 Roberto Mariotti1†
Roberto Mariotti1† Alice Fornasiero2,3†
Alice Fornasiero2,3† Soraya Mousavi1
Soraya Mousavi1 Nicolò G.M. Cultrera1
Nicolò G.M. Cultrera1 Federico Brizioli1
Federico Brizioli1 Saverio Pandolfi1
Saverio Pandolfi1 Valentina Passeri1Martina Rossi1Gabriele Magris2,3Simone Scalabrin4
Valentina Passeri1Martina Rossi1Gabriele Magris2,3Simone Scalabrin4 Davide Scaglione4
Davide Scaglione4 Gabriele Di Gaspero2
Gabriele Di Gaspero2 Pierre Saumitou-Laprade5Philippe Vernet5
Pierre Saumitou-Laprade5Philippe Vernet5 Fiammetta Alagna6
Fiammetta Alagna6 Michele Morgante2,3
Michele Morgante2,3 Luciana Baldoni1*
Luciana Baldoni1*- 1CNR - Institute of Biosciences and Bioresources (IBBR), Perugia, Italy
- 2Institute of Applied Genomics, Udine, Italy
- 3Department of Agricultural, Food, Environmental and Animal Sciences, University of Udine, Udine, Italy
- 4IGA Technology Services, Udine, Italy
- 5University of Lille, CNRS, UMR 8198 - Evo-Eco-Paleo, F-59000, Lille, France
- 6ENEA - Trisaia Research Centre, Rotondella, Italy
The genetic control of self-incompatibility (SI) has been recently disclosed in olive. Inter-varietal crossing confirmed the presence of only two incompatibility groups (G1 and G2), suggesting a simple Mendelian inheritance of the trait. A double digest restriction associated DNA (ddRAD) sequencing of a biparental population segregating for incompatibility groups has been performed and high-density linkage maps were constructed in order to map the SI locus and identify gene candidates and linked markers. The progeny consisted of a full-sib family of 229 individuals derived from the cross ‘Leccino’ (G1) × ‘Dolce Agogia’ (G2) varieties, segregating 1:1 (G1:G2), in accordance with a diallelic self-incompatibility (DSI) model. A total of 16,743 single nucleotide polymorphisms was identified, 7,006 in the female parent ‘Leccino’ and 9,737 in the male parent ‘Dolce Agogia.’ Each parental map consisted of 23 linkage groups and showed an unusual large size (5,680 cM in ‘Leccino’ and 3,538 cM in ‘Dolce Agogia’). Recombination was decreased across all linkage groups in pollen mother cells of ‘Dolce Agogia,’ the parent with higher heterozygosity, compared to megaspore mother cells of ‘Leccino,’ in a context of a species that showed exceptionally high recombination rates. A subset of 109 adult plants was assigned to either incompatibility group by a stigma test and the diallelic self-incompatibility (DSI) locus was mapped to an interval of 5.4 cM on linkage group 18. This region spanned a size of approximately 300 Kb in the olive genome assembly. We developed a sequence-tagged site marker in the DSI locus and identified five haplotypes in 57 cultivars with known incompatibility group assignment. A combination of two single-nucleotide polymorphisms (SNPs) was sufficient to predict G1 or G2 phenotypes in olive cultivars, enabling early marker-assisted selection of compatible genotypes and allowing for a rapid screening of inter-compatibility among cultivars in order to guarantee effective fertilization and increase olive production. The construction of high-density linkage maps has led to the development of the first functional marker in olive and provided positional candidate genes in the SI locus.
Introduction
In cultivated olive (Olea europaea subsp. europaea var. europaea), the cross breeding activities have been delayed by the particularly long generation time (Santos-Antunes et al., 2005), the extended juvenile phase, the high demanding nursery practices, such as the forcing of seedling growth (Rugini et al., 2016) and the time and space needed for plant growing (Picheny et al., 2017). In olive, breeding programs last about 30 years on average (Lavee et al., 2014; Rallo et al., 2016) and have been limited to the empirical selection of a few sporadic intraspecific crosses (Rallo et al., 2008), or to clonal selection (Manaï et al., 2007; Gomes et al., 2008; Trapero et al., 2013; Mousavi et al., 2019), while the timing for the selection of new cultivars in other fruit crops has been greatly reduced, also by the application of new efficient genomic tools (Biscarini et al., 2017; Laurens et al., 2018; Cai et al., 2019). However, the importance of olive cultivation at worldwide level and the new challenges posed by the ongoing climate change, are leading to an ever increasing demand for new cultivars (Gutierrez et al., 2009; Urban, 2015; Bosso et al., 2016).
One of the current limitations to olive productivity is represented by its complex self- and inter-incompatibility system (Saumitou-Laprade et al., 2017a; Alagna et al., 2019), a barrier that may seriously curb yield and restrict the varietal choice for planting to only a few inter-compatible or self-fertile varieties. Fruit set deficiencies due to ineffective pollination are generally underestimated by the farmers, however, it has been demonstrated that supplemental pollination may significantly increase olive production (Ayerza and Coates, 2004), indicating the importance of an effective pollination design of olive orchards.
In olive as well as in other species of the Oleaceae family, such as Phillyrea (Phillyrea angustifolia) and ash (Fraxinus excelsior), an incompatibility system known as diallelic self‐incompatibility (DSI) has been described (Saumitou-Laprade et al., 2010; Vernet et al., 2016; Saumitou-Laprade et al., 2018). It has been hypothesized that two alleles at the DSI locus exist in cultivated olives, S and s, with S dominant over s, with only two possible genotypic combinations (Ss and ss), corresponding to G1 and G2 incompatibility groups, respectively, and by stigma test analysis it was never found G1xG1 or G2xG2 compatibility (Saumitou-Laprade et al., 2017a), where G1xG2 crosses always generate G1:G2 = 1:1 balanced progenies. All olive cultivars seem to be self-incompatible, even if pseudo-self-fertility might occur for some cultivars in particular conditions (Alagna et al., 2019).
Although the DSI mechanism in olive is known, many important aspects remain to be clarified, such as the location of the incompatibility locus on the genome, the identification of candidate genes controlling this trait, and markers closely linked to incompatibility. The availability of such information will allow for a systematic screening of olive cultivars to identify their group of incompatibility through genotyping with linked markers.
Olive is a diploid species (2n = 2x = 46) with a genome size of approximately 1.4 Gb (Cruz et al., 2016), with a mean C-value of 1.59 pg (1.56 Gb), where more than 30% sequences are represented by tandem repeats (Barghini et al., 2014). Up to now, only intraspecific crosses have been used for olive mapping, and very early studies were performed with dominant markers (de la Rosa et al., 2003; Wu et al., 2004; Aabidine et al., 2010; Khadari et al., 2010). Recently, more dense maps have been produced by the use of codominant markers, such as diversity arrays technology (DArT) (Domínguez-García et al., 2012; Atienza et al., 2014), simple-sequence repeat (SSR) (Sadok et al., 2013), and single-nucleotide polymorphism (SNP) markers (Marchese et al., 2016; İpek et al., 2017; Unver et al., 2017).
SNPs are sequence-tagged markers widely used for association and genetic mapping due to their wide distribution along the genome, high-throughput genotyping, and ease to score (Vezzulli et al., 2008; Deulvot et al., 2010; Lou et al., 2017). Molecular markers linked to the traits of interest can be identified through different strategies, such as genetic linkage mapping based on biparental populations (Curtolo et al., 2017; Ji et al., 2018; Zheng et al., 2018; Sapkota et al., 2019), or through genome-wide association studies (GWAS), based on unrelated individuals (Khan et al., 2013; Nicolas et al., 2016; Elsadr et al., 2019). The generation of high-resolution linkage maps, a prerequisite for gene positional cloning, allows the genetic dissection of quantitative trait loci, assists in comparisons of synteny, and provides marker order for anchoring sequence scaffolds or physical contigs to linkage groups. In perennial fruit crops, the availability of markers tightly linked to traits under selection may strongly facilitate the progress of breeding programs (Minamikawa et al., 2018).
Linkage analysis can be performed on multi-generation families derived by the cross, back-cross, or selfing of homozygous or heterozygous lines. In fruit trees, the use of these progenies is hindered by the lack, in most cases, of homozygous genotypes and by the long generation time (Bai et al., 2007; Jaillon et al., 2007). For this reason, full-sib F1 families deriving from inter- or intra-specific varietal crosses of highly heterozygous parents are generally used in tree species (Bartholomé et al., 2015) and linkage analysis is conducted separately for each parent using a two-way pseudo-testcross mapping strategy (Grattapaglia and Sederoff, 1994). Pseudo-testcross mapping has been carried out in many fruit crops, such as apricot (Irisarri et al., 2019), peach (Zeballos et al., 2016), oil palm (Bai et al., 2018), clementine (Ollitrault et al., 2012), grapevine (Zhu et al., 2018), apple (Di Pierro et al., 2016), and forest trees, like poplar (Zhigunov et al., 2017) and oak (Bodénès et al., 2016). A number of markers closely linked to important simple or complex traits has been identified with this strategy (Zhu et al., 2015; Zhai et al., 2016). Marker-trait associations, either for qualitative or quantitative trait loci (QTL), should allow to predict in advance the final breeding value of genotypes, permitting to discard unwanted genotypes immediately after seed germination and to only grow individuals that will later display the traits of interest (Edge-Garza et al., 2015; Migicovsky and Myles, 2017).
In the present work, a F1 progeny derived from the cross of two highly heterozygous and completely self-incompatible cultivars, ‘Leccino’ × ‘Dolce Agogia,’ respectively belonging to G1 and G2 groups of incompatibility (Saumitou-Laprade et al., 2017b), was genotyped by ddRAD sequencing technology in order to construct linkage maps. This crossing was performed because parents show different phenotypes for numerous important agronomical traits. In particular, ‘Dolce Agogia’ is a vigorous cultivar, with an alternate bearing, resistant to some of the most dangerous olive diseases, such as Verticillium dahliae and Spilocaea oleagina (Iannotta and Scalercio, 2012; Arias-Calderón et al., 2015), whereas ‘Leccino’ has medium vigor and constant bearing, is susceptible or only partially resistant to the pathogens indicated above (Salman, 2017), but it was recently reported as tolerant to Xylella fastidiosa (Giampetruzzi et al., 2016), the most devastating emerging plant pathogen for the Mediterranean agriculture (Suffert et al., 2009). The maps generated in the present work, represent a significant improvement over the previous ones (Marchese et al., 2016; İpek et al., 2017; Unver et al., 2017), because they include a higher number of SNP markers with a uniform genome distribution and can, therefore, serve as saturated maps for trait mapping. We used the ‘Leccino’ map to identify the location of the DSI locus, to select sequence scaffolds of ‘Leccino’ that were anchored to the DSI by linked markers, and to develop and validate SNP markers for the incompatibility trait.
