 Yu Takahashi1*†
Yu Takahashi1*† Hiroaki Sakai2†
Hiroaki Sakai2† Hirotaka Ariga1
Hirotaka Ariga1 Shota Teramoto3
Shota Teramoto3 Takashi L. Shimada4,5Heesoo Eun2
Takashi L. Shimada4,5Heesoo Eun2 Chiaki Muto1
Chiaki Muto1 Ken Naito1Norihiko Tomooka1
Ken Naito1Norihiko Tomooka1- 1Research Center of Genetic Resources, National Agriculture and Food Research Organization, Tsukuba, Japan
- 2Research Center of Advanced Analysis, National Agriculture and Food Research Organization, Tsukuba, Japan
- 3Institute of Crop Science, National Agriculture and Food Research Organization, Tsukuba, Japan
- 4Graduate School of Horticulture, Chiba University, Matsudo, Japan
- 5Plant Molecular Science Center, Chiba University, Inage-ku, Japan
To increase food production under the challenges presented by global climate change, the concept of de novo domestication—utilizing stress-tolerant wild species as new crops—has recently gained considerable attention. We had previously identified mutants with desired domestication traits in a mutagenized population of the legume Vigna stipulacea Kuntze (minni payaru) as a pilot for de novo domestication. Given that there are multiple stress-tolerant wild legume species, it is important to establish efficient domestication processes using reverse genetics and identify the genes responsible for domestication traits. In this study, we identified VsPSAT1 as the candidate gene responsible for decreased hard-seededness, using a Vigna stipulacea isi2 mutant that takes up water from the lens groove. Scanning electron microscopy and computed tomography revealed that the isi2 mutant has lesser honeycomb-like wax sealing the lens groove than the wild-type, and takes up water from the lens groove. We also identified the pleiotropic effects of the isi2 mutant: accelerating leaf senescence, increasing seed size, and decreasing numbers of seeds per pod. While doing so, we produced a V. stipulacea whole-genome assembly of 441 Mbp in 11 chromosomes and 30,963 annotated protein-coding sequences. This study highlights the importance of wild legumes, especially those of the genus Vigna with pre-existing tolerance to biotic and abiotic stresses, for global food security during climate change.
1 Introduction
Owing to the effects of climate change on food production, de novo domestication of stress-tolerant wild species has recently gained considerable attention (Fernie and Yan, 2019; Gasparini et al., 2021; Razzaq et al., 2021). Feasibility studies have been conducted in which domestication-related traits using new genetic technologies, such as genome editing or reverse genetic approach to mutagenesis, have been introduced in wild grass and vegetable species (Shapter et al., 2013; Dorn et al., 2015; Zsögön et al., 2018; Yu et al., 2021). In legumes, wild species of the genus Vigna Savi are candidates for de novo domestication, as they demonstrate remarkable tolerance to biotic and abiotic stresses, including salinity, drought, high/low pH, and flooding (Tomooka et al., 2014). We previously reported the first step of domesticating Vigna stipulacea Kuntze by ethyl methanesulfonate mutagenesis followed by phenotype screening (Takahashi et al., 2019). This species inhabits mainly South Asia and has a short growing period, heat tolerance, and resistance to multiple biotic stresses (Tomooka et al., 2011; van Zonneveld et al., 2020). Its seeds are edible, and it is mainly locally cultivated as pasture but sometimes as food. However, only a few farmers cultivate V. stipulacea because of the high labor caused by the strong behavior of pod-shattering and hard-seededness, which needs to be released for uniform germination (Tomooka et al., 2011). To improve these traits, we screened and obtained mutants with reduced pod-shattering and with decreased hard-seededness (Takahashi et al., 2019). As there are multiple stress-tolerant wild legumes, it is important to establish a domestication process with new genetic technologies. However, this process requires the identification of genes relevant to desired domestication-related traits.
One such trait is seed dormancy, defined by Foley (2001) as “the temporary failure of a viable seed to germinate, after a specific length of time and in a particular set of environmental conditions that allow germination after the restrictive state has been terminated by either natural or artificial conditions (Simpson, 1990).” It is an adaptive trait for the temporal control of germination in response to variable external environments. There are several types of seed dormancy including physical, mechanical, and chemical inhibition by the outer layers of the embryo, inability to germinate because of an undifferentiated or immature embryo, and repression of germination by metabolic restraints (Bewley et al., 2013). Among these, physical seed dormancy, identified in 18 angiosperms, is characterized by water impermeability of seed or fruit coats (Gama-Arachchige et al., 2013). Hard-seededness, the primary cause of physical seed dormancy in legumes, is provided by a palisade layer of lignified macrosclerids, impregnated with water-repellent phenolic and suberin-like compounds. Hard-seededness is overcome by opening water gap structures, such as the lens in legumes (Steinbrecher and Leubner-Metzger, 2018). Moreover, temperature- and moisture-dependent stimuli cause irreversible opening of the water gap.
Major domesticated legumes have decreased hard-seededness which has been associated with one to six loci, depending on species, during domestication (Smýkal et al., 2014). One of the genes controlling hard-seededness in soybeans (Glycine max (L.) Merr.) is GmHs1-1, which encodes a calcineurin-like transmembrane metallophosphoesterase associated with seed coat calcium content (Sun et al., 2015). In the common bean (Phaseolus vulgaris L.), a 5-bp frameshift in the gene encoding pectin acetylesterase-8-2 (PAE8-2) has been reported as a candidate causal variant that accelerates lens-specific water uptake by expanding microcracks within the lens groove (Soltani et al., 2021). However, it is unclear whether such single mutations are sufficient to decrease hard-seededness in wild species. In barrel clover (Medicago truncatula Gaertn.), a loss of function mutation in a class II KNOTTED-like homeobox (KNOX) gene, KNOX4, forms dysfunctional palisade cuticular layer due to altered composition of lipid monomer in the seed coat, leading to decreased hard-seededness (Chai et al., 2016).
We have previously identified three V. stipulacea mutants that decrease hard-seededness—increased seed imbibition 1 (isi1), isi2, and isi3—from ethyl methanesulfonate mutagenesis (Takahashi et al., 2019). The isi1 mutant begins taking up water through the entire seed coat within minutes of watering whereas the isi2 mutant takes up water through the seed’s lens, requiring a week before 80% of seeds complete water uptake. We also identified VsCESA7, which encodes a putative ortholog of cellulose synthase A catalytic subunit 7 in Arabidopsis (AtCESA7/IRX3/MUR10, AT5G17420), as a candidate gene for the isi1 phenotype (Takahashi et al., 2019) using whole-genome sequencing, which facilitated bulked segregant analysis. However, it was challenging to perform bulked segregant analysis in our previous study because the V. stipulacea genome was fragmented, with 2,102 scaffolds. In such cases, it is important to obtain chromosome-level assemblies.
In the present study, we report another bulked segregant analysis that allowed us to identify a gene responsible for the isi2 phenotype. We constructed a chromosome-level genome assembly, with annotation of 30,963 protein-coding genes. We then resequenced the bulked segregant derived from wild-type and the isi2 individuals, to identify VsPSAT1, a putative ortholog of Arabidopsis PSAT1, encoding phospholipid sterol acyltransferase 1.
2 Materials and methods
2.1 Plants
We used V. stipulacea (accession JP252948 in NARO Genebank) and its ethyl methanesulfonate-induced isi2 mutant (Takahashi et al., 2019). Plants were cultivated in 5-L pots with seven biological replicates in a single greenhouse maintained at 25°C or higher. Plants were scored for days to flowering, height 46 days after sowing, largest terminal leaflet width and length, shoot dry weight, seeds per pod, pod length, 100-seed weight, and water uptake in 20 seeds per individual following storage at 4°C for 1, 6, and 24 months after harvest. We counted the number of seeds taking up water at 1, 2, 3, 7, 10, 14, 21, and 28 days after watering. Data for one-month-old seeds and six-month-old seeds were published in our previous study (Takahashi et al., 2019).
We used the SPAD chlorophyll meter (SPAD-502, KONICA MINOLTA, INC., Tokyo, Japan) to measure early senescence. Fully expanded third terminal leaflets were detached from plants cultivated for one month in 5-L pots with 5 biological replicates in a single greenhouse maintained at 30°C or lower, then incubated on the water surface in 9-cm Petri dishes for 28 days under a cycle of 12 h of light at 25°C and 12 h of darkness at 22°C.
2.2 X-Ray computed tomography
Water entry points were identified using the method of Soltani et al. (2021). Seeds were submerged in Lugol solution containing 33 g/L of I2 (092-05422, FUJIFILM Wako Pure Chemical, Tokyo, Japan) and 66 g/L of KI (160-03952, FUJIFILM Wako Pure Chemical) as a contrasting agent for 6 h at 25°C, followed by rinsing with water and drying with a paper towel. Seeds were imaged using an inspeXio SMX-225CT FPD HR (Shimadzu Corporation, Kyoto, Japan). Each scan included 1200 projections, averaging two frames over 360° (pixel detector resolution 3000 × 3000), at 4 frames per second. Finally, 1024 × 1024-pixel slices were computed at a final spatial resolution of 15 μm. Tube voltages of 75 kV and tube currents of 100 μA were used. Metal filters were not used during imaging. Reconstructed CT volumes were visualized using VG Studio MAX 3.2 software (Volume Graphics, Heidelberg, Germany). Regions with strong signal intensity were assumed to be Lugol solution-permeated.
2.3 Scanning electron microscopy
Seed surface morphologies were analyzed using a JEOL JSM-5610 instrument (LV, Tokyo, Japan). Samples were coated using a fully automated sputter coater (JFC-1600, JEOL Ltd., Tokyo, Japan) with fine-grained platinum for 100 s at 80 mA. The hilum side and the lens groove were photographed at 50X and 500X magnifications, respectively.
2.4 Optical microscopy
Before drying, mature seeds were sectioned at 200 μm using a Plant Microtome MTH-1 (Nippon Medical & Chemical Instruments Co., Ltd., Osaka, Japan). Sections were stained with 0.01% toluidine blue O and observed under an ECLIPSE Ci (OLYMPUS, Tokyo, Japan).
2.5 Whole-genome sequencing and annotation
Genomic DNA from wild-type plants was extracted using the NucleoBond HMW DNA extraction kit (Macherey-Nagel, Düren, Germany) and sequenced using the PacBio Sequel II platform (Pacific Biosciences, Menlo Park, CA, USA). HiFi reads were created using the ccs and lima tools (https://github.com/PacificBiosciences/pbbioconda) with “–min-passes 5 –min-rq 0.995” and “–min-score 26 –min-length 10000” options, respectively. De novo assembly of the HiFi reads was performed using Canu ver. 2.0 (Koren et al., 2017), followed by the removal of haplotigs using Purge_Dups (Guan et al., 2020) with default settings.
