencing
The flax SDG biosynthetic pathway is relatively well-known (Figure 1). To determine whether or not random EMS mutations were also induced in other SDG-related biosynthetic genes of the mutant lines, Ampliseq sequencing of 11 genes, including UGT74S1, was performed using the Ion PGM™ system (Life Technologies/Thermo Fischer, CA, USA), as reported by Nishio et al. (2015). The plant material used consisted of a total of 84 M4 plants from 14 lines in six families previously shown to carry homozygous mutations (Supplementary Table 5). Nine CDC Bethune plants were used as positive control. DNA was extracted from each individual plant using the magnetic bead based Mag-Bind Plant DNA Plus 96 extraction kit (Omega Bio-tek) as described earlier and quantified using a Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) following manufacturer's instructions.
Custom Target Gene Panel Design
A custom Ion AmpliSeq™ panel targeting 11 genes, including six dirigent proteins (DIR 1–DIR 6), two PINO reductases (PLR 1 and 2), reported to be involved in SECO biosynthesis (Halls et al., 2005; Nakatsubo et al., 2008; Gasper et al., 2016) and three UGTs (UGT74S1, UGT74S3, and UGT74S4) recently shown to play different roles in SECO glucosylation (Ghose et al., 2014, Fofana et al., 2017) was designed using Ion AmpliSeq Designer (IAD) software v.2.0.3 (Thermo Fisher Scientific, CA, USA). For each target, the 50 bp upstream and downstream of the genes were added to cover the complete coding sequence. The custom panel was designed in such a way that 11,226 of the 11,248 bp potential targeted regions were covered, accounting for 99.6% coverage (22 bp missing) and included 49 primer pairs divided into two pools of 27 and 22 primer pairs each, respectively (Supplementary Table 6). The targeted 11,248 bp genomic regions were uploaded as a reference BED file sequence. This Ampliseq custom panel was used to screen the 84 EMS mutagenized and nine wild type CDC Bethune plants.
Library Preparation and Sequencing
Genomic DNA and library preparation were performed following Life Technology/Thermo Fisher recommendations. First, pooled barcoded libraries were prepared by pooling equal amount of DNA from three to eight M4 plants from each line per family. Twenty nanograms of genomic DNA was amplified through two multiplex PCR reactions using 27 and 22 sets of Ampliseq primers, respectively (Supplementary Table 6). A total of 12 pooled DNA libraries (11 from mutant lines and one from CDC Bethune) were prepared using the Ion Ampliseq™ Library kit 2.0 (Thermo Fisher Scientific, Carlsbad, CA, USA) according to manufacturer's instructions. Second, individual libraries were prepared from individual M4 plants in each of the lines from which mutations were detected in the pooled libraries. All libraries were quantified using the qPCR based Ion library quantitation kit (Thermo Fisher Scientific), diluted to 30 pM and subjected to sequencing template preparation using the Ion PGM™ Hi-Q™ Chef Kit and the Ion Chef Instrument (Thermo Fisher Scientific) following manufacturer's protocols. Ion 316 and 314 chips were used for loading and sequencing samples of the pooled and individual libraries, respectively. The sequencing was performed using an Ion Torrent Personal Genome Machine (PGM) (Thermo Fisher Scientific). For each run, four libraries were pooled together on each 316-chip, for a minimum of 500× coverage of the 49 amplicons, whereas five individual libraries were pooled together on each 314-chip to achieve a minimum of 100× coverage. All sequencing runs were performed using the Ion PGM HiQ Sequencing kit (Thermo Fisher Scientific) with 850 flows.
Mapping Sequencing Data and Variant Call
The Ampliseq sequence data was processed using the Ion Suite Software v 5.04 and the Ion Server, and mapped to the flax Ampliseq panel targeted genomic regions uploaded as the reference BED file. Variants were called using Torrent Variant Caller plug-in software set to default parameters under high stringency (minimum allele frequency 0.1, minimum SNP quality 10 and minimum coverage of 5 for SNP identification; minimum allele frequency 0.1, minimum SNP quality 10 and a minimum coverage of 10 for INDEL identification) as performed by Nishio et al. (2015). The selected variant positions were detected, genotyped and reported as wild type, heterozygous or homozygous. For each variant, its mutational effect was predicted using the phyre 2 investigator software as previously reported (Ghose et al., 2015).
Results
Targeted Amplicons of UGT74S1
The two genomic amplicons containing the UGT74S1 gene were successfully amplified and sequenced from the 2016 plants. Amplicon 1 included 55 nucleotides of the 5′UTR, the 651 nucleotides of exon 1, and 274 nucleotides of the single intron. Amplicon 2 covered the last 93 nucleotides of the intron, the 756 nucleotides of exon 2, and 248 nucleotides of the 3′UTR. It is worth mentioning that only a portion of the intron, represented by 367 out of the 739 nucleotides of the intronic region was amplified and sequenced (Figure 2).
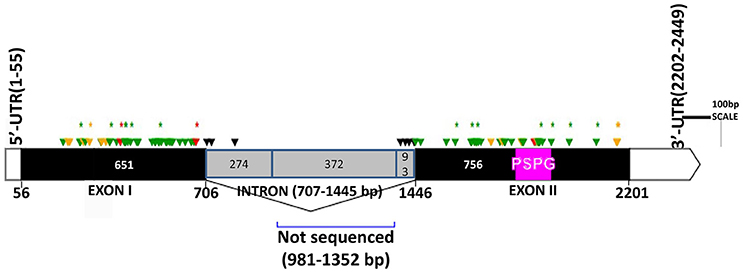
Figure 2. Distribution of EMS-induced mutations within the flax UGT74S1. UTRs are indicated by blank boxes, exons by solid black boxes, and the intron by a gray box. The PSPG region within exon 2 is colored in magenta. Green arrows, missense mutations; yellow arrows, sense mutations; red arrows, nonsense mutations; black arrows, mutations in intron; asterisks (*) indicate position of homozygous mutations only; Numbers are in bp and the starts and ends of exons are indicated in bp.
EMS-Induced Mutations Detected in UGT74S1
Following SNP variants call and filtrations, a total of 80 positions displayed SNP variants, including 40 positions in exon 1, 7 intronic positions, and 33 positions in exon 2 (Table 2). Only 3.12% of the calls were missing for the 80 SNP positions of the 2016 individuals. Since EMS mutagenesis usually causes transition mutations, GC content was determined in the amplicons. Exon 1 had a GC content of 54% and a higher proportion of SNP positions than exon 2 which had a GC content of 46% (Table 2). In the M2 lines sequenced, heterozygosity at SNP positions was observed four times more frequently than homozygosity, showing that most of the EMS-induced mutations were not fixed in the M2 population. Among the exonic mutations, 22.5% were homozygous, thus fixed in these plants. Because not all substitutions have the same mutational effects on gene functionality, the mutational effect for each mutation was determined. Ten mutated amino acids at 12 positions showed high to very high mutational effects, while another 16 amino acid changes were found with low mutational effects at 16 positions (Table 3). Interestingly, two of the mutations with high mutational effects appeared to be nonsense homozygous mutations, thereby expected to have a deleterious effect on SECO glucosylation. Altogether, 73 SNPs were observed in the exonic regions in 93 single plants originating from different M2 families (Figure 2, Table 3). Seven of the 93 M2 plants had two exonic SNPs each (labeled with * in Table 3). Because six SNP positions had both a homozygous and a heterozygous SNP variants, 79 variants were identified at the 73 exonic SNP positions and, of those, 55 were missense, six were nonsense, and 18 were sense mutations. Only 18 of the 79 SNP variants were homozygous including 13 of the missense, 2 of the nonsense, and 3 sense mutations (Table 3). The mutation rate in the exonic regions was calculated to be 1 per 28 kb [i.e., (651 + 756 bp) × 1996/100 = 28,084 bp/1 or 1 mutation in 28 kb]. Eight of the missense mutations and one nonsense mutation from 12 M2 plants were located in the PSGP region spanning amino acids 334–377 (Figure 2).
