 Juan Li
Juan Li Xi Wang1†
Xi Wang1† Xinxin Miao
Xinxin Miao Lin Li
Lin Li- 1National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China
- 2Hubei Hongshan Laboratory, Wuhan, China
Tassel branch number is a key trait that contributes greatly to grain yield in maize (Zea mays). We obtained a classical mutant from maize genetics cooperation stock center, Teopod2 (Tp2), which exhibits severely decreased tassel branch. We conducted a comprehensive study, including phenotypic investigation, genetic mapping, transcriptome analysis, overexpression and CRISPR knock-out, and tsCUT&Tag of Tp2 gene for the molecular dissection of Tp2 mutant. Phenotypic investigation showed that it is a pleiotropic dominant mutant, which is mapped to an interval of approximately 139-kb on Chromosome 10 harboring two genes Zm00001d025786 and zma-miR156h. Transcriptome analysis showed that the relative expression level of zma-miR156h was significantly increased in mutants. Meanwhile, overexpression of zma-miR156h and knockout materials of ZmSBP13 exhibited significantly decreased tassel branch number, a similar phenotype with Tp2 mutant, suggesting that zma-miR156h is the causal gene of Tp2 and targets ZmSBP13 gene. Besides, the potential downstream genes of ZmSBP13 were uncovered and showed that it may target multiple proteins to regulate inflorescence structure. Overall, we characterized and cloned Tp2 mutant, and proposed a zma-miR156h-ZmSBP13 model functioning in regulating tassel branch development in maize, which is an essential measure to satisfy the increasing demands of cereals.
1 Introduction
Maize (Zea mays) is a staple crop that is experiencing a steady increase in both planting area and crop yields due to the rising population, diet shifts, and increasing biofuel consumption (Tilman et al., 2011). However, to ensure food security, increasing crop yields through sustainable means is more viable than expanding the land used for food production (Godfray et al., 2010; Phalan et al., 2011). Consequently, breeders are continually selecting cultivars that own optimal plant architecture to increase maize grain yield while minimizing land usage. To this end, breeders currently focus on studying morphological traits such as leaf, root, stem, and inflorescence morphology, which constitute maize plant architecture, for selective breeding. By doing so, they aim to identify and select ideal maize plant architecture that will maximize yield.
The Poaceae family is one of the largest angiosperm families and has evolved complex inflorescence morphologies, such as branches and spikelets (Zhang and Yuan, 2014). In maize, the inflorescence structures initially derive from inflorescence meristem (IM) which is responsible for initiating lateral primordia (Wang et al., 2022). IM is transferred from shoot apical meristem (SAM), which initiates leaf development during the reproductive transition. After SAM converts into IM, IM initiates three axillary meristems (AMs): branch meristems (BMs), spikelet meristems (SMs), or both, which further initiate floral meristems (FMs) (Koppolu and Schnurbusch, 2019). Specifically, BMs in the male inflorescence develop into long branches. Tassel with more branches contributes to prolonged pollination time and improved seed setting rate. However, more tassel branch numbers will consume more energy to produce pollen, thus reducing the absorption of nutrients by ear, and then affecting corn yield. Studies have found that smaller tassels with a few branches are beneficial to the increase of yield during modern maize breeding (Wang et al., 2020).
Maize, a monoecious crop, has a unique inflorescence structure consisting of primary and secondary paracladia (Vegetti and Anton, 1995). The development of inflorescence is a complex process, regulated by numerous genes. Several genes affecting tassel branch trait have been cloned in maize based on mutant analysis. For example, FASCIATED EAR 4 (FEA4) affects the transition from SAM to IM, and its mutations cause more tassel branches (Pautler et al., 2015). The genes ramosa1 (ra1), ramosa2 (ra2), ramosa3 (ra3) and RAMOSA1 ENHANCER LOCUS2 (rel2), play important roles in regulating inflorescence branching and result in increased tassel branch number (TBN) (Vollbrecht et al., 2005; Satoh-Nagasawa et al., 2006; Gallavotti et al., 2010). In contrast, unbranched2 (ub2), unbranched3 (ub3) and tasselsheath4 (tsh4), which belong to the SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) family, regulate the differentiation of lateral primordia and have mutants that exhibit a significant reduction in TBN (Chuck et al., 2014). KNOTTED1 (KN1) plays a crucial role in stem cell maintenance and organ initiation in shoot meristems, independent of the CLV-WUS pathway. kn1 mutants in B73 background produce tassel primordia with fewer AMs, leading to fewer TBN (Kerstetter et al., 1997). BLH12 and BLH14, two cofactors of KN1, function redundantly in maintenance of axillary meristems and tassel branch patterning (Tsuda et al., 2017). Phytohormones also play significant roles in plant development and growth, including TBN. For example, SPARSE INFLORESCENCE1 (SPI1) and VANISHING TASSEL2 (VT2) participate in auxin biosynthesis, while BARREN INFLORESCENCE1 (BIF1) and BARREN INFLORESCENCE4 (BIF4) are involved in auxin signaling (Gallavotti et al., 2008; Phillips et al., 2011; Galli et al., 2015). Additionally, nutrition transport and epigenetic regulation are also verified to affect tassel morphology development (Chatterjee et al., 2014; Forestan et al., 2018).
The juvenile-to-adult phase transition is a critical biological process that signifies the transformation of SAM from the vegetative to the inflorescence stage. MicroRNA156 (miR156) is a class of conserved endogenous microRNAs that regulate gene expression post-transcriptionally in plants. It has been demonstrated that miR156 is involved in multiple essential biological processes, including the maintenance of the juvenile phase of plants and flower organ development (Wang and Wang, 2015). In Arabidopsis, prolonged juvenile vegetative periods and delayed flowering were observed as a result of constitutive miR156 expression (Wu and Poethig, 2006). Similarly, overexpression of OsmiR156b or OsmiR156h in rice caused a prolonged juvenile vegetative period and delayed flowering (Xie et al., 2006). In maize, the Corngrass1 (Cg1) mutant exhibited a prolonged juvenile stage and retention of juvenile traits in the adult reproductive phase due to the overexpression of two tandem miR156 genes in the meristem and lateral organs (Chuck et al., 2007). miR156 is a class of microRNAs that are well-conserved and target a specific subset of SPLs in plants. These microRNAs define an age-dependent flowering regulatory pathway, known as the miR156-SPL modules, and have been verified to play important roles in multiple developmental processes in plants, such as the transition from the vegetative phase to the reproductive stage and the development of flower organs (Wang et al., 2009). In rice, mutation of Ideal Plant Architecture1 (IPA1), which is targeted by miR156, led to altered plant architecture, including panicle branches (Jiao et al., 2010). Thus, miR156-SPL modules coordinately regulate plant growth and development traits in crops.
The maize mutant Teopod2 (Tp2) has been extensively studied for over a century due to its dominant phenotype affecting tassel development. However, the genetic basis of Tp2 and its interactions with other genes involved in tassel development are still unclear. In this study, we successfully cloned Tp2 and mapped it to Chromosome 10, which encodes zma-miR156h, a paralog of Cg1. Overexpression of zma-miR156h resulted in the similarity of Tp2 and Cg1 phenotypes. Differential expression analysis and prediction of targeted genes identified several significantly downregulated SPL genes, including ub2, ub3, tsh4 and ZmSBP13/23/27/29. The phenotypic investigation of CRISPR-edited ZmSBP13 mutants provided conclusive evidence of its involvement in tassel development. The observed decrease in TBN clearly indicates that ZmSBP13 is a target gene of zma-miR156h. Motif analysis of ZmSBP13-targeted DNA sequences indicated that it binds to typical regulatory sites involved in inflorescence architecture. These findings enhance our understanding of tassel branch development in maize and suggest that precise manipulation of miR156-SPL modules has significant potential to improve plant architecture and crop quality in modern breeding programs.
