 Shenghui Zhou
Shenghui Zhou Baiqiang YanFei LiJinpeng ZhangJing ZhangHuihui MaWeihua LiuYuqing LuXinming YangXiuquan LiXu Liu*Lihui Li*
Baiqiang YanFei LiJinpeng ZhangJing ZhangHuihui MaWeihua LiuYuqing LuXinming YangXiuquan LiXu Liu*Lihui Li*- National Key Facility for Crop Gene Resources and Genetic Improvement, Institute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing, China
Agropyron cristatum, which is a wild grass of the tribe Triticeae, grows widely in harsh environments and provides many desirable genetic resources for wheat improvement. However, unclear interspecific phylogeny and genome-wide variation has limited the utilization of A. cristatum in the production of superior wheat varieties. In this study, by sequencing the transcriptome of the representative tetraploid A. cristatum Z559 and the common wheat variety Fukuhokomugi (Fukuho), which are often used as parents in a wide cross, their phylogenetic relationship and interspecific variation were dissected. First, 214,854 transcript sequences were assembled, and 3,457 orthologous genes related to traits of interest were identified in A. cristatum. Second, a total of 72 putative orthologous gene clusters were used to construct phylogenetic relationships among A. cristatum, Triticeae and other genomes. A clear division between A. cristatum and the other Triticeae species was revealed. Third, the sequence similarity of most genes related to traits of interest is greater than 95% between A. cristatum and wheat. Therefore, using the 5% mismatch parameter for A. cristatum, we mapped the transcriptome sequencing data to wheat reference sequences to discover the variations between A. cristatum and wheat and 862,340 high-quality variants were identified. Additionally, compared with the wheat A and B genomes, the P and D genomes displayed an obviously larger variant density and a longer evolutionary distance, suggesting that A. cristatum is more distantly related to the wheat D genome. Finally, by using Kompetitive Allele Specific PCR array (KASPar) technology, 37 of 53 (69.8%) SNPs were shown to be genuine in Z559, Fukuho, and additional lines with seven different P chromosomes, and function of the genes in which these SNPs are located were also determined. This study provides not only the first insights into the phylogenetic relationships between the P genome and Triticeae but also genetic resources for gene discovery and specific marker development in A. cristatum, and this information will be vital for future wheat-breeding efforts. The sequence data have been deposited in the Sequence Read Archive (SRA) database at the NCBI under accession number SRP090613.
Introduction
Wheat (Triticum aestivum L., 2n = 6x = 42, genomes AABBDD) is one of the most important crops. Improving the productivity, adaptation, quality, and nutritional value of wheat to meet the demand for cereal crops remains a major challenge in modern agriculture. However, the narrow genetic basis of wheat has become a bottleneck for wheat improvement, which has led to a surge of interest in exploring natural biodiversity as a source of novel alleles to deliver high-performing wheat varieties (Dubcovsky and Dvorak, 2007). Wild wheat relatives harbor superior agronomic traits, and hybridization is considered an effective measure to enrich the genetic base that has been widely used in wheat-breeding programmes (Bevan et al., 2017).
Agropyron is a perennial genus of the tribe Triticeae, which is commonly called the crested wheatgrass complex, and is built upon one basic P genome, including 3 ploidy levels, diploid, tetraploid, and hexaploid (Dewey, 1984). Most Agropyron species, as excellent sources of forage, have mainly been used as forage crops with high economic value and have also served as a tertiary gene pool for wheat improvement (Dewey, 1984). The tetraploid crested wheatgrass Agropyron cristatum (L.) Gaertn. (2n = 4x = 28; genomes PPPP) is the most common member of the Agropyron genus. A. cristatum is native to Europe and Asia, especially in low-temperature grasslands and sands regions in Eurasian, and has short broad spikes that taper at the top, small seeds, and short stature (Chen et al., 2013) (Figure 1). A. cristatum not only provides protein as a forage source but also possesses several desirable traits for wheat improvement, such as more tillers, spikelets, and florets, resistance against powdery mildew, barley yellow dwarf virus, leaf rust, stripe rust, and stem rust and tolerance against salinity, drought, and low temperatures. In the early 1990s, the inter-generic hybridization of wheat cv. Fukuhokomugi (Fukuho) and A. cristatum accession Z559 was successfully completed via a wide cross and embryo rescue (Li, 1995), and then, many additional lines with excellent characteristics, including disomic substitution lines, translocation lines, and introgression lines, were produced (Zhang et al., 2015b; Li et al., 2016). Several of these lines have been used in wheat-breeding programmes, such as Pubing3504 and Pubing3228, which exhibit an increased number of spikelets and florets (Chen et al., 2012). Although their growth characteristics and utilization in wheat-breeding programmes have been extensively investigated, little is known regarding the phylogenetic relationship and genetic diversity between A. cristatum and wheat.

Figure 1. Morphological characterization of A. cristatum. (A) The A. cristatum growing in the natural environment. (B) Spike morphological characterization. (C) Spikelet morphological characterization.
Molecular markers are important not only for studies regarding evolutionary processes, novel functional gene discovery and development of conservation strategies but also for effectively using genetic resources in breeding programmes. In total, 34 polymorphic markers in A. cristatum and wheat have been identified from 48 A. cristatum-specific sequences obtained from degenerate oligonucleotide-primed PCR (DOP-PCR) products (Han et al., 2017). Additionally, Wu et al. (2010) developed three sequence-characterized amplified region (SCAR) markers that were specific for the P genome of A. cristatum. However, attempts to understand the genetic diversity and phylogenetic relationships between Z559 and Fukuho using these molecular markers have been limited. The identification of single-nucleotide polymorphisms (SNPs) via next-generation sequencing (NGS) is efficient and cost-effective. In addition, because SNPs are biallelic and are the most abundant genetic variations, SNP are evenly distributed at higher frequencies throughout the genome of most plant species (Allen et al., 2011). Therefore, SNP markers have become increasingly useful for developing high-density genetic maps, conducting genome-wide association mapping, marker-assisted selection (MAS) and genomic selection (GS) studies and assessing genetic diversity. Winfield et al. (2016) used a wheat NimbleGen array (Winfield et al., 2012) to direct the capture and targeted re-sequencing of the wheat exome and identified a large number of SNPs from 43 bread wheat accessions and wheat relatives. In addition, 218 genome-wide wheat/Ambylopyrum muticum introgressions were detected and characterized using these SNP markers and the Axiom genotyping array (King et al., 2017).
NGS has facilitated the large-scale discovery of SNPs in various model and non-model plant species. SNP discovery from NGS data was also combined with bulked segregant analysis for the fine mapping of genes in polyploid wheat (Trick et al., 2012; Ramirez-Gonzalez et al., 2015; Bassi et al., 2016). RNA-sequencing (RNA-Seq) has also recently become a popular technique because it is cost-effective, does not rely on a reference genome and can contribute to transcriptional analysis, gene discovery, phylogenetic analysis, and molecular marker development (Meena et al., 2016). In a previous study, de novo transcriptome assembly and unigene functional annotation were conducted in A. cristatum, and gene resources related to stress resistance and inflorescence development and specific to A. cristatum within the tribe Triticeae were identified (Zhang et al., 2015a). However, the phylogenetic relationships, genetic diversity and development of SNP molecular markers in A. cristatum and wheat, especially in inter-generic hybrid parents used in our lab (wheat cv. Fukuho and A. cristatum accession Z559), have been poorly investigated.
In this study, a pipeline was used to dissect phylogenetic relationship and interspecific variation between A. cristatum and wheat (Figure 2). Transcriptome sequencing was performed using Illumina HiSeq 2500 in Z559 and Fukuho. Sequencing data of Z559 were assembled and annotated, and candidate genes related to traits of interest were identified. Furthermore, to better understand the evolutionary history of A. cristanum (P genome), a phylogenetic tree for A. cristatum, Triticeae, and some other genomes was constructed using single-copy orthologous genes. Then, according to the sequence similarity, the trimmed sequencing data were mapped against the wheat reference genome to discover and characterize variants, reveal the genetic diversity at the transcriptome level, and create large subsets of markers for A. cristatum and wheat.