To construct a chromosome-level assembly of the wild-type genome, we performed RAD-seq using 384 F2 plants derived from a cross between wild-type and another accession JP252958. Genomic DNA was extracted from young leaves using the CTAB method, digested with BglII and EcoRI, ligated with Y-shaped adaptors, amplified using polymerase chain reaction (PCR) with KAPA HiFi HS ReadyMix (Kapa Biosystems Inc., Wilmington, MA, USA), and size-selected using E-Gel size select (Life Technologies, Carlsbad, CA, USA). Approximately 450-bp library fragments were retrieved. Further details of the library preparation have been previously described (Sakaguchi et al., 2015). Sequencing was performed using the paired-end 151-bp mode of HiSeq X (Illumina Inc., San Diego, CA, USA). Quality filtering was performed using Trimmomatic ver. 0.39 (Bolger et al., 2014). Filtered sequence reads were mapped to contigs using BWA-MEM ver. 0.7.17 (Li and Durbin, 2009) with the “-q 20 -F 0×100” option. Single nucleotide polymorphisms (SNPs) were called using Stacks ver. 2.53 (Rochette et al., 2019) using default settings. Missing genotypes were imputed using ABHGenotypeR (Furuta et al., 2017). We removed SNPs for which genotypes were missing in more than 30% of the 384 samples or allele frequencies deviated from Hardy–Weinberg equilibria (p < 1.0E-10). The genetic map was constructed using OneMap software (Margarido et al., 2007) with the “max.rf=0.14” option (for linkage grouping) and the Kosambi map function. SNPs were ordered using three algorithms: “rec,” “rcd,” and “ug.” Chromosome sequences were reconstructed by aligning contigs on the genetic map according to the order of the SNPs.
Protein-coding genes were identified as previously described (Takahashi et al., 2019). For ab initio- and transcriptome-based gene prediction, we used the RNA-Seq data published by Takahashi et al. (2019). We combined protein sequence data deposited in UniProt (UniProt Consortium, 2021) with those of 10 Vigna species downloaded from the Vigna genome server (Sakai et al., 2016): V. angularis; Vigna exilis Tateishi and Maxted; Vigna indica T. M. Dixit, K. V. Bhat, and S. R. Yadav; Vigna marina (Burm. f.) Merr.; Vigna minima (Roxb.) Ohwi and Ohashi; Vigna mungo (L.) Hepper; Vigna riukiuensis (Ohwi) Ohwi and Ohashi; Vigna trilobata (L.) Verdc.; Vigna vexillata (L.) A. Rich.; and Vigna sp. (NI1135 in Meise Botanic Garden, Belgium). These data were used to create a non-redundant set of sequences using MMseqs2 (Steinegger and Söding, 2017) with the “–min-seq-id 0.99 -c 0.99 –cov-mode 0” options. We annotated transposable elements using the Extensive de novo Transposable Element Annotator with default settings (Ou et al., 2019).
2.6 Detecting presence variations
V. stipulacea and V. angularis genomes were aligned one-to-one using LAST (Frith and Noé, 2014). Furthermore, unaligned regions (≥100 bp) in one species where flanking sequences (≥1000 bp for each end) were aligned with another species with no large gaps (>100 bp) (PVs candidates) were extracted. PV coordinates were determined by aligning each PV and flanking sequences of the two species using MAFFT (Katoh and Standley, 2013).
2.7 Genetic analysis
F2 plants were produced by crossing the wild-type with the isi2 mutant. In total, 288 F2 plants were cultivated in 1-L pots; DNA was extracted from their young leaves, and water uptake in 20 one-month-old seeds 10 days after watering and 20-seed weight (to estimate 100-seed weight) were measured. Five plants exhibiting the wild-type phenotype that did not produce sufficient seeds were excluded from the evaluation of seed weight. A total of 163 F3 plants derived from 11 F2 plants with recombination among the SNP positions 31,842,848, 32,352,997, and 32,970,540 nt on chromosome 4 were cultivated in 0.2-L pots; DNA was extracted from their young leaves, and water uptake in 10 2-week-old seeds 28 days after watering, 10-seed weights (to calculate 100-seed weights), and numbers of seeds per pod for three pods were measured.
2.8 Bulked segregant analysis
We adopted the MutMap strategy (Abe et al., 2012) to identify candidate SNPs for the isi2 phenotype. Two DNA pools of 215 wild-type phenotypes and 73 isi2 phenotypes in the F2 plants from the wild-type and the isi2 mutant were sequenced using the paired-end 151-bp mode of HiSeq X (Illumina). Sequence reads were quality-filtered using Trimmomatic and mapped to the genome assembly using BWA-MEM. SNPs were identified using GenotypeGVCFs of GATK ver. 4.2.1.0 (Van der Auwera and O’Connor, 2020) and filtered with the “QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0” options of VariantFiltration implemented in GATK. The retained variants were used to calculate SNP indices. Each variant was functionally annotated with the SnpEff ver. 4.3t (Cingolani et al., 2012) using annotated gene structures.
2.9 Amplicon sequencing
The genotypes for the SNPs at 31,842,848 nt, 32,352,997 nt, and 32,970,540 nt on chromosome 4 were determined for 288 F2 and 163 F3 plants. Primers used for PCR and Sanger sequencing are shown in Table 1. For Sanger sequencing, we amplified template DNA using AmpliTaq Gold 360 Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) followed by sequencing with BigDye Terminator v3.1 (Thermo Fisher Scientific) and an ABI Genetic Analyzer 3130xl (Thermo Fisher Scientific) according to the manufacturer’s protocols.
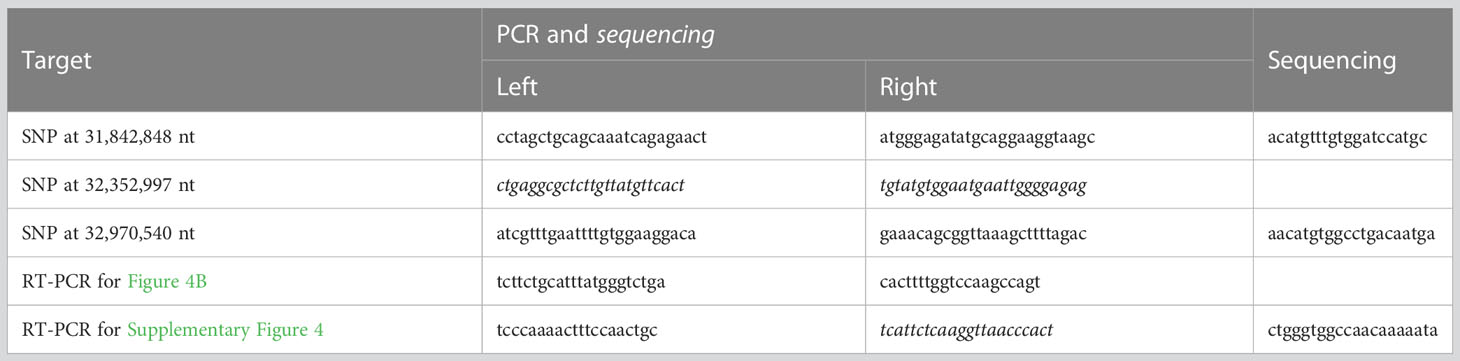
Table 1 PCR and sequencing primers.
2.10 Reverse transcription (RT)-PCR
Young pods of approximately 5 mm in length 3 days after flowering and unexpanded third leaves were harvested. Total RNA was extracted using the RNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Vigst.04G198600 cDNA was amplified using the PrimeScript One Step RT-PCR Kit Ver. 2 (TAKARA Bio Inc., Shiga, Japan) with the primers shown in Table 1. DNA extracted from unexpanded third leaves was used as a negative control. Electrophoresis was performed using 2% agarose gels.
2.11 Orthology analysis
To identify Vigst.04G198600 orthologs in the genomes of closely related legume species, we obtained protein sequences from 11 Vigna species from the Vigna genome server as described above, and Phytozome (for V. unguiculata), as well as those of P. vulgaris, G. max, and Glycine soja Siebold and Zucc. from Phytozome (Goodstein et al., 2012). We ran Orthofinder ver. 2.5.4 (Emms and Kelly, 2019) using default settings to identify the single orthogroup including Vigst.04G198600. We measured collinearity between V. stipulacea and each of the legume species using MCScanX and confirmed that all genes in the orthogroup were syntenic with Vigst.04G198600. To construct a phylogenetic tree of orthologs, we created codon alignments and generated a neighbor-joining tree using the Nei–Gojobori method in MEGA ver. 10 (Kumar et al., 2018). Amino-acid sequences were aligned using the Clustal Omega multiple sequence alignment tool (http://www.ebi.ac.uk/Tools/msa/clustalo/).
3 Results
3.1 Decreased hard-seededness of isi2
Seeds preserved at 4°C for one, six, and twenty-four months after harvest were submerged in water. In wild-type V. stipulacea, water uptake at one month after watering was 1% in 1-month-old seeds, 11% in 6-month-old seeds, and 28% in 2-year-old seeds. These results indicate that water uptake was low in young seeds but higher in older wild-type seeds (Figure 1A, Supplementary Table 1). In contrast, water uptake in isi2 mutant seeds was consistently higher at 96–98% a month after submersion regardless of the storage period but tended to be higher for older seeds until 3 days of submersion (Figure 1A, Supplementary Table 1).