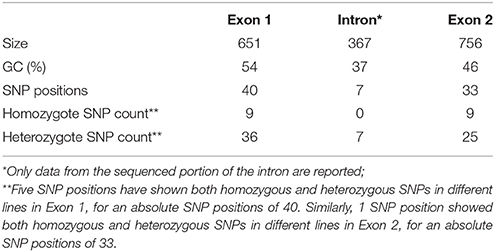
Table 2. Summary of EMS-induced mutations in UGT74S1 gene of a mutagenized flax population.
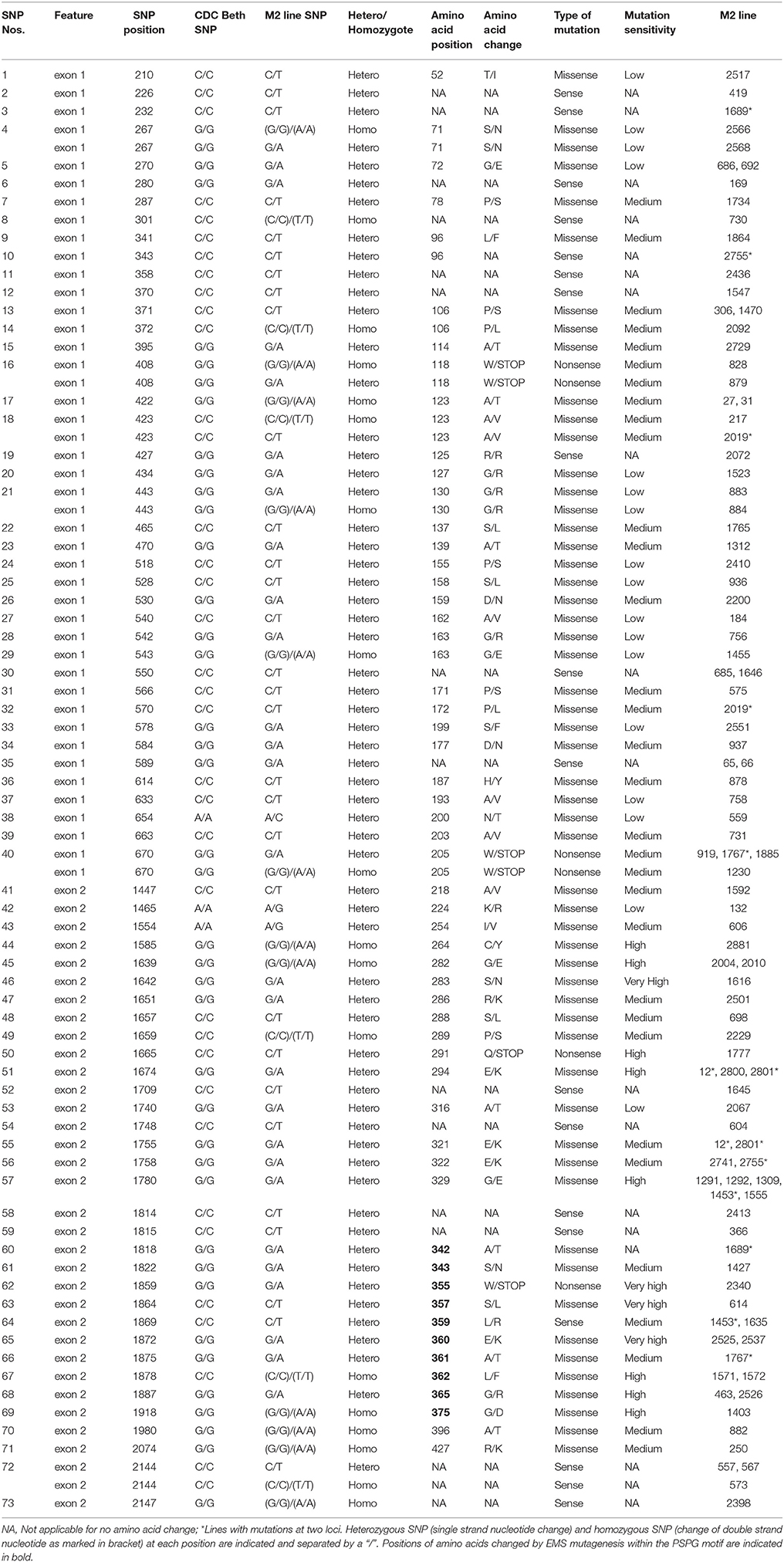
Table 3. Details of 79 exonic mutations observed in UGT74S1 of 93 M2 flax plants.
Effect of UGT74S1 Mutations on SDG Content in Bulked M2 Seeds
To establish a genotype to phenotype relationship, the effect of each mutation on SDG lignan production was assessed in planta by determining the total SDG lignan content in the original mature bulked M2 seeds in comparison with that of the wild type CDC Bethune. UPLC chromatograms of non-hydrolyzed oligomeric lignan polymers from the M2 family 1230 carrying a homozygous nonsense mutation in UGT74S1 displayed a narrower peak whereas that of the M2 family 2340 that carried a heterozygous nonsense mutation showed additional peaks overlapping the main peak (Supplementary Figure 2A). Variations were also observed in the profiles of SDG and other phenolic acid glycosides present in the lignan macromolecule (Supplementary Figure 2B). By determining the total hydrolyzed SDG lignan content in the original M2 bulked mature seeds from each of the 69 M2 families, we observed 21 with reduced SDG content, eight of which displayed almost no SDG. In contrast, 43 families had a higher SDG content compared to CDC Bethune (Figure 3). In many cases, no direct correlation could be established between the mutation and alteration of the lignan profile. In fact, some M2 families such as 828, 919, 1230, and 2340, carrying homozygous nonsense mutations showed increased SDG lignan content compared to the wild type. Similarly, eight M2 families (882, 1403, 1571, 1572, 2092, 2229, 2566, and 2881) carrying homozygote missense mutations showed higher or inconsistent lignan content (Figure 3). These unexpected observations prompted us to re-assess and re-validate the genotype of single M2 plants derived from each of the 69 M2 families after re-sampling within the original seed lots.