Materials and Methods
Plant Material and Deoxyribonucleic Acid Extraction
A full-sib F1 progeny of 229 individuals was generated from the cross between ‘Leccino,’ as female parent, and ‘Dolce Agogia,’ as male parent. The progeny, thereafter referred to as Le×DA, is represented by a first set of 16-year-old seedlings (amounting to 155 individuals) and by a second set of 5 years old seedlings (74 individuals) grown in an experimental field, for a total of 229 F1 genotypes. The progeny showed high phenotypic variability, in particular for the length of the juvenile phase, plant vigor, tree habit, fruit bearing, and cutting’s rooting ability (Hedayati et al., 2015). DNA was extracted from leaves using the DNeasy Plant Mini Kit (Qiagen).
Parentage Analysis of the Progeny
‘Leccino,’ ‘Dolce Agogia,’ and the Le×DA progeny were genotyped with a set of SSR markers for confirming the parentage (Baldoni et al., 2009). PCRs were performed as previously reported (Mousavi et al., 2017) and the amplified fragments were separated on an ABI 3130 Genetic Analyzer capillary sequencer (Applied Biosystems, Foster City, CA). Alleles were called using the GeneMapper 3.7 software (Applied Biosystems, Foster City, CA). Parentage analysis was performed using CERVUS version 3.0.7 program (Kalinowski et al., 2007) to sort out seedlings derived from open pollination.
Phenotyping for Incompatibility Groups of the Le×DA Progeny
Those offspring that have reached the mature phase and started blooming (109 out of 229, all belonging to the first set of 16-year-old seedlings), were phenotyped for the incompatibility group. Data on 91 individuals were previously reported (Saumitou-Laprade et al., 2017a), all the others were de novo phenotyped following the same protocol previously applied. In order to minimize errors, phenotyping of all individuals was repeated on previously clonally propagated plants grown in a separate field. Before blooming, flowering twigs of each tree were protected from open pollination with blossom bags. At full blooming, bagged twigs were collected and 20 open flowers per tree were used for the analysis. Sepals and petals were removed from 10 flowers and pistils were placed in two separate plates (one for each of the two pollen donors) onto Brewbacker and Kwack medium. The stigmas of five pistils in one plate were pollinated with pollen of ‘Leccino’ (G1) and the other five with pollen of ‘Dolce Agogia’ (G2). From the remaining 10 flowers, pollen was collected and used for pollinating ‘Leccino’ and ‘Dolce Agogia’ stigmas. The same protocol applied by Saumitou-Laprade et al. (2017a) was used to observe the growth of pollen tubes on the stigmata.
Double Digest Restriction Associated Deoxyribonucleic Acid Sequencing of Parents and Progeny
Genomic DNA was digested with SphI and MboI restriction enzymes, following a double digest restriction associated DNA sequencing (ddRADseq), according to the method proposed by Peterson et al. (2012) and modified by Scaglione et al. (2015). Fragments were added to a ligation reaction containing barcoded adapters, pooled, and then fractioned by agarose gel-electrophoresis. DNA in the size range between 350 and 600 bp was purified using a QIAquick Gel Extraction kit (Qiagen, Venlo, Netherlands). Enrichment PCR was performed with PCR primers that incorporate Illumina hybridization/sequencing sites and index sequences for combinatorial multiplexing. Quality, quantity, and reproducibility of libraries were assessed using a Caliper instrument (DNA High Sensitivity chip). Sequencing was carried out on an Illumina HiSeq 2500 instrument, generating 125-bp paired-end reads.
Single-Nucleotide Polymorphism Calling
Reads were aligned using Bowtie 2 software (Langmead and Salzberg, 2012) with default parameters against a whole-genome assembly of ‘Leccino’ (Muleo et al., 2016). The reference consisted of 509,032 scaffolds, with N50 length of 10,037 bp, amounting to a total of 1.429 giga base pairs (Gbp). Alignments with mapping quality < 4 were removed. Segregating sites were identified using the software Stacks (Catchen et al., 2011) and a bounded SNP model with alpha = 0.05 and upper error (epsilon) of 0.1. Genotypes were called with a minimum coverage of eight reads. Heterozygous genotypes were called within a read coverage ratio of 0.15-0.85 for reference and alternate alleles. Segregating loci were retained if genotypes were called in > 150 progeny.
Genetic Mapping
Genetic maps were generated with the double pseudo-test-cross approach for each parent using SNP genotypic data and phenotypic binary data for the incompatibility group. Linkage groups were obtained using the R/qtl module of the R statistical package with a logarithm of odds (LOD) threshold > 10 and a recombination rate of 0.20. Markers were first ordered using MSTMAP (Wu et al., 2008) with default parameters. A Perl implementation of the SMOOTH program (van Os et al., 2005) was used to remove errors within haplotypes. Markers were re-ordered using MSTMAP. Marker distances were calculated using the Kosambi function {map distance in centi-Morgan (cM) equals ¼ ln [(1 + 2r)/(1 − 2r)], where r is the observed recombination frequency}. Markers with distorted segregation were identified using a χ2 test (α = 0.05).
Comparative Genomic Analyses
Scaffolds of ‘Leccino’ that were anchored to the DSI locus by flanking markers were aligned with the genome assembly of the wild olive (Olea europaea var. sylvestris, GCA_002742605.1) using (B)LASTZ. Gene annotation was performed using the highest hit in blastp alignments with the NCBI protein database.
Development of a Sequence-Tagged Site Marker for Diallelic Self-Incompatibility
PCR primers were designed on the sequence of scaffold_6030 using the program Primer3 version 4.0 (forward 5’-3’: TTTTGGGTGCGAATTGTCCA, reverse 5’-3’: AGGCCACTGTATTTCTAACTCG) for the amplification of a 476-bp fragment spanning two adjacent SNPs that co-segregate with DSI. PCRs were performed using 25 ng of template DNA and Q5 High-Fidelity DNA polymerase (New England Biolabs). The thermal profile consisted of 98°C for 30 s, followed by 35 cycles at 98°C for 10 s, 60°C for 20 s, and 72°C for 30 s, and a final extension at 72°C for 2 min. Amplicon size was checked by 1% agarose gel electrophoresis. Amplicons of the expected size showing unique bands were sequenced using the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and an ABI PRISM 3130 XL Genetic Analyzer (Applied Biosystems, Foster City, CA). The obtained sequences were aligned using BioEdit 7.1.7 (www.mbio.ncsu.edu/BioEdit/bioedit.html), to identify polymorphisms. In order to phase single-nucleotide substitutions in heterozygous individuals and to obtain full sequences from individuals carrying heterozygous small indels, amplicons were cloned by using pGEM-T Easy Vector (Promega) and Escherichia coli XL1 blue strain. DNA from different colonies for each genotype was amplified and sequenced as described above.
Results
Genetic Maps
The parentage analysis allowed to validate the origin of 95% of the 241 seedlings for a total of 229 true-to-type F1 genotypes. The remaining 12 individuals were excluded from further analyses. A total of 16,743 segregating loci were identified in the Le×DA progeny by ddRAD sequencing. The parental maps consisted of 23 linkage groups, including 9,737 RAD loci in ‘Dolce Agogia’ and 7,006 RAD loci in ‘Leccino’ (Table 1). Linkage groups were numbered consistently with the chromosome numbering of the wild olive genome assembly (Unver et al., 2017). The male parental map (‘Dolce Agogia’) included 1,829 genetic bins (i.e., positions on the genetic map with a unique segregation pattern) for a total length of 3,538 cM. The female parental map (‘Leccino’) included 2,311 genetic bins for a total length of 5,680 cM. The average distance between adjacent genetic bins was 2.46 cM in ‘Leccino’ and 1.93 cM in ‘Dolce Agogia’.
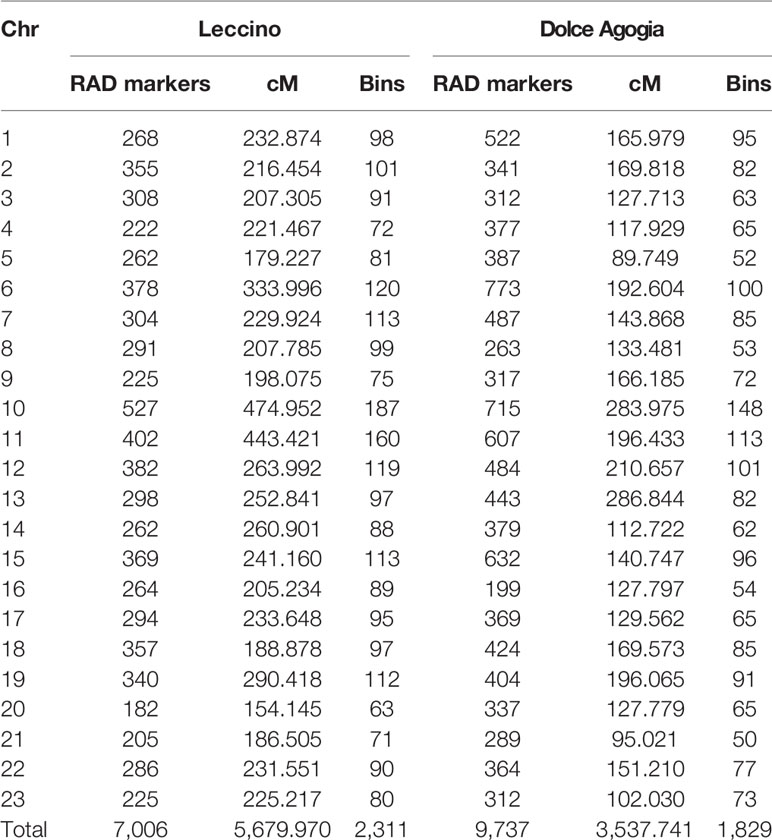
Table 1 Statistics of the parental linkage maps. Chromosome number (Chr), restriction associated DNA (RAD) markers, length (cM), and bins are reported for ‘Leccino’ and ‘Dolce Agogia’.
All linkage groups of the ‘Leccino’ map were consistently longer that those of the ‘Dolce Agogia’ map, presumably as a result of a consistently higher recombination rate between homologous chromosomes in megaspore mother cells of ‘Leccino’ than in pollen mother cells of ‘Dolce Agogia.’ The shorter map obtained in ‘Dolce Agogia’ was not explained by runs of homozygosity at the chromosome termini compared to ‘Leccino,’ except for the upper 34 cM of Linkage Group (LG) 16 in ‘Leccino,’ which lacked segregating sites in ‘Dolce Agogia.’ The longer map obtained in ‘Leccino’ was, on the other hand, not explained by the presence of isolated markers with high likelihood of genotypic errors, which usually locally inflate genetic distances due to inconsistent genotype calls compared to neighboring markers. It was noteworthy that the suppression of recombination was observed in the parent with the higher level of heterozygosity, as revealed by the larger number of segregating sites.
