 Alexander A. Myburg
Alexander A. Myburg Steven G. Hussey
Steven G. Hussey Jack P. Wang
Jack P. Wang Nathaniel R. Street
Nathaniel R. Street Eshchar Mizrachi
Eshchar Mizrachi- 1Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Hatfield, South Africa
- 2Forest Biotechnology Group, Department of Forestry and Environmental Resources, North Carolina State University, Raleigh, NC, United States
- 3Umeå Plant Science Center, Department of Plant Physiology, Umeå University, Umeå, Sweden
Fast-growing forest plantations are sustainable feedstocks of plant biomass that can serve as alternatives to fossil carbon resources for materials, chemicals, and energy. Their ability to efficiently harvest light energy and carbon from the atmosphere and sequester this into metabolic precursors for lignocellulosic biopolymers and a wide range of plant specialized metabolites make them excellent biochemical production platforms and living biorefineries. Their large sizes have facilitated multi-omics analyses and systems modeling of key biological processes such as lignin biosynthesis in trees. High-throughput ‘omics’ approaches have also been applied in segregating tree populations where genetic variation creates abundant genetic perturbations of system components allowing construction of systems genetics models linking genes and pathways to complex trait variation. With this information in hand, it is now possible to start using synthetic biology and genome editing techniques in a bioengineering approach based on a deeper understanding and rational design of biological parts, devices, and integrated systems. However, the complexity of the biology and interacting components will require investment in big data informatics, machine learning, and intuitive visualization to fully explore multi-dimensional patterns and identify emergent properties of biological systems. Predictive systems models could be tested rapidly through high-throughput synthetic biology approaches and multigene editing. Such a bioengineering paradigm, together with accelerated genomic breeding, will be crucial for the development of a new generation of woody biorefinery crops.
Introduction
Compared to herbaceous plants, forest trees afford numerous of advantages to plant biologists interested in studying growth and development. Most obviously, trees produce vast quantities of wood comprising multiple cells types produced from the meristematic cambial initials of the vascular cambium. In the context of developmental studies of secondary growth, greater size represents greater spatial resolution to profile stages of development during the formation of secondary phloem and xylem as recently demonstrated for aspen (Obudulu et al., 2016; Sundell et al., 2017) and Norway spruce (Jokipii-Lukkari et al., 2017). Such studies have been instrumental in identifying the individual genes, proteins, metabolites, and pathways comprising the molecular components of biological processes such as wood formation. Many wood formation genes have been targeted using transgenic approaches to demonstrate the ability to modify wood properties (Chang et al., 2018). Unfortunately, compared to greenhouse results, some of these single gene modifications have had adverse effects on growth or altered transgene effect when tested in field trials (Leplé et al., 2007; Voelker et al., 2010). Such outcomes suggest a need for greater understanding of the interaction of systems components with intrinsic and extrinsic factors that differ between the greenhouse and field (Beckers et al., 2016).
In the context of industrial application, woody biomass is a renewable, carbon-neutral source of lignocellulosic materials for construction, pulp, bioenergy and, increasingly, for advanced biomaterials such as nanocellulose (Kunaver et al., 2016; Thomas et al., 2018). These and other biorefinery products, including novel biopolymers and biochemicals, are key to the emerging plant-based bio (Vanholme et al., 2013a; Van de Wouwer et al., 2018). Most woody biomass traits of commercial value are genetically complex. As such, it is challenging to devise strategies determining which genes contribute to these complex phenotypes and to associate these with causal mechanisms. In contrast to genomic selection that can be used as a “black box” route to improve complex traits, bioengineering approaches require systems-level understanding to elucidate the molecular basis of complex traits. “Multi-omics” analyses, together with extensive transgenic perturbation of a biological pathway and mathematical modeling of pathway dynamics, have recently been successfully applied to generate the most detailed systems biology model yet, of lignin biosynthesis, in a forest tree (Wang et al., 2018). The large amount of genetic diversity retained in tree breeding programs has also facilitated the use of natural variation to dissect complex traits (Mizrachi and Myburg, 2016; Mizrachi et al., 2017). Such systems genetics approaches pose opportunities for rapid advances in genomic breeding and provide diverse genetic backgrounds for genetic engineering of biomass traits.
Synthetic biology (“SynBio”) aims to reduce biologically complex systems into discrete functional components that can be combined in numerous ways to create efficient and novel biological products or properties (for a synopsis of SynBio development, see Cameron et al., 2014). This transdisciplinary field draws on engineering principles and standardization of the fabrication process (i.e., gene circuit assembly) (Heinemann and Panke, 2006). Not only can synthetic biology incorporate existing genetic components (biological “parts”) across the three Domains of life, but chemical gene and whole-genome synthesis, directed evolution, model-informed protein engineering, and synthetic regulatory machinery design enable the creation of xenobiological systems. The long-lived nature of trees and their vast reserves of carbon-rich sink tissues present both a challenge and opportunity for accumulating and storing synthetically produced compounds (Wilkerson et al., 2014; Mottiar et al., 2016). Synthetically modified trees could serve as renewable factories for the production of large quantities of custom-designed biomass and biochemicals that do not exist naturally in trees.
We present a brief review of recent progress and our perspective on the application of systems biology approaches to understand complex tree biology, from molecular to organismal level crucial for sustainable production of woody biomass. We propose a bioengineering paradigm based on rational design, drawing on systems-level understanding of biological processes – and the application of synthetic biology and genome editing technologies – to engineer such processes in combination with accelerated, genome-assisted breeding of forest trees.
Systems Biology: Integrative Understanding of Biological Processes in Forest Trees
Systems modeling of biological processes requires extensive perturbation of systems components to produce the comprehensive experimental data required for modeling (Figure 1). The lack of extensive mutant collections in forest trees has necessitated the use of transgenic approaches to perturb genes. This has limited such studies to a relatively small number of genes, typically in the same biological pathway. Lignin biosynthesis has emerged as one of the most well-studied biological processes in forest trees, largely due to the interest in reducing lignin content or altering lignin composition to reduce the recalcitrance of woody biomass during industrial processing (Pilate et al., 2002; Leplé et al., 2007; Voelker et al., 2010; Mansfield et al., 2012; Bryan et al., 2016; Saleme et al., 2017). The first comprehensive, systems-level analysis of the lignin biosynthetic pathway was performed in Arabidopsis (Vanholme et al., 2012). This resulted in the discovery of additional components of the biological process, in particular, a novel component in the form of a caffeoyl shikimate esterase (CSE) that hydrolyzes caffeoyl shikimate into caffeate and, when mutated, leads to a fourfold increase in glucose release from cellulose during saccharification without pretreatment in Arabidopsis (Vanholme et al., 2013b). In poplar downregulated for CSE, 60% more glucose was released from wood without pretreatment, as a consequence of the lower lignin amount and the higher cellulose content (Saleme et al., 2017).