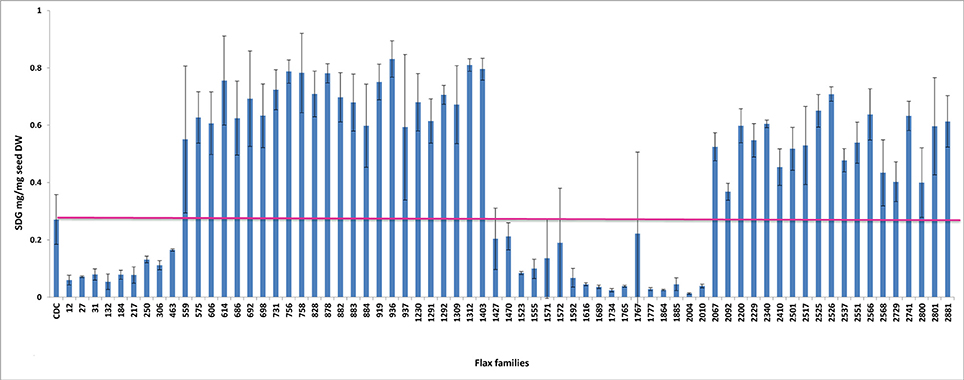
Figure 3. Diversity in the spectrum of SDG production in wild type Bethune (CDC) and 69 EMS mutant lines. The pink line indicates the CDC Bethune threshold line for comparison.
Validation of EMS-Induced Mutations in M2 Plants
To validate some of the EMS-induced mutations identified in UGT74S1 by amplicon resequencing, plants from 28 M2 families (Supplementary Table 3) were genotyped by KASP assays (LGC Genomics, UK) using markers targeting 20 EMS-induced SNP mutations. Ten of the 20 targeted mutations were thus validated in 21 plants from 15 M2 families, but the other 10 from 13 M2 families were not (Table 4). Among the validated plants, 17 were heterozygous in 13 families and four plants from four different families were homozygous. After validation of EMS-induced mutations in the M2 plants, the stability of the mutations was determined in single seed descent M3 plants derived from KASP-validated M2 plants. KASP assays showed that all M3 plants derived from homozygous M2 plants remained homozygous whereas M3 plants derived from a heterozygous M2 plants were either heterozygous or segregated (Table 4).
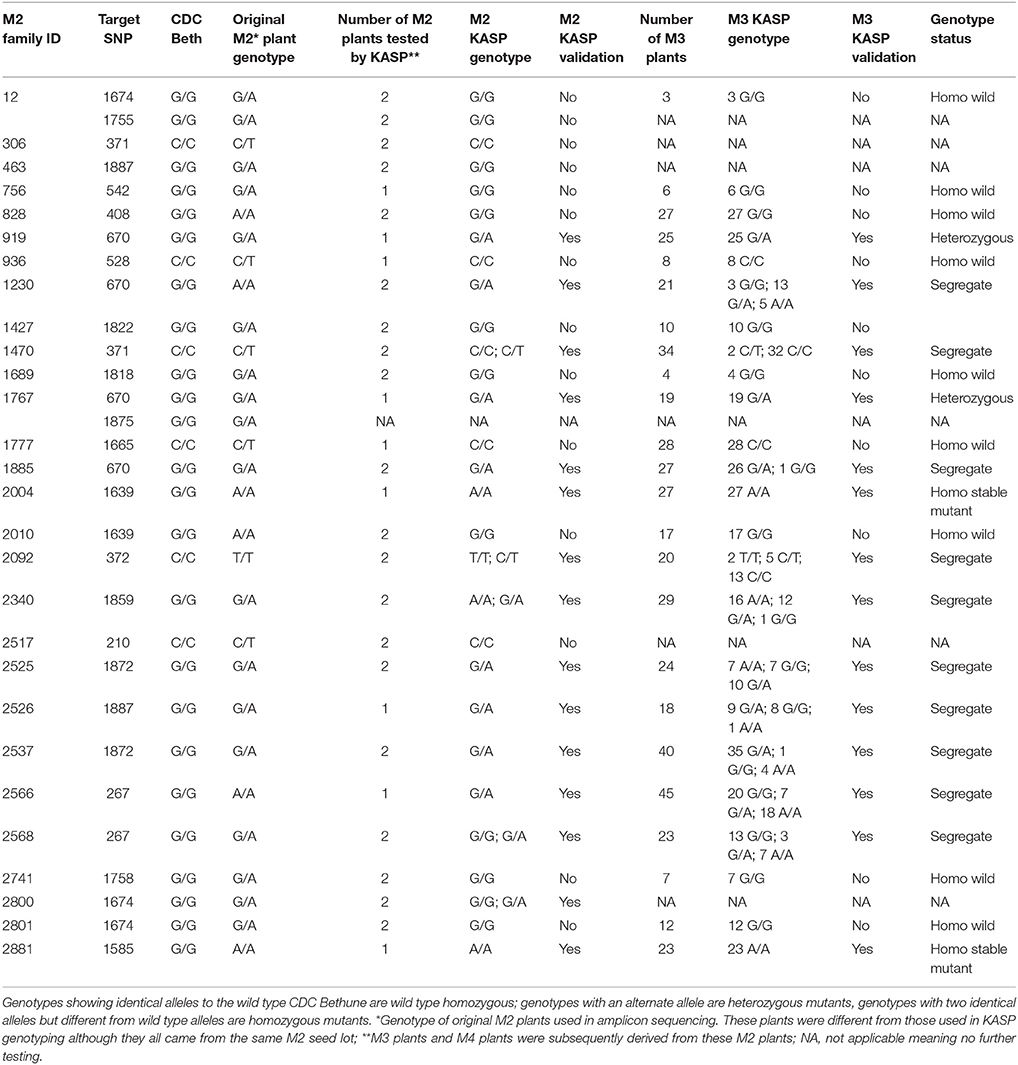
Table 4. Mutation stability and segregation among seed from single flax bolls.
Stability of EMS-Induced Mutation Effect on SDG Content in M4 Seeds Derived from Single Seed Descent of M3 Plants
By phenotyping the M4 seeds derived from individual M3 plants for SDG, we identified knock-out mutants devoid of SDG (Figure 4). However, most of the heterozygous and homozygous missense mutations showed a wide range of SDG content, some having SDG content greater than CDC Bethune and others, such as M4 plants from family 2004 (homozygous missense mutation) were complete knock-outs (Figure 5). Surprisingly, disappearance of the SDG peak did not result in the appearance of SECO peaks in any of the knock-out lines. Instead, the mass spectra analysis of peak #10 revealed the presence of SMG in these knock-out lines but not in the others (Figure 5). Interestingly, EMS mutations led to variations in the levels of the phenol acid glucosides encountered in the lignan macromolecule (Supplementary Figure 3).
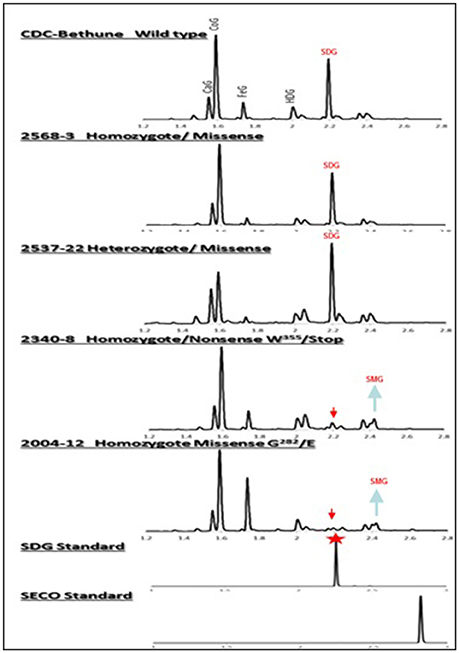
Figure 4. UPLC chromatograms of hydrolyzed lignan complex from M4 seed derived from M3 single seed descent lines and seed from CDC Bethune showing the diversity in SDG profile. The peaks of phenolic acid glucosides and SDG are shown on the top. CaG, caffeic glucoside; CoG, coumaric glucoside; FeG, ferulic glucoside; HDG, herbacetin diglucoside. The red arrows indicate the disappearance of SDG peaks in homozygote nonsense (#2340) and missense (#2004) M4 lines compared to CDC Bethune. Note the equal and higher SDG peaks in the seed from homozygous (#2568) and heterozygous (#2537) lines carrying missense mutations. SDG peak from the SDG standard is indicated by the red star.
