 Sonia Sánchez-Campos1†
Sonia Sánchez-Campos1† Guillermo Domínguez-Huerta1,2†
Guillermo Domínguez-Huerta1,2† Luis Díaz-Martínez2
Luis Díaz-Martínez2 Diego M. Tomás1
Diego M. Tomás1 Jesús Navas-Castillo1
Jesús Navas-Castillo1 Enrique Moriones1
Enrique Moriones1 Ana Grande-Pérez2*
Ana Grande-Pérez2*- 1Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora,” Consejo Superior de Investigaciones Científicas-Universidad de Málaga, Estación Experimental “La Mayora,” Algarrobo-Costa, Málaga, Spain
- 2Instituto de Hortofruticultura Subtropical y Mediterránea “La Mayora,” Consejo Superior de Investigaciones Científicas-Universidad de Málaga, Área de Genética, Facultad de Ciencias, Campus de Teatinos, Málaga, Spain
Geminiviruses (family Geminiviridae) possess single-stranded circular DNA genomes that are replicated by cellular polymerases in plant host cell nuclei. In their hosts, geminivirus populations behave as ensembles of mutant and recombinant genomes, known as viral quasispecies. This favors the emergence of new geminiviruses with altered host range, facilitating new or more severe diseases or overcoming resistance traits. In warm and temperate areas several whitefly-transmitted geminiviruses of the genus Begomovirus cause the tomato yellow leaf curl disease (TYLCD) with significant economic consequences. TYLCD is frequently controlled in commercial tomatoes by using the dominant Ty-1 resistance gene. Over a 45 day period we have studied the diversification of three begomoviruses causing TYLCD: tomato yellow leaf curl virus (TYLCV), tomato yellow leaf curl Sardinia virus (TYLCSV) and tomato yellow leaf curl Malaga virus (TYLCMaV, a natural recombinant between TYLCV and TYLCSV). Viral quasispecies resulting from inoculation of geminivirus infectious clones were examined in plants of susceptible tomato (ty-1/ty-1), heterozygous resistant tomato (Ty-1/ty-1), common bean, and the wild reservoir Solanum nigrum. Differences in virus fitness across hosts were observed while viral consensus sequences remained invariant. However, the complexity and heterogeneity of the quasispecies were high, especially in common bean and the wild host. Interestingly, the presence or absence of the Ty-1 allele in tomato did not lead to differences in begomovirus mutant spectra. However, the fitness decrease of TYLCSV and TYLCV in tomato at 45 dpi might be related to an increase in CP (Coat protein) mutation frequency. In Solanum nigrum the recombinant TYLCMaV, which showed lower fitness than TYLCSV, at 45 dpi actively explored Rep (Replication associated protein) ORF but not the overlapping C4. Our results underline the importance of begomovirus mutant spectra during infections. This is especially relevant in the wild reservoir of the viruses, which has the potential to maintain highly diverse mutant spectra without modifying their consensus sequences.
Introduction
Animal and plant RNA viruses exist as viral quasispecies that adapt to changing environments by evolving rapidly (Domingo et al., 2006, 2012; Roossinck and Schneider, 2006). Viral quasispecies are defined as distributions of nonidentical but related genomes subjected to a continuous process of genetic variation, competition, selection and genetic drift, and which act as a unit of selection (Domingo et al., 2012). Their high genetic diversity in host populations is partially explained by the low replication fidelity of viral RNA-dependent DNA (RdDp) or RNA polymerases (RdRp), which lack proofreading activity (Steinhauer et al., 1992; Domingo and Holland, 1997). Eukaryotic single-stranded DNA (ssDNA) viruses do not encode DNA polymerases (Gutierrez, 2000; Hanley-Bowdoin et al., 2000) and replicate their genomes using unknown host cell polymerases. High mutation frequencies of approximately 10−4 mutations per nucleotide as well as substitution rates similar or even higher than those observed for RNA viruses have been found in ssDNA viruses such as animal circoviruses (family Circoviridae) (Firth et al., 2009; Harkins et al., 2014; Sarker et al., 2014) and parvoviruses (family Parvoviridae) (López-Bueno et al., 2006), or plant geminiviruses (family Geminiviridae) (Isnard et al., 1998; Sanz et al., 1999; Ge et al., 2007; Duffy and Holmes, 2008, 2009; Urbino et al., 2008; van der Walt et al., 2008; Harkins et al., 2009) and nanoviruses (family Nanoviridae) (Grigoras et al., 2010; Stainton et al., 2015)
Observed genetic diversity within mutant spectra at a given time during viral evolution is the result of natural selection, genetic drift and migration over the continuous generation of viral variants. The heterogeneous composition of the mutant ensemble enables viral emergence and pathogenesis (Coffin, 1996; Domingo et al., 2006, 2012), thus complicating prevention and treatment of viral diseases (Perales et al., 2012). Antiviral or immune-escape mutants present in small proportions of viral populations, for example in human immunodeficiency virus 1 (HIV-1) (Coffin, 1995; Nájera et al., 1995; Henn et al., 2012), hepatitis B virus (HBV) or hepatitis C virus (HCV) (Domingo et al., 2012; Bittar et al., 2013) infections, may be selected and become dominant in quasispecies following antiviral treatment, thus making disease control more difficult (Perales et al., 2014). A widely employed strategy to control plant viruses is the use of host genetic resistance (Boiteux et al., 1993; Monci et al., 2005; Fuchs, 2008; García-Cano et al., 2010). However, given the dynamic nature of viral populations, resistant mutants can emerge and result in epidemics.
Viral emergence requires a sufficient degree of genetic variation in the reservoir host to allow successful infection of the new host after transmission (Parrish et al., 2008; Holmes and Grenfell, 2009; Elena et al., 2011, 2014). Once the virus adapts to the new host, the emergent virus may spread and reach epidemic levels. Thus, knowledge of the genetic structure of host viral populations, and the factors that determine their evolution and host adaptation, are essential for designing robust control strategies for plant viral diseases (Acosta-Leal et al., 2011). The genetic diversity observed in several RNA plant virus quasispecies appears to be host dependent, indicating that virus-host interactions are responsible for the maintenance of variability in mutant spectra (Schneider and Roossinck, 2000, 2001). This diversity might influence viral adaptability to new hosts. It remains to be determined, however, to what extent hosts can play a similar role in shaping quasispecies genetic variability in ssDNA plant viruses.
Begomoviruses (genus Begomovirus, family Geminiviridae) are emerging pathogens transmitted by the whitefly Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae). They are responsible for severe losses of economically important crops such as tomato (Solanum lycopersicum), cotton (Gossypium spp.) or cassava (Manihot esculenta) in tropical, subtropical and temperate regions (Parrish et al., 2008; Rojas and Gilbertson, 2008; Hanssen et al., 2010; Navas-Castillo et al., 2011). The tomato yellow leaf curl disease (TYLCD) is one of the most devastating diseases affecting tomato worldwide (Czosnek and Laterrot, 1997; Díaz-Pendón et al., 2010; Navas-Castillo et al., 2011). TYLCD is caused by a complex of at least 11 different begomoviruses, which produce similar symptoms in infected tomato plants but can differ in host range (Czosnek and Laterrot, 1997; Díaz-Pendón et al., 2010; Navas-Castillo et al., 2011). Most TYLCD-associated viruses have a monopartite genome with coding regions present in both virion (V) and complementary (C) sense strands separated by an intergenic non-coding region (IR). A total of six partially overlapping genes are encoded by monopartite TYLCD-associated virus genomes: CP (coat protein, open reading frame or ORF V1), V2 (movement-like protein, precoat protein), Rep (replication associated protein, C1), TrAP (transcriptional activator protein, C2), REn (replication enhancer protein, C3), and C4 (involved in systemic movement and a pathogenesis factor) (Czosnek, 2007). TYLCD epidemics have occurred in the Mediterranean Basin since the late 1980s (Czosnek, 2007; Navas-Castillo et al., 2011). In Spain, outbreaks of the disease in the early 1990s were associated with the Spanish (ES) strain of tomato yellow leaf curl Sardinia virus (TYLCSV) (Noris et al., 1994; Navas-Castillo et al., 2011). The subsequent introduction in the late 1990s and early 2000s of the type (also known as Israel, IL) (currently accepted as type member) and Mild (Mld) strains of tomato yellow leaf curl virus (TYLCV) (Noris et al., 1994; Morilla et al., 2004) resulted in new sources of genetic variation, and the emergence of recombinant species such as tomato yellow leaf curl Malaga virus (TYLCMaV), derived from a genetic exchange between isolates of TYLCSV and TYLCV (Navas-Castillo et al., 1997). Although 99% of the TYLCMaV genome sequence is shared with its corresponding parental virus sequences, the virus significantly differs in host range (Morilla et al., 2004; García-Andrés et al., 2007). Thus, TYLCMaV can infect common bean (Phaseolus vulgaris) and the wild host Solanum nigrum. In contrast, TYLCSV cannot infect common bean (Monci et al., 2002) and the accumulation of TYLCV is strongly impaired in S. nigrum (Sánchez-Campos et al., 1999, 2000). None of the latter three viruses induce disease in tomato cultivars carrying the dominant Ty-1 resistance allele, which has been shown to limit the accumulation of TYLCV, TYLCSV and, to a lesser extent, TYLCMaV (García-Andrés et al., 2006, 2009).
Knowing how hosts affect begomovirus diversity is important to understand the emergence of new begomovirus variants and to assist in designing more durable control strategies. To this end we have monitored the genetic variability of TYLCV, TYLCSV, and TYLCMaV begomovirus mutant spectra for 45 days after the inoculation of susceptible and Ty-1 resistant tomato, common bean and the wild reservoir S. nigrum with single sequence variants of each virus. Our results show that the host can have an important role in driving the fitness and diversity of begomovirus mutant spectra. Differences in absolute fitness of begomovirus in the different hosts existed without any modifications in their consensus sequences. However, quasispecies with high complexity and heterogeneity existed in all four hosts, especially in common bean and the wild host. Interestingly, the presence or absence of the Ty-1 resistance allele in tomato did not lead to differences in begomovirus mutant spectra. However, in tomato CP mutation frequency of TYLCSV and TYLCV increased at 45 dpi, which may be related to their fitness decrease in this host. In the wild host TYLCMaV displayed lower fitness than in the other hosts and actively explored Rep ORF but not the overlapping C4. Our findings underline the great complexity and heterogeneity of begomovirus mutant spectra and that differences might exist between host plants. This is particularly relevant for the wild reservoir, which hosts a highly diverse begomovirus population containing potentially emergent variants in spite of the stability of their quasispecies consensus sequences.
Materials and Methods
Plants, Virus Source, and Viral Inoculation
Homozygous TYLCD susceptible (ty-1/ty-1) and heterozygous TYLCD resistant (Ty-1/ty-1) tomato quasi-isogenic lines (Monci et al., 2002) (kindly provided by M. J. Díez, Universidad Politécnica de Valencia, Spain), S. nigrum (IHSM “La Mayora” germplasm bank) and common bean cv Donna (Nunhems, Haelen, The Netherlands) were used as experimental hosts. Plants at the three-leaf growth stage were inoculated with Agrobacterium tumefaciens LBA4404 containing infectious clones, and therefore single sequence variants, of isolates [ES:72:97] of the Mld strain of TYLCV (GenBank accession number AF071228) (García-Andrés et al., 2009), [ES:Mur1:92] of the ES strain of TYLCSV (Z25751) (Navas-Castillo et al., 1999), and [ES:421:99] of TYLCMaV (AF271234) (Noris et al., 1994). These isolates were derived from infected tomato plants except for TYLCMaV, which was derived from an infected common bean plant. TYLCMaV is the result of a genetic exchange between TYLCV-Mld and TYLCSV-ES, with recombinant genome sequences that share 99% nucleotide identity with that of the parental virus sequences. As depicted in Figure 1 in TYLCMaV, the 3′ half of the IR and the virus sense V2 and the CP ORFs derive from TYLCSV, whereas the remainder 5′ half of the IR and the Rep, C4, TrAP and most of REn ORFs derive from TYLCV, with the 3′ terminal 40 nucleotides of the REn ORF derived from TYLCSV (Monci et al., 2002). Viral infectious clones of TYLCSV, TYLCV or TYLCMaV were used to inoculate three plants each of a TYLCD-susceptible tomato, a quasi-isogenic resistant tomato containing the Ty-1 allele in heterozygosis, common bean and S. nigrum (Figure 1). Liquid cultures of A. tumefaciens were adjusted to an OD600 of 1.00 and 20 μl were inoculated to each plant by the stem-puncture method as described elsewhere (Monci et al., 2002). After agroinoculation, plants were maintained for 45 days in a growth chamber (26°C during the day and 18°C at night, 70% relative humidity, with a 16 h photoperiod at 250 μmol s−1m−2 photosynthetically active radiation). Systemic virus infection of agroinoculated plants was confirmed on young non-inoculated leaves by tissue-print hybridization using a mixture of digoxigenin (DIG)-labeled probes specific to TYLCV and TYLCSV (Monci et al., 2002) that also recognizes TYLCMaV.