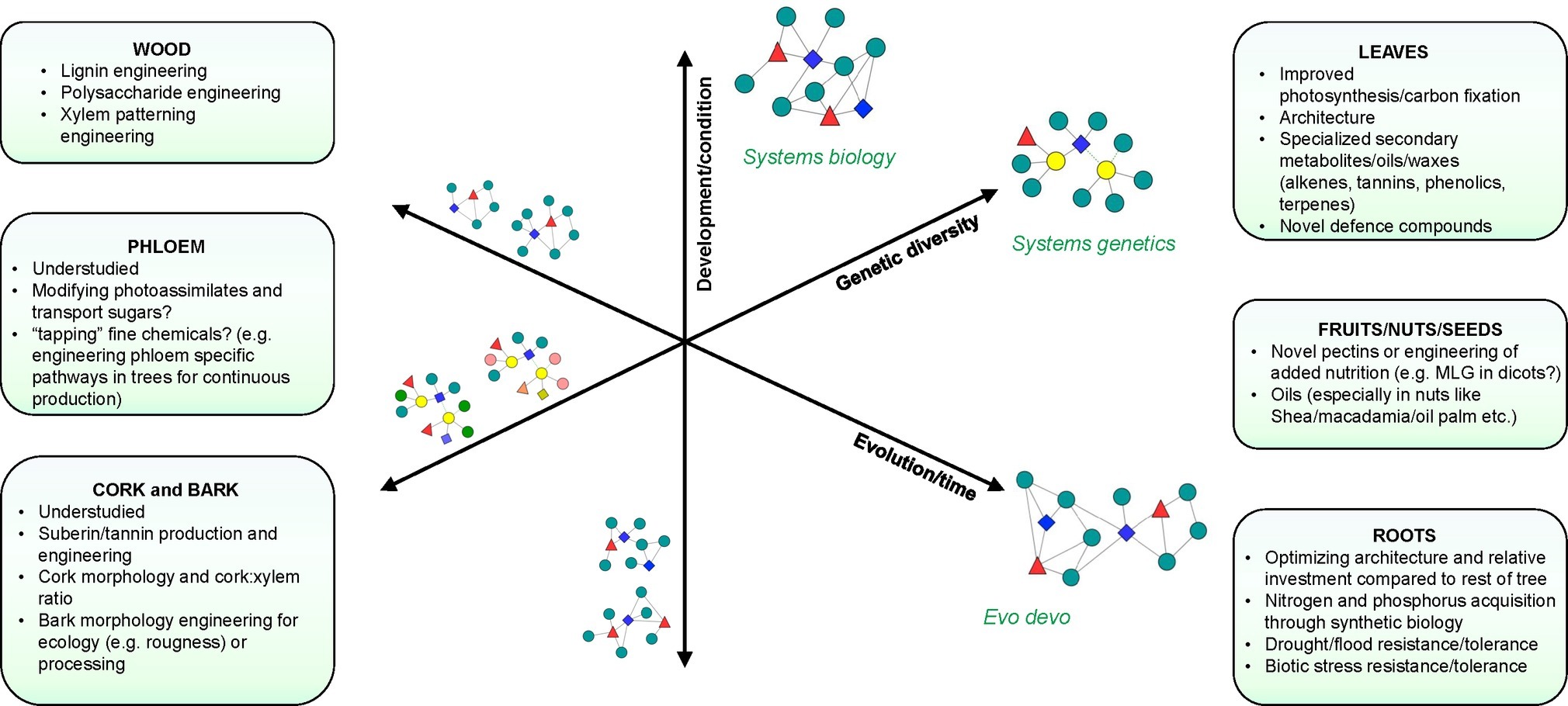
Figure 1. Systems approaches to study biological processes in trees. Axes represent different dimensions or types of perturbation that can be used to dissect biological processes. Network components (colored nodes and shapes) represent the study of different biological layers (DNA, RNA, protein, metabolite, etc.) using integrative approaches that take into account development/condition (Systems biology), genetic diversity (Systems genetics), and evolution (Evo-devo). Yellow nodes in “systems genetics” indicate the connection to complex traits that can be established in population-wide studies. Networks on the left represent intermediate integration steps of components that vary along these axes. Networks on the right represent integrated networks that can be constructed from each type of perturbation. Boxes show target traits and processes associated with different tissues or organs where systems approaches may provide new opportunities for research.
The most extensive analysis of systems biology in forest trees has been done on the lignin biosynthetic pathway in differentiating xylem of Populus trichocarpa (Wang et al., 2018). Transgenic trees systematically perturbed in the expression of lignin genes were produced, and quantitative data on genomics, transcriptomics, fluxomics, biochemical, chemical, and cellular analyses were integrated to construct a mathematical description of the pathway. The lignin model calculates how changing expression of any pathway gene or gene combination affects protein abundance, metabolic-flux, and phenotypic traits, including lignin content and composition, tree growth, wood density, and saccharification, for sugar release. The model predicts improvements in any of these traits individually or in combinations, through modifying the expression of specific lignin genes. The lignin pathway is amenable to modeling because major pathway components have been identified and methodologies for their quantification have been developed (Shi et al., 2010; Vanholme et al., 2010, 2019; Wang et al., 2014, 2018). Genomic sequence information (Goujon et al., 2003; Raes et al., 2003; Tuskan et al., 2006; Myburg et al., 2014; Carocha et al., 2015) defined the gene families of the known proteins that are components of the lignin biosynthetic pathway. Some enzymes are encoded by single genes and others are members of large gene families (Goujon et al., 2003; Shi et al., 2010), while other members of the families are not associated with lignification in time or place. This highlights the importance of identifying those family members that are bona fide components of the systems model of wood lignification. The abundance of lignin enzymes in vivo has been measured in P. trichocarpa to determine the quantitative relationship between transcripts and proteins, and the enzymes in the pathway have been purified and characterized biochemically and genetically so reliable kinetics data could be obtained for substrates and inhibitors (Wang et al., 2014).