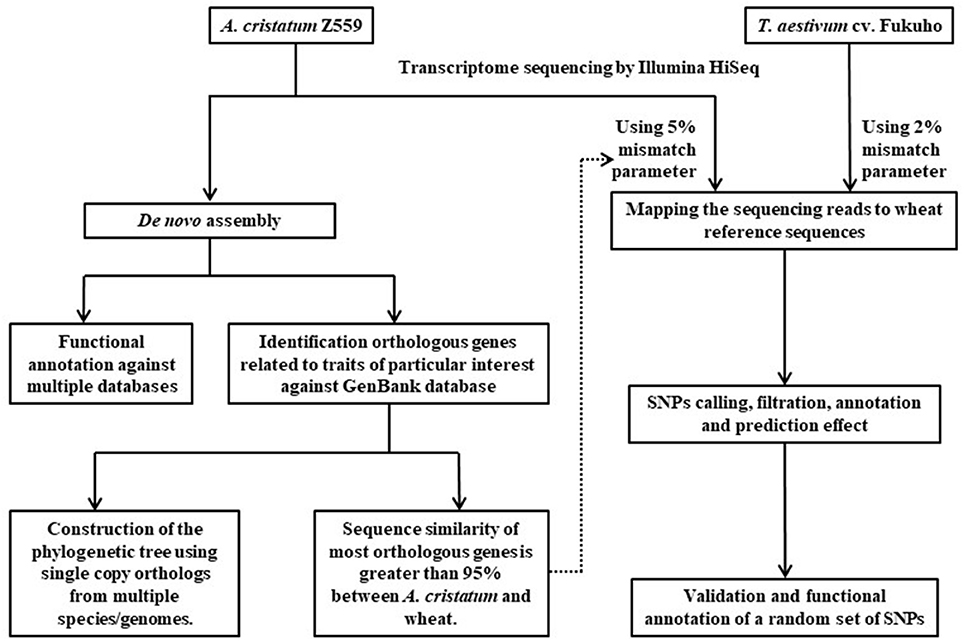
Figure 2. Pipeline used to dissect phylogenetic relationship and interspecific variation between A. cristatum and wheat in this study.
Materials and Methods
Plant Materials
The A. cristatum accession Z559 (2n = 4x = 28, PPPP, from Xinjiang, China) has been previously described (Zhang et al., 2015a). T. aestivum cv. Fukuho was used as a recipient parent for the wide cross between wheat and A. cristatum (Li, 1995).
RNA Extraction and Sequencing
The seedlings of A. cristatum Z559 were planted in the greenhouse. Then, A. cristatum Z559 using tillers planting and Fukuho were cultivated in the same experimental field of the Chinese Academy of Agricultural Sciences (E116.33, N39.96), Beijing, China. The type of soil was sandy clay loam. Sowing date was October 1 of 2015 and experimental field was fertilized with 112 kg ha−1 of a N-P-K mixture (16–20–14%) plus 170 kg ha−1 of urea. Plants were watered as needed. Unfertilized caryopses and young healthy leaves were collected from five different plants at growth stage 54 (Zadoks et al., 1974) and immediately snap-frozen in liquid nitrogen, ground into powder, and then extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's recommendations. The quantity and integrity of the total RNA were assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and 1% agarose gel electrophoresis. Equal amounts of total RNA from each tissue were pooled for Z559 and Fukuho. Only the total RNA samples with RIN values ≥8 were used for constructing the cDNA libraries. All libraries were sequenced using the Illumina HiSeq 2500 platform with a paired-end read length of 150 bp. The library construction and sequencing were performed by the Novogene Corporation (Beijing, China).
Data Pre-processing, De novo Assembly, and Annotation
The raw sequence reads were cleaned by removing the RNA adapters and trimming the low-quality bases (Q < 20) with a minimum read length of 36 bases using Trimmomatic (version 0.36) (Bolger et al., 2014). The trimmed reads were then de novo assembled using the Trinity package (version 2.4.0) with the default parameters and a minimum transcript length setting of 200 nt (Haas et al., 2013). The clustering of the transcripts was performed using the CD-HIT-EST software (version 4.6.6) (Li and Godzik, 2006) to reduce the unavoidable redundancy produced by the Trinity assembly. Many low-quality transcripts and potential assembly errors can occur during assembly. Therefore, all reads were mapped back to transcripts using Bowtie (version 1.2.0) (Langmead et al., 2009). The fragments per kilobase of transcript per million mapped reads (FPKM) value of the transcripts was calculated using RSEM (version 1.3.0) (Li and Dewey, 2011), and transcripts with FPKM values <1 were also removed. The remaining transcripts were then used to perform the downstream analysis.
The filtered transcripts were analyzed by TransDecoder (version v3.0.0) (Grabherr et al., 2011) to determine their cDNA coding sequence (CDS) and protein sequences. For the annotation, all CDSs were aligned using BLAST (version 2.5.0+) (Altschul et al., 1990) to the Non-Redundant (NR) database, Pfam-A database (version 31.0) (Finn et al., 2014), SwissProt database (release 2017_04), and Eukaryotic Orthologous Groups (KOG) database (with E-value cut-off of 1e-5) (Tatusov et al., 2003). The protein sequence with the best hit was considered the optimal annotation. The results were loaded into Trinotate (version 3.0) (Grabherr et al., 2011) SQLite database to generate a comprehensive annotation report.
Ortholog Identification and Phylogenetic Analysis
Genes potentially related to traits of particular interest were searched against GenBank sequence database (http://www.ncbi.nlm.nih.gov/genbank/) using an in-house Perl script. All traits of particular interest were more concerned in wheat breeding programmes and classified into three major categories. These categories were as follows: (i) agronomic traits, which included kernels per spike, seed weight, tiller, plant height, photosynthetic efficiency, stem toughness, and growth period-related traits; (ii) biotic stress-related traits, which included fungal/virus disease and insect pests resistance-related traits; and (iii) abiotic stress-related traits, which included cold, heat, drought and salt stress, fertilizer and water use efficiency, seed storage and adaptation-related traits (Tables S1, S2 and Files S1, S2). The clustering of genes was performed using the CD-HIT-EST software to obtain the representative genes. OrthoMCL (version 2.0.9) was used to identify the orthologous genes in A. cristatum (Li L. et al., 2003). A sequence similarity analysis was also performed using BLAST with the default parameters to determine the sequence identity.
The agronomic traits and biotic and abiotic stress-related genes identified above in A. cristatum were used to find orthologous genes in other genomes, e.g., Triticum aestivum (A, B, and D genomes were separated), Triticum urartu, Triticum turgidum (A and B genomes were separated), Aegilops tauschii, A. tauschii, Secale cereale, Brachypodium distachyon, Sorghum bicolor, Zea mays, Setaria italica, Oryza sativa, and Arabidopsis thaliana. The cDNA sequences of these genomes were downloaded from EnsemblPlants (www.plants.ensembl.org), and OrthoMCL was used to identify orthologous gene pairs. Putative orthologs with a BLAST score ratio of the second best-hit to the first best-hit greater than 0.8 were discarded, which can yield single-copy genes in one genome. Gene clusters that are single copy in one genome and highly conserved in all genomes were aligned by MUSCLE (Edgar, 2004). The alignment was subjected to MEGA7 (Kumar et al., 2016) to construct the phylogenetic tree using the Neighbour-Joining method (Saitou and Nei, 1987). The bootstrap consensus tree inferred from 500 replicates is taken to represent the evolutionary history of the analyzed taxa (Felsenstein, 1985). Branches corresponding to partitions reproduced in less than 50% of bootstrap replicates were collapsed. Evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004) and are in units of the number of base substitutions per site. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. All positions containing gaps and missing data were eliminated.