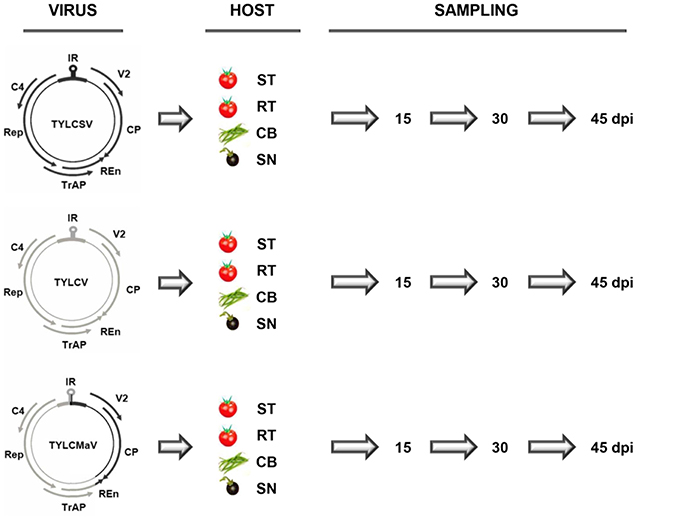
Figure 1. Time-course infections of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) infectious clones in susceptible tomato, resistant tomato, common bean and the wild reservoir Solanum nigrum. Genomic organization of the viruses showing the two open reading frames (ORFs), V2, and CP, on the sense strand, and four ORFs, Rep, TrAP, REn, and C4, on the complementary sense strand. They are separated by an intergenic non-coding region (IR) that contains the origin of replication in a region that adopts a stem-loop structure. Infectious clones and therefore single sequence variants of TYLCSV (black), TYLCV (gray) and recombinant TYLCMaV were used to inoculate three plants each of test hosts: susceptible (ST) and resistant (RT, carrying the Ty-1 resistance allele in heterozygosis) quasi-isogenic tomato plants, common bean (CB) and the wild reservoir S. nigrum (SN). Young apical non-inoculated leaves were collected at the indicated days post inoculation (dpi).
DNA Extraction, Virus Amplification, Cloning, and Sequencing
To monitor quasispecies in each infected plant, young apical leaves were harvested at 15, 30, and 45 dpi (Figure 1) and lyophilized. DNA was extracted following the Edwards' procedure (Edwards et al., 1991) with some modifications. From each sample, 8 mg of dried tissue were ground in a 1.5 ml Eppendorf tube with liquid nitrogen, and 400 μl of extraction buffer [200 mM Tris-HCl, 200 mM NaCl, 25 mM EDTA, SDS 0.5% (w/v), pH 7.5, 0.1% β-mercaptoethanol] added. After vortexing at 37°C for 2 min extracts were centrifuged at 12,000 × g at room temperature for 5 min. The supernatants were transferred to a new tube and mixed with 1 volume of phenol:chloroform (1:1). The aqueous phase was transferred to a new tube and mixed with 1 volume of isopropanol. After 2 min at room temperature, nucleic acids were recovered by centrifugation at 12,000 × g at room temperature for 5 min. The pellet was washed with 70% ethanol, dried and resuspended in 200 μl of RTE buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, 0.1% RNase A) by incubation at 65°C for 10 min. Viral DNA from total DNA extracts was amplified by rolling-circle amplification (RCA) with ϕ 29 DNA polymerase according to Inoue-Nagata et al. (Inoue-Nagata et al., 2004) using the TempliPhi DNA Amplification Kit (GE Healthcare). For all samples, 400 to 500 ng/μl of the amplification product measured by UV-spectrophotometry and gel electrophoresis were obtained. Full-lenght genomic consensus sequences were obtained by direct sequencing (Macrogen Inc., South Korea) of RCA products amplified from total DNA extractions. Primers used to sequence consensus sequences of TYLCSV, TYLCV, and TYLCMaV are indicated in Table 1. Sanger sequencing was preferred over NGS due to the difficulty of the latter to discriminate between the error of the technology and the error rate of the viruses, which is paramount for quasispecies analysis.
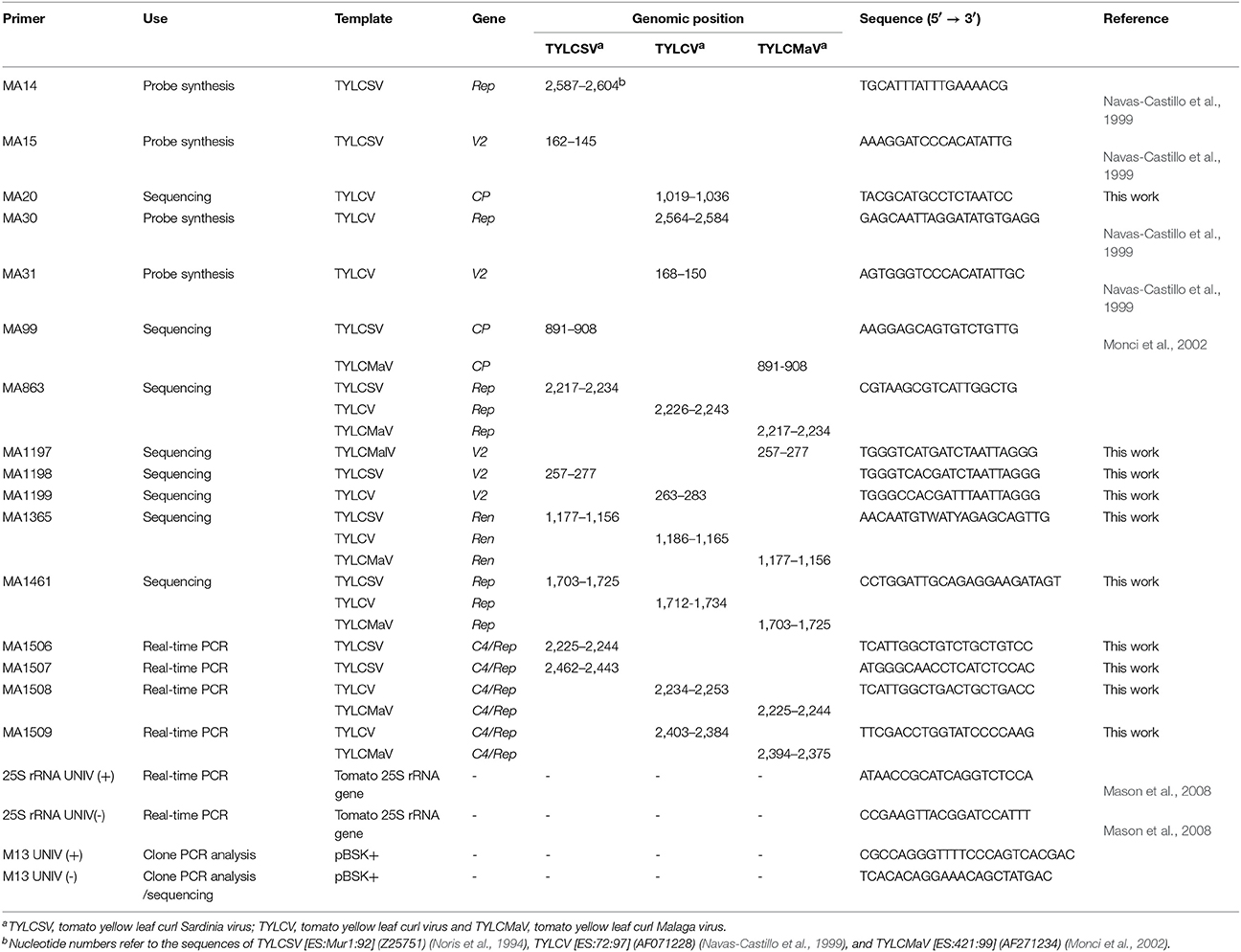
Table 1. Primers used in this study for probe synthesis, sequencing, qPCR and clone analysis.
RCA products were digested with single cutter (in viral genome) restriction enzyme BamHI to obtain the linear full-length DNA (c. 2.7 kbp) that was subsequently cloned into pBluescript II SK (+) (Stratagene). The product obtained was transformed into Escherichia coli DH5α by electroporation. Transformed colonies were analyzed by PCR using the universal M13 primers (Table 1). Only those clones containing inserts representing the 2.7 kbp full-length genome size were selected for the study. Amplification of molecular clones was also performed using RCA before sequencing. A total of 18–22 clones were sequenced per mutant spectrum from a single plant. Primers MA863 and M13 UNIV (-) were used for all virus isolates (Table 1) to sequence the 5′-end of Rep and C4 ORFs, and the intergenic region (IR) on the C strand. On the virus sense strand the sequenced region comprises the 5′-end of the V2 ORF and the complete CP ORF. Primers MA1197, MA1198, and MA1199 were used for sequencing molecular clones of TYLCMaV, TYLCSV, and, TYLCV, respectively, whereas primer MA1365 was used for all of them.
Sequence Analysis and Mutant Spectra Characterization
EditSeq, SeqBuilder and SeqMan software (DNAStar Inc., USA) were used for sequence assembly and analysis. Mutations were scored relative to the consensus sequence of each mutant spectrum. To analyze virus quasispecies genetic complexity (mutational composition of the ensemble) mutation frequencies were calculated by dividing the number of different mutations (repeated mutations in each quasispecies were computed only once) by the total number of nucleotides sequenced (Domingo et al., 2006). Virus quasispecies genetic complexity was also assessed by analyzing the average genetic distance (d), defined as to the average number of mutations per site between any pair of sequences chosen at random from the population. Genetic distances were estimated for each genomic region by Kimura's two-parameter method (Kimura, 1980) in MEGA version 4 (Tamura et al., 2007) in the different host-begomovirus combinations at 15, 30, and 45 dpi. Standard error was calculated by the bootstrap method with 1,000 repeats (Nei and Kumar, 2000).
Mutant spectra heterogeneity was determined using the normalized Shannon entropy (SE) calculation according to the formula − [Σi (pi x lnpi)]/lnN], where pi is the frequency of each sequence in the mutant spectrum and N is the total number of sequences compared (Volkenstein, 1994). SE values range from 0 (all sequences are identical) to 1 (all sequences are different).
To analyze the effect of selection on mutant spectra the rate of substitutions per synonymous (dS) and nonsynonymous (dNS) site was estimated using the Pamilo-Bianchi-Li method (Pamilo and Bianchi, 1993) implemented in MEGA. The structure-genetic (SG) matrix of Feng et al. (1984) was used to obtain the acceptability values of amino acid changes found in each genomic coding region. The values range from 0 (drastic amino acid changes) to 6 (synonymous replacements) and take into account structural similarities and probabilities of amino acid changes. The probability of occurrence of amino acid replacements found in quasispecies was calculated according to the PAM-250 substitution matrix (Feng and Doolittle, 1996).
Mutations in overlapping genes were only considered for the computation of mutation types or the estimation of d, mutation frequency or Shannon entropy of each ORF but not of the entire sequenced zone. Also, since the Rep and C4 ORFs are not in frame, neither V2 and CP, the resulting amino acid changes in the overlapping sequence were considered for the dNS/dS calculation.
The statistical significance of differences in ORF mutation frequencies between samples was evaluated by a generalized linear model with logit link function assuming a binary distribution, and followed by post hoc analysis of pairwise comparisons with least square (LSD) correction to test for differences between treatments, using IBM SPSS Statistics v. 22 software (P < 0.05 was considered statistically significant). To check for a random distribution of mutations in the overlapping Rep and C4 a Wald–Wolfowitz runs test was conducted.
Viral Fitness Determination
To determine absolute fitness the accumulation ability of a virus genotype in a given host, that is, the number of viral genomes produced after a given time (per unit of total DNA extracted from the plant tissue) in apical leaves was measured. Absolute fitness was approximated as the Malthusian growth rate per day, m, calculated according to the formula logQ, where Q is the number of pg of begomovirus ssDNA per 100 ng of total plant DNA (Lalić et al., 2011), which is estimated from the qPCR determination of the accumulated viral load in the apical part of each plant being monitored during the 15 days that have elapsed between each collection time (15, 30, and 45 dpi).
Quantification of viral DNA in test plants was carried out on total DNA extracts previously analyzed by agarose gel electrophoresis and UV-spectrophotometry (ND-1,000 spectrophotometer, NanoDrop, Fisher Thermo). Quantification was done in triplicate with a final volume of 20 μl with SYBR Premix Ex Taq Perfect Real Time (Takara), 0.125 μM of specific primers and 10 ng of total DNA. An amplicon of 169 bp within the Rep ORF of TYLCV and TYLCMaV was amplified with primers MA1508 and MA1509 (Table 1). In the case of TYLCSV, a fragment of 240 bp also in Rep was amplified with primers MA1506 and MA1507. PCR amplification was performed in an iCycler PCR System (BioRad) and consisted of an initial denaturation step at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 34 s. Specificity of the amplified products was assessed by melting curve analysis. Data from total DNA plant extracts were normalized to the 25S ribosomal RNA gene amplified using primers 25S rRNA UNIV (+ and –) (Mason et al., 2008; Table 1). To obtain the total number of ssDNA molecules, values were multiplied by two as each standard dsDNA molecule (a phagemid harbors the infectious clone) contains two ssDNAs (virion sense and complementary sense).
For the absolute quantification of TYLCV-like viruses, standard curves were prepared using pBluescript II SK (+) containing full-length viral genomes. Phagemids were serially diluted tenfold in 5 ng/μl of DNA extracted from either non-inoculated tomato, common bean or S. nigrum plants, to obtain 102 to 109 viral genome copies per μl. For each PCR system (primer pair and background host DNA), standard curves were obtained by linear regression analysis of the threshold cycle (Cq) values of three standard dilution replicates over the Log of the number of genome copies present in each sample.
A generalized linear model was fitted to the Malthusian growth rate and the log value of viral load data. Post hoc analysis of pairwise comparisons via Bonferroni correction were used to test for differences between hosts at 45 dpi (P < 5.56 × 10−3) or along the time course of viral infections (P < 0.010 or 6.25 × 10−3).