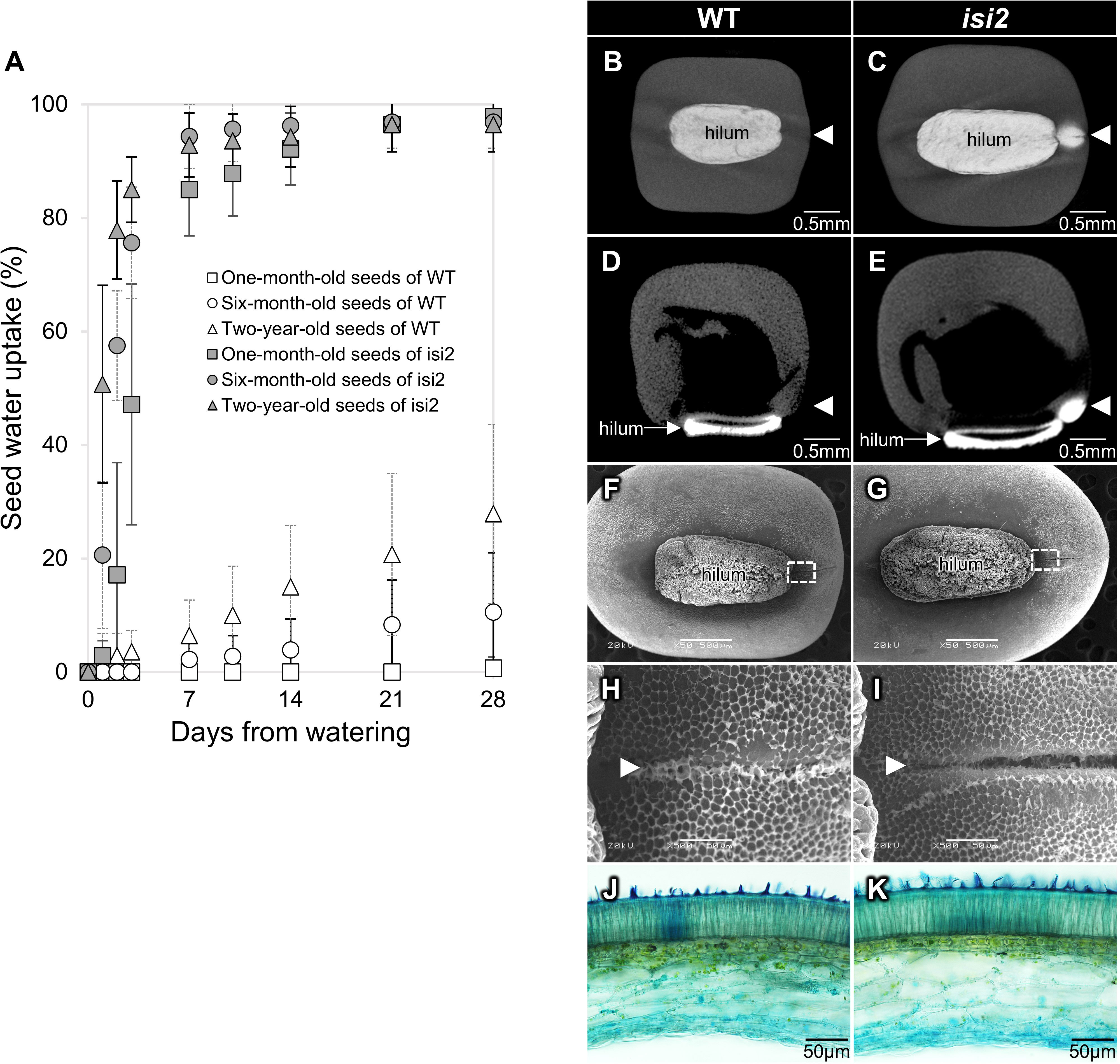
Figure 1 Characterization of the isi2 mutant. Seed water uptake over time as a function of storage period (A). White shapes indicate the wild-type and gray shapes indicate the isi2 mutant. Squares, one-month-old seeds; circles, six-month-old seeds; triangles, two-year-old seeds. N=7. 3D-rendered images facing the hilum (B, C) and longitudinal sections of centers (D, E) from computed tomography (CT) scans of wild-type (B, D) and the isi2 mutant (C, E) seeds six hours after submergence in a contrasting agent. White areas indicate the presence of the agent. The white arrowheads indicate the lens. 50X images facing the hilum with a 500 μm scale bar (F, G), and 500X images of the dotted square photographs F and G with a 50 μm scale bar (H, I) from scanning electron microscopy (SEM) of the surfaces of the wild-type (F, H) and isi2 mutant (G, I) seeds. White arrowheads indicate the lens groove. Cross sections of coats observed under an optical microscope of wild-type (J) and the isi2 mutant (K) seeds.
In addition, CT scanning identified water entry at the lens in the isi2 mutant (Figure 1B, C). Longitudinal sections showed entry into the cotyledon through the lens (Figure 1D, E). SEM revealed that the seed coat microstructure of isi2 was different from that of the wild-type. The mutant had decreased honeycomb-like epicuticular wax deposition in the lens groove compared to the wild-type; however, no differences were observed in the entire seed coat (Figure 1F-I, Supplementary Figure 1). Similarly, optical microscopy revealed no reproducible differences in seed coat cross sections between the mutant and the wild-type (Figure 1J, K). Three biological replicates are shown in the Supplementary Figure 2.
3.2 Whole-genome sequencing and annotation
We sequenced the wild-type genome and obtained 20.62 Gbp of HiFi reads. De novo assembly generated 823 contigs with a total length of 413.48 Mbp and an N50 length of 11.87 Mbp (Table 2). We constructed a genetic map of the 11 chromosomes based on 1,163 SNP markers derived from RAD-seq and anchored 44 contigs to it. These anchored contigs spanned 387.57 Mbp, covering 88% of the estimated genome size (441.54 Mbp). The repetitive fraction of the genome was 38% (156.62 Mbp), smaller than that of the V. angularis genome (45%). The most abundant class of repetitive elements was Gypsy type LTR-retrotransposons (7.5%), followed by unknown (6.8%) and Copia type (6.4%) LTR-retrotransposons.
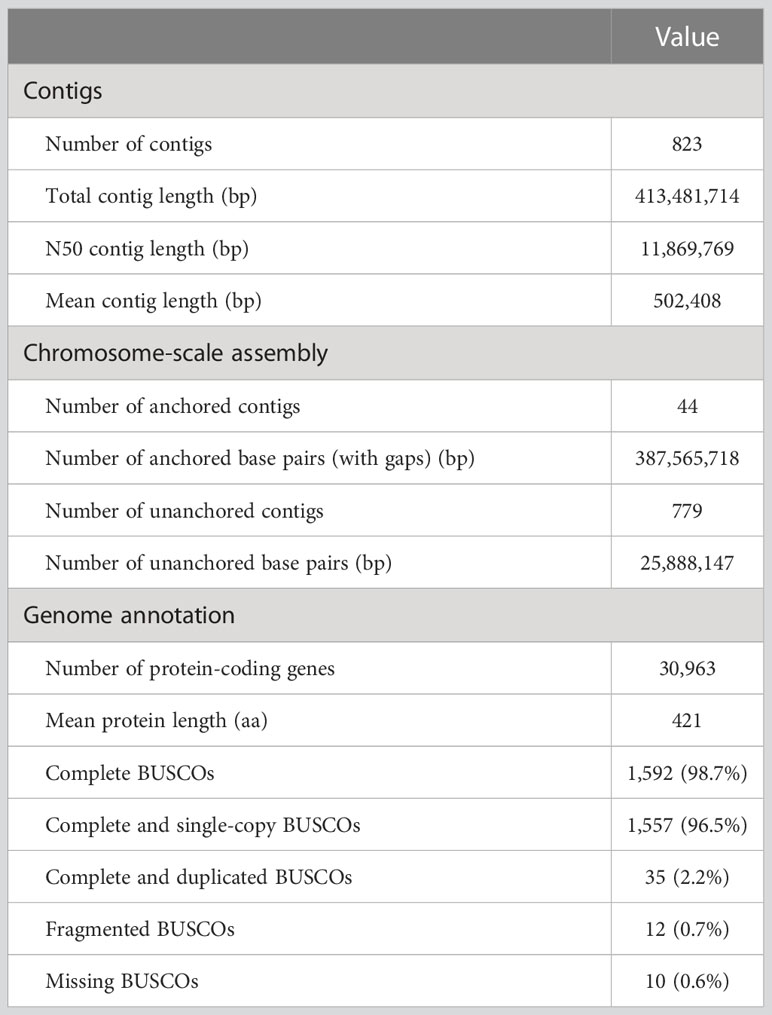
Table 2 Vigna stipulacea genome assembly statistics.
We identified 30,963 protein-coding genes by combining ab initio-based, transcriptome-based, and protein-based approaches (Table 2). BUSCO analysis of the embryophyta_odb10 dataset showed 98.7% completeness in the predicted proteins, slightly higher than our previous version (95.8%; Takahashi et al., 2019).
In addition, we assessed synteny between the genomes of V. stipulacea and azuki bean (Vigna angularis (Willd.) Ohwi et H. Ohashi) and between V. stipulacea and P. vulgaris. The genomes of V. stipulacea and V. angularis were highly syntenic across all 11 chromosomes. We detected 669 synteny blocks between V. stipulacea and V. angularis with 23,574 (76%) of the 30,963 genes overlapping. In contrast, the genomes of V. stipulacea and P. vulgaris had several structural differences, including translocations and inversions (Figure 2).
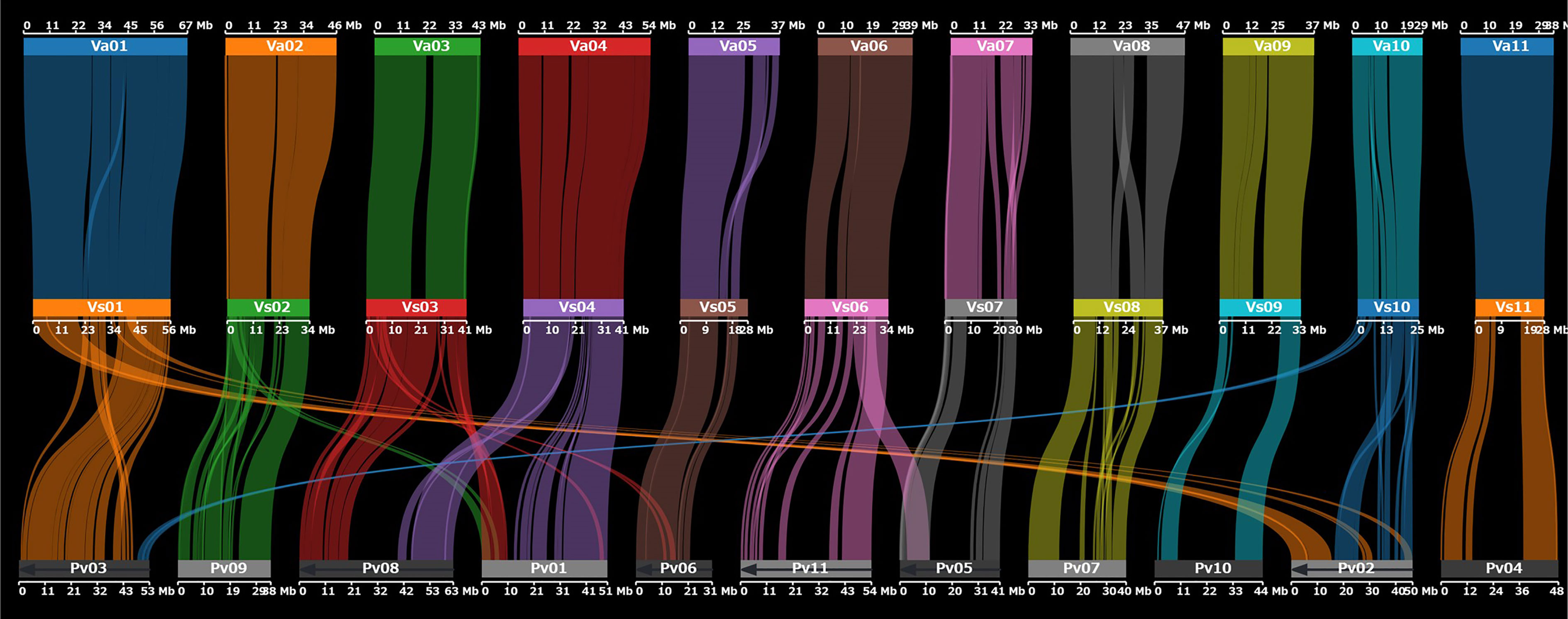
Figure 2 Dot plot showing chromosomal synteny between V. stipulacea (middle) and V. angularis (top) and between V. stipulacea and P. vulgaris (bottom).