SNP Discovery in the Transcriptomes of A. cristatum and Wheat
The raw reads were aligned to the wheat reference genome sequence TGACv1 (Clavijo et al., 2017) using the STAR tool (version 2.5.3a) (Dobin et al., 2013), including the 2-pass STAR method with a minimum intron length of 20 bp, a maximum intron length of 20 kb the default settings for the other parameters and 5 and 2% maximum mismatches for A. cristatum and wheat, respectively. The creation of the GTF annotation file and the quantification of the genes and isoforms were performed using StringTie (version 1.3.3b) (Pertea et al., 2015) from a mapping bam file. Duplicate markings, split “N” Trimming and reassigning mapping qualities were successively performed to filter the mapping results. The haplotype caller in the GATK variant pipeline was used to call SNPs between A. cristatum and wheat from the genome-mapped alignments. The variant filtration was performed with a total coverage >6 and an absolute value of allele frequency difference >0.9 between A. cristatum and wheat to obtain high-quality SNPs. The SnpEff (version 4.3k) pipeline was used to annotate and predict the effects of the SNPs based on their genomic locations and predict their coding effects (Cingolani et al., 2012), providing a simple assessment of the putative impact of the variant, i.e., high (frame shifts, addition/deletion of stop codons, etc.), moderate (codon change/deletion/insertion, etc.), low (synonymous changes, etc.), and modifier (changes outside coding regions, etc.). The definitions of the putative impact of the variants are listed in the Table S3 with brief explanations. The visualization of the data at the genome-wide level (TGACv1 map) was performed using the software Circos (version 0.69-3) (Krzywinski et al., 2009).
SNP Marker Validation
Young leaves of Z559, Fukuho and their additional lines involving seven different P chromosomes were used for DNA extraction, PCR amplification, and SNP marker validation. In total, 53 SNP markers were validated using the Kompetitive Allele Specific PCR (KASP) array. The SNP contextual sequences that did not cross the intron in the wheat genome were obtained and used to design the primers. Based on the 53 SNP loci contextual sequence, two allele-specific primers (one for each SNP allele) and one common (reverse) primer were designed for each KASP assay using a tool provided by LGC Genomics (www.lgcgenomics.com) (Table S4). The KASP assays were designed by LGC Genomics and carried out according to the company's protocol (http://lgcgenomics.com).
Results
Sample Sequencing
Two transcriptome libraries were generated from the pooled RNA-extracts from two tissues (leaves and young spikes) of Z559 and Fukuho. The sequencing produced paired-end reads with a length of 150 base pairs (bp). A summary of the transcriptome sequencing data is presented in Table 1. The Illumina sequencing generated 64,254,354 and 42,700,222 sequence reads in Z559 and Fukuho, respectively. After filtering the low-quality reads, 99.98% of the sequencing reads (64,243,343 reads for Z559 and 42,692,045 reads for Fukuho) were retained, and a downstream analysis was performed. GC content (52%) in the sequences of Z559 and Fukuho was identical (Table 1). The sequence data have been deposited in the Sequence Read Archive (SRA) database at the NCBI under the accession number SRP090613.

Table 1. Summary of the RNA-Seq data.
A. cristatum Z559 Transcriptome Assembly and Gene Annotation
A de novo assembly of Z559 was performed using the high-quality filtered reads. The assembly produced a total of 214,854 transcripts ranging in length from 201 to 13,610 bp with a mean size of 611.22 bp, and the size of N50 was 792 bp (Table 2, Figure 3).

Table 2. Statistics of the A. cristatum transcriptome assembly and annotation.
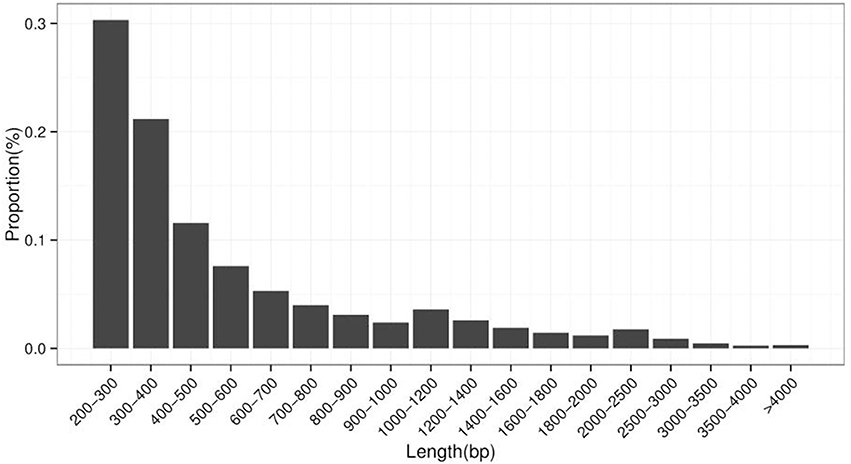
Figure 3. Transcript length distribution in A. cristatum.
Of the assembled transcripts, 78,912 (36.7%) transcripts with coding sequences were identified using TransDecoder software (Grabherr et al., 2011). Of the predicted transcripts, 57,183 transcripts were found to have homologs in the NR database. The number of transcripts with Pfam domain assignments was 38,208, and 55,157 transcripts had significant matches in the SwissProt database. In addition, 18,073 transcripts were associated with GO terms, and 43,487 transcripts had matches in the KOG database. Altogether, 66,346 transcripts had at least one hit in these databases (Table 2).
Identification of Orthologous Genes and Phylogenetic Analysis
To further understand and better use the genetic diversity of A. cristatum, genes potentially related to agronomic and biotic and abiotic stress-related traits were identified in A. cristatum. In total, 9,354 genes related to these traits were downloaded from GenBank (Table S1). After filtering the highly homologous genes, 4,841 representative genes remained (Tables S1, S2 and Files S1, S2). Furthermore, 3,457 genes orthologous to these representative genes were identified in our assembled Z559 transcripts (Table S1).
Of the 3,457 genes identified above, a total of 72 putative orthologous gene clusters were identified after carefully filtering with one copy in one genome and conversed in all genomes. Phylogenetic relationships were then constructed based on the alignment of these 72 orthologs from A. cristatum transcriptome data and cDNA data from other genomes (Figure 4). Specifically, A. cristatum and other Triticeae (Poaceae), including T. aestivum and their ancestral species T. urartu, A. tauschii and T. turgidum, Hordeum vulgare, and S. cereale, clustered into a sister clade. However, four major clades existed within Triticeae, and the analysis also showed a clear division between A. cristatum and the other Triticeae species, indicating the P genome should possess rich genetic variation that can be used for wheat genetic improvement. The results also supported the point that hybridization between Agropyron Gaertn. and wheat is difficult to achieve (Dewey, 1984). Moreover, the evolutionary distance indicated that the P genome was more distantly related to the wheat D genome than to the A and B genomes (Figure 4).

Figure 4. Phylogenetic relationships between A. cristatum and other genomes. Numbers at each node are the bootstrap values, shown as percentages. The branch lengths are the same as the evolutionary distances.
Variant Calling and Effects between A. cristatum and T. aestivum
In this study, A. cristatum transcript sequences assembled using Trinity were compared with the published wheat CDS database using a blastn search. Sequence identity averages of 94.2% were found between the wheat and A. cristatum transcript sequences (Figure 5). The highest peak of the distribution indicates a 97.6% identity between the A. cristatum transcriptome and the CDS of wheat (Figure 5). Overall, the transcript sequences of A. cristatum show highly conserved wheat transcript sequences, and most of the transcript sequences (72.5%) (Figure 5) and genes related traits of interest (83.3%) have ≥95% sequence identity to the CDS of wheat. Thus, the A. cristatum transcriptome is closely related to the wheat transcriptome.