Results
Host-Virus Interactions Determine Differences in Viral Fitness
To evaluate how the host affects the behavior of the different begomoviruses first we measured the viral load of the three begomoviruses in the four hosts. Under the conditions studied, no effective amplification was possible from young apical non-inoculated leaves for TYLCSV in common bean or for TYLCV in S. nigrum. As shown in Figure 2, it was clear that the presence of the Ty-1 resistance allele repressed accumulation of TYLCSV, and TYLCV to some extent, whereas no such repression was observed for TYLCMaV. Significantly higher TYLCV accumulation was observed in common bean at 45 dpi than in susceptible (510-fold; P = 0.005) or resistant (1 × 103-fold; P = 0.000) tomato plants. Also, significantly more TYLCSV molecules accumulated in the wild reservoir S. nigrum at 45 dpi than in susceptible (60-fold, P = 0.005) or resistant (3.6 × 106-fold, P = 0.0004) tomatoes (Figure 2). Thus, TYLCV and TYLCSV infections in common bean and S. nigrum, respectively, resulted in the generation of larger virus populations suggesting higher viral fitness compared to tomato. Our results also suggest better adaptation of TYLCMaV to common bean (with an accumulation level similar to TYLCV) than to S. nigrum (much lower accumulation than TYLCSV).
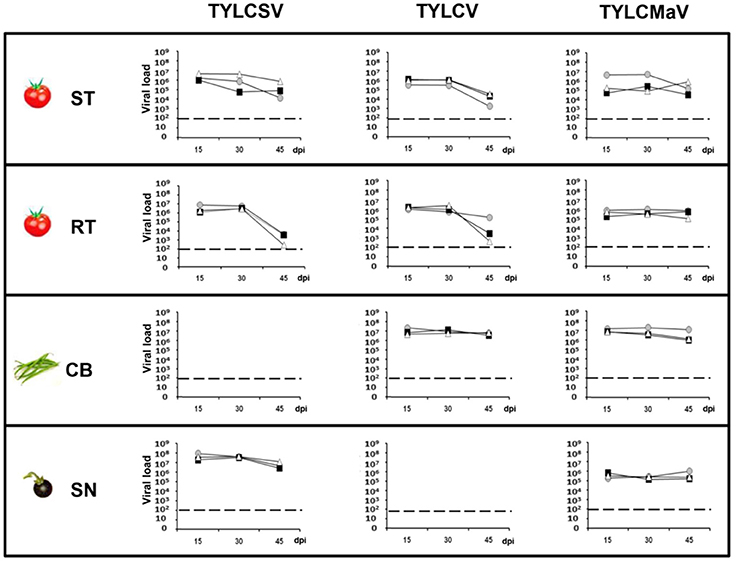
Figure 2. Absolute quantification of begomovirus ssDNA molecules accumulated during time-course analyses of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) infections in susceptible tomato (ST), resistant tomato (RT), common bean (CB) and the wild reservoir Solanum nigrum (SN). Begomovirus DNA molecules present in the apical leaves of plants infected with infectious begomovirus clones were quantified by qPCR. Where possible, begomovirus ssDNA levels were monitored in three individual systemically infected plants (as judged by hybridization analysis) for each virus/host combination. Apical leaf samples were collected at 15, 30, and 45 days post inoculation (dpi). Graphs show viral loads (number of ssDNA genomes per ng of total DNA) in apical leaf samples from individual test plants, indicating average values from three technical qPCR replicates for each DNA extract. Viral DNA values were normalized to the 25S rRNA gene. No viral accumulation was detected at any time point for TYLCSV/common bean or TYLCV/S. nigrum infections either by hybridization or qPCR. Horizontal dotted lines indicate the threshold for qPCR detection. Standard error ranged from 0.01 to 0.001% of the quantified molecules.
To better understand the effect of the different hosts on the three begomoviruses absolute fitness was assessed. Since we used single-sequence variants as inoculum the Malthusian growth rate (m) during viral infection time-courses was calculated as an estimate for absolute fitness (Lalić et al., 2011). The results shown in Figure 3 indicate differences in fitness across hosts during time-course infections. The presence of the Ty-1 allele significantly restricted the growth rate of TYLCSV (P = 0.004) in apical leaves at 45 dpi, and of both TYLCSV and TYLCV during the time-course infection (P = 0.000). However, as previously shown (Monci et al., 2002), the presence of the Ty-1 resistance allele in tomato had no evident effect on TYLCMaV fitness at 45 dpi (P > 5.56 × 10−3) or during the infection (P > 6.25 × 10−3). Despite being a recombinant of the former two viruses, TYLCMaV maintained similar fitness throughout the whole experiment in all four hosts (P > 6.25 × 10−3 or 0.010). Moreover, viral fitness in common bean and in S. nigrum was sustained along the experiment (P >0.010). In summary, these results suggest better performance of TYLCV and TYLCSV in common bean and S. nigrum, respectively, whereas TYLCMaV accumulates similarly well in all hosts, including the resistant tomato.
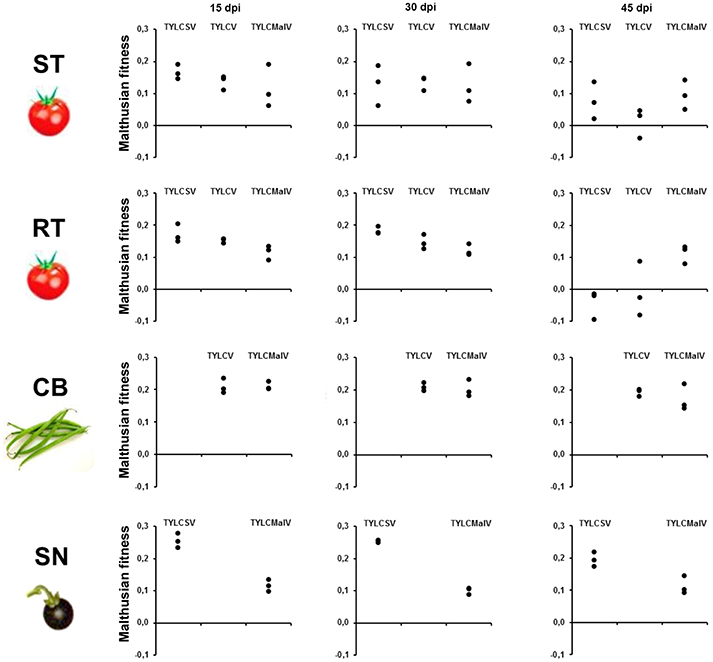
Figure 3. Absolute fitness of begomovirus during time-course analyses of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) infections in susceptible tomato (ST), resistant tomato (RT), common bean (CB) and the wild reservoir Solanum nigrum (SN). Absolute fitness of begomovirus DNA molecules present in the apical leaves of plants infected with infectious begomovirus clones was approximated by calculating the Malthusian growth rate per day, m, calculated according to the formula logQ, where Q is the number of pg of begomovirus ssDNA per 100 ng of total plant DNA. Three individual systemically infected plants (as judged by hybridization analysis) for each virus/host combination were monitored. Apical leaf samples (non-inoculated) were taken at 15, 30 and 45 days post inoculation (dpi). No viral detection was possible at any time point for TYLCSV/common bean or TYLCV/S. nigrum infections either by hybridization or qPCR.
High Genetic Complexity and Heterogeneity of TYLCD-Associated Begomovirus Quasispecies
Next we analyzed the quasispecies complexity and heterogeneity of the three begomoviruses during the time-course infections. Specifically, viral quasispecies complexity refers to the composition of the mutant spectrum, which can be described both by mutation frequency and average genetic distance (d), whereas viral quasispecies heterogeneity refers to the proportion of different viral genomes in the mutant spectrum, as measured by Shannon entropy (Domingo et al., 2006). DNA samples from one infected plant per virus/host combination at each time point, plus a replicate sample of each combination at 45 dpi, were subjected to rolling circle amplification (RCA) to specifically amplify circular DNA using the high-fidelity φ29 DNA polymerase with a reported error rate of 3–6 × 10−6 (Esteban et al., 1993; Nelson et al., 2002). As a measure to reduce amplification bias we amplified 3–7 × 108 input viral molecules. Sequences obtained for each begomovirus region were compared to the consensus sequence to determine mutation frequencies. Despite large variation among viruses, the highest mutation frequency values calculated for the whole sequenced region were detected in common bean and in S. nigrum (Figure 4A). When analyzed by genomic region, the mutation frequency of the quasispecies varied from <0.616 × 10−4 to 8.91 × 10−4 mutations/nt (mut/nt) (Table 2). The most complex region was V2 (e.g., 8.91 × 10−4 for TYLCV in resistant tomato, 8.6 × 10−4 and 8.0 × 10−4 mut/nt in common bean for TYLCV and TYLCMaV, respectively, or 8.4 × 10−4 mut/nt in S. nigrum for TYLCSV). In contrast, the least complex mutant spectra were found in the Rep and C4 genomic regions with values generally below 3 × 10−4 mut/nt (Table 2). Higher complexity values were found in common bean or S. nigrum than in tomato for CP or IR in all viral species. Thus, CP mutation frequency of TYLCV in common bean was higher than in resistant tomato at 15 dpi (P = 0.025), and also higher at 30 dpi than in susceptible (P = 0.034) and resistant tomato (P = 0.034). Only in TYLCV, CP (P = 0.030) and V2 (P = 0.045) regions showed more complexity in resistant tomato than in common bean at 45 dpi. Additionally, our results suggest that CP and IR were differentially enriched in mutations respect to other regions within a particular quasispecies. In S. nigrum at 45 dpi the complexity of CP (P = 0.001) and IR (P = 0.045) of TYLCSV was higher than that of both Rep and C4 regions. Also in S. nigrum CP region of TYLCMaV was more complex than Rep, C4, and V2 at 15 (P = 0.045) and 30 dpi (P = 0.005). In susceptible tomato at 45 dpi the mutation frequency was higher in CP than in C4 (P = 0.025) for TYLCSV and in resistant tomato CP was more complex than Rep (P = 0.001) and C4 (P = 0.014) for TYLCV.
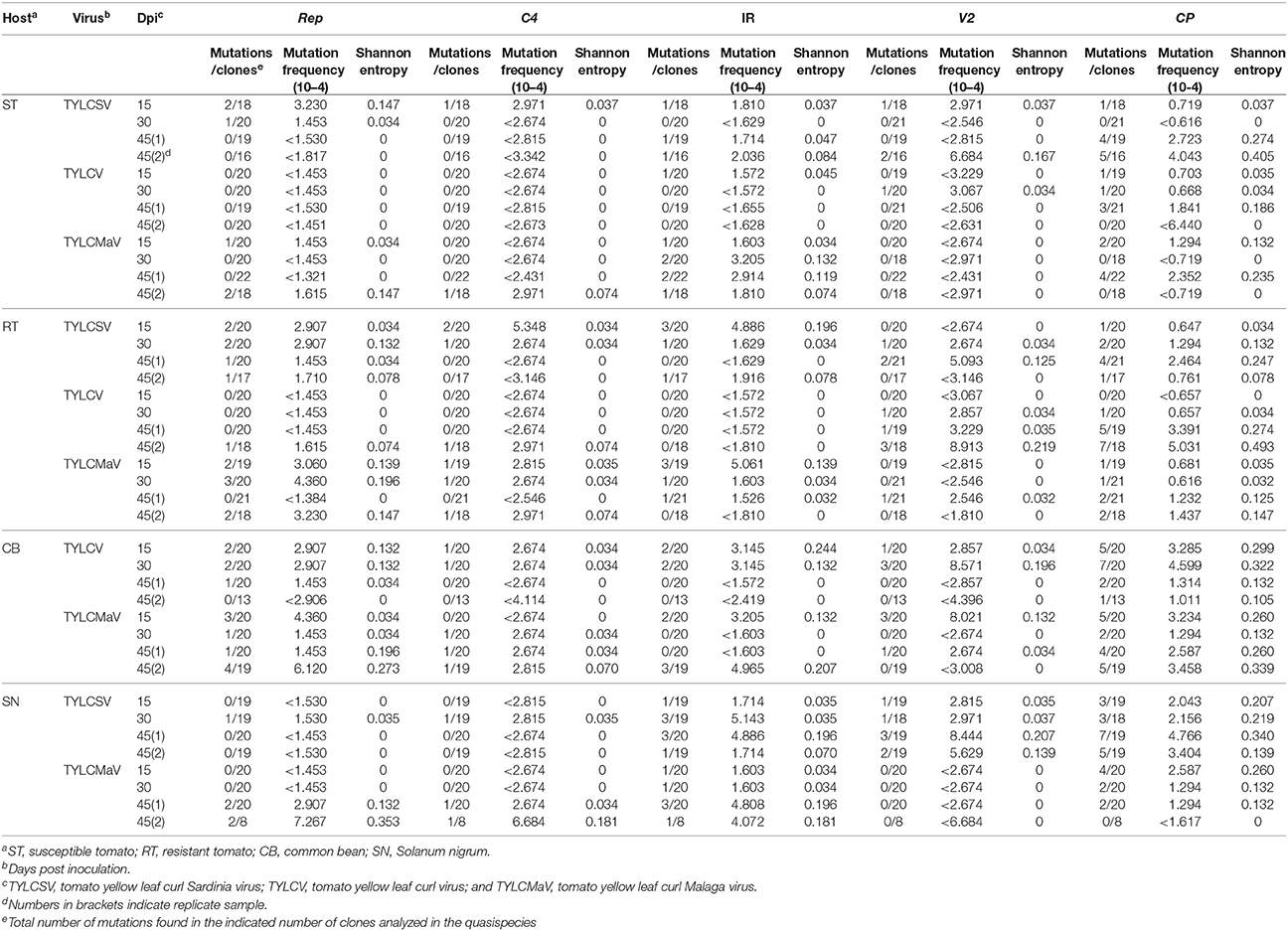
Table 2. Complexity and heterogeneity of tomato yellow leaf curl disease-associated begomovirus populations evolved from single viral sequences in three crop species and a wild host.