The systems analysis of lignin biosynthesis in P. trichocarpa has provided valuable information on the formation and function of lignin in forest trees, such as where genetic regulation is significantly different from non-woody species (e.g., Arabidopsis). For example, in the case of 4-coumarate CoA ligase (4CL), Arabidopsis has a single gene and enzyme for 4CL, while P. trichocarpa has two distinct 4CL genes, one encoding a protein with a regulatory function (Chen et al., 2014). Similar regulatory functions have been demonstrated for genes upstream of lignin biosynthesis in the shikimate pathway (Xie et al., 2018) supporting the 4CL finding. In silico simulations furthermore predicted the consequence on flux and wood traits for every possible combination of multigene perturbations where expression of lignin genes was either upregulated, downregulated, or remained at the wild-type level. Systems modeling therefore provides a unique opportunity to guide multi-trait engineering and breeding strategies to create novel tree feedstocks optimized for fast growth and conversion to energy and materials. The models also inform identification of alleles or combinations of alleles to breed superior forest tree varieties (Wang et al., 2018).
Systems Genetics: Dissecting Systems Components and Interactions Underlying Complex Traits
While systems biology studies have advanced our understanding of developmental and stress response pathways (especially in identifying key regulators within these processes), they frequently are based on severe phenotypes that represent an aggregate of a cascade of cellular and physiological reactions to the adverse effect of the perturbation. Furthermore, such studies do not resolve the molecular basis or mechanisms of trait variation among individuals within populations. Revealing how complex trait variation arises from genetic polymorphisms affords opportunities to discover novel approaches to inducing trait variation in addition to advancing understanding of the evolutionary processes and the mechanistic basis of variation in complex traits (Figure 1).
Complex traits are the emergent outcome of actions and interactions of hundreds to thousands of genes, functioning within pathways in combination with environmental effects. Variation at any biological layer from genome to trait can contribute to the final outcome. Full understanding of complex phenotypes requires approaches that can model these individual actions and interactions at the population level. Systems genetics provides a clearer path linking the genome to complex phenotypes by deducing the mechanisms by which genetic variation impacts variation in these intermediate biological layers (e.g., transcript, protein, and/or metabolite) to ultimately impact the complex trait (Civelek and Lusis, 2014; Feltus, 2014). Systems genetics has benefited substantially from recent sequencing technology improvements that have enabled population-wide genome resequencing and global transcript profiling via mRNA and small RNA sequencing. While SNP genotyping typically provides adequate genetic resolution for controlled crosses (QTL analysis using pedigrees), genome-wide association studies (GWAS) using natural populations often mandate whole-genome re-sequencing due to the rapid decay of LD in many tree species (McKown et al., 2014; Zhang et al., 2018). There have now been a number of studies in trees mapping the genetic architecture of gene expression variation (eQTLs) in tree pedigrees (Kirst et al., 2004, 2005; Drost et al., 2010) and natural populations (Porth et al., 2013; Mähler et al., 2017; Zhang et al., 2018); however, there is only one reported true systems genetics study (Mizrachi et al., 2017) in addition to genetical genomics approaches (Street et al., 2006; Drost et al., 2015).
These studies have reported contrasting results between pedigrees and natural populations, suggesting that the genetic background is an important consideration, especially for extrapolating findings to a wider context. Due to developments in sequencing technologies, there has been greater focus on eQTL studies; however, variation in protein (allelic) structural variation, epigenomic variation (Cheung et al., 2017), protein abundance (Consoli et al., 2002), and post translational modifications (Cesnik et al., 2016) or metabolome variation (Morreel et al., 2006; Joseph et al., 2014; Matsuda et al., 2015) fit equally well and indeed would be as responsible for variation, within the systems genetics framework. While systems genetics studies can reveal, for example, genomic loci linked to gene expression variation contributing to trait variation, it can be problematic to place these within a biological framework to understand how these novel mechanisms influence the trait. One approach to overcoming this barrier can be integration of systems biology studies, such as developmental studies, to provide context to these novel genes. Combining systems genetics and systems biology co-expression networks is also a powerful approach as biological inference can be aided by considering the neighborhood of genes connected to a novel candidate (Mähler et al., 2017).
To date, one level of complexity that has not been considered in eQTL and systems genetics studies is transcriptional plasticity. There is now extensive knowledge of alternative splicing and transcript usage, which can vary among tissues, during development or among genotypes (Bao et al., 2013; Xu et al., 2014; Zhao et al., 2014). Mapping expression at the transcript level will provide greater resolution to the link between genome and phenotype. However, fine-scale expression analysis at the transcript level remains challenging, mainly because most tissue samples contain a complex mix of transcripts and splicing variants from different cell types or cells at different stages of development. There is equally a need for improvement in network inference methods, with an understanding that no single current method is adequate to capture the range of interactions present in biological networks (Marbach et al., 2012) and with few tools available to facilitate aggregate inference approaches (Schiffthaler et al., 2018). Similarly, no studies have yet integrated genome-wide assays of DNA modifications or accessibility despite increasing evidence of the additional insight such information brings. There is also a paucity of large-scale transcription factor binding or protein-protein interaction data for plants in general, which further limits comprehensive understanding.