2 Materials and methods
2.1 Plant materials and trait measurements
The F1 population was generated by crossing the maize inbred line Mo17 as the female parent with Tp2/Tp2 as the male parent during the winter season in Hainan, China. The F1 seeds were then grown in the winter of 2016 in Hainan, China and allowed to undergo self-fertilization to produce the F2 population. The agronomic traits, including plant height, ear height, length and width of ear leaf, and tassel branch number, were assessed for each individual after the flowering stage. A total of 177 individuals from two F2 families were subjected to chi-square test. One segregating F2 family consisting of 89 individuals was utilized for BSR-Seq (Bulked segregant RNA-Seq) (Liu et al., 2012) and MMAPPR (mutation mapping analysis pipeline for pooled RNA-seq) (Hill et al., 2013) analyses. Three OE lines, one CRISPR lines, and their respective negative controls were planted at the experimental station of Huazhong Agricultural University in Wuhan, China, and were managed according to local agronomic practices.
2.2 RNA isolation and gene expression analysis
To perform BSR-Seq and MMAPPR, total RNAs were extracted from two pooled samples of leaf tissues from 20 mutant individuals exhibiting single tassel branch number and 20 wild type individuals showing multiple tassel branch numbers, respectively. To detect differentially expressed genes, total RNAs were extracted from 2mm tassels of homozygous wild-type and homozygous mutants, respectively, and each set was prepared with two biological replicates. Additionally, miRNAs were extracted from the SAM of 10 homozygous wild-type and 10 homozygous mutants, respectively, to obtain the expression level of microRNA. The Direct-zol RNA Miniprep Kit (ZYMO RESEARCH) was used for total RNA and miRNA extraction according to the manufacturer’s instructions.
The raw PE (pair-end) sequencing data of mRNA-seq were first preprocessed using fastp (Chen et al., 2018) software to remove short reads (<72 bp) and low-quality base pair (q>20). over 81% clean reads were successfully mapped to the B73 RefGen_v4 using Tophat (version 2.1.1) (Supplementary Table S1). The expression level was calculated using Cufflinks (version 2.2.1) with FPKM (fragments per kilo bases of exon per million fragments mapped). Cuffdiff was then used to perform a differential expression analysis between wild type and Tp2 mutant, using a base of |log2(Fold change)| ≥1 and false discovery rate (FDR) <0.001. To investigate the functions of the differential expression genes, GO enrichment analysis was performed using agriGO v2.0 (Tian et al., 2017).
To evaluate the expression level of small RNAs, the raw reads were first subjected to adaptor sequence and short read removal using Cutadapt (version 1.9.1). Over 92.6% of the clean reads (Supplementary Table S2) were retained and mapped to the Chr10.fa from the B73 RefGen_v4 reference genome. 80% of the clean data were randomly sampled 100 times, and the expression levels were calculated with RPM (reads of exon model per million mapped reads) using ShortStack (version 3.8.5). Expression differences were compared by extracting the RPM values of the zma-mir156h gene in the mutant and wild-type samples.
2.3 Quantitative real-time PCR
To validate the results of RNA-seq and miRNA-seq, we performed quantitative real-time PCR (qRT-PCR) to determine the expression levels of Zm00001d025786 and zma-miR156h. For Zm00001d025786, cDNA synthesis was conducted using the HiScript® II 1st Strand cDNA Synthesis Kit, and RT-PCR was carried out using AceQ® Universal SYBR qPCR Master Mix according to the manufacturer’s instructions with three technical replicates. For zma-miR156h, cDNA synthesis was conducted using the miRNA 1st Strand cDNA Synthesis Kit (stem-loop method), and RT-PCR was carried out using the miRNA Universal SYBR qPCR Master Mix according to the manufacturer’s instructions with three technical replicates. The primers for Zm00001d025786 and zma-miR156h were designed and listed in Supplementary Table S3. We used ubiquitin as an internal control for normalization, and relative expression levels were evaluated using the 2^-ΔΔCT method.
2.4 Vector construction and plant transformations
To construct the overexpression vector, the pre-MIR156h sequence was amplified using specific primers listed in Supplementary Table S4. The resulting PCR product was then cloned into the vector pCombia3300-3xflag, and subsequently transformed into the maize inbred line B73 by Beijing BomeiXingao Technology Co., Ltd. The CR-sbp13 vector construction and plant transformation were carried out by Wimi Biotechnology Co., Ltd (Changzhou, China).
2.5 tsCUT&Tag experiment
A tsCUT&Tag experiment was conducted for the identification of regulatory network following the guideline of our previous study (Wu et al., 2022). The raw reads from two replicates were subjected to trimming using Fastqc (version 0.11.5). The clean reads were then mapped to the B73 RefGen_v4 genome using Bowtie2 (version 2.4.1) with the parameters “–no-mixed –no-discordant”. Duplicated reads were removed using SAMtools (version 1.9), and peak calling was performed using MACS2 (version 2.2.7.1) with parameters “-g 2.2e+9 -p 1e-5”. Finally, peak genes were obtained using the BEDTools (version 2.27) windows command. Additionally, motif analysis was performed using MEME-ChIP (Machanick and Bailey, 2011).
3 Results
3.1 Tp2 affects multiple traits of maize development
Tp2 is a classical mutant from Maize Genetics Cooperation Stock Center that extends the duration of the juvenile vegetative phase of maize development. In normal maize plants, tillers that grow in axillary positions of juvenile leaves are produced in low numbers, ranging from zero to a few (Chuck et al., 2007). However, Tp2 mutants exhibit an increased number of phytomers that produce ears and initiate numerous tillers in the axils of each leaf, resulting in a larger number of leaves that retain a slender juvenile morphology (middle) (Figures 1A, B). While Tp2/+ plants initiate tassels later than their wild-type siblings, the mutant tassel arrests differentiation at the same time as, or shortly before, the primary meristem of a wild-type tassel completes its development, resulting in the formation of a single tassel branch (Figure 1C). To assess the impact of the mutation on other agronomic traits, such as plant height (PH), ear height (EH), ear leaf length (ELL), and ear leaf width (ELW), we measured these traits in homozygous wild-type and homozygous mutant plants. An independent sample Student’s t-test was performed to evaluate the significance of the difference between the two counterpart genotypes. The phenotypic statistics revealed significant differences for most agronomically important traits, including plant height, ear height, ear leaf length, and ear leaf width, when compared to the wild type (Supplementary Figures S1A, B). Furthermore, the TBN phenotypes of Tp2/Tp2 and Tp2/+ and their response to variation in gene dose indicate that Tp2 is characterized by gain-of-function mutations. To better understand the period during which the mutant tassels exhibit abnormal differentiation, we examined the growth process of young ears ranging from 1-3mm in size. Our observations suggest that the differentiation of mutant tassels begins to deviate from normalcy around 2mm, as revealed by microscopic analysis (Figure 1D).

Figure 1 Vegetative and inflorescence traits. In each, the wild type is on the left, Tp2/Tp2 is on the middle and Tp2/+ is on the right. (A) Whole plant during vegetative development. (B) Excised mature leaf blades. (C) Mature tassels. (D) 1-3mm tassel of wild type and Tp2/Tp2. Scale bars= 10 cm in (A–C) 0.5 mm in (D).
3.2 Genetic analysis and mapping of the Tp2 mutant
To explore the genetic basis of TBN variation in the Tp2 mutant, the maize inbred Mo17 was used as the female parent to cross with Tp2 mutant as the male parent, which generated two segregating F2 families comprising 177 plants. The segregation ratio of single tassel branch to multiple tassel branches in the two F2 family were consistent with a 3:1 ratio (Supplementary Table S5), thereby supporting the hypothesis that a single, dominant gene is responsible for the TBN phenotype.
To identify the causal gene of Tp2 mutant, we bulked RNA samples from mutant and non-mutant individuals of a F2 population into two separate pools (wild-type pool and mutant pool) and performed RNA-Seq. We used BSR-Seq and MMAPPR to delimit the candidate interval of the causal gene. BSR-Seq and MMAPPR mapping analysis pipeline identified a genomic interval of approximately 14 Mb, between 119 and 133 Mb on chromosome 10 as the location of the Tp2 mutant (Figures 2A–C). We selected seven SNP markers (Supplementary Table S6) within the candidate interval (119 Mb to 133 Mb) to genotype a large segregating population, including approximately 5,000 individuals. Based on the genotypes and phenotypes of the progeny families derived from recombinants, Tp2 was localized between the markers P5 (129.2 Mb) and P6 (132 Mb) (Figure 3A). To further fine-map Tp2, we genotyped the segregating population and the progeny families derived from recombinants using five newly developed markers (Supplementary Table S6). Based on the genotypic and phenotypic data, we narrowed down the location of Tp2 to an approximately 139-kb region flanked by markers P9 and P10, in which only two genes (Zm00001d025786 and zma-miR156h) are located (Figure 3A).