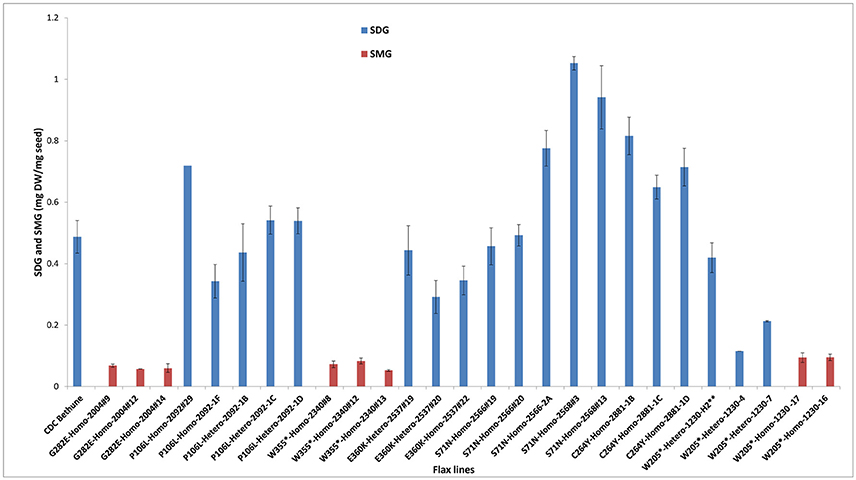
Figure 5. Variations in SDG and SMG lignan contents in M3 and M4 seeds from EMS mutagenized flax lines following lignan complex hydrolysis. SDG and SMG UPLC peaks were monitored using mass spectra and quantified. EMS lines are named as EMS-induced amino acid changed followed by the homo or heterozygous status and the line number. Single asterisk (*) indicates premature stop codon. The profile of M3 seeds derived from M2 line 1230-H2 is indicated by a double asterisk (**). Lignan was extracted from CDC Bethune and from M4 seeds derived from their respective M3 plants.
UGT74S1 Is Unique in Controlling SECO Glucosylation into SDG
To ascertain whether mutations occurred in other lignan-related biosynthetic genes that may have contributed to the observed altered lignan phenotypes, plants with altered lignan profiles were subjected to targeted Ampliseq gene sequencing of 11 genes known to play diverse roles in SDG lignan biosynthesis (Figure 1). For each gene sequenced, a high percentage of aligned bases (99.2%) was achieved, with an average coverage depth of 5,340×. Of the six M4 flax families used in the Ampliseq sequencing experiment, all plants from lines in four of the families were re-confirmed with the mutations previously detected in UGT74S1 by amplicon sequencing and confirmed by KASP genotyping. No loss-of-function mutation was found in any plants of the lines derived from families 2566 and 2568. A mutation with low sensitivity was detected only in plants from lines #2004-9 and #2004-14 at SNP position 1,537 of UGT74S3 that caused the P471S missense mutation with potential neutral effect. No mutation was detected in UGT74S4 in any of the plants. Similarly, no mutation was found in any of the three UGTs (UGT74S1, UGT74S3, and UGT74S4) of plants derived from CDC Bethune. In contrast, three mutations (Y35H, K38H, R40Q) and one (L15S) with low mutational sensitivity were detected in DIR 4 and DIR 5, respectively, in M4 plants of lines# 2004-9 and 2004-14 (Figure 6, Table 5, Supplementary Table 7).
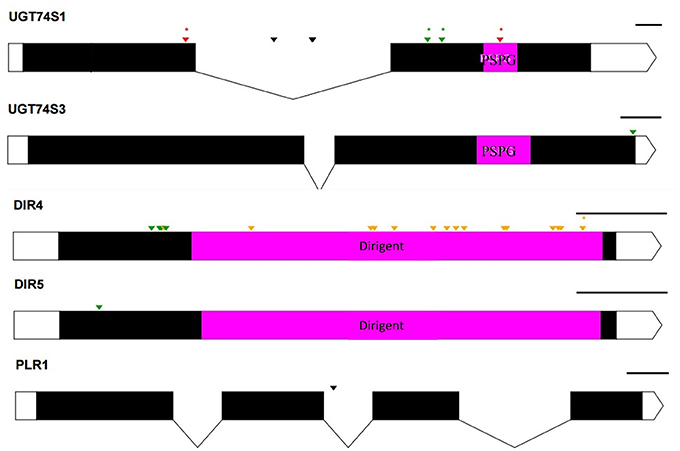
Figure 6. Distribution of EMS-induced mutations on five genes of the 11 custom gene panel submitted to Ampliseq sequencing. UTRs are indicated by white boxes, exons by solid black boxes, and introns by gaps with downward arrows. The PSPG region within exon 2 of UGT74S1 and UGT74S3 is colored in magenta. The magenta box in DIR genes indicates DIR protein domains. The PSPG and dirigent regions are conserved domains for UGT and dirigent proteins, respectively. Green arrows, missense mutations; orange arrows, sense mutations; red arrows, nonsense mutation; black arrows, mutations in introns; asterisks (*) indicate position of homozygous mutations only; The black lines on the right top of each gene indicate the gene drawing scale.
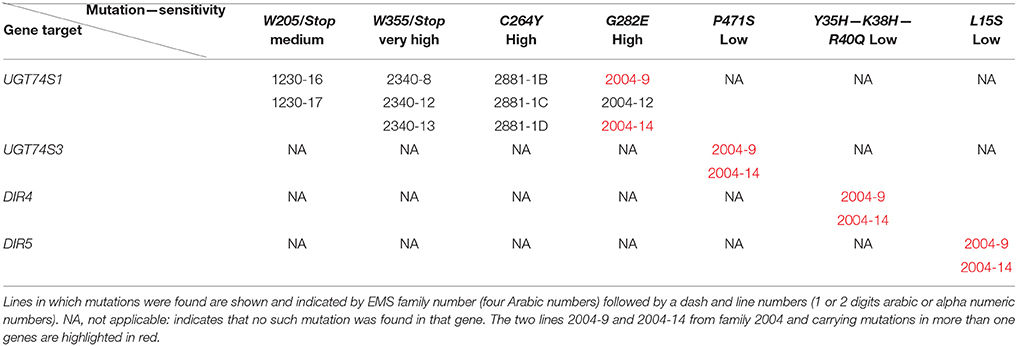
Table 5. Mutations confirmed in UGT74S1 or identified in other genes by Ampliseq sequencing.
