 Peter Hempel
Peter Hempel Annette Hohe
Annette Hohe Conny Tränkner
Conny Tränkner- 1Leibniz Institute of Vegetable and Ornamental Crops, Erfurt, Germany
- 2Faculty of Landscape Architecture, Horticulture and Forestry, University of Applied Sciences Erfurt, Erfurt, Germany
The ornamental crop species Hydrangea macrophylla exhibits diploid and triploid levels of ploidy and develops lacecap (wild type) or mophead inflorescences. In order to characterize a H. macrophylla germplasm collection, we determined the inflorescence type and the 2C DNA content of 120 plants representing 43 cultivars. We identified 78 putative diploid and 39 putative triploid plants by flow cytometry. In our collection 69 out of 98 flowering plants produced lacecap inflorescences, whereas 29 plants developed mophead inflorescences. Surprisingly, 12 cultivars included diploid as well as triploid plants, while 5 cultivars contained plants with different inflorescence types. We genotyped this germplasm collection using 12 SSR markers that detected 2–7 alleles per marker, and identified 51 different alleles in this collection. We detected 62 distinct fingerprints, revealing a higher genetic variation than the number of cultivars suggested. Only one genotype per cultivar is expected due to the vegetative propagation of Hydrangea cultivars; however we identified 25 cultivars containing 2–4 different genotypes. These different genotypes explained the variation in DNA content and inflorescence type. Diploid and triploid plants with the same cultivar name were exclusively mix-ups. We therefor assume, that 36% of the tested plants were mislabeled. Based on the “Wädenswil” pedigree, which includes 31 of the tested cultivars, we predicted cultivar-specific fingerprints and identified at least 21 out of 31 cultivars by SSR marker-based reconstruction of the “Wädenswil” pedigree. Furthermore, we detected 4 putative interploid crosses between diploid and triploid plants in this pedigree. These interploid crosses resulted in diploid or/and triploid offspring, suggesting that crosses with triploids were successfully applied in breeding of H. macrophylla.
Introduction
Hydrangea macrophylla (THUNB.) SER. is an economically important crop of the upmarket segment of ornamentals. H. macrophylla cultivars are famous for their rich foliage and impressively large, colorful inflorescences. Inflorescences can be divided into the lacecap (wild type) and the mophead (hortensia) type, according to the position and number of decorative flowers in the inflorescence. The lacecap inflorescence develops mainly non-decorative flowers, while several decorative flowers are present at the margin of the inflorescence. In contrast, the mophead inflorescence develops more decorative flowers, which are distributed over the complete inflorescence (Uemachi and Okumura, 2012). H. macrophylla originates from Japan and has been bred to create attractive ornamental pot plants and for landscaping in temperate regions worldwide. As for many ornamental crops, Hydrangea plants are commercially propagated through clones via cuttings. Mostly, the method of clone breeding is applied, where new varieties are selected in the F1 generation, followed by vegetative propagation. Thus, most cultivars possess a distinctive, predominantly heterozygous genotype. In addition, some varieties were obtained by mutant selection (Guérin, 2002). The fluctuation of established Hydrangea varieties is slow. For instance, varieties like ‘Mariesii’ (1879), ‘Madame Emile Mouillère’ (1909), ‘Libelle’ (1964) or ‘Blaumeise’ (1979) are still on the market.
Several famous lacecap cultivars such as ‘Libelle’ and ‘Blaumeise’ were derived from a systematic breeding program performed at the Experimental Station for Fruit Production, Viticulture and Horticulture in Wädenswil, Switzerland (nowadays Agroscope Changins-Wädenswil Research Station, ACW). This program had started in 1952 by crossing the mophead cultivar ‘Tödi’ with a lacecap wild type. It ended in 1987 after the release of 26 lacecap cultivars. In 1990, the complete pedigree of these lacecap cultivars was published (Meier, 1990). This pedigree, subsequently named “Wädenswil”, is a unique documentation of 36 years of systematic Hydrangea breeding. Noteworthy, all “Wädenswil” lacecap cultivars are based on one lacecap ancestor. The inflorescence type is a monogenic, dominant-recessive trait (Uemachi and Okumura, 2012). Thus, we propose that all “Wädenswil” lacecap cultivars carry at least one lacecap allele from the wild type ancestor.
The “Wädenswil” pedigree includes diploid and triploid cultivars (Zonneveld, 2004; Jones et al., 2007). Diploid H. macrophylla varieties contain 2n = 2x = 36 chromosomes, while triploid varieties possess 2n = 3x = 54 chromosomes. Thereby, the 2C DNA content of H. macrophylla ranges from 3.85 to 4.97 pg for diploid varieties and from 6.48 to 7.27 pg for triploid varieties (Cerbah et al., 2001; Zonneveld, 2004; Jones et al., 2007; Gürtler et al., 2013; Alexander, 2017). Triploid hydrangeas develop larger organs and floral structures than diploids (Alexander, 2017). Furthermore, they are regarded to be more robust against biotic and abiotic stresses and are desired in breeding for cultivar selection. However, little is known about breeding triploid hydrangeas. As observed in other species, triploids can be generated through somatic fusion, sexual hybridization between reduced and unreduced gametes of diploid parents or sexual hybridization between a diploid and a tetraploid parent (Wang et al., 2016). To our knowledge, natural tetraploids of H. macrophylla have not been identified until now, although tetraploid H. macrophylla plants can be generated by artificially induced autoploidization as shown for the diploid cultivars ‘Adria’ and ‘Libelle’ (Gürtler et al., 2013). Recently, the production of triploid hydrangeas via unreduced male gametes was described (Alexander, 2017). Crosses between the diploid cultivars ‘Princess Juliana’ and ‘Trophee’ as well as ‘Zaunkönig’ and ‘Princess Juliana’ resulted in triploid and diploid F1 plants, while the reciprocal cross and a cross with ‘Zaunkönig’ as male parent resulted in only diploid offspring. Alexander (2017) hypothesized that the triploids resulted from sexual autopolyploidy, which was most likely caused by genotype-specific production of unreduced male gametes.