Synthetic Biology: A New Bioengineering Paradigm for Forest Trees
SynBio has made its greatest advancements in prokaryotes and single-celled eukaryotes such as yeast, but plant synthetic biology is catching up (Patron et al., 2015; Schaumberg et al., 2016; de Lange et al., 2018; Hanson and Jez, 2018; Pouvreau et al., 2018). In addition to a large number of modifications made by conventional transgenic approaches (Chang et al., 2018), there have been some notable successes in the synthetic modification of trees using single-gene strategies such as the introduction of chemically labile ester linkages into the lignin backbone of poplar trees through the xylem-specific expression of an exogenous feruloyl-coenzyme A monolignol transferase (FMT) from Angelica sinensis (Wilkerson et al., 2014). Future strategies will attempt to evaluate far more complex designs, relying to a large extent on the ability to assemble DNA fragments idempotently (that is, the flexibility to assemble basic parts with increasing complexity using a universal method and without having to re-modify each intermediate). There is currently a scarcity of freely available standardized biological parts suitable for plant biology akin to the International Genetically Engineered Machines (iGEM) BioBrick parts collection1. Encouragingly, the Phytobrick synthetic biology standard with a universal lexicon for plant gene elements coupled to powerful Type IIS idempotent assembly methods (Engler et al., 2014; Patron et al., 2015) is fast gaining traction, with an increasing number of Phytobricks now included in the iGEM Standard Registry2. Tree biologists and biotechnologists should adapt to this conceptual framework as soon as possible to keep up with synthetic biology development in annual crops. We recently developed an open access synthetic panel of 221 secondary cell wall-related Eucalyptus grandis transcription factors and 65 promoter sequences in partnership with the Department of Energy Joint Genome Institute, most of which were designed as Phytobricks (Hussey et al., in preparation). Accessibility of high-throughput DNA synthesis services will ensure that a growing number of standardized parts become available under open material transfer agreements.
One considerable challenge to tree synthetic biology is precise spatiotemporal control of complex multigene constructs, especially in woody tissues where inducing gene expression with external agents is impractical. Such constructs must function optimally in a tissue of interest, be resistant to eukaryotic silencing mechanisms such as RNAi or epigenetic silencing, be somatically stable such that somatic mutations that disrupt a synthetic construct should not be selectively favored, take into account compartmentalized plant cell biology, and have built-in biosafety mechanisms preventing transgene escape. Furthermore, synthetic gene circuits should consist of composable parts (de Lange et al., 2018) that individually encode defined and transferrable functions (a property known as modularity) and that function independently of endogenous processes to avoid unwanted interference, a property known as orthogonality. Designer transcription factors based on zinc finger, TALE, and dCas9 technologies targeting endogenous or synthetic promoters (Liu and Stewart, 2016) are ideal orthogonal synthetic tools that allow considerable transgene regulation flexibility but may require extensive testing and optimization.
Currently, thousands of iterations of multigene constructs can be produced by robotics-assisted DNA Foundry services. However, it is not feasible to transform and phenotype thousands of transgenic trees for iterative design-build-test-learn cycles envisioned for plant synthetic biology (Pouvreau et al., 2018). Early synthetic designs will therefore have to be tested in a simplified system (or “chassis” in synthetic biology terminology) until optimized constructs can be evaluated in target tree species. This will necessitate the development of experimental chassis derived from the target tree species that are easier to transform and phenotype en masse, such as protoplasts, cell cultures, or agroinfiltrated leaves, or “lab rat” genotypes that perform well in tissue culture and have high transformation rates (Figure 2). In vitro tracheary element transdifferentiation approaches relying on hormonal induction (Fukuda and Komamine, 1980; Kubo et al., 2005; Saito et al., 2017), or the VND7 inducible system (Yamaguchi et al., 2010; Goué et al., 2013), for example, could be used to induce secondary cell wall formation in suspension culture cells or explant tissues of a target tree species and thus evaluate the phenotypes of many cell wall-modifying constructs before semi-optimized constructs are tested in a multicellular tree model. Successful constructs may either be directly introduced into an elite genotype of the target species or rapidly crossed into elite breeding material after co-transformation of parental genotypes with an early flowering construct (Figure 2).
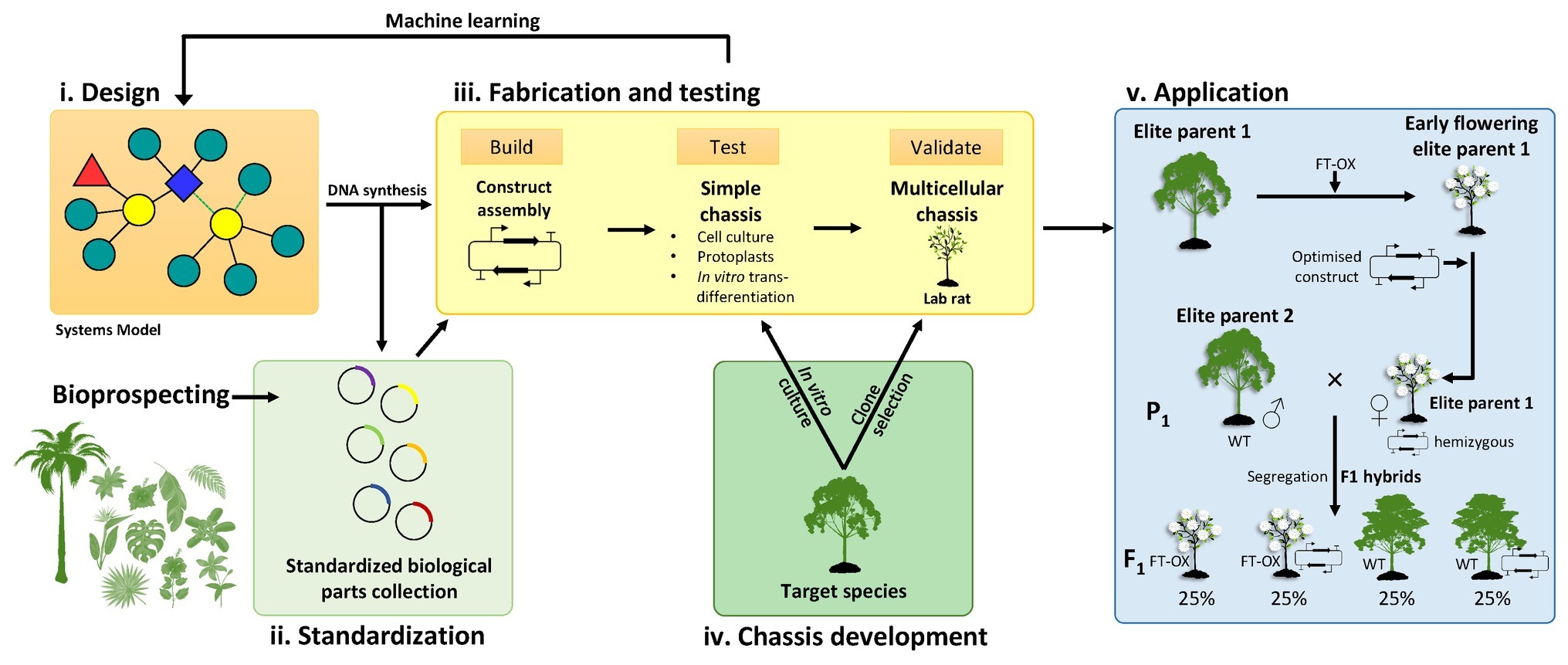