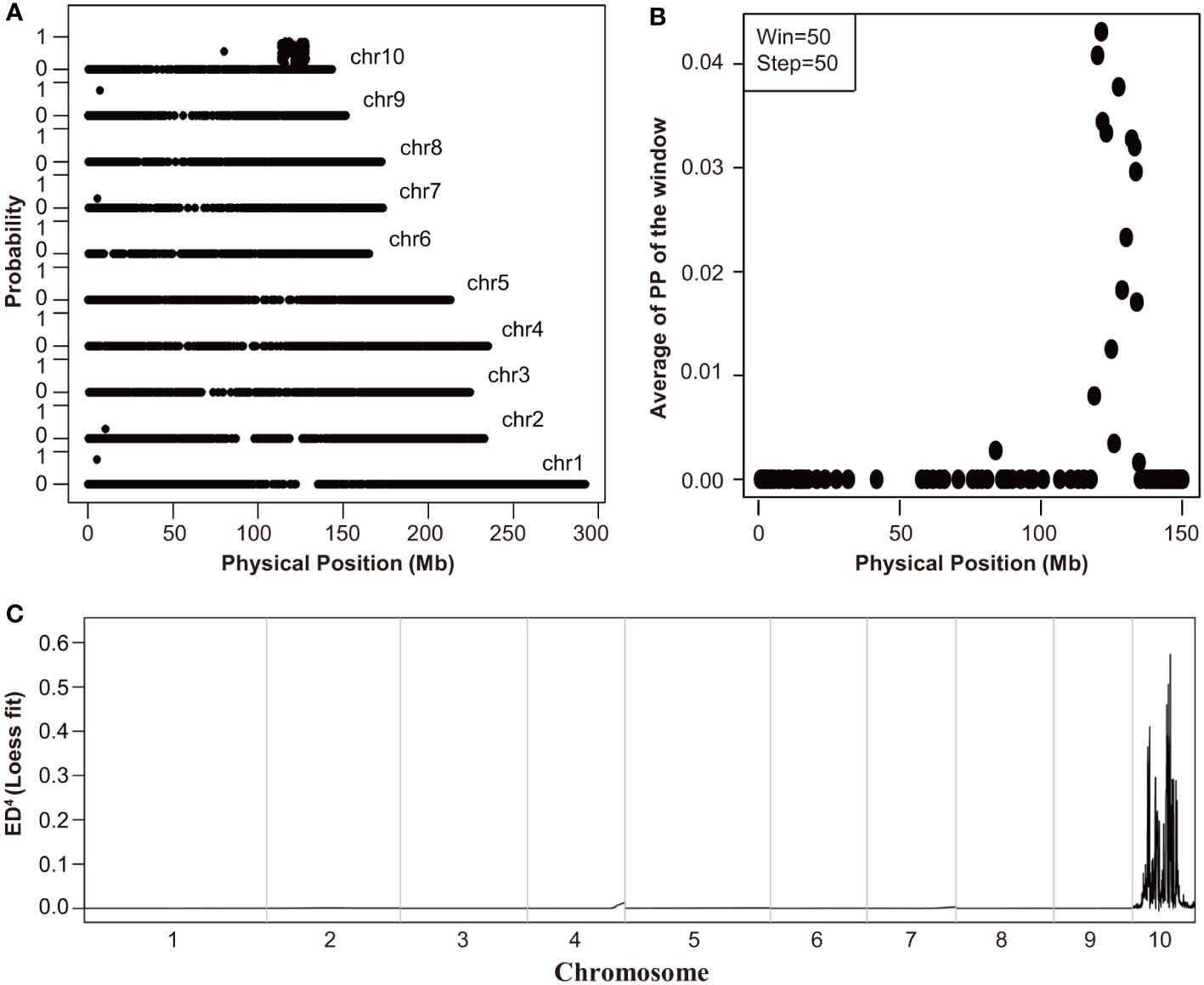
Figure 2 The initial mapping of Tp2. (A, B) The mapping results of BSR-Seq. The physical position of each SNP marker was plotted versus the probability of each SNP marker being in complete linkage disequilibrium with the causal gene (A) and the Chromosome 10 was scanned by using a window containing 50 SNPs with a step size of 50 SNPs (B). (C) The mapping results of MMAPPR. Loess fit curve calculated using the ED4 (Euclidean distance raised to the fourth power) scores across the genome.
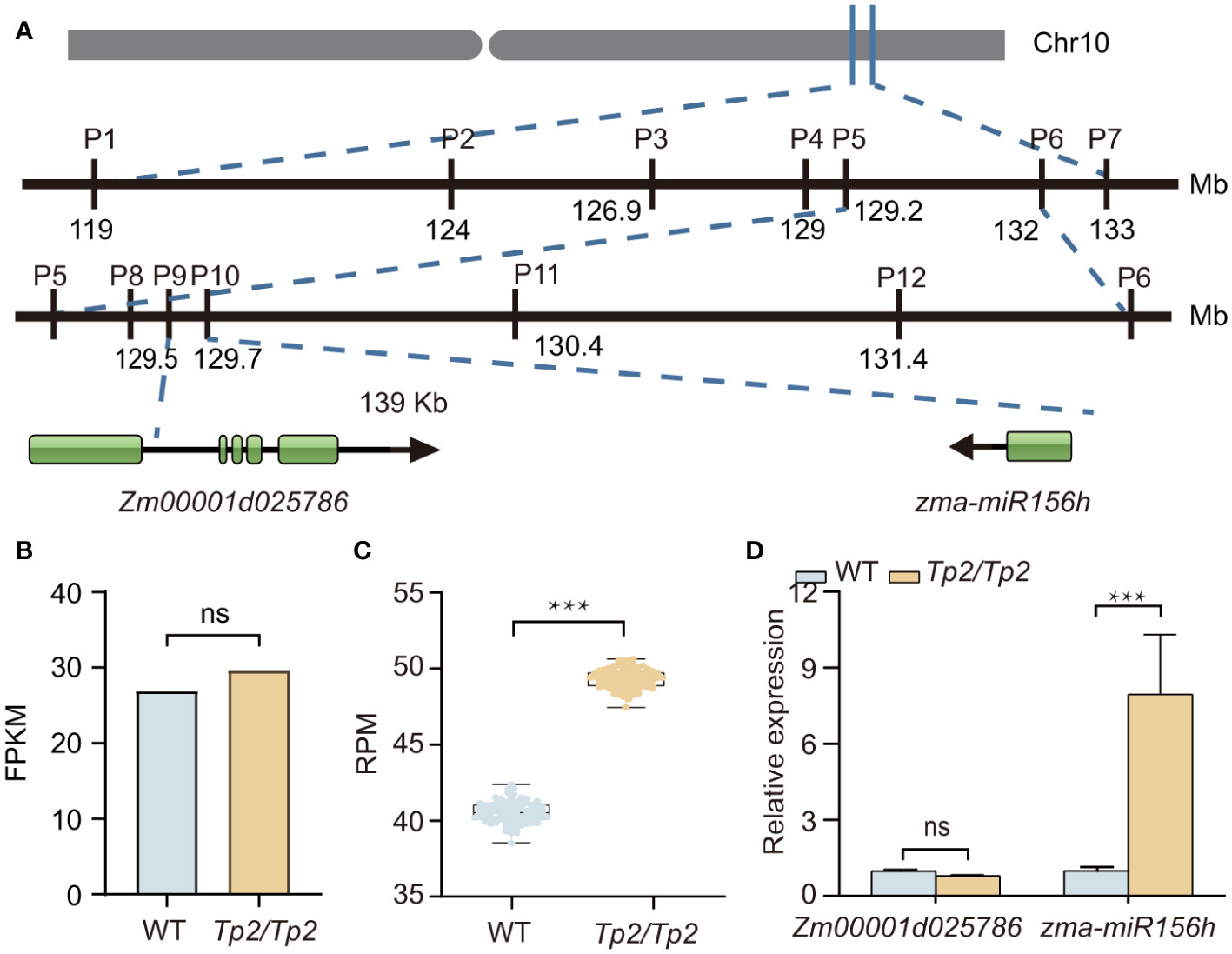
Figure 3 Fine mapping and candidate genes analysis for Tp2. (A) Fine mapping of Tp2 on maize chromosome 10. The physical positions of molecular markers (according to the B73_V4) used for fine mapping of the Tp2 locus. (B) Transcription levels of Zm00001d025786 estimated by mRNA-seq in WT (wild type) and the Tp2/Tp2 mutant from the about 2mm tassel tissue. (C) Transcription levels of zma-miR156h estimated by miRNA-Seq in WT and the Tp2/Tp2 mutant from the SAM tissues. (D) Relative expression levels of Zm00001d025786 and zma-miR156h in homozygous wild-type and homozygous mutant via qRT-PCR assay, respectively. P value was determined by Student’s t-test. ***P < 0.001, “ns” means it is no significant difference.