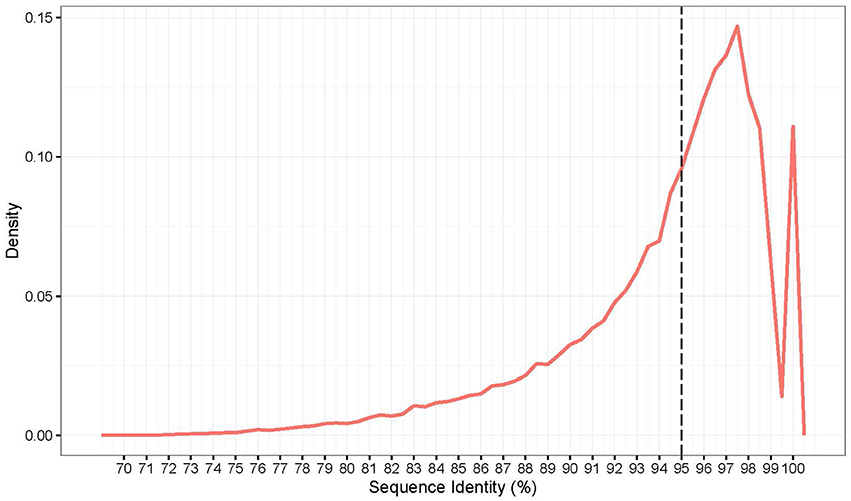
Figure 5. Density distribution of A. cristatum transcript sequences identity compared to the wheat.
To identify local genomic variability, the Z559 and Fukuho transcriptome sequencing data were mapped to a wheat reference sequence (Figure 6A). The average gene density and transcriptional density were calculated along all chromosomes in the wheat genome. In total, 268,200 and 184,423 transcriptional regions were identified in Z559 and Fukuho, respectively. In addition, 112,765 protein-coding genes were predicted with functional support and the entire gene structure. The average gene density and transcriptional density sharply decreased from centromeres to telomeres in the whole chromosomal region in both Z559 and Fukuho (Figures 6C–E).
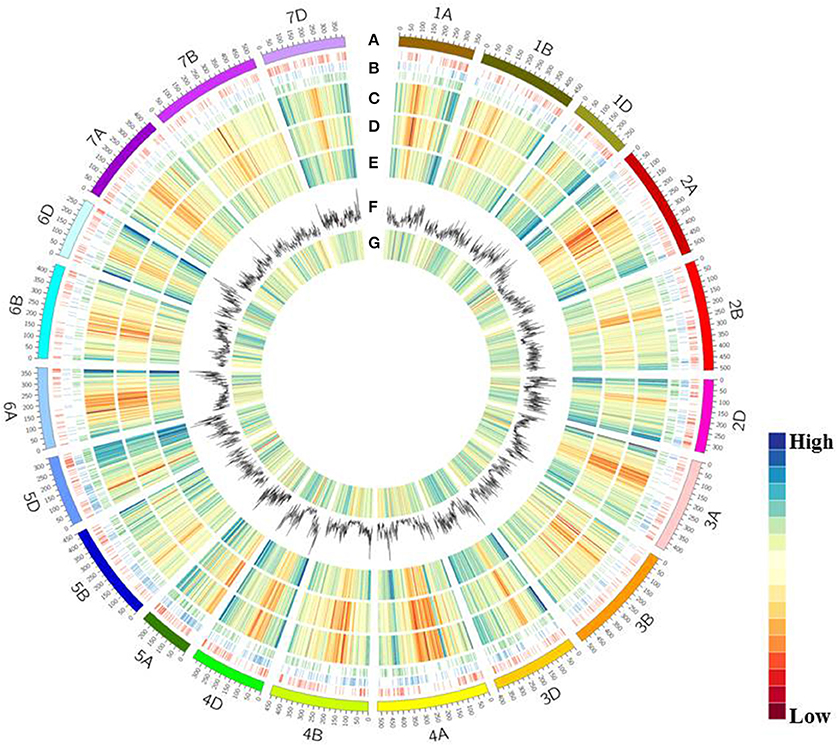
Figure 6. CIRCOS visualization of different data at the wheat genome-wide level. (A) Karyotype of the wheat genome. (B) Location of homologous genes, including SNPs related to traits. Agronomic traits (red), biotic stress (blue) abiotic stress (green). (C) Gene content density distribution; Gene density was calculated in a 3-Mb window. (D) Transcriptional density distribution in wheat. Transcript regions density was calculated in 3-Mb window intervals. (E) Transcriptional density distribution in A. cristatum. Transcript region density was calculated in 3-Mb window intervals. (F) Variant distribution by chromosome. SNP density was calculated in 3-Mb window intervals. (G) Variant density in transcript regions. Variant density was calculated in transcript regions at 3-Mb window intervals.
A total of 862,340 high-quality variants were identified, including 817,970 SNPs and 44,370 InDels, in the transcripts of Fukuho and Z559, which were spread across the wheat genome (Figures 6F, 7A). The number of variants in each chromosome was not directly proportional to the chromosome length and gene number. Homologous group 2 chromosomes contained the most variants, whereas homologous group 6 chromosomes contained the fewest variants (Figure 7A). The density of the SNPs per 1 Mb of the genetic region on each chromosome is shown in Figure 7B. Overall, 59.28 SNPs/Mb were found on average across the wheat genome. Importantly, in each homologous group, the highest SNP density was observed on the D genome (77.88 SNPs/Mb), followed by the B genome (53.26 SNPs/Mb), and A genome (53.59 SNPs/Mb) (Figure 7B). Interestingly, the number of variants in the telomeric regions was the highest in the whole chromosome (Figure 6F), but the variant density in the transcriptional regions was relatively lower in the telomeric and centromeric regions than that in the other chromosomal regions (Figure 6G).
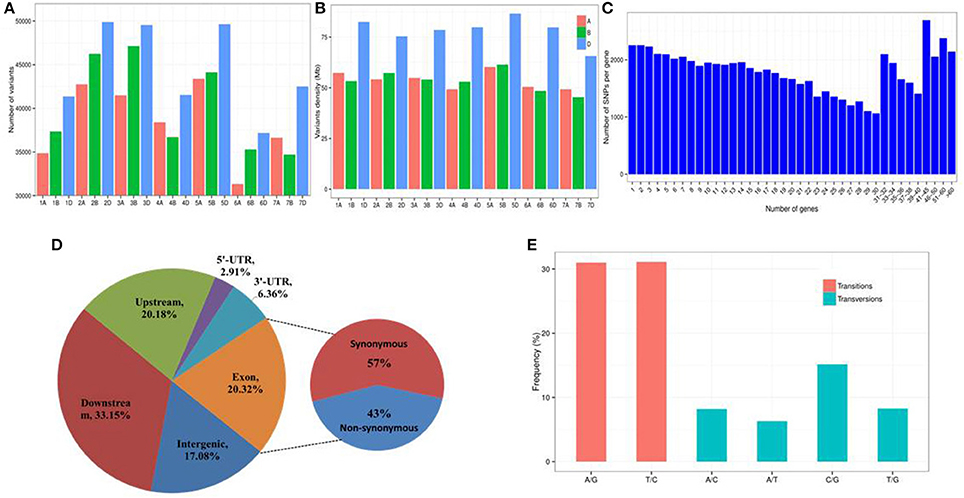
Figure 7. Characterization of SNPs between A. cristatum and wheat. (A) Distribution of SNPs in the wheat genome. (B) Density distribution of SNPs in the wheat genome. Density was measured by averaging the number of SNPs per 1 Mb of the wheat genomic region. (C) Distribution of the number of SNPs per gene. (D) Frequency of different substitution types in the identified SNPs. (E) Distribution of SNPs in different genomic regions.
Overall, 70,456 annotated genes (62.48%) contained one or more variants (Figure 7C). Of these genes, 5,036 genes had high impact variants, 42,415 genes had low impact variants, 37,902 genes had moderate impact variants and 65,967 genes had modifier variants. The average number of SNPs per gene was 21.8, while 44.4% of the genes had fewer than 20 SNPs. Interestingly, 2,143 genes harbored more than 60 SNPs (Figure 7C), which implied that these genes exhibited high diversity. Thus, these 2,143 genes might be particularly amenable to artificial selection and are helpful for understanding the genetic diversity of Triticeae. The distribution of the discovered variants within various genomic features is shown in Figure 7D and Table S5. The number of variants in the intergenic, downstream, and upstream regions of the genes is 376,552 (17.08%), 730,523 (33.15%), and 444,836 (20.18%), respectively.