Figure 2. A proposed bioengineering paradigm based on forest tree synthetic biology. (i) Systems biology models inform the initial design of synthetic multigene constructs. Individual components (DNA parts) are sourced from existing or novel genetic resources (e.g., bioprospecting), synthesized, and submitted to biological parts collections as standardized Phytobricks (ii). The fabrication and testing phase (iii) involves high-throughput idempotent construct assembly followed by transformation and testing in a simplified chassis such as a cell culture derived from the target tree species (iv). Construct expression and phenotypic data are then integrated into a machine learning model to optimize the construct design, an iterative process that produces a reduced number of semi-optimized constructs for validation in a genotype of the target species selected to perform favorably in laboratory conditions and conducive to transformation (“lab rat”). Such validation may include greenhouse trials involving juvenile trees or mature trees in field trials. (v) Successful constructs may be introduced into a preferred elite parental genotype for intraspecific or interspecific hybrid breeding. A possible avenue to rapidly mobilize synthetic gene constructs into more diverse genetic backgrounds would be to introduce an early flowering construct (such as overexpression of the FLOWERING LOCUS T gene; FT-OX) into a number of elite parental genotypes. Such genotypes can then be transformed with the optimized synthetic gene constructs and used as female parents in crosses with non-transgenic (wild-type, WT) parents to produce F1 progeny segregating for both constructs. If the two constructs are on different chromosomes, approximately 25% of the progeny should be WT for growth and flowering but contain the synthetic gene construct. Early flowering parental genotypes can be propagated in vitro to be transformed with various synthetic constructs and, if unrelated, different parental genotypes could be crossed for transgene stacking.
Future Perspective
Most fundamental discoveries, including proof-of-concept cell wall and growth modifications (and even extrapolations on biomass processing efficiency), are still derived from the analysis of Arabidopsis inflorescence stems, which remains a poor representation of large tree stems comprised mainly of wood. Of priority in the short term is testing genetic perturbations as much as possible in a woody model such as Populus and, if possible, directly in target species of interest. Several priorities must be met here, such as enhancing the transformation efficiency of commercial species or genotypes, capacity for large-scale transformation experiments, as well as (crucially) field trials confirming greenhouse phenotypes in mature trees. Large consortia and industry collaborations, as well as engagement and an improvement in the regulatory landscape, must be met for this to be truly realized.
Also in the short to medium term, the convergence of high-resolution technologies that capture genomics, epigenomics, and other cell ‘omics’, phenomics, and environment (including microbiome) data, as well as computational modeling of the interactions of these, requires transdisciplinary innovations and probably the application of artificial intelligence methodology. Combined with genome editing (with broader scale synthetic biology applications), this makes the field of forest biotechnology ripe for a new wave of creativity, especially in thinking of the tree itself as a living biorefinery and as a stable and continuous producer of specialized high-value compounds or polymers in sustainably harvestable tissues and organs such as leaves, secondary phloem, and bark. Higher resolution knowledge of metabolite precursors, tissue-specific pathway engineering, and knowledge of novel high-value derivatives that can be discovered using bioprospecting methods and produced in trees has the potential for a new generation of relatively low volume, but high value, forest products.
How far does the application of these technologies go? Given the long rotation times of forest trees as harvestable biomass crops, it is unlikely (and indeed not essential) that we will see movement toward a “bottom up” approach that builds on a synthetic minimal tree genome. It is much more important to optimize the precise introduction of complex regulatory circuits and metabolic pathways that remain stable through breeding generations, a nascent field of research in itself. Such a bioengineering paradigm, combined with advanced genomic breeding approaches and accelerated flowering technologies, may empower rapid development of woody biomass crops tailored for diverse biorefinery, biomaterials, and timber construction products. In many forest-growing countries, an advanced forest products industry will be one of the cornerstones of the bioeconomy and key to achieving global sustainable development goals.
Author Contributions
All authors contributed to the drafting and editing of the manuscript.
Funding
This study was supported by National Research Foundation of South Africa (Research Funding, NRF Bioinformatics and Functional Genomics Programme grant UID 97911), Department of Science and Technology of South Africa, (Research Funding) Technology Innovation Agency (TIA) of South Africa (Research Support), Sappi and Mondi (Forestry Industry Partners and Research Support), and University of Pretoria (Research Facilities and Support). NRS is supported by the Trees for the Future (T4F) project (Sweden).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors NS.