Discussion
Establishing phenotype to genotype relationship and assigning functions to variant alleles are of interest to plant breeders and biologists. Using chemically induced mutations and TILLING as a reverse genetics tool, gene and allele functions have been elucidated in several biosynthetic pathways in plants (Slade and Knauf, 2005; Till et al., 2007; Xin et al., 2008; Dahmani-Mardas et al., 2010; Uchida et al., 2011; Rawat et al., 2012; Chantreau et al., 2013; Dhaliwal et al., 2015; Galindo-González et al., 2015). In the current study, we mined an EMS mutagenized flaxseed population and used an NGS-based reverse genetics platform to identify flax lines carrying mutations in the lignan glucosylation gene UGT74S1 and to determine allele-specific mutational effects on SDG lignan production in planta. The study demonstrated that EMS-induced mutations in UGT74S1 resulted in alteration of SDG accumulation in seeds and, as such, it constitutes the first report of non-transgenic flax germplasm with altered lignan content.
Whereas, TILLING terminology is relatively new, generating mutant alleles in flax using EMS is not (Green and Marshall, 1984; Rowland et al., 1995). The novelty of the chemical mutagenesis in the plant reverse genetics field resides in the much larger screening throughput capacity that we can now achieve (Sikora et al., 2011; Vicente-Dólera et al., 2014). In the present report, 1996 M2 families were screened by reverse genetics and, as expected, only G to A or C to T transition mutations were observed (Sikora et al., 2011). The calculated mutation frequency of one mutation per 28 kb is close to the range recently reported rate of one mutation per 30–49 kb in flax treated with different doses of EMS (Chantreau et al., 2013). A high proportion of 91% of the SNP variation was located in exons, in agreement with their higher G/C content which are more prone to EMS alkylation causing mainly transition mutations (Sikora et al., 2011). In this study, 22.5% of exonic mutations were homozygous, with potential loss (Uchida et al., 2011) or gain-of-function effects (Bailly et al., 2014). Indeed, whereas DNA mutations can affect any of the amino acids, not all substitutions have the same mutational effects on gene functionality (Rogers et al., 2000). Moreover, depending on the amino acids, homozygous missense mutations showing low to medium mutational effects can be expected to be neutral or hypermorphic. In the latter, the mutation may cause an increased contribution of the mutated gene to the original function (Lu et al., 2012).
By genotyping three generations of plants, the identified EMS-induced mutations were heritable, stable, or segregating, as reported in other plants (Uauy et al., 2009). Through phenotyping we showed that nonsense and missense mutations in UGT74S1 coding regions altered SDG lignan and phenolic acid glucoside profiles in flax seed. In a previous study, we showed that site-directed mutagenesis of targeted amino acids within the PSPG (W334–Q374) motif of UGT74S1 abolished completely or altered the SECO glucosylation in vitro (Ghose et al., 2015). In agreement with these findings, the EMS treatment randomly mutagenized nine amino acids (A342F, S343N, W355Stop, S357L, E360K, A361T, L362F, G365R, and G375D) within the PSPG in planta and, as previously observed in vitro, homozygous nonsense EMS-induced mutation of W355 to stop codon completely abolished SDG formation in planta, but retained trace amounts of SMG. Homozygous nonsense mutation of W205 to a stop codon outside the PSGP as well as the homozygous missense G282E mutation also led to loss-of-function of UGT74S1 for SECO glucosylation into SDG, but again a small amount of SMG remained. In a recent study, we reported that UGT74S1 is the key player in controlling SECO glucosylation into SDG, while also showing that UGT74S3 and UGT74S4, a pair of duplicated genes most closely related to UGT74S1, were able to glucosylate SECO into SMG at low efficiency but unable to synthesize SDG from SMG (Fofana et al., 2017). These findings suggested that these two paralogs may play roles in other biological processes other than SDG formation from SECO (Fofana et al., 2017). This was corroborated herein where we showed that plants from four UGT74S1 EMS lines lacking mutations in UGT74S3 and UGT74S4 failed to produce SDG but could still produce trace amounts of SMG. This clearly demonstrates occurrence of low background activity of UGT74S3 and UGT74S4 catalyzing trace SMG formation despite UGT74S1 being knocked out, in agreement with our previous report (Fofana et al., 2017). Moreover, some residual glucosylation activity may also be associated with each amino acid substitution effect within the mutated UGT74S1, as reported by in vitro site-directed mutagenesis data (Ghose et al., 2015). Additionally, the pool of phenolic acid glucosides was found to be increased in all mutant lines, as compared to the lignan component of the polymer. Additional biochemical studies using the phenolic acids encountered in the lignan macromolecule as substrates are required to characterize the exact role(s) of these two UGT paralogs in planta.
Targeted gene panel sequencing by Ampliseq methodology confirmed the mutations detected in UGT74S1 by amplicon re-sequencing and KASP assays. It also identified the P471S mutation in UGT74S3 from UGT74S1 mutant lines G282E-Homo-2004-9 and G282E-Homo-2004-14, each showing small amounts of SMG, thereby demonstrating the residual activity from UGT74S4 and/or the residual glucosylation activity associated with the G282E substitution in the mutated UGT74S1. The M4 plants from lines 2004-9 and 2004-14 also carried mutations, albeit with low effects, in both DIR4 and DIR5 genes at positions Y35H, K38H, R40Q, and L15S, but still produced trace amounts of SMG. This observation demonstrates that SECO was produced in these lines and that DIR4 and DIR5 are not rate limiting in the pinoresinol formation. Considering that not all the flax genome was scanned in the current study, one may wonder whether some transcription factors controlling lignan biosynthetic genes may have also been affected by EMS. Whereas, this assumption cannot be totally ruled, it seems to be highly unlikely or these mutations, if any, may be of very low or neutral effect because the lignan phenotypes observed in the lines herein described are in agreement with that from the in vitro data (Ghose et al., 2015), in the absence of implication of any transcription factors.
In conclusion, this study found EMS mutagenesis to be successful in altering SECO glucosylation toward SDG formation in planta. The wide range of lignan profiles observed from the current investigation constitutes a useful non-GMO genetic resource for flax breeders in developing new cultivars with high SDG content or new cultivars with SMG as a new trait in flax. It also offers plant biologists and natural health product scientists an opportunity to better understand the behavior of plants carrying the new trait in field environments. Using Ampliseq sequencing of UGT74S1 along with 10 other genes potentially playing a role in the SDG production, we validated our SNP detection approach and provided further evidence that UGT74S3 and UGT74S4 are not critical for SDG production in planta and that UGT74S1 is unique in controlling SDG formation from SECO.
Author Contributions
BF: Conception, coordination, design, experiments, data analysis, interpretation, and writing of the manuscript; KG: Performed experiments, data analysis, interpretation, drafting and revision of the manuscript; JM: UPLC and mass spectrometry data acquisition and analysis of lignan extracts, revision of manuscript; DM and AS: EMS population nursery; SC: coordination, administration, and revision of the manuscript; MD and GR: EMS mutagenesis and M2 population generation, revision and proof reading of the manuscript. All authors read, commented and approved the manuscript.
Funding
This study was conducted as part of the Total Utilization Flax Genomics (TUFGEN) project funded by Genome Canada/Genome Prairie with financial contribution from the Flax Council of Canada to BF.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Colin Stetson and all summer students at the Charlottetown Research and Development Centre, Charlottetown, PEI, for their help during the course of this project.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01638/full#supplementary-material
Supplementary Figure 1. Reverse genetics of 1996 EMS-mutagenized M2 flax lines targeting UGT74S1 gene. (A) genomic region of UGT74S1 used for targeted amplicon resequensing; (B) Schematic representation of the workflow adopted for SNP variants detection in the EMS flax population.