Approximately 204,285 (9.27%) SNPs located in the 5′- or 3′-UTR regions were identified, and 20.32% of the variants were present in coding regions in which the ratio of non-synonymous to synonymous variants was ~0.75 (163,226/220,482) (Figure 7D, Table S5), suggesting that the transcriptional regions have been under purifying selection.
The frequency of substitution transitions (62.09%; A/G: 30.99% and T/C: 31.10%) was much higher than the frequency of transversions (37.91%; A/C: 8.19%, A/T: 6.30%, C/G: 15.16, and T/G: 8.26%) (Figure 7E). The proportion of A/G transitions (30.99%) was similar to that of T/C transitions (31.10%). The frequency of each type of transversion was ~7%, except for the C/G transitions, which had a frequency of 15.16% (Figure 7E). The ratio of transitions to transversions (ti/tv) was 1.64:1.
Validation of SNPs Using the KASP Genotyping Assay
Fifty-three SNPs between Z559 and Fukuho identified in this study were validated using a KASP genotyping assay, in which an allele-specific primer was used to discriminate between single-nucleotide substitutions (Table S4). Thirty-seven (69.8%) of the 53 SNP loci were successfully confirmed in Z559 and Fukuho and additional lines with 7 different P chromosomes. Among these, 6, 3, 5, 1, 8, and 8 SNPs were located on the 1P, 2P, 3P, 4P, 5P, and 7P chromosomes, respectively. In addition, 1 SNP located on both 2P and 5P, 2 SNPs located on both 5P and 6P, 1 SNP located on 6P and 7P, and 1 SNP located on 5P, 6P and 7P were also determined, and 1 SNP was undetermined (Table S4). The functional information of genes in which SNPs are located was shown in Table 3 and these KASP markers could be used to follow specific genes of interest.
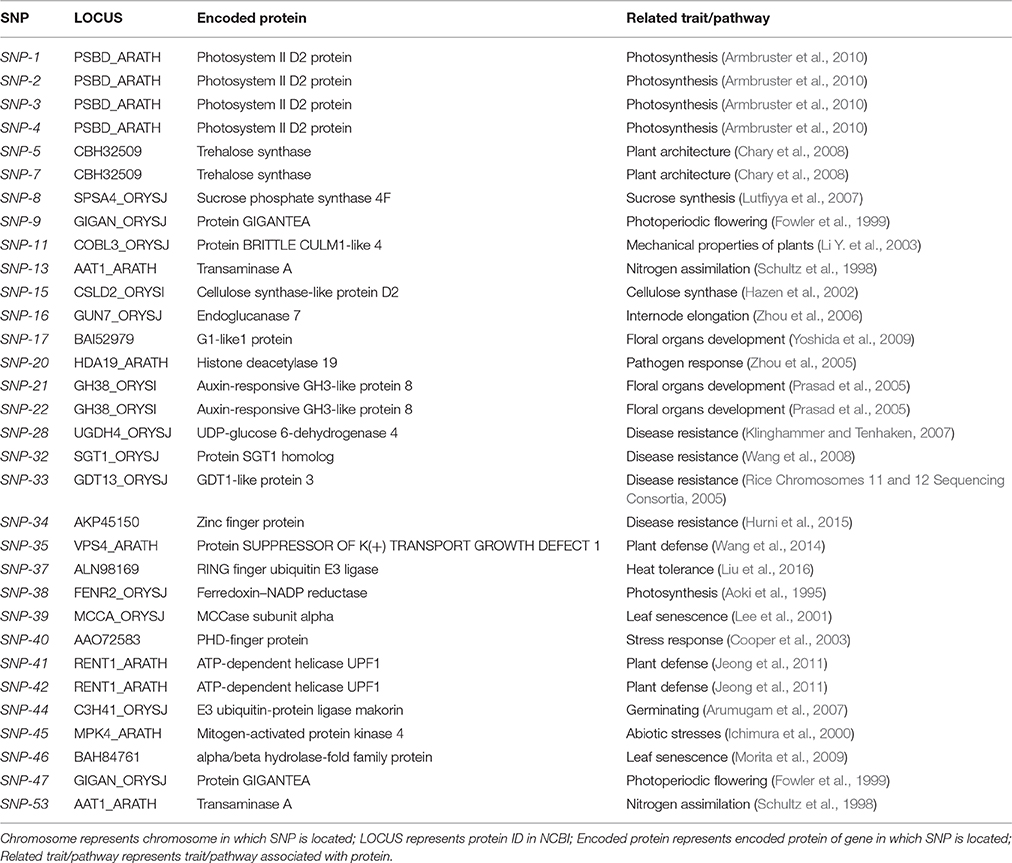
Table 3. Functional information of genes in which SNPs are located.
Discussion
An Effective Research Method to Develop Markers and Analyse the Genetic Diversity of the Wild Relatives of Wheat
Although the potential of using wild relatives to improve wheat has been recognized for a long time, the available genetic diversity remains largely underexploited. To utilize the full potential of these genes, it is important to understand the genetic diversity in the wild wheat relatives at a molecular level, increase the number of genome-specific molecular markers and identify the loci underlying the traits of interest (Hajjar and Hodgkin, 2007). Due to the large genome complexity and sequence redundancy, reference genomes are not currently available for most wild relatives of wheat. Transcriptome RNA sequencing is an effective strategy for identifying polymorphisms in transcribed regions of the genome. Tanwar et al. (2017) performed a transcriptome sequencing of two guar varieties to develop genomic resources. Potential SSRs and SNPs were identified and 20 SSRs were validated using wet laboratory analysis. Nigam et al. (2017) reported a transcriptome sequencing of closest of wild relative to Cajanus cajan. And unigenes and SSR markers were identified in this study. Huang et al. (2016) used Illumina high-throughput deep transcriptome sequencing to characterize of two agriculturally important Hemarthria materials. After assembly, unigenes were obtained and annotated and SNPs and SSRs were identified. Some markers were randomly selected to validate the identified markers. However, the low representation of wild wheat relatives in the SNP design process may limit the utility of these platforms in wheat-alien introgression breeding (Wulff and Moscou, 2014). High-quality reference genome assemblies are available for Chinese Spring wheat (Clavijo et al., 2017), which provides a basis for solving this problem. In this study, an effective research method was used to develop genus-specific markers and explore the genetic diversity of wild wheat relatives compared to wheat. First, de novo assembly, gene annotation and discovery of genes related to agronomic traits and biotic and abiotic stresses in A. cristatum were performed. The comparative genomics analysis revealed that the highest peak value in the sequence identity distribution is 97.6%, and most genes have an identity that is greater than 95% between the A. cristatum transcriptome and wheat (Figure 5). The above-mentioned information suggests that there is a close genetic relationship between A. cristatum and wheat, which is consistent with previously reported results (Zhang et al., 2015a). Genes with a low similarity between rye sequences and their closest matches in the Triticum genome have been shown to have a higher probability of being repressed or deleted in the allopolyploid genome (Khalil et al., 2015). Therefore, molecular markers developed based on conserved expressed sequences with a high similarity could be applied in wheat improvement because of their high transferability. Based on the concept of discovering variations and revealing genetic diversity between A. cristatum and Fukuho for wheat improvement, transcriptome sequencing data were mapped to a wheat reference sequence with a 5% mismatch parameter for Z559, which ensures that the sequencing reads of most genes can be mapped to the wheat reference genome. Based on the mapped results, the discovery of variants and the development of genus-specific markers were performed smoothly and efficiently. The effective research method used in this study can be applied in other studies to investigate genus-specific markers in other wild relatives of wheat.