Despite the high synteny between V. stipulacea and V. angularis, precise comparisons based on the whole-genome alignment revealed several structural variations. We detected 6,298 presence variations (PVs) in the V. stipulacea genome, with a total length of 5,094,208 bp, whereas the V. angularis genome harbored 8,090 PVs with a total length of 9,822,523 bp. Of the 5,094,208 bp of PVs in V. stipulacea, 3,154,013 (62%) were annotated as repetitive elements whereas 27 protein-coding genes were annotated in the PV regions.
3.3 Mapping of the isi2 candidate loci
Upon testing the genetic factor, we concluded that the isi2 phenotype was caused by a mutation in a single locus according to the law of independence. We defined the isi2 phenotype as one in which more than 80% of seeds exhibited water uptake within one to four weeks after watering. Among F2 (WT × isi2) plants, 215 showed the wild-type phenotype and 73 showed the isi2 phenotype, consistent with the expected 3:1 Mendelian ratio (216:72; p = 0.89, based on the Chi-square test; Supplementary Table 2).
For bulked segregant analysis, we pooled F2 (WT × isi2) plants into wild-type and isi2 phenotypes and sequenced each of them. We identified 61,664 SNPs across 11 chromosomes with an average depth of 64.6 for the wild-type pool and 70.8 for the mutant pool. Of these, 9 located between 31,842,848 nt and 35,015,640 nt were completely or nearly fixed with non-reference alleles (Figure 3, Supplementary Figure 3, Table 3). Of the nine SNPs, two were located in the coding regions of Vigst.04G193900 and Vigst.04G214200 and were predicted to cause single-residue substitutions whereas one was located in the splice donor site of intron 10 of Vigst.04G198600 (Figure 4A, Table 3). The other six were located in intergenic regions or introns and were not further characterized.
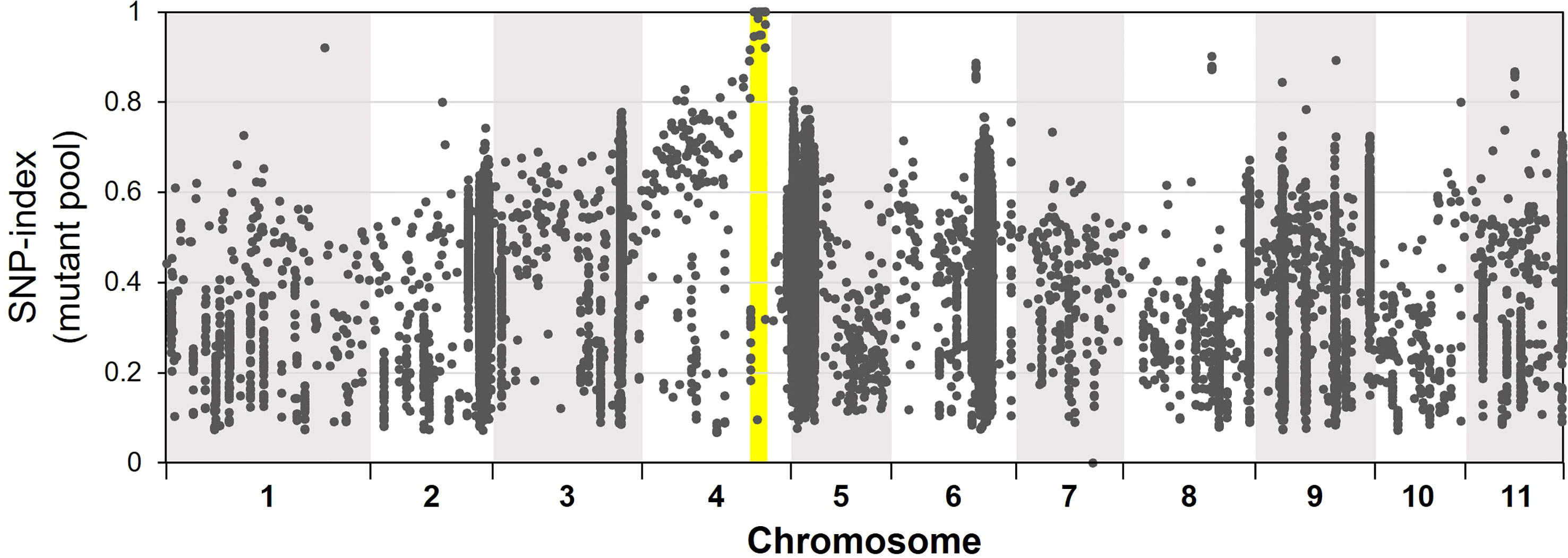
Figure 3 Frequencies of mutant alleles (SNP index) in the mutant pool of the F2 (WT × isi2) plants. The x-axis indicates physical positions across chromosomes and the y-axis indicates mutant allele frequencies for each SNP. Yellow highlighting indicates the locus fixed with mutant alleles.
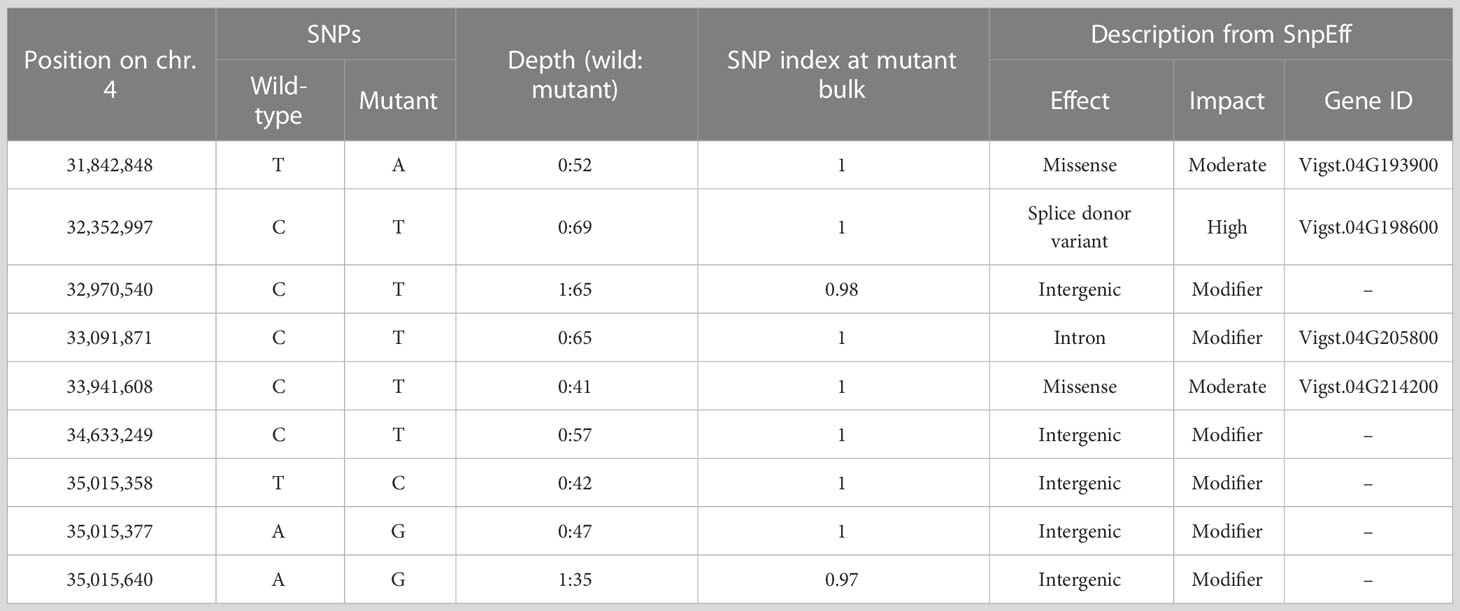
Table 3 SNP loci having zero or one wild allele in the mutant bulk of F2 (WT × isi2) plants.