It is unknown whether interploid crosses between diploid and triploid hydrangeas can be used to develop triploid varieties. As observed in other species, triploids tend to be sterile and seedless due to meiotic errors (Sattler et al., 2016; Wang et al., 2016). Due to this, triploids are usually excluded from systematic breeding programs. However, DNA content data of “Wädenswil” cultivars (Zonneveld, 2004; Jones et al., 2007) revealed, that triploid hydrangeas have been developed in breeding to a considerable extent. Apparently interploid crosses between diploid and triploid genotypes have been successfully performed, although probably unwittingly. In order to prove this assumption, we collected 1–6 plants of 43 H. macrophylla cultivars from different sources. We determined their DNA content and inflorescence type to characterize this germplasm collection. We found several cultivars that included diploid and triploid plants or plants that showed either the lacecap or mophead inflorescence type. In order to clarify these mixes, we performed an SSR marker analysis. Within this study, we present (i) a systematic DNA content screening of 120 plants that represent in total 43 cultivars, including nearly all “Wädenswil” cultivars, (ii) a genotype identification based on SSR marker fingerprints, and (iii) a cultivar-specific genotype identification based on the molecular reconstruction of the “Wädenswil” pedigree.
Materials and Methods
Plant Material
We studied 1–6 plants of 43 cultivars, resulting in 120 plants (Table 1). This collection included 1–5 plants of 26 “Wädenswil” lacecap cultivars and 1–6 plants of ‘Tödi’, ‘Bodensee’, ‘Enziandom’, ‘Glärnisch’, and ‘Hörnli’, which were used as crossing partners to develop “Wädenswil” cultivars (Meier, 1990). No plants were found for the crossing partners ‘Speer’, Nr. 1703, Nr. 1192, and Nr. 254. In addition, we included 11 cultivars with 1 to 6 plants, which were already available in Europe before the “Wädenswil” breeding program started (Möhring et al., 1956). Finally, we included ‘Bela’, which was assumed to be a spontaneous, somatic mophead mutant of the “Wädenswil” cultivar ‘Blaumeise’ (Guérin, 2002). Forty-seven plants were provided by the Botanical Garden Pirna-Zuschendorf of the Dresden University of Technology, a member of the German Gene Bank for Ornamental Plants. Seventy-three plants were provided by Kötterheinrich-Hortensienkulturen.
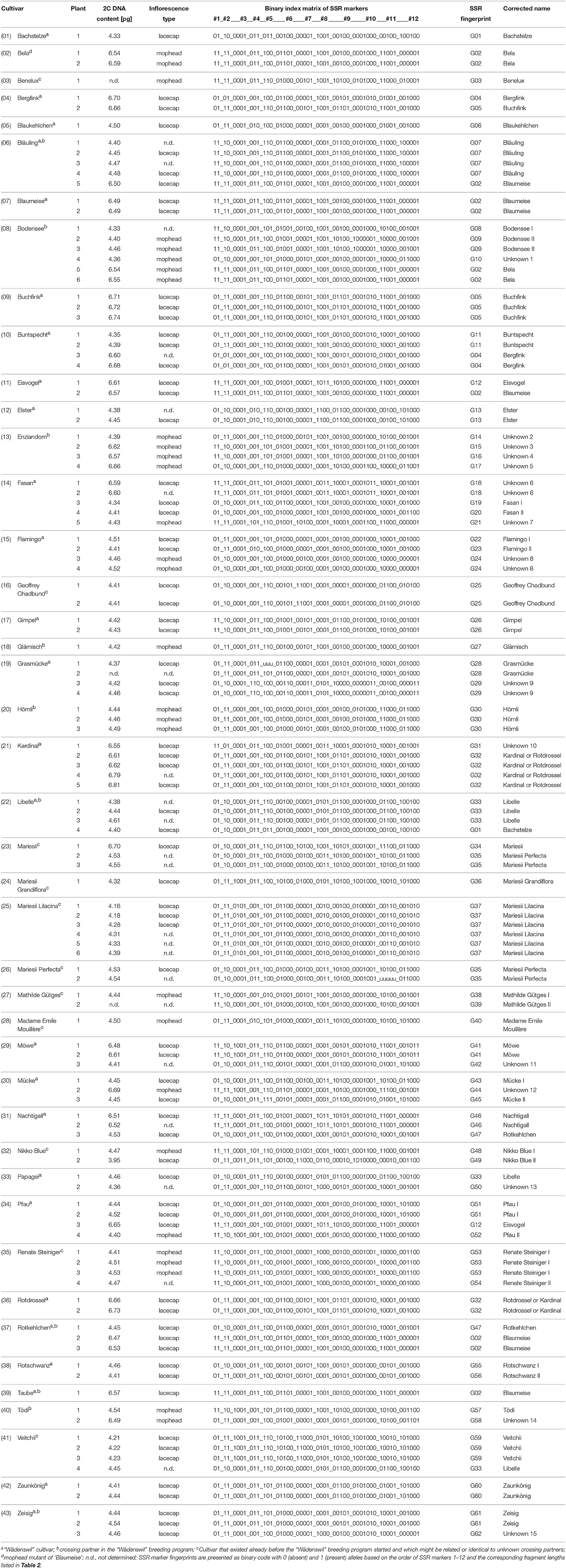
Table 1. DNA content, inflorescence type and SSR marker fingerprint of 120 H. macrophylla plants.
Plants were cultivated in 17 cm pots filled with Einheitserde® CL Hortensien blau on tables in a frost-free greenhouse of the Leibniz Institute of Vegetable and Ornamental Crops in Erfurt, Germany, without additional light supply. Plants were fertilized with Universol® blue 0.1% (Everris International BV) and irrigated as necessary. At the beginning of July all plants were pruned.