Distant Relationship and Abundant Genetic Diversity between A. cristatum and Wheat
Although the wheat-A. cristatum derivatives represent an attractive source of value-added traits (Zhang et al., 2015b; Li et al., 2016), the genetic relationship and variants, which represent the fundamental key to breeding success and provide a basis for breeders to select varieties with a constantly improving yield performance (Bedő and Láng, 2015), of A. cristatum compared to wheat at the molecular level remain unknown. This study showed that A. cristatum had abundant genetic resources and diversity compared to wheat from the various aspects described below. First, phylogenetic analysis indicated that A. cristatum is most distantly related to wheat among Triticeae and also showed a clear division between A. cristatum and the other Triticeae species, suggesting the P genome possesses rich genetic variation that can be used for wheat genetic improvement (Figure 4). Second, 3,457 genes associated with agronomic traits and biotic and abiotic stress in A. cristatum were identified by searching for their homologs in four different genomes (A. thaliana, rice, maize, and wheat) (Table S1), and their homologous gene variations were widely distributed in the wheat genome (Figure 6B). In our previous study, 532 genes were associated with biotic stress, 176 genes were associated with abiotic stress and 89 genes with a large spike phenotype were identified in A. cristatum (Zhang et al., 2015a). Uncovering novel functional genes paves the way for the efficient mining of the gene pools of wild relatives to improve wheat. Third, 817,970 SNP markers exhibited extremely high allelic variations in wild A. cristatum compared to wheat. The SNP density in the wheat D genome was obviously larger than that in the A and B genomes (Figure 7B), indicating that A. cristatum is more distantly related to the D genome. This conclusion is also supported by the phylogenetic analysis between P and the wheat A, B, and D genomes (Figure 4). These results were the first to reveal the evolutionary relatedness between A. cristatum and the wheat A, B, and D genomes and explained that the wheat A genome and B genome had a higher number of chromosome arrangements with A. cristatum chromosomes than the D genome (Badaeva et al., 2007; Li et al., 2016). Interestingly, the variant density in the transcriptional regions was relatively lower in the telomeric and centromeric regions than that in the other chromosomal regions (Figure 6G). This result may explain the phenomenon in which the intercalary translocation lines were difficult to explore in the spontaneous or induced wheat-alien chromosome translocation lines (Jiang et al., 1993), but further research studies are necessary to clarify the molecular mechanisms. Furthermore, the analysis of the evolutionary relationship and genetic diversity of A. cristatum compared with wheat, including gene pools related to agronomic traits and biotic and abiotic stress, abundant genetic variation and specific expression genes, provided a good basis for the utilization of A. cristatum genomic target regions for the restoration of genetic diversity in future wheat-breeding efforts.
Future Applications of SNP Markers
Many types of molecular markers were used in wheat-alien introgression breeding to identify and characterize alien chromosome/chromosome-arm additions and substitution lines. SNP markers provide another means of assessing genetic variation and are abundant and easily obtained via high-throughput sequencing (Vatanparast et al., 2016). Consequently, as a genotyping SNP polymorphism technology, KASP SNP markers are becoming popular and are used in large-scale projects (He et al., 2014; Semagn et al., 2014). Recently, more and more studies have used SNP molecular markers to study alien gene transfer in wheat and detect wheat/wild relative introgressions when they occur (Tiwari et al., 2014, 2015; Winfield et al., 2016; King et al., 2017). In our study, the highest throughput variants, including 817,970 SNPs, between A. cristatum and wheat using transcriptome sequencing were identified. Additionally, 37 of the 53 SNPs (69.8%) identified in this study have been verified using KASP genotyping SNP technology. SNP markers developed from transcriptome sequencing are suitable for the development of functional markers that are tightly linked to traits of interest, the large-scale screening of progenies of wild hybrids and the production of lines with introgressed genes of interest and minimum unwanted chromatin (Rey et al., 2015). In addition, SNP markers discovered through transcriptome sequencing are currently used to design genotyping arrays for several important crop plants containing thousands of markers spread throughout the genome to analyse large numbers of samples (Ganal et al., 2012; Sim et al., 2012; Houston et al., 2014; Humble et al., 2016). Therefore, an array using the SNPs discovered in this study can also be developed to detect alien genetic transfer in wheat-A. cristatum derivative lines and simplify wheat-alien introgression breeding. Overall, the development of SNP molecular markers in this study can increasingly contribute to incorporating the value-added trait genes from wild relatives to wheat in breeding programmes.
Author Contributions
SZ, LL, and XL designed the study. SZ conducted the bioinformatics analyses and wrote the manuscript. BY and FL performed the experiments. JpZ, JZ, XqL, and XY prepared the samples for sequencing. HM, YL, and WL contributed to editing the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by grants from the National Natural Science Foundation of China (Grant No. 31471493), the National Science and Technology Support Program of China (Grant No. 2013BAD01B02), and the National Key Research and Development Program of China (Grant No. 2016YFD0102000).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01644/full#supplementary-material
Table S1. Statistical results of the functionally characterized genes downloaded from GenBank and identified in A. cristatum.
Table S2. List of functional genes downloaded from GenBank.
Table S3. A list of effects, with some explanations and putative impacts of the variants.
Table S4. Primer sequences and chromosomes of SNP markers for validation by KASP assay.
Table S5. The distribution of the discovered variants in various genomic features.
File S1. Protein sequences of functional genes downloaded from GenBank (FASTA format).
File S2. Detailed information on functional genes downloaded from GenBank (GenBank format).
References
Allen, A. M., Barker, G. L. A., Berry, S. T., Coghill, J. A., Gwilliam, R., Kirby, S., et al. (2011). Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol. J. 9, 1086–1099. doi: 10.1111/j.1467-7652.2011.00628.x
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Aoki, H., Tanaka, K., and Ida, S. (1995). The genomic organization of the gene encoding a nitrate-inducible ferredoxin-NADP+ oxidoreductase from rice roots. Biochim. Biophys. Acta. 1229, 389–392. doi: 10.1016/0005-2728(95)00032-E
Armbruster, U., Zuhlke, J., Rengstl, B., Kreller, R., Makarenko, E., Ruhle, T., et al. (2010). The Arabidopsis thylakoid protein PAM68 is required for efficient D1 biogenesis and photosystem II assembly. Plant Cell 22, 3439–3460. doi: 10.1105/tpc.110.077453
Arumugam, T. U., Davies, E., Morita, E. H., and Abe, S. (2007). Sequence, expression and tissue localization of a gene encoding a makorin RING zinc-finger protein in germinating rice (Oryza sativa L. ssp. Japonica) seeds. Plant Physiol. Biochem. 45, 767–780. doi: 10.1016/j.plaphy.2007.07.006
Badaeva, E. D., Dedkova, O. S., Gay, G., Pukhalskyi, V. A., Zelenin, A. V., Bernard, S., et al. (2007). Chromosomal rearrangements in wheat: their types and distribution. Genome 50, 907–926. doi: 10.1139/G07-072
Bassi, F. M., Ghavami, F., Hayden, M. J., Wang, Y., Forrest, K. L., Kong, S., et al. (2016). Fast-forward genetics by radiation hybrids to saturate the locus regulating nuclear–cytoplasmic compatibility in Triticum. Plant Biotechnol. J. 14, 1716–1726. doi: 10.1111/pbi.12532
Bedő, Z., and Láng, L. (2015). “Wheat breeding: current status and bottlenecks,” in Alien Introgression in Wheat: Cytogenetics, Molecular Biology, and Genomics, eds M. Molnár-Láng, C. Ceoloni, and J. Doležel (Cham: Springer International Publishing), 77–101.