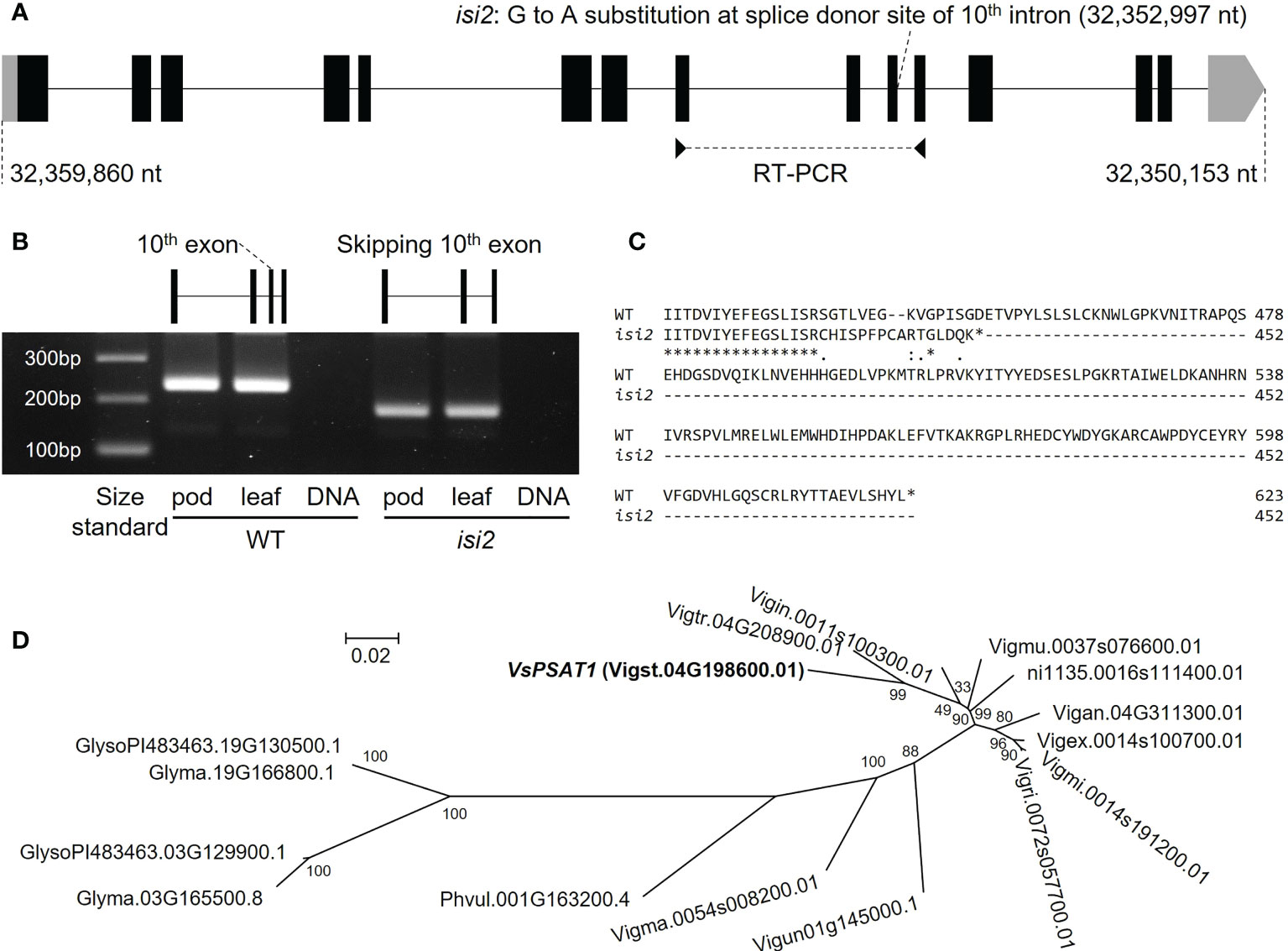
Figure 4 VsPSAT1 transcripts and putative orthologs. (A) Schematic showing differences between wild-type and isi2 alleles. Boxes indicate exons; lines indicate introns. Coding DNA sequences (CDS) are filled with black. A G>A substitution at the splice donor site between exon 10 and intron 10 is present at 32,352,997 nt. (B) RT-PCR products. Primer pairs were designed from the sequences of exons 8 and 11. The schematic shows the skipping of exon 10 in the isi2 mutant. (C) Predicted amino-acid sequences of wild-type and mutant VsPSAT1. Asterisks indicate conserved residues. Colons indicate similar residues; periods indicate weakly similar residues. Hyphens indicate gaps introduced to optimize the alignment. (D) Phylogeny of putative PSAT1 orthologs in the tribe Phaseoleae. The operational taxonomic units are amino-acid sequences.
To determine which of the three SNPs is responsible for the isi2 phenotype, we genotyped 288 F2 (WT × isi2) and 163 F3 (WT × isi2) plants for the T>A SNP at 31,842,848 nt, the C>T SNP at 32,352,997 nt, and the C>T SNP at 32,970,540 nt on chromosome 4. Complete linkage was found only for the C>T SNP at 32,352,997 nt, whereas recombination was observed between this SNP and both of the other two SNPs (Table 4, Supplementary Table 2). Data from 163 F3 (WT × isi2) plants derived from 11 F2 (WT × isi2) plants with recombination among the three SNPs were consistent with the F2 data (Table 5, Supplementary Table 3). Thus, the C>T SNP at 32,352,997 nt, located in the splice donor site of the 10th intron in Vigst.04G198600, was the single remaining candidate for the isi2 phenotype.
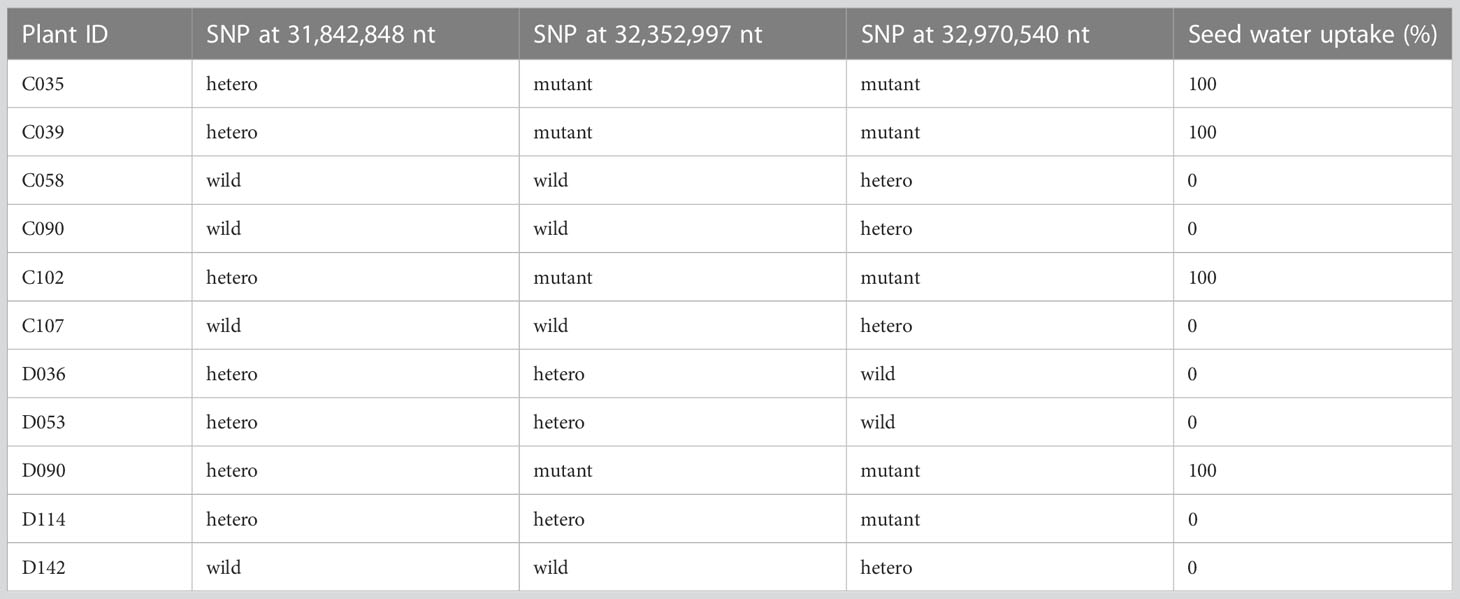
Table 4 Genotypes and phenotypes of recombinants among SNPs in F2 (WT × isi2) plants.
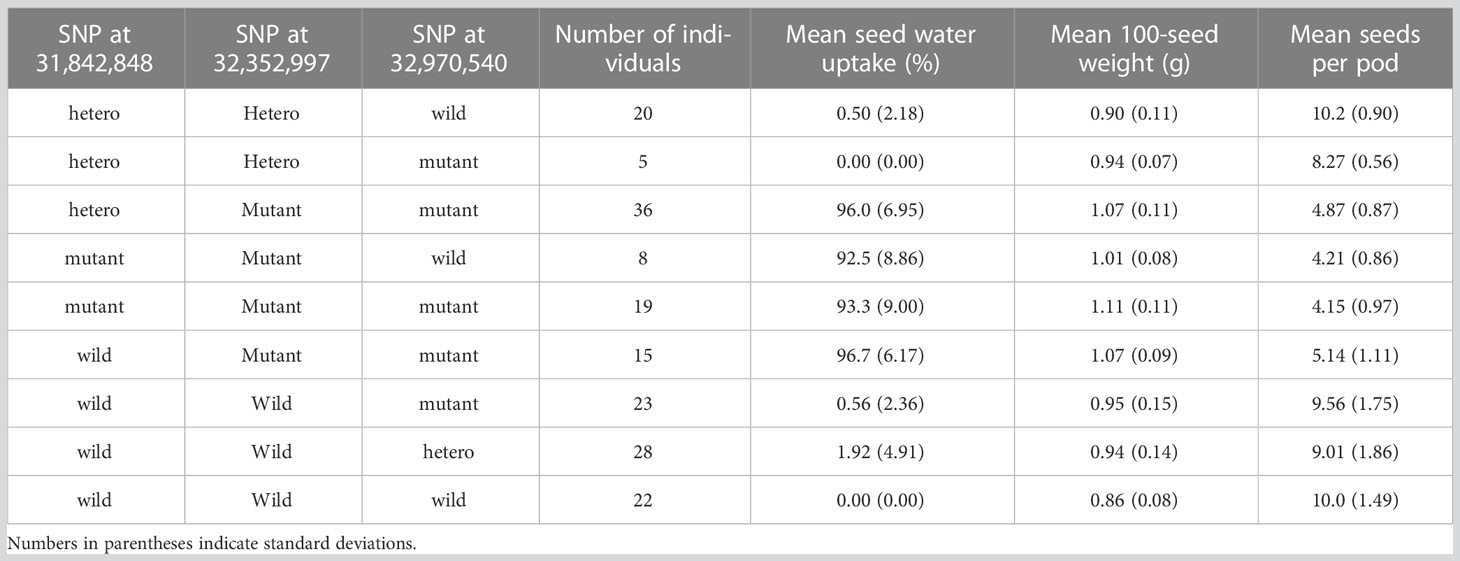
Table 5 Genotypes and phenotypes of recombinants among SNPs in F3 (WT × isi2) plants.
3.4 VsPSAT1 in the isi2 mutant and other legumes
A BLAST search identified Vigst.04G198600 as a putative ortholog of the A. thaliana gene encoding phospholipid sterol acyltransferase 1 (AtPSAT1/ERP1, AT1G04010). Therefore, we renamed it VsPSAT1. Querying the V. stipulacea genome with the amino-acid sequence of AtPSAT1 identified only VsPSAT1. The predicted VsPSAT1 protein is 623 residues in length, with an overall identity of 76% with AtPSAT1. The conserved LCAT-like domain (Banas et al., 2005; Lara et al., 2018) has a total of 41 residues in six regions, of which 5 residues in 3 regions were replaced by strongly similar residues, and all 3 residues of the catalytic triad Ser-Asp-His were identical (Figure 5).