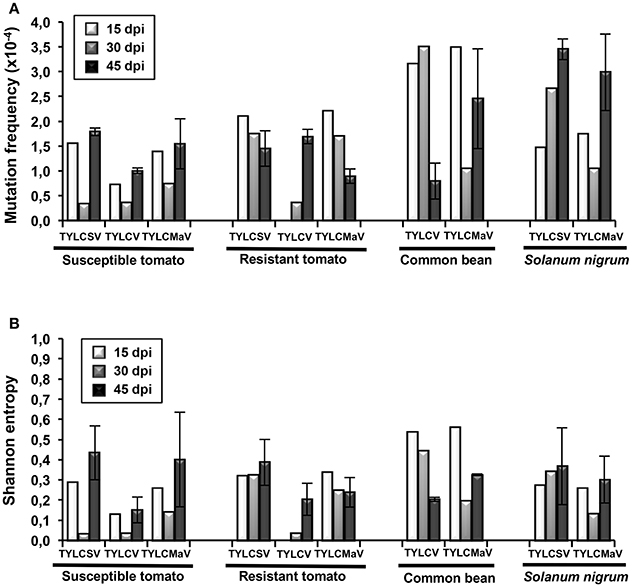
Figure 4. Complexity and heterogeneity of begomovirus quasispecies of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) in quasi-isogenic susceptible (ty-1/ty-1) and resistant (Ty-1/ty-1) tomato, common bean, and Solanum nigrum plants. DNA from young apical leaf samples of the same infected plant per virus-host combination collected at 15, 30, and 45 days post-inoculation (dpi), and of a replicate plant of each virus-host combination at 45 dpi, was amplified by rolling circle amplification (RCA) and the viral DNA products cloned. Sequences of 1,437 nucleotides for TYLCSV, 1,438 nucleotides for TYLCV, and 1,429 nucleotides for TYLCMaV from molecular clones (approximately 20 per quasispecies) were aligned and compared to their consensus sequences. Only mutations (base substitutions and indels) relative to the consensus sequence of each quasispecies were computed. (A) Mutation frequencies (mutations per nucleotide) were calculated for each quasispecies. (B) Normalized Shannon entropy estimated for each begomovirus quasispecies at the indicated d of experimental evolution. The formula − [Σi (pi x lnpi)/lnN] was used, where pi is the frequency of each sequence in the mutant spectrum and N is the total number of sequences compared.
Quasispecies genetic complexity was also assessed by analyzing average genetic distance (d) values. As shown in Table 3, d values varied from 0.00012 to 0.00173. The maximum genetic distance values for the four coding regions and the IR were found in the V2 region (especially for common bean and S. nigrum). High d values were also observed for C4 in some cases (e.g., in resistant tomato for TYLCSV at 15 dpi or in common bean for TYLCV at 30 dpi).
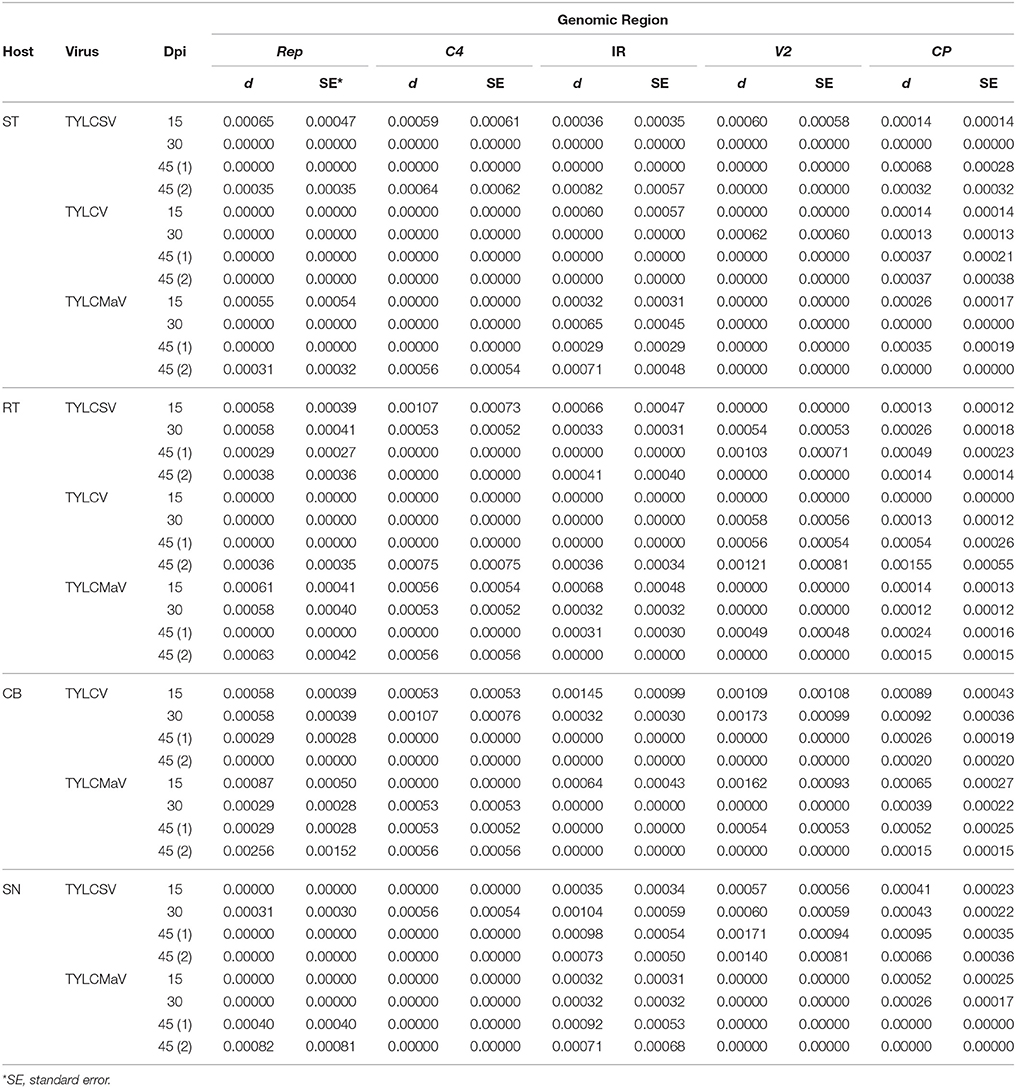
Table 3. Average pairwise genetic distances (d) estimated by Kimura's two-parameter method for begomovirus Rep, C4, V2, and CP coding regions, and the noncoding IR, after 45 days of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) infection in susceptible (ST) and resistant tomato (RT), common bean (Phaseolus vulgaris, CB) and Solanum nigrum (SN) hosts.
Begomovirus mutant spectra heterogeneity was evaluated by calculating the normalized Shannon entropy of viral quasispecies. Shannon entropy values calculated for the whole sequenced region were highest in common bean at 15 dpi and no difference between hosts were detected a later times post infection (Figure 4B). Susceptible tomato begomovirus quasispecies were the least heterogeneous, followed by those of resistant tomato and S. nigrum. Analyzed by genomic region, as shown in Table 2, Shannon entropy values ranged from 0 to 0.340. The CP region showed the highest heterogeneity in all four hosts and viruses, although this was especially pronounced in common bean and S. nigrum. Conversely, the lowest Shannon entropy values in the four hosts were detected in the C4 genomic region, except for TYLCSV at 15 dpi in resistant tomato. Therefore, heterogeneity of quasispecies was high in all host/begomovirus combinations, with the most heterogeneous populations exhibited by common bean and S. nigrum. Notably, the wild host S. nigrum contained the most heterogeneous populations at later infection time points for the viruses that were able to infect it. In general, mutation frequency, d and Shannon entropy values at 45 dpi were similar for replicates 1 and 2 for each virus/host combination.
Altogether, our results support the idea that begomovirus infections are characterized by the generation of highly complex and heterogeneous ssDNA mutant spectra in the host species tested, especially in common bean and the wild reservoir S. nigrum. V2 region supported the most complex spectra, CP was the most heterogeneous, while C4 exhibited both the least complexity and least heterogeneity. Interestingly, despite the high genetic complexity and heterogeneity of TYLCD-associated begomovirus quasispecies, no changes were detected in their whole genomic consensus sequences in any of the four hosts at any time point during infection.
Analysis of Mutations Suggests that the CP Region is the Most Susceptible to Change
Mutation data, comprising 128 base substitutions and 16 indels (complete list shown in Table 4), were pooled together irrespective of sampling time, virus or host. Mutation types and their frequency (%) relative to the number of changes found in a given genomic region or the whole sequenced zone are indicated in Table 5. Independent analysis of Rep, C4, IR, V2, and CP regions, shows that the most abundant mutations were base substitutions (86.7–100%), with indels only absent in C4. The IR contained the highest fraction of insertions, representing 11.1% of all mutations in the region. On the other hand, the highest fraction of deletions was detected in Rep (5.3% of total mutations). Of the base substitutions, the most common transition was C → T, which was highly enriched in C4 and V2 regions. G → A was the next most common transition, although it was less common in the IR and absent from C4. With regards to transversions, G → T was the most abundant. Conversely, A → T and A → C and G → T transversions were exceptionally frequent in the IR compared to the other four regions. Interestingly, the transition:transversion ratio was close to 1 in all regions. Transition:transversion ratios in DNA-based organisms (Begun et al., 2007; Hodgkinson and Eyre-Walker, 2010) as well as in RNA and dsDNA viruses (Duchêne et al., 2015) are over 2. While it has not been shown for ssDNA viruses, lower ratios might suggest that in all the begomovirus regions analyzed, especially in the IR and CP (with ratios of 0.9), either negative selection is removing transitions or transversions are favored. Nonsynonymous changes were more frequent than synonymous mutations in all regions, ranging from 65.4 to 79.2% of total mutations in Rep and V2, respectively.

Table 4. Nucleotide and amino acid changes found in genomic regions of begomovirus mutant clones.
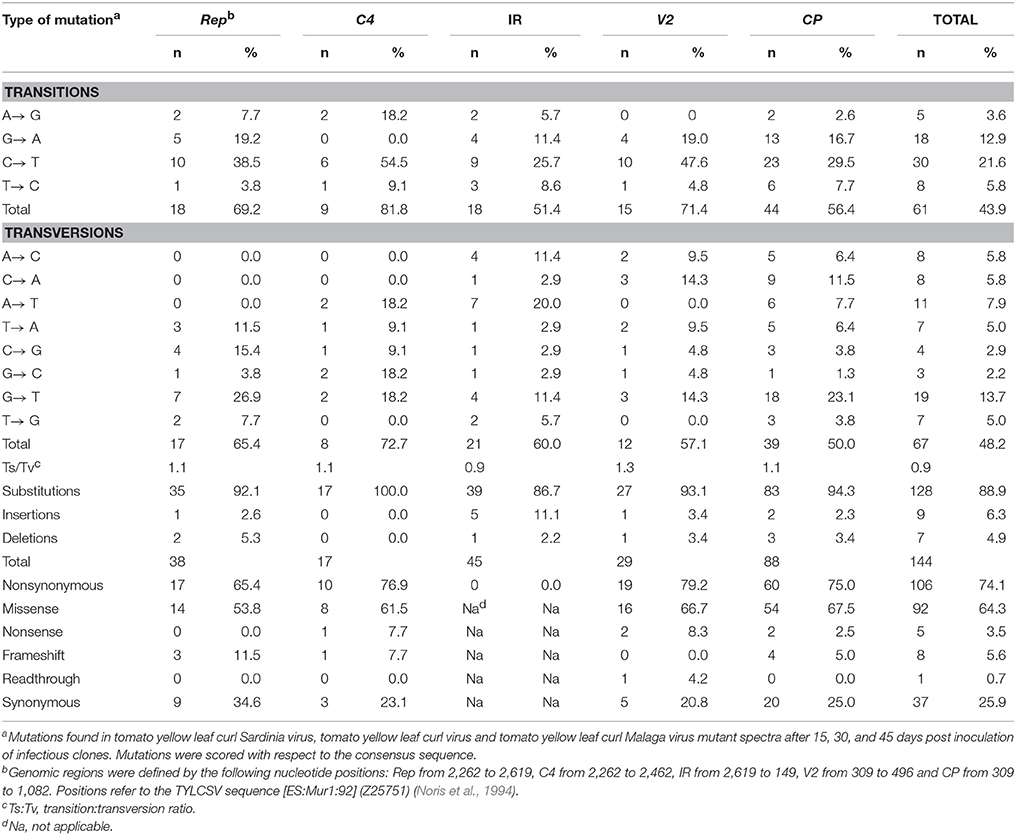
Table 5. Number and relative frequency (%) of the different types of mutations found in begomovirus mutant spectra.
Next, we studied the frequency and acceptability of the amino acid changes found in the different gene products under study. Firstly, amino acid changes were analyzed using the SG matrix as described previously (Feng et al., 1984) generating values from 0 to 6, with 0 indicating the most drastic amino acid changes and 6 the least, and their relative frequencies shown in Figure 5A. The results show that amino acid changes in Rep were poorly tolerated, since 83.9% of changes have acceptability values above 3, with 38.7% having a value of 6 (synonymous changes). Although the overall degree of amino acid change was similar in all regions, the higher relative frequency of more pronounced changes (with values of 2 or less) found in the CP might indicate that this protein is more tolerant to drastic amino acid changes. We analyzed the probability of amino acid substitution occurrence using a PAM-250 substitution matrix (Feng and Doolittle, 1996). The results shown in Figure 5B indicate that the highest frequency of probable amino acid replacements occurred in Rep and that unlikely amino acid changes were most prevalent in the CP protein. Altogether the results indicate that the Rep protein tends to be more conserved and that the CP protein is more amenable to change.