Bevan, M. W., Uauy, C., Wulff, B. B. H., Zhou, J., Krasileva, K., and Clark, M. D. (2017). Genomic innovation for crop improvement. Nature 543, 346–354. doi: 10.1038/nature22011
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chary, S. N., Hicks, G. R., Choi, Y. G., Carter, D., and Raikhel, N. V. (2008). Trehalose-6-phosphate synthase/phosphatase regulates cell shape and plant architecture in Arabidopsis. Plant Physiol. 146, 97–107. doi: 10.1104/pp.107.107441
Chen, D., Zhang, J. P., Wang, J. S., Yang, X. M., Liu, W. H., Gao, A. N., et al. (2012). Inheritance and availability of high grain number per spike in two wheat germplasm lines. J. Integr. Agric. 11, 1409–1416. doi: 10.1016/S2095-3119(12)60140-3
Chen, S. Y., Ma, X., Zhang, X. Q., Huang, L. K., and Zhou, J. N. (2013). Genetic diversity and relationships among accessions of five crested wheatgrass species (Poaceae: Agropyron) based on gliadin analysis. Genet. Mol. Res. 12, 5704–5713. doi: 10.4238/2013.November.18.19
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6, 80–92. doi: 10.4161/fly.19695
Clavijo, B. J., Venturini, L., Schudoma, C., Accinelli, G. G., Kaithakottil, G., Wright, J., et al. (2017). An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 27, 885–896. doi: 10.1101/gr.217117.116
Cooper, B., Clarke, J. D., Budworth, P., Kreps, J., Hutchison, D., Park, S., et al. (2003). A network of rice genes associated with stress response and seed development. Proc. Natl. Acad. Sci. U.S.A 100, 4945–4950. doi: 10.1073/pnas.0737574100
Dewey, D. R. (1984). “The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae,” in Gene Manipulation in Plant Improvement, ed J. P. Gustafson (Boston, MA: Springer), 209–279.
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dubcovsky, J., and Dvorak, J. (2007). Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316, 1862–1866. doi: 10.1126/science.1143986
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fowler, S., Lee, K., Onouchi, H., Samach, A., Richardson, K., Morris, B., et al. (1999). GIGANTEA: a circadian clock-controlled gene that regulates photoperiodic flowering in Arabidopsis and encodes a protein with several possible membrane-spanning domains. EMBO J. 18, 4679–4688. doi: 10.1093/emboj/18.17.4679
Ganal, M. W., Polley, A., Graner, E.-M., Plieske, J., Wieseke, R., Luerssen, H., et al. (2012). Large SNP arrays for genotyping in crop plants. J. Biosci. 37, 821–828. doi: 10.1007/s12038-012-9225-3
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hajjar, R., and Hodgkin, T. (2007). The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica 156, 1–13. doi: 10.1007/s10681-007-9363-0
Han, H., Liu, W., Lu, Y., Zhang, J., Yang, X., Li, X., et al. (2017). Isolation and application of P genome-specific DNA sequences of Agropyron Gaertn. in Triticeae. Planta 245, 425–437. doi: 10.1007/s00425-016-2616-1
Hazen, S. P., Scott-Craig, J. S., and Walton, J. D. (2002). Cellulose synthase-like genes of rice. Plant Physiol. 128, 336–340. doi: 10.1104/pp.010875
He, C., Holme, J., and Anthony, J. (2014). SNP genotyping: the KASP assay. Crop Breed. Methods Protoc. 1145, 75–86. doi: 10.1007/978-1-4939-0446-4_7
Houston, R. D., Taggart, J. B., Cézard, T., Bekaert, M., Lowe, N. R., Downing, A., et al. (2014). Development and validation of a high density SNP genotyping array for Atlantic salmon (Salmo salar). BMC Genomics 15:90. doi: 10.1186/1471-2164-15-90
Huang, X., Yan, H. D., Zhang, X. Q., Zhang, J., Frazier, T. P., Huang, D. J., et al. (2016). De novo transcriptome analysis and molecular marker development of two Hemarthria species. Front. Plant Sci. 7:496. doi: 10.3389/fpls.2016.00496
Humble, E., Thorne, M. A. S., Forcada, J., and Hoffman, J. I. (2016). Transcriptomic SNP discovery for custom genotyping arrays: impacts of sequence data, SNP calling method and genotyping technology on the probability of validation success. BMC Res. Notes 9:418. doi: 10.1186/s13104-016-2209-x
Hurni, S., Scheuermann, D., Krattinger, S. G., Kessel, B., Wicker, T., Herren, G., et al. (2015). The maize disease resistance gene Htn1 against northern corn leaf blight encodes a wall-associated receptor-like kinase. Proc. Natl. Acad. Sci. U.S.A. 112, 8780–8785. doi: 10.1073/pnas.1502522112
Ichimura, K., Mizoguchi, T., Yoshida, R., Yuasa, T., and Shinozaki, K. (2000). Various abiotic stresses rapidly activate Arabidopsis MAP kinases ATMPK4 and ATMPK6. Plant J. 24, 655–665. doi: 10.1046/j.1365-313x.2000.00913.x
Jeong, H. J., Kim, Y. J., Kim, S. H., Kim, Y. H., Lee, I. J., Kim, Y. K., et al. (2011). Nonsense-mediated mRNA decay factors, UPF1 and UPF3, contribute to plant defense. Plant Cell Physiol. 52, 2147–2156. doi: 10.1093/pcp/pcr144
Jiang, J., Friebe, B., and Gill, B. S. (1993). Recent advances in alien gene transfer in wheat. Euphytica 73, 199–212. doi: 10.1007/BF00036700
Khalil, H. B., Ehdaeivand, M.-R., Xu, Y., Laroche, A., and Gulick, P. J. (2015). Identification and characterization of rye genes not expressed in allohexaploid triticale. BMC 16:1. doi: 10.1186/s12864-015-1480-x
King, J., Grewal, S., Yang, C. Y., Hubbart, S., Scholefield, D., Ashling, S., et al. (2017). A step change in the transfer of interspecific variation into wheat from Amblyopyrum muticum. Plant Biotechnol. J. 15, 217–226. doi: 10.1111/pbi.12606
Klinghammer, M., and Tenhaken, R. (2007). Genome-wide analysis of the UDP-glucose dehydrogenase gene family in Arabidopsis, a key enzyme for matrix polysaccharides in cell walls. J. Exp. Bot. 58, 3609–3621. doi: 10.1093/jxb/erm209
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lee, R. H., Wang, C. H., Huang, L. T., and Chen, S. C. (2001). Leaf senescence in rice plants: cloning and characterization of senescence up-regulated genes. J. Exp. Bot. 52, 1117–1121. doi: 10.1093/jexbot/52.358.1117
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H. H., Lv, M. J., Song, L. Q., Zhang, J. P., Gao, A. N., Li, L. H., et al. (2016). Production and identification of wheat-Agropyron cristatum 2P translocation lines. PLoS ONE 11:e0145928. doi: 10.1371/journal.pone.0145928
Li, L. (1995). Cytogenetics and self-fertility of hybrids between Triticum aestivum L. and Agropyron cristatum (L.) Gaertn. Chin. Sci. Abstr. Ser. B. 4, 43.
Li, L., Stoeckert, C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, Y., Qian, Q., Zhou, Y., Yan, M., Sun, L., Zhang, M., et al. (2003). BRITTLE CULM1, which encodes a COBRA-like protein, affects the mechanical properties of rice plants. Plant Cell 15, 2020–2031. doi: 10.1105/tpc.011775
Liu, J., Zhang, C., Wei, C., Liu, X., Wang, M., Yu, F., et al. (2016). The RING finger ubiquitin E3 ligase OsHTAS enhances heat tolerance by promoting H2O2-induced stomatal closure in rice. Plant Physiol. 170, 429–443. doi: 10.1104/pp.15.00879
Lutfiyya, L. L., Xu, N., D'Ordine, R. L., Morrell, J. A., Miller, P. W., and Duff, S. M. (2007). Phylogenetic and expression analysis of sucrose phosphate synthase isozymes in plants. J. Plant Physiol. 164, 923–933. doi: 10.1016/j.jplph.2006.04.014
Meena, S., Kumar, S. R., Venkata Rao, D. K., Dwivedi, V., Shilpashree, H. B., Rastogi, S., et al. (2016). De novo sequencing and analysis of lemongrass transcriptome provide first insights into the essential oil biosynthesis of aromatic grasses. Front. Plant Sci. 7:1129. doi: 10.3389/fpls.2016.01129
Morita, R., Sato, Y., Masuda, Y., Nishimura, M., and Kusaba, M. (2009). Defect in non-yellow coloring 3, an alpha/beta hydrolase-fold family protein, causes a stay-green phenotype during leaf senescence in rice. Plant J. 59, 940–952. doi: 10.1111/j.1365-313X.2009.03919.x
Nigam, D., Saxena, S., Ramakrishna, G., Singh, A., Singh, N. K., and Gaikwad, K. (2017). De novo assembly and characterization of Cajanus scarabaeoides (L.) Thouars transcriptome by paired-end sequencing. Front. Mol. Biosci. 4:48. doi: 10.3389/fmolb.2017.00048
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). String tie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Prasad, K., Parameswaran, S., and Vijayraghavan, U. (2005). OsMADS1, a rice MADS-box factor, controls differentiation of specific cell types in the lemma and palea and is an early-acting regulator of inner floral organs. Plant J. 43, 915–928. doi: 10.1111/j.1365-313X.2005.02504.x
Ramirez-Gonzalez, R. H., Segovia, V., Bird, N., Fenwick, P., Holdgate, S., Berry, S., et al. (2015). RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 13, 613–624. doi: 10.1111/pbi.12281
Rey, E., Molnár, I., and Doležel, J. (2015). “Genomics of wild relatives and alien introgressions,” in Alien Introgression in Wheat: Cytogenetics, Molecular Biology, and Genomics, eds M. Molnár-Láng, C. Ceoloni, and J. Doležel. (Cham: Springer International Publishing), 347–381.