Figure 5 Protein sequence alignment of V. stipulacea and A. thaliana PSAT1. Asterisks indicate conserved residues. Colons indicate similar residues; periods indicate weakly similar residues. Hyphens indicate gaps introduced to optimize the alignment. Six conserved regions of LCAT-like proteins are highlighted in gray, and the conserved catalytic triad (Ser-Asp-His) is highlighted in black.
We performed RT-PCR to determine whether the SNP at the splice donor site affected the mRNA sequence of VsPSAT1. The region between the exons 8 and 11 in the isi2 mutant was approximately 50 bp shorter than that in the wild-type (Figure 4B). Sequencing revealed that exon 10 had been skipped (Supplementary Figure 4), leading to a frameshift and an ectopic stop codon that truncated the protein from 623 to 452 residues (Figure 4C).
We next searched for putative orthologs in species of the tribe Phaseoleae Bronn ex DC., specifically other Vigna species, P. vulgaris, and G. max. The diploids (Vigna species and P. vulgaris) had one copy and the palaeopolyploid soybean had two copies (Figure 4D), without any significant degradation in the conservation of the LCAT-like proteins, including the catalytic triad (Supplementary Figure 5).
3.5 Pleiotropic effects of the isi2 mutant
As the A. thaliana AtPSAT1 mutant exhibits early leaf senescence due to overaccumulation of sterol esters (Bouvier-Navé et al., 2010), we investigated whether this phenotype occurs in the isi2 mutant plants. Browning was observed at isi2 leaflet margins four weeks after detached leaves were floated in water (Figure 6A, B). The SPAD value, indicating chlorophyll content, was significantly (p < 0.05, t-test) different between the wild-type (27.3; SD = 1.37) and isi2 mutant (20.5; SD = 1.81); however, it was not significantly different between both intact leaves (Supplementary Table 4).
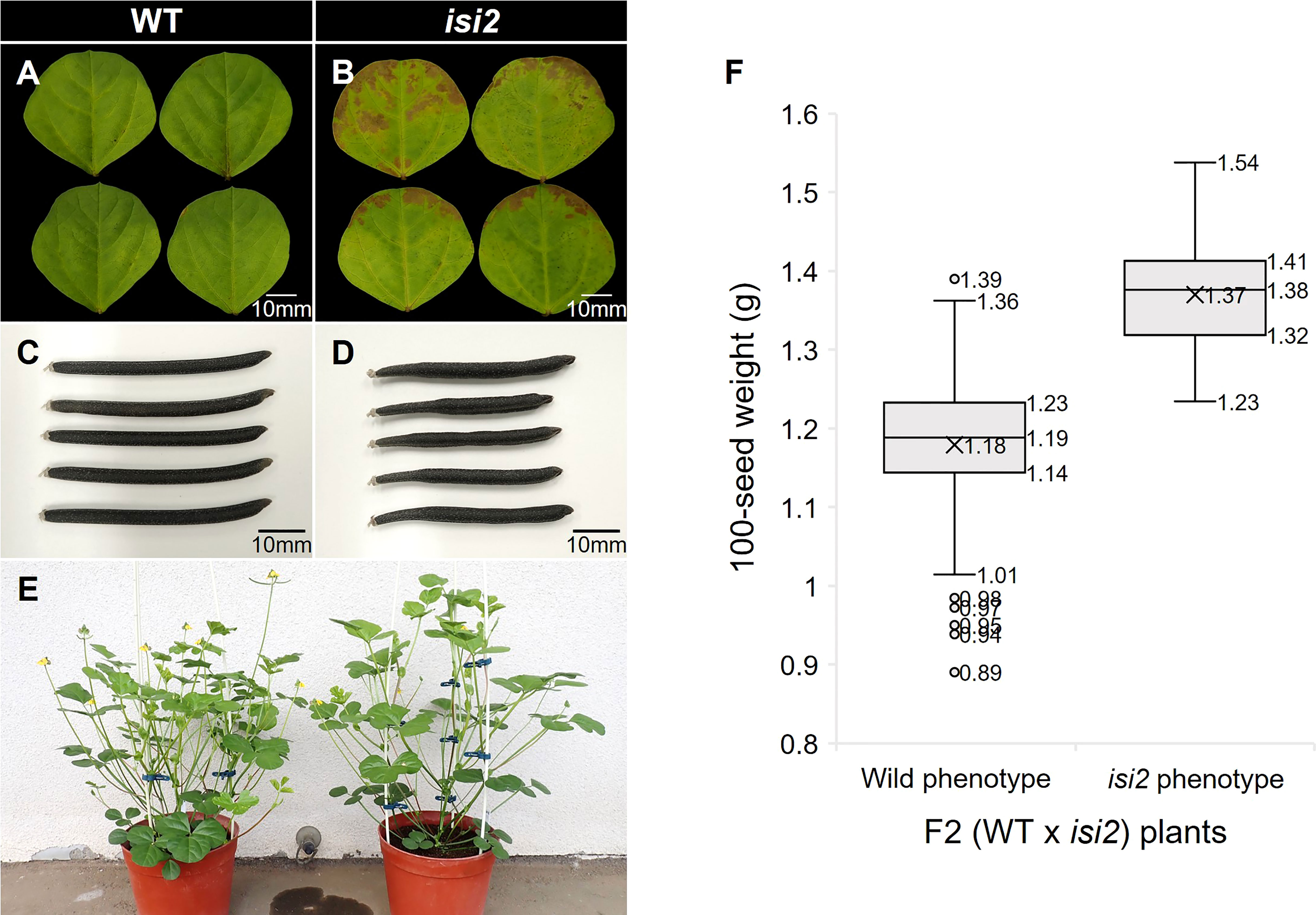
Figure 6 Pleiotropic effects of the isi2 mutant. (A, B) Fully expanded third terminal leaflets were detached from plants and incubated on the water surface for 28 days. N=5. (C, D) Mature seed pods of the wild-type and isi2 mutant, respectively. (E) Plants 50 days after sowing. Left, wild-type; right, isi2 mutant. (F) Box-and-whisker plots of 100-seed weights of F2 (WT × isi2) plants divided into the wild (N=210) and mutant (N=73) phenotypes. Top, middle, and bottom lines in the boxes indicate the 1st, 2nd, and 3rd quartiles, respectively. Crosses indicate means. Top and bottom lines of the whiskers indicate maximum and minimum values, respectively. Plots indicate outliers.
We next evaluated the isi2 mutant for eight traits to elucidate whether it showed pleiotropic effects and found that days to flowering increased by 3 days, the largest leaflet width increased by 7%, the number of seeds per pod decreased by 50%, the pod length decreased by 23%, and the 100-seed weight increased by 37%, compared to the wild-type (Figure 6C-E, Table 6). No significant differences were observed in height, largest leaf length, and shoot dry weight between the wild-type and isi2 mutant (p > 0.05, t-test).
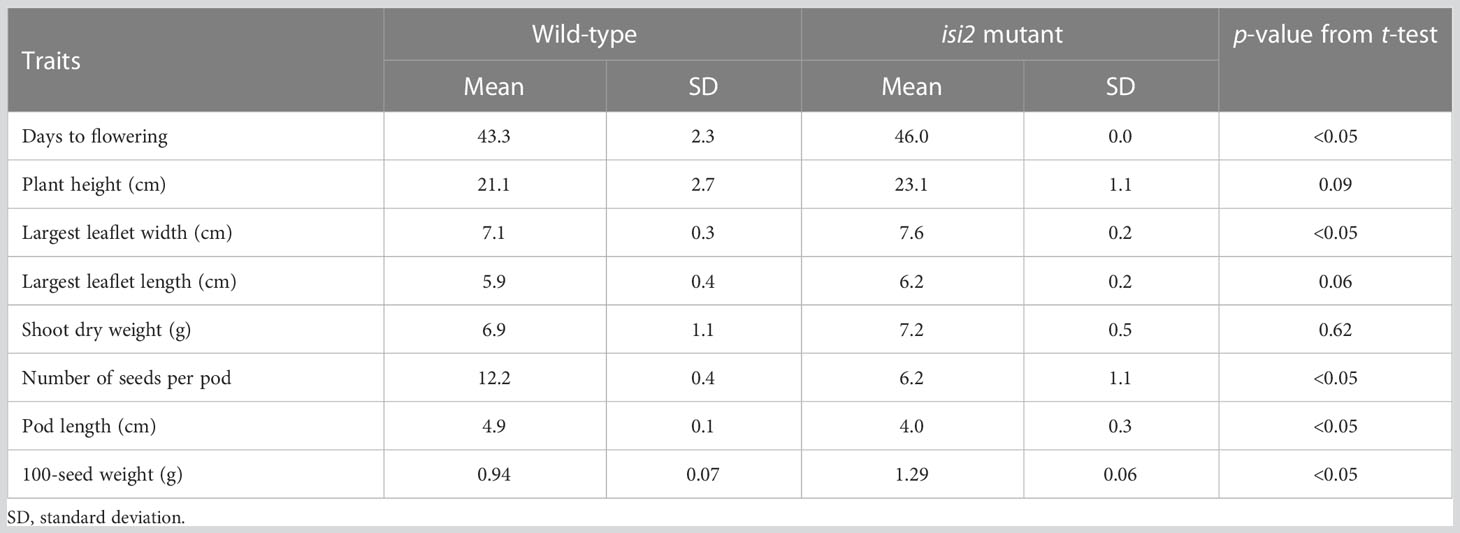
Table 6 Wild-type and isi2 mutant phenotypes for eight agronomic traits (N=7).
We next determined if the isi2 VsPSAT1 mutation was genetically linked with the 100-seed weight and number of seeds per pod. The mean 100-seed weight in 73 isi2 F2 (WT × isi2) plants was 1.38 g, significantly higher than that of 210 wild-type F2 (WT × isi2) plants (1.19 g; p < 0.05, t-test; Figure 6, Supplementary Table 2). In addition to seed size, the number of seeds per pod was tightly linked to the isi2 phenotype in 163 F3 (WT × isi2) plants derived from 11 F2 (WT × isi2) plants with recombination among the 3 SNPs (Table 5). However, the linkage of 100-seed weight and number of seeds per pod to the isi2 phenotype was not complete on an individual basis, owing to high variability (Supplementary Table 3).