Phenotyping
The inflorescence type was recorded in 2016 and 2017, when the plants were in full bloom. Hydrangea plants show a distinct inflorescence type: If decorative flowers are present only at the periphery of the inflorescences, then the plant is recorded as lacecap. If all non-decorative flowers are covered by decorative flowers, then the plant is recorded as mophead.
Flow Cytometry
2C DNA content was determined by flow cytometry according to Dolezel et al. (2007) with slight modifications. We used Pisum sativum L. ‘Ctirad’ with a 2C DNA content of 9.09 pg as internal standard. About 0.3 and 0.9 cm2 of young leaves of sample and standard were chopped with a razor blade for 30–60 s in a plastic petri dish containing 1 ml Galbraith's buffer (45 mM MgCl2, 20 mM MOPS, 30 mM sodium citrate, 0.1% (v/v) Triton X-100, pH 7) freshly supplemented with 50 μg/ml propidium iodide, 50 μg/ml RNAse A and 1% (w/v) PVP 25. The homogenate was mixed by pipetting, passed through a 30 μm CellTrics filter (Partec) and analyzed using a Partec CyFlow Space analyzer with a 488 nm blue solid state laser at a flow rate of 0.1 μl/s. Data analysis was performed using the software FloMax version 2.70 (Quantum Analysis GmbH). For each sample-standard-mixture, about 10,000 nuclei were analyzed, which yielded about 5,000 nuclei per sample. High quality peaks were determined at CV < 4%. The 2C DNA content of each sample was calculated in relation to the 2C DNA content of the standard as follows: 2C DNA content sample = mean fluorescence value of sample * 9.09 pg / mean fluorescence value of P. sativum.
Chromosome Counting
Based on flow cytometric data, 3 diploid and 2 triploid plants were selected for chromosome counts. Root tips were incubated in 2 mM 8-hydroxyquinoline for 3 h at room temperature and fixed in 3:1 ethanol:acetic acid for at least 24 h. Subsequently, the root tips were washed 10 min in aqua dest. and macerated for 40 min at 37°C in an enzymatic solution containing 4% cellulose R10 (Duchefa), 1% pectolyase Y-23 (Duchefa), 75 mM KCl and 7.5 mM Na2EDTA at pH 4. Afterwards, the root tips were washed in aqua dest., transferred onto a slide, covered with 6 μl 45% acetic acid and carefully squeezed under a cover slip. Then, the cover slip was removed by freezing the slide in liquid nitrogen. After drying the slides, chromosomes were stained with DAPI using VECTASHIELD® HardSet™ Antifade Mounting Medium with DAPI (VECTOR Laboratories, Inc.) according to the manufacturer's protocol. Chromosomes were visualized using a fluorescence microscope with 360 nm excitation and 460 nm emission filter. Chromosome counts were made from 5 metaphase cells per examined cultivar.
DNA Extraction and Marker Analysis
Genomic DNA was extracted from young leaves. About 100 mg shock-frozen leaf samples were homogenized using Precellys®24 with the Cryolys® cooling module (Bertin Technologies S.A.S.). DNA was isolated using the DNeasy® Plant Mini Kit (QIAGEN) according to the manufacturer's protocol. DNA was eluted with aqua dest. DNA concentration was measured using the NanoDrop 2000c (Thermo Scientific).
We used 11 Simple Sequence Repeat (SSR) markers that were already used to estimate the genetic diversity for the genus Hydrangea (Rinehart et al., 2006; Reed and Rinehart, 2007). The characterization of these SSR loci based on 114 H. macrophylla taxa was published by Reed and Rinehart (2007). In addition, we developed one SSR marker based on an RNAseq contig sequence published by Chen et al. (2015). These SSR markers detected between 2 and 7 alleles per locus in our germplasm collection, on average 4.25 alleles per locus. The marker information and corresponding primer sequences are given in Table 2. All primers were obtained from Metabion International AG. Polymerase chain reaction (PCR) assays were done in a total volume of 12.5 μl containing 5 ng DNA, 1x PCR buffer including MgCl2 (Metabion International AG), 0.2 mM dNTPs (Metabion International AG), 0.2 μM unlabeled forward and reverse primers, additionally 0.004 μM primer labeled with IRD700 or 0.006 μM primer labeled with IRD800 (Metabion International AG), and 0.02 U mi-Taq DNA polymerase (Metabion International AG). The PCR conditions were 3 min at 94 °C, 35 cycles of 30 s at 94 °C, 30 s at 60 °C and 30 s at 72 °C, and finally 5 min at 72 °C. PCR fragment lengths were determined by polyacrylamide gel electrophoresis according to Borchert and Gawenda (2010). For this, we mixed 5 μl IRD 700 PCR product, 5 μl IRD 800 PCR product and 90 μl pararosaniline loading dye and separated PCR fragments together with a 50–700 bp sizing standard (LI-COR Biosciences) on a 6.5% KBPlus gel matrix (LI-COR Biosciences) using a LI-COR 4300 DNA analyzer (LI-COR Biosciences). Data analysis was performed with the software program SAGA 3.3 (LI-COR Biosciences). PCR fragment lengths were determined in relation to the 50–700 bp sizing standard (LI-COR Biosciences). For each genotype, SSR marker alleles were recorded in a binary code as 1 (present) and 0 (absent).