Structural Variation Between Parental Genomes and Segregation Distortion
We used 2,305 RAD loci segregating from both parents with an allelic status “ABxCD” for pairing and aligning linkage groups between parental maps. The alignment of the two parental maps did not show inter-chromosomal translocations. Two large inversions were detected in LG 1, involving a segment of about 30 cM, and in LG 8, involving a segment of about 12 cM. We did not detect suppression of recombination around each inversion in any of the parental maps and we mapped segregating sites within the inverted regions in each parental map. In both cases, the parental genomes were therefore homozygous for alternative structural variants, but the haplotypes carrying the inversion were ancient enough to have accumulated segregating sites. Minor rearrangements in marker order were identified on LGs 2, 4, 5, 7, and 17. Extended regions with distorted segregation (α = 0.05) were detected on several linkage groups in both maps, with a higher frequency being observed in ‘Dolce Agogia’ map (Figure 1). Localized distortion was also detected at markers flanking the lower side of the incompatibility locus (see below) with an excess of marker alleles in phase with the incompatibility recessive allele.
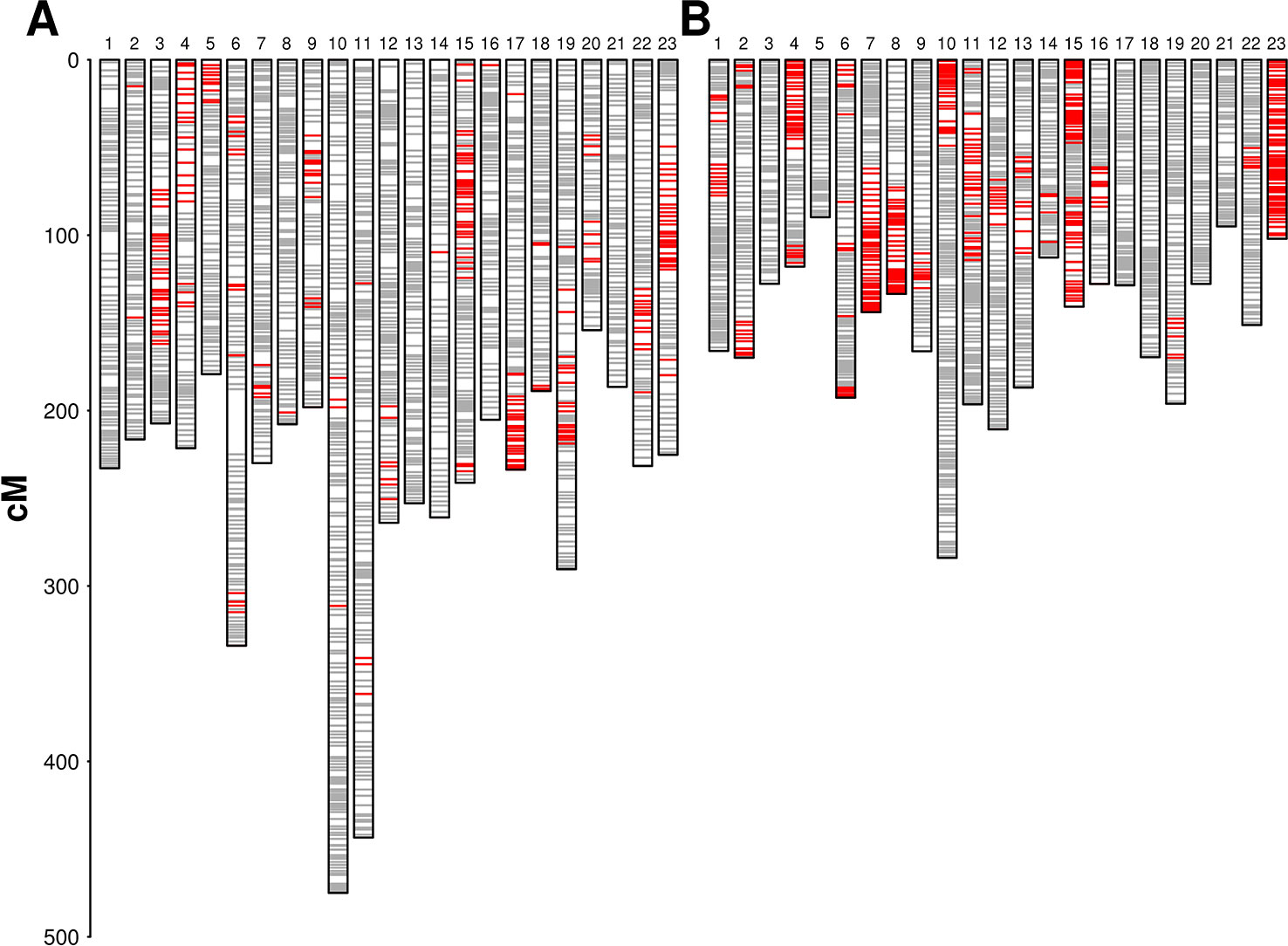
Figure 1 Parental genetic maps: diagrams show linkage groups in ‘Leccino’ (panel A) and ‘Dolce Agogia’ (panel B). Markers are plotted in gray. Markers with distorted segregation (α = 0.05) are plotted in red. LGs are not oriented with respect to the conventional orientation of chromosomes in the wild olive genome assembly.
Identification of the Incompatibility Locus
A stigma test was performed on 106 adult Le×DA individuals. The incompatibility group phenotyping assigned 56 individuals to the G1 group, 47 to the G2 group, and discarded 3 individuals due to uncertain phenotype. Segregation of the phenotype followed the expected 1:1 ratio (χ2 = 0.79). A self-pollination test of ‘Leccino’ and ‘Dolce Agogia’ showed no growth of pollen tubes on the stigmas, confirming their complete self-incompatibility.
The incompatibility locus was mapped as a Mendelian trait in the ‘Leccino’ map. The locus was located on LG 18 within an interval of 5.4 cM. Within this interval, the RAD markers on scaffold_6030 (sizing 42,057 bp) co-segregated with the trait in all 103 individuals of the progeny with a reliable incompatibility group assignment (Figure 2). The upper border of the locus was supported by a recombination event observed in one individual between the incompatibility group and two RAD markers located in scaffold_63515 (sizing 3,877 bp) and in scaffold_26872 (sizing 32,989 bp). The lower border of the genetic interval was supported by five crossing-over events observed in the progeny between the incompatibility group trait and a RAD marker in scaffold_7600 (sizing 34,918 bp) and by even more crossing-overs with a RAD marker in scaffold_13712 (sizing 37,403 bp). All these scaffolds of ‘Leccino’ aligned to a region between coordinates 8,500,000 and 9,100,000 of chromosome 18 in the genome assembly of a wild olive (Unver et al., 2017) (Figure 3).
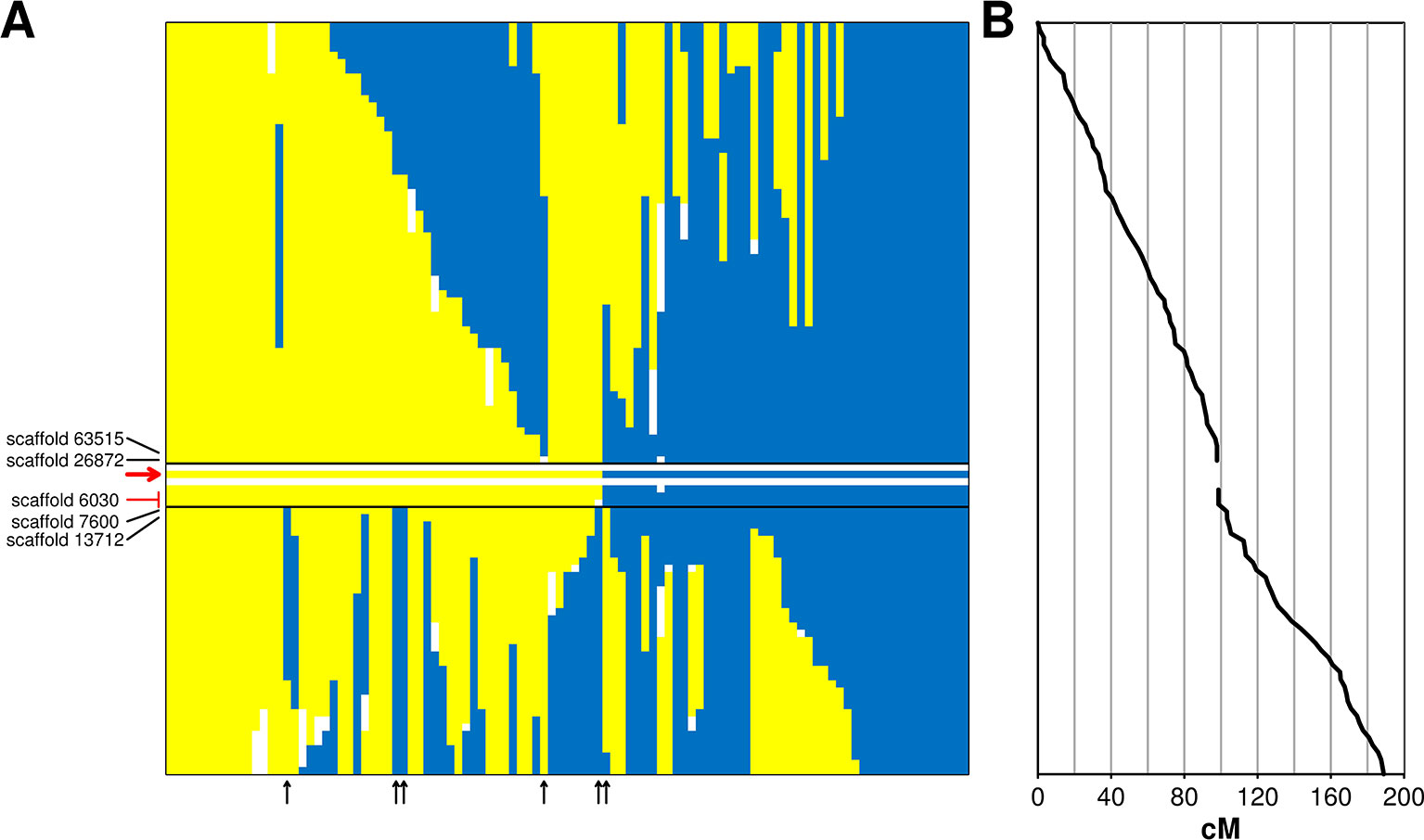
Figure 2 Panel (A): genetic interval controlling the incompatibility system on linkage group 18 of cv. Leccino. The diallelic self-incompatibility (DSI) locus is delimited by black lines, the red arrow points to the DSI phenotype. Each vertical bar shows native and recombinant chromosomes of ‘Leccino’. The homolog carrying the S allele is shown in yellow, the homolog carrying the s allele is shown in blue and the same color indicate the G1 and G2 phenotypes, respectively. Blanks indicate missing genotypes. Flanking restriction associated DNA (RAD) markers and corresponding scaffolds are shown in black. Co-segregating RAD markers are shown in red and the corresponding scaffold is black. Vertical black arrows indicate the diagrams of recombinant chromosomes in the seedlings that defined the genetic borders of the DSI locus. Panel (B): genetic distance from the top of LG 18.