Footnotes
References
Bao, H., Li, E., Mansfield, S. D., Cronk, Q. C., El-Kassaby, Y. A., and Douglas, C. J. (2013). The developing xylem transcriptome and genome-wide analysis of alternative splicing in Populus trichocarpa (black cottonwood) populations. BMC Genomics 14:359. doi: 10.1186/1471-2164-14-359
Beckers, B., Op De Beeck, M., Weyens, N., Van Acker, R., Van Montagu, M., Boerjan, W., et al. (2016). Lignin engineering in field-grown poplar trees affects the endosphere bacterial microbiome. Proc. Natl. Acad. Sci. USA 113, 2312–2317. doi: 10.1073/pnas.1523264113
Bryan, A. C., Jawdy, S., Gunter, L., Gjersing, E., Sykes, R., Hinchee, M. A., et al. (2016). Knockdown of a laccase in Populus deltoides confers altered cell wall chemistry and increased sugar release. Plant Biotechnol. J. 14, 2010–2020. doi: 10.1111/pbi.12560
Cameron, D. E., Bashor, C. J., and Collins, J. J. (2014). A brief history of synthetic biology. Nat. Rev. Microbiol. 12, 381–390. doi: 10.1038/nrmicro3239
Carocha, V., Soler, M., Hefer, C., Cassan-Wang, H., Fevereiro, P., Myburg, A. A., et al. (2015). Genome-wide analysis of the lignin toolbox of Eucalyptus grandis. New Phytol. 206, 1297–1313. doi: 10.1111/nph.13313
Cesnik, A. J., Shortreed, M. R., Sheynkman, G. M., Frey, B. L., and Smith, L. M. (2016). Human proteomic variation revealed by combining RNA-Seq proteogenomics and global post-translational modification (G-PTM) search strategy. J. Proteome Res. 15, 800–808. doi: 10.1021/acs.jproteome.5b00817
Chang, S., Mahon, E. L., Mackay, H. A., Rottmann, W. H., Strauss, S. H., Pijut, P. M., et al. (2018). Genetic engineering of trees: progress and new horizons. In Vitro Cell Dev. Biol. Plant 54, 341–376. doi: 10.1007/s11627-018-9914-1
Chen, H. C., Song, J., Wang, J. P., Lin, Y. C., Ducoste, J., Shuford, C. M., et al. (2014). Systems biology of lignin biosynthesis in Populus trichocarpa: heteromeric 4-coumaric acid:coenzyme A ligase protein complex formation, regulation, and numerical modeling. Plant Cell 26, 876–893. doi: 10.1105/tpc.113.119685
Cheung, W. A., Shao, X., Morin, A., Siroux, V., Kwan, T., Ge, B., et al. (2017). Functional variation in allelic methylomes underscores a strong genetic contribution and reveals novel epigenetic alterations in the human epigenome. Genome Biol. 18:50. doi: 10.1186/s13059-017-1173-7
Civelek, M., and Lusis, A. J. (2014). Systems genetics approaches to understand complex traits. Nat. Rev. Genet. 15, 34–48. doi: 10.1038/nrg3575
Consoli, L., Lefevre, A., Zivy, M., De Vienne, D., and Damerval, C. (2002). QTL analysis of proteome and transcriptome variations for dissecting the genetic architecture of complex traits in maize. Plant Mol. Biol. 48, 575–581. doi: 10.1023/A:1014840810203
de Lange, O., Klavins, E., and Nemhauser, J. (2018). Synthetic genetic circuits in crop plants. Curr. Opin. Biotechnol. 49, 16–22. doi: 10.1016/j.copbio.2017.07.003
Drost, D. R., Benedict, C. I., Berg, A., Novaes, E., Novaes, C. R. D. B., Yu, Q., et al. (2010). Diversification in the genetic architecture of gene expression and transcriptional networks in organ differentiation of Populus. Proc. Natl. Acad. Sci. USA 107, 8492–8497. doi: 10.1073/pnas.0914709107
Drost, D. R., Puranik, S., Novaes, E., Novaes, C. R., Dervinis, C., Gailing, O., et al. (2015). Genetical genomics of Populus leaf shape variation. BMC Plant Biol. 15:166. doi: 10.1186/s12870-015-0557-7
Engler, C., Youles, M., Gruetzner, R., Ehnert, T. M., Werner, S., Jones, J. D., et al. (2014). A golden gate modular cloning toolbox for plants. ACS Synth. Biol. 3, 839–843. doi: 10.1021/sb4001504
Feltus, F. A. (2014). Systems genetics: a paradigm to improve discovery of candidate genes and mechanisms underlying complex traits. Plant Sci. 223, 45–48. doi: 10.1016/j.plantsci.2014.03.003
Fukuda, H., and Komamine, A. (1980). Establishment of an experimental system for the tracheary element differentiation from single cells isolated from the mesophyll of Zinnia elegans. Plant Physiol. 65, 57–60. doi: 10.1104/pp.65.1.57
Goué, N., Mortimer, J. C., Nakano, Y., Zhang, Z., Josserand, M., Ohtani, M., et al. (2013). Secondary cell wall characterization in a BY-2 inductive system. Plant Cell Tissue Organ Cult. 115, 223–232. doi: 10.1007/s11240-013-0354-7
Goujon, T., Sibout, R., Eudes, A., Mackay, J., and Joulanin, L. (2003). Genes involved in the biosynthesis of lignin precursors in Arabidopsis thaliana. Plant Physiol. Biochem. 41, 677–687. doi: 10.1016/S0981-9428(03)00095-0
Hanson, A. D., and Jez, J. M. (2018). Synthetic biology meets plant metabolism. Plant Sci. 273, 1–2. doi: 10.1016/j.plantsci.2018.04.004
Heinemann, M., and Panke, S. (2006). Synthetic biology--putting engineering into biology. Bioinformatics 22, 2790–2799. doi: 10.1093/bioinformatics/btl469
Jokipii-Lukkari, S., Sundell, D., Nilsson, O., Hvidsten, T. R., Street, N. R., and Tuominen, H. (2017). NorWood: a gene expression resource for evo-devo studies of conifer wood development. New Phytol. 216, 482–494. doi: 10.1111/nph.14458
Joseph, B., Atwell, S., Corwin, J. A., Li, B., and Kliebenstein, D. J. (2014). Meta-analysis of metabolome QTLs in Arabidopsis: trying to estimate the network size controlling genetic variation of the metabolome. Front. Plant Sci. 5:461. doi: 10.3389/fpls.2014.00461
Kirst, M., Basten, C. J., Myburg, A. A., Zeng, Z. B., and Sederoff, R. R. (2005). Genetic architecture of transcript-level variation in differentiating xylem of a Eucalyptus hybrid. Genetics 169, 2295–2303. doi: 10.1534/genetics.104.039198