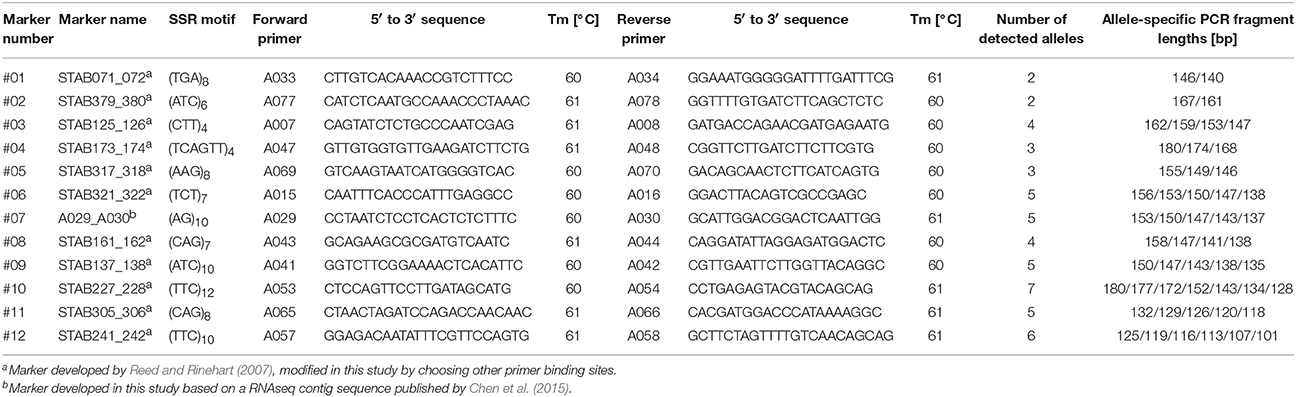
Table 2. SSR markers used in this study.
Results
Variability of Phenotype and Ploidy Level
We studied 120 plants that belonged to 43 cultivars according to their labels (Table 1). These cultivars included all 26 “Wädenswil” lacecap cultivars as well as ‘Tödi’, ‘Bodensee’, ‘Enziandom’, ‘Glärnisch’, and ‘Hörnli’, which were used as crossing partners to develop “Wädenswil” lacecap cultivars (Meier, 1990). Furthermore, this collection included 11 cultivars available prior to the “Wädenswil” breeding program and also ‘Bela’, an assumed spontaneous mophead mutant of ‘Blaumeise’ (Guérin, 2002). All cultivars were represented by 1–6 plants from different sources.
Firstly, we recorded the inflorescence type. In total, 69 plants developed lacecap inflorescences, while 29 plants produced mophead inflorescences (Table 1). The inflorescence type of the remaining 22 plants was not determined, because these plants did not flower. Interestingly, 5 cultivars (namely ‘Fasan’, ‘Mücke’, ‘Pfau’, ‘Flamingo’, ‘Nikko Blue’) contained plants that produced either lacecap or mophead inflorescences (Figure 1). These varying inflorescence types may be the result of mixed plants or spontaneous mutations at the inflorescence locus.

Figure 1. Examples of plants that varied in inflorescence type, 2C DNA content or SSR marker fingerprint.
Secondly, we determined the 2C DNA content of all plants by flow cytometry, in order to identify diploid and triploid plants. In total, we detected 78 plants, whose 2C DNA content ranged from 3.95 to 4.61 pg with an average of 4.42 ± 0.10 pg. For 39 plants, we measured 2C DNA contents from 6.47 to 6.81 pg, on average 6.61 ± 0.09 pg (Table 1). We counted 36 chromosomes for ‘Bläuling’, ‘Bodensee’, and ‘Libelle’, whose 2C DNA content ranged from 4.40 to 4.45 pg. In contrast, 54 chromosomes were counted for ‘Blaumeise’ and ‘Bela’ with 2C DNA contents of 6.50 and 6.59 pg, respectively. In addition, Jones et al. (2007) reported 36 chromosomes for ‘Veitchii’ and 54 chromosomes for ‘Nachtigall’ and ‘Taube’, whose 2C DNA contents was determined with 4.22, 6.51, and 6.57 pg, respectively. Thus, our Hydrangea collection included 78 putatively diploid and 39 putatively triploid plants. However, aneuploids cannot be excluded, because flow cytometry allows only an approximation of chromosome numbers. Surprisingly, 12 out of 43 cultivars included diploid as well as triploid plants.
SSR Fingerprinting Identified up to Four Different Genotypes Within One Cultivar
Cultivars of H. macrophylla are vegetatively propagated. Therefore, all clones of a cultivar have the same genotype and should display the same chromosome number and inflorescence type. In order to test whether these varying plants have identical genotypes, we performed an SSR marker analysis using 12 polymorphic SSR markers. These markers detected 2–7 different alleles per marker and gave in total 51 different alleles within the 120 plants (Table 1). Using these 12 SSR markers, we identified 18 cultivars whose plants showed a unique, cultivar-specific SSR marker fingerprint. However, 25 out of 43 cultivars comprised 2–4 different genotypes based on the SSR marker fingerprints. In total, we detected 62 distinct genotypes (Table 1), although only 43 genotypes (one genotype per cultivar) were expected.
Our 12 SSR markers were unable to differentiate between ‘Blaumeise’ and ‘Bela’ nor ‘Kardinal’ and ‘Rotdrossel’. This result was expected for ‘Blaumeise’ and ‘Bela’, because ‘Bela’ is a mophead mutant of ‘Blaumeise’, which we confirmed now at molecular level. It is unlikely that any of our used SSR markers is able to detect this specific mutation. Thus, ‘Blaumeise’ and ‘Bela’ have an identical SSR marker fingerprint, but ‘Blaumeise’ develops lacecap inflorescences whereas ‘Bela’ produces mophead inflorescences. In contrast, ‘Kardinal’ and ‘Rotdrossel’ should display different genotypes as they originated from different crosses. The screened plants of ‘Kardinal’ and ‘Rotdrossel’ may belong to the same genotype and the other cultivar is missing in our collection. Alternatively, the marker number could be too low to detect genotype-specific polymorphisms. The probability that ‘Kardinal’ and ‘Rotdrossel’ show the identical fingerprint is P = 2.291E-06 based on the frequency of the 12 different SSR marker fingerprints within the triploid genotype pool analyzed in this study. Furthermore, all plants of ‘Kardinal’ and ‘Rotdrossel’ have a very similar plant phenotype. To confirm the hypothesis that all plants of ‘Kardinal’ and ‘Rotdrossel’ belong to the same genotype, more plants and markers should be analyzed.