To mine the candidate genes for Tp2, we calculated gene abundance from transcriptome data. Since the tassel of Tp2/Tp2 begin to appear abnormal at about 2mm, SAM is the key niche for the growth and development of the aerial part. Therefore, the RNA library from 2mm tassel and the SAM of 2-week-old seedling of homozygous wild-type and homozygous mutant genotypes were used for mRNA sequencing and miRNA sequencing, respectively. For the data of mRNA sequencing, the Spearman Correlation coefficients are at least 81% between two biological replicates (Supplementary Figures S2A, B), and the DEG analysis revealed a total of 58 genes were up-regulated and 43 genes were down-regulated (Supplementary Figure S2C; Supplementary Table S7). To further explore the functions of the DEGs, we performed gene ontology (GO) enrichment analysis (p-value < 0.01). The significantly enriched biological process such as plant organ development and formation, regulation of organ morphogenesis, flower development (Supplementary Figure S3). These findings show that these DEGs are related with regulating maize inflorescences and organ morphogenesis.
Notably, the expression level of Zm00001d025786 had no difference between wild type and mutant pools (Figure 3B). Further miRNA sequencing between mutant and wild-type showed that mainly 20–24 nucleotide noncoding RNAs were enriched (Supplementary Figure S4), indicating that the high quality of sequencing data. Further analysis of miRNA sequencing data showed the expression level of zma-miR156h in Tp2/Tp2 is up-regulated (Figure 3C). To validate the results of the transcriptome sequencing, quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) was performed. The expression levels of Zm00001d025786 and zma-miR156h genes were consistent with the results of transcriptome sequencing (Figure 3D). Thus, zma-miR156h was considered as the candidate gene of Tp2 mutant
3.3 zma-miR156h-OE and CR-sbp13 plants produce similar phenotypes
MicroRNA156 plays vital roles in maize development and reproduction. Vegetative phase change in maize is regulated by miR156, a microRNA that promotes the expression of the juvenile phase and represses the expression of the adult phase. To confirm that overexpression of zma-miR156h is the cause of the Tp2 phenotype, we generated transgenic maize lines using the B73 as the background, in which a 115-bp precursor sequence of zma-miR156h was overexpressed by using the ubiquitin promoter (Supplementary Figure S5). We observed conspicuous single tassel branch in zma-miR156h-OE compared with wild-type. Additionally, in place of tassel branches, long bract leaves were present, clustered at the base of the tassel in zma-miR156h-OE#1 (Figure 4A). The relative expression level of miR156 was compared between the OE-plants and their control. Compared with the wild type counterpart, miR156 were up-regulated in the OE lines. The expression levels and phenotypic results of OE#1 and OE#2 revealed that the pleiotropic phenotype in OE-plants was positively correlated with the expression level of miR156 (Supplementary Figure S6), which was related to the regulation of multiple pathways by miR156. The overexpression of zma-miR156h in maize plants produced similar phenotypes to Tp2 mutants, indicating that the Tp2 mutant phenotype is due to overexpression of zma-miR156h.
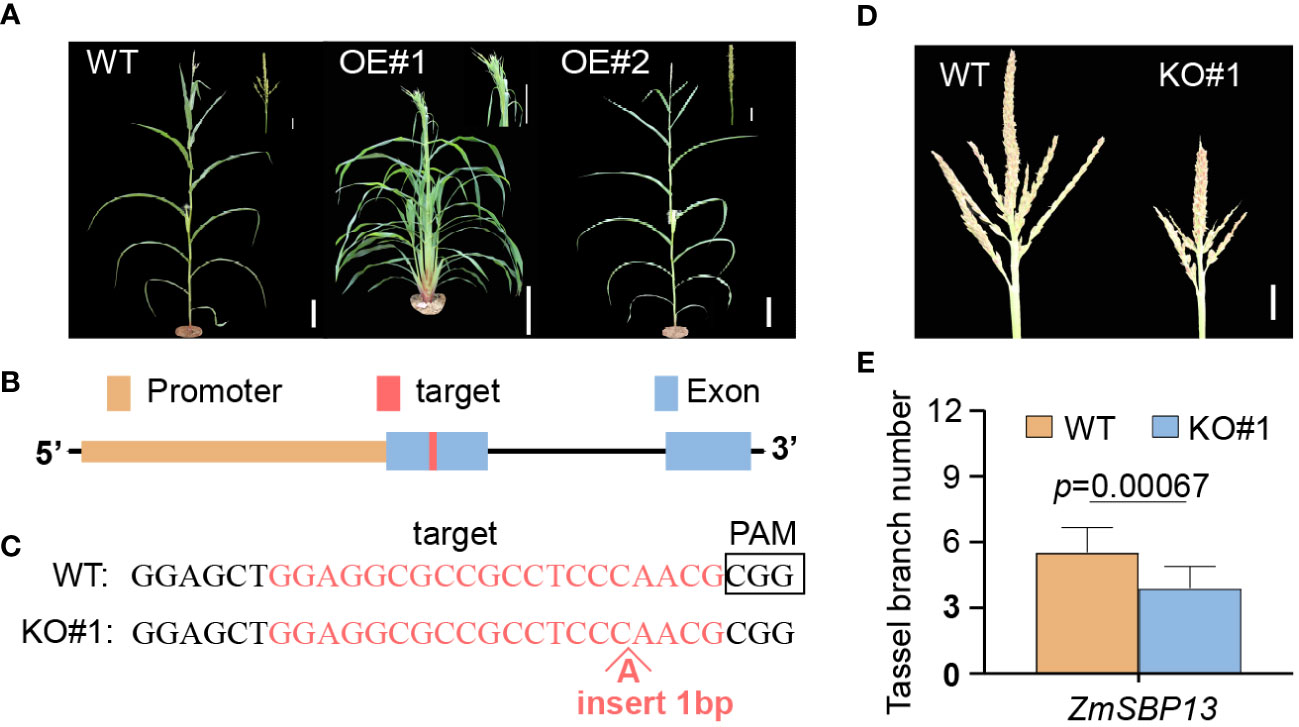
Figure 4 Characters of transgenic zma-miR156h-OE and CR-sbp13 plants. (A) Plant morphology from wild-type (left) and overexpression transgenic (OE#1 and OE#2) plants after the flowering stage. (B) Gene structure of ZmSBP13 and the target site in the first exon of ZmSBP13 for CRISPR/Cas9 editing. (C) Sequences of a homozygous knockout line with insertion in target site (KO#1). The wild-type sequence is shown at the Top. Target site and protospacer-adjacent motif (PAM) sequences are highlighted and boldface fonts, respectively. (D) Morphologies of wild-type and knockout line. (E) Phenotypic comparison of wild-type and knockout line. The data is shown as means ± SD; Scale bars= 10 cm in (A) 20 mm in (D) P value was determined by Student’s t test.