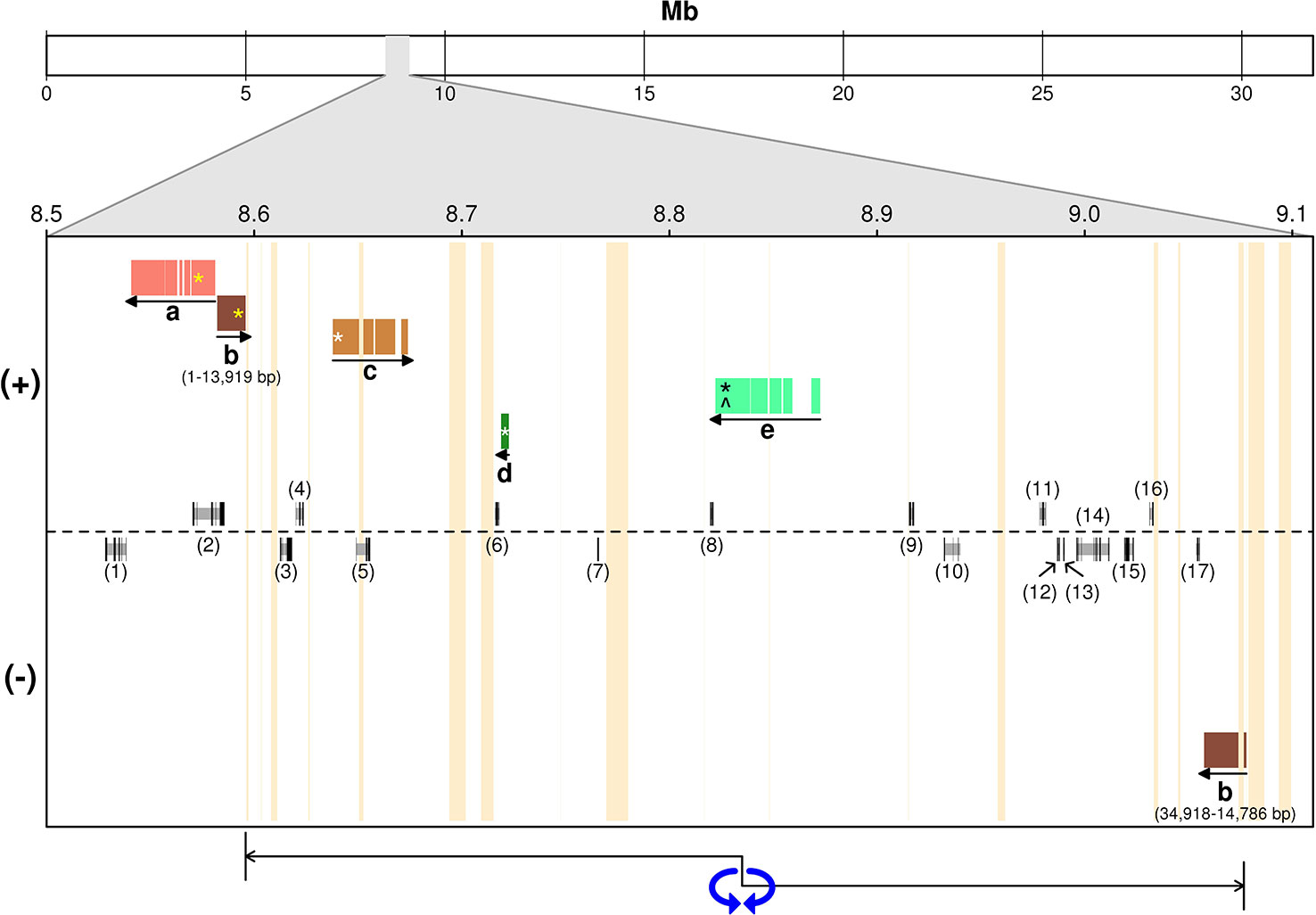
Figure 3 Alignment of ‘Leccino’ sequence scaffolds in the diallelic self-incompatibility (DSI) locus against the assembly of a wild olive genome (Unver et al., 2017). (+) and (−) indicate the strand of the wild olive genome sequence. Letters in the figure replace scaffolds IDs: a, scaffold_13712; b, scaffold_7600; c, scaffold_26872; d, scaffold_ 63515; e, scaffold_6030. The models above and below the dotted line indicate gene prediction in the wild olive genome on either strand. Numbers between parentheses identify the gene models described in Supplementary Table S1. Vertical orange bars indicate sequence gaps in wild olive assembly. Horizontal colored bars indicate the regions of alignment. Yellow asterisks on “a” and “b” indicate the position within each scaffold of the flanking restriction associated DNA (RAD) markers on one side the genetic locus. Asterisks indicate the position of RAD markers within each scaffold sequence that anchored that scaffold to the genetic map. White asterisks on “c” and “d” indicate the position within each scaffold of the flanking RAD markers on the other side the genetic locus. The black asterisk and the circumflex on “e” indicate the positions of co-segregating RAD and sequence-tagged site (STS) markers, respectively. The double-head arrow below indicates the region of order inconsistency between ‘Leccino’ map and the wild olive genome assembly.
Alignment of Diallelic Self-Incompatibility Markers and Scaffolds With the Wild Olive Genome Assembly and Candidate Genes in the Region
The projection of ‘Leccino’ scaffolds containing the RAD flanking markers from either side of the DSI locus onto the assembly of chromosome 18 identified a seemingly short physical interval of a few dozen Kbs, comprised between scaffolds 13712 and 7600, on one side, and scaffold 26872 on the other side (Figure 3). The region around scaffolds 13712, 7600, 26872 in the wild olive assembly, corresponding to the chromosomal interval between 8.5 and 8.7 Mb, encodes five predicted proteins (Supplementary Table S1, IDs 1 to 5). However, the inconsistent physical position of scaffold_7600, which contains a RAD marker co-segregating with DSI and aligns outside of the interval defined by the flanking markers, and the split alignment of the initial 14 Kb of scaffold_7600 on one side of the locus and the remaining 12 Kb of scaffold_7600 on the opposite strand at the other side of the locus suggest that the physical interval might be substantially longer. A 450-kb inversion, indicated by a double-head arrow in Figure 3, or an assembly error in the wild olive genome between two flanking sequence gaps would reconcile the genetic marker order in the ‘Leccino’ map and the assembly of scaffold_7600 and would define a physical interval for the DSI locus in the wild olive assembly from coordinates 8,720,000 to 9,080,000, with inverted orientation (220 Kb-580 Kb relative coordinates in Figure 3). Under this hypothesis, the DSI region in the assembly of wild olive would encompass 12 additional predicted proteins, bringing the total number of candidate genes to 17 (Supplementary Table S1, IDs 6 to 17). RNA-Seq data also showed that other regions outside of the predicted gene models are transcribed in the physical interval of wild olive.
Validation of Sequence-Tagged Site Markers Linked to the Incompatibility Group
We amplified and sequenced a PCR fragment (hereafter referred to as Oe-DSI-locus-fragment-A), spanning two SNPs on scaffold_6030 that showed co-segregation with RAD loci in ‘Leccino’ map. The sequencing results of 165 genotypes, which included cultivars and progenies, allowed to identify only two polymorphic sites at positions 63 and 283 bp of the Oe-DSI-locus-fragment-A, which define unambiguously the two groups of incompatibility (Table 2 and Supplementary Table S2). In the parental cultivars Leccino and Dolce Agogia, the amplified fragment showed four alleles differentiated by eleven SNPs (Table 3). The inheritance of four haplotypes segregating from ‘Leccino’ (S-A/s-a) and ‘Dolce Agogia’ (s-b/s-c) and the linkage of the S-A haplotype with the genetic determinant of the G1 phenotype was confirmed by analyzing the Oe-DSI-locus-fragment-A in the Le×DA progeny (Figure 4). Combining the stigma test results with the genotypic data, the dominant allele S-A and three recessive ones, s-a, s-b, s-c, were identified.
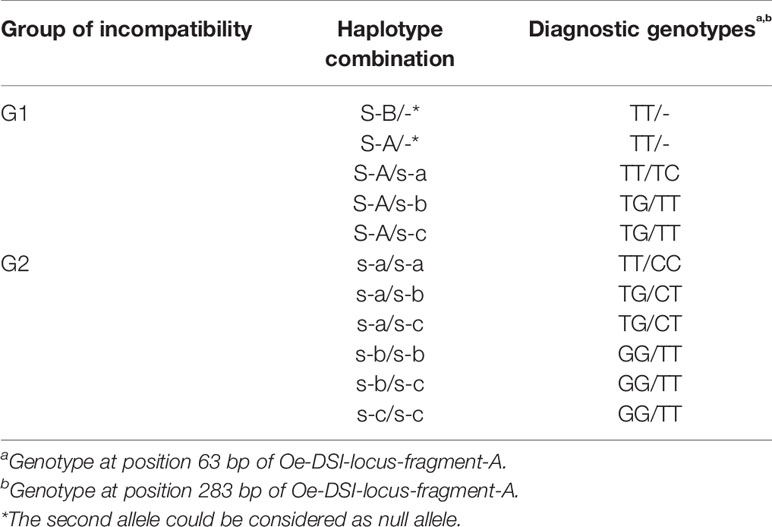
Table 2 Incompatibility group phenotypes and corresponding haplotype combinations of Oe-DSI-locus-fragment-A observed in the analyzed olive cultivars.