For 13 genotypes, we detected up to 3 alleles at various marker loci. Three alleles at one locus indicate triploidy, which we confirmed for 11 out of 13 genotypes by flow cytometry. However, the genotypes G25 and G45 showed 3 alleles at marker loci 7 and 5, respectively, but 2C DNA contents of 4.41 and 4.45 pg, which suggests diploidy. Since we also detected these alleles independently in other plants, they were not rejected as unspecific PCR fragments, but might indicate duplicated chromosomal regions.
Reconstruction of the “Wädenswil” Pedigree on Molecular Level
All plants of a cultivar that varied in their inflorescence type or DNA content displayed different genotypes. Moreover, different genotypes were also detected between plants showing the same inflorescence type and ploidy level, e.g., ‘Bergfink’ or ‘Eisvogel’ (Table 1), therefore several plants must be labeled incorrectly. In order to identify the correct plant of a cultivar, we reconstructed the “Wädenswil” pedigree at molecular level. Based on the published “Wädenswil” pedigree (Meier, 1990), which is shown with slight modifications in Figure 2, we started the molecular reconstruction from the initial cross W31. We followed the inheritance of marker alleles from known parental genotypes to descendants or by predicting putative parental genotypes from known descendant genotypes based on two criteria: If a marker locus of a descendent is heterozygous, both parents (diploid or triploid) must contain at least one of these alleles. If a marker locus of a descendent is homozygous, both parents must contain at least this certain allele. For example, ‘Libelle’ was derived from cross W31 and is parent in cross W45 and W74. We detected two genotypes for ‘Libelle’, showing the SSR marker fingerprints G01 and G33, respectively. Only the allele configuration of G33 fitted into cross W31 with alleles derived from ‘Tödi’ (fingerprint G57), into cross W45 as parent of ‘Bläuling’ (fingerprint G07) and into cross W74 as back-cross parent of ‘Elster’ and ‘Bachstelze’, while several marker alleles of G01 failed. G01 was also detected as SSR marker fingerprint of ‘Bachstelze’. Thus, we assume that ‘Libelle’ with fingerprint G01 was misnamed and is actually ‘Bachstelze’. In cross W45, none of the four different ‘Enziandom’ genotypes contributed to alleles of ‘Bläuling’. We assume that either the correct ‘Enziandom’ is missing in our collection or that ‘Enziandom’ was not the crossing parent in W45. Furthermore, we were able to predict some allele configurations of the parental genotype based on our criteria. This predicted genotype can be used to identify the correct crossing partner in W45. Using this method, all cross combinations were analyzed. The complete molecular reconstruction of the “Wädenswil” pedigree is shown in Figure 2. We were able to assign the genotypes of 18 out of 26 “Wädenswil” cultivars. For ‘Fasan’, ‘Rotschwanz’, ‘Flamingo’, and ‘Mücke’ we could reduce the number of genotypes to two possible candidates, which both fitted into the pedigree. The SSR marker fingerprints of ‘Kardinal’ and ‘Rotdrossel’ fitted to both pedigree positions. Here, more plants and markers must be analyzed to identify the cultivar-specific genotype and fingerprint. In contrast, none of the SSR marker fingerprints, which were detected for different plants of ‘Enziandom’, ‘Papagei’, and ‘Taube’, fitted to the corresponding predicted allele configuration in the “Wädenswil” pedigree. Thus, the right genotypes of these three cultivars are missing in our collection.
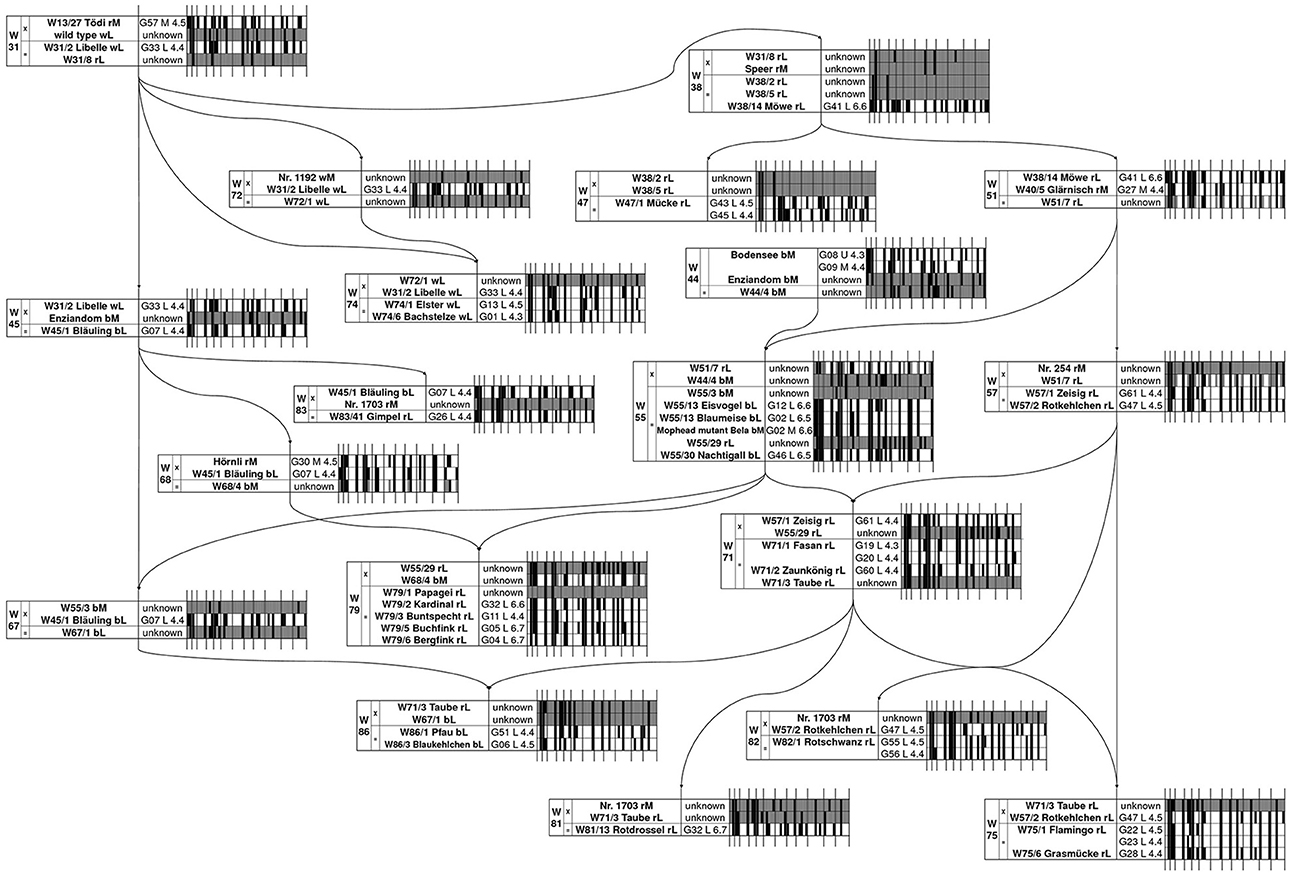
Figure 2. Reconstruction of the “Wädenswil” pedigree of lacecap cultivars of H. macrophylla according to Meier (1990) based on 12 SSR markers. The barcode tables show schematically the fingerprints of the SSR markers 1–12 as given in the binary index matrix in Table 1. The detected 51 SSR marker alleles are given as small bars as alleles • present, ◦ absent, or allele configuration unknown at this position. Wxx/x, Wädenswil cross number/plant number; Gxx_M/L/U_x.x, SSR fingerprint _ inflorescence type _ 2C DNA content in pg; M, mophead; L, lacecap; U, unknown inflorescence type; r, red/pink; b, blue; w, white flower color.