Kirst, M., Myburg, A. A., De Leon, J. P., Kirst, M. E., Scott, J., and Sederoff, R. (2004). Coordinated genetic regulation of growth and lignin revealed by quantitative trait locus analysis of cDNA microarray data in an interspecific backcross of Eucalyptus. Plant Physiol. 135, 2368–2378. doi: 10.1104/pp.103.037960
Kubo, M., Udagawa, M., Nishikubo, N., Horiguchi, G., Yamaguchi, M., Ito, J., et al. (2005). Transcription switches for protoxylem and metaxylem vessel formation. Genes Dev. 19, 1855–1860. doi: 10.1101/gad.1331305
Kunaver, M., Anzlovar, A., and Zagar, E. (2016). The fast and effective isolation of nanocellulose from selected cellulosic feedstocks. Carbohydr. Polym. 148, 251–258. doi: 10.1016/j.carbpol.2016.04.076
Leplé, J. C., Dauwe, R., Morreel, K., Storme, V., Lapierre, C., Pollet, B., et al. (2007). Downregulation of cinnamoyl-coenzyme A reductase in poplar: multiple-level phenotyping reveals effects on cell wall polymer metabolism and structure. Plant Cell 19, 3669–3691. doi: 10.1105/tpc.107.054148
Liu, W., and Stewart, C. N. Jr. (2016). Plant synthetic promoters and transcription factors. Curr. Opin. Biotechnol. 37, 36–44. doi: 10.1016/j.copbio.2015.10.001
Mähler, N., Wang, J., Terebieniec, B. K., Ingvarsson, P. K., Street, N. R., and Hvidsten, T. R. (2017). Gene co-expression network connectivity is an important determinant of selective constraint. PLoS Genet. 13:e1006402. doi: 10.1371/journal.pgen.1006402
Mansfield, S. D., Kang, K. Y., and Chapple, C. (2012). Designed for deconstruction–poplar trees altered in cell wall lignification improve the efficacy of bioethanol production. New Phytol. 194, 91–101. doi: 10.1111/j.1469-8137.2011.04031.x
Marbach, D., Costello, J. C., Küffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., et al. (2012). Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804. doi: 10.1038/nmeth.2016
Matsuda, F., Nakabayashi, R., Yang, Z., Okazaki, Y., Yonemaru, J., Ebana, K., et al. (2015). Metabolome-genome-wide association study dissects genetic architecture for generating natural variation in rice secondary metabolism. Plant J. 81, 13–23. doi: 10.1111/tpj.12681
Mckown, A. D., Klapste, J., Guy, R. D., Geraldes, A., Porth, I., Hannemann, J., et al. (2014). Genome-wide association implicates numerous genes underlying ecological trait variation in natural populations of Populus trichocarpa. New Phytol. 203, 535–553. doi: 10.1111/nph.12815
Mizrachi, E., and Myburg, A. A. (2016). Systems genetics of wood formation. Curr. Opin. Plant Biol. 30, 94–100. doi: 10.1016/j.pbi.2016.02.007
Mizrachi, E., Verbeke, L., Christie, N., Fierro, A. C., Mansfield, S. D., Davis, M. F., et al. (2017). Network-based integration of systems genetics data reveals pathways associated with lignocellulosic biomass accumulation and processing. Proc. Natl. Acad. Sci. USA 114, 1195–1200. doi: 10.1073/pnas.1620119114
Morreel, K., Goeminne, G., Storme, V., Sterck, L., Ralph, J., Coppieters, W., et al. (2006). Genetical metabolomics of flavonoid biosynthesis in Populus: a case study. Plant J. 47, 224–237. doi: 10.1111/j.1365-313X.2006.02786.x
Mottiar, Y., Vanholme, R., Boerjan, W., Ralph, J., and Mansfield, S. D. (2016). Designer lignins: harnessing the plasticity of lignification. Curr. Opin. Biotechnol. 37, 190–200. doi: 10.1016/j.copbio.2015.10.009
Myburg, A. A., Grattapaglia, D., Tuskan, G. A., Hellsten, U., Hayes, R. D., Grimwood, J., et al. (2014). The genome of Eucalyptus grandis. Nature 510, 356–362. doi: 10.1038/nature13308
Obudulu, O., Bygdell, J., Sundberg, B., Moritz, T., Hvidsten, T. R., Trygg, J., et al. (2016). Quantitative proteomics reveals protein profiles underlying major transitions in aspen wood development. BMC Genomics 17:119. doi: 10.1186/s12864-016-2458-z
Patron, N. J., Orzaez, D., Marillonnet, S., Warzecha, H., Matthewman, C., Youles, M., et al. (2015). Standards for plant synthetic biology: a common syntax for exchange of DNA parts. New Phytol. 208, 13–19. doi: 10.1111/nph.13532
Pilate, G., Guiney, E., Holt, K., Petit-Conil, M., Lapierre, C., Leple, J. C., et al. (2002). Field and pulping performances of transgenic trees with altered lignification. Nat. Biotechnol. 20, 607–612. doi: 10.1038/nbt0602-607
Porth, I., Klapste, J., Skyba, O., Friedmann, M. C., Hannemann, J., Ehlting, J., et al. (2013). Network analysis reveals the relationship among wood properties, gene expression levels and genotypes of natural Populus trichocarpa accessions. New Phytol. 200, 727–742. doi: 10.1111/nph.12419
Pouvreau, B., Vanhercke, T., and Singh, S. (2018). From plant metabolic engineering to plant synthetic biology: the evolution of the design/build/test/learn cycle. Plant Sci. 273, 3–12. doi: 10.1016/j.plantsci.2018.03.035
Raes, J., Rohde, A., Christensen, J., Van De Peer, Y., and Boerjan, W. (2003). Genome-wide characterization of the lignification toolbox in Arabidopsis. Plant Physiol. 133, 1051–1071. doi: 10.1104/pp.103.026484
Saito, M., Nurani, A. M., Kondo, Y., and Fukuda, H. (2017). Tissue culture for xylem differentiation with Arabidopsis leaves. Methods Mol. Biol. 1544, 59–65. doi: 10.1007/978-1-4939-6722-3_5
Saleme, M. L. S., Cesarino, I., Vargas, L., Kim, H., Vanholme, R., Goeminne, G., et al. (2017). Silencing CAFFEOYL SHIKIMATE ESTERASE affects lignification and improves saccharification in poplar. Plant Physiol. 175, 1040–1057. doi: 10.1104/pp.17.00920
Schaumberg, K. A., Antunes, M. S., Kassaw, T. K., Xu, W., Zalewski, C. S., Medford, J. I., et al. (2016). Quantitative characterization of genetic parts and circuits for plant synthetic biology. Nat. Methods 13, 94–100. doi: 10.1038/nmeth.3659