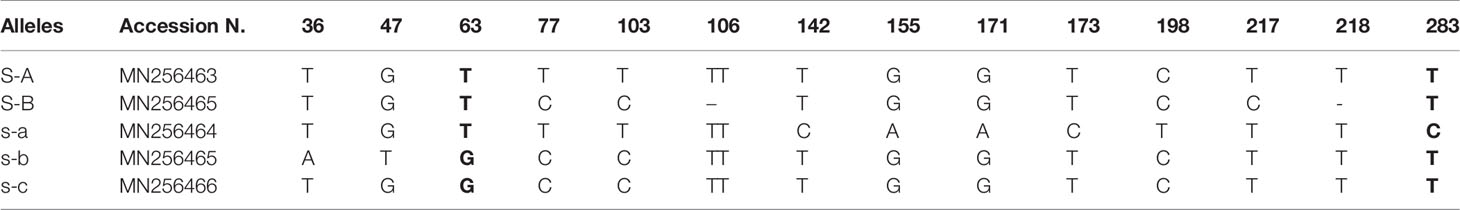
Table 3 Haplotypes of Oe-DSI-locus-fragment-A and position (base pair distance from the forward primer) of the polymorphisms [single-nucleotide polymorphisms (SNPs) and indels] identified. SNP combinations that identify uniquely each incompatibility group are indicated in bold.
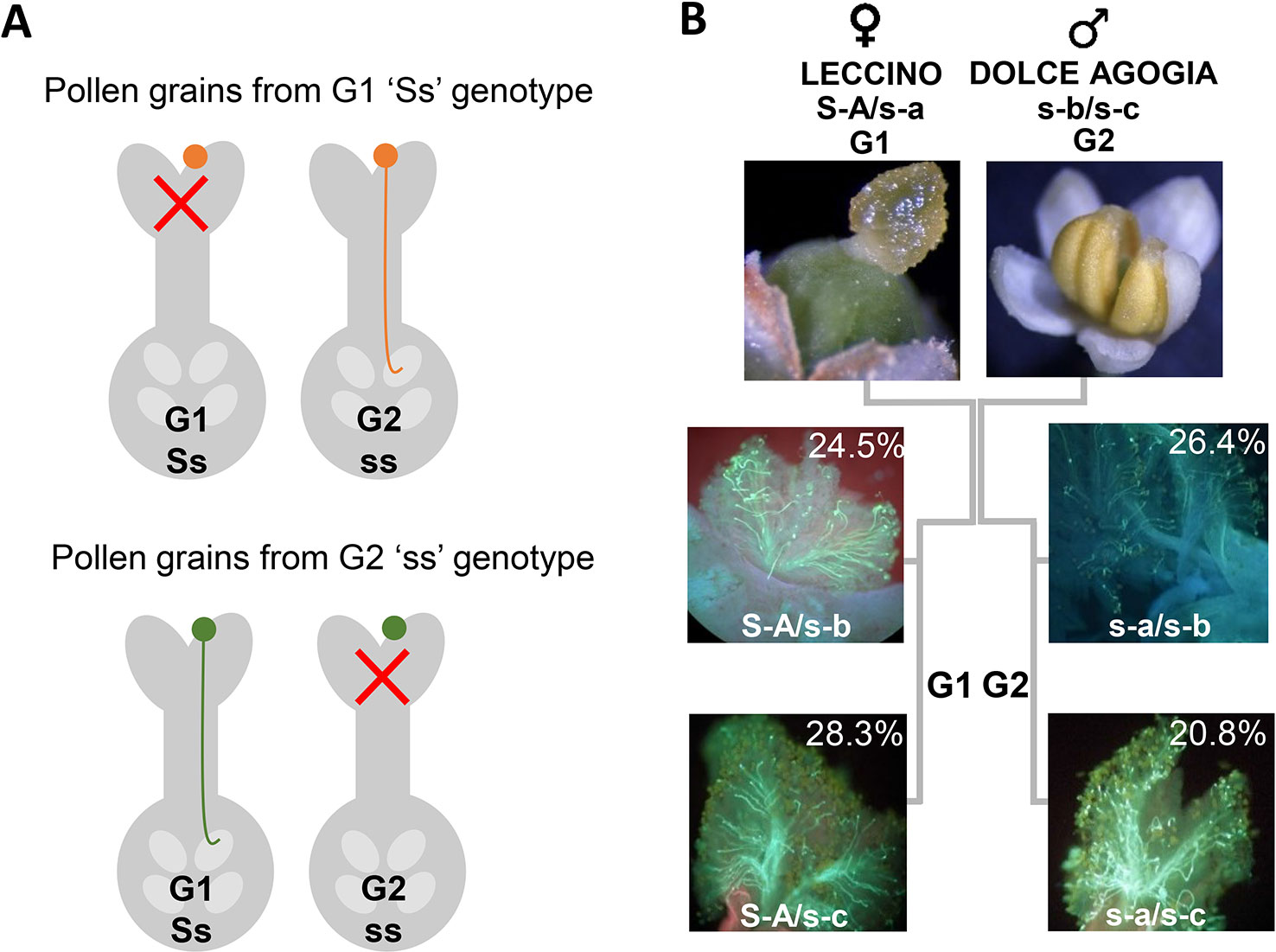
Figure 4 (A) Graphic illustration of the sporophytic diallelic self-incompatibility (DSI) system in olive. (B) Schematic representation of ‘Leccino’ (S-A/s-a) and ‘Dolce Agogia’ (s-b/s-c) allele combinations and segregation of four haplotypes in the Le×DA progenies.
When the same fragment was amplified in 57 olive cultivars with known incompatibility group assignment, an additional haplotype was found, the S-B allele, which represents a new dominant allele distinguishing G1 cultivars (Supplementary Table S2). The five haplotypes carried a total of 12 SNPs and 2 small indels (Table 3). In the 20 G1 cultivars, the dominant S-A allele was detected 13 times and S-B was present in apparent homozygous state in six out of seven cases and in one case with the haplotype s-b, detected by cloning. Both dominant alleles were found in combination with s-a, s-b, and s-c recessive alleles. The presence of only one dominant allele in some cultivars could be due to homozygosity or to the inability to amplify the second allele (null allele). Among the 37 G2 cultivars, the most frequent recessive haplotype was s-b, also showing a high percentage of homozygosity (Supplementary Table S2). A skewed geographical distribution was observed for some alleles between Italy and Spain, the two most represented countries in our data set, such the S-B allele, mainly present in Spanish cultivars, and s-c, only present within the Italian ones (Supplementary Figure S1).
Discussion
Species with a long juvenile phase require many years to reach the first flowering and fruit setting (Purba et al., 2001; Flachowsky et al., 2011; Yang et al., 2016), delaying the generation of crossbreed, F2, and backcross populations and slowing down the process of genetic improvement. In these cases, the availability of genomic tools to assist the selection of new genotypes becomes mandatory in order to guide the choice of parental combinations, to select genotypes carrying the traits of interest, and to introgress specific alleles into new varieties. Markers linked to the traits under selection, identified either through genetic mapping approaches or by means of genome-wide association studies, represent the most powerful tools for breeding in woody perennial crops, offering new opportunities to develop early selection strategies and new ways to integrate variation from different sources (Varshney et al., 2005; Montanari et al., 2013; Bink et al., 2014; Kole et al., 2015; Muranty et al., 2015).
The use of high-throughput sequencing technologies is speeding up the genotyping of mapping populations, providing an unprecedented high number of markers that allows the construction of dense genetic maps (Kujur et al., 2015; Liu et al., 2017). In this work, we have used ddRAD sequencing for genotyping 7,006 and 9,737 segregating sites in a biparental population, which largely exceeded the number of genetic bins that can be resolved in a progeny of 229 individuals. Olive tree is known to be a highly heterozygous species with very high levels of nucleotide diversity observed even in cultivated varieties (Gros-Balthazard et al., 2019). Here we show that the species is characterized also by very high levels of recombination as attested by the lengths of the two parental genetic maps. We observed a significant difference in the length between the male Dolce Agogia (3,538 cM) and the female Leccino (5,680 cM) maps. Heterochiasmy, i.e., the presence of different crossover frequencies in male and female meiosis, has frequently been observed in plants, without a fixed trend of higher frequency in male or female meiosis (Lenormand and Dutheil, 2005). In Arabidopsis, for example, a dramatically higher crossing over rate (575 cM vs. 332 cM) is observed in male than in female meiosis (Giraut et al., 2011). Theory would predict that haploid selection determines heterochiasmy, with the sex experiencing more intense selection during the haploid phase showing lower recombination (Lenormand and Dutheil, 2005). In a highly heterozygous and obligately outcrossing species such as olive, we can expect stronger selection among male gametophytes that could explain the observed difference in genetic map length. An alternative explanation could be provided by a high frequency of chromosomal inversions or other chromosomal rearrangements that suppress recombination. The identification of two large inversions for which the two parental varieties are homozygous for alternative alleles makes us believe that in such a highly heterozygous species, each individual accession may be heterozygous for a much larger set of inversions. As ‘Dolce Agogia’ appears to be more heterozygous than ‘Leccino,’ the lower recombination frequency observed could be explained by a higher frequency of heterozygous chromosomal rearrangements that suppress recombination.
In this case, the generation of markers did not represent a limiting factor for trait mapping, whose resolution was rather limited by the size of the progeny subject to phenotyping. Phenotyping of reproductive traits requires adult plants and, in the case of DSI group assignment, is also labor intensive. We mapped the DSI locus to a 5.4 cM genetic interval, which is estimated to correspond to a physical distance of approximately 300 Kb in a chromosome-scale assembly of a wild olive genome (Unver et al., 2017). Gene prediction and gene annotation in the wild olive assembly did not provide any obvious functional candidate, except for transcription factors that might be involved in gynoecium development regulation. Two candidate genes encode proteins putatively related to flower development: the ortholog of Arabidopsis STYLISH 1 encoding a binding protein with nuclear localization that promotes formation of stylar and stigmatic tissues and proliferation of stylar xylem (Gomariz-Fernández et al., 2017; Min et al., 2019), and FAR1 related sequence 5, which is expressed in hypocotyls, inflorescences stems and flowers (Li et al., 2017; Ma and Li, 2018). However, the prediction of proteins with uncharacterized function in the same region, the presence of non-annotated transcribed regions, the expected intraspecific presence/absence variation between genomes, make it necessary to proceed with the complete assembly of the two ‘Leccino’ haplotypes across the entirety of the DSI locus.
While we confirmed the monogenic nature of the DSI system in olive, as postulated by Saumitou-Laprade et al. (2017a), five haplotypes were identified using a sequence-tagged site (STS) marker in the locus and, via linkage mapping and association mapping, we demonstrated that two of them are in phase with the dominant genetic determinant of the G1 incompatibility group. The STS haplotypes were linked to the DSI genetic determinant, but they do not correspond to the DSI alleles. It was noteworthy that the cumulative frequency of the STS haplotypes in phase with the dominant S allele was roughly half the cumulative frequency of haplotypes in phase with the s allele, although the accessions analyzed in this paper are not a natural population. It is possible to speculate that this condition may reflect a balancing selection for maintaining G1 and G2 genotypes in a cultivated population at frequencies that maximize pollination rate.