Differing Ploidy Levels Within a Cultivar Are due to Mix-up
We detected 12 out of 43 cultivars that included diploid and triploid plants. To prove, whether these diploids were derived from triploid precursors, we compared the allele configurations of triploid and diploid plants with the same cultivar name. If a chromosomal re-organization took place, all alleles of diploid plants would be present in the corresponding triploid precursor. However, none of the 12 cultivars that included diploid and triploid plants fulfilled this condition. Instead, we were able to assign several of these plants to other cultivars based on their SSR marker fingerprint, which then matched perfectly with the corresponding ploidy level (Table 1). Thus, all differing plants were mislabeled.
Interploid Crosses Within the “Wädenswil” Pedigree
In total, we detected at least 45 diploid and 15 triploid genotypes based on our marker analysis. 25 diploid and 7 triploid genotypes were attached to the “Wädenswil” pedigree (Figure 2). Our analysis identified cross W51 as an interploid cross between triploid ‘Möwe’ and diploid ‘Glärnisch’. Furthermore, the predicted genotypes of W44/4 and W55/29 contained up to 3 alleles at various marker loci, which suggests polyploidy. W44/4 and W55/29 were used as crossing partners in crosses W55, W71, and W79, which reveals 3 further interploid crosses in the “Wädenswil” pedigree. These crosses resulted in diploid or/and triploid offspring. Based on these findings, we assume that interploid crosses with triploids were successfully applied in Hydrangea breeding, resulting in diploid and triploid offspring.
In addition, crosses W38, W44, and W81 also gave triploid offspring. The mechanism that caused triploidy remains unclear, because most of the crossing partners and their DNA content are unknown.
Discussion
By genotyping a set of 120 H. macrophylla plants with 12 SSR markers, we identified 25 out of 43 cultivars that included 2–4 plants with different SSR marker fingerprints that varied also with regard to DNA content and inflorescence type. In total, we detected at least 62 distinct genotypes instead of 43 expected ones. Approximately 36% of these plants carried a wrong cultivar name. Based on the published “Wädenswil” pedigree of lacecap hydrangeas, we were able to predict cultivar-specific SSR marker fingerprints and retrieved 18 out of 26 “Wädenswil” cultivars. Thus, genotyping allowed reducing the total number of plants to the extent of almost 52%, while 19 extra genotypes were identified in our germplasm collection.
Our study confirmed that genotyping with SSR markers is a powerful tool to identify and differentiate between cultivar-specific genotypes in clonally propagated plant species. Plants with the same genotype can be reduced to decrease the number of plants in germplasm collections. In contrast, plants with differing genotypes can be separated in the case of mix-ups, and can be assigned to the correct cultivar. This would be beneficial especially for gene banks. Gene banks conserve and document genetic resources and provide material and data to maintain genetic- and biodiversity (www.croptrust.org). The ability to sort and correct stock using SRR markers would be incredibly beneficial for gene banks. Around the world, about 1,750 gene banks hold more than 7.4 million seeds or plant tissues from thousands of crop species (Gruber, 2017). In October 2016, Hydrangea collections became part of the national Gene Bank for Ornamental Plants in Germany (Spellerberg, 2017). However, plant conservation is expensive and storage capacities are limited. To organize gene banks efficiently, redundancies in plant collections and misnamed plants must be avoided.