4 Discussion
In this study, we identified VsPSAT1 as the gene likely mutated to cause the isi2 phenotype of V. Stipulacea plants, increasing water uptake through the lens groove. We hypothesize that VsPSAT1 functions to close this gap, probably by producing sterol esters. We also identified the pleiotropic effects of the isi2 mutant: accelerating leaf senescence, increasing seed size, and decreasing numbers of seeds per pod. We assembled the genome of V. stipulacea and revealed the differences between the phenotypes of the wild-type and the isi2 mutant in detail, which we had only approximately evaluated in our previous study (Takahashi et al., 2019). Moreover, we revealed using SEM and CT that the isi2 mutant has lesser honeycomb-like wax sealing the lens groove than the wild-type, and takes up water from the lens groove. In addition, the isi2 mutant had pleiotropic effects on other agronomic traits, including organ gigantism in leaves and seeds.
In the present study, we show that VsPSAT1 is likely the ortholog of AtPSAT1/ERP1 (AT1G04010), whose product catalyzes the transacylation of acyl groups from phospholipids to multiple sterols to produce sterol esters, preventing excessive sterol accumulation. Null AtPSAT1 mutants have ~30% higher sterol levels in leaves than the wild-type (Shimada et al., 2019). Because sterols are important contributors to cuticular wax in alfalfa (Medicago sativa L.) (Lee and Suh, 2015), the loss of VsPSAT1 function readily explains the isi2 lens-groove phenotype. However, this phenotype conflicts with that of the Arabidopsis psat1 mutant, whose seeds exhibit reduced water uptake (Shimada et al., 2021). Abnormal columellar structure in the second layer of the outer integument (oi2) of the seed coat appears to suppress seed water uptake in the Arabidopsis psat1 mutant (Shimada et al., 2021), and sterols are not detected in the cuticular wax of A. thaliana (Lee and Suh, 2015). Moreover, the columella is absent in the seed coat of V. stipulacea, and the oi2 layer has honeycomb-like epicuticular wax deposition. Therefore, we hypothesize that differences in seed water uptake between isi2 and psat1 mutants are due to differences in seed coat structure and cuticular wax composition, as legumes produce water-impermeable seed coats for physical seed dormancy whereas A. thaliana does not. These findings suggest that as PSAT1 genes are conserved in other wild Vigna species, they may act as potential targets for the de novo domestication of these species. However, further experiments are required to verify gene function by transformation and chemical analysis.
For de novo domestication using reverse genetics or genome editing, it is better to identify multiple genes influencing the same domestication trait, allowing identifications from small, mutagenized populations. Genetic strategies to decrease legume hard-seededness can be divided into two categories: permeabilization of the seed coat and permanent opening of the water gap. In the former case, the genes include VsCESA7 from the isi1 mutant in our previous study (Takahashi et al., 2019) and GmHs1-1 from soybeans (Sun et al., 2015). In the latter case, the genes include VsPSAT1 from this study and PAE-8-2 from common beans (Soltani et al., 2021). Of the two categories, counteracting hard-seededness by the permanent opening of the water gap may be more favorable because the isi2 mutant may provide higher environmental adaptability. In addition, although reduced hard-seededness is necessary for uniform germination, it allows rapid water uptake into the seed immediately after sowing, which causes flooding injury that disrupts soybean seed tissue (Nakayama and Komatsu, 2008), promoting both preharvest germination of pods and fungal infection of seeds in mungbean (Vigna radiata (L.) Wilczek), which is closely related to V. stipulacea (Humphry et al., 2005). The isi1 mutant that we previously identified is similar to soybeans, in that it begins to rapidly take up water through the entire seed coat, whereas the isi2 mutant is similar to common beans in that it exhibits slower water uptake through the lens (Takahashi et al., 2019). In the current global environment with sporadic rainfall and drying cycles, VsPSAT1 of the isi2 mutant and PAE-8-2 of common bean, which contribute to slow water uptake from the lens-specific, are likely more useful genes than VsCesA7 and soybean GmHs1-1. In contrast, VsCesA7 and GmHs1-1 are more likely to provide more uniform germination and higher productivity when managed under appropriate environments.
Pleiotropic effects must be considered when selecting target genes for de novo domestication. In the present study, the isi2 mutant showed both agronomically positive traits (decreased hard-seededness and increased seed size) and agronomically negative traits (decreased seed number). Larger seeds are better at lifting the soil and germinating in mungbean and produce plants with a higher shoot and root biomass in soybean (Kluyver et al., 2013; Limede et al., 2018), although the tradeoff between seed size and number is a well-known problem in plant breeding (Dwivedi et al., 2021). The increased seed size and decreased seed number in isi2 mutants are likely caused by the mutation in VsPSAT1, as the SNP and 100-seed weight or the number of seeds per pod were tightly linked in the segregated generations. Moreover, the tight but incomplete linkage likely may have resulted from errors because seed weight is sensitive to environmental factors, and a large number of F2 and F3 plants were cultivated in small pots, which inhibit vegetative growth. Other studies suggest that quantitative or qualitative changes in cuticular wax affect plant growth, including organ size (Zhang et al., 2005; Zhang et al., 2007; Nobusawa et al., 2013; Fukuda et al., 2022; Kajino et al., 2022). In the mungbean, quantitative trait loci (QTLs) for hard-seededness and seed size colocalize (Humphry et al., 2005). We did not discuss seed yield because we did not evaluate the number of pods per plant because complex efforts are needed to estimate seed yield due to its long-lasting flowering and pod-shattering. However, seed yield is one of the most important traits and we will evaluate it in the future. In addition to those traits, the isi2 mutant also showed early leaf senescence as observed in AtPSAT1 mutants (Bouvier-Navé et al., 2010). As early leaf senescence shortens the maturation period and prevents green stem disorder in soybeans (Yamatani et al., 2021), we view this phenotype as an agronomically positive trait. Although we detected a significant difference between wild-type and isi2 mutants for days to flowering, this trait was not linked to an SNP on VsPSAT1 in F2 and F3 plants (data not shown). To date, few studies have described the pleiotropic effects of domestication-related genes in detail. However, we believe that it is important to catalog both positive and negative pleiotropic effects for better selection of appropriate target genes for de novo domestication. Therefore, transformation experiments are needed to overcome the limitations of the present study, such as incomplete linkage.
In addition, our chromosome-level assembly of V. stipulacea greatly accelerated our identification of a candidate SNP. The bulked segregant analyses revealed a single clear peak in allele frequency. Moreover, combined with whole-genome sequencing, we identified two candidate genes involved in hard-seededness: VsCESA7 from our previous study (Takahashi et al., 2019) and VsPSAT1 in this study. As we previously identified a mutant with non-shattering seed pods (Takahashi et al., 2019), we plan to do another bulked segregant analysis to identify candidate SNPs. We will continue screening for other important traits, including larger seed size and faster growth. As new technologies become available, de novo domestication will likely be significantly more rapid and easier than classical breeding to develop climate-adapted crops. We believe that wild legumes, especially those of the genus Vigna with pre-existing tolerance to biotic and abiotic stresses, are key to global food security during climate change.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author contributions
YT, KN, and NT conceived the study. YT and HS planned the study. CM and YT cultivated plants. HS performed assembly, annotation, BSA, and phylogenetic analysis. HS and KN constructed the genetic map. HE and YT performed SEM. ST and YT performed CT. AH and YT performed RT-PCR. YT performed other experiments. YT, HS, and KN wrote the manuscript with input from TS, ST, AH, HE, and NT. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Japan Society for the Promotion of Science KAKENHI [grant numbers 19KT0016, 21KK0272], the Genebank Project of the National Agriculture and Food Research Organization, and JST CREST, Japan [grant number JPMJCR17O1].