In the set of cultivars analyzed in this paper, we found only two dominant S-A and S-B alleles that confer the G1 phenotype and no cultivar was observed carrying both dominant alleles. It was also noteworthy that some haplotypes were more frequent in groups of cultivars typical of specific geographic locations. For instance, the allele s-c showed 18% frequency in the sample and was almost exclusively present in varieties of Central Italy, leading to an excess of homozygous genotypes in that geographic area, whereas the S-B haplotype was mostly present in cultivars from the Iberian Peninsula. Variation of haplotype frequencies at DSI-linked markers in geographically distant populations may simply reflect genetic drift or may indicate adaptive evolution changing the frequency of DSI alleles. Further analyses will help to clarify these issues and to identify additional untapped variation at the DSI locus.
From an evolutionary point of view, the maintenance of a homomorphic DSI in a hermaphrodite species like O. europaea is unexpected and remains to be explained. Indeed, the SI systems are susceptible to rapid invasion by new self-incompatibility alleles, which should experience a strong negative frequency-dependent advantage. In Oleaceae species in which DSI was identified, such invasion was not observed (Saumitou-Laprade et al., 2010; Vernet et al., 2016; Saumitou-Laprade et al., 2017a; Saumitou-Laprade et al., 2017b; Saumitou-Laprade et al., 2018). The homomorphic DSI system seems to “resist” and this is an intriguing finding (Barrett, 2019). In the absence of obvious evolutionary constraints that could prevent the selection of new SI specificities, the molecular constraint appears to be the best candidate. Therefore, the molecular characterization of the S locus region in Olea and its comparison with other Oleaceae species are of strong interest. The present work is opening the way and should contribute efficiently to solve the evolutionary paradox of the stable homomorphic DSI in Oleaceae.
The STS marker identified in the present work represents a new tool for large-scale screening of thousands of olive accessions from their traditional areas of cultivation across the Mediterranean shores and from new growing areas and will allow to determine their incompatibility group. In addition, it represents a starting point for the identification of the genetic determinant of such a peculiar incompatibility system. A comprehensive understanding of the genetic control of DSI can offer great opportunities to characterize cultivars for their incompatibility group, increase the olive production and guide the orchard plantation design with optimal spatial distribution of inter-compatible varieties.
Conclusions
This work provides markers for a fast and reliable genotyping of olive cultivars for their incompatibility group, offering great opportunities to rapidly screen and identify inter-compatible varieties, planning inter-varietal crosses, and reducing the time for seedling selection. It also represents the initial effort for the identification of the genetic determinants of incompatibility, a starting point for understanding the molecular mechanisms underlying the DSI system in olive.
The genetic maps of ‘Leccino’ and ‘Dolce Agogia’ will also serve to identify Mendelian loci or QTLs responsible for other important traits that segregate in the Le×DA progeny. Information yet to be gained on the number and location of the genetic determinants of those traits, along with the DSI-linked ddRAD and STS markers developed in this paper, may pave the way to the application of genomics-assisted breeding in olive.
Data Availability Statement
Raw reads of ddRAD-Seq have been deposited in Short Read Archive under the BioProject number PRJNA594490. The matrix of genotypic calls has been deposited in the figshare repository with the DOI 10.6084/m9.figshare.11352068.
Author Contributions
RM, PS-L, PV, MM and LB conceived the study. SP, RM, NC and LB provided the plant material. RM, SM, MR, PS-L, PV and FA performed the stigma tests. RM, NC, VP, MR, AF, GM, SS, DS, GDG and MM performed the molecular, sequencing and bioinformatic analyses. FB, LB, RM, SM and VP wrote the first draft of the manuscript. PS-L, PV, DS, GDG and MM contributed to the writing and revision of the manuscript. All the authors agreed on the final version of this work.
Funding
The research was supported by the European Union’s Horizon 2020 Research and Innovation Program Marie Sklodowska-Curie - Before Project (Grant Agreement No 645595), by the EU projects “OLIVE4CLIMATE – LIFE” (LIFE15 CCM/IT/000141) and by the Rural Development Program of Umbria Region, 2014-2020 – Measure 16.2.1, INNO.V.O. - Development of alternative varieties to face the new challenges of olive growing, SIAN n. 84250258245.
Conflict of Interest
The authors declared that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Istituto Tecnico Agrario “Ciuffelli” (ISIS) Todi (PG) for hosting the Le×DA progeny in the field and for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01760/full#supplementary-material
Supplementary Figure S1 | Geographical distribution of DSI alleles in the Italian and Spanish analyzed varieties.
References
Aabidine, A. Z. E., Charafi, J., Grout, C., Doligez, A., Santoni, S., Moukhli, A., et al. (2010). Construction of a genetic linkage map for the olive based on AFLP and SSR markers. Crop Sci. 50 (6), 2291–2302. doi: 10.2135/cropsci2009.10.0632
Alagna, F., Caceres, M. E., Pandolfi, S., Collani, S., Mousavi, S., Mariotti, R., et al. (2019). The paradox of self-fertile varieties in the context of self-incompatible genotypes in olive. Front. Plant Sci. 10, 725. doi: 10.3389/fpls.2019.00725
Arias-Calderón, R., León, L., Bejarano-Alcázar, J., Belaj, A., De la Rosa, R., Rodríguez-Jurado, D. (2015). Resistance to Verticillium wilt in olive progenies from open-pollination. Sci. Hortic. 185, 34–42. doi: 10.1016/j.scienta.2015.01.015
Atienza, S. G., de la Rosa, R., León, L., Martín, A., Belaj, A. (2014). Identification of QTL for agronomic traits of importance for olive breeding. Mol. Breed. 34, 725–737. doi: 10.1007/s11032-014-0070-y
Ayerza, R., Coates, W. (2004). Supplemental pollination–increasing olive (Olea europaea) yields in hot, arid environments. Exp. Agric. 40, 481–491. doi: 10.1017/S0014479704002133
Bai, W.-N., Zeng, Y.-F., Zhang, D.-Y. (2007). Mating patterns and pollen dispersal in a heterodichogamous tree, Juglans mandshurica (Juglandaceae). New Phytol. 176, 699–707. doi: 10.1111/j.1469-8137.2007.02202.x
Bai, B., Wang, L., Zhang, Y. J., Lee, M., Rahmadsyah, R., Alfiko, Y., et al. (2018). Developing genome-wide SNPs and constructing an ultrahigh-density linkage map in oil palm. Sci. Rep. 8, 691. doi: 10.1038/s41598-017-18613-2
Baldoni, L., Cultrera, N. G., Mariotti, R., Ricciolini, C., Arcioni, S., Vendramin, G. G., et al. (2009). A consensus list of microsatellite markers for olive genotyping. Mol. Breed. 24, 213–231. doi: 10.1007/s11032-009-9285-8
Barghini, E., Natali, L., Cossu, R. M., Giordani, T., Pindo, M., Cattonaro, F., et al. (2014). The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol. Evol. 6, 776–791. doi: 10.1093/gbe/evu058
Barrett, S. C. (2019). ‘A most complex marriage arrangement’: recent advances on heterostyly and unresolved questions. New Phytol. 224, 1051–1067. doi: 10.1111/nph.16026
Bartholomé, J., Mandrou, E., Mabiala, A., Jenkins, J., Nabihoudine, I., Klopp, C. S., et al. (2015). High-resolution genetic maps of Eucalyptus improve Eucalyptus grandis genome assembly. New Phytol. 206, 1283–1296. doi: 10.1111/nph.13150
Bink, M. C. A. M., Jansen, J., Madduri, M., Voorrips, R. E., Durel, C.-E., Kouassi, A. B., et al. (2014). Bayesian QTL analyses using pedigreed families of an outcrossing species, with application to fruit firmness in apple. Theor. Appl. Genet. 127, 1073–1090. doi: 10.1007/s00122-014-2281-3
Biscarini, F., Nazzicari, N., Bink, M., Arús, P., Aranzana, M. J., Verde, I., et al. (2017). Genome-enabled predictions for fruit weight and quality from repeated records in European peach progenies. BMC Genomics 18 (1), 432. doi: 10.1186/s12864-017-3781-8
Bodénès, C., Chancerel, E., Ehrenmann, F., Kremer, A., Plomion, C. (2016). High-density linkage mapping and distribution of segregation distortion regions in the oak genome. DNA Res. 23, 115–124. doi: 10.1093/dnares/dsw001
Bosso, L., Di Febbraro, M., Cristinzio, G., Zoina, A., Russo, D. (2016). Shedding light on the effects of climate change on the potential distribution of Xylella fastidiosa in the Mediterranean basin. Biol. Invasions 18, 1759–1768. doi: 10.1007/s10530-016-1118-1
Cai, L., Quero-García, J., Barreneche, T., Dirlewanger, E., Saski, C., Iezzoni, A. (2019). A fruit firmness QTL identified on linkage group 4 in sweet cherry (Prunus avium L.) is associated with domesticated and bred germplasm. Sci. Rep. 9 (1), 5008. doi: 10.1038/s41598-019-41484-8
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W., Postlethwait, J. H. (2011). Stacks: building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genet. 1, 171–182. doi: 10.1534/g3.111.000240
Cruz, F., Julca, I., Gómez-Garrido, J., Loska, D., Marcet-Houben, M., Cano, E., et al. (2016). Genome sequence of the olive tree, Olea europaea. Gigascience 5, 29. doi: 10.1186/s13742-016-0134-5
Curtolo, M., Cristofani-Yaly, M., Gazaffi, R., Takita, M. A., Figueira, A., Machado, M. A. (2017). QTL mapping for fruit quality in Citrus using DArTseq markers. BMC Genomics 18, 289. doi: 10.1186/s12864-017-3629-2
de la Rosa, R., Angiolillo, A., Guerrero, C., Pellegrini, M., Rallo, L., Besnard, G., et al. (2003). A first linkage map of olive (Olea europaea L.) cultivars using RAPD, AFLP, RFLP and SSR markers. Theor. Appl. Genet. 106, 1273–1282. doi: 10.1007/s00122-002-1189-5
Deulvot, C., Charrel, H., Marty, A., Jacquin, F., Donnadieu, C., Lejeune-Hénaut, I., et al. (2010). Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genomics 11, 468. doi: 10.1186/1471-2164-11-468
Di Pierro, E. A., Gianfranceschi, L., Di Guardo, M., Koehorst-van Putten, H. J., Kruisselbrink, J. W., Longhi, S., et al. (2016). A high-density, multi-parental SNP genetic map on apple validates a new mapping approach for outcrossing species. Hortic. Res. 3, 16057. doi: 10.1038/hortres.2016.57
Domínguez-García, M. C., Belaj, A., de la Rosa, R., Satovic, Z., Heller-Uszynska, K., Kilian, A., et al. (2012). Development of DArT markers in olive (Olea europaea L.) and usefulness in variability studies and genome mapping. Sci. Hortic. 136, 50–60. doi: 10.1016/j.scienta.2011.12.017
Edge-Garza, D. A., Luby, J. J., Peace, C. (2015). Decision support for cost-efficient and logistically feasible marker-assisted seedling selection in fruit breeding. Mol. Breed. 35, 223. doi: 10.1007/s11032-015-0409-z
Elsadr, H., Sherif, S., Banks, T., Somers, D., Jayasankar, S. (2019). Refining the genomic region containing a major locus controlling fruit maturity in peach. Sci. Rep. 9, 7522. doi: 10.1038/s41598-019-44042-4
Flachowsky, H., Roux, P.-M. L., Peil, A., Patocchi, A., Richter, K., Hanke, M.-V. (2011). Application of a high-speed breeding technology to apple (Malus × domestica) based on transgenic early flowering plants and marker-assisted selection. New Phytol. 192, 364–377. doi: 10.1111/j.1469-8137.2011.03813.x
Giampetruzzi, A., Morelli, M., Saponari, M., Loconsole, G., Chiumenti, M., Boscia, D., et al. (2016). Transcriptome profiling of two olive cultivars in response to infection by the CoDiRO strain of Xylella fastidiosa subsp. pauca. BMC Genomics 17, 475. doi: 10.1186/s12864-016-2833-9
Giraut, L., Falque, M., Drouaud, J., Pereira, L., Martin, O. C., Mézard, C. (2011). Genome-wide crossover distribution in Arabidopsis thaliana meiosis reveals sex-specific patterns along chromosomes. PloS Genet. 7, e1002354. doi: 10.1371/journal.pgen.1002354
Gomariz-Fernández, A., Sánchez-Gerschon, V., Fourquin, C., Ferrándiz, C. (2017). The role of SHI/STY/SRS genes in organ growth and carpel development is conserved in the distant eudicot species Arabidopsis thaliana and Nicotiana benthamiana. Front. Plant Sci. 8, 814. doi: 10.3389/fpls.2017.00814
Gomes, S., Martins-Lopes, P., Lima-Brito, J., Meirinhos, J., Lopes, J., Martins, A., et al. (2008). Evidence for clonal variation in ‘Verdeal-Transmontana’ olive using RAPD, ISSR and SSR markers. J. Hortic. Sci. Biotechnol. 83, 395–400. doi: 10.1080/14620316.2008.11512397
Grattapaglia, D., Sederoff, R. (1994). Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137, 1121–1137.