Hydrangea is a vegetatively propagated crop and each cultivar possesses a specific genotype that can be represented by only one plant. However, a strong mix-up of plants was detected in our plant collection, which partially represents two independent Hydrangea collections. Several mix-ups were clearly detectable by phenotype, for instance differing inflorescence types, but most were not obvious due to phenotypic similarity resulting from the close relationship of some Hydrangea cultivars. For example, the siblings ‘Blaumeise’, ‘Eisvogel’, and ‘Nachtigall’ are half-siblings of ‘Rotkehlchen’, while ‘Elster’ and ‘Bachstelze’ are backcross progenies of ‘Libelle’ (Meier, 1990). If the cultivar-specific differences are too small and outstanding characteristics are missing, cultivar identification by phenotype alone is critical and mix-ups can occur especially for old cultivars out of commercial production. Redundancies and mix-ups were also observed in germplasm collections of other species (Hokanson et al., 1998; Lund et al., 2003). Thus, we strongly recommend to document plants not only by phenotype but also by genotype, in order to identify mixed, to eliminate wrong and to reduce redundant plants. Furthermore, we propose to do plant descriptions only after genotype identification and to add these phenotypic and genotypic data to a public accessible data base.
For genotyping, the marker system and number of markers have to be individually chosen for each species according to the genetic diversity within the germplasm collection. In our plant collection, 12 SSR markers, which detected in total 51 different alleles, distinguished 62 out of putative 64 genotypes. SSR markers are very suitable to detect duplicates or misnamed plants (Hokanson et al., 1998; Lund et al., 2003). They are convenient for the genotyping of germplasm collections due to their locus-specific, co-dominant inheritance, high reproducibility and utilization in high throughput. Moreover, they are relatively cheap and data handling is easy. However, only a low number of loci are considered and the information about genetic diversity remains low. In times of next generation sequencing, resequencing of whole genomes and genotyping-by-sequencing are increasingly widespread and affordable (Lin et al., 2014; Cheng et al., 2016; Varshney et al., 2017). Although highly interesting with regard to genome-wide association analysis and pre-breeding research, genotyping-by-sequencing might be over-dimensioned for simple genotype identification. The number of markers used in this study is relatively low. However, genotype differentiation depends not only on the number of markers but also on the number of detected alleles and the allele frequencies within the germplasm collection. For systematic genotyping of Hydrangea gene bank collections, the marker number has to be established and this depends on the genetic diversity of the plant collection. This study provides a foundation for future Hydrangea genotype analyses using SSR markers. Although SSR markers seem to be old-fashioned in times of next generation sequencing, they fulfill the requirements of easy and reliable genotype identification nicely.
Based on the reconstruction of the “Wädenswil” pedigree, we identified four putative interploid crosses, which produced diploid or/and triploid progenies even in top-select quality. Thereby, triploids were used as seed and pollen parents. Crosses with triploids are considered to be difficult due to meiotic errors. Triploids are often sterile as observed for citrus, banana and watermelon (Wang et al., 2016), or they produce aneuploid gametes as observed in maize. Crosses between triploid and diploid maize plants resulted in aneuploid offspring with differing chromosome numbers and insufficient phenotype compared to the diploid and triploid parental plants (McClintock, 1929). In contrast to maize, cross experiments of triploid and tetraploid rose plants indicated that triploid roses were able to produce haploid and diploid male and female gametes (Van Huylenbroeck et al., 2005). The 2C DNA contents of verified “Wädenswil” cultivars gave no significant indication for aneuploidy, although we have DNA content variations in flow cytometric measurements in the range of ± 1 chromosome. However, this variation might be caused by natural variations in DNA content (Cerbah et al., 2001) or the technical error of flow cytometric measurements. In addition, all studied hydrangeas showed a normal, very attractive phenotype, which might be unlikely for aneuploid plants. The plants with SSR marker fingerprint G25 and G45 showed 3 alleles using the markers 7 and 5, respectively, although the corresponding plants were assumed to be diploid based on the corresponding 2C DNA content. All alleles were independently detected also in other plants and therefore cannot be excluded as unspecific PCR fragments. Three alleles in a nearly diploid background might indicate aneuploidy concerning single chromosomes. But before speculating about the generation of aneuploid hydrangeas, the corresponding chromosome numbers of these plants have to be determined and the identity of the 3 putative alleles must be verified in subsequent studies.
The “Wädenswil” pedigree gives clear indications, that triploid hydrangeas can be used for systematic breeding. Nevertheless, the generation of triploid, aneuploid and maybe tetraploid hydrangeas and their suitability in breeding programs requires further analyses including systematic cross experiments with various diploid and triploid varieties as well as cytogenetic analyses of meiosis.
Author Contributions
PH designed and performed experiments and analyzed the data, AH conceived the study and rose funding, CT supervised the study and wrote the manuscript. All authors revised the data, read and approved the final manuscript.
Funding
This study was supported by funds of the Landwirtschaftliche Rentenbank grant no. 28RF40011, the Ministries of Consumer Protection, Food and Agriculture of the Federal Republic of Germany, the Ministry for Science, Research and Culture of the State of Brandenburg and the Thuringian Ministry of Infrastructure and Agriculture.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Katja Krüger, Janett Grimmer, and Jörg Krüger for technical assistance. We are grateful to Jörg Fuchs for the initial flow cytometry setup. We thank Frauke Engel, Matthias Riedel, Kristina Werner, and Anne Behrend for enlightening discussions and Helen Brabham for proofreading the manuscript. We are grateful to Kötterheinrich-Hortensienkulturen and the Botanical Garden Pirna-Zuschendorf for providing plant material.