Supplementary Figure 2. UPLC chromatograms of non-hydrolyzed (A) and hydrolyzed (B) lignan extracted from bulked seeds of wild type CDC Bethune and M2 families 1230 and 2340, identified to carry nonsense mutations in UGT74S1. (A) lignan macromolecule profiles for the wild type CDC Bethune, M2 family 1230 predicted to have a premature stop codon at amino acid 205, and M2 family 2340, heterozygous for SNP-1859 and predicted to encode a wild type allele of UGT74S1 gene and a mutant one with a premature stop codon at amino acid 355. Note the narrower width of the lead peak in family 1230 compared to CDC Bethune as indicated by the two arrows and additional peaks on the lead peak in family 2340 as indicated by stars compared to CDC Bethune and M2 family 1230. The patterns of both M2 families also displayed additional and higher peaks (indicated by stars) in other parts of the chromatograms compared to CDC Bethune. (B) UPLC chromatograms of the hydrolyzed lignan complex from CDC Bethune, M2 families 1230 and 2340 showing peak height variations for SDG and phenolic acid glucosides encountered in the complex lignan polymers.
Supplementary Figure 3. Summary of mutational effects on lignan glucoside species production in UGT74S1 flax mutant lines. Number of stars represents the relative amount of lignan species; HCA, hydroxycinnamic acid.
Abbreviations
SECO, Secoisolariciresinol; SDG, Secoisolariciresinol diglucoside; SMG, Secoisolariciresinol monoglucoside; PINO, Pinolariciresinol; ENL, Enterolactone; END, Enterodiol; UGT, Uridine glucosyl transferases; PSPG, Plant secondary product glucosyl transferase; EMS, Ethyl methane sulphonate; KASP, Kompetitive allele specific PCR; SNP, Single nucleotide polymorphism; UTR, Untranslated region; CDS, Coding DNA sequence; ER, Estrogen receptor; VCF, Variant call format.
References
Adolphe, J. L., Whiting, S. J., Juurlink, B. H. J., Thorpe, L. U., and Alcorn, J. (2010). Health effects with consumption of the flax lignan secoisolariciresinol diglucoside. Br. J. Nutr. 103, 929–938. doi: 10.1017/S0007114509992753
Bailly, A. E., Wang, B., Zwiewka, M., Pollmann, S., Schenck, D., Luthen, H., et al. (2014). Expression of TWISTED DWARF1 lacking its in-plane membrane anchor leads to increased cell elongation and hypermorphic growth. Plant J. 77, 108–118. doi: 10.1111/tpj.12369
Buck, K., Zaineddin, A. K., Vrieling, A., Heinz, J., Linseisen, J., Flesch-Janys, D., et al. (2011). Estimated enterolignans, lignan-rich foods, and fibre in relation to survival after postmenopausal breast cancer. Br. J. Cancer 105, 1151–1157. doi: 10.1038/bjc.2011.374
Caputi, L., Malnoy, M., Goremykin, V., Nikiforova, S., and Martens, S. (2012). A genome-wide phylogenetic reconstruction of family 1 UDP-glycosyltransferases revealed the expansion of the family during the adaptation of plants to life on land. Plant J. 69, 1030–1042. doi: 10.1111/j.1365-313X.2011.04853.x
Chantreau, M., Grec, S., Gutierrez, L., Dalmais, M., Pineau, C., Demailly, H., et al. (2013). PT-flax (phenotyping and Tilling of flax) development of a flax (Linum usitatissimum L.) mutant population and tilling platform for forward and reverse genetics. BMC Plant Biol. 13:159. doi: 10.1186/1471-2229-13-159
Clavel, T., Henderson, G., Alpert, C. A., Philippe, C., Rigotter-Gois, L., Dore, J., et al. (2005). Intestinal bacteria communities that produce active estrogen like compounds enterodiol and enterolactone in humans. Appl. Environ. Microbiol. 71, 6077–6085. doi: 10.1128/AEM.71.10.6077-6085.2005
Clavel, T., Henderson, G., Engest, W., Dore, J., and Blaut, M. (2006). Phylogeny of human intestinal bactaria that activate the dietary lignans secoisolariciresinol diglucoside. FEMS Microbiol. Ecol. 55, 471–478. doi: 10.1111/j.1574-6941.2005.00057.x
Clavel, T., Lippman, R., Gavini, F., Dore, J., and Blaut, M. (2007). Clostridium saccharogumia sp. nov and Lactonifactor longoViformis gen. nov., sp nov., two novel human faecal bacteria involved in the conversion of the dietary phytoestrogen secoisolariciresinol diglucoside. Syst. Appl. Microbiol. 30, 16–26. doi: 10.1016/j.syapm.2006.02.003
Comai, L., and Henikoff, S. (2006). TILLING: practical single-nucleotide mutation discovery. Plant J. 45, 684–694. doi: 10.1111/j.1365-313X.2006.02670.x
Dahmani-Mardas, F., Troadec, C., Boualem, A., Lévêque, S., Alsadon, A. A., Aldoss, A. A., et al. (2010). Engineering melon plants with improved fruit shelf life using the TILLING approach. PLoS ONE 5:e15776. doi: 10.1371/journal.pone.0015776
Dhaliwal, A. K., Mohan, A., Sidhu, G., Maqbool, R., and Gill, K. S. (2015). An ethylmethane sulfonate mutant resource in pre-green revolution hexaploid wheat. PLoS ONE 10:e0145227. doi: 10.1371/journal.pone.0145227
Dribnenki, J. C. P., Green, A. G., and Atlin, G. N. (1996). Linola™ 989 low linolenic flax. Can. J. Plant Sci. 76, 329–331. doi: 10.4141/cjps96-057
Duguid, S. D., Rashid, K. Y., and Kenaschuk, E. O. (2014). Prairie Thunder flax. Can. J. Plant Sci. 94, 445–449. doi: 10.4141/cjps2013-195
During, A., Debouche, C., Raas, T., and Larondelle, Y. (2012). Among plant lignans, pinoresinol has the strongest anti-inflammatory properties in human intestinal Caco-2 cells. J. Nutr. 142, 1798–1805. doi: 10.3945/jn.112.162453
EFSA (European Food Safety Authority) (2007). Opinion of the scientific panel on contaminants in the food chain on a request from the commission related to cyanogenic compounds as undesirable substances in animal feed. EFSA J. 434, 1–67. doi: 10.2903/j.efsa.2007.434
Fofana, B., Ghose, K., McCallum, J., You, F. M., and Cloutier, S. (2017). UGT74S1 is the key player in controlling secoisolariciresinol diglucoside (SDG) formation in flax. BMC Plant Biol. 17:35. doi: 10.1186/s12870-017-0982-x
Fofana, B., Ragupathy, R., and Cloutier, S. (2010). “Flax lipids: classes, biosynthesis, genetics and the promise of applied genomics for understanding and altering of fatty acids,” in Lipids: Categories, Biological Functions and Metabolism, Nutrition, and Health, ed P. L. Gilmore (New York, NY: Nova Science Publishers Inc.), 71–98.