Acknowledgments
We are grateful to Ms. Motoyoshi, Ms. Asano, and Ms. Yamamoto for taking good care of the plants and their materials. We would like to thank Editage (www.editage.com) for English-language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1119625/full#supplementary-material
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Banas, A., Carlsson, A. S., Huang, B., Lenman, M., Banas, W., Lee, M., et al. (2005). Cellular sterol ester synthesis in plants is performed by an enzyme (phospholipid:sterol acyltransferase) different from the yeast and mammalian acyl-CoA:sterol acyltransferases. J. Biol. Chem. 280, 34626–34634. doi: 10.1074/jbc.M504459200
Bewley, J. D., Bradford, K. J., Hilhorst, H. W. M., Nonogaki, H. (2013). “Dormancy and the control of germination,” in Seeds (New York: Springer). doi: 10.1007/978-1-4614-4693-4_6
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouvier-Navé, P., Berna, A., Noiriel, A., Compagnon, V., Carlsson, A. S., Banas, A., et al. (2010). Involvement of the phospholipid sterol acyltransferase1 in plant sterol homeostasis and leaf senescence. Plant Physiol. 152, 107–119. doi: 10.1104/pp.109.145672
Chai, M., Zhou, C., Molina, I., Fu, C., Nakashima, J., Li, G., et al. (2016). A class II KNOX gene, KNOX4, controls seed physical dormancy. Proc. Natl. Acad. Sci. U.S.A. 113, 6997–7002. doi: 10.1073/pnas.1601256113
Cingolani, P., Platts, A., le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. doi: 10.4161/fly.19695
Dorn, K. M., Fankhauser, J. D., Wyse, D. L., Marks, M. D. (2015). A draft genome of field pennycress (Thlaspi arvense) provides tools for the domestication of a new winter biofuel crop. DNA Res. 22, 121–131. doi: 10.1093/dnares/dsu045
Dwivedi, S. L., Reynolds, M. P., Ortiz, R. (2021). Mitigating tradeoffs in plant breeding. iScience 24, 102965. doi: 10.1016/j.isci.2021.102965
Emms, D. M., Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238. doi: 10.1186/s13059-019-1832-y
Fernie, A. R., Yan, J. (2019). De novo domestication: an alternative route toward new crops for the future. Mol. Plant 12, 615–631. doi: 10.1016/j.molp.2019.03.016
Foley, M. E. (2001). Seed dormancy: an update on terminology, physiological genetics, and quantitative trait loci regulating germinability. Weed Sci. 49, 305–317. doi: 10.1614/0043-1745(2001)049[0305:SDAUOT]2.0.CO;2
Frith, M. C., Noé, L. (2014). Improved search heuristics find 20,000 new alignments between human and mouse genomes. Nucleic Acids Res. 42, e59. doi: 10.1093/nar/gku104
Fukuda, N., Oshima, Y., Ariga, H., Kajino, T., Koyama, T., Yaguchi, Y., et al. (2022). ECERIFERUM 10 encoding an enoyl-coa reductase plays a crucial role in osmotolerance and cuticular wax loading in arabidopsis. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.898317
Furuta, T., Ashikari, M., Jena, K. K., Doi, K., Reuscher, S. (2017). Adapting genotyping-by-sequencing for rice F2 populations. G3 (Bethesda) 7, 881–893. doi: 10.1534/g3.116.038190
Gama-Arachchige, N. S., Baskin, J. M., Geneve, R. L., Baskin, C. C. (2013). Identification and characterization of ten new water gaps in seeds and fruits with physical dormancy and classification of water-gap complexes. Ann. Bot. 112, 69–84. doi: 10.1093/aob/mct094
Gasparini, K., Moreira, J. D. R., Peres, L. E. P., Zsögön, A. (2021). De novo domestication of wild species to create crops with increased resilience and nutritional value. Curr. Opin. Plant Biol. 60, 102006. doi: 10.1016/j.pbi.2021.102006
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Guan, D., McCarthy, S. A., Wood, J., Howe, K., Wang, Y., Durbin, R. (2020). Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898. doi: 10.1093/bioinformatics/btaa025
Humphry, M. E., Lambrides, C. J., Chapman, S. C., Aitken, E. A. B., Imrie, B. C., Lawn, R. J., et al. (2005). Relationships between hard-seededness and seed weight in mungbean (Vigna radiata) assessed by QTL analysis. Plant Breed. 124, 292–298. doi: 10.1111/j.1439-0523.2005.01084.x
Kajino, T., Yamaguchi, M., Oshima, Y., Nakamura, A., Narushima, J., Yaguchi, Y., et al. (2022). KLU/CYP78A5, a cytochrome P450 monooxygenase identified via fox hunting, contributes to cuticle biosynthesis and improves various abiotic stress tolerances. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.904121
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kluyver, T. A., Charles, M., Jones, G., Rees, M., Osborne, C. P. (2013). Did greater burial depth increase the seed size of domesticated legumes? J. Exp. Bot. 64, 4101–4108. doi: 10.1093/jxb/ert304
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lara, J. A., Burciaga-Monge, A., Chávez, A., Revés, M., Lavilla, R., Arró, M., et al. (2018). Identification and characterization of sterol acyltransferases responsible for steryl ester biosynthesis in tomato. Front. Plant Sci. 9. doi: 10.3389/fpls.2018.00588
Lee, S. B., Suh, M. C. (2015). Advances in the understanding of cuticular waxes in arabidopsis thaliana and crop species. Plant Cell Rep. 34, 557–572. doi: 10.1007/s00299-015-1772-2
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Limede, A. C., da Silva Oliveira, C. E., Zoz, A., Steiner, F., Zoz, T. (2018). Effects of seed size and sowing depth in the emergence and morphophysiological development of soybean cultivated in sandy texture soil. Aust. J. Crop Sci. 12, 93–98. doi: 10.21475/ajcs.18.12.01.pne765
Margarido, G. R. A., Souza, A. P., Garcia, A. A. F. (2007). OneMap: software for genetic mapping in outcrossing species. Hereditas 144, 78–79. doi: 10.1111/j.2007.0018-0661.02000.x
Nakayama, N., Komatsu, S. (2008). Water uptake by seeds in yellow-seeded soybean (Glycine max (L.) Merrill) cultivars with contrasting imbibition behaviors. Plant Prod. Sci. 11, 415–422. doi: 10.1626/pps.11.415
Nobusawa, T., Okushima, Y., Nagata, N., Kojima, M., Sakakibara, H., Umeda, M. (2013). Synthesis of very-long-chain fatty acids in the epidermis controls plant organ growth by restricting cell proliferation. PloS Biol. 11, e1001531. doi: 10.1371/journal.pbio.1001531
Ou, S., Su, W., Liao, Y., Chougule, K., Agda, J. R. A., Hellinga, A. J., et al. (2019). Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275. doi: 10.1186/s13059-019-1905-y
Razzaq, A., Saleem, F., Wani, S. H., Abdelmohsen, S. A. M., Alyousef, H. A., Abdelbacki, A. M. M., et al. (2021). De-novo domestication for improving salt tolerance in crops. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.681367
Rochette, N. C., Rivera-Colón, A. G., Catchen, J. M. (2019). Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754. doi: 10.1111/mec.15253
Sakaguchi, S., Sugino, T., Tsumura, Y., Ito, M., Crisp, M. D., Bowman, D. M. J. S., et al. (2015). High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genet. Genomes 11, 121. doi: 10.1007/s11295-015-0944-0
Sakai, H., Naito, K., Takahashi, Y., Sato, T., Yamamoto, T., Muto, I., et al. (2016). The vigna genome server, ‘VigGS’: a genomic knowledge base of the genus vigna based on high-quality, annotated genome sequence of the azuki bean, vigna angularis (Willd.) ohwi & ohashi. Plant Cell Physiol. 57, e2. doi: 10.1093/pcp/pcv189
Shapter, F. M., Cross, M., Ablett, G., Malory, S., Chivers, I. H., King, G. J., et al. (2013). High-throughput sequencing and mutagenesis to accelerate the domestication of microlaena stipoides as a new food crop. PloS One 8, e82641. doi: 10.1371/journal.pone.0082641
Shimada, T. L., Shimada, T., Okazaki, Y., Higashi, Y., Saito, K., Kuwata, K., et al. (2019). HIGH STEROL ESTER 1 is a key factor in plant sterol homeostasis. Nat. Plants 5, 1154–1166. doi: 10.1038/s41477-019-0537-2
Shimada, T. L., Ueda, T., Hara-Nishimura, I. (2021). Excess sterol accumulation affects seed morphology and physiology in arabidopsis thaliana. Plant Signal. Behav. 16, 1872217. doi: 10.1080/15592324.2021.1872217
Simpson, G. M. (1990). Seed dormancy in grasses. Cambridge Univ. Press New York. p, 297. doi: 10.1017/CBO9780511721816
Smýkal, P., Vernoud, V., Blair, M. W., Soukup, A., Thompson, R. D. (2014). The role of the testa during development and in establishment of dormancy of the legume seed. Front. Plant Sci. 5. doi: 10.3389/fpls.2014.00351
Soltani, A., Walter, K. A., Wiersma, A. T., Santiago, J. P., Quiqley, M., Chitwood, D., et al. (2021). The genetics and physiology of seed dormancy, a crucial trait in common bean domestication. BMC Plant Biol. 21, 58. doi: 10.1186/s12870-021-02837-6
Steinbrecher, T., Leubner-Metzger, G. (2018). Tissue and cellular mechanics of seeds. Curr. Opin. Genet. Dev. 51, 1–10. doi: 10.1016/j.gde.2018.03.001
Steinegger, M., Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Sun, L., Miao, Z., Cai, C., Zhang, D., Zhao, M., Wu, Y., et al. (2015). GmHs1-1, encoding a calcineurin-like protein, controls hard-seededness in soybean. Nat. Genet. 47, 939–943. doi: 10.1038/ng.3339
Takahashi, Y., Sakai, H., Yoshitsu, Y., Muto, C., Anai, T., Pandiyan, M., et al. (2019). Domesticating vigna stipulacea: a potential legume crop with broad resistance to biotic stresses. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01607
Tomooka, N., Naito, K., Kaga, A., Sakai, H., Isemura, T., Ogiso-Tanaka, E., et al. (2014). Evolution, domestication and neo-domestication of the genus vigna. Plant Genet. Resour. 12, S168–S171. doi: 10.1017/S1479262114000483
Tomooka, N., Pandiyan, M., Senthil, N. (2011). Conservation of leguminous crops and their wild relatives in Tamil nadu, India. Annu. Rep. Explor. Introduction Plant Genet. Resour. 27, 111–127.
UniProt: the universal protein knowledgebase (2021). Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Van der Auwera, G. A., O’Connor, B. D. (2020). Genomics in the Cloud (Sebastopol, CA: O’Reilly Media, Inc).
van Zonneveld, M., Rakha, M., Tan, S. Y., Chou, Y. Y., Chang, C. H., Yen, J. Y., et al. (2020). Mapping patterns of abiotic and biotic stress resilience uncovers conservation gaps and breeding potential of vigna wild relatives. Sci. Rep. 10, 2111. doi: 10.1038/s41598-020-58646-8
Yamatani, H., Heng, T., Yamada, T., Kusaba, M., Kaga, A. (2021). Identification and characterization of an early leaf senescence gene ELS1 in soybean. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.784105
Yu, H., Lin, T., Meng, X., Du, H., Zhang, J., Liu, G., et al. (2021). A route to de novo domestication of wild allotetraploid rice. Cell 184, 1156–1170.e14. doi: 10.1016/j.cell.2021.01.013
Zhang, J. Y., Broeckling, C. D., Blancaflor, E. B., Sledge, M. K., Sumner, L. W., Wang, Z. Y. (2005). Overexpression of WXP1, a putative medicago truncatula AP2 domain-containing transcription factor gene, increases cuticular wax accumulation and enhances drought tolerance in transgenic alfalfa (Medicago sativa). Plant J. 42, 689–707. doi: 10.1111/j.1365-313X.2005.02405.x
Zhang, J. Y., Broeckling, C. D., Sumner, L. W., Wang, Z. Y. (2007). Heterologous expression of two medicago truncatula putative ERF transcription factor genes, WXP1 and WXP2, in arabidopsis led to increased leaf wax accumulation and improved drought tolerance, but differential response in freezing tolerance. Plant Mol. Biol. 64, 265–278. doi: 10.1007/s11103-007-9150-2
Keywords: de novo domestication, genus Vigna, legume, physical seed dormancy, wild species, hard-seededness
Citation: Takahashi Y, Sakai H, Ariga H, Teramoto S, Shimada TL, Eun H, Muto C, Naito K and Tomooka N (2023) Domesticating Vigna stipulacea: Chromosome-Level genome assembly reveals VsPSAT1 as a candidate gene decreasing hard-seededness. Front. Plant Sci. 14:1119625. doi: 10.3389/fpls.2023.1119625
Received: 09 December 2022; Accepted: 27 March 2023;
Published: 17 April 2023.
Edited by:
Leonor Morais-Cecilio, University of Lisbon, PortugalReviewed by:
Susana Araújo, Technology and Innovation Campus, PortugalSzymon Kubala, Polish Academy of Sciences, Poland
Magdalena Woloszynska, Wroclaw University of Environmental and Life Sciences, Poland
Copyright © 2023 Takahashi, Sakai, Ariga, Teramoto, Shimada, Eun, Muto, Naito and Tomooka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Takahashi, dGFrYWhhc2hpMDEyNkBhZmZyYy5nby5qcA==
†These authors have contributed equally to this work