Rice Chromosomes 11 and 12 Sequencing Consortia (2005). The sequence of rice chromosomes 11 and 12, rich in disease resistance genes and recent gene duplications. BMC Biol. 3:20. doi: 10.1186/1741-7007-3-20
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Schultz, C. J., Hsu, M., Miesak, B., and Coruzzi, G. M. (1998). Arabidopsis mutants define an in vivo role for isoenzymes of aspartate aminotransferase in plant nitrogen assimilation. Genetics 149, 491–499.
Semagn, K., Babu, R., Hearne, S., and Olsen, M. (2014). Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): overview of the technology and its application in crop improvement. Mol. Breed. 33, 1–14. doi: 10.1007/s11032-013-9917-x
Sim, S.-C., Durstewitz, G., Plieske, J., Wieseke, R., Ganal, M. W., Van Deynze, A., et al. (2012). Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS ONE 7:e40563. doi: 10.1371/journal.pone.0040563
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tanwar, U. K., Pruthi, V., and Randhawa, G. S. (2017). RNA-Seq of guar (Cyamopsis tetragonoloba, L. Taub.) leaves: de novo transcriptome assembly, functional annotation and development of genomic resources. Front. Plant Sci. 8:91. doi: 10.3389/fpls.2017.00091
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinform. 4:41. doi: 10.1186/1471-2105-4-41
Tiwari, V. K., Wang, S., Danilova, T., Koo, D. H., Vrana, J., Kubalakova, M., et al. (2015). Exploring the tertiary gene pool of bread wheat: sequence assembly and analysis of chromosome 5Mg of Aegilops geniculata. Plant J. 84, 733–746. doi: 10.1111/tpj.13036
Tiwari, V. K., Wang, S., Sehgal, S., Vrána, J., Friebe, B., Kubaláková, M., et al. (2014). SNP discovery for mapping alien introgressions in wheat. BMC Genomics 15:273. doi: 10.1186/1471-2164-15-273
Trick, M., Adamski, N. M., Mugford, S. G., Jiang, C. C., Febrer, M., and Uauy, C. (2012). Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol. 12:14. doi: 10.1186/1471-2229-12-14
Vatanparast, M., Shetty, P., Chopra, R., Doyle, J. J., Sathyanarayana, N., and Egan, A. N. (2016). Transcriptome sequencing and marker development in winged bean (Psophocarpus tetragonolobus; Leguminosae). Sci. Rep. 6:29070. doi: 10.1038/srep29070
Wang, F., Shang, Y., Fan, B., Yu, J. Q., and Chen, Z. (2014). Arabidopsis LIP5, a positive regulator of multivesicular body biogenesis, is a critical target of pathogen-responsive MAPK cascade in plant basal defense. PLoS Pathog. 10:e1004243. doi: 10.1371/journal.ppat.1004243
Wang, Y., Gao, M., Li, Q., Wang, L., Wang, J., Jeon, J. S., et al. (2008). OsRAR1 and OsSGT1 physically interact and function in rice basal disease resistance. Mol. Plant Microbe Interact. 21, 294–303. doi: 10.1094/MPMI-21-3-0294
Winfield, M. O., Allen, A. M., Burridge, A. J., Barker, G. L., Benbow, H. R., Wilkinson, P. A., et al. (2016). High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206. doi: 10.1111/pbi.12485
Winfield, M. O., Wilkinson, P. A., Allen, A. M., Barker, G. L., Coghill, J. A., Burridge, A., et al. (2012). Targeted re-sequencing of the allohexaploid wheat exome. Plant Biotechnol. J. 10, 733–742. doi: 10.1111/j.1467-7652.2012.00713.x
Wu, M., Zhang, J. P., Wang, J. C., Yang, X. M., Gao, A. N., Zhang, X. K., et al. (2010). Cloning and characterization of repetitive sequences and development of SCAR markers specific for the P genome of Agropyron cristatum. Euphytica 172, 363–372. doi: 10.1007/s10681-009-0033-2
Wulff, B. B., and Moscou, M. J. (2014). Strategies for transferring resistance into wheat: from wide crosses to GM cassettes. Front. Plant Sci. 5:692. doi: 10.3389/fpls.2014.00692
Yoshida, A., Suzaki, T., Tanaka, W., and Hirano, H. Y. (2009). The homeotic gene long sterile lemma (G1) specifies sterile lemma identity in the rice spikelet. Proc. Natl. Acad. Sci. U.S.A. 106, 20103–20108. doi: 10.1073/pnas.0907896106
Zadoks, J. C., Chang, T. T., and Konzak, C. F. (1974). A decimal code for the growth stages of cereals. Weed Res. 14, 415–421. doi: 10.1111/j.1365-3180.1974.tb01084.x
Zhang, J., Liu, W. H., Han, H. M., Song, L. Q., Bai, L., Gao, Z. H., et al. (2015b). De novo transcriptome sequencing of Agropyron cristatum to identify available gene resources for the enhancement of wheat. Genomics 106, 129–136. doi: 10.1016/j.ygeno.2015.04.003
Zhang, J., Zhang, J. P., Liu, W. H., Han, H. M., Lu, Y. Q., Yang, X. M., et al. (2015a). Introgression of Agropyron cristatum 6P chromosome segment into common wheat for enhanced thousand-grain weight and spike length. Theor. Appl. Genet. 128, 1827–1837. doi: 10.1007/s00122-015-2550-9
Zhou, C. H., Zhang, L., Duan, J., Miki, B., and Wu, K. Q. (2005). HISTONE DEACETYLASE19 is involved in jasmonic acid and ethylene signaling of pathogen response in Arabidopsis. Plant Cell 17, 1196–1204. doi: 10.1105/tpc.104.028514
Keywords: wheat, wild relatives, phylogenetic relationship, interspecific variation, SNP, RNA-Seq
Citation: Zhou S, Yan B, Li F, Zhang J, Zhang J, Ma H, Liu W, Lu Y, Yang X, Li X, Liu X and Li L (2017) RNA-Seq Analysis Provides the First Insights into the Phylogenetic Relationship and Interspecific Variation between Agropyron cristatum and Wheat. Front. Plant Sci. 8:1644. doi: 10.3389/fpls.2017.01644
Received: 25 June 2017; Accepted: 07 September 2017;
Published: 21 September 2017.
Edited by:
Chengdao Li, Murdoch University, AustraliaReviewed by:
Filippo Maria Bassi, ICARDA, MoroccoGeorge Fedak, Agriculture and Agri-Food Canada, Canada
Copyright © 2017 Zhou, Yan, Li, Zhang, Zhang, Ma, Liu, Lu, Yang, Li, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xu Liu, bGl1eHUwMUBjYWFzLmNu
Lihui Li, bGlsaWh1aUBjYWFzLmNu