Gachon, C. M., Langlois-Meurinne, M., and Saindrenan, P. (2005). Plant secondary metabolism glycosyltransferases: the emerging functional analysis. Trends Plant Sci. 10, 542–549. doi: 10.1016/j.tplants.2005.09.007
Galindo-González, L., Pinzón-Latorre, D., Bergen, E. A., Jensen, D. C., and Deyholos, M. K. (2015). Ion Torrent sequencing as a tool for mutation discovery in the flax (Linum usitatissimum L.) genome. Plant Methods 11:19. doi: 10.1186/s13007-015-0062-x
Gasper, R., Effenberg, P., Terlele, I., Kolesinki, P., Terlecka, B., Hofmann, E., et al. (2016). Dirigent protein mode of action revealed by the crystal structure of AtDIR6. Plant Phys. 172, 2165–2175. doi: 10.1104/pp.16.01281
Ghose, K., McCallum, J., Kirby, C. W., Sweeney-Nixon, M., and Fofana, B. (2015). Histidine 352 (His352) and Tryptophan 355 (Trp355) are essential for flax UGT74S1 glucosylation activity toward secoisolariciresinol. PLoS ONE 10:e116248. doi: 10.1371/journal.pone.0116248
Ghose, K., Selvaraj, K., McCallum, J., Kirby, C. W., Sweeney-Nixon, M., Cloutier, S. J., et al. (2014). Identification and functional characterization of a flax UDP-glycosyltransferase glucosylating secoisolariciresinol (SECO) into secoisolariciresinol monoglucoside (SMG) and diglucoside (SDG). BMC Plant Biol. 14:82. doi: 10.1186/1471-2229-14-82
Green, A. G., and Marshall, D. R. (1984). Isolation of induced mutants of linseed (Linum usitatissimum L.) having reduced linolenic acid content. Euphytica 33, 321–328. doi: 10.1007/BF00021128
Halls, S. C., Davin, L. B., Kramer, D. M., and Lewis, N. G. (2005). Kinetic study of coniferyl alcohol radical binding to the (+)-pinoresinol forming dirigent protein. Biochemistry 43, 2587–2595. doi: 10.1021/bi035959o
Kim, H. J., Ono, E., Morimoto, K., Yamagaki, T., Pkazawa, A., Kobayashi, A., et al. (2009). Metabolic engineering of lignan biosynthesis in Forsythia cell culture. Plant Cell Physiol. 50, 2200–2209. doi: 10.1093/pcp/pcp156
Kosińska, A., Penkacik, K., Wiczkowski, W., and Amarowicz, R. (2011). Presence of caffeic acid in flaxseed lignan macromolecule. Plant Foods Hum. Nutr. 66, 270–274. doi: 10.1007/s11130-011-0245-1
Landete, J. M., Arqués, J., Medina, M., Gaya, P., De La Rivas, B., and Mu-oz, R. (2015). Bioactivation of phytoestrogens: intestinal bacteria and health. Crit. Rev. Food Sci. Nutr. 56, 1826–1843. doi: 10.1080/10408398.2013.789823
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, X., Yuan, J.-P., Xu, S.-P., Wang, J.-H., and Liu, X. (2008). Separation and determination of secoisolariciresinol diglucoside oligomers and their hydrolysates in the flaxseed extract by high-performance liquid chromatography. J. Chromatogr. A 1185, 223–232. doi: 10.1016/j.chroma.2008.01.066
Lu, Y. H., Arnaud, D., Belcram, H., Falentin, C., Rouault, P., Piel, N., et al. (2012). A dominant point mutation in a RINGv E3 ubiquitin ligase homoeologous gene leads to cleistogamy in Brassica napus. Plant Cell 24, 4875–4891. doi: 10.1105/tpc.112.104315
McCallum, C. M., Comai, L., Greene, E. A., and Henikoff, S. (2000). Targeting Induced Local Lesions IN Genomes (TILLING) for plant functional genomics. Plant Phys. 123, 439–442. doi: 10.1104/pp.123.2.439
Mukker, J. K., Singh, R. S. P., Muir, A. D., Krol, E. S., and Alcorn, J. (2015). Comparative pharmacokinetics of purified flaxseed and associated mammalian lignans in male Wistar rats. Br. J. Nutr. 113, 749–757. doi: 10.1017/S0007114514004371
Nakatsubo, T., Mizutani, M., Suzuki, S., Hattori, T., and Umezawa, T. (2008). Characterization of Arabidopsis thaliana pinoresinol reductase, a new type of enzyme involved in lignan biosynthesis. J. Biol. Chem. 283, 15550–15557. doi: 10.1074/jbc.M801131200
Nishio, S. Y., Hayashi, Y., Watanabe, M., and Usami, S. (2015). Clinical application of a custom AmpliSeq library and ion torrent PGM sequencing to comprehensive mutation screening for deafness genes. Genet. Test Mol. Biomark. 4, 209–217. doi: 10.1089/gtmb.2014.0252
Ntiamoah, C., and Rowland, G. G. (1997). Inheritance and characterization of two low linolenic acid EMS-induced McGregor mutant flax (Linum usitatissimum L.). Can. J. Plant Sci. 77, 353–358. doi: 10.4141/P96-137
Osmani, S. A., Bak, S., and Møller, B. L. (2009). Substrate specificity of plant UDP-dependent glycosyltransferases predicted from crystal structures and homology modeling. Phytochemistry 70, 325–347. doi: 10.1016/j.phytochem.2008.12.009
Pan, J. Y., Chen, S. L., Yang, M. H., Wu, J., Sinkkonen, J., and Zoud, K. (2009). An update on lignans: natural products and synthesis. Nat. Prod. Rep. 26, 1251–1292. doi: 10.1039/b910940d
Pavelek, M., Tejklova, E., and Bjelkva, M. (2015). “Flax and linseed,” in Industrial Crops: Breeding for Bioenergy and Bioproducts, Handbook of Plant Breeding, eds V. M. V. Cruz and D. A. Dietig (New York, NY: Springer), 233–264. doi: 10.1007/978-1-4939-1447-0_11
Possemiers, S., Bolca, S., Eeckhaut, E., Depypere, H., and Verstraete, W. (2007). Metabolism of isoflavones, lignans and prenylflavonoids by intestinal bacteria: producer phenotyping and relation with intestinal community. FEMS Microbiol. Ecol. 61, 372–383. doi: 10.1111/j.1574-6941.2007.00330.x
Rawat, N., Sehgal, S. K., Joshi, A., Rothe, N., Wilson, D. L., McGraw, N., et al. (2012). A diploid wheat TILLING resource for wheat functional genomics. BMC Plant Biol. 12:205. doi: 10.1186/1471-2229-12-205
Rogers, E. E., Eide, D. J., and Guerinot, M. L. (2000). Altered selectivity in an Arabidopsis metal transporter. Proc. Natl. Acad. Sci. U.S.A. 97, 12356–12360. doi: 10.1073/pnas.210214197
Rowland, G. G. (1991). An EMS-induced low-linolenic-acid mutant in McGregor flax (Linum usitatissimum L.). Can. J. Plant Sci. 71, 393–396. doi: 10.4141/cjps91-054
Rowland, G. G., Hormis, Y. A., and Rashid, K. Y. (2002). CDC Bethune flax. Can. J. Plant Sci. 82, 101–102. doi: 10.4141/P01-066
Rowland, G. G., McHughen, A., Gusta, L. V., Bhatty, R. S., MacKenzie, S. L., and Taylor, D. C. (1995). The application of chemical mutagenesis and biotechnology to the modification of linseed (Linum usitatissimum L.). Euphytica 85, 317–321. doi: 10.1007/BF00023961