Studies have shown that miR156 regulates the spatiotemporal changes of plants by repressing SPL genes (Wang et al., 2009; Wu et al., 2009). To further elucidate the molecular bases of the defects in plant architecture caused by overexpression of zma-miR156h, we analyzed the target genes of zma-miR156h based on the maize cDNA library (zea mays, cDNA, AGPv4) using the web-based plant small RNA target analysis tool psRNATarget (Dai and Zhao, 2011). A total of 61 potential miR156-targeting genes were obtained (Supplementary Table S8). Furthermore, the functional annotation of 43 down-regulated genes in Tp2 mutant showed that 7 genes were annotated to encode SBP-domain protein, and all of them were target genes of miR156 (Supplementary Table S9). Among them, tsh4, ub2 and ub3 had already been reported to affect the development of tassel branch in previous studies, while the other four SPL genes (ZmSBP13, ZmSBP23, ZmSBP27, ZmSBP29) have not been researched yet.
To further investigate the relationship between SPL genes and zma-miR156h, we chose ZmSBP13 gene to generate knockout transgenic line (CR-sbp13) using CRISPR-Cas9 (Figures 4B, C). Using a PCR-based genotyping procedure, we identified an independent sbp13 knockout line with a 1bp insertion in the ZmSBP13 gene body in the T1 generation. Phenotypic and statistical analyses showed that TBN was decreased in these CR-sbp13 lines compared with WT plants (Figures 4D, E). The CR-sbp13 transgenic lines produced similar phenotypes with zma-miR156h-OE, indicating that ZmSBP13 gene, as the target of zma-mir156h, was inhibited by zma-mir156h, which led to the decrease of the tassel branches number.
3.4 ZmSBP13 may target multiple genes to regulate inflorescence structure
To investigate the molecular mechanism in regulating TBN, we clarified the potential target genes of ZmSBP13 using tsCUT&Tag assay. The Spearman Correlation coefficients for tsCUT&Tag data of two replicates is 0.98 (Supplementary Figure S7A). And ZmSBP13-bound signals were more prominent at promoter regions compared with negative control (Figure 5A; Supplementary Figures S7B, C). The number of overlapping detectable target genes for ZmSBP13 were 2631 (Figure 5B; Supplementary Table S10), demonstrating a good reproducibility of the data from tsCUT&Tag. To reveal the proteins that potentially interact with ZmSBP13, we performed motif analysis of ZmSBP13-targeted DNA sequences obtained from tsCUT&Tag results. Notably, the motif analysis showed that it is the typical binding site for the RAMOSA1, DOF, GROWTH REGULATING FACTOR (GRF4) and GLYMA proteins which control inflorescence architecture (Figure 5C), indicating that ZmSBP13 may interact with multiple proteins to regulate inflorescence structure.
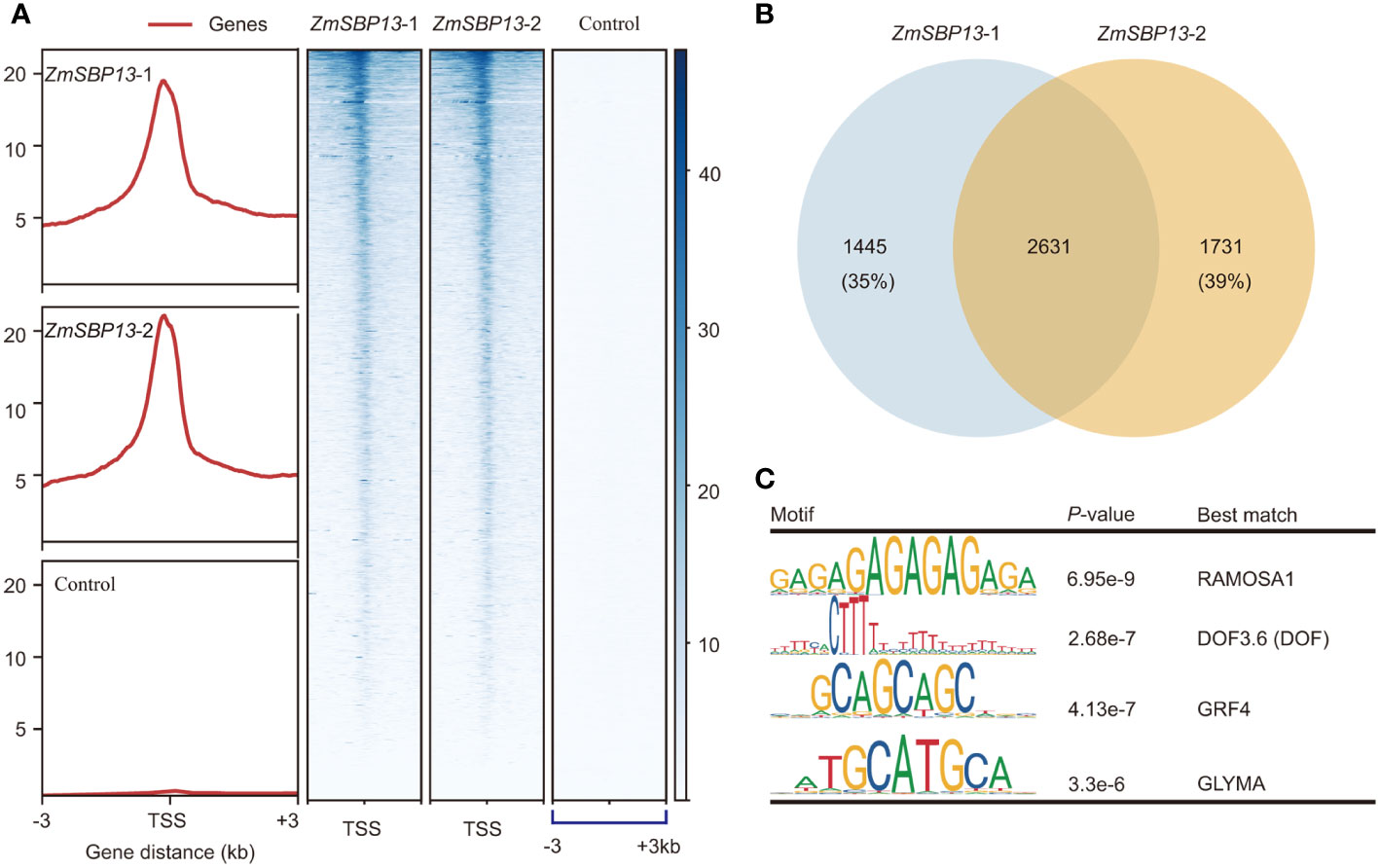
Figure 5 ZmSBP13-bound target genes. (A) Heatmap of the binding sites of ZmSBP13 at the positions − 3.0 kb upstream to + 3.0 kb downstream relative to transcription start site (TSS). (B) Overlapped target genes between ZmSBP13 tsCUT&Tag replicates. (C) ZmSBP13 bound motifs identified by MEME. The motif sequence is shown in the left column, the P value is shown in middle column and the corresponding proteins are shown in the right column.
4 Discussion
Tp2 had been verified that exhibits different phenotypes under different genetic backgrounds, indicating that gene dosage contributes to the observed variation (Poethig, 1988). This is further supported by our findings that overexpression of zma-miR156h produced different phenotypes (Figure 4A). Another dominant mutant, Cg1, exhibited more severe phenotypes compared to several Teopod mutants (Chuck et al., 2007). Cg1 encodes two tandem miR156 genes (miR156b/c) that are overexpressed in the meristem and lateral organs, while overexpression of zma-miR156h produced similar phenotypes to Cg1. Both mutants result from overproduction and misexpression of mature miR156, highlighting the importance of suitable expression of miRNA156 throughout the plant’s life cycle. Teopod1 and Teopod3 also exhibited similar phenotypes to Tp2, including an increase in the number of vegetative phytomers and phytomers producing ears, tillers, and prop roots, a decrease in the size of leaves, internodes, ear, and tassel, and transformation of reproductive structures into vegetative ones (Poethig, 1988). Additionally, glossy15 (gl15) also affected the juvenile-to-adult phase transition but appeared to affect only epidermal phase transition traits, such as epidermal waxes, and acted downstream of Cg1, Tp1, and Tp2 (Moose and Sisco, 1994). Gl15 encodes a member of the AP2-domain family and functions in maintaining juvenile epidermal traits by negatively regulating microRNA172, which has been shown to promote juvenile-to-adult phase transition (Moose and Sisco, 1996; Lauter et al., 2005).