Gros-Balthazard, M., Besnard, G., Sarah, G., Holtz, Y., Leclercq, J., Santoni, S., et al. (2019). Evolutionary transcriptomics reveals the origins of olives and the genomic changes associated with their domestication. Plant J. 100, 143–157. doi: 10.1111/tpj.14435
Gutierrez, A. P., Ponti, L., Cossu, Q. A. (2009). Effects of climate warming on Olive and olive fly (Bactrocera oleae (Gmelin)) in California and Italy. Clim. Change 95, 195–217. doi: 10.1007/s10584-008-9528-4
Hedayati, V., Mousavi, A., Razavi, K., Cultrera, N., Alagna, F., Mariotti, R., et al. (2015). Polymorphisms in the AOX2 gene are associated with the rooting ability of olive cuttings. Plant Cell Rep. 34, 1151–1164. doi: 10.1007/s00299-015-1774-0
Iannotta, N., Scalercio, S. (2012). “Susceptibility of cultivars to biotic stresses,” in Olive germplasm and olive cultivation, table olive and olive oil industry in Italy, id. I. Muzzalupo (Rijeka, Croatia: InTech), 81–106.
İpek, A., İpek, M., Ercişli, S., Tangu, N. A. (2017). Transcriptome-based SNP discovery by GBS and the construction of a genetic map for olive. Funct. Integr. Genomics 17, 493–501. doi: 10.1007/s10142-017-0552-1
Irisarri, P., Zhebentyayeva, T., Errea, P., Pina, A. (2019). Inheritance of self-and graft-incompatibility traits in an F1 apricot progeny. PloS One 14, e0216371. doi: 10.1371/journal.pone.0216371
Jaillon, O., Aury, J.-M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Ji, F., Wei, W., Liu, Y., Wang, G., Zhang, Q., Xing, Y., et al. (2018). Construction of a SNP-based high-density genetic map using genotyping by sequencing (GBS) and QTL analysis of nut traits in Chinese chestnut (Castanea mollissima Blume). Front. Plant Sci. 9, 816. doi: 10.3389/fpls.2018.00816
Kalinowski, S. T., Taper, M. L., Marshall, T. C. (2007). Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment: CERVUS LIKELIHOOD MODEL. Mol. Ecol. 16, 1099–1106. doi: 10.1111/j.1365-294X.2007.03089.x
Khadari, B., El Aabidine, A. Z., Grout, C., Sadok, I. B., Doligez, A., Moutier, N., et al. (2010). A genetic linkage map of olive based on amplified fragment length polymorphism, intersimple sequence repeat and simple sequence repeat markers. J. Am. Soc Hortic. Sci. 135, 548–555. doi: 10.21273/JASHS.135.6.548
Khan, M. A., Zhao, Y., (Frank), and Korban, S. S. (2013). Identification of genetic loci associated with fire blight resistance in Malus through combined use of QTL and association mapping. Physiol. Plant 148, 344–353. doi: 10.1111/ppl.12068
Kole, C., Muthamilarasan, M., Henry, R., Edwards, D., Sharma, R., Abberton, M., et al. (2015). Application of genomics-assisted breeding for generation of climate resilient crops: progress and prospects. Front. Plant Sci. 6, 563. doi: 10.3389/fpls.2015.00563
Kujur, A., Upadhyaya, H. D., Shree, T., Bajaj, D., Das, S., Saxena, M. S., et al. (2015). Ultra-high density intra-specific genetic linkage maps accelerate identification of functionally relevant molecular tags governing important agronomic traits in chickpea. Sci. Rep. 5, 9468. doi: 10.1038/srep09468
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357. doi: 10.1038/nmeth.1923
Laurens, F., Aranzana, M. J., Arus, P., Bassi, D., Bink, M., Bonany, J., et al. (2018). An integrated approach for increasing breeding efficiency in apple and peach in Europe. Hortic. Res. 5 (1), 11. doi: 10.1038/s41438-018-0016-3
Lavee, S., Avidan, B., Ben-Ari, G. (2014). Trends in breeding new olive varieties in Israel for quality and economic management. Agric. Sci. 5, 701. doi: 10.4236/as.2014.58073
Lenormand, T., Dutheil, J. (2005). Recombination difference between sexes: a role for haploid selection. PloS Biol. 3, e63. doi: 10.1371/journal.pbio.0030063
Li, Y., Lu, J., Chang, Y., Tang, W., Yang, Q. (2017). Comparative analysis of tree peony petal development by transcriptome sequencing. Acta Physiol. Plant 39 (10), 216. doi: 10.1007/s11738-017-2520-8
Liu, H., Cao, F., Yin, T., Chen, Y. (2017). A highly dense genetic map for Ginkgo biloba constructed using sequence-based markers. Front. Plant Sci. 8, 1041. doi: 10.3389/fpls.2017.01041
Lou, H., Dong, L., Zhang, K., Wang, D. W., Zhao, M., Li, Y., et al (2017). High‐throughput mining of E‐genome‐specific SNP s for characterizing Thinopyrum elongatum introgressions in common wheat. Mol. Ecol. Resour. 17, 1318–1329. doi: 10.1111/1755-0998.12659
Ma, L., Li, G. (2018). FAR1-related sequence (FRS) and FRS-related factor (FRF) family proteins in Arabidopsis growth and development. Front. Plant Sci. 9, 692. doi: 10.3389/fpls.2018.00692
Manaï, H., Haddada, F. M., Trigui, A., Daoud, D., Zarrouk, M. (2007). Compositional quality of virgin olive oil from two new Tunisian cultivars obtained through controlled crossings. J. Sci. Food Agric. 87, 600–606. doi: 10.1002/jsfa.2732
Marchese, A., Marra, F. P., Caruso, T., Mhelembe, K., Costa, F., Fretto, S., et al. (2016). The first high-density sequence characterized SNP-based linkage map of olive (Olea europaea L. subsp. europaea) developed using genotyping by sequencing. Aust. J. Crop Sci. 10, 857. doi: 10.21475/ajcs.2016.10.06.p7520
Migicovsky, Z., Myles, S. (2017). Exploiting wild relatives for genomics-assisted breeding of perennial crops. Front. Plant Sci. 8 (8), 460. doi: 10.3389/fpls.2017.00460
Min, Y., Bunn, J. I., Kramer, E. M. (2019). Homologs of the STYLISH gene family control nectary development in Aquilegia. New Phytol. 221 (2), 1090–1100. doi: 10.1111/nph.15406
Minamikawa, M. F., Takada, N., Terakami, S., Saito, T., Onogi, A., Kajiya-Kanegae, H., et al. (2018). Genome-wide association study and genomic prediction using parental and breeding populations of Japanese pear (Pyrus pyrifolia Nakai). Sci. Rep. 8, 11994. doi: 10.1038/s41598-018-30154-w
Montanari, S., Saeed, M., Knäbel, M., Kim, Y., Troggio, M., Malnoy, M., et al. (2013). Identification of pyrus single nucleotide polymorphisms (SNPs) and evaluation for genetic mapping in European pear and interspecific pyrus hybrids. PloS One 8, e77022. doi: 10.1371/journal.pone.0077022
Mousavi, S., Mariotti, R., Bagnoli, F., Costantini, L., Cultrera, N. G., Arzani, K., et al. (2017). The eastern part of the Fertile Crescent concealed an unexpected route of olive (Olea europaea L.) differentiation. Ann. Bot. 119, 1305–1318. doi: 10.1093/aob/mcx027
Mousavi, S., Stanzione, V., Mencuccini, M., Baldoni, L., Bufacchi, M., Mariotti, R. (2019). Biochemical and molecular profiling of unknown olive genotypes from central Italy: determination of major and minor components. Eur. Food Res. Technol. 245, 83–94. doi: 10.1007/s00217-018-3142-0
Muleo, R., Morgante, M., Cattonaro, F., Scalabrin, S., Cavallini, A., Natali, L., et al. (2016). “Genome sequencing, transcriptomics, and proteomics,” in The Olive Tree Genome (Cham: Springer), 141–161.