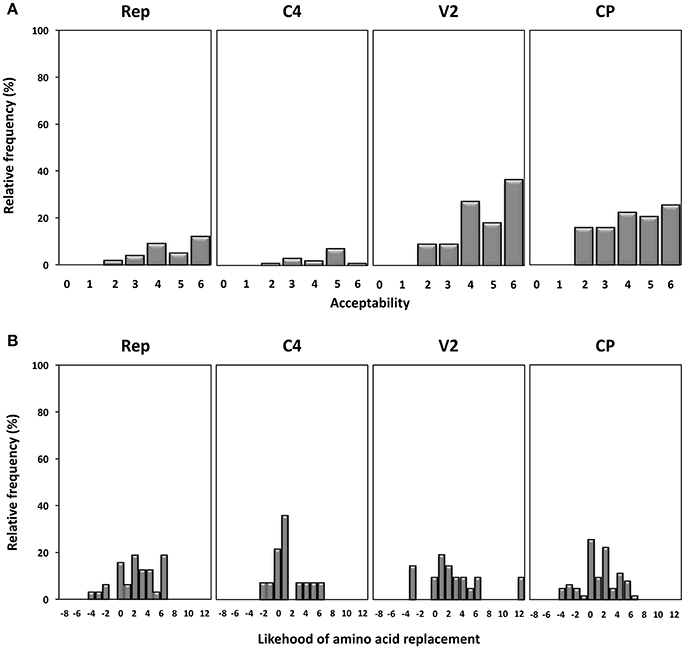
Figure 5. Acceptability and occurrence probability of the amino acid replacements observed in begomovirus quasispecies of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) in quasi-isogenic susceptible (ty-1/ty-1) and resistant (Ty-1/ty-1) tomato, common bean, and Solanum nigrum plants. (A) Acceptability values of amino acid changes found in each genomic coding region determined according to the structure-genetic (SG) matrix of Feng et al. (Feng et al., 1984). The values range from 0 (drastic amino acid changes) to 6 (synonymous replacements). (B) Probability of occurrence of amino acid replacements calculated according to the PAM-250 substitution matrix.
Host Influences the Sequence Space Explored by Begomoviruses
Sequence variations explored during begomovirus infections seemed to be affected by specific virus-host interactions. Sequence space distribution of the mutations detected in our time-course assays is summarized in Figure 6, which shows the location of the mutations found in begomovirus quasispecies by assay time point, host and begomovirus (TYLCSV, TYLCV, or TYLCMaV). Considering the genomic regions explored at each sampling time, we observed that the tomato Ty-1 resistance allele did not significantly influence mutation frequency for any ORF, since no significant differences between susceptible tomato and resistant tomato were found at any time point (generalized linear model assuming binary distribution and using logit link function, P ≥ 0.05). However, differences between hosts were observed with respect to where mutations occurred over time. Thus, for example, mutation frequency of the CP increased for TYLCSV in susceptible tomato between 15 and 45 dpi (P = 0.048) and between 30 and 45 dpi (P = 0.003), and for TYLCV in resistant tomato between 15 and 45 dpi (P = 0.001). This increase in CP mutation frequency might be related to the decrease in fitness in tomato. In S. nigrum, TYLCMaV (replicates 1 and 2 at 45 dpi) showed a significant increase in the number of mutations in Rep between 15 and 45 dpi (P = 0.044) and between 30 and 45 dpi (P = 0.005) but not in the overlapping C4 ORF. A random distribution of mutations was found both in Rep and C4 (runs test, P > 0.05) so mutational hotspots were ruled out. This result suggests an active exploration of sequence space in Rep but not in C4 for the recombinant virus. No parallel mutations were found in replicate samples at 45 dpi. Our results suggest that begomoviruses explore different sequence space depending on host species, although further studies are needed to confirm this including additional replicates (at 15 and 30 dpi). They also suggest that the presence of the Ty-1 gene in tomato does not affect how begomoviruses move in sequence space at each time point. Furthermore, it is interesting to note that begomovirus consensus sequences were completely invariant throughout the infection time-courses regardless of mutant spectra or host, and despite the large number of mutations found.
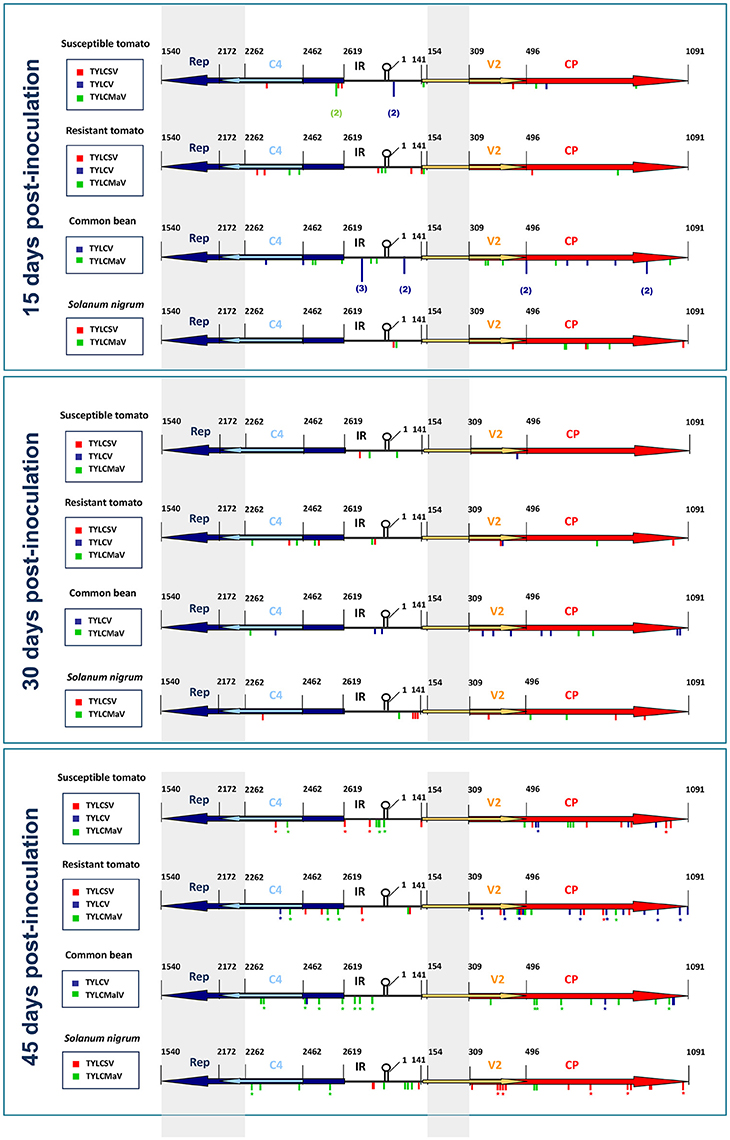
Figure 6. Schematic representation of the distribution of mutations found in quasispecies of time-course infections of tomato yellow leaf curl Sardinia virus (TYLCSV), tomato yellow leaf curl virus (TYLCV) and tomato yellow leaf curl Malaga virus (TYLCMaV) in quasi-isogenic susceptible (ST, ty-1/ty-1) and resistant (RT, Ty-1/ty-1) tomato lines, common bean (CB) and Solanum nigrum (SN) at 15, 30, and 45 days after inoculation of single sequence variants. Sequenced regions of quasispecies molecular clones include 357 nts of the 5′-end of Rep, 200 nts of the 5′-end of C4 ORF, the IR, 187 nts of the 5′-end of the V2 ORF and the complete CP ORF. Nucleotide positions refer to the TYLCSV sequence [ES:Mur1:92] (Z25751) (Noris et al., 1994). Consensus sequences of the quasispecies were determined for whole viral genomic sequences. Mutations detected in separate quasispecies in the same host/time combination are indicated by the number of times they appeared. Mutations found in replicate samples are denoted with an asterisk.
Direction of Selective Forces Depends on Host and is not Influenced by Ty-1
To assess begomovirus mutant spectra adaptation to hosts, we studied the direction of selective forces on their coding regions. To this end the average rate of synonymous substitutions per synonymous site (dS) and of nonsynonymous substitutions per nonsynonymous site (dNS) for each mutant spectrum were estimated as reported previously (Pamilo and Bianchi, 1993). Where possible, the dNS/dS ratio was also calculated. The results in Table 6 show that dNS/dS ratios of begomovirus quasispecies evolving in tomato ranged from 0.149 to 0.616 in Rep, V2, and CP. Ratios calculated for TYLCSV and TYLCMaV were below 1, suggesting that all three regions were under negative selection, both in susceptible and resistant tomato. However, dNS/dS ratios ranged from 0.357 to 1.842 in common bean and from 0.464 to 1.778 in S. nigrum, showing that, in these hosts, selective forces acted differentially depending on the coding region. In fact, we observed positive selection (dNS/dS > 1) in the CP region of both common bean, at 15 and 45 dpi for TYLCV and TYLCMaV infections, respectively, and S. nigrum, at 45 and 15 dpi for TYLCSV and TYLCMaV infections, respectively. The results on dNS/dS ratios of replicates 1 and 2 at 45 dpi of each virus/host combination were similar, although additional replicates would add more robustness to our conclusions. Altogether, these results suggest differing selective constraints of begomovirus quasispecies in different hosts. Also, it seems that Ty-1 resistance does not alter the way begomoviruses are selected in tomato, as purifying selection dominates in all the coding regions analyzed. Our data also support positive selection of virus variants in the CP region in common bean and S. nigrum, whereas Rep, C4, and V2 coding regions are under purifying selection. Positive selection of CP region variants in common bean and S. nigrum together with higher virus fitness and diversity, and differential mutation distribution occur without alterations to begomovirus consensus sequences. These results suggest that the mutant spectrum plays an important role in begomovirus infections in each host.
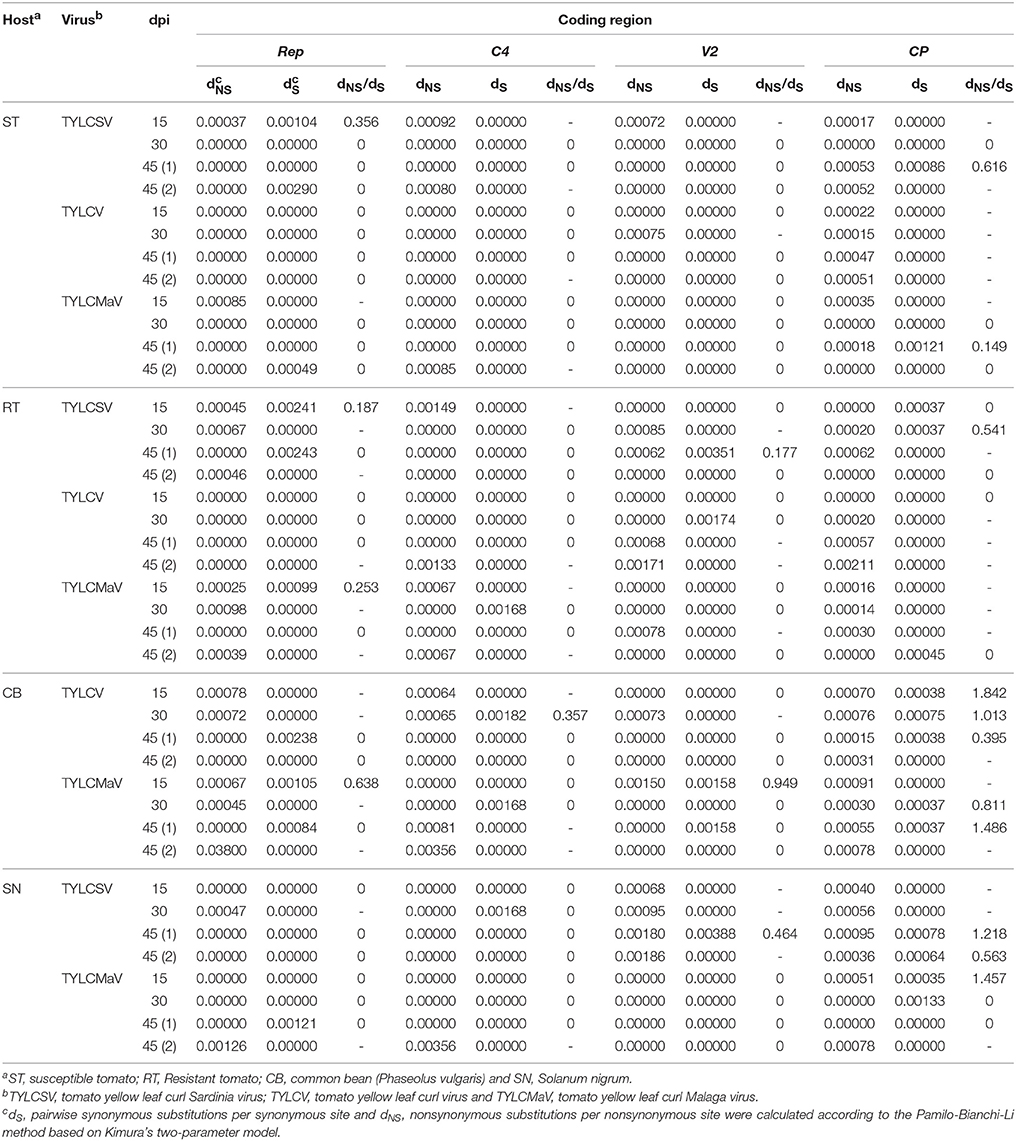
Table 6. Analysis of diversity at nonsynonymous and synonymous positions in four coding regions of begomovirus time-course infections.