Rowland, I. R., Wiseman, H., Sanders, T. A., Adlercreutz, H., and Bowey, E. A. (2000). Interindividual variation in metabolism of soy isoflavones and lignans: influence of habitual diet on equol production by the gut flora. J. Nutr. 36, 27–32. doi: 10.1207/S15327914NC3601_5
Saastamoinen, M., Pihlava, J. M., Eurola, M., Klemola, A., Jauhiainen, L., and Hietaniemi, V. (2013). Yield, SDG lignan, cadmium, lead, oil and protein contents of linseed (Linum usitatissimum L.) cultivated in trials and at different farm conditions in the south-western part of Finland. Agric. Food Sci. 22, 296–306. Available online at: https://journal.fi/afs/article/view/7355
Satake, H., Koyama, T., Bahabadi, S. E., Matsumoto, E., Ono, E., and Murata, J. (2015). Essences in metabolic engineering of lignan biosynthesis. Metabolites 5, 270–290. doi: 10.3390/metabo5020270
Setchell, K. D. R., Brown, N. M., Zimmer-Nechemias, L., Wolfe, B., and Heubi, J. E. (2014). Metabolism of secoisolariciresinol-diglycoside the dietary precursor to the intestinally derived lignan enterolactone in humans. Food Funct. 5, 491–501. doi: 10.1039/C3FO60402K
Setchell, K. D. R., and Cole, S. J. (2006). Method of defining equol-producer status and its frequency among vegetarians. J. Nutr. 136, 2188–2193. Available online at: http://jn.nutrition.org/content/136/8/2188.full
Sikora, P., Chawade, A., Larson, M., Olson, J., and Olson, O. (2011). Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int. J. Plant Genomics 2011:314829. doi: 10.1155/2011/314829
Simopoulos, A. P. (2008). The importance of omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. 233, 674–688. doi: 10.3181/0711-MR-311
Slade, A. J., and Knauf, V. C. (2005). TILLING moves beyond functional genomics into crop improvement. Transgenic Res. 14, 109–115. doi: 10.1007/s11248-005-2770-x
Struijs, K., Vincken, J. P., Doeswijk, T. G., Voragen, A. G., and Gruppen, H. (2009). The chain length of lignan macromolecule from flaxseed hulls is determined by the incorporation of coumaric acid glucosides and ferulic acid glucosides. Phytochemistry 70, 262–269. doi: 10.1016/j.phytochem.2008.12.015
Thompson, L. U., Boucher, B. A., Liu, Z., Cotterchio, M., and Kreiger, N. (2006). Phytoestrogen content of foods consumed in Canada including isoflavones lignans and coumestan. Nutr. Cancer 54, 184–201. doi: 10.1207/s15327914nc5402_5
Till, B. J., Cooper, J., Tai, T. H., Colowit, P., Greene, E. A., Henikoff, S., et al. (2007). Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 7:19. doi: 10.1186/1471-2229-7-19
Touré, A., and Xueming, X. (2010). Flaxseed lignans: source, biosynthesis, metabolism, antioxidant activity, bio-active components, and health benefits. Compr. Rev. Food Sci. Food Saf. 9, 261–269. doi: 10.1111/j.1541-4337.2009.00105.x
Uauy, C., Paraiso, F., Colasuonno, P., Tran, R. K., Tsai, H., Berardi, S., et al. (2009). A modified TILLING approach to detect induced mutations in tetraploid and hexaploid wheat. BMC Plant Biol. 9:115. doi: 10.1186/1471-2229-9-115
Uchida, N., Sakamoto, T., Kurata, T., and Tasak, M. (2011). Identification of EMS-Induced causal mutations in a non-reference Arabidopsis thaliana accession by whole genome sequencing. Plant Cell Physiol. 52, 716–722. doi: 10.1093/pcp/pcr029
Udani, J. K., Brown, D. J., Tan, M. O. C., and Hardy, M. (2013). Pharmacokinetics and bioavailability of plant lignan 7-hydroxymatairesinol and effects on serum enterolactone and clinical symptoms in postmenopausal women: a single-blinded, parallel, dose-comparison study. J. Am. Coll. Nutr. 32, 428–435. doi: 10.1080/07315724.2013.849578
Vicente-Dólera, N., Troadec, C., Moya, M., del Río-Celestino, M., Pomares-Viciana, T., Bendahmane, A., et al. (2014). First TILLING platform in Cucurbita pepo: a new mutant resource for gene function and crop improvement. PLoS ONE 9:e112743. doi: 10.1371/journal.pone.0112743
Wang, J., and Hou, B. (2009). Glycosyltransferases: key players involved in the modification of plant secondary metabolites. Front. Biol. China 4, 39–46. doi: 10.1007/s11515-008-0111-1
Wang, Y., Fofana, B., Roy, M., Ghose, K., Yao, X., Nixon, M.-S., et al. (2015). Flaxseed lignan secoisolariciresinol diglucoside improves insulin sensitivity through upregulation of GLUT4 expression in diet-induced obese mice. J. Funct. Foods 18, 1–9. doi: 10.1016/j.jff.2015.06.053
Wang, Z., Hobson, N., Galindo, L., Zhu, S., Shi, D., McDill, J., et al. (2012). The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 72, 461–473. doi: 10.1111/j.1365-313X.2012.05093.x
Webb, A. L., and McCullough, M. L. (2005). Dietary lignans: potential role in cancer prevention. Nutr. Cancer 51, 117–131. doi: 10.1207/s15327914nc5102_1
Witte, S., Moco, S., Vervoort, J., Matern, U., and Martens, S. (2009). Recombinant expression and functional characterisation of regiospecific flavonoid glycosyltransferases from Hieracium pilosella L. Planta 229, 1135–1146. doi: 10.1007/s00425-009-0902-x
Xin, Z., Li Wang, M., Barkley, N. A., Burow, G., Franks, C., Pederson, G., et al. (2008). Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 8:103. doi: 10.1186/1471-2229-8-103
Keywords: flax, lignan, UGT74S1, EMS, SNPs, secoisolariciresinol, reverse genetics, forward genetics
Citation: Fofana B, Ghose K, Somalraju A, McCallum J, Main D, Deyholos MK, Rowland GG and Cloutier S (2017) Induced Mutagenesis in UGT74S1 Gene Leads to Stable New Flax Lines with Altered Secoisolariciresinol Diglucoside (SDG) Profiles. Front. Plant Sci. 8:1638. doi: 10.3389/fpls.2017.01638
Received: 13 May 2017; Accepted: 06 September 2017;
Published: 21 September 2017.
Edited by:
José Manuel Pérez-Pérez, Universidad Miguel Hernández de Elche, SpainReviewed by:
Ravi Maruthachalam, Indian Institute of Science Education and Research, Thiruvananthapuram, IndiaChristopher Cullis, Case Western Reserve University, United States
Copyright © 2017 Fofana, Ghose, Somalraju, McCallum, Main, Deyholos, Rowland and Cloutier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bourlaye Fofana, Ym91cmxheWUuZm9mYW5hQGFnci5nYy5jYQ==
†Present Address: Kaushik Ghose, Department of Plant and Soil Science, Texas Tech University, Lubbock, TX, United States