References
Alexander, L. (2017). Production of triploid Hydrangea macrophylla via unreduced gamete breeding. HortScience 52, 221–224. doi: 10.21273/HORTSCI11358-16
Borchert, T., and Gawenda, I. (2010). Development and application of high-throughput amplified fragment length polymorphism technique in Calluna vulgaris (Ericaceae). Electr. J. Biotech. 13, 7–8. doi: 10.2225/vol13-issue2-fulltext-3
Cerbah, M., Mortreau, E., Brown, S., Siljak-Yakovlev, S., Bertrand, H., and Lambert, C. (2001). Genome size variation and species relationships in the genus Hydrangea. Theor. Appl. Genet. 103, 45–51. doi: 10.1007/s001220000529
Chen, H., Lu, C., Jiang, H., and Peng, J. (2015). Global transcriptome analysis reveals distinct aluminum-tolerance pathways in the Al-accumulating species Hydrangea macrophylla and marker identification. PLoS ONE 10:e0144927. doi: 10.1371/journal.pone.0144927
Cheng, F., Wu, J., Cai, C., Fu, L., Liang, J., Borm, T., et al. (2016). Genome resequencing and comparative variome analysis in a Brassica rapa and Brassica oleracea collection. Sci. Data 3:160119. doi: 10.1038/sdata.2016.119
Dolezel, J., Greilhuber, J., and Suda, J. (2007). Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2:2233. doi: 10.1038/nprot.2007.310
Gürtler, S., Klocke, E., and Schrader, O. (2013). Mutagenese und Polyploidisierung zur Schaffung neuer genetischer Variabilität bei der Hortensie (Hydrangea macrophylla). J. Cultiv. Plants 65, 273–284. doi: 10.5073/JfK.2013.07.03
Hokanson, S., Szewc-McFadden, A., Lamboy, W., and McFerson, J. (1998). Microsatellite (SSR) markers reveal genetic identities, genetic diversity and relationships in a Malus × domestica Borkh. core subset collection. Theor. Appl. Genet. 97, 671–683. doi: 10.1007/s001220050943
Jones, K. D., Reed, S. M., and Rinehart, T. A. (2007). Analysis of ploidy level and its effects on guard cell length, pollen diameter, and fertility in Hydrangea macrophylla. Hortscience 42, 483–488. Available online at: http://handle.nal.usda.gov/10113/17762
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220–1226. doi: 10.1038/ng.3117
Lund, B., Ortiz, R., Skovgaard, I., Waugh, R., and Andersen, S. (2003). Analysis of potential duplicates in barley gene bank collections using re-sampling of microsatellite data. Theor. Appl. Genet. 106, 1129–1138. doi: 10.1007/s00122-002-1130-y
Meier, F. (1990). Tellerhortensien-Züchtungen. Wädenswil: Eidgenössische Forschungsanstalt für Obst-, Wein- und Gartenbau.
Möhring, H. K., Kuhlen, H., and Bosse, G. (1956). Die Hortensien: ihre Geschichtliche Entwicklung, Systematik, Anatomie und Morphologie, Züchterische Bearbeitung, Sortenentwicklung und Kultur im Erwerbsgartenbau. Aachen: Verlag Deutsche Gärtnerbörse.
Reed, S. M., and Rinehart, T. A. (2007). Simple sequence repeat marker analysis of genetic relationships within Hydrangea macrophylla. J. Am. Soc. Hort. Sci. 132, 341–351. Available online at: http://handle.nal.usda.gov/10113/7485
Rinehart, T. A., Scheffler, B. E., and Reed, S. M. (2006). Genetic diversity estimates for the genus Hydrangea and development of a molecular key based on SSR. J. Amer. Soc. Hort. Sci. 131, 787–797. Available online at: http://handle.nal.usda.gov/10113/21586
Sattler, M. C., Carvalho, C. R., and Clarindo, W. R. (2016). The polyploidy and its key role in plant breeding. Planta 243, 281–296. doi: 10.1007/s00425-015-2450-x
Spellerberg, B. (2017). Aktuelle Entwicklungen bei der Deutschen Genbank Zierpflanzen. Julius-Kühn-Archiv 457, 15–17. doi: 10.5073/jka.2017.457.001
Uemachi, T., and Okumura, A. (2012). The inheritance of inflorescence types in Hydrangea macrophylla. J. Jap. Soc. Hort. Sci. 81, 263–268. doi: 10.2503/jjshs1.81.263
Van Huylenbroeck, J., Leus, L., and van Bockstaele, E. (2005). Interploidy crosses in roses: use of triploids. Acta Hort. 690, 109–112. doi: 10.17660/ActaHortic.2005.690.15
Varshney, R. K., Saxena, R. K., Upadhyaya, H. D., Khan, A. W., Yu, Y., Kim, C., et al. (2017). Whole-genome resequencing of 292 pigeonpea accessions identifies genomic regions associated with domestication and agronomic traits. Nat. Genet. 49, 1082–1088. doi: 10.1038/ng.3872
Wang, X., Cheng, Z.-M., Zhi, S., and Xu, F. (2016). Breeding Triploid Plants: a Review. Czech, J. Genet. Plant Breed. 52, 41–54. doi: 10.17221/151/2015-CJGPB
Keywords: genotype identification, gene bank, interploid crosses, microsatellite, SSR, fingerprint, ornamental crop
Citation: Hempel P, Hohe A and Tränkner C (2018) Molecular Reconstruction of an Old Pedigree of Diploid and Triploid Hydrangea macrophylla Genotypes. Front. Plant Sci. 9:429. doi: 10.3389/fpls.2018.00429
Received: 06 November 2017; Accepted: 20 March 2018;
Published: 18 April 2018.
Edited by:
Jianjun Chen, University of Florida, United StatesReviewed by:
Humira Sonah, Laval University, CanadaAna Pina, Centro de Investigación y Tecnología Agroalimentaria de Aragón (CITA), Spain
Yi Li, University of Connecticut, United States
Copyright © 2018 Hempel, Hohe and Tränkner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Conny Tränkner, dHJhZW5rbmVyQGVyZnVydC5pZ3pldi5kZQ==