Muranty, H., Troggio, M., Sadok, I. B., Rifaï, M. A., Auwerkerken, A., Banchi, E., et al. (2015). Accuracy and responses of genomic selection on key traits in apple breeding. Hortic. Res. 2, 15060. doi: 10.1038/hortres.2015.60
Nicolas, S. D., Péros, J.-P., Lacombe, T., Launay, A., Le Paslier, M.-C., Bérard, A., et al. (2016). Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L) diversity panel newly designed for association studies. BMC Plant Biol. 16, 74. doi: 10.1186/s12870-016-0754-z
Ollitrault, P., Terol, J., Chen, C., Federici, C. T., Lotfy, S., Hippolyte, I., et al. (2012). A reference genetic map of C. clementina hort. ex Tan.; citrus evolution inferences from comparative mapping. BMC Genomics 13, 593. doi: 10.1186/1471-2164-13-593
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PloS One 7, e37135. doi: 10.1371/journal.pone.0037135
Picheny, V., Casadebaig, P., Trépos, R., Faivre, R., Da Silva, D., Vincourt, P., et al. (2017). Using numerical plant models and phenotypic correlation space to design achievable ideotypes. Plant Cell Environ. 40, 1926–1939. doi: 10.1111/pce.13001
Purba, A. R., Flori, A., Baudouin, L., Hamon, S. (2001). Prediction of oil palm (Elaeis guineensis, Jacq.) agronomic performances using the best linear unbiased predictor (BLUP). Theor. Appl. Genet. 102, 787–792. doi: 10.1007/s001220051711
Rallo, L., Barranco, D., de la Rosa, R., León, L. (2008). ‘Chiquitita’ Olive. HortScience 43, 529–531. doi: 10.21273/HORTSCI.43.2.529
Rallo, L., Barranco, D., de la Rosa, R., León, L. (2016). New olive cultivars and selections in Spain: results after 25 years of breeding. Acta Hortic. 1199, 21–26. doi: 10.17660/ActaHortic.2018.1199.4
Rugini, E., Cristofori, V., Silvestri, C. (2016). Genetic improvement of olive (Olea europaea L.) by conventional and in vitro biotechnology methods. Biotechnol. Adv. 34, 687–696. doi: 10.1016/j.biotechadv.2016.03.004
Sadok, I. B., Celton, J.-M., Essalouh, L., Aabidine, A. Z. E., Garcia, G., Martinez, S., et al. (2013). QTL mapping of flowering and fruiting traits in olive. PloS One 8, e62831. doi: 10.1371/journal.pone.0062831
Salman, M. (2017). Biological control of Spilocaea oleagina, the causal agent of olive leaf spot disease, using antagonistic bacteria. J. Plant Pathol. 99, 741–744. doi: 10.4454/jpp.v99i3.3958
Santos-Antunes, F., León, L., de la Rosa, R., Alvarado, J., Mohedo, A., Trujillo, I., et al. (2005). The length of the juvenile period in olive as influenced by vigor of the seedlings and the precocity of the parents. HortScience 40, 1213–1215. doi: 10.21273/HORTSCI.40.5.1213
Sapkota, S., Chen, L.-L., Yang, S., Hyma, K. E., Cadle-Davidson, L., Hwang, C.-F. (2019). Construction of a high-density linkage map and QTL detection of downy mildew resistance in Vitis aestivalis-derived ‘Norton’. Theor. Appl. Genet. 132, 137–147. doi: 10.1007/s00122-018-3203-6
Saumitou-Laprade, P., Vernet, P., Vassiliadis, C., Hoareau, Y., de Magny, G., Dommee, B., et al. (2010). A self-incompatibility system explains high male frequencies in an androdioecious plant. Science 327, 1648–1650. doi: 10.1126/science.1186687
Saumitou-Laprade, P., Vernet, P., Vekemans, X., Billiard, S., Gallina, S., Essalouh, L., et al. (2017a). Elucidation of the genetic architecture of self-incompatibility in olive: evolutionary consequences and perspectives for orchard management. Evol. Appl. 10 (9), 867–880. doi: 10.1111/eva.12457
Saumitou-Laprade, P., Vernet, P., Vekemans, X., Castric, V., Barcaccia, G., Khadari, B., et al. (2017b). Controlling for genetic identity of varieties, pollen contamination and stigma receptivity is essential to characterize the self-incompatibility system of Olea europaea L. Evol. Appl. 10, 860–866. doi: 10.1111/eva.12498
Saumitou-Laprade, P., Vernet, P., Dowkiw, A., Bertrand, S., Billiard, S., Albert, B., et al. (2018). Polygamy or subdioecy? the impact of diallelic self-incompatibility on the sexual system in Fraxinus excelsior (Oleaceae). Proc. R. Soc. B. Biol. Sci. 285, 20180004. doi: 10.1098/rspb.2018.0004
Scaglione, D., Fornasiero, A., Cattonaro, F., Mesa, K., Pinto, C., Shiny, P., et al. (2015). A RAD-based linkage map of kiwifruit (Actinidia chinensis Pl.) and map position of genetic determinants of traits of interest to breeders. Acta Hortic. 1096, 191–198. doi: 10.17660/ActaHortic.2015.1096.20
Suffert, F., Latxague, É., Sache, I. (2009). Plant pathogens as agroterrorist weapons: assessment of the threat for European agriculture and forestry. Food Secur. 1, 221–232. doi: 10.1007/s12571-009-0014-2
Trapero, C., Serrano, N., Arquero, O., Del Río, C., Trapero, A., López-Escudero, F. J. (2013). Field resistance to Verticillium wilt in selected olive cultivars grown in two naturally infested soils. Plant Dis. 97, 668–674. doi: 10.1094/PDIS-07-12-0654-RE
Unver, T., Wu, Z., Sterck, L., Turktas, M., Lohaus, R., Li, Z., et al. (2017). Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. 114, E9413–E9422. doi: 10.1073/pnas.1708621114
Urban, M. C. (2015). Accelerating extinction risk from climate change. Science 348, 571–573. doi: 10.1126/science.aaa4984
van Os, H., Stam, P., Visser, R. G., van Eck, H. J. (2005). SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor. Appl. Genet. 112, 187–194. doi: 10.1007/s00122-005-0124-y
Varshney, R. K., Graner, A., Sorrells, M. E. (2005). Genomics-assisted breeding for crop improvement. Trends Plant Sci. 10, 621–630. doi: 10.1016/j.tplants.2005.10.004
Vernet, P., Lepercq, P., Billiard, S., Bourceaux, A., Lepart, J., Dommée, B., et al. (2016). Evidence for the long-term maintenance of a rare self-incompatibility system in Oleaceae. New Phytol. 210, 1408–1417. doi: 10.1111/nph.13872
Vezzulli, S., Troggio, M., Coppola, G., Jermakow, A., Cartwright, D., Zharkikh, A., et al. (2008). A reference integrated map for cultivated grapevine (Vitis vinifera L.) from three crosses, based on 283 SSR and 501 SNP-based markers. Theor. Appl. Genet. 117, 499–511. doi: 10.1007/s00122-008-0794-3
Wu, S.-B., Collins, G., Sedgley, M. (2004). A molecular linkage map of olive (Olea europaea L.) based on RAPD, microsatellite, and SCAR markers. Genome 47, 26–35. doi: 10.1139/g03-091
Wu, Y., Bhat, P. R., Close, T. J., Lonardi, S. (2008). Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PloS Genet. 4 (10), e1000212. doi: 10.1371/journal.pgen.1000212
Yang, S., Fresnedo-Ramírez, J., Wang, M., Cote, L., Schweitzer, P., Barba, P., et al. (2016). A next-generation marker genotyping platform (AmpSeq) in heterozygous crops: a case study for marker-assisted selection in grapevine. Hortic. Res. 3, 16002. doi: 10.1038/hortres.2016.2
Zeballos, J. L., Abidi, W., Giménez, R., Monforte, A. J., Moreno, M. Á., Gogorcena, Y. (2016). Mapping QTLs associated with fruit quality traits in peach [Prunus persica (L.) Batsch] using SNP maps. Tree Genet. Genomes 12, 37. doi: 10.1007/s11295-016-0996-9
Zhai, H., Feng, Z., Li, J., Liu, X., Xiao, S., Ni, Z., et al. (2016). QTL analysis of spike morphological traits and plant height in winter wheat (Triticum aestivum L.) using a high-density SNP and SSR-based linkage map. Front. Plant Sci. 7, 1617. doi: 10.3389/fpls.2016.01617
Zheng, Y., Xu, F., Li, Q., Wang, G., Liu, N., Gong, Y., et al. (2018). QTL mapping combined with bulked segregant analysis identify SNP markers linked to leaf shape traits in Pisum sativum using SLAF sequencing. Front. Genet. 9, 615. doi: 10.3389/fgene.2018.00615
Zhigunov, A. V., Ulianich, P. S., Lebedeva, M. V., Chang, P. L., Nuzhdin, S. V., Potokina, E. K. (2017). Development of F1 hybrid population and the high-density linkage map for European aspen (Populus tremula L.) using RADseq technology. BMC Plant Biol. 17, 180. doi: 10.1186/s12870-017-1127-y
Zhu, Y., Yin, Y., Yang, K., Li, J., Sang, Y., Huang, L., et al. (2015). Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (Juglans regia L.). BMC Genomics 16, 614. doi: 10.1186/s12864-015-1822-8
Keywords: genetic map, Olea europaea, double digest restriction associated deoxyribonucleic acid sequencing, self-incompatibility, functional markers
Citation: Mariotti R, Fornasiero A, Mousavi S, Cultrera NGM, Brizioli F, Pandolfi S, Passeri V, Rossi M, Magris G, Scalabrin S, Scaglione D, Di Gaspero G, Saumitou-Laprade P, Vernet P, Alagna F, Morgante M and Baldoni L (2020) Genetic Mapping of the Incompatibility Locus in Olive and Development of a Linked Sequence-Tagged Site Marker. Front. Plant Sci. 10:1760. doi: 10.3389/fpls.2019.01760
Received: 27 August 2019; Accepted: 16 December 2019;
Published: 28 January 2020.
Edited by:
Inaki Hormaza, Institute of Subtropical and Mediterranean Hortofruticultura La Mayora (IHSM), SpainReviewed by:
Juan De Dios Alché, Experimental Station of Zaidín (EEZ), SpainCarlos Romero, Polytechnic University of Valencia, Spain
Copyright © 2020 Mariotti, Fornasiero, Mousavi, Cultrera, Brizioli, Pandolfi, Passeri, Rossi, Magris, Scalabrin, Scaglione, Di Gaspero, Saumitou-Laprade, Vernet, Alagna, Morgante and Baldoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciana Baldoni, bHVjaWFuYS5iYWxkb25pQGliYnIuY25yLml0
†These authors have contributed equally to this work.