RNA-seq analysis revealed several differentially expressed genes associated with maize inflorescence development in Tp2 mutants. Among them, genes such as ub2, ub3, tsh4, ra3, and silky1 have been previously demonstrated to affect maize inflorescence development (Ambrose et al., 2000; Satoh-Nagasawa et al., 2006; Chuck et al., 2014). Additionally, MADS-box and SPL family genes, including ZmMADS8/14/18/31 and ZmSBP13/23/27/29 were significantly differentially expressed between Tp2 mutants and wild type materials (Supplementary Table S7). These genes have been reported to play important roles in inflorescence development in maize and other species (Heuer et al., 2000; Heuer et al., 2001; Schilling et al., 2018). Thiamine biosynthesis1 (Thi1) also exhibited significant up-regulated expression in Tp2 mutants, whose paralogue thiamine biosynthesis2 has been shown to affect maintenance of SAM and inflorescence development in maize (Supplementary Table S7) (Woodward et al., 2010). Furthermore, two gibberellin synthesis genes, dwarf plant3 and gibberellin 20-oxidase1, were significantly down-regulated in Tp2 mutants, possibly explaining relatively decreased plant height phenotype of Tp2 (Supplementary Table S7) (Winkler and Helentjaris, 1995; Voorend et al., 2016). Thus, dissection of Tp2 mutants provides insights into the understanding of inflorescence and plant architecture development in maize.
Maize tassel branch trait is regulated by multiple genes and involved in various regulatory networks. The CLV-WUS feedback pathway plays an essential role in maintaining and determining the SAM. THICK TASSEL DWARF1 (TD1), FASCIATED EAR2 (FEA2) and ZmCLE7 are some of the primary genes involved in this pathway (Taguchi-Shiobara et al., 2001; Bommert et al., 2005; Liu et al., 2021). The ramosa pathway, centered around the REL2/RA1 transcriptional repressor complex, plays an antagonizes role in the formation of indeterminate branches during maize inflorescence development. This pathway includes ra2 and ra3 (Gallavotti et al., 2010). The miR156-SPL pathway also plays a crucial role in tassel branch development by regulating Inflorescence bracts and axillary meristem growth, including ub2, ub3, tsh4, and ZmSBP13 (identified in this study). Phytohormones, such as auxin, also play a vital role in regulating tassel developments, with SPI1, VT2, BIF1 and BIF4 genes having been identified in this regard (Gallavotti et al., 2008; Phillips et al., 2011; Galli et al., 2015). Epigenetic regulation also has been shown to affect tassel morphology development with histone deacetylase 108 (HDA108) playing a role in overall plant architecture and inflorescence patterning and fertility (Forestan et al., 2018). Crosstalks between different pathways can help to fully understand the mechanism of tassel development. Several genes, including ra3, dwarf plant3, GA20ox1 and thi1 exhibited significantly differentially expressed in Tp2 mutants, providing an avenue for understanding the crosstalks between the miR156-SPL pathway and other pathways in tassel development.
During the process of modern maize breeding, the plant architecture of maize was significantly changed for adaptation to high planting density, including more compact architecture with reduced relative ear height and upper leaf angle, smaller tassel with decreased tassel branch number. In this study, we identified new miR156-SPL modules functioning in plant architecture development in maize. Overexpression of zma-miR156h, as well as knockout of ZmSBP13, exhibited decreased tassel branch number in various degrees. Therefore, the accurate regulation of miR156-SPL modules exhibited huge potential for plant architecture improvement during modern maize breeding.
Other than maize, the miR156-SPL modules are widely conserved in various plant species and plays crucial roles in multiple developmental processes throughout the entire life cycle. Overexpression of miR156 in plants, such as Arabidopsis, rice, tomato, among others, has been shown to cause prolonged juvenile vegetative periods and delayed flowering (Wu and Poethig, 2006; Xie et al., 2006). In rice, Ideal Plant Architecture1 (IPA1) encodes the SPL family transcription factor, OsSPL14, which has been identified as a key regulator of plant architecture. Point mutations in the miRNA156 recognition site led to decreased tiller number and increased plant height and panicle branches due to the repression of IPA1 mRNA degradation (Jiao et al., 2010; Miura et al., 2010; Lu et al., 2013). Additionally, miR156-SPL modules also functioned in grain filling (Wang et al., 2012), branch angle controlling (Ishii et al., 2013; Zhu et al., 2013), fruit ripening (Manning et al., 2006), biotic and abiotic stress responses (Stone et al., 2005; Padmanabhan et al., 2013; Stief et al., 2014). Subtle manipulation of miR156-SPL modules has partially contributed to plant architecture improvement, yield and quality elevation in rice, and will continually make great contributions during modern crop breeding processes including maize, wheat and so on.
Based on our results and previous studies, we proposed a miR156-SPL model regulating tassel branch development in maize (Figure 6): Overexpression of zma-miR156h in Tp2 mutant, repressed the expression of series of SPL transcriptional factors, including ZmSBP13, ZmSBP23/27/29, as well as UB2, UB3 and TSH4, which positively regulating a large number of target genes, therefore, contributing to decreased tassel branch number trait in maize. Especially, a new identified SPL gene, ZmSBP13 affected tassel development by targeting many important genes in maize, for example indeterminate spikelet1 (ids1), KNOTTED1 (KN1), ZmBLH12 and so on (Supplementary Table S10).
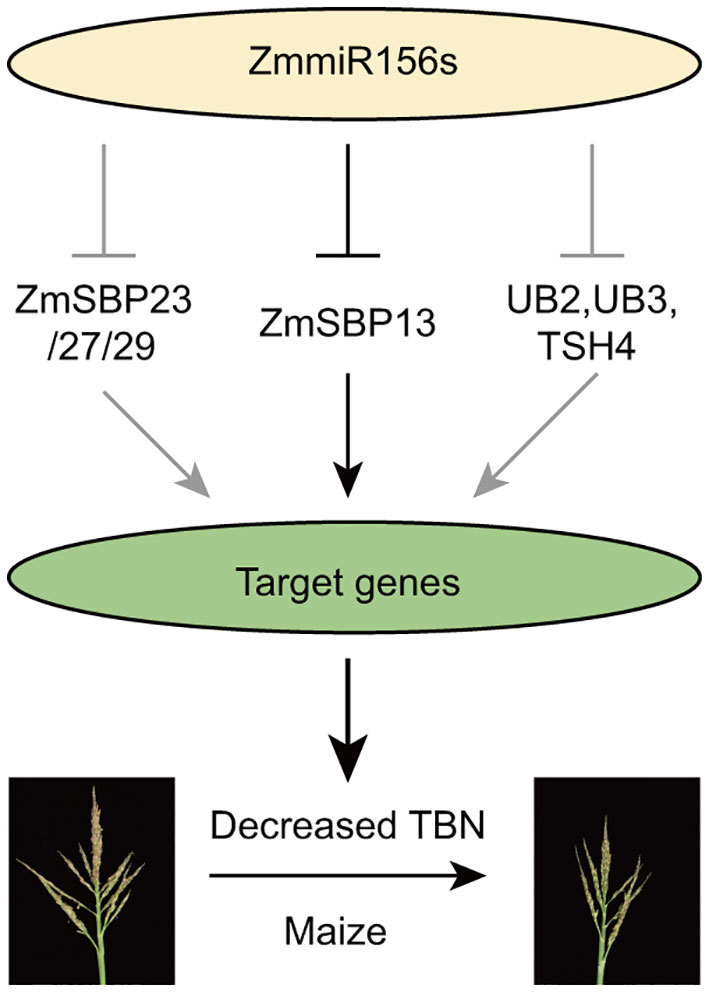
Figure 6 A proposed model for miRNA156-SPL to regulate the tassel branch.