Discussion
Single-stranded DNA viruses belonging to Parvoviridae, Geminiviridae, and Nanoviridae have been shown to be as variable as RNA viruses. Moreover, ssDNA virus populations, composed of mutant spectra, have shown strong adaptive capacities similar to those observed in RNA virus populations (Isnard et al., 1998; Nishizawa et al., 1999; López-Bueno et al., 2006; Urbino et al., 2008; Grigoras et al., 2010). The origin of this variability, however, remains unknown for these viruses. Quasispecies-like evolution combined with invariance of the Rep, C4, and IR genomic consensus sequences has been previously described for the begomovirus tomato yellow leaf curl China virus (TYLCCNV) (Ge et al., 2007) in naturally infected tomato, and in tomato and Nicotiana benthamiana plants experimentally infected with TYLCCNV clones. Here we have looked at the evolution of the whole genomic consensus sequence and mutant spectra variability of the Rep, C4, IR, V2, and CP ORFs of three TYLCD-associated begomoviruses in species of the family Solanaceae family, that is, susceptible tomato, resistant tomato carrying the Ty-1 allele and the wild reservoir S. nigrum, as well as to common bean, a species of the distantly-related family Fabaceae.
In this work we noticed differences in the fitness of viral isolates between hosts. We used daily Malthusian growth rates (m) to estimate viral fitness since differences in growth rate reflect fitness to a great extent and also because single sequence variants were used to infect hosts (Lalić et al., 2011). Further, because monopartite begomoviruses are restricted to the phloem and replicate only in companion cells perturbations in copy numbers are reduced. Thus, at the last assay time point (45 dpi) fitness in tomato of either TYLCSV or TYLCV was lower than in S. nigrum or common bean, respectively. This was clearly evident in tomato plants bearing the Ty-1 resistance allele, where negative growth rate values were obtained for both TYLCSV and TYLCV, in contrast to TYLCMaV. The fitness of this latter virus remained stable over time in resistant tomato and no significant differences were seen between hosts. Indeed, the statistics support that there are no significant differences in TYLCMaV accumulation between susceptible and resistant tomato. It is interesting that in genotypes with the resistance gene the accumulation of TYLCMaV does not decrease much, compared with the other two TYLCD-associated viruses tested. Its better adaptation in resistant genotypes can have important ecological consequences Therefore, our results corroborate that TYLCMAV is a virus with resistance-breaking properties. This was already observed previously (Monci et al., 2002) and quoted subsequently (García-Arenal and McDonald, 2003). How TYLCMaV, is able to overcome the growth restriction conferred by the Ty-1 gene, despite the genome sequences of the recombinant virus sharing 99% identity with the corresponding parental virus sequences, is an interesting open question. An improved combination of coding and/or nonconding regions may have been selected after a recombination event in TYLCMaV that better overcame Ty-1 resistance allele limitations. The Ty-1 allele has been shown to impair the accumulation of begomoviruses involved in TYLCD (García-Andrés et al., 2009), although, similar to the results obtained here, it has been shown to have little effect on TYLCMaV (Monci et al., 2002). Ty-1 encodes a γ type RNA-directed RNA polymerase (RDRP) (Verlaan et al., 2013), with Ty-1 resistance involving enhanced transcriptional gene silencing against TYLCV, as revealed by the enrichment of siRNAs directed against CP and REn, and cytosine methylation of the CP promoter region (Butterbach et al., 2014). We did not detect differences in the complexity or heterogeneity of the three begomovirus mutant spectra in the presence or absence of the Ty-1 allele for any of the genomic regions analyzed in this study. Nevertheless, in resistant tomato, our data show that a high mutation frequency value was observed for the C4 region of TYLCSV, the most restricted virus (Monci et al., 2002), suggesting exploration of TYLCSV for better adapted variants to overcome restriction imposed by this host. As C4 has been shown to be a mild suppressor of gene silencing (Luna et al., 2012) and a viral determinant in overcoming the genetic resistance of some wild tomato species (Tomás et al., 2011), the role of this region in permitting viral accumulation in the presence of Ty-1 merits further study. Since the sequence of the TYLCMaV isolate used in this work only differs in 7 and 15 nucleotide changes to the corresponding sequences of the parental TYLCV and TYLCSV viruses, respectively, it seems reasonable to hypothesize that the key to overcoming Ty-1 resistance resides in this particular combination of nucleotide changes, with those detected in the C4 ORF being prime candidates. Alternatively, an optimized combination of genome fragments to overcome Ty-1 resistance might be achieved in this virus driven by a recombination event then supporting the importance of modular evolution to adapt to a fluctuating environment (Botstein, 1980; Stavrinides and Guttman, 2004; Belabess et al., 2016).
The presence of putative low fitness variants in mutant spectra could be the result of compensatory mutations to maintain RNA secondary structure. Further, they may be maintained by complementation or by reduced purifying selection due to a release from the constraint of whitefly transmission in the conducted experiment. Our analysis of mutant spectra identified insertions, deletions, nonsense mutations and readthrough mutations. Such changes often have deleterious or lethal effects on proteins. Others may affect secondary structure or fall in regulatory elements needed to maintain structure for function like some of the mutations found in the IR adjacent to or within a regulatory element (see Table 4 and Figure 7). Among mutations found in TYLCSV, TYLCV and TYLCMaV during infection time-courses in the four hosts (included in Table 4) we highlight the following. The nonsense mutation Q81Stop (C395T) found in the TYLCV V2 protein in infected common bean lacks residues C84 and C86 that have been shown to be essential for PTGS suppression and interaction with the tomato SGS3 protein (Zrachya et al., 2007; Glick et al., 2008). The Rep missense mutation C2466T in TYLCMaV-infected resistant tomato, results in an E52K (C2466T) amino acid change between the RCR-1 and RCR-2 DNA binding motifs involved in the specific binding of Rep to DNA (Jupin et al., 1995). The TYLCSV CP V135I and M136I (G711A and G716A, respectively) mutations are in the critical 129–152 region required for effective capsid assembly and transmissibility by Bemisia tabaci, the virus vector (Noris et al., 1998; Hallan and Gafni, 2001; Caciagli et al., 2009), although in our experimental design the latter is not a limiting factor. The TYLCMaV CP R18C (C360T) amino acid change is adjacent to R19L, a change which has been shown to deeply impact both TYLCV capsid assembly, as well as CP protein interaction with the tomato nuclear receptor karyopherin α1 and the GroEL protein of B. tabaci bacterial endosymbiont (Yaakov et al., 2011). CP mutations Q76E, Y78H, K80T, and R82C (C540G, T546C, A568C, and C585T, respectively) of TYLCV, and Q79H, R80L and D82V (G545T, G547T, and A553T) of TYLCMaV would affect the putative zinc finger motif, thus potentially impairing the ssDNA binding capacity of this protein.
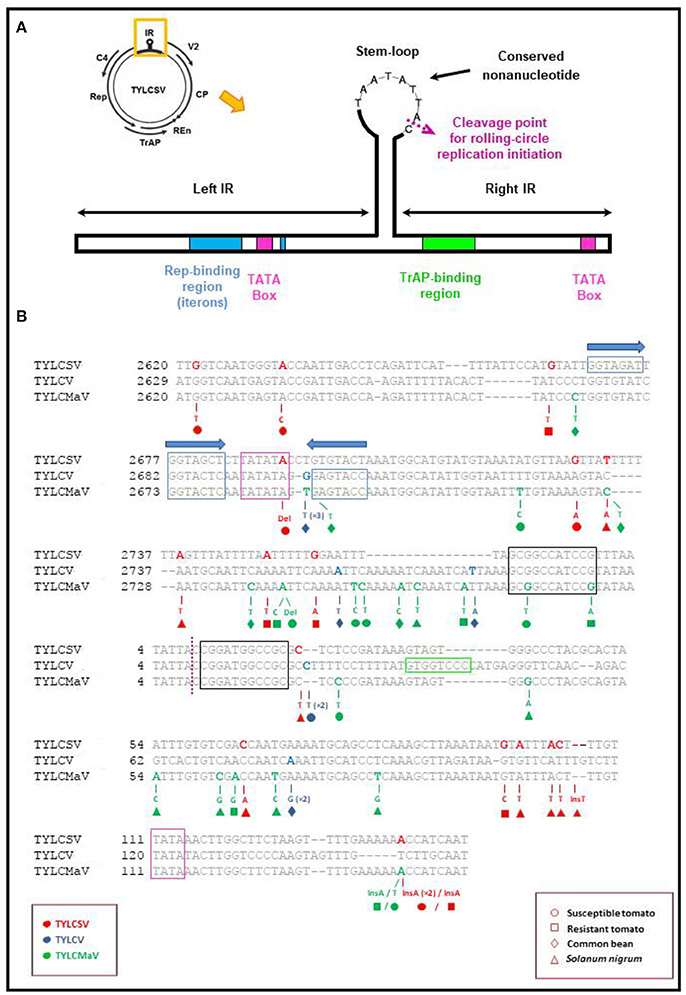
Figure 7. Localization of mutations found in the Intergenic region (IR) of begomovirus quasispecies. (A) Schematic representation of the intergenic region of tomato yellow leaf curl-like viruses showing the location of the replication associated (Rep) and the transcription activator (TrAP) protein binding regions, TATA boxes and ERE-like motifs (Bañuelos-Hernández et al., 2012). The nonanucleotide sequence conserved in all geminiviruses and the cleavage site for the initiation of rolling-circle replication in the stem-loop are also shown. (B) Representation of the nucleotide mutations found in clones obtained from susceptible (°) and resistant tomato (□), common bean (♢) and Solanum nigrum (Δ) plants inoculated with TYLCSV (red), TYLCV (blue) or TYLCMaV (green) infectious clones. Nucleotide numbers on the left-hand side of sequences refer to GenBank accession numbers Z25751 (TYLCSV), AF071228 (TYLCV) and AF271234 (TYLCMaV). Note that Ins141 appeared twice in TYLCSV-infected susceptible tomato at 45 dpi (replicate 1).
It could be argued that the probability of sequencing genomes with low frequency is expected to be extremely low because of a potential RCA bias (only 0.05% available genomes amplified with RCA) (Gallet et al., 2017) and because of the limited number of sequenced genomes (about 20). In this sense, as a measure to reduce the amplification bias, 3–7 × 108 viral genomes were used as input for RCA, which means that roughly 1.5–3.5 × 105 molecules may have served as template for amplification so no bottlenecks were created at this step. Notwithstanding, there is a previous report with the same experimental nanovirus system (Grigoras et al., 2009) in which the relative quantity of the eight segments of FBNSV, measured by qPCR in total DNA of infected plant tissue, was similar. In addition, the same relative abundance was found in qPCR analysis after performing RCA of the same total DNA or after restriction of the same RCA product with a single-cut enzyme, which ruled out a bias in the RCA. Finally, the analysis of 20 clones per quasispecies may appear insufficient, but assuming each clone is present with a relative frequency of at least 5% within the quasispecies, it means that we might be detecting at least a portion of the most frequent viral variants that are high fitness variants.
Here we have shown that, after the inoculation of a single sequence variant, complex populations arise with a high number of different begomovirus genomes accumulating within each individual plant. We estimate that an average of 2 × 1011 begomovirus genomic variants coexist per gram of fresh tissue in apical tomato leaves. Tenfold higher-numbers are reached in common bean and the wild reservoir, S. nigrum. This allows begomovirus quasispecies to explore intensively and efficiently sequence space conferring them an enormous capacity to adapt and overcome selective constraints imposed by the host such as the innate or adaptive immune response or post transcriptional gene silencing (Vanitharani et al., 2005; Chen et al., 2010; Raja et al., 2010). The presence of high viral loads of highly diverse geminiviruses is of particular importance for viral emergence. In the infection time-courses, TYLCMaV was able to infect and accumulate sustained high titers within every individual plant species tested, even in the resistant tomato. We showed that quasispecies complexity is especially high in common bean and S. nigrum. Common bean is usually employed as a transition host between tomato crops in field rotations, for example in Spain (Sánchez-Campos et al., 1999), which might favor diversification of the begomovirus population between tomato crops. S. nigrum is a common wild reservoir in growing areas (García-Andrés et al., 2006), therefore its presence can ensure the maintenance of begomovirus diversity for new epidemics. Importantly, mutations arising in wild reservoirs could be transmitted to crop plants by the insect vector B. tabaci, potentially resulting in the emergence and spread of new better fit variants. TYLCMaV can efficiently infect the Ty-1-resistant tomato frequently used to contain TYLCD epidemics, as well as the wild host S. nigrum and common bean (Noris et al., 1994). Therefore, the high viral diversity of TYLCMaV infections might favor the appearance of ecologically-better adapted emergent variants.