Schiffthaler, B., Serrano, A., Delhomme, N., and Street, N. R. (2018). Seidr: a toolkit for calculation of crowd networks. bioRxiv [Preprint]. doi: 10.1101/250696
Shi, R., Sun, Y. H., Li, Q., Heber, S., Sederoff, R., and Chiang, V. L. (2010). Towards a systems approach for lignin biosynthesis in Populus trichocarpa: transcript abundance and specificity of the monolignol biosynthetic genes. Plant Cell Physiol. 51, 144–163. doi: 10.1093/pcp/pcp175
Street, N. R., Skogstrom, O., Sjodin, A., Tucker, J., Rodriguez-Acosta, M., Nilsson, P., et al. (2006). The genetics and genomics of the drought response in Populus. Plant J. 48, 321–341. doi: 10.1111/j.1365-313X.2006.02864.x
Sundell, D., Street, N. R., Kumar, M., Mellerowicz, E. J., Kucukoglu, M., Johnsson, C., et al. (2017). AspWood: high-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell 29, 1585–1604. doi: 10.1105/tpc.17.00153
Thomas, B., Raj, M. C., B, A. K., H, R. M., Joy, J., Moores, A., et al. (2018). Nanocellulose, a versatile green platform: from biosources to materials and their applications. Chem. Rev. 118, 11575–11625. doi: 10.1021/acs.chemrev.7b00627
Tuskan, G. A., Difazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., et al. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313, 1596–1604. doi: 10.1126/science.1128691
Van de Wouwer, D., Boerjan, W., and Vanholme, B. (2018). Plant cell wall sugars: sweeteners for a bio-based economy. Physiol. Plant. 164, 27–44. doi: 10.1111/ppl.12705
Vanholme, R., Cesarino, I., Rataj, K., Xiao, Y., Sundin, L., Goeminne, G., et al. (2013b). Caffeoyl shikimate esterase (CSE) is an enzyme in the lignin biosynthetic pathway in Arabidopsis. Science 341, 1103–1106. doi: 10.1126/science.1241602
Vanholme, R., De Meester, B., Ralph, J., and Boerjan, W. (2019). Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 56, 230–239. doi: 10.1016/j.copbio.2019.02.018
Vanholme, R., Demedts, B., Morreel, K., Ralph, J., and Boerjan, W. (2010). Lignin biosynthesis and structure. Plant Physiol. 153, 895–905. doi: 10.1104/pp.110.155119
Vanholme, B., Desmet, T., Ronsse, F., Rabaey, K., Van Breusegem, F., De Mey, M., et al. (2013a). Towards a carbon-negative sustainable bio-based economy. Front. Plant Sci. 4:174. doi: 10.3389/fpls.2013.00174
Vanholme, R., Storme, V., Vanholme, B., Sundin, L., Christensen, J. H., Goeminne, G., et al. (2012). A systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell 24, 3506–3529. doi: 10.1105/tpc.112.102574
Voelker, S. L., Lachenbruch, B., Meinzer, F. C., Jourdes, M., Ki, C., Patten, A. M., et al. (2010). Antisense down-regulation of 4CL expression alters lignification, tree growth, and saccharification potential of field-grown poplar. Plant Physiol. 154, 874–886. doi: 10.1104/pp.110.159269
Wang, J. P., Matthews, M. L., Williams, C. M., Shi, R., Yang, C., Tunlaya-Anukit, S., et al. (2018). Improving wood properties for wood utilization through multi-omics integration in lignin biosynthesis. Nat. Commun. 9:1579. doi: 10.1038/s41467-018-03863-z
Wang, J. P., Naik, P. P., Chen, H. C., Shi, R., Lin, C. Y., Liu, J., et al. (2014). Complete proteomic-based enzyme reaction and inhibition kinetics reveal how monolignol biosynthetic enzyme families affect metabolic flux and lignin in Populus trichocarpa. Plant Cell 26, 894–914. doi: 10.1105/tpc.113.120881
Wilkerson, C. G., Mansfield, S. D., Lu, F., Withers, S., Park, J. Y., Karlen, S. D., et al. (2014). Monolignol ferulate transferase introduces chemically labile linkages into the lignin backbone. Science 344, 90–93. doi: 10.1126/science.1250161
Xie, M., Muchero, W., Bryan, A. C., Yee, K., Guo, H. B., Zhang, J., et al. (2018). A 5-enolpyruvylshikimate 3-phosphate synthase functions as a transcriptional repressor in Populus. Plant Cell 30, 1645–1660. doi: 10.1105/tpc.18.00168
Xu, P., Kong, Y. M., Song, D. L., Huang, C., Li, X., and Li, L. G. (2014). Conservation and functional influence of alternative splicing in wood formation of Populus and Eucalyptus. BMC Genomics 15:780. doi: 10.1186/1471-2164-15-780
Yamaguchi, M., Goue, N., Igarashi, H., Ohtani, M., Nakano, Y., Mortimer, J. C., et al. (2010). VASCULAR-RELATED NAC-DOMAIN6 and VASCULAR-RELATED NAC-DOMAIN7 effectively induce transdifferentiation into xylem vessel elements under control of an induction system. Plant Physiol. 153, 906–914. doi: 10.1104/pp.110.154013
Zhang, J., Yang, Y., Zheng, K., Xie, M., Feng, K., Jawdy, S. S., et al. (2018). Genome-wide association studies and expression-based quantitative trait loci analyses reveal roles of HCT2 in caffeoylquinic acid biosynthesis and its regulation by defense-responsive transcription factors in Populus. New Phytol. 220, 502–516. doi: 10.1111/nph.15297
Keywords: synthetic biology (synbio), systems biology, systems genetics, woody biomass, biorefinery, bioeconomy, lignin biosynthesis, wood formation
Citation: Myburg AA, Hussey SG, Wang JP, Street NR and Mizrachi E (2019) Systems and Synthetic Biology of Forest Trees: A Bioengineering Paradigm for Woody Biomass Feedstocks. Front. Plant Sci. 10:775. doi: 10.3389/fpls.2019.00775
Edited by:
Wout Boerjan, Flanders Institute for Biotechnology, BelgiumReviewed by:
Kelly Mayrink, University of Florida, United StatesDaniel Conde, University of Florida, United States
Copyright © 2019 Myburg, Hussey, Wang, Street and Mizrachi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander A. Myburg, emFuZGVyLm15YnVyZ0B1cC5hYy56YQ==; emFuZGVyLm15YnVyZ0BmYWJpLnVwLmFjLnph