In summary, we characterized and functionally analyzed a classical maize dominant mutant, Teopod2 (Tp2), and identified a microRNA, zma-miR156h, which regulates multiple agronomic traits in maize by inhibiting the expression of ZmSBP13, and the ZmSBP13 may interact with multiple proteins to regulate inflorescence structure in maize. Nevertheless, there are still some unresolved questions, such as the relationships between ZmSBP13 and other known SPL family genes (ub2, ub3 and tsh4). ub2, ub3 and tsh4 had been verified functioning redundantly in tassel development (Chuck et al., 2014). Whether there are functional redundancies between ZmSBP13 and three known SPL genes, as well as other unknown SPL genes, should be addressed and dissected by future genetic analyses. Moreover, overexpression of miRNA156 in various species exhibiting multiple phenotypes, including plant architecture, inflorescence structure, and yield. Realizing accurate regulation of the miRNA156-SPL module remains a significant challenge, as well as an essential measure to satisfy the increasing demands of cereals.
Data availability statement
The datasets presented in this study can be found in National Center for Biotechnology Information. The names of the accession number(s) can be found below: SRR23581318, SRR23581319, SRR23581320, SRR23581321, SRR23581322.
Author contributions
LL and JL designed the experiments. JL conducted experiments with the assistance of XW. JL generated the datasets and performed data analysis with the assistance of JW. JL and XW wrote the manuscript; LL and JL reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by National Natural Science Foundation of China (32261143463), Hainan Yazhou Bay Seed Lab (B21HJ8102), the major Program of Hubei Hongshan laboratory (2021hszd008), and Huazhong Agricultural University Scientific & Technological Self-innovation Foundation (2021ZKPY001).
Acknowledgments
We appreciate the high-performance computing platform at National Key Laboratory of Crop Genetic Improvement in Huazhong Agricultural University. We also thank Maize Genetics Cooperation Stock Center for the generosity of sharing the Tp2 mutant.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2023.1183697/full#supplementary-material
References
Ambrose, B. A., Lerner, D. R., Ciceri, P., Padilla, C. M., Yanofsky, M. F., Schmidt, R. J. (2000). Molecular and genetic analyses of the silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579. doi: 10.1016/s1097-2765(00)80450-5
Bommert, P., Lunde, C., Nardmann, J., Vollbrecht, E., Running, M., Jackson, D., et al. (2005). Thick tassel dwarf1 encodes a putative maize ortholog of the arabidopsis CLAVATA1 leucine-rich repeat receptor-like kinase. Development 132, 1235–1245. doi: 10.1242/dev.01671
Chatterjee, M., Tabi, Z., Galli, M., Malcomber, S., Buck, A., Muszynski, M., et al. (2014). The boron efflux transporter ROTTEN EAR is required for maize inflorescence development and fertility. Plant Cell 26, 2962–2977. doi: 10.1105/tpc.114.125963
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Chuck, G. S., Brown, P. J., Meeley, R., Hake, S. (2014). Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc. Natl. Acad. Sci. U.S.A. 111, 18775–18780. doi: 10.1073/pnas.1407401112
Chuck, G., Cigan, A. M., Saeteurn, K., Hake, S. (2007). The heterochronic maize mutant Corngrass1 results from overexpression of a tandem microRNA. Nat. Genet. 39, 544–549. doi: 10.1038/ng2001
Dai, X., Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Forestan, C., Farinati, S., Rouster, J., Lassagne, H., Lauria, M., Dal Ferro, N., et al. (2018). Control of maize vegetative and reproductive development, fertility, and rRNAs silencing by HISTONE DEACETYLASE 108. Genetics 208, 1443–1466. doi: 10.1534/genetics.117.300625
Gallavotti, A., Barazesh, S., Malcomber, S., Hall, D., Jackson, D., Schmidt, R. J., et al. (2008). Sparse inflorescence1 encodes a monocot-specific YUCCA-like gene required for vegetative and reproductive development in maize. Proc. Natl. Acad. Sci. U.S.A. 105, 15196–15201. doi: 10.1073/pnas.0805596105
Gallavotti, A., Long, J. A., Stanfield, S., Yang, X., Jackson, D., Vollbrecht, E., et al. (2010). The control of axillary meristem fate in the maize ramosa pathway. Development 137, 2849–2856. doi: 10.1242/dev.051748
Galli, M., Liu, Q., Moss, B. L., Malcomber, S., Li, W., Gaines, C., et al. (2015). Auxin signaling modules regulate maize inflorescence architecture. Proc. Natl. Acad. Sci. U.S.A. 112, 13372–13377. doi: 10.1073/pnas.1516473112
Godfray, H. C. J., Beddington, J. R., Crute, I. R., Haddad, L., Lawrence, D., Muir, J. F., et al. (2010). Food security: the challenge of feeding 9 billion people. Science 327, 812–818. doi: 10.1126/science.1185383
Heuer, S., Hansen, S., Bantin, J., Brettschneider, R., Kranz, E., Lörz, H., et al. (2001). The maize MADS box gene ZmMADS3 affects node number and spikelet development and is co-expressed with ZmMADS1 during flower development, in egg cells, and early embryogenesis. Plant Physiol. 127, 33–45. doi: 10.1104/pp.127.1.33
Heuer, S., Lörz, H., Dresselhaus, T. (2000). The MADS box gene ZmMADS2 is specifically expressed in maize pollen and during maize pollen tube growth. Sex Plant Reprod. 13, 21–27. doi: 10.1007/PL00009838
Hill, J. T., Demarest, B. L., Bisgrove, B. W., Gorsi, B., Su, Y.-C., Yost, H. J. (2013). MMAPPR: mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 23, 687–697. doi: 10.1101/gr.146936.112
Ishii, T., Numaguchi, K., Miura, K., Yoshida, K., Thanh, P. T., Htun, T. M., et al. (2013). OsLG1 regulates a closed panicle trait in domesticated rice. Nat. Genet. 45, 462–465. doi: 10.1038/ng.2567
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544. doi: 10.1038/ng.591
Kerstetter, R. A., Laudencia-Chingcuanco, D., Smith, L. G., Hake, S. (1997). Loss-of-function mutations in the maize homeobox gene, knotted1, are defective in shoot meristem maintenance. Development 124, 3045–3054. doi: 10.1242/dev.124.16.3045
Koppolu, R., Schnurbusch, T. (2019). Developmental pathways for shaping spike inflorescence architecture in barley and wheat. J. Integr. Plant Biol. 61, 278–295. doi: 10.1111/jipb.12771
Lauter, N., Kampani, A., Carlson, S., Goebel, M., Moose, S. P. (2005). microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc. Natl. Acad. Sci. U.S.A. 102, 9412–9417. doi: 10.1073/pnas.0503927102
Liu, L., Gallagher, J., Arevalo, E. D., Chen, R., Skopelitis, T., Wu, Q., et al. (2021). Enhancing grain-yield-related traits by CRISPR–Cas9 promoter editing of maize CLE genes. Nat. Plants 7, 287–294. doi: 10.1038/s41477-021-00858-5
Liu, S., Yeh, C.-T., Tang, H. M., Nettleton, D., Schnable, P. S. (2012). Gene mapping via bulked segregant RNA-seq (BSR-seq). PloS One 7, e36406. doi: 10.1371/journal.pone.0036406
Lu, Z., Yu, H., Xiong, G., Wang, J., Jiao, Y., Liu, G., et al. (2013). Genome-wide binding analysis of the transcription activator IDEAL PLANT ARCHITECTURE1 reveals a complex network regulating rice plant architecture. Plant Cell 25, 3743–3759. doi: 10.1105/tpc.113.113639