Positive selection in the CP region was observed in all three begomovirus quasispecies evolving in common bean and S. nigrum, but not in tomato. This finding is unexpected because capsid proteins are involved in vector recognition and particle formation (Noris et al., 1998), and are therefore usually more resistant to selective pressures, as evidenced by lower dNS/dS ratios in other viruses (Chare and Holmes, 2004). However, our results indicate that under the conditions studied begomovirus CP region incorporated nonsynonymous mutations and accepted more drastic amino acid changes than other genomic regions. Purifying selection might occur in nature during the transmission process, which was not included in our experimental design, as CP strongly determines insect recognition (Noris et al., 1998). Nevertheless, under the same experimental conditions, the absence of positive selection in tomato suggests that additional selective constraints on quasispecies development exist in tomato that are absent from the other hosts. Another reason why positive selection in the CP region is surprising is because we showed that quasispecies consensus sequences did not change in any of the hosts. It has been suggested that rates of mutation fixation can be higher when viruses are under positive selection, such as when they are adapting to a new host or to cell culture, thus allowing beneficial mutations to become dominant in a short period of time (Carrigan and Knox, 1990; Fares et al., 2001; Acosta-Leal et al., 2011; Domingo et al., 2012). It is worth mentioning that the host change between the plant from which the viral clones were isolated, where they were probably better adapted, and the test plants could constitute such an environmental change that would lead to the evolution and novel exploration of the sequence space. However, in the course of this experiment, we have not found evidence of begomovirus adaptation to different hosts by changing the consensus sequence. Successive passaging of the viruses through the hosts for a longer period of time would have allowed fixation of novel mutations and might have revealed adaptive evolution and perhaps changes in the consensus sequence. Nevertheless, our work suggests that differences in fitness across hosts could be influenced by the distribution of mutations in the mutant spectra. We have shown in previous studies the remarkable continuity of the TYLCSV consensus sequence over an 8-year period (Sánchez-Campos et al., 2002). In this work we have shown that high genetic diversity is generated in all four hosts, especially in common bean and the wild reservoir S. nigrum after infection with single sequence variants of three TYLCD-associated begomoviruses in spite of the invariance of the viral consensus sequences. Therefore, our results imply that quasispecies diversity need to be addressed to understand the adaptive potential of these economically important emergent viruses in their different hosts in order to design more durable control strategies.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
SS-C, DT, EM, JN-C, and AG-P: conceived and designed the experiments; SS-C, GD-H, DT, and LD-M: performed the experiments; SS-C, GD-H, LD-M, and AG-P: analyzed the data; SS-C, GD-H, LD-M, and AG-P: wrote the main manuscript text and prepared figures. All authors reviewed the manuscript.
Funding
This work was supported by grants P09-CVI-5428 and P10-CVI-6561 to AG-P, and grant P10-AGR-6516 to EM, from the Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain; and grants AGL2013-48913-C02 and AGL2016-75819-C2-2-R to JN-C, and EM, from the Ministerio de Economía y Competitividad, and Ministerio de Economía, Industria y Competitividad, Spain, respectively, with assistance from the European Regional Development Fund (ERDF) and the European Social Fund (ESF); and a JAE-DOC postdoctoral contract to SS-C, from the Consejo Superior de Investigaciones Científicas (CSIC) with assistance from ESF; and grants to Research Group AGR-214 to JN-C, EM, and CVI-264 to AG-P, from the Consejería de Economía, Innovación y Ciencia of the Junta de Andalucía with assistance from the ERDF and ESF; and grant from the Primer Plan Propio de Investigación y Transferencia de la Universidad de Málaga to AG-P. LD-M was supported by Junta de Andalucía grant P10-CVI-6561. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank M. J. Díez (Dept. de Biotecnología, Universidad Politécnica de Valencia, Spain) for providing the near isogenic tomato lines used in this study, Rafael Fernández-Muñoz for assistance with statistical analysis, Esteban Domingo for critically reading the manuscript, and John Robert Pearson for scientific English editing of the manuscript.
References
Acosta-Leal, R., Duffy, S., Xiong, Z., Hammond, R. W., and Elena, S. F. (2011). Advances in plant virus evolution: translating evolutionary insights into better disease management. Phytopathology 101, 1136–1148. doi: 10.1094/PHYTO-01-11-0017
Bañuelos-Hernández, B., Mauricio-Castillo, J. A., Cárdenas-Conejo, Y., Guevara-González, R. G., and Argüello-Astorga, G. R. (2012). A new strain of tomato severe leaf curl virus and a unique variant of tomato yellow leaf curl virus from Mexico. Arch. Virol. 157, 1835–1841. doi: 10.1007/s00705-012-1358-z
Begun, D. J., Holloway, A. K., Stevens, K., Hillier, L. W., Poh, Y. P., Hahn, M. W., et al. (2007). Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 5:e310. doi: 10.1371/journal.pbio.0050310
Belabess, Z., Peterschmitt, M., Granier, M., Tahiri, A., Blenzar, A., and Urbino, C. (2016). The non-canonical tomato yellow leaf curl virus recombinant that displaced its parental viruses in southern Morocco exhibits a high selective advantage in experimental conditions. J. Gen. Virol. 97, 3433–3445. doi: 10.1099/jgv.0.000633
Bittar, C., Jardim, A. C., Yamasaki, L. H., Carareto, C. M., Pinho, J. R., Lemey, P., et al. (2013). On hepatitis C virus evolution: the interaction between virus and host towards treatment outcome. PLoS ONE 8:e62393. doi: 10.1371/journal.pone.0062393
Boiteux, L. S., Nagata, T., Dutra, W. P., and Fonseca, M. E. N. (1993). Sources of resistance to tomato spotted wilt virus (TSWV) in cultivated and wild species of Capsicum. Euphytica 67, 89–94. doi: 10.1007/bf00022729
Botstein, D. (1980). A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 354, 484–490.
Butterbach, P., Verlaan, M. G., Dullemans, A., Lohuis, D., Visser, R. G., Bai, Y., et al. (2014). Tomato yellow leaf curl virus resistance by Ty-1 involves increased cytosine methylation of viral genomes and is compromised by cucumber mosaic virus infection. Proc. Natl. Acad. Sci. U.S.A. 111, 12942–12947. doi: 10.1073/pnas.1400894111
Caciagli, P., Medina Piles, V., Marian, D., Vecchiati, M., Masenga, V., Mason, G., et al. (2009). Virion stability is important for the circulative transmission of tomato yellow leaf curl sardinia virus by Bemisia tabaci, but virion access to salivary glands does not guarantee transmissibility. J. Virol. 83, 5784–5795. doi: 10.1128/JVI.02267-08
Carrigan, D. R., and Knox, K. K. (1990). Identification of interferon-resistant subpopulations in several strains of measles virus: positive selection by growth of the virus in brain tissue. J. Virol. 64, 1606–1615.
Chare, E. R., and Holmes, E. C. (2004). Selection pressures in the capsid genes of plant RNA viruses reflect mode of transmission. J. Gen. Virol. 85(Pt 10), 3149–3157. doi: 10.1099/vir.0.80134-0
Chen, H., Zhang, Z., Teng, K., Lai, J., Zhang, Y., Huang, Y., et al. (2010). Up-regulation of LSB1/GDU3 affects geminivirus infection by activating the salicylic acid pathway. Plant J. 62, 12–23. doi: 10.1111/j.1365-313X.2009.04120.x
Coffin, J. M. (1995). HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267, 483–489.
Czosnek, H. (ed.). (2007). Tomato yellow leaf curl virus disease,” in Management, Molecular Biology, Breeding for Resistance (Jerusalem: Springer), 1–447.
Czosnek, H., and Laterrot, H. (1997). A worldwide survey of tomato yellow leaf curl viruses. Arch. Virol. 142, 1391–1406.
Díaz-Pendón, J. A., Cañizares, M. C., Moriones, E., Bejarano, E. R., Czosnek, H., and Navas-Castillo, J. (2010). Tomato yellow leaf curl viruses: ménage à trois between the virus complex, the plant and the whitefly vector. Mol. Plant Pathol. 11, 441–450. doi: 10.1111/j.1364-3703.2010.00618.x
Domingo, E., and Holland, J. J. (1997). RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51, 151–178. doi: 10.1146/annurev.micro.51.1.151
Domingo, E., Martín, V., Perales, C., Grande-Pérez, A., García-Arriaza, J., and Arias, A. (2006). Viruses as quasispecies: biological implications. Curr. Top. Microbiol. Immunol. 299, 51–82. doi: 10.1007/3-540-26397-7_3
Domingo, E., Sheldon, J., and Perales, C. (2012). Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76, 159–216. doi: 10.1128/MMBR.05023-11
Duchêne, S., Ho, S. Y., and Holmes, E. C. (2015). Declining transition/transversion ratios through time reveal limitations to the accuracy of nucleotide substitution models. BMC Evol. Biol. 15:36. doi: 10.1186/s12862-015-0312-6
Duffy, S., and Holmes, E. C. (2008). Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 82, 957–965. doi: 10.1128/JVI.01929-07
Duffy, S., and Holmes, E. C. (2009). Validation of high rates of nucleotide substitution in geminiviruses: phylogenetic evidence from East African cassava mosaic viruses. J. Gen. Virol. 90(Pt 6), 1539–1547. doi: 10.1099/vir.0.009266-0
Edwards, K., Johnstone, C., and Thompson, C. (1991). A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 19, 1349.
Elena, S. F., Bedhomme, S., Carrasco, P., Cuevas, J. M., de la Iglesia, F., Lafforgue, G., et al. (2011). The evolutionary genetics of emerging plant RNA viruses. Mol. Plant Microbe Interact. 24, 287–293. doi: 10.1094/MPMI-09-10-0214
Elena, S. F., Fraile, A., and García-Arenal, F. (2014). Evolution and emergence of plant viruses. Adv. Virus Res. 88, 161–191. doi: 10.1016/B978-0-12-800098-4.00003-9
Esteban, J. A., Salas, M., and Blanco, L. (1993). Fidelity of phi 29 DNA polymerase. Comparison between protein-primed initiation and DNA polymerization. J. Biol. Chem. 268, 2719–2726.
Fares, M. A., Moya, A., Escarmís, C., Baranowski, E., Domingo, E., and Barrio, E. (2001). Evidence for positive selection in the capsid protein-coding region of the foot-and-mouth disease virus (FMDV) subjected to experimental passage regimens. Mol. Biol. Evol. 18, 10–21.
Feng, D. F., and Doolittle, R. F. (1996). Progressive alignment of amino acid sequences and construction of phylogenetic trees from them. Meth. Enzymol. 266, 368–382.
Feng, D. F., Johnson, M. S., and Doolittle, R. F. (1984). Aligning amino acid sequences: comparison of commonly used methods. J. Mol. Evol. 21, 112–125.
Firth, C., Charleston, M. A., Duffy, S., Shapiro, B., and Holmes, E. C. (2009). Insights into the evolutionary history of an emerging livestock pathogen: porcine circovirus 2. J. Virol. 83, 12813–12821. doi: 10.1128/JVI.01719-09
Fuchs, M. (2008). “Plant resistance to viruses: engineered resistance,” in Encyclopedia of Virology, 3rd Edn, eds B.W.J. Mahy and M.H.V.v. Regenmortel (Oxford: Academic Press), 156–164.
Gallet, R., Fabre, F., Michalakis, Y., and Blanc, S. (2017). The number of target molecules of the amplification step limits accuracy and sensitivity in ultra deep sequencing viral population studies. J. Virol. 91:e00561-17. doi: 10.1128/JVI.00561-17
García-Andrés, S., Accotto, G. P., Navas-Castillo, J., and Moriones, E. (2007). Founder effect, plant host, and recombination shape the emergent population of begomoviruses that cause the tomato yellow leaf curl disease in the Mediterranean basin. Virology 359, 302–312. doi: 10.1016/j.virol.2006.09.030
García-Andrés, S., Monci, F., Navas-Castillo, J., and Moriones, E. (2006). Begomovirus genetic diversity in the native plant reservoir Solanum nigrum: evidence for the presence of a new virus species of recombinant nature. Virology 350, 433–442. doi: 10.1016/j.virol.2006.02.028
García-Andrés, S., Tomás, D. M., Navas-Castillo, J., and Moriones, E. (2009). Resistance-driven selection of begomoviruses associated with the tomato yellow leaf curl disease. Virus Res. 146, 66–72. doi: 10.1016/j.virusres.2009.08.012
García-Arenal, F., and McDonald, B. A. (2003). An analysis of the durability of resistance to plant viruses. Phytopathology 93, 941–952. doi: 10.1094/PHYTO.2003.93.8.941
García-Cano, E., Navas-Castillo, J., Moriones, E., and Fernández-Muñoz, R. (2010). Resistance to Tomato chlorosis virus in wild tomato species that impair virus accumulation and disease symptom expression. Phytopathology 100, 582–592. doi: 10.1094/PHYTO-100-6-0582
Ge, L., Zhang, J., Zhou, X., and Li, H. (2007). Genetic structure and population variability of tomato yellow leaf curl China virus. J. Virol. 81, 5902–5907. doi: 10.1128/JVI.02431-06
Glick, E., Zrachya, A., Levy, Y., Mett, A., Gidoni, D., Belausov, E., et al. (2008). Interaction with host SGS3 is required for suppression of RNA silencing by tomato yellow leaf curl virus V2 protein. Proc. Natl. Acad. Sci. U.S.A. 105, 157–161. doi: 10.1073/pnas.0709036105
Grigoras, I., Timchenko, T., Grande-Pérez, A., Katul, L., Vetten, H. J., and Gronenborn, B. (2010). High variability and rapid evolution of a nanovirus. J. Virol. 84, 9105–9117. doi: 10.1128/JVI.00607-10
Grigoras, I., Timchenko, T., Katul, L., Grande-Pérez, A., Vetten, H. J., and Gronenborn, B. (2009). Reconstitution of authentic nanovirus from multiple cloned DNAs. J. Virol. 83, 10778–10787. doi: 10.1128/JVI.01212-09
Gutierrez, C. (2000). DNA replication and cell cycle in plants: learning from geminiviruses. EMBO J. 19, 792–799. doi: 10.1093/emboj/19.5.792
Hallan, V., and Gafni, Y. (2001). Tomato yellow leaf curl virus (TYLCV) capsid protein (CP) subunit interactions: implications for viral assembly. Arch. Virol. 146, 1765–1773. doi: 10.1007/s007050170062
Hanley-Bowdoin, L., Settlage, S. B., Orozco, B. M., Nagar, S., and Robertson, D. (2000). Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation. Crit. Rev. Biochem. Mol. Biol. 35, 105–140. doi: 10.1016/s0735-2689(99)00383-4
Hanssen, I. M., Lapidot, M., and Thomma, B. P. (2010). Emerging viral diseases of tomato crops. Mol. Plant Microbe Interact. 23, 539–548. doi: 10.1094/MPMI-23-5-0539
Harkins, G. W., Delport, W., Duffy, S., Wood, N., Monjane, A. L., Owor, B. E., et al. (2009). Experimental evidence indicating that mastreviruses probably did not co-diverge with their hosts. Virol. J. 6:104. doi: 10.1186/1743-422X-6-104
Harkins, G. W., Martin, D. P., Christoffels, A., and Varsani, A. (2014). Towards inferring the global movement of beak and feather disease virus. Virology 450–451, 24–33. doi: 10.1016/j.virol.2013.11.033
Henn, M. R., Boutwell, C. L., Charlebois, P., Lennon, N. J., Power, K. A., Macalalad, A. R., et al. (2012). Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 8:e1002529. doi: 10.1371/journal.ppat.1002529
Hodgkinson, A., and Eyre-Walker, A. (2010). Human triallelic sites: evidence for a new mutational mechanism? Genetics 184, 233–241. doi: 10.1534/genetics.109.110510
Holmes, E. C., and Grenfell, B. T. (2009). Discovering the phylodynamics of RNA viruses. PLoS Comput. Biol. 5:e1000505. doi: 10.1371/journal.pcbi.1000505
Inoue-Nagata, A. K., Albuquerque, L. C., Rocha, W. B., and Nagata, T. (2004). A simple method for cloning the complete begomovirus genome using the bacteriophage phi29 DNA polymerase. J. Virol. Methods 116, 209–211. doi: 10.1016/j.jviromet.2003.11.015
Isnard, M., Granier, M., Frutos, R., Reynaud, B., and Peterschmitt, M. (1998). Quasispecies nature of three maize streak virus isolates obtained through different modes of selection from a population used to assess response to infection of maize cultivars. J. Gen. Virol. 79(Pt 12), 3091–3099.
Jupin, I., Hericourt, F., Benz, B., and Gronenborn, B. (1995). DNA replication specificity of TYLCV geminivirus is mediated by the amino-terminal 116 amino acids of the Rep protein. FEBS Lett. 362, 116–120.
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120.
Lalić, J., Cuevas, J. M., and Elena, S. F. (2011). Effect of host species on the distribution of mutational fitness effects for an RNA virus. PLoS Genet. 7:e1002378. doi: 10.1371/journal.pgen.1002378
López-Bueno, A., Villarreal, L. P., and Almendral, J. M. (2006). Parvovirus variation for disease: a difference with RNA viruses? Curr. Top. Microbiol. Immunol. 299, 349–370. doi: 10.1007/3-540-26397-7_13
Luna, A. P., Morilla, G., Voinnet, O., and Bejarano, E. R. (2012). Functional analysis of gene-silencing suppressors from tomato yellow leaf curl disease viruses. Mol. Plant Microbe Interact. 25, 1294–1306. doi: 10.1094/MPMI-04-12-0094-R
Mason, G., Caciagli, P., Accotto, G. P., and Noris, E. (2008). Real-time PCR for the quantitation of Tomato yellow leaf curl Sardinia virus in tomato plants and in Bemisia tabaci. J. Virol. Methods 147, 282–289. doi: 10.1016/j.jviromet.2007.09.015
Monci, F., García-Andrés, S., Maldonado, J. A., and Moriones, E. (2005). Resistance to monopartite begomoviruses associated with the bean leaf crumple disease in phaseolus vulgaris controlled by a single dominant gene. Phytopathology 95, 819–826. doi: 10.1094/PHYTO-95-0819
Monci, F., Sánchez-Campos, S., Navas-Castillo, J., and Moriones, E. (2002). A natural recombinant between the geminiviruses Tomato yellow leaf curl Sardinia virus and Tomato yellow leaf curl virus exhibits a novel pathogenic phenotype and is becoming prevalent in Spanish populations. Virology 303, 317–326. doi: 10.1006/viro.2002.1633
Morilla, G., Krenz, B., Jeske, H., Bejarano, E. R., and Wege, C. (2004). Tete a tete of tomato yellow leaf curl virus and tomato yellow leaf curl sardinia virus in single nuclei. J. Virol. 78, 10715–10723. doi: 10.1128/JVI.78.19.10715-10723.2004
Nájera, I., Holguín, A., Quiñones-Mateu, M. E., Muñoz-Fernandez, M. A., Nájera, R., López-Galíndez, C., et al. (1995). Pol gene quasispecies of human immunodeficiency virus: mutations associated with drug resistance in virus from patients undergoing no drug therapy. J. Virol. 69, 23–31.
Navas-Castillo, J., Fiallo-Olivé, E., and Sánchez-Campos, S. (2011). Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 49, 219–248. doi: 10.1146/annurev-phyto-072910-095235
Navas-Castillo, J., Sánchez-Campos, S., Díaz, J. A., Sáez-Alonso, E., and Moriones, E. (1997). First report of tomato yellow leaf curl virus-Is in Spain: coexistence of two different geminiviruses in the same epidemic outbreak. Plant Dis. 81, 1461–1461. doi: 10.1094/pdis.1997.81.12.1461b
Navas-Castillo, J., Sánchez-Campos, S., Díaz, J. A., Sáez-Alonso, E., and Moriones, E. (1999). Tomato yellow leaf curl virus-Is causes a novel disease of common bean and severe epidemics in tomato in Spain. Plant Dis. 83, 29–32.
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press.
Nelson, J. R., Cai, Y. C., Giesler, T. L., Farchaus, J. W., Sundaram, S. T., Ortiz-Rivera, M., et al. (2002). TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques (Suppl), 44–47.
Nishizawa, T., Okamoto, H., Tsuda, F., Aikawa, T., Sugai, Y., Konishi, K., et al. (1999). Quasispecies of TT virus (TTV) with sequence divergence in hypervariable regions of the capsid protein in chronic TTV infection. J. Virol. 73, 9604–9608.
Noris, E., Hidalgo, E., Accotto, G. P., and Moriones, E. (1994). High similarity among the tomato yellow leaf curl virus isolates from the west Mediterranean basin: the nucleotide sequence of an infectious clone from Spain. Arch. Virol. 135, 165–170.
Noris, E., Vaira, A. M., Caciagli, P., Masenga, V., Gronenborn, B., and Accotto, G. P. (1998). Amino acids in the capsid protein of tomato yellow leaf curl virus that are crucial for systemic infection, particle formation, and insect transmission. J. Virol. 72, 10050–10057.
Pamilo, P., and Bianchi, N. O. (1993). Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol. Biol. Evol. 10, 271–281.
Parrish, C. R., Holmes, E. C., Morens, D. M., Park, E. C., Burke, D. S., Calisher, C. H., et al. (2008). Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol. Mol. Biol. Rev. 72, 457–470. doi: 10.1128/MMBR.00004-08
Perales, C., Beach, N. M., Sheldon, J., and Domingo, E. (2014). Molecular basis of interferon resistance in hepatitis C virus. Curr. Opin. Virol. 8, 38–44. doi: 10.1016/j.coviro.2014.05.003
Perales, C., Iranzo, J., Manrubia, S. C., and Domingo, E. (2012). The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 20, 595–603. doi: 10.1016/j.tim.2012.08.010
Raja, P., Wolf, J. N., and Bisaro, D. M. (2010). RNA silencing directed against geminiviruses: post-transcriptional and epigenetic components. Biochim. Biophys. Acta 1799, 337–351. doi: 10.1016/j.bbagrm.2010.01.004
Rojas, M., and Gilbertson, R. (2008). “Emerging plant viruses: a diversity of mechanisms and opportunities,” in Plant Virus Evolution, ed M. Roossinck (Berlin;Heidelberg: Springer), 27–51.
Roossinck, M. J., and Schneider, W. L. (2006). Mutant clouds and occupation of sequence space in plant RNA viruses. Curr. Top. Microbiol. Immunol. 299, 337–348. doi: 10.1007/b137531
Sánchez-Campos, S., Díaz, J. A., Monci, F., Bejarano, E. R., Reina, J., Navas-Castillo, J., et al. (2002). High genetic stability of the begomovirus tomato yellow leaf curl Sardinia virus in southern Spain over an 8-year period. Phytopathology 92, 842–849. doi: 10.1094/PHYTO.2002.92.8.842
Sánchez-Campos, S., Navas-Castillo, J., Camero, R., Soria, C., Díaz, J. A., and Moriones, E. (1999). Displacement of tomato yellow leaf curl virus (TYLCV)-Sr by TYLCV-Is in tomato epidemics in Spain. Phytopathology 89, 1038–1043. doi: 10.1094/PHYTO.1999.89.11.1038
Sánchez-Campos, S., Navas-Castillo, J., Monci, F., Díaz, J. A., and Moriones, E. (2000). Mercurialis ambigua and Solanum luteum: two newly discovered natural hosts of tomato yellow leaf curl geminiviruses. Eur. J. Plant Pathol. 106, 391–394. doi: 10.1023/a:1008758622582
Sanz, A. I., Fraile, A., Gallego, J. M., Malpica, J. M., and García-Arenal, F. (1999). Genetic variability of natural populations of cotton leaf curl geminivirus, a single-stranded DNA virus. J. Mol. Evol. 49, 672–681.
Sarker, S., Patterson, E. I., Peters, A., Baker, G. B., Forwood, J. K., Ghorashi, S. A., et al. (2014). Mutability dynamics of an emergent single stranded DNA virus in a naive host. PLoS ONE 9:e85370. doi: 10.1371/journal.pone.0085370
Schneider, W. L., and Roossinck, M. J. (2000). Evolutionarily related Sindbis-like plant viruses maintain different levels of population diversity in a common host. J. Virol. 74, 3130–3134. doi: 10.1128/JVI.74.7.3130-3134.2000
Schneider, W. L., and Roossinck, M. J. (2001). Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 75, 6566–6571. doi: 10.1128/JVI.75.14.6566-6571.2001
Stainton, D., Martin, D. P., Muhire, B. M., Lolohea, S., Halafihi, M., Lepoint, P., et al. (2015). The global distribution of Banana bunchy top virus reveals little evidence for frequent recent, human-mediated long distance dispersal events. Virus Evol. 1:vev009. doi: 10.1093/ve/vev009
Stavrinides, J., and Guttman, D. S. (2004). Mosaic evolution of the severe acute respiratory syndrome coronavirus. J. Virol. 78, 76–82. doi: 10.1128/JVI.78.1.76-82.2004
Steinhauer, D. A., Domingo, E., and Holland, J. J. (1992). Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 122, 281–288.
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Tomás, D. M., Cañizares, M. C., Abad, J., Fernández-Muñoz, R., and Moriones, E. (2011). Resistance to Tomato yellow leaf curl virus accumulation in the tomato wild relative Solanum habrochaites associated with the C4 viral protein. Mol. Plant Microbe Interact. 24, 849–861. doi: 10.1094/MPMI-12-10-0291
Urbino, C., Thebaud, G., Granier, M., Blanc, S., and Peterschmitt, M. (2008). A novel cloning strategy for isolating, genotyping and phenotyping genetic variants of geminiviruses. Virol. J. 5:135. doi: 10.1186/1743-422X-5-135
van der Walt, E., Martin, D. P., Varsani, A., Polston, J. E., and Rybicki, E. P. (2008). Experimental observations of rapid Maize streak virus evolution reveal a strand-specific nucleotide substitution bias. Virol. J. 5:104. doi: 10.1186/1743-422X-5-104
Vanitharani, R., Chellappan, P., and Fauquet, C. M. (2005). Geminiviruses and RNA silencing. Trends Plant Sci. 10, 144–151. doi: 10.1016/j.tplants.2005.01.005
Verlaan, M. G., Hutton, S. F., Ibrahem, R. M., Kormelink, R., Visser, R. G., Scott, J. W., et al. (2013). The Tomato Yellow Leaf Curl Virus resistance genes Ty-1 and Ty-3 are allelic and code for DFDGD-class RNA-dependent RNA polymerases. PLoS Genet. 9:e1003399. doi: 10.1371/journal.pgen.1003399
Yaakov, N., Levy, Y., Belausov, E., Gaba, V., Lapidot, M., and Gafni, Y. (2011). Effect of a single amino acid substitution in the NLS domain of Tomato yellow leaf curl virus-Israel (TYLCV-IL) capsid protein (CP) on its activity and on the virus life cycle. Virus Res. 158, 8–11. doi: 10.1016/j.virusres.2011.02.016
Keywords: geminivirus, begomovirus, tomato yellow leaf curl virus, ssDNA virus quasispecies, viral fitness, begomovirus mutant spectra, host adaptation, emergent viruses
Citation: Sánchez-Campos S, Domínguez-Huerta G, Díaz-Martínez L, Tomás DM, Navas-Castillo J, Moriones E and Grande-Pérez A (2018) Differential Shape of Geminivirus Mutant Spectra Across Cultivated and Wild Hosts With Invariant Viral Consensus Sequences. Front. Plant Sci. 9:932. doi: 10.3389/fpls.2018.00932
Received: 07 February 2018; Accepted: 11 June 2018;
Published: 02 July 2018.
Edited by:
Ricardo Flores, Instituto de Biología Molecular y Celular de Plantas (IBMCP), SpainReviewed by:
Yong Liu, Hunan Academy of Agricultural Sciences (CAAS), ChinaJose Trinidad Ascencio-Ibáñez, North Carolina State University, United States
Copyright © 2018 Sánchez-Campos, Domínguez-Huerta, Díaz-Martínez, Tomás, Navas-Castillo, Moriones and Grande-Pérez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Grande-Pérez, YWdyYW5kZUB1bWEuZXM=
†These authors have contributed equally to this work.