Machanick, P., Bailey, T. L. (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697. doi: 10.1093/bioinformatics/btr189
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Miura, K., Ikeda, M., Matsubara, A., Song, X.-J., Ito, M., Asano, K., et al. (2010). OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549. doi: 10.1038/ng.592
Moose, S. P., Sisco, P. H. (1994). Glossy15 controls the epidermal juvenile-to-Adult phase transition in maize. Plant Cell 6, 1343–1355. doi: 10.1105/tpc.6.10.1343
Moose, S. P., Sisco, P. H. (1996). Glossy15, an APETALA2-like gene from maize that regulates leaf epidermal cell identity. Genes Dev. 10, 3018–3027. doi: 10.1101/gad.10.23.3018
Padmanabhan, M. S., Ma, S., Burch-Smith, T. M., Czymmek, K., Huijser, P., Dinesh-Kumar, S. P. (2013). Novel positive regulatory role for the SPL6 transcription factor in the n TIR-NB-LRR receptor-mediated plant innate immunity. PloS Pathog. 9, e1003235. doi: 10.1371/journal.ppat.1003235
Pautler, M., Eveland, A. L., LaRue, T., Yang, F., Weeks, R., Lunde, C., et al. (2015). FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell 27, 104–120. doi: 10.1105/tpc.114.132506
Phalan, B., Onial, M., Balmford, A., Green, R. E. (2011). Reconciling food production and biodiversity conservation: land sharing and land sparing compared. Science 333, 1289–1291. doi: 10.1126/science.1208742
Phillips, K. A., Skirpan, A. L., Liu, X., Christensen, A., Slewinski, T. L., Hudson, C., et al. (2011). Vanishing tassel2 encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize. Plant Cell 23, 550–566. doi: 10.1105/tpc.110.075267
Poethig, R. S. (1988). Heterochronic mutations affecting shoot development in maize. Genetics 119, 959–973. doi: 10.1093/genetics/119.4.959
Satoh-Nagasawa, N., Nagasawa, N., Malcomber, S., Sakai, H., Jackson, D. (2006). A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 441, 227–230. doi: 10.1038/nature04725
Schilling, S., Pan, S., Kennedy, A., Melzer, R. (2018). MADS-box genes and crop domestication: the jack of all traits. J. Exp. Bot. 69, 1447–1469. doi: 10.1093/jxb/erx479
Stief, A., Altmann, S., Hoffmann, K., Pant, B. D., Scheible, W.-R., Bäurle, I. (2014). Arabidopsis miR156 regulates tolerance to recurring environmental stress through SPL transcription factors. Plant Cell 26, 1792–1807. doi: 10.1105/tpc.114.123851
Stone, J. M., Liang, X., Nekl, E. R., Stiers, J. J. (2005). Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to fumonisin B1. Plant J. 41, 744–754. doi: 10.1111/j.1365-313X.2005.02334.x
Taguchi-Shiobara, F., Yuan, Z., Hake, S., Jackson, D. (2001). The fasciated ear2 gene encodes a leucine-rich repeat receptor-like protein that regulates shoot meristem proliferation in maize. Genes Dev. 15, 2755–2766. doi: 10.1101/gad.208501
Tian, T., Liu, Y., Yan, H., You, Q., Yi, X., Du, Z., et al. (2017). agriGO v2.0: a GO analysis toolkit for the agricultural community 2017 update. Nucleic Acids Res. 45, W122–W129. doi: 10.1093/nar/gkx382
Tilman, D., Balzer, C., Hill, J., Befort, B. L. (2011). Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. U.S.A. 108, 20260–20264. doi: 10.1073/pnas.1116437108
Tsuda, K., Abraham-Juarez, M.-J., Maeno, A., Dong, Z., Aromdee, D., Meeley, R., et al. (2017). KNOTTED1 cofactors, BLH12 and BLH14, regulate internode patterning and vein anastomosis in maize. Plant Cell 29, 1105–1118. doi: 10.1105/tpc.16.00967
Vegetti, A., Anton, A. M. (1995). Some evolution trends in the inflorescence of poaceae. Flora 190, 225–228. doi: 10.1016/S0367-2530(17)30655-2
Vollbrecht, E., Springer, P. S., Goh, L., Buckler, E. S., Martienssen, R. (2005). Architecture of floral branch systems in maize and related grasses. Nature 436, 1119–1126. doi: 10.1038/nature03892
Voorend, W., Nelissen, H., Vanholme, R., De Vliegher, A., Van Breusegem, F., Boerjan, W., et al. (2016). Overexpression of GA20-OXIDASE1 impacts plant height, biomass allocation and saccharification efficiency in maize. Plant Biotechnol. J. 14, 997–1007. doi: 10.1111/pbi.12458
Wang, J.-W., Czech, B., Weigel, D. (2009). miR156-regulated SPL transcription factors define an endogenous flowering pathway in arabidopsis thaliana. Cell 138, 738–749. doi: 10.1016/j.cell.2009.06.014
Wang, B., Lin, Z., Li, X., Zhao, Y., Zhao, B., Wu, G., et al. (2020). Genome-wide selection and genetic improvement during modern maize breeding. Nat. Genet. 52, 565–571. doi: 10.1038/s41588-020-0616-3
Wang, H., Wang, H. (2015). The miR156/SPL module, a regulatory hub and versatile toolbox, gears up crops for enhanced agronomic traits. Mol. Plant 8, 677–688. doi: 10.1016/j.molp.2015.01.008
Wang, S., Wu, K., Yuan, Q., Liu, X., Liu, Z., Lin, X., et al. (2012). Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 44, 950–954. doi: 10.1038/ng.2327
Wang, C., Yang, X., Zhang, Y., Shen, C., Shi, J., Xia, C., et al. (2022). Barley FASCIATED EAR genes determine inflorescence meristem size and yield traits. Crop J. doi: 10.1016/j.cj.2022.10.001
Winkler, R. G., Helentjaris, T. (1995). The maize Dwarf3 gene encodes a cytochrome P450-mediated early step in gibberellin biosynthesis. Plant Cell 7, 1307–1317. doi: 10.1105/tpc.7.8.1307
Woodward, J. B., Abeydeera, N. D., Paul, D., Phillips, K., Rapala-Kozik, M., Freeling, M., et al. (2010). A maize thiamine auxotroph is defective in shoot meristem maintenance. Plant Cell 22, 3305–3317. doi: 10.1105/tpc.110.077776
Wu, L., Luo, Z., Shi, Y., Jiang, Y., Li, R., Miao, X., et al. (2022). A cost-effective tsCUT&Tag method for profiling transcription factor binding landscape. J. Integr. Plant Biol. 64, 2033–2038. doi: 10.1111/jipb.13354
Wu, G., Park, M. Y., Conway, S. R., Wang, J.-W., Weigel, D., Poethig, R. S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in arabidopsis. Cell 138, 750–759. doi: 10.1016/j.cell.2009.06.031
Wu, G., Poethig, R. S. (2006). Temporal regulation of shoot development in arabidopsis thalianaby miR156 and its target SPL3. Development 133, 3539–3547. doi: 10.1242/dev.02521
Xie, K., Wu, C., Xiong, L. (2006). Genomic organization, differential expression, and interaction of SQUAMOSA promoter-Binding-Like transcription factors and microRNA156 in rice. Plant Physiol. 142, 280–293. doi: 10.1104/pp.106.084475
Zhang, D., Yuan, Z. (2014). Molecular control of grass inflorescence development. Annu. Rev. Plant Biol. 65, 553–578. doi: 10.1146/annurev-arplant-050213-040104
Keywords: Teopod2, tassel branch number, genetic mapping, functional analysis, zma-miR156h
Citation: Li J, Wang X, Wei J, Miao X, Shang X and Li L (2023) Genetic mapping and functional analysis of a classical tassel branch number mutant Tp2 in maize. Front. Plant Sci. 14:1183697. doi: 10.3389/fpls.2023.1183697
Received: 10 March 2023; Accepted: 16 May 2023;
Published: 02 June 2023.
Edited by:
Rodomiro Ortiz, Swedish University of Agricultural Sciences, SwedenReviewed by:
Qingyu Wu, Chinese Academy of Agricultural Sciences (CAAS), ChinaVignesh Muthusamy, Indian Agricultural Research Institute (ICAR), India
Copyright © 2023 Li, Wang, Wei, Miao, Shang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Li, aHphdWxpbGluQG1haWwuaHphdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship