 Serge Yaacoub1
Serge Yaacoub1 Elton Vannoy1
Elton Vannoy1 Stefanyda Maslova2
Stefanyda Maslova2 Abigail Haffey1
Abigail Haffey1 Khatereh Khorsandi1
Khatereh Khorsandi1 Natasha Sheybani2,3
Natasha Sheybani2,3 Dalia Haydar1*
Dalia Haydar1*- 1Center for Cancer and Immunology Research, Children’s National Hospital, Washington, DC, United States
- 2Department of Biomedical Engineering, University of Virginia, Charlottesville, VA, United States
- 3Focused Ultrasound Cancer Immunotherapy Center, University of Virginia, Charlottesville, VA, United States
Chimeric Antigen Receptor T (CAR-T) cell therapy offers substantial promise for the treatment of brain malignancies, yet its clinical translation remains limited. Tumors such as Glioblastoma Multiforme (GBM), Diffuse Intrinsic Pontine Glioma (DIPG), and Medulloblastoma (MB) are associated with poor prognoses and exhibit limited responsiveness to conventional treatment modalities, including radiotherapy, chemotherapy, and surgical resection. The application of CAR-T cell therapy in these contexts faces significant challenges, primarily in terms of efficient cellular trafficking into the tumor microenvironment and access to heterogeneous tumor regions. Furthermore, CAR-T cell persistence, defined by the long-term survival and functionality of infused cells, remains a critical hurdle in achieving durable therapeutic responses and preventing tumor relapses. This review aims to address the two predominant barriers, trafficking and persistence, by discussing the underlying mechanisms that limit CAR-T cell efficacy in brain tumors, reviewing current strategies aimed at overcoming these challenges, and evaluating novel approaches to enhance the effectiveness of CAR-T therapies in this setting.
1 Introduction
Chimeric Antigen Receptor (CAR) T-cell therapies have revolutionized cancer treatment, particularly for hematological malignancies, thus significantly improving patient outcomes compared to traditional chemotherapeutic approaches (1). In recent years, adoptive immunotherapy has demonstrated remarkable promise, culminating in FDA approval for several anti-CD19 CAR therapies, including Kymriah for refractory or relapsed follicular lymphoma, Yescarta for large B-cell lymphoma, Tecartus for mantle cell lymphoma, Breyanzi for large B-cell lymphoma, and Carvykti and Abecma for multiple myeloma (2). These achievements underscore the transformative potential of CAR-T cell therapies and have since catalyzed efforts to apply these innovative approaches to solid tumors, particularly those affecting the brain (3).
However, central nervous system (CNS) tumors present distinct challenges that limit the effectiveness of CAR-T cell therapies (4). The solid tumor microenvironment (TME) in the brain is complex, characterized by immune-suppressive tumor cells, dense extracellular matrices, antigen heterogeneity, and limited trafficking of immune cells (5, 6). These barriers have significantly hindered the progress of CAR T cell therapies for brain tumors, despite promising preclinical findings and numerous clinical trials conducted in both adult and pediatric populations (7). While these trials (Table 1) have demonstrated safety and feasibility, particularly with intra-tumoral delivery, therapeutic responses have been less robust compared to those observed in hematological malignancies.
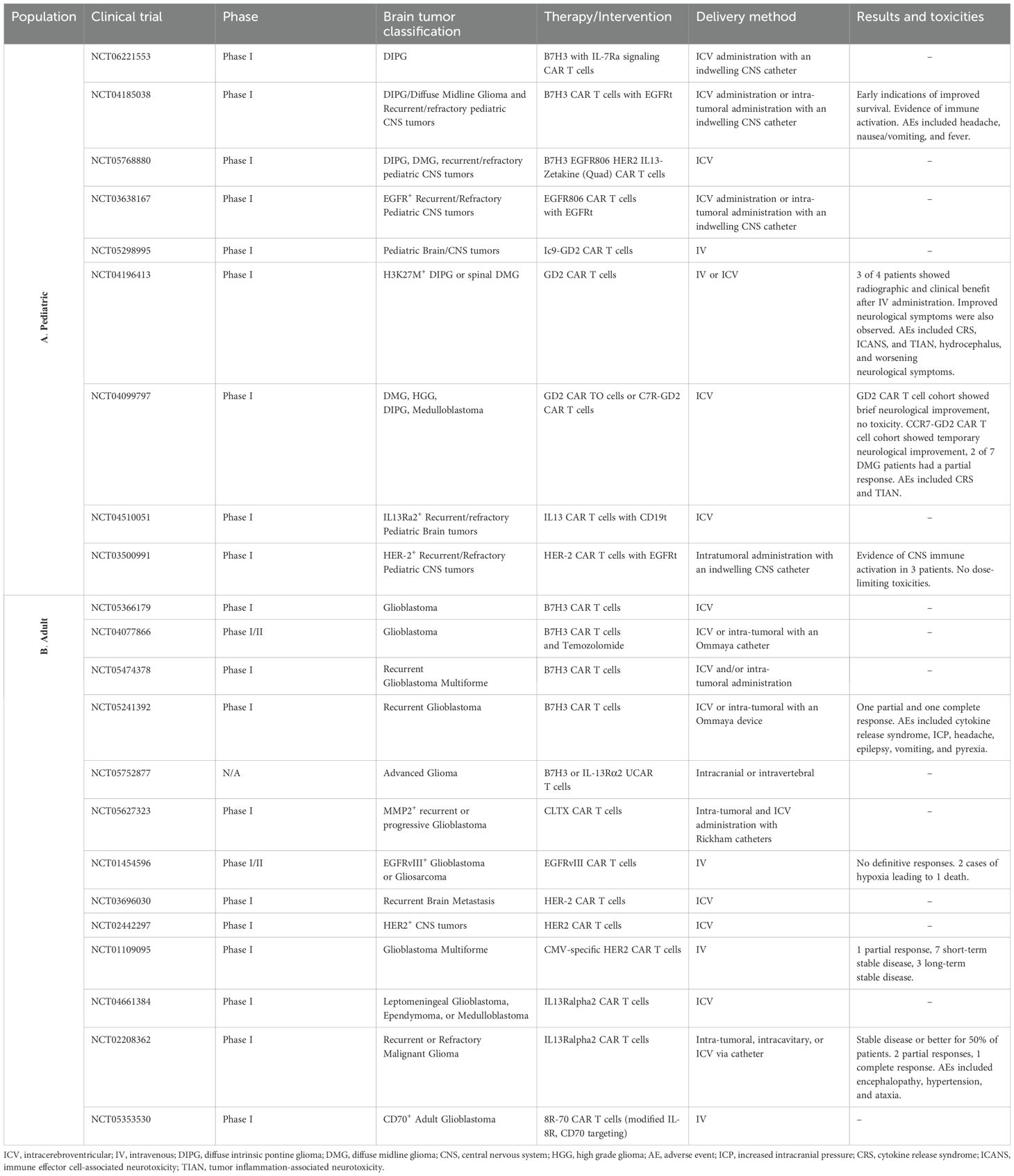
Table 1. List of clinical trials pertaining to CAR-T therapies for brain tumors in the adult and pediatric populations (A- Pediatric Population; B- Adult Population).
This review will focus on two critical hurdles: (1) the inefficiency of CAR T cell trafficking to the tumor site and the complexities of the CNS TME that impede immune cell infiltration, and (2) the lack of CAR T cell persistence and long-term functionality required for sustained tumor control. We will explore the underlying mechanisms that drive these challenges, evaluate current strategies aimed at overcoming them, and discuss emerging approaches to enhance the therapeutic potential of CAR-T cell therapies for brain malignancies. To our knowledge, this is the first comprehensive review to systematically map the full spectrum of barriers undermining CAR T cell efficacy in solid CNS tumors. Unlike previous reviews, our work uniquely integrates both preclinical and clinical data accumulated over several years, with a specific focus on enhancing CAR T cell persistence and delivery in the brain. While we discuss well-established challenges (including heterogeneous antigen expression, the immunosuppressive tumor microenvironment, anatomical constraints, and safety concerns), we also highlight novel technologies and delivery strategies that have not yet been widely incorporated into brain tumor cell therapy. Additionally, we offer our own perspective on how these emerging tools could be leveraged to overcome current limitations. By synthesizing recent clinical trial data alongside cutting-edge preclinical approaches, we propose targeted, stage-specific solutions to improve the endurance and efficacy of CAR T cells in treating CNS tumors.
2 CAR-T cells versus other adoptive T-cell therapies
Adoptive T-cell therapies include various approaches such as T-cell receptor (TCR)-engineered T cells, tumor-associated antigen (TAA)-specific T cells, and CAR-T cells, each utilizing distinct mechanisms of action (Figure 1) (8, 9):
a. TCR-Engineered T Cells: These therapies harness the T-cell receptor’s natural ability to recognize tumor antigens presented on the cell surface by the Major Histocompatibility Complex (MHC) (10). This MHC-dependent mechanism enables targeting intracellular antigens that are processed and displayed (11). However, the need for intact antigen presentation machinery and specific human leukocyte antigen (HLA) haplotypes limits their efficacy, particularly in solid tumors like brain cancers, where antigen presentation is often impaired (12).
b. TAA-Specific T Cells: Derived from tumor-infiltrating lymphocytes (TILs) or peripheral T cells, TAA-specific T cells target tumor-associated antigens that are naturally recognized by the immune system (13, 14). While these cells can target a broad range of antigens, their effectiveness is reduced by the immunosuppressive tumor microenvironment and the low frequency of tumor-reactive T cells (15, 16).
c. CAR T Cells: CAR T cell therapy represents a significant advancement in adoptive immunotherapy, where T cells are genetically engineered to recognize and kill tumor cells (17). This is achieved by introducing synthetic receptors that consist of an extracellular antigen-binding domain (often from antibodies), a transmembrane domain, and an intracellular signaling domain (17). CAR-T cells uniquely recognize antigens in an MHC-independent manner, allowing them to target surface antigens directly without the need for MHC-mediated antigen presentation (9). Additionally, CAR T cells can be engineered with co-stimulatory domains, cytokine secretion modules, and synthetic circuits to enhance their efficacy, persistence, and resistance to immune suppression (18) (Figure 2).
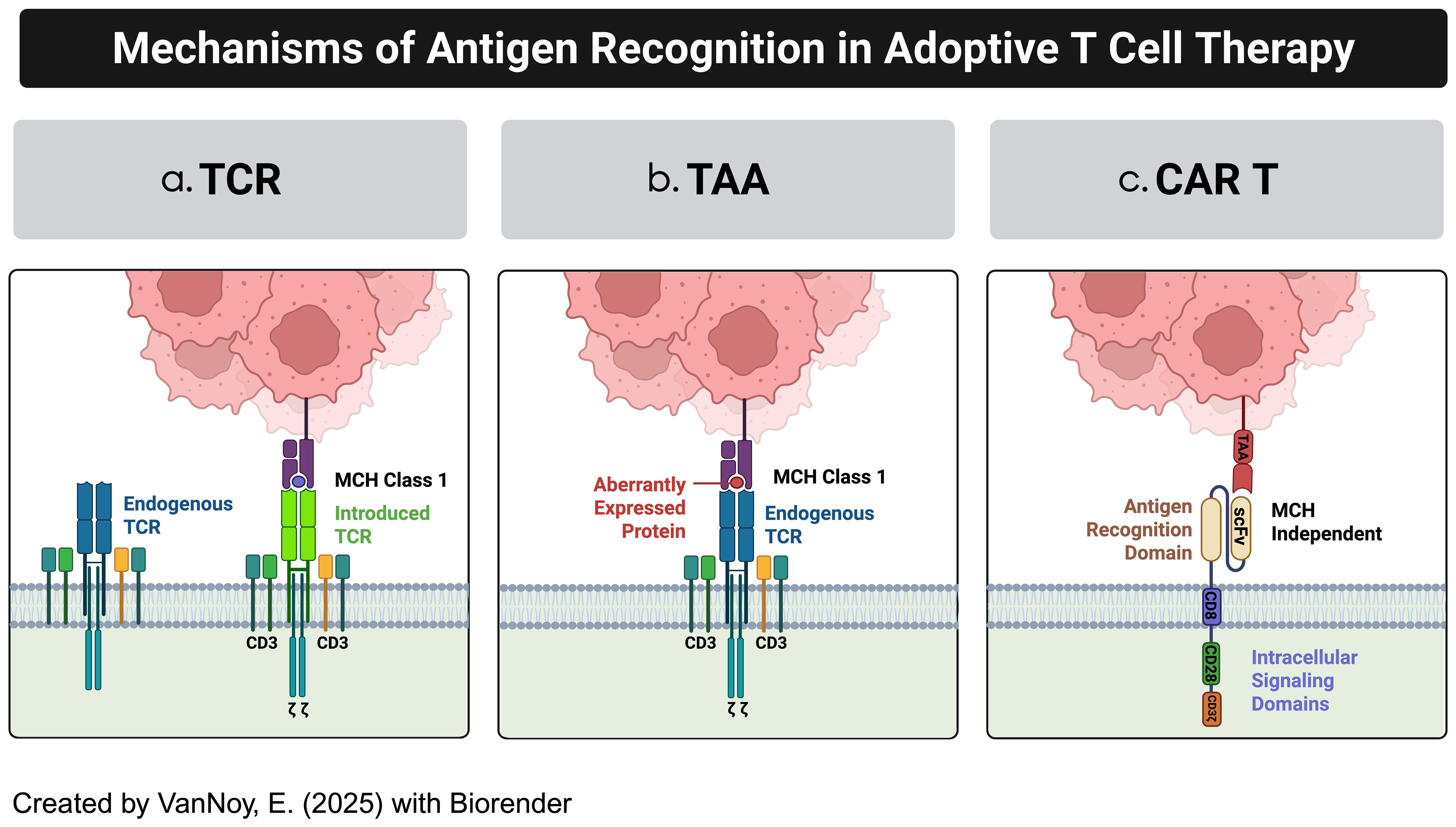
Figure 1. Examples of adoptive T-cell technologies. (a) Modified TCR cytotoxic T-cells (CTLs) (b) Tumor-associated antigen-specific T cells (TAA-T cells) (c) Chimeric antigen receptor modified T cells.
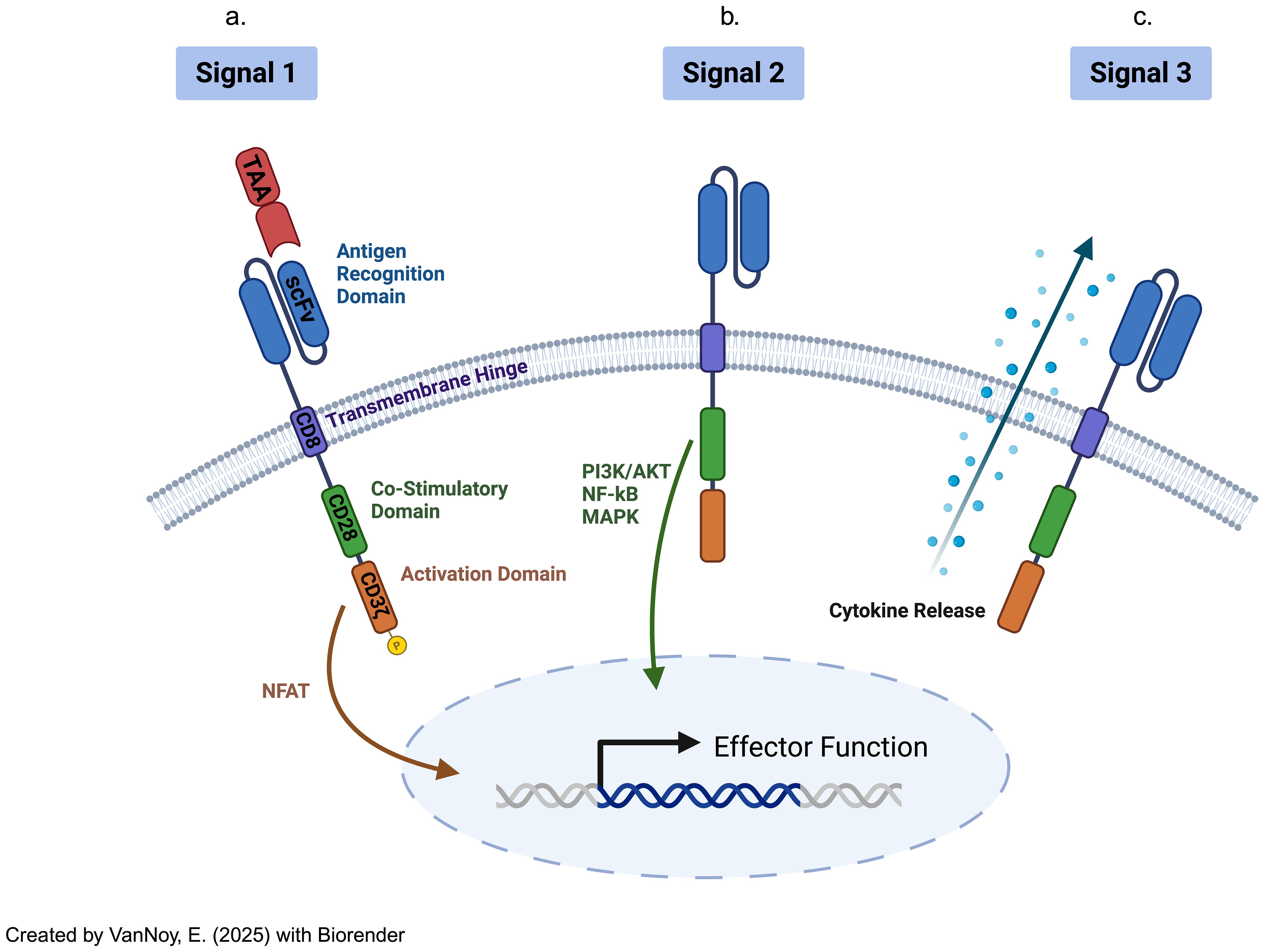
Figure 2. Chimeric antigen receptor-modified T cells have distinct CAR receptors with their own transduction pathways, separate from the TCR-mediated pathways, virtue of their variant extracellular, transmembrane, costimulatory, and intracellular activation domains. The signaling pathway is initiated upon CAR-T cells (a) binding to the extracellular ligands (signal 1: "activation"). Then, (b) co-stimulation (through signal 2) functions as a necessary co-activation signal, prior to (c) cytokine release (signal 3) that supports eventual CAR-T cell differentiation and ultimate activation .
Hence, CAR-T cell therapy stands apart from TCR-engineered and TAA-specific T-cell therapy by offering MHC-independent antigen recognition, enabling direct targeting of extracellular antigens (8, 11, 19). While TCR-based therapies are limited to intracellular antigens and require MHC presentation, CAR-T cells can target a broader range of surface antigens. Furthermore, the extensive engineering capabilities of CAR-T cells, such as incorporating co-stimulatory domains and cytokine modules, provide significant advantages for overcoming challenges like antigen heterogeneity and immune evasion in solid tumors (1, 20).
3 Challenges for effective CAR T cell therapy in CNS tumors
This section examines the key challenges limiting the success of CAR T cell therapies for CNS tumors, focusing on the anatomical barriers posed by tumor location within the brain, the immunosuppressive TME, and the lack of persistence of CAR T cells in the CNS, all of which hinder effective therapeutic outcomes (Figure 3). For clarification, we define pertinent and repetitive terms as the following:
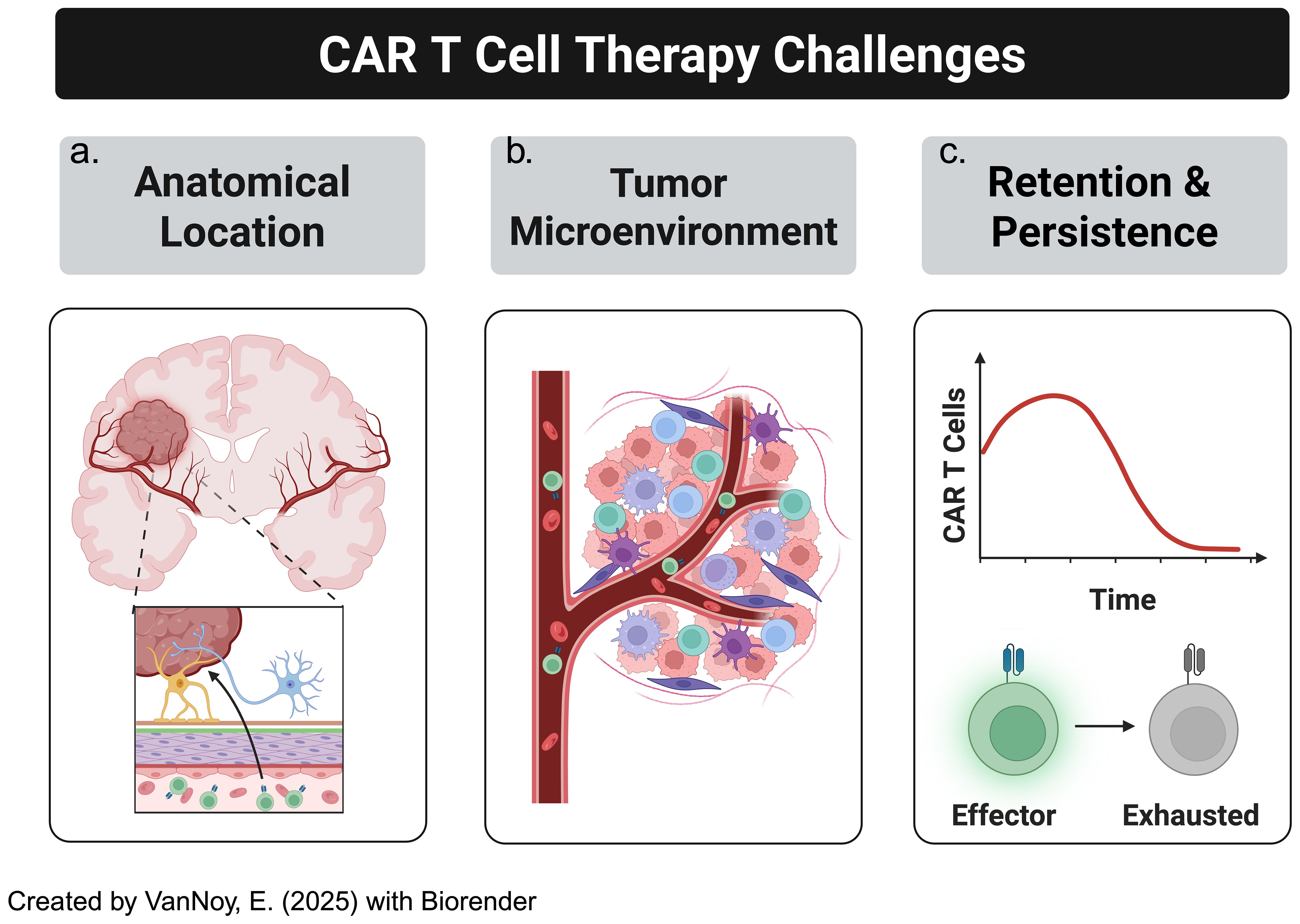
Figure 3. CAR-T cell therapy in solid tumors, particularly brain tumors, has several challenges to overcome. This includes (a) location: solid brain tumors are much less accessible than other malignancies, especially hematological malignancies, (b) "cold" or "immunosuppressive" tumor microenvironments, (c) blunted CAR-T cell persistence.
CNS (Central Nervous System) Tumors: Neoplastic lesions originating in the brain or spinal cord—including primary and metastatic forms—which face delivery and immunological challenges.
TME/TIME (Tumor/tumor immune microenvironment): The local cellular (e.g., myeloid cell populations) and non-cellular (e.g., cytokines, growth factors) milieu within the tumor periphery. It is a major player in dictating immune response efficacy to endogenous and exogenous agents.
BBB (Blood-brain barrier): A highly selective barrier that shields the brain from pathogens, toxins, and certain therapeutics, through a dynamic vascular and endothelial framework.
BTB (Blood-tumor barrier): The vascular interface surrounding central nervous system tumors that exhibits heterogeneous permeability to drugs and immune-cell infiltration, contrary to the traditional blood-brain barrier (BBB).
3.1 Anatomical tumor location within the CNS
The anatomical location of tumors within the CNS significantly impacts the effectiveness of CAR T cell therapy, primarily due to interactions with the blood-brain barrier (BBB), which regulates immune cell access to tumor sites (21, 22) (Table 2). The BBB is a selective barrier that restricts the passage of molecules between the bloodstream and the brain, protecting the brain from harmful agents. After the development of the tumor mass with its own vascular supply secondary to cancerous angiogenesis, addressing the “blood-tumor barrier” (BTB) becomes an additional challenge (23). Despite the unregulated nature of the BTB, with its relatively more permissive nature, the underlying BBB intermingled with the BTB forms an additional safety net preventing the free-flowing passage of different cellular and non-cellular agents (24).
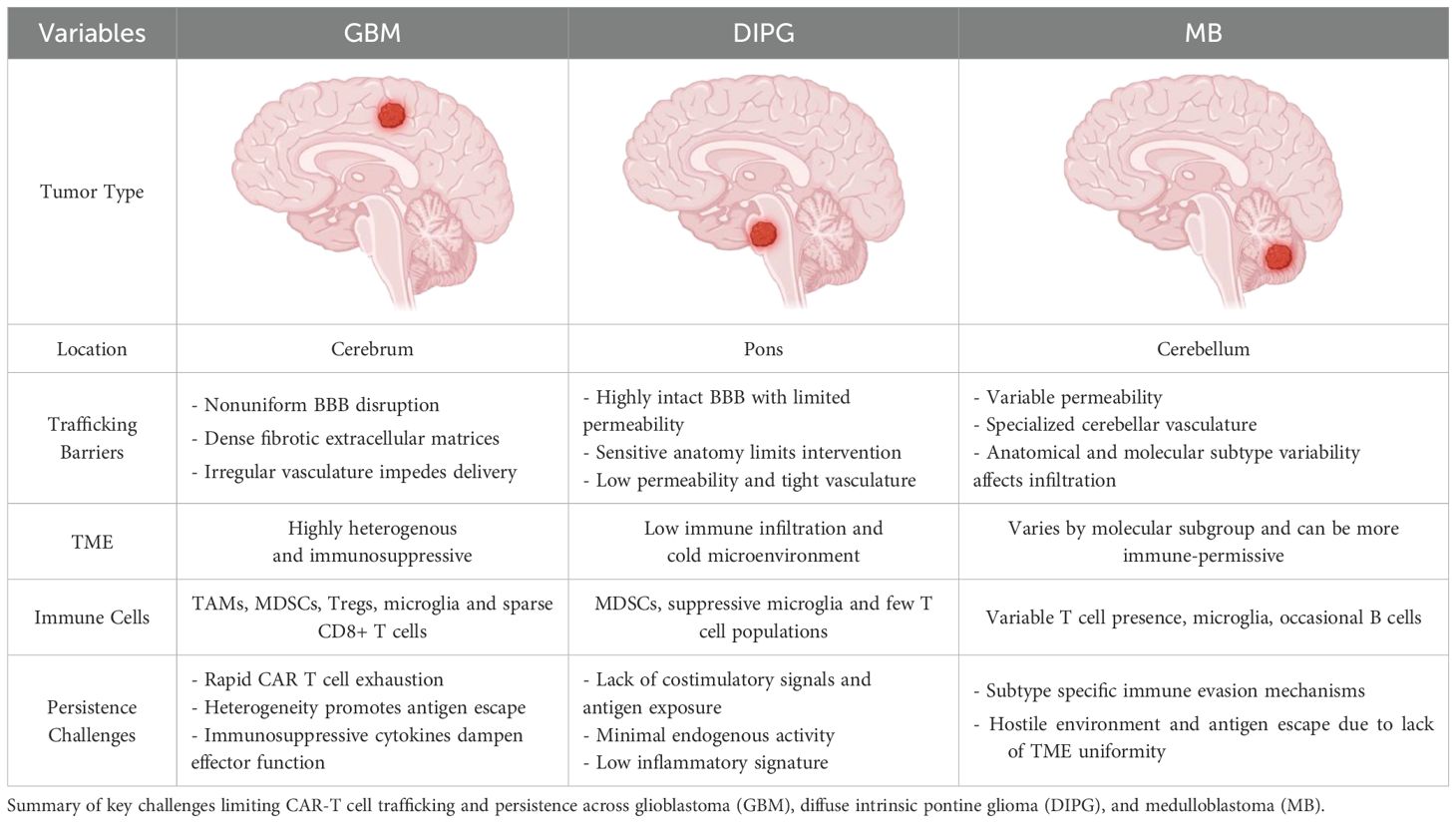
Table 2. Barriers to CAR T cell trafficking and persistence across pediatric brain tumors.
1. Glioblastoma (GBM): GBM, one of the most aggressive and prevalent primary brain tumors, is associated with significant BBB/BTB disruption, particularly in areas exhibiting angiogenesis (the formation of new blood vessels) stimulated by vascular endothelial growth factor (VEGF) (25, 26). In fact, strategies to address the VEGF-induced angiogenic nature of GBM include anti-VEGF agents that can help traffic CAR-T cells to the site of action (27). Nevertheless, the disruption of the BBB/BTB in such regions increases vascular permeability, potentially facilitating the delivery of therapeutic agents, including CAR T cells (28, 29). This may enhance CAR T cell infiltration in specific areas of the tumor (29, 30).
However, the disruption and metamorphosis of the BBB into the BTB is not uniform across the entire tumor. In more aggressive or deeply infiltrative regions of the tumor, the BTB may remain intact, limiting CAR T cell access (4). Furthermore, the immunosuppressive TME and dense extracellular matrices presents additional challenges for CAR T cell function, even in areas where BTB permeability is increased (15, 16). Therefore, while certain areas of GBM may allow CAR T cell penetration, the heterogeneous BBB/BTB disruption complicates comprehensive tumor targeting (31). Another aspect of BTB heterogeneity is the temporal nature of its evolution; the BTB is not static, but changes dynamically, frequently coordinated by tumoral strategies to evade endogenous and exogenous immunological agents (32). This aspect of BTB variability is especially key in aggressive brain malignancies such as GBM (33, 34).
2. Diffuse Intrinsic Pontine Glioma (DIPG): DIPG, located in the brainstem, presents unique challenges for CAR T cell therapy (35). Unlike GBM, which shows more widespread BTB/BBB disruption, DIPG generally maintains an intact personalized BTB in the brainstem (36). This intact barrier restricts the ability of systemically administered CAR T cells to infiltrate the tumor, as the BTB prevents their entry into the tumor core (35, 37). This impermeability categorizes DIPG as a rather resistant brain tumor as even chemotherapeutic agents fail to achieve effective results (38). The anatomical location of the brainstem also adds another layer of complexity to managing DIPG, as it controls vital functions such as respiration and heart rate (33). Therapies designed to breach the BBB thus risk damaging these critical regions of the brain (38). In addition, even with intrathecal delivery of CAR T cells into cerebrospinal fluid (CSF), the integrity of the BBB in the brainstem remains a major challenge, limiting the effectiveness of the therapy (39).
3. Medulloblastoma (MB): Medulloblastoma, particularly the aggressive Sonic Hedgehog (SHH), Group 3 and 4 subtypes, typically arises in the cerebellum and presents additional challenges for CAR T cell therapy (40). These tumors often display varying degrees of BTB disruption; the WNT subtype, a relatively less aggressive variant of MB, has a more permeable BTB, whereby, in some regions, compromised BTB integrity may allow for more efficient CAR T cell infiltration (41). However, in the more aggressive MB subtypes, the BTB remains intact, preventing CAR T cells from effectively reaching the tumor (42). Therefore, the prognostic outlook of each MB subtype is different. This is further complicated by discordant respective therapeutic responses to treatment interventions which can variably impart further resistance onto the BTB. Moreover, the anatomical position of the cerebellum within the posterior fossa, and surrounded by specialized vasculature, also complicates therapeutic CAR T cell delivery (43). Therefore, the ability of these immune cells to penetrate the tumor core is restricted by the extent of varying BTB disruption in different areas of MB which constitutes a distinct challenge to therapeutic strategies (41).
4. Ependymoma (EPN): EPN are rare tumors arising from ependymal cells lining the ventricles or spinal cord; they present specific challenges based on their location within the CNS (44). Ependymomas are typically surrounded by CSF, which further complicates CAR T cell access, particularly if its BTB remains intact in certain regions (45). Furthermore, EPNs located near the brainstem or in the spinal cord may face additional anatomical barriers, such as restricted access to systemic immune therapies (46). Even when BTB disruption occurs, the immunosuppressive TME may further limit CAR T cell efficacy (5, 47). Thus, the nature and behavior of EPN have wide implications onto therapeutic responses to CAR-T cell therapy.
5. Atypical Teratoid Rhabdoid Tumor (ATRT): ATRT is an aggressive pediatric CNS tumor that often occurs in the posterior fossa, including the cerebellum and brainstem (48). The unique anatomical location of ATRT presents challenges for CAR T cell delivery, as reaching these tumors is dictated by both BBB/BTB integrity and complex vascular structures that limit access of immune cells (49). In cases where ATRT displays significant angiogenesis, certain regions of the tumor may exhibit increased vascular permeability, potentially improving the likelihood of CAR T cell infiltration (50). However, the deep-seated location of these tumors, combined with the potential for elevated intracranial pressure or other delivery challenges, can hinder the effectiveness of CAR T cell therapy (51).
3.2 Tumor microenvironment
TMEs play a crucial role in influencing the efficacy of CAR T cell therapy in CNS tumors (5, 52, 53). It encompasses a complex network consisting of cancer cells, stromal cells, immune cells, vasculature, and extracellular matrix (ECM) components, all of which interact to dictate tumor progression and modulate immune responses (54). One of the most significant challenges in CNS tumor therapy is the heterogeneity of the TME, which can exist not only between various tumor types but also within different regions of the same tumor; this diversity complicates the development of universal CAR T cell therapies, as the distinct TME characteristics of each tumor can either enhance or obstruct CAR T cell efficacy (52).
1. Glioblastoma (GBM): GBM is characterized by highly heterogeneous TME: it contains various immunosuppressive factors, including tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs), which contribute to an environment that dampens T cell activation and function (55). Additionally, the ECM in GBM is often dense and fibrotic, impeding CAR T cell infiltration and migration toward the tumor (56). Hypoxic regions within the tumor core further limit CAR T cell survival and activity (57). Notably, different tumor regions exhibit distinct immune profiles: the tumor periphery may show some signs of immune activation, while the core is more likely to be dominated by suppressive factors such as transforming growth factor-beta (TGF-β), which inhibits CAR T cell function, thus posing a challenge for CAR T cells to uniformly target and eliminate all tumor cells (58).
2. Diffuse Intrinsic Pontine Glioma (DIPG): DIPG is characterized by an inert or “cold” TME, which subsists of immature blood vessels, a dense ECM, and severe hypoxia, all of which significantly limit the effectiveness of CAR T cell therapies (59). The DIPG TME also contains inhibitory immune cells, such as regulatory T-cells (Tregs) and myeloid-derived suppressive cells (MDSCs), which suppress antitumor immune responses (60). The limited vascularization and resultant hypoxic nature of DIPG hinders CAR T cell infiltration, as the lack of an adequate blood supply prevents efficient delivery of CAR T cells to the tumor site (61). Moreover, the inert TME may impair T cell priming and activation, further limiting CAR T cell efficacy (59). Additionally, DIPG often contains glioma stem cells, which are notoriously resistant to immune attack and can secrete additional immunosuppressive factors that evade immune surveillance (60).
3. Medulloblastoma (MB): MB is characterized by several subtypes, including WNT, SHH, group 3, and group 4 (62). Each of these subtypes has a heterogeneous TME profile which varies depending on the respective molecular underpinnings (63). For example, Group 3 MB tumors are typically infiltrated by immune-suppressive cells, such as Tregs and MDSCs, which hinder CAR T cell activity (64). Moreover, the ECM in MB can differ in composition: group 4 MB tumors often have a more fibrotic ECM that obstructs CAR T cell infiltration. In contrast, Sonic Hedgehog (SHH)-type MB tumors may have a less immunosuppressive environment but still possess substantial immune evasion mechanisms that prevent effective CAR T cell function (63). A significant challenge in MB is the lack of uniformity in the TME across tumor regions as tumor cells in areas with dense ECM or high levels of immunosuppressive cytokines can effectively shield themselves from CAR T cells, limiting the therapy’s ability to target and eliminate these cells (5, 65).
4. Ependymoma (EPN): EPN occurs in both the intracranial and spinal cord regions and exhibits TME heterogeneity depending on the tumor location and subtype (66). Intracranial EPN, particularly arising from the ventricular system, tends to have high vascularity but also feature a dense ECM that may obstruct CAR T cell migration (46, 67). These tumors are often associated with elevated levels of TGF-β, interleukin-10 (IL-10), and VEGF, thus creating an immunosuppressive TME that can inhibit CAR T cell activity (68, 69). Spinal EPN, on the other hand, may have a more limited vascular supply, resulting in poorer CAR T cell infiltration (66). The inflammatory cytokines present in the TME can also exacerbate the challenges of CAR T cell penetration, as immune cells such as neutrophils and macrophages may create a hostile environment that hinders CAR T cell engagement and tumor cell killing (66).
5. Atypical Teratoid/Rhabdoid Tumor (ATRT): ATRT features a heterogeneous TME that also varies by tumor location, as is the case with its other pediatric brain tumor counterparts (69–72). For tumors located in the posterior fossa, including the brainstem, challenges arise due to dense ECM and hypoxic conditions typical of ATRT (57). Like other CNS tumors, ATRT tumors harbor MDSCs, Tregs, and macrophages that contribute to the immunosuppressive microenvironment, actively inhibiting T cell activation and limiting CAR T cell efficacy (73). Additionally, ATRT tumors often exhibit genomic instability, leading to the secretion of immune-modulating factors such as IL-6 and interferons, which may further impair CAR T cell function (74–77).
The heterogeneity of the TME across different CNS tumors thus significantly limits the efficacy of CAR T cell therapy. This is accomplished through several mechanisms:
a. Immunosuppressive Cells: The presence of Tregs, MDSCs, macrophages, and myeloid cells that release immunosuppressive cytokines (e.g., TGF-β, IL-10, IL-6, VEGF) can actively suppress CAR T cell function, diminishing their ability to mount an effective antitumor response (78).
b. Physical Barriers: The dense ECM, fibrosis, and hypoxia within the TME can act as physical barriers that prevent CAR T cells from infiltrating the tumor or migrating to the areas where they are most needed (5). This is particularly evident in tumors with dense stromal components, such as GBM and ATRT (48, 49, 55).
c. Tumor Heterogeneity: Different regions of the same tumor can have varying levels of immune suppression, ECM density, and vascularization.; for example, while one region may exhibit enhanced vascular permeability, another may have an intact BBB and a suppressive immune landscape, complicating the design of universal CAR T cell therapies (1, 40, 52, 79).
d. Glioma Stem Cells and Immune Evasion: Some CNS tumors, such as DIPG and GBM, contain glioma stem cells (GSCs) that are resistant to immune attack (80, 81). These cells secrete factors that enable them to evade immune detection, further hindering the effectiveness of CAR T cells (5).
e. Altered Cytokine and Growth Factor Production: Tumors such as EPN and ATRT may produce altered levels of cytokines and growth factors that shift the TME toward an environment that actively resists CAR T cell action (82). These factors can attract suppressive immune cells or directly interfere with CAR T cell activity (83).
Therefore, the heterogeneity of the TME across different CNS tumor types is a critical factor that limits the efficacy of CAR T cell therapy. Variations in immune cell populations, ECM composition, cytokine profiles, and vascularization create both physical and immunosuppressive barriers to CAR T cell function (39, 52). Moreover, the diversity in TME characteristics complicates the development of therapies that can effectively target and eliminate tumors across these complex and varied microenvironments (5, 54). Addressing these architectural and TME-related challenges will be essential for improving the therapeutic potential of CAR T cells in CNS tumors.
3.3 CAR T cell retention and persistence in CNS tumors
The persistence and retention of CAR T cells within the CNS are critical factors in determining the therapeutic efficacy of CAR T cell therapy for CNS tumors (84). Sustained CAR T cell activity and their ability to continuously target tumor cells are essential for achieving durable tumor control and preventing recurrence (85). However, clinical studies have consistently highlighted suboptimal CAR T cell persistence, often associated with tumor relapses or disease progression; this challenge is largely attributed to the immune-privileged nature of the brain and spinal cord, as well as the immunosuppressive and complex characteristics of the TME (63, 86). For instance, in trials investigating CAR-T cells in GBM, which included anti-GD2 (NCT04196413) and anti-EGFRvIII (NCT02209376) CARs, detection was only sustained at the post-infusion stage, thus exhibiting short-lived persistence, with exhaustion and loss of function contributing to tumor recurrence (37, 87). Similarly, in DIPG trials (NCT03500991 and NCT03618381) targeting HER2, GD2, and EGFR, CAR T cells were initially detected following infusion, but persistence was limited, particularly in brainstem tumors (88, 89). Factors such as the intact or partially disrupted BBB and the highly immunosuppressive TME restricted their long-term activity with limited adoptive T cell durability and tumor progression (21, 24, 30). Moreover, a CAR-T trial involving MB, which investigated GD2 CAR T cells (NCT04099797), demonstrated detectable activity in early phases but showed poor long-term persistence, especially in Group 3 MB, where the TME is dominated by immunosuppressive elements (90, 91). Trials for CAR-T cell interventions in ATRT and EPN are limited given the rarity of these malignancies, but preclinical studies and anecdotal data suggest that CAR T cells are transiently detectable post-infusion despite experiencing rapid attrition due to immune evasive mechanisms and the dense ECM further exacerbating CAR-T cell persistence (48, 66, 69, 92, 93).
Pre-clinical and clinical experiences thus highlight that the challenges related to CAR T cell persistence stem from several complex and interrelated factors, including T cell exhaustion, immune evasion mechanisms within the TME, the presence of physical barriers like the BBB, and the immunosuppressive cytokine milieu characteristic of various CNS tumors. These variables are summarized below:
a. Immunosuppressive TME: As detailed in the previous section, the immunosuppressive TME in many CNS tumors presents a significant barrier to CAR T cell persistence (84). Tumors such as GBM, DIPG, and MB often contain suppressive immune cells, including Tregs, MDSCs, and macrophages, which not only limit CAR T cell efficacy but also drive T cell exhaustion (94). Additionally, the secretion of immune-suppressive cytokines such as TGF-β, IL-10, and IL-6 actively inhibits CAR T cell expansion and persistence, reducing long-term survival of therapeutic T cells at the tumor site (86, 95, 96).
b. Immune Privilege and BBB: The CNS is considered an immune-privileged site largely due to the restrictive nature of the BBB, which limits the entry of immune cells, including CAR T cells, from the systemic circulation into the brain and spinal cord (21–23, 30, 97). This limited and often delayed infiltration hinders the persistence of adoptively transferred T cells at the tumor site (4). While some CNS tumors may partially disrupt the BBB, as evidenced by detectable CAR T cell infiltration following systemic administration, the BBB and the blood-tumor barrier (BTB) remain significant physical obstacles which impede uniform CAR T cell access to the tumor, particularly to its core, thus challenging CAR-T cell retention at the site of action, ultimately restricting sustained CAR T cell activity within the tumor microenvironment (98).
c. Tumor Heterogeneity: Tumors such as GBM, MB, and DIPG exhibit intra-tumoral heterogeneity, where different regions of the tumor may possess varying immune cell populations, vascularization, and hypoxic zones (99). This heterogeneity creates regions where CAR T cell infiltration is severely limited. Even in areas where CAR T cells successfully penetrate, they often become rapidly exhausted after initial activity, compromising their persistence and cytotoxic function across the entire tumor mass (100) Moreover, tumors can adapt to immune pressure by altering antigen expression or upregulating immunosuppressive ligands, further impeding CAR T cell survival and efficacy within the TME (101).
In addition to these challenges, antigen heterogeneity across CNS tumors presents a major barrier to effective CAR T cell therapy. Targeting a single antigen or epitope often fails to eradicate all tumor cells due to variable levels of antigen expression within and between tumors (15, 93, 102). In some cases, subpopulations of tumor cells may completely lack expression of the targeted antigen, enabling immune escape even in the presence of active CAR T cells. Additionally, tumors may express truncated or mutated forms of antigens that are not recognized by the CAR, further diminishing therapeutic efficacy. Antigen downregulation following selective immune pressure contributes to inconsistent therapeutic outcomes and tumor relapses. For instance, in GBM, heterogeneous expression of antigens such as EGFRvIII has led to incomplete tumor targeting and subsequent relapses after EGFRvIII-directed CAR T cell therapy. Similarly, in medulloblastoma, the differential antigenic expression of markers such as HER2 or B7-H3 across tumor subpopulations creates reservoirs of antigen-negative cells that can evade immune clearance. These issues are especially problematic in solid tumors, which tend to exhibit greater genetic instability and more hostile microenvironments compared to hematological malignancies, where malignant cells generally maintain more stable antigen profiles (103).
d. CAR T Cell Exhaustion and Phenotypic Changes: CAR T cells frequently experience exhaustion due to sustained exposure to tumor antigens and immune-suppressive factors within the TME (94). This exhaustion is characterized by the upregulation of inhibitory receptors, such as programmed death-1 (PD-1), T cell immunoglobulin and mucin-domain-containing protein 3 (TIM-3), and lymphocyte activation gene 3 (LAG-3), which diminish CAR T cell functionality and persistence (104). Additionally, epigenetic remodeling plays a significant role in driving exhaustion by inducing stable, heritable changes in gene expression that reinforce the exhausted phenotype (105). Furthermore, CAR T cells often differentiate into terminally differentiated subsets, such as effector T cells, or into less effective memory subsets, both of which exhibit reduced proliferative capacity and impaired long-term antitumor activity (106). These phenotypic and epigenetic changes collectively undermine CAR T cell persistence and therapeutic efficacy in CNS tumors (84, 95).
e. Cytokine Environment and Interleukin (IL) Signaling: The cytokine milieu in the CNS TME plays a pivotal role in determining the survival and persistence of CAR T cells (107). In CNS tumors, high levels of IL-10, TGF-β, and IL-6 are often secreted, contributing to immune suppression and CAR T cell attrition (108). The lack of supportive cytokines, such as IL-2, which promotes T cell survival, can further contribute to the reduced persistence of CAR T cells in the CNS, leading to an early termination of the therapeutic response (109).
Therefore, the limited persistence of CAR T cells in CNS tumors remains a major barrier to successful clinical outcomes (110). Factors such as immunosuppressive mechanisms in the TME, physical barriers like the BBB, and tumor heterogeneity contribute to the early exhaustion and loss of CAR T cells, leading to disease progression or tumor relapse (111). Overcoming these challenges requires a deeper understanding of how the TME impacts CAR T cell engraftment and persistence, as well as the development of strategies to enhance CAR T cell survival and functionality in the CNS.
4 Strategies for overcoming current challenges
Several pre-clinical studies and clinical trials have focused on potential strategies to improve CAR T cell design, manufacturing processes, and combination therapies, as well as incorporating additional engineering and genetic modifications to overcome challenges related to persistence and exhaustion (112–114). In this section, we will break down these approaches and provide additional perspectives on novel potential solutions to address these critical obstacles (Table 3).
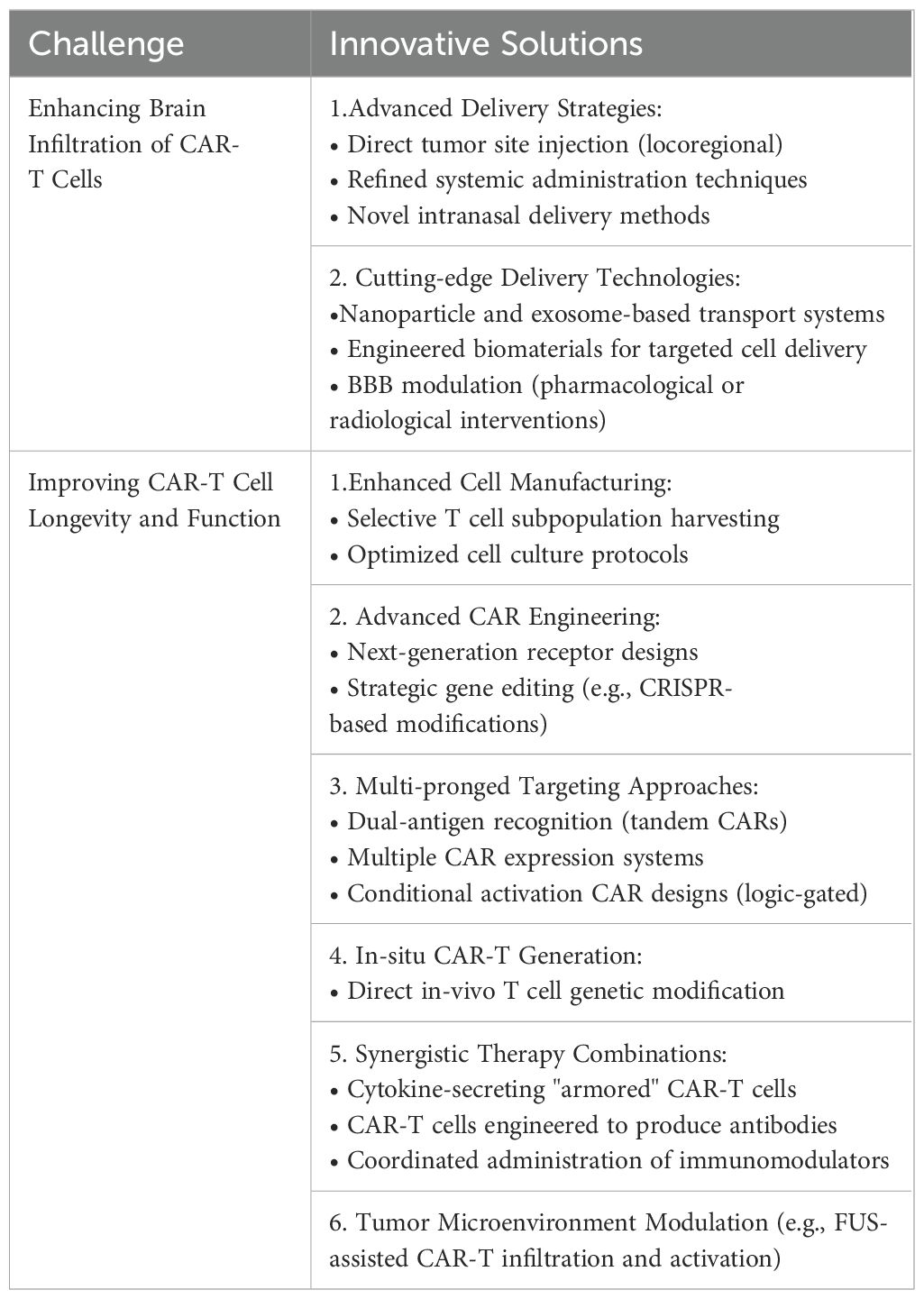
Table 3. Summary of challenges and respective solutions to CAR-T cell therapy in the context of malignant primary brain tumors.
4.1 Mechanisms to overcome trafficking hurdles
4.1.1 Modulating method of CAR-T delivery
Optimizing the route of CAR-T cell administration is a critical factor in enhancing therapeutic efficacy for CNS tumors; both locoregional and systemic injection modalities have been investigated, each with distinct advantages and limitations depending on tumor characteristics and the physiological barriers present in the CNS (Figure 4) (115).
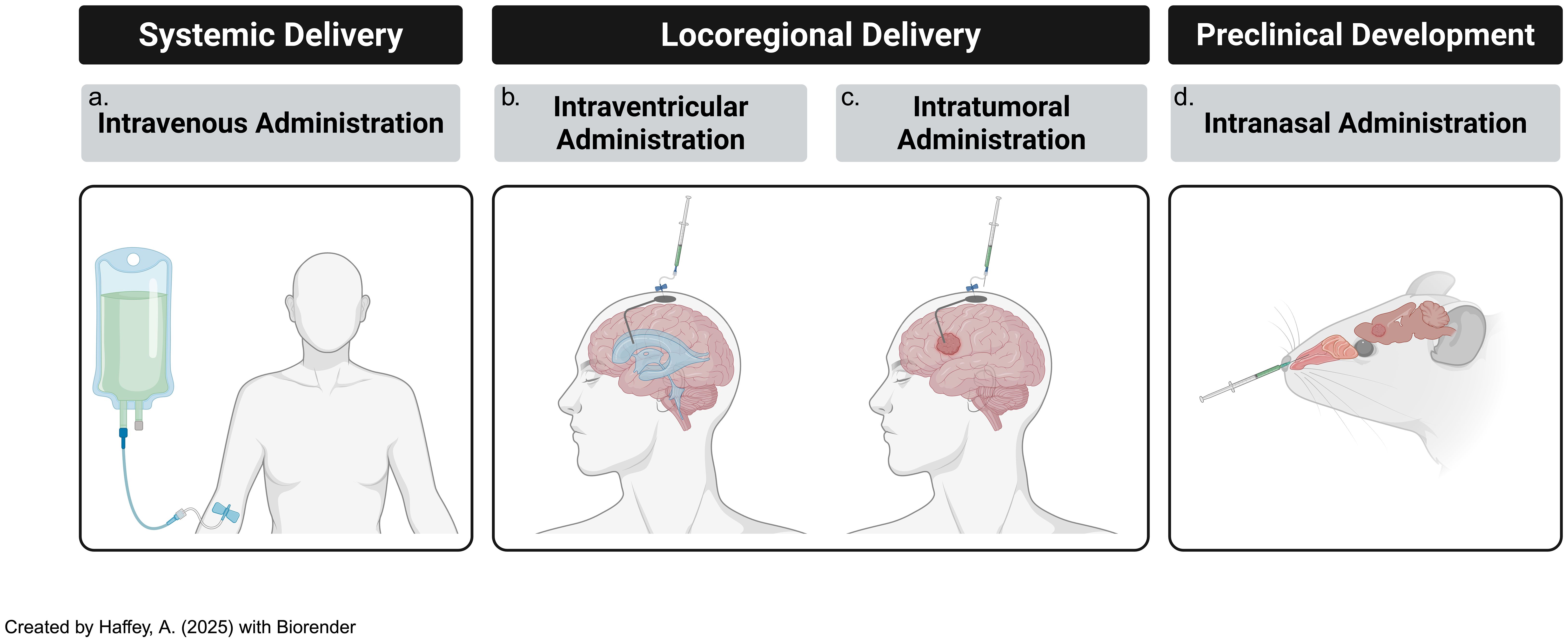
Figure 4. Modes of delivery of adoptive t-cells, including CAR-Ts, subsists of several tested paths [including (a) systemic & (b) locoregional routes] and (c) novel, developing techniques such as intranasal routes of administration. All three modes of delivery are consistently updated with novel iterations that attempt to ameliorate previous challenges..
Locoregional Administration: Intraventricular and intrathecal routes of CAR-T cell delivery have shown promise in preclinical models for targeting CNS tumors; for instance, intraventricular CAR-T cell infusion in group 3 MB mouse models resulted in significantly improved therapeutic outcomes and reduced systemic toxicity compared to intravenous delivery (116). The key benefit of locoregional administration is its ability to bypass the BBB, allowing direct delivery of CAR-T cells to the tumor site, thus minimizing off-target effects and systemic adverse events (117). However, its major limitation lies in restricted spatial targeting, which may not effectively address disseminated disease or micro metastases (118, 119). Additionally, given their invasive nature, procedures like intraventricular cannula implantation or repeated intrathecal injections may pose challenges for long-term patient management.
Systemic Administration: Intravenous infusion of CAR-T cells provides the advantage of widespread distribution, potentially reaching distant tumor sites and micro-metastases that locoregional approaches may miss; however, the BBB remains a major hurdle in ensuring that CAR-T cells reach brain tumors (4). Despite this challenge, systemic delivery offers several benefits, including simpler administration in outpatient settings, reduced reliance on invasive procedures, and greater likelihood of treating diffuse or residual tumors that are less amenable to locoregional targeting (120).
Intranasal Delivery: Another emerging approach is intranasal administration of CAR-T cells, which constitutes a direct pathway to the brain, thereby bypassing the BBB to a certain extent, and facilitating greater infiltration of CAR-T cells into the brain to treat malignant or autoimmune diseases (121). This approach can minimize risk of systemic exposure due to direct brain access but also has potential for more feasible maintenance therapy convenience, in addition to enhanced patient comfort and compliance.
While preclinical studies such as those incorporating HER-2 targeting CAR-T cells and non-engineered cytotoxic lymphocytes (CTLs) in murine GBM models have demonstrated that intranasal delivery of immune cells can lead to migration within brain regions affected by disease, this method is still in its early stages, and requires rigorous testing to ensure safety and efficacy, particularly given the heterogeneous nature of BBB disruption across various CNS tumor variants (21).
4.1.2 Leveraging novel delivery platforms
Leveraging Nanocarrier Systems: Nanotechnology presents a promising strategy for enhancing the delivery of CAR-T cell therapy, particularly in overcoming the challenges of the blood-brain barrier (BBB) and improving tumor targeting (122). By encapsulating CAR-T cells or related therapeutic agents in nanoparticles, it is possible to enhance BBB penetration and deliver the cells more efficiently to tumor sites (123–125). Nanoparticles can also carry immunomodulatory agents, such as RNA vaccines or immune checkpoint inhibitors, which can improve CAR-T cell function and persistence within the tumor microenvironment (126–128). Additionally, magnetic nanoparticles have been studied in combination with CAR-T cells, demonstrating reduced toxicity and improved tumor targeting when CAR-T cells are attached to the nanoparticles (129).
A form of nanocarrier technology includes exosomes, which are naturally occurring extracellular vesicles (EVs) secreted by cells containing a multitude of molecular mediators unique to their source of production (130). EV research in the field of cancer therapeutics has yielded a plethora of data points delineating the potential of their use in diseases such as Non-Hodgkin Lymphoma (NHL) as diagnostic, prognostic, and therapeutic tools (131). The latter includes incorporating Chimeric Antigen Receptors; studies have revealed various promising but precarious therapeutic endpoints of combining EVs harboring targets like those of CAR-T cells. Nevertheless, they have shown promise as substitutes to CAR-T cells given the possibility of loading EVs with CARs on their surface to target and avert tumor growth (131). Notwithstanding these innovative strategies, EVs themselves harbor potential for ameliorating CAR-T cell therapy, whereby cellular CAR constructs or other acellular therapeutic agents are delivered directly to brain tumors via EVs (132). Exosomes have also been shown to cross the BBB, making them an effective vehicle for delivering CAR-T cell–derived therapeutic cargo in a controlled and targeted manner (133). One key advantage of using exosomes is their ability to modulate the tumor microenvironment (TME), which can enhance CAR-T cell efficacy (134). By utilizing these innovative delivery methods, it may be possible to improve CAR-T cell therapy outcomes and reduce off-target effects, ultimately enhancing the therapeutic potential for CNS tumors (130). Moreover, they can be loaded with surface markers that serve as CAR-T “backpacks” to delivery therapeutic payloads (e.g., loaded with cytokines) that coincide with CAR-T cell action at the tumor site (131). Furthermore, interventions such as chemically grafting molecular moieties like short-chain fatty acids (SCFAs) and tryptophan metabolites, which are microbial products that have a specialized traversal paths across the BBB, onto payload nanocarriers such as EVs, can forge the way forward towards more innovative strategies of ameliorating difficulties associated with delivering CAR-T-supportive agents to the CNS (135).
While each method incorporating nanocarrier modalities to improve immunotherapeutic strategies and attenuate obstacles offers unique benefits, their effectiveness will ultimately depend on addressing the complexities of tumor microenvironments and BBB permeability (99, 134). Continued innovation and further research in this field could substantially improve the outcomes of CAR-T cell therapy for brain tumors.
Utilizing Biomaterial Formulations for CAR-T Cell Delivery: Biomaterials present an innovative approach to enhancing CAR-T cell therapy by providing a platform for delivering CAR-T cells and supporting adjunct agents, thus improving their persistence and function within the tumor microenvironment (TME) (136–138). Hydrogel-based microstructures have been explored as scaffolds for co-delivering therapeutic agents, including small molecules (e.g., chemotherapeutic drugs) and cytokines (e.g., IL-15) to enhance CAR-T cell activity (139–141). These hydrogels are biocompatible, biodegradable, and capable of controlled release, allowing for sustained delivery of agents directly at the tumor site (142). In preclinical studies, CAR-T cells targeting melanoma marker chondroitin sulfate proteoglycan 4 (CSPG4) were combined with hydrogels containing IL-15 and platelets (141). The addition of cytokines in the hydrogel formulation improved CAR-T cell persistence and proliferation, potentially overcoming the challenges imposed by the TME (141).
Hydrogel microstructures are also beneficial for bypassing dense tumor barriers, facilitating better CAR-T cell infiltration and expansion within the tumor (143). These innovations are particularly valuable for treating brain tumors, where the TME is characterized by barriers such as the BBB and a dense extracellular matrix (ECM) that restrict drug and cell delivery (144). The clinical feasibility of hydrogel-based formulations is dependent on their safe implantation and compatibility with patient-specific tumor characteristics. For example, a collagenase nanogel biomaterial equipped with CXCR4 antagonism was shown to efficiently help traffic CAR-T cells to the site of pancreatic solid tumors in a pancreatic tumor model through overcoming traditional ECM (extracellular matrix) barriers, thus facilitating a “CAR-T/collagenase nanogel backpack” delivery system (145). Furthermore, fibrin-based hydrogels have been tested in glioblastoma models and were shown to improve CAR-T cell outcomes compared to direct injections (146). Fibrin’s biocompatibility, along with its ability to support CAR-T cell growth and modify the immunosuppressive TME, makes it a promising candidate for clinical application, especially in pediatric brain tumors where the BBB and dense ECM create significant challenges for drug and cell delivery (147). Therefore, further incorporating biomaterial usage in the realm of brain tumor therapeutics may be key in overcoming key challenges in immunotherapeutic strategy implementation.
Chemical Manipulation of the BBB: As previously noted, the BBB is a formidable barrier to therapeutic delivery, including CAR-T cells (23). Various methods have thus been explored to transiently disrupt the BBB to improve the delivery of therapeutic agents. Strategies such as intra-arterial administration of mannitol have been frequently attempted, but with notable adverse effects. More recently, agents like xNEO100 (a purified form of P-OH, or perillyl alcohol) have shown promise in enhancing BBB permeability (148). This agent has been investigated in-vivo for treating GBM and demonstrated the ability to reversibly permeabilize the BBB by loosening endothelial tight junction proteins (149). Compared to mannitol, NEO100 is more efficient, requiring lower concentrations and achieving better BBB disruption in a dose- and time-dependent manner (150). One clinical implication of using NEO100 is its potential combination with CAR-T cell therapy to improve targeting and immune cell recruitment, such as CD8+ T cells, within the brain tumor microenvironment (151). In addition, mouse models have shown that co-administration of NEO100 with PD-1 inhibitors and CAR-T cells enhances both delivery and therapeutic efficacy (148). Such combination therapies could improve CAR-T cell localization to brain tumors and boost the overall immune response, leading to better patient outcomes; however, careful monitoring of systemic toxicity, particularly in combination with immune checkpoint inhibitors, is essential to ensuring patient safety (152).
Applying Focused Ultrasound (FUS): FUS is a non-invasive, non-ionizing technique that transiently opens the BBB, facilitating improved delivery of therapeutic agents, including CAR-T cells, to brain tumors (153). This process is enhanced through the introduction of microbubbles (µB) that, when exposed to acoustic waves, create mechanical oscillations that disrupt the tight junctions of the BBB, allowing for deeper penetration of therapeutic agents (Figure 5) (154, 155). Clinical trials have demonstrated the potential of FUS in various brain tumor types, including GBM, DIPG, and brain metastases (156–162). For instance, at the University of Maryland Medical Center, MRI-guided FUS was used to open the BBB and improve the delivery of chemotherapeutic agents to GBM (163), while in DIPG, FUS enhanced the delivery of doxorubicin (164). Additionally, FUS has been shown to increase the penetration of trastuzumab into the brain in patients with brain metastasis (165). Early clinical trials combining FUS with CAR-T cells for pediatric patients with aggressive brain tumors such as DIPG and MB have shown encouraging results, with improved delivery of CAR-T cells and other therapeutic agents like immune checkpoint inhibitors to the tumor site (155, 166–169). This approach has the potential to increase survival rates and enhance CAR-T cell therapy efficacy in pediatric populations. However, challenges persist, such as optimizing treatment parameters (e.g., sonication intensity and duration) to minimize tissue damage. Moreover, evaluating the long-term effects of repeated FUS treatments on brain tissue will be essential in optimizing parameters (166). Despite these challenges, FUS has significant potential to revolutionize CAR-T cell therapy for brain tumors by improving BBB permeability and ensuring that CAR-T cells can effectively reach and treat tumors within the brain.
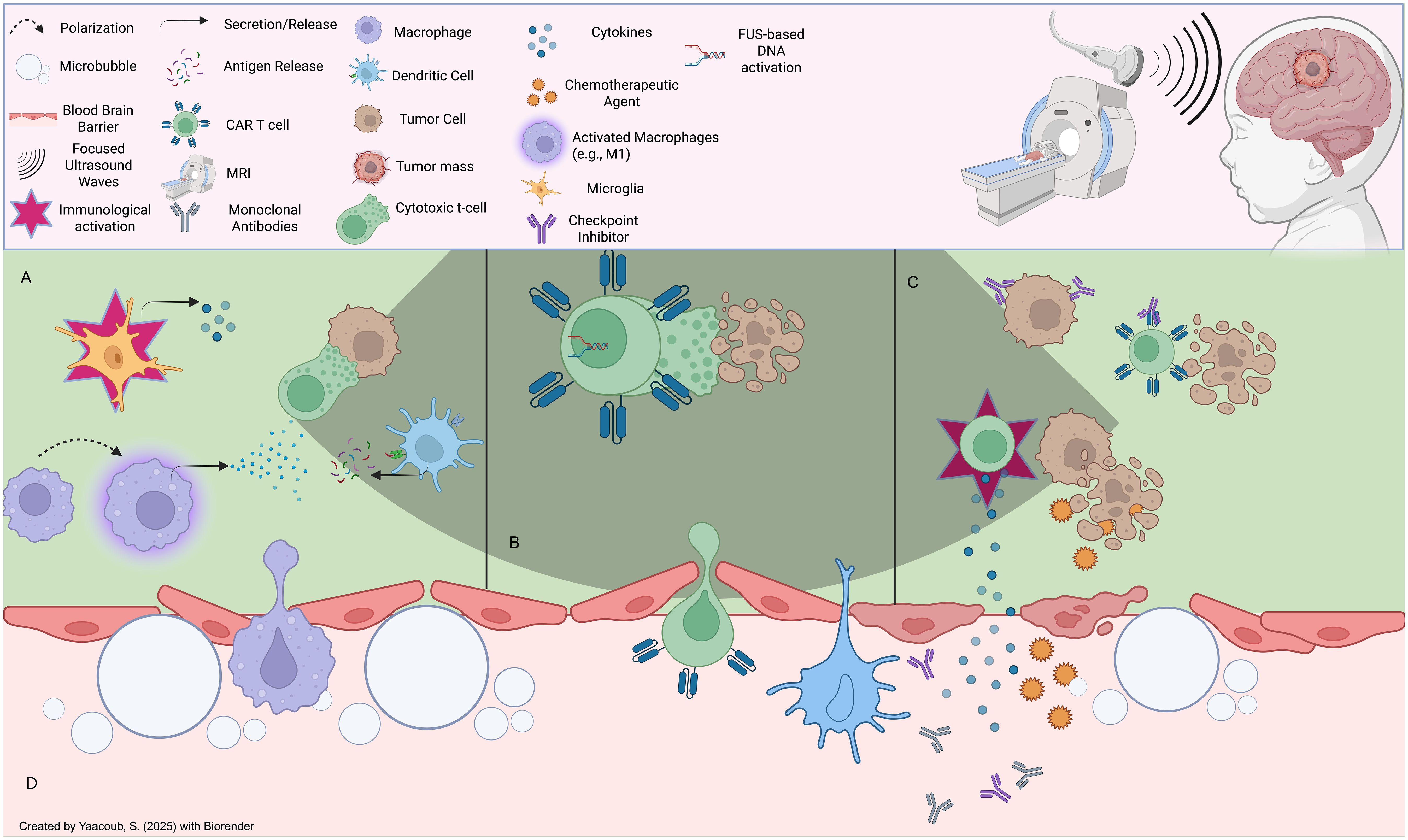
Figure 5. Effects of focused ultrasound on the tumor microenvironment: (A) FUS induces immunomodulatory effects onto immune cells in the TME, such as microglial activation, macrophage repolarization (e.g., M2 to M1), and antigenic presentation of tumor neoantigens onto immune cells (eg., T cells), (B) FUS can be leveraged to induce acousto-genetic, controlled CAR-T cell activation to limit unwanted pro-inflammatory effects (e.g., adverse effects onto host tissues) (C) FUS-mediated BBB opening can mediate CAR-T cell infiltration post-systemic administration intravenously, in addition to entry of other therapeutic agents such as endogenous or extrinsic cytokine and antibody-based therapies that can induce anti-tumor effects. (D) FUS induces reversible BBB disruption through microbubble-based cavitation effects which can loosen tight junctions; this can lead to the infiltration of a plethora of endogenous immune cells as well as extemal therapeutic agents.
Therefore, overcoming the trafficking hurdles associated with CAR-T cell therapy for CNS tumors involves optimizing injection modalities, leveraging nanotechnology and exosomes, and employing both chemical and physical techniques to disrupt the BBB. These strategies present promising opportunities to enhance the efficacy of CAR-T cell therapy. Ongoing research and innovation in these areas are essential for improving treatment outcomes for patients with brain tumors.
4.2 Strategies to enhance CAR-T cell persistence
The long-term effectiveness of CAR-T cell therapy relies on the persistence and functionality of engineered T cells at the tumor site (113). However, CAR-T cells often struggle to maintain activity, particularly within an immunosuppressive TME (52). To overcome these challenges, strategies have been developed to enhance CAR-T cell persistence, including optimizing the T cell source, refining cell culture conditions, modifying CAR constructs, and utilizing genetic engineering techniques (Figure 6). The overarching nuances in CAR-T cell synthesis in the traditional pipeline include, but are not limited to, patient-specific variability, the phenotypes of T cell subsets derived from peculiar sources (autologous vs other), the transduction success of CAR constructs, culturing conditions, and CAR construct designs (113). Moreover, on a larger scale, the feasibility of administering CAR-T cells to patient populations requires near-perfect standards; this includes product consistency, cryopreservation impact, product turnaround time, Good Manufacturing Practice (GMP) standards, and scalability and cost implications. For the scope of this manuscript, we will focus on a handful of all these variables.
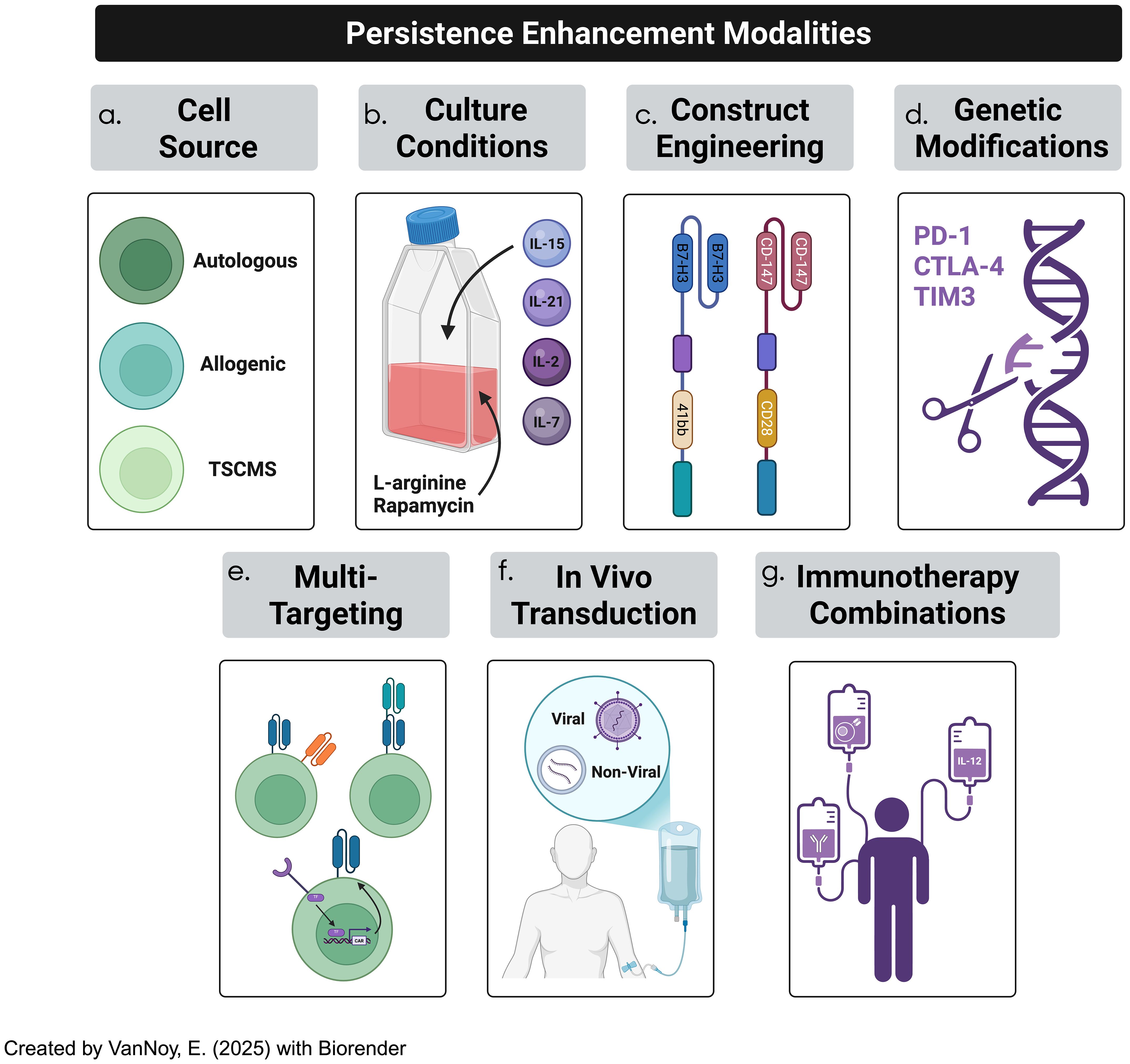
Figure 6. CAR-T cell therapy efficacy in solid tumor treatment has been often hindered by lack of persistence due to a plethora of factors. Several strategies to overcome persistence challenges occurs through (a) cell-source optimization, (b) culturing conditions, (c) optimizing CAR constructs, (d) genetic modification of CAR constructs, (e) adopting multi-targeted techniques, (f) employing in-vivo T cell transduction into CAR-T cells, and (g) incorporating immunotherapeutic combinations.
4.2.1 T cell source optimization
The persistence of CAR-T cells is influenced by their origin, which can be either autologous (derived from the patient’s own T cells) or allogeneic (sourced from a healthy donor) (86). Autologous CAR-T cells are favored in most clinical settings due to their lower risk of immune rejection and graft-versus-host disease (GvHD), but they are often associated with reduced long-term persistence in the tumor microenvironment (170). This persistence is hindered by factors such as T cell exhaustion and tonic signaling (94, 171). Additionally, the logistical complexities and high costs associated with autologous therapies present significant challenges (172). Allogeneic CAR-T cells, on the other hand, offer advantages such as cost-efficiency and scalable manufacturing, as they can be pre-sourced and stored for use in multiple patients; however, the risk of GvHD and immune rejection due to the HLA mismatch remains a key concern (173, 174). Advances in genetic engineering, including the knockout of T-cell receptors (TCRs) or HLA modification, are being explored to mitigate these risks, although such modifications must be carefully optimized to minimize off-target effects (175). Recent advancements have also highlighted the potential of using stem memory T-cells (TSCMs), a rare but highly durable subset of T cells that exhibit self-renewal capacity and can persist long-term; these T cells are less prone to differentiation and exhaustion compared to conventional T cells, making them ideal candidates for CAR-T cell therapies targeting solid tumors (176).
4.2.2 Optimizing T cell culture conditions
Cell culture conditions play a pivotal role in determining the functionality, expansion, and persistence of CAR-T cells (177). During the ex-vivo expansion phase, the cytokine milieu and culture environment both influence the differentiation and longevity of the engineered T cells (109). Traditional expansion protocols often rely on IL-2 for cell proliferation; however, IL-2 promotes differentiation into effector T cells, which are typically less persistent and more prone to exhaustion (177). Recent studies have suggested that the use of IL-7, IL-15, and IL-21 in culture can promote a less differentiated and longer-lasting phenotype, characterized by a stem cell memory phenotype that enhances both persistence and anti-tumor efficacy (178, 179). These cytokines are more effective at maintaining a population of central memory T-cells (TCM) and stem cell memory T-cells (TSCM), which are associated with improved long-term survival and robust anti-tumor responses (179). Additionally, metabolic programming has emerged as a critical aspect of CAR-T cell persistence: T cells undergoing aerobic glycolysis (the Warburg effect) tend to differentiate into effector cells, which are short-lived and less effective (180). By promoting oxidative phosphorylation and inhibiting glycolysis, the metabolic reprogramming of T cells can support a memory-like phenotype and enhance their durability (181). In particular, the use of mTOR inhibitors like rapamycin and L-arginine supplementation has been shown to improve metabolic fitness and support long-term persistence (182).
4.2.3 Engineering CAR constructs for enhanced persistence
The design of the CAR construct itself is critical to enhancing T cell persistence; the extracellular binding domain, transmembrane domain, and intracellular signaling domains all play pivotal roles in shaping the function and longevity of CAR-T cells (17). The single-chain variable fragment (scFv) dictates antigen specificity and binding affinity, both of which have considerable impact on tonic signaling and desired specific targeting (17). The hinge and transmembrane domains also have pivotal implications on how the CAR-T engages antigens such that T-cell fitness and epitope targeting is heavily influenced (183). Moreover, the selection of intracellular binding domains, including the co-stimulatory and primary activation domains, has been shown to alter the metrics of CAR-T cell effectiveness; this is embodied by improved short-term efficacy, offset by diminished persistence, and vice-versa (183). Finally, CAR constructs have been developed over the years such that various (five) generations exist, each of which has driven different groups to study their relative effectiveness.
Notably, the inclusion of co-stimulatory signaling domains such as 4-1BB and CD28 has profound implications for T cell persistence (184). While CD28 provides rapid expansion and potent short-term effector functions, it can also induce early exhaustion; in contrast, 4-1BB-based CARs promote a more sustained T cell activation and a memory phenotype, which is associated with improved long-term persistence and anti-tumor efficacy (185). Further optimization of the CAR construct can reduce the likelihood of tonic signaling, which can lead to premature activation and exhaustion of CAR-T cells (86, 96). The design of the scFv (single-chain variable fragment), including the spacer and hinge regions, must be carefully adjusted to prevent tonic signaling without compromising CAR-T cell activation (186). Moreover, incorporating transient CAR expression systems, where CAR expression is controlled based on external signals, could provide a more balanced activation response and prevent premature exhaustion (187).
4.2.4 Genetic modifications to enhance CAR-T cell longevity
The non-durable effects of CAR-T cells in CNS tumor treatment have led to increasing interest in genetic modifications of CAR-T cells to improve their longevity and functional persistence (188). One promising approach is the genetic knockout of immune checkpoints such as PD-1, CTLA-4, and TIM-3; these inhibitory receptors are generally upregulated in response to chronic antigen exposure, leading to T cell exhaustion (189). By knocking out or inhibiting these molecules, researchers can potentiate CAR-T cell function and enhance persistence (113). CRISPR/Cas9 technology has enabled precise, targeted gene editing in CAR-T cells, allowing for the deletion of specific genes involved in immune regulation (190). For example, knocking out PD-1 has shown promise in preclinical glioma studies by improving CAR-T cell activity, particularly in immune-suppressive tumor microenvironments (191). However, the risk of off-target effects and unintended mutations necessitates careful monitoring and validation before these approaches can be translated into clinical practice. Additionally, another emerging strategy is the manipulation of DNA methylation pathways to modulate T cell exhaustion; for instance, the reduction of DNMT3a expression, a key DNA methyltransferase, has been shown to sustain a relatively undifferentiated phenotype in CAR-T cells, leading to better persistence and anti-tumor responses (192). This modification, coupled with epigenetic reprogramming, offers a novel way to prolong the effectiveness of CAR-T therapies.
4.2.5 Multi-targeting approaches
To address the challenges of tumor heterogeneity and antigen escape in brain tumors, multi-targeting strategies are emerging as a promising approach in CAR-T cell therapy (193). These approaches aim to enhance the specificity and efficacy of CAR-T cells by enabling them to recognize and target multiple tumor-associated antigens simultaneously. By diversifying the targets, multi-targeting strategies reduce the risk of antigen escape and improve the likelihood of complete tumor eradication, especially in heterogeneous tumor populations like those found in CNS malignancies (194).
a. Tandem CARs: These combine two or more CAR constructs into a single molecule, allowing simultaneous targeting of multiple antigens, thus increasing the likelihood of tumor elimination, even in cases of antigen escape (195).
b. Dual and Multi-CAR Constructs: Dual CARs express two distinct CARs targeting different antigens, while multi-CAR constructs target several antigens (196). These strategies broaden the range of tumor recognition, enhancing efficacy and preventing relapses.
c. Synthetic Notch (synNotch) CARs: SynNotch CARs enable conditional activation based on antigen recognition, reducing off-tumor toxicity and ensuring T cell activation only in the presence of specific antigens, enhancing safety and specificity (197, 198).
d. Co-activation Systems: These systems require the recognition of two or more antigens for CAR-T cells to be activated, ensuring precise targeting of tumor cells while minimizing damage to healthy tissues (199).
e. Logic-Gated CARs: These integrate multiple antigen signals, activating CAR-T cells only when specific combinations are recognized, improving control over T cell activation and reducing off-tumor effects (200).
f. Split-CARs: Split-CARs require the presence of two separate components expressed on different T cells, ensuring activation only when both signals converge at the tumor site, and reducing off-target toxicity (201).
Incorporating these multi-targeting designs into CAR-T cell therapies offers a promising strategy to improve efficacy and safety, addressing key obstacles in treating CNS tumors. In summary, the enhancement of CAR-T cell persistence requires a comprehensive approach that includes optimizing the T cell source, refining culture conditions, engineering CAR constructs, and incorporating genetic modifications. By addressing these factors, CAR-T cell therapies can be made more durable and effective, particularly for treating challenging pediatric brain tumors, where sustained persistence is essential for achieving long-term therapeutic success.
4.2.6 In vivo CAR-T cell transduction
In vivo CAR-T cell transduction represents an innovative strategy to enhance CAR-T cell therapy for CNS tumors (202). This strategy remains in a relatively infantile stage of development, but has great implications on manufacturing bottlenecks, scalability/accessibility concerns, and alleviated obstacles such as potential for shorter window times of repeated dosing and rapid treatment indices. This method modifies T cells directly within the body, avoiding the need for ex vivo expansion (203). Techniques such as viral vectors (lentivirus, adenovirus) or non-viral methods like CRISPR-Cas9 and mRNA-based systems can be used to introduce CAR constructs into T cells circulating in the bloodstream or within the tumor microenvironment (204). This approach has the potential to simplify CAR-T cell production and reduce the time required for therapy. In vivo transduction may also enable better control over the persistence and expansion of CAR-T cells within the body, enhancing the durability of therapeutic responses (205).
4.2.7 Implementing immunotherapy combinations
Advances in CAR-T cell therapy underscore the importance of synergistic combinations to enhance the persistence, expansion, and efficacy of CAR-T cells (206). Combining CAR-T cells with pharmacological agents or radiological techniques that improve cell trafficking, enhance penetration, or mitigate the effects of the immunosuppressive TME presents a promising strategy for improving therapeutic outcomes (207, 208).
4.2.7.1 CAR-T and monoclonal antibodies
Combining CAR-T cell therapy with monoclonal antibodies targeting immune checkpoints offers a promising strategy to overcome the challenges posed by immune evasion mechanisms (189). Key immune checkpoints, including PD-1, PD-L1, CTLA-4, TIM-3, TIGIT, and LAG-3, are critical regulators of T-cell exhaustion and immune suppression, making them valuable targets for combination therapies (209, 210). Checkpoint inhibitors such as PD-1 and PD-L1 blockers (nivolumab, pembrolizumab) and CTLA-4 inhibitors (ipilimumab) have shown clinical success in a range of cancers; combining these agents with CAR-T cells aims to relieve T-cell exhaustion, enhance anti-tumor immunity, and improve CAR-T cell persistence within the TME (209, 210). Preclinical studies have shown that combining CAR-T cells targeting EGFRvIII with anti-PD-1 or anti-PD-L1 antibodies results in significant tumor regression in GBM models, indicating that checkpoint blockade can potentiate CAR-T cell activity (211). Additionally, emerging studies investigating the role of targeted anti-CD47 agents in oncological diseases constitute valuable attempts at inducing immunomodulatory changes capable of potentiating accompanying immunotherapeutic strategies such as CAR-T therapies (212). However, safety concerns associated with systemic administration of checkpoint inhibitors and other monoclonal antibody agents, which have been shown to occasionally lead to significant toxicity and undesirable immunological adverse sequelae, are paramount, which underscores the need for additional investigations and an open mind to alternative approaches (213).
One promising strategy is engineering CAR-T cells to express checkpoint inhibitors on their surface, thereby overcoming the limitations of systemic delivery (214). CAR-T cells engineered to secrete PD-1 blocking antibodies have demonstrated improved anti-tumor efficacy and reduced exhaustion in solid tumor models, including glioblastoma; this approach not only enhances CAR-T cell proliferation and cytotoxicity but also ensures sustained activity within the immunosuppressive TME (215). In addition to PD-1 and PD-L1, other immune checkpoints like TIGIT and LAG-3 are gaining attention for their role in immune suppression (216). TIGIT inhibits T-cell activation by binding to its ligand, CD155, while LAG-3 downregulates T-cell function through its interaction with MHC class II molecules (217). Combining blockade of TIGIT and PD-1, or TIGIT and LAG-3, has been shown to improve CAR-T cell efficacy in preclinical studies of solid tumors, supporting the use of combination strategies that target multiple immune checkpoints (218). Furthermore, antibodies targeting other immune-regulatory pathways, such as the inhibitory receptor TIM-3 or the co-inhibitory molecule VISTA, are being explored in combination with CAR-T therapy (218). These molecules are involved in regulating T-cell activity in tumors and could provide additional avenues to enhance CAR-T cell persistence and efficacy.
Overall, the integration of checkpoint inhibitors, both through systemic administration and engineering CAR-T cells to express them, represents an exciting direction for improving the persistence, expansion, and therapeutic efficacy of CAR-T cells in treating CNS tumors and other solid cancers. Continued research into other immune-regulatory pathways will likely lead to more refined combination strategies, further advancing CAR-T cell therapy for challenging malignancies.
4.2.7.2 CAR-T and cytokine modulation
Cytokine support plays a crucial role in the activation, proliferation, and long-term persistence of CAR-T cells; cytokines IL-2, IL-15, IL-12, and others have been extensively studied for their ability to modulate immune responses and enhance the therapeutic efficacy of CAR-T cells (219, 220). While cytokines can significantly improve CAR-T cell function, their systemic administration often comes with significant risks, including toxicity (221). As a result, innovative approaches are being developed to localize cytokine delivery to the TME, minimizing systemic side effects while maximizing therapeutic benefits (221).
IL-2 and IL-15: Expanding and Sustaining CAR-T Cells: IL-2 has long been used to promote T-cell expansion due to its potent stimulatory effects; however, its use is often limited by the risk of systemic toxicity, including vascular leak syndrome, which can lead to serious complications (222). To overcome this challenge, researchers have focused on engineering “armored” CAR-T cells capable of secreting cytokines locally within the TME (223). This localized delivery helps modulate the immune response at the tumor site while avoiding systemic side effects. Although IL-2 remains valuable for expanding CAR-T cells, alternatives like IL-15 are becoming increasingly popular for their ability to promote CAR-T cell expansion and persistence without the same level of toxicity (224). IL-15 is particularly promising because it supports long-lasting T-cell immunity, improves T-cell generation, and enhances the durability of CAR-T cell therapies (224). The incorporation of IL-15 or IL-15 super agonists in CAR-T cell constructs has shown great promise in preclinical models, particularly for solid tumors, where the TME often limits the persistence and effectiveness of CAR-T cells (225).
IL-12: Modulating the TME for Improved CAR-T Function: IL-12 has garnered attention as a potent immune-modulatory cytokine that enhances anti-tumor immunity by promoting the differentiation of Th1 cells and activating macrophages, dendritic cells, and NK cells; importantly, it also plays a critical role in driving a pro-inflammatory TME, which is favorable for CAR-T cell function (226). In preclinical models of glioblastoma, the combination of CAR-T cells with IL-12 demonstrated improved tumor infiltration, reduced PD-1 expression on T cells, and enhanced anti-tumor cytotoxicity (227). Importantly, IL-12 has been shown to induce a TME more conducive to CAR-T cell survival, thus addressing one of the major challenges in solid tumors—immune suppression (228). By secreting IL-12 locally, CAR-T cells can enhance their anti-tumor effects, reduce T-cell exhaustion, and further improve persistence within the hostile TME of CNS tumors (229).
T-Cell Engagers: Targeting Cytokines and Cytokine Receptors: Beyond traditional cytokine supplementation, recent studies have explored the development of T-cell engagers that combine CAR-T cells with co-expressed cytokines or cytokine receptors. These innovative designs enable CAR-T cells to release cytokines in a controlled, localized manner within the TME, enhancing their activation and persistence without triggering widespread systemic effects (230). For instance, CAR-T cells engineered to express a high-affinity IL-2 receptor (CD25) can leverage the use of IL-2 present in the TME, leading to sustained expansion and prolonged persistence of CAR-T cells at the tumor site. This localized approach to cytokine delivery has the potential to overcome one of the key barriers in CAR-T cell therapy for solid tumors—persistence.
As the understanding of the TME evolves, the development of more sophisticated cytokine-based strategies will be essential for advancing CAR-T cell therapies. Targeted modulation of the TME through cytokines will likely become a cornerstone of combination therapy approaches, enhancing the immune response and ultimately improving patient outcomes, particularly for pediatric brain cancers and other challenging solid tumors.
4.2.7.3 CAR T cells and FUS
As introduced in previous sections, FUS is an innovative technique that temporarily disrupts the BBB, significantly improving the delivery of CAR-T cells to the tumor site (231). By overcoming the BBB, FUS facilitates CAR-T cell infiltration into CNS tumors (232, 233). Beyond aiding CAR-T cell penetration, FUS also plays a crucial role in modulating the TME through inducing sterile inflammation, recruiting immune cells, and activating microglia, all of which enhance the anti-tumor immune response (234) (Figure 6). When combined with checkpoint inhibitors, such as anti-PD-1, FUS helps reverse immune suppression within the TME, further boosting CAR-T cell efficacy (157, 166). Studies have shown that this combination significantly improves CAR-T cell activity and enhances tumor control (168). Moreover, FUS can disrupt the tumor’s ECM, through reducing tumor compactness via loosening the dense structure of the ECM, thus improving access for CAR-T cells (235). The enhanced tissue permeability following FUS treatment is thus crucial for improving CAR-T cell delivery and overall therapeutic efficacy.
Another promising development involves combining FUS with gene-modified CAR-T cells engineered to respond to FUS-induced activation (187). This strategy enables more localized and controlled activation of CAR-T cells at the tumor site, minimizing systemic toxicity and enhancing their therapeutic potential (187). Preclinical studies have demonstrated that this combination not only improves tumor control but also extends survival, offering a novel approach to treating brain tumors and other solid cancers (236). FUS can also be combined with other immunomodulatory agents or therapeutic compounds to further enhance CAR-T cell activity (156, 231).
The synergy between CAR-T cells and FUS addresses the critical challenge of BBB penetration while enhancing CAR-T cell access and infiltration by targeting the ECM. By combining these innovative approaches, this strategy holds significant promise for improving the treatment of brain tumors, offering new hope for patients with these challenging cancers.
4.2.7.4 CAR T cells and other emerging therapeutic modalities
Other emerging therapeutic modalities also have the potential to transform the landscape of CAR-T cell therapy:
Gene Editing for TME Modulation: Advances in gene editing technologies such as CRISPR-Cas9 allow for precise modifications of the CAR-T cell genome to improve functionality in solid tumors (188). CAR-T cells can be edited to knock out genes that promote immune suppression, such as PD-1, or to express pro-inflammatory cytokines and chemokines that recruit other immune cells to the tumor site (214, 237, 238). In addition, gene editing can be used to manipulate the TME directly, improving tumor responsiveness to CAR-T cells (239).
Oncolytic Viruses: Oncolytic viruses (OVs) that selectively infect and kill tumor cells while stimulating anti-tumor immunity are emerging as powerful adjuncts to CAR-T cell therapy (240). These viruses can enhance CAR-T cell infiltration into tumors, modulate the TME, and induce immune responses (241). When used in combination, OVs can synergize with CAR-T cells by triggering tumor cell death, releasing tumor antigens, and promoting a more immunostimulatory environment (242).
Radiation Therapy: While traditionally used as a standalone modality, radiation therapy can enhance CAR-T cell efficacy by inducing immunogenic cell death and promoting antigen release (243). Combining low-dose radiation with CAR-T therapy has been shown to enhance CAR-T cell activity in solid tumors by inducing favorable changes in the TME, such as upregulation of tumor-associated antigens and increased immune cell infiltration (244).
CAR T Cell Manufacturing: Innovations in automated and closed-system platforms have streamlined CAR-T cell production, improving scalability, reducing costs, and minimizing contamination risks (245). Advances in viral transduction, including lentiviral and retroviral vector engineering, have improved gene transfer efficiency, reduced production times, and enhanced safety profiles (246). Additionally, next-generation CAR-T cells with enhanced functionality and allogeneic “off-the-shelf” therapies are accelerating clinical applications, making CAR-T therapy more accessible and efficient (103). Adopting immunotherapy combinations involving CAR-T cell therapy with monoclonal antibodies, cytokines, FUS, and other innovative therapies therefore holds great promise for overcoming the limitations of CAR-T cells in treating solid tumors, particularly in the context of the immunosuppressive brain tumor microenvironment. These combination strategies aim to enhance CAR-T cell persistence, modulate the TME, and potentiate anti-tumor immunity, potentially leading to improved therapeutic outcomes for patients with brain malignancies. However, clinical translation of these combination therapies will require rigorous evaluation of safety, efficacy, and potential long-term side effects in human patients.
5 Future perspectives
5.1 Tracking CAR-T cells
CAR-T cell therapy has revolutionized cancer treatment, especially for hematologic malignancies, but its translation to brain tumors and other solid cancers presents unique challenges. In this paper, we outlined a plethora of possible solutions that can be put forth to ameliorate obstacles in the face of CAR-T cell therapy in pediatric brain tumors (Table 4). One of the critical barriers is the lack of straightforward biomarkers to monitor CAR-T cells, unlike traditional chemical agents where serum concentration is often used as an indicator of efficacy and toxicity (247). Moreover, the personalized nature of autologous CAR-T cell therapy results in heterogeneous responses, making it challenging to predict therapeutic effects and manage potential toxicities (119). Consequently, there is a growing need for effective, non-invasive techniques to track CAR-T cells in vivo and evaluate their therapeutic impact.
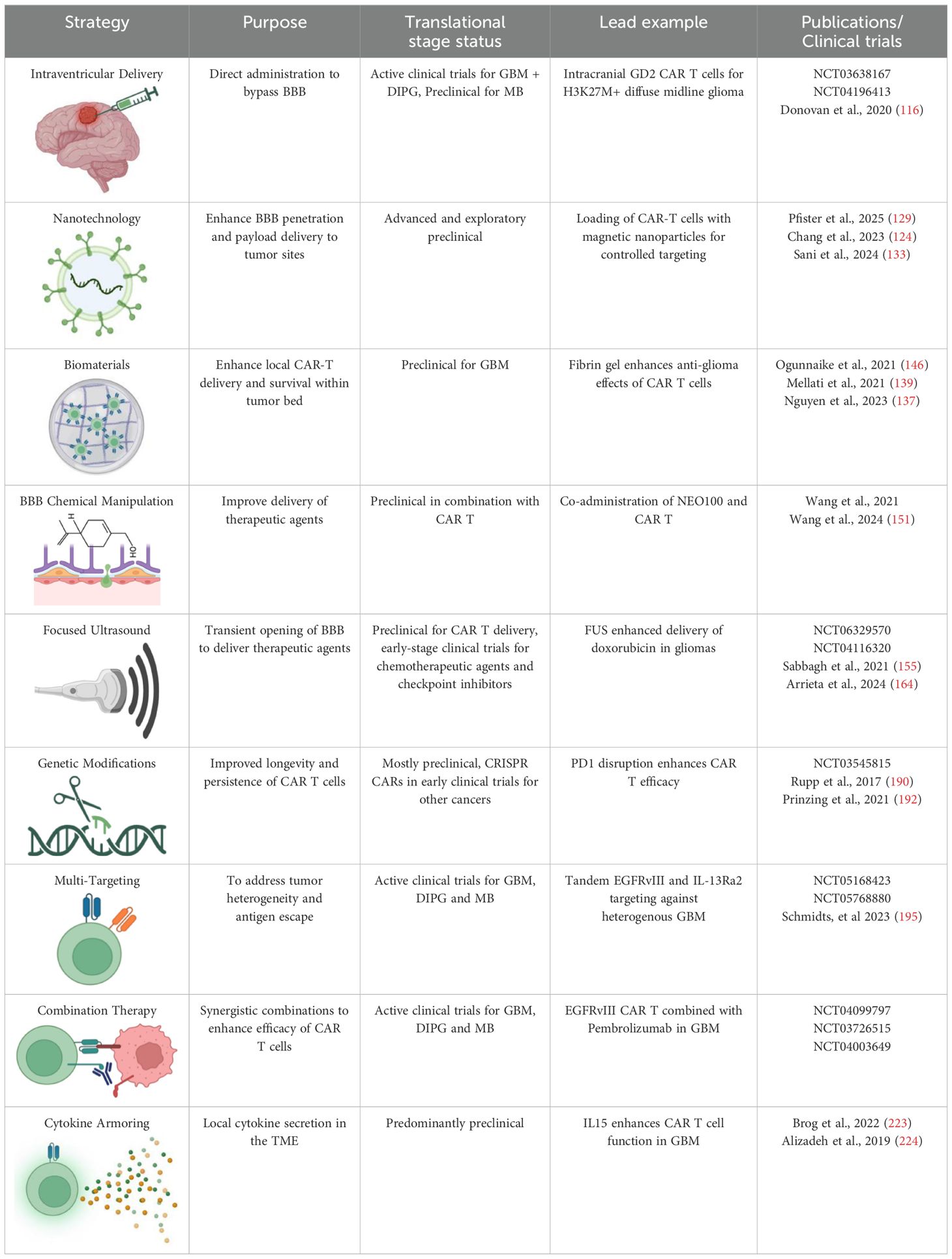
Table 4. Current therapeutic strategies to enhance CAR T Cell efficacy: An overview of emerging therapeutic strategies and their current stage of development aimed at overcoming challenges in treating pediatric brain tumors.
Non-invasive imaging techniques have therefore emerged as a promising solution for real-time monitoring of CAR-T cells (248). These techniques involve either direct labeling of CAR-T cells (ex-vivo marking and subsequent labeling) or indirect labeling, where the cells are genetically modified to express a reporter gene (249). Direct labeling, while useful in some cases, faces limitations such as dilution during cell division, reducing the tracking efficacy over time (250). This issue complicates long-term monitoring, particularly in clinical settings. On the other hand, indirect labeling, which involves incorporating radiolabeled reporter genes into CAR-T cells, enables more efficient tracking (249). This method allows monitoring of cell expansion over time, although it remains technically challenging for routine clinical application.
In addition to traditional labeling methods, alternative technologies such as nanoparticle-based techniques and microfluidic platforms are being explored to enhance the efficiency and precision of CAR-T cell tracking (251, 252). These techniques promise to overcome the limitations of current labeling methods by providing higher sensitivity and more accurate imaging, facilitating the assessment of CAR-T cell migration, distribution, and functional patterns (252). As research progresses, these imaging techniques will likely become invaluable for evaluating the in vivo behavior of CAR-T cells, improving patient-specific treatment strategies and optimizing therapy outcomes.
Various imaging modalities have also been explored for monitoring labeled CAR-T cells. Common techniques include CT, PET, and PET/CT, which are well-established for assessing tumor responses (253). For CAR-T cell imaging, specific criteria must be met to ensure the successful translation of these methods into clinical practice. The reporter molecules used in these imaging systems must be highly specific to the CAR-T cells, non-immunogenic, and sensitive enough to detect low quantities of the cells (254). Additionally, these reporters should have minimal expression profiles in normal tissues to reduce background noise and allow precise tracking of CAR-T cells, especially when administered to the brain (255). The ability of these reporters to cross the BBB and not alter the CAR-T cells’ functionality is also a critical consideration. Therefore, clinical success of these imaging strategies will depend on the integration of these criteria, influencing how CAR-T cell therapies are monitored in clinical trials and how toxicity and efficacy are evaluated in brain tumor models.
5.2 Toxicities of CAR T-cell therapy in CNS tumors
CAR T-cell therapy has shown transformative potential in treating various malignancies, including CNS tumors. However, its clinical application, particularly in brain tumors, is accompanied by significant toxicities that require careful management (256, 257). These toxicities, primarily associated with cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), pose unique challenges due to the delicate and enclosed nature of the CNS (258).
CRS: Cytokine release syndrome is a systemic inflammatory response triggered by CAR T-cell activation and proliferation, which releases large amounts of cytokines (259). CRS has been well documented as a dose-limiting toxicity in hematological cancers, but its manifestation in CNS tumors differs (260). In CNS solid tumors, the onset of CRS is typically delayed, occurring as CAR T cells gradually infiltrate the tumor and undergo full activation (256). This delayed onset contrasts with the more immediate inflammatory response seen in hematological malignancies, where tumor cells are disseminated throughout the body, leading to quicker systemic inflammation (260).
While CRS is less frequent and less severe in patients with solid tumors, including CNS malignancies, it remains a significant concern. The compromised vasculature of brain tumors, which is impacted by the BBB/BTB, limits CAR T-cell infiltration and may slow down the onset of CRS (261, 262). However, when CRS does occur, it can still be severe, requiring immediate intervention (256). The typical clinical management strategies for CRS include the administration of tocilizumab, an IL-6 receptor inhibitor, which has shown efficacy in mitigating cytokine release without significantly impairing CAR T-cell activity. In cases of severe CRS, corticosteroids may also be employed, although their use can reduce CAR T-cell effectiveness and inhibit tumor clearance (260).
Recent studies suggest that the use of pre-treatment cytokine levels as biomarkers could help predict the likelihood of CRS and guide the management of patients at higher risk (263). This approach could improve patient stratification and allow for more personalized treatment regimens, minimizing the incidence of severe CRS.
ICANS: A critical and unique toxicity in the context of CAR T-cell therapy for CNS tumors is ICANS (256). ICANS is characterized by a range of neurological symptoms, including encephalopathy, cerebral edema, seizures, and aphasia (264). The CNS-specific nature of this toxicity presents unique clinical challenges, as it can be difficult to distinguish between tumor progression and the onset of ICANS, especially in patients with brain tumors. This diagnostic challenge complicates the management of these patients, as physicians must differentiate between true tumor growth and neurotoxic effects induced by CAR T-cell treatment.
ICANS occurs because of CAR T-cell infiltration into the brain, where localized inflammation causes neurotoxicity (264, 265). Localized delivery of CAR T-cells, such as through intracranial or intraventricular infusion, is being explored as a strategy to mitigate systemic side effects and reduce the risk of ICANS (117, 266). However, this approach does not eliminate the risk of neurotoxic effects. Indeed, even localized CAR T-cell therapy can lead to severe neurotoxicity in some patients, particularly if the CAR T-cells trigger an intense inflammatory response within the confined environment of the CNS (265).
Management of ICANS typically involves the use of corticosteroids to suppress inflammation; however, corticosteroid use can impair CAR T-cell functionality, presenting a difficult balance between managing toxicity and maintaining therapeutic efficacy (267). Tocilizumab, commonly used to manage CRS, is also occasionally used to address ICANS, but its effectiveness in the context of neurotoxicity is still under investigation (257). In addition to corticosteroids and IL-6 inhibitors, emerging strategies are being explored to mitigate neurotoxicity without compromising CAR T-cell activity. These include modifying CAR T-cell constructs to reduce the release of pro-inflammatory cytokines, as well as incorporating specific immunosuppressive agents that target neuroinflammation while preserving CAR T-cell function (257, 258).
Clinical Management Strategies and Emerging Approaches: The clinical management of CAR T-cell therapy toxicities in CNS tumors is evolving as the field advances. In addition to corticosteroids and tocilizumab, other therapies, such as Janus kinase (JAK) inhibitors, are being explored to manage cytokine-driven inflammation with potentially fewer impacts on CAR T-cell efficacy (268). Moreover, pre-treatment screening for biomarkers that predict the risk of CRS and ICANS is an active area of research (265). Several studies have indicated that baseline levels of cytokines such as IL-6 and IL-1, as well as tumor burden and CAR T-cell expansion kinetics, may serve as predictive markers for adverse events (263, 265).
In the realm of CAR T-cell engineering, significant progress is being made to minimize toxicities while enhancing therapeutic efficacy (269). Advances in novel CAR design modifications, such as dual-target CAR T-cells, are designed to target both the tumor and the immune microenvironment, show promise in reducing off-target effects and improving specificity (269). Additionally, research into “safety switches” that allow for controlled elimination of CAR T-cells in the event of severe toxicities is gaining traction to mitigate risks associated with both CRS and ICANS (267, 270).
5.3 Current clinical landscape and translational bottlenecks in CAR T cell therapy for CNS tumors
Significant strides have been made in advancing CAR T cell therapy for CNS tumors, with early clinical trials demonstrating feasibility and initial safety. However, major biological, regulatory, and logistical challenges continue to hinder the broad and effective clinical implementation of these therapies.
In Table 1, we summarize a selection of notable early-phase clinical trials (primarily Phase 1) evaluating CAR T cell therapy in CNS tumors. These include trials targeting GD2 (e.g., NCT04196413, NCT04099797), HER2 (NCT03500991, NCT03596073), B7-H3 (NCT04815307), and IL13Rα2 (NCT02208362). Across these trials, common primary endpoints include safety, tolerability, and dose-limiting toxicities, while secondary endpoints involve CAR T cell persistence, tumor response by imaging (RANO criteria), and survival outcomes.
Although these trials confirm that intracranial and locoregional delivery of CAR T cells is generally safe and feasible, clinical outcomes remain modest. For instance:
● In NCT02208362, IL13Rα2 CAR T cells delivered intratumorally in GBM showed transient radiographic responses, but tumor regrowth occurred within months, and CAR T cells were no longer detectable after peak expansion.
● GD2-targeting trials (NCT04196413) in diffuse midline glioma demonstrated the ability of CAR T cells to traffic to CSF and tumor sites, yet persistence remained limited and correlated with transient responses.
● In NCT03500991, HER2 CAR T cells infused intracranially in pediatric CNS tumors showed no dose-limiting toxicities, but antitumor activity was minimal, highlighting challenges in potency and durability.
These findings collectively underscore key biological limitations, including suboptimal trafficking, low CAR T cell persistence, and limited immune activation in the hostile CNS tumor microenvironment.
Beyond biologic barriers, translational and regulatory bottlenecks also constrain progress:
● Autologous manufacturing is slow, often taking 3–4 weeks. This is unfeasible for rapidly progressing CNS malignancies like glioblastoma or DIPG.
● Product heterogeneity (e.g., variable transduction efficiency, inconsistent memory phenotypes) introduces uncertainty in clinical outcomes.
● There is a lack of standardized management protocols for treatment-related neurotoxicity, especially ICANS, which may manifest differently in patients with preexisting CNS pathology.
● Preclinical models are frequently immunodeficient and do not recapitulate human tumor–immune interactions, hampering predictive accuracy of efficacy and safety.
● GMP and regulatory hurdles, including requirements for clean-room manufacturing, release testing, and quality control, create bottlenecks, particularly in resource-limited settings or academic institutions.
● Accessibility and equity issues remain, as most trials are conducted at specialized centers, leaving patients in underserved regions with limited options.
Regulatory challenges in pediatric CAR T trials are significant due to stricter safety requirements, longer follow-up mandates, and the need for age-specific dosing and neurodevelopmental risk assessments. These factors often delay trial approval and limit patient enrollment. Given the rarity of pediatric CNS tumors, trials tend to be small and underpowered. Streamlining regulatory processes and creating pediatric-focused development frameworks will be key to accelerating safe and effective CAR T therapies for children.
To overcome these barriers, a multi-pronged and coordinated global approach is needed. Strategies may include the development of allogeneic or off-the-shelf CAR T products, decentralized manufacturing platforms, improvement in preclinical immunocompetent CNS tumor models, and harmonization of regulatory pathways to reduce delays while maintaining safety standards. Integration of biomarker-driven adaptive trials and real-time translational feedback loops can further accelerate refinement and deployment of effective CAR T cell therapies for CNS tumors.
5.4 Clinical translation
CAR T-cell therapy holds tremendous potential in revolutionizing cancer treatment, especially for CNS solid tumors that have historically shown minimal to no long-term responses to traditional therapies. The transition of various CAR T-cell models from preclinical studies to clinical application, however, faces significant challenges that require a multifaceted approach.
One of the major obstacles to successful clinical translation is the failure of preclinical models to adequately replicate the TME and immune system dynamics of humans. Non-syngeneic (non-immunocompetent) murine models, which are commonly used in preclinical research, are limited in their ability to mimic the complexity of human immune interactions. These models fall short in accounting for the full spectrum of immune mediators, immune checkpoints, and cytokine environments that influence CAR T-cell behavior in humans. While syngeneic (immunocompetent) murine models are more effective in providing a closer approximation of human immune responses, they present their own challenges. Particularly developing CAR T-cell therapies targeting tumor-specific antigens that are not expressed on normal host tissues thus requires careful optimization to avoid potential off-target effects.
Moreover, translating CAR T-cell therapies from animal models to human patients involves addressing several other critical considerations, such as optimizing CAR construct design, improving T-cell manufacturing processes, and overcoming regulatory hurdles. The combination of mono- or combination therapies, which may target multiple antigens or modulate the immune microenvironment, holds promise for improving the efficacy of CAR T-cells in solid CNS tumors. However, ensuring that these therapies are both effective and safe will require extensive research into the identification of appropriate antigens, the design of CAR constructs with improved specificity, and the development of scalable and standardized protocols for T-cell production.
In summary, while CAR T-cell therapy represents a promising breakthrough for treating resistant CNS solid tumors, achieving successful clinical translation demands concerted efforts to refine preclinical models, improve CAR T-cell engineering, and address logistical and regulatory challenges. These combined efforts will be essential in making CAR T-cell therapy a viable, standardized treatment option for CNS cancers.
6 Discussion and conclusion
As it stands, the lack of success of CAR T cell therapy in brain malignancies highlights the urgent need for innovative therapeutic strategies. However, the unique challenges of CNS tumors complicate this translation. The relevant variables include inadequate tumor infiltration, immune suppression within the TME, the restrictive nature of the BBB, and systemic toxicities associated with immune activation. Overcoming these obstacles requires a multidisciplinary approach that integrates biological insights with cutting-edge engineering advancements.
A range of innovative solutions has emerged to address these barriers. Locoregional delivery systems, such as intra-tumoral injections, micro-injectable hydrogels, and implantable scaffolds, can facilitate direct CAR-T cell administration to tumor sites while minimizing systemic exposure. Moreover, the integration of FUS acts as a promising modality for temporarily disrupting the BBB and subsequently enhancing CAR-T cell delivery to CNS tumors. Beyond improving trafficking, FUS can also modulate the TME by inducing sterile inflammation, activating microglia, and recruiting immune cells, thus augmenting CAR-T cell activity.
Advancements in CAR T cell engineering are also pivotal. Gene-editing technologies like CRISPR-Cas9 enable precise modifications to enhance CAR-T cell functionality, including knocking out inhibitory receptors such as PD-1, optimizing metabolic pathways for improved cell fitness, and designing CARs that secrete pro-inflammatory cytokines to reshape the TME. Furthermore, combining CAR-T cells with immune checkpoint inhibitors has shown synergistic potential in reversing T cell exhaustion and boosting therapeutic efficacy.
Meanwhile, ongoing manufacturing innovations are grappling with key bottlenecks in scalability, cost, and quality control. For instance, circRNAs have been explored as potential candidates to alleviate concerns of safety and precision in the context of CAR-T cell therapy (271). Moreover, non-viral gene delivery methods, such as electroporation and nanoparticle-based transfection, are reducing reliance on viral vectors, thereby lowering production costs and enabling precise engineering. Moreover, dynamic bioreactors, feeder-free culture systems, and automation-driven workflows are optimizing CAR-T cell expansion and functionality while expediting production timelines to improve patient accessibility.
Looking forward, these advancements pave the way for more effective and personalized treatment strategies. Real-time imaging technologies, including immunoPET and MRI, will enable dynamic monitoring of CAR-T cell behavior, allowing clinicians to track trafficking, proliferation, and efficacy within the CNS and adjust therapies in real time. Additionally, synergistic approaches, such as combining CAR-T cells with oncolytic viruses or integrating innovative delivery systems like FUS and nanotechnology, are promising strategies to overcome TME-induced resistance and improving therapeutic outcomes. Additionally, the potential incorporation of CAR technology into other cell lines such as macrophages and Natural Killer (NK) will help further define and characterize the obstacles ahead of recombinant adoptive cellular therapies (272).
In conclusion, CAR-T cell therapy for brain tumors has made significant progress, yet critical hurdles remain. Addressing challenges such as trafficking, TME suppression, and persistence through multidisciplinary innovation and engineering is vital for expanding CAR-T cell efficacy in CNS malignancies. With ongoing advancements in gene editing, manufacturing, and combination therapies, CAR-T cell therapy is poised to transform the treatment landscape, offering renewed hope for patients and families affected by these devastating diseases.
Author contributions
SY: Writing – original draft, Investigation, Visualization, Software, Conceptualization, Supervision, Writing – review & editing, Methodology. SM: Writing – review & editing, Writing – original draft. EV: Writing – review & editing, Writing – original draft, Visualization. AH: Writing – review & editing, Writing – original draft, Visualization. KK: Writing – original draft. NS: Writing – original draft, Writing – review & editing. DH: Validation, Visualization, Supervision, Investigation, Conceptualization, Writing – review & editing, Funding acquisition, Resources, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This was supported by NCI R00CA256262, DOD CA230573, and Lilabean Foundation award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dagar G, Gupta A, Masoodi T, Nisar S, Merhi M, Hashem S, et al. Harnessing the potential of CAR-T cell therapy: progress, challenges, and future directions in hematological and solid tumor treatments. J Transl Med. (2023) 21:449. doi: 10.1186/s12967-023-04292-3
2. Katz B-Z and Herishanu Y. Therapeutic targeting of CD19 in hematological Malignancies: past, present, future and beyond. Leuk Lymphoma. (2014) 55:999–1006. doi: 10.3109/10428194.2013.828354
3. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C, et al. Current progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. (2019) 15:2548–60. doi: 10.7150/ijbs.34213
4. Del Baldo G, Del Bufalo F, Pinacchio C, Carai A, Quintarelli C, De Angelis B, et al. The peculiar challenge of bringing CAR-T cells into the brain: Perspectives in the clinical application to the treatment of pediatric central nervous system tumors. Front Immunol. (2023) 14:1142597. doi: 10.3389/fimmu.2023.1142597
5. Quail DF and Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
6. Chen Q, Lu L, and Ma W. Efficacy, safety, and challenges of CAR T-cells in the treatment of solid tumors. Cancers (Basel). (2022) 14:5983. doi: 10.3390/cancers14235983
7. Patel U, Abernathy J, Savani BN, Oluwole O, Sengsayadeth S, and Dholaria B. CAR T cell therapy in solid tumors: A review of current clinical trials. EJHaem. (2022) 3:24–31. doi: 10.1002/jha2.356
8. Wachsmann TLA, Wouters AK, Remst DFG, Hagedoorn RS, Meeuwsen MH, van Diest E, et al. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology. (2022) 11:2033528. doi: 10.1080/2162402X.2022.2033528
9. Tassilo LA and Wachsmann AK. Wouters DFGRRSHMHME van DJLJKJHFF, Heemskerk MHM. Comparing CAR and TCR engineered T cell performance as a function of tumor cell exposure. Oncoimmunology. (2022) 11:2033528. doi: 10.1080/2162402X.2022.2033528
10. Baulu E, Gardet C, Chuvin N, and Depil S. TCR-engineered T cell therapy in solid tumors: State of the art and perspectives. Sci Adv. (2023) 9:eadf3700. doi: 10.1126/sciadv.adf3700
11. He Q, Jiang X, Zhou X, and Weng J. Targeting cancers through TCR-peptide/MHC interactions. J Hematol Oncol. (2019) 12:139. doi: 10.1186/s13045-019-0812-8
12. Ma P, Jiang Y, Zhao G, Wang W, Xing S, Tang Q, et al. Toward a comprehensive solution for treating solid tumors using T-cell receptor therapy: A review. Eur J Cancer. (2024) 209:114224. doi: 10.1016/j.ejca.2024.114224
13. Hont AB, Powell AB, Sohai DK, Valdez IK, Stanojevic M, Geiger AE, et al. The generation and application of antigen-specific T cell therapies for cancer and viral-associated disease. Mol Ther. (2022) 30:2130–52. doi: 10.1016/j.ymthe.2022.02.002
14. Kinoshita H, Cooke KR, Grant M, Stanojevic M, Cruz CR, Keller M, et al. Outcome of donor-derived TAA-T cell therapy in patients with high-risk or relapsed acute leukemia post allogeneic BMT. Blood Adv. (2022) 6:2520–34. doi: 10.1182/bloodadvances.2021006831
15. DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front Immunol. (2020) 11:1402. doi: 10.3389/fimmu.2020.01402
16. Zhang X, Zhao L, Zhang H, Zhang Y, Ju H, Wang X, et al. The immunosuppressive microenvironment and immunotherapy in human glioblastoma. Front Immunol. (2022) 13:1003651. doi: 10.3389/fimmu.2022.1003651
17. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
18. Zheng Z, Li S, Liu M, Chen C, Zhang L, and Zhou D. Fine-tuning through generations: advances in structure and production of CAR-T therapy. Cancers (Basel). (2023) 15:3476. doi: 10.3390/cancers15133476
19. Anikeeva N, Panteleev S, Mazzanti NW, Terai M, Sato T, and Sykulev Y. Efficient killing of tumor cells by CAR-T cells requires greater number of engaged CARs than TCRs. J Biol Chem. (2021) 297:101033. doi: 10.1016/j.jbc.2021.101033
20. Kuehn BM. The promise and challenges of CAR-T gene therapy. JAMA. (2017) 318:2167–9. doi: 10.1001/jama.2017.15605
21. Mo F, Pellerino A, Soffietti R, and Rudà R. Blood-brain barrier in brain tumors: Biology and clinical relevance. Int J Mol Sci. (2021) 22:12654. doi: 10.3390/ijms222312654
22. Daneman R and Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. (2015) 7:a020412. doi: 10.1101/cshperspect.a020412
23. Steeg PS. The blood-tumour barrier in cancer biology and therapy. Nat Rev Clin Oncol. (2021) 18:696–714. doi: 10.1038/s41571-021-00529-6
24. Doolittle ND, Muldoon LL, Culp AY, and Neuwelt EA. Delivery of chemotherapeutics across the blood-brain barrier: challenges and advances. Adv Pharmacol. (2014) 71:203–43. doi: 10.1016/bs.apha.2014.06.002
25. Kringel R, Lamszus K, and Mohme M. Chimeric antigen receptor T cells in glioblastoma-current concepts and promising future. Cells. (2023) 12:1770. doi: 10.3390/cells12131770
26. Sabahi M, Salehipour A, Bazl MSY, Rezaei N, Mansouri A, and Borghei-Razavi H. Local immunotherapy of glioblastoma: A comprehensive review of the concept. J Neuroimmunol. (2023) 381:578146. doi: 10.1016/j.jneuroim.2023.578146
27. Dong X, Ren J, Amoozgar Z, Lee S, Datta M, Roberge S, et al. Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. J Immunother Cancer. (2023) 11:e005583. doi: 10.1136/jitc-2022-005583
28. Kim HJ, Park JW, and Lee JH. Genetic architectures and cell-of-origin in glioblastoma. Front Oncol. (2020) 10:615400. doi: 10.3389/fonc.2020.615400
29. Maggs L, Cattaneo G, Dal AE, Moghaddam AS, and Ferrone S. CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci. (2021) 15:662064. doi: 10.3389/fnins.2021.662064
30. van Tellingen O, Yetkin-Arik B, de Gooijer MC, Wesseling P, Wurdinger T, and de Vries HE. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist Update. (2015) 19:1–12. doi: 10.1016/j.drup.2015.02.002
31. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro Oncol. (2018) 20:184–91. doi: 10.1093/neuonc/nox175
32. Arvanitis CD, Ferraro GB, and Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. (2020) 20:26–41. doi: 10.1038/s41568-019-0205-x
33. Grimm SA and Chamberlain MC. Brainstem glioma: A review. Curr Neurol Neurosci Rep. (2013) 13:346. doi: 10.1007/s11910-013-0346-3
34. Haydar D, Ibañez-Vega J, and Krenciute G. T-cell immunotherapy for pediatric high-grade gliomas: new insights to overcoming therapeutic challenges. Front Oncol. (2021) 11:718030. doi: 10.3389/fonc.2021.718030
35. Aziz-Bose R and Monje M. Diffuse intrinsic pontine glioma: molecular landscape and emerging therapeutic targets. Curr Opin Oncol. (2019) 31:522–30. doi: 10.1097/cco.0000000000000577
36. Chaves C, Declèves X, Taghi M, Menet M-C, Lacombe J, Varlet P, et al. Characterization of the blood–brain barrier integrity and the brain transport of SN-38 in an orthotopic xenograft rat model of diffuse intrinsic pontine glioma. Pharmaceutics. (2020) 12:399. doi: 10.3390/pharmaceutics12050399
37. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603:934–41. doi: 10.1038/s41586-022-04489-4
38. El-Khouly FE, Veldhuijzen van Zanten SEM, Santa-Maria Lopez V, Hendrikse NH, Kaspers GJL, Loizos G, et al. Diagnostics and treatment of diffuse intrinsic pontine glioma: where do we stand? J Neurooncol. (2019) 145:177–84. doi: 10.1007/s11060-019-03287-9
39. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, and Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res. (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
40. Ferreras C, Fernández L, Clares-Villa L, Ibáñez-Navarro M, Martín-Cortázar C, Esteban-Rodríguez I, et al. Facing CAR T cell challenges on the deadliest paediatric brain tumours. Cells. (2021) 10:2940. doi: 10.3390/cells10112940
41. Phoenix TN, Patmore DM, Boop S, Boulos N, Jacus MO, Patel YT, et al. Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell. (2016) 29:508–22. doi: 10.1016/j.ccell.2016.03.002
42. Lin YJ, Mashouf LA, and Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol. (2022) 13:817296. doi: 10.3389/fimmu.2022.817296
43. Roussel MF and Hatten ME. Chapter 8 - cerebellum: development and medulloblastoma. In: Dyer MA, editor. Cancer and Development. Academic Press (2011). p. 235–82. Current Topics in Developmental Biology. (USA: Academic Press) doi: 10.1016/B978-0-12-380916-2.00008-5
44. Baloyannis SJ and Baloyannis IS. The fine structure of ependymomas. CNS Oncol. (2014) 3:49–59. doi: 10.2217/cns.13.64
45. Wang X, Huynh C, Urak R, Weng L, Walter M, Lim L, et al. The cerebroventricular environment modifies CAR T cells for potent activity against both central nervous system and systemic lymphoma. Cancer Immunol Res. (2021) 9:75–88. doi: 10.1158/2326-6066.CIR-20-0236
46. Korones DN. Pediatric ependymomas: Something old, something new. Pediatr Hematol Oncol J. (2023) 8:114–20. doi: 10.1016/j.phoj.2023.04.002
47. Pal K and Sheth RA. Engineering the tumor immune microenvironment through minimally invasive interventions. Cancers (Basel). (2023) 15:196. doi: 10.3390/cancers15010196
48. Julian S, Rechberger CLN, and Daniels DJ. Atypical teratoid rhabdoid tumor (ATRT): disease mechanisms and potential drug targets. Expert Opin Ther Targets. (2022) 26:187–92. doi: 10.1080/14728222.2022.2040017
49. Tran S, Plant-Fox AS, Chi SN, and Narendran A. Current advances in immunotherapy for atypical teratoid rhabdoid tumor (ATRT). Neurooncol Pract. (2023) 10:322–34. doi: 10.1093/nop/npad005
50. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, and Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. (2019) 290:60–84. doi: 10.1111/imr.12773
51. Alva E, Rubens J, Chi S, Rosenberg T, Reddy A, Raabe EH, et al. Recent progress and novel approaches to treating atypical teratoid rhabdoid tumor. Neoplasia. (2023) 37:100880. doi: 10.1016/j.neo.2023.100880
52. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, and Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncol. (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
53. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. (2020) 181:1643–1660.e17. doi: 10.1016/j.cell.2020.05.007
54. Anderson NM and Simon MC. The tumor microenvironment. Curr Biol. (2020) 30:R921–5. doi: 10.1016/j.cub.2020.06.081
55. Sharma P, Aaroe A, Liang J, and Puduvalli VK. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol Adv. (2023) 5:vdad009. doi: 10.1093/noajnl/vdad009
56. Mohiuddin E and Wakimoto H. Extracellular matrix in glioblastoma: opportunities for emerging therapeutic approaches. Am J Cancer Res. (2021) 11(8):3742–54.
57. Chen Z, Han F, Du Y, Shi H, and Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. (2023) 8:70. doi: 10.1038/s41392-023-01332-8
58. Piwocka O, Piotrowski I, Suchorska WM, and Kulcenty K. Dynamic interactions in the tumor niche: how the cross-talk between CAFs and the tumor microenvironment impacts resistance to therapy. Front Mol Biosci. (2024) 11:1343523. doi: 10.3389/fmolb.2024.1343523
59. Messiaen J, Jacobs SA, and De Smet F. The tumor micro-environment in pediatric glioma: friend or foe? Front Immunol. (2023) 14:1227126. doi: 10.3389/fimmu.2023.1227126
60. Chen Y, Zhao C, Li S, Wang J, and Zhang H. Immune microenvironment and immunotherapies for diffuse intrinsic pontine glioma. Cancers (Basel). (2023) 15:602. doi: 10.3390/cancers15030602
61. Tykocki T. Diffuse intrinsic pontine glioma and chimeric antigen receptor T-cell therapy: an emerging frontier. World Neurosurg. (2025) 194:123579. doi: 10.1016/j.wneu.2024.123579
62. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers. (2019) 5:11. doi: 10.1038/s41572-019-0063-6
63. van Bree NFHN and Wilhelm M. The tumor microenvironment of medulloblastoma: an intricate multicellular network with therapeutic potential. Cancers (Basel). (2022) 14:5009. doi: 10.3390/cancers14205009
64. Kurdi M, Mulla N, Malibary H, Bamaga AK, Fadul MM, Faizo E, et al. Immune microenvironment of medulloblastoma: The association between its molecular subgroups and potential targeted immunotherapeutic receptors. World J Clin Oncol. (2023) 14:117–30. doi: 10.5306/wjco.v14.i3.117
65. Zhang J and Wang T. Immune cell landscape and immunotherapy of medulloblastoma. Pediatr Investig. (2021) 5:299–309. doi: 10.1002/ped4.12261
66. Zhang Q, Cheng S, Wang Y, Wang M, Lu Y, Wen Z, et al. Interrogation of the microenvironmental landscape in spinal ependymomas reveals dual functions of tumor-associated macrophages. Nat Commun. (2021) 12:6867. doi: 10.1038/s41467-021-27018-9
67. Lester A and McDonald KL. Intracranial ependymomas: molecular insights and translation to treatment. Brain Pathol. (2020) 30:3–12. doi: 10.1111/bpa.12781
68. Hwang EI, Hanson D, Filbin MG, and Mack SC. Why haven’t we solved intracranial pediatric ependymoma? Current questions and barriers to treatment advances. Neoplasia. (2023) 39:100895. doi: 10.1016/j.neo.2023.100895
69. Griesinger AM, Riemondy K, Eswaran N, Donson AM, Willard N, Prince EW, et al. Multi-omic approach identifies hypoxic tumor-associated myeloid cells that drive immunobiology of high-risk pediatric ependymoma. iScience. (2023) 26:107585. doi: 10.1016/j.isci.2023.107585
70. Paassen I, Williams J, Ríos Arceo C, Ringnalda F, Mercer KS, Buhl JL, et al. Atypical teratoid/rhabdoid tumoroids reveal subgroup-specific drug vulnerabilities. Oncogene. (2023) 42:1661–71. doi: 10.1038/s41388-023-02681-y
71. Hua T, Xue Y, Sarker DB, Kiran S, Li Y, and Sang Q-XA. Modeling human brain rhabdoid tumor by inactivating tumor suppressor genes in induced pluripotent stem cells. Bioact Mater. (2024) 31:136–50. doi: 10.1016/j.bioactmat.2023.08.009
72. Gastberger K, Fincke VE, Mucha M, Siebert R, Hasselblatt M, and Frühwald MC. Current molecular and clinical landscape of ATRT – the link to future therapies. Cancer Manag Res. (2023) 15:1369–93. doi: 10.2147/CMAR.S379451
73. Disch A, Melcher V, Walter C, Interlandi M, Moreno N, de Faria FW, et al. ATRT-16. Mechanisms of myeloid cell-induced resistance in AT/RT. Neuro Oncol. (2022) 24:i6–6. doi: 10.1093/neuonc/noac079.015
74. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald M, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors—a reinvestigation and current consensus. Neuro Oncol. (2020) 22:613–24. doi: 10.1093/neuonc/noz235
75. Lobón-Iglesias M-J, Andrianteranagna M, Han Z-Y, Chauvin C, Masliah-Planchon J, Manriquez V, et al. Imaging and multi-omics datasets converge to define different neural progenitor origins for ATRT-SHH subgroups. Nat Commun. (2023) 14:6669. doi: 10.1038/s41467-023-42371-7
76. Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell. (2016) 29:379–93. doi: 10.1016/j.ccell.2016.02.001
77. Merk DJ, Tsiami F, Hirsch S, Walter B, Haeusser LA, Maile JD, et al. Functional screening reveals genetic dependencies and diverging cell cycle control in atypical teratoid rhabdoid tumors. Genome Biol. (2024) 25:301. doi: 10.1186/s13059-024-03438-w
78. Najem H, Khasraw M, and Heimberger AB. Immune microenvironment landscape in CNS tumors and role in responses to immunotherapy. Cells. (2021) 10:2032. doi: 10.3390/cells10082032
79. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, and Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. (2018) 15:325–40. doi: 10.1038/nrclinonc.2018.29
80. Rodriguez SMB, Staicu GA, Sevastre AS, Baloi C, Ciubotaru V, Dricu A, et al. Glioblastoma stem cells—Useful tools in the battle against cancer. Int J Mol Sci. (2022) 23:4602. doi: 10.3390/ijms23094602
81. Prager BC, Bhargava S, Mahadev V, Hubert CG, and Rich JN. Glioblastoma stem cells: driving resilience through chaos. Trends Cancer. (2020) 6:223–35. doi: 10.1016/j.trecan.2020.01.009
82. Landskron G, de la Fuente M, Thuwajit P, Thuwajit C, and Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. (2014) 2014. doi: 10.1155/2014/149185
83. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, and Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
84. Kong Y, Tang L, You Y, Li Q, and Zhu X. Analysis of causes for poor persistence of CAR-T cell therapy. Vivo Front Immunol. (2023) 14:1063454. doi: 10.3389/fimmu.2023.1063454
85. Wittibschlager V, Bacher U, Seipel K, Porret N, Wiedemann G, Haslebacher C, et al. CAR T-cell persistence correlates with improved outcome in patients with B-cell lymphoma. Int J Mol Sci. (2023) 24:5688. doi: 10.3390/ijms24065688
86. López-Cantillo G, Urueña C, Camacho BA, and Ramírez-Segura C. CAR-T cell performance: how to improve their persistence? Front Immunol. (2022) 13:878209. doi: 10.3389/fimmu.2022.878209
87. Tang OY, Tian L, Yoder T, Xu R, Kulikovskaya I, Gupta M, et al. PD1 expression in EGFRvIII-directed CAR T cell infusion product for glioblastoma is associated with clinical response. Front Immunol. (2022) 13:872756. doi: 10.3389/fimmu.2022.872756
88. Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Künkele A, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. (2021) 27:1544–52. doi: 10.1038/s41591-021-01404-8
89. Albert CM, Pinto NR, Taylor M, Wilson A, Rawlings-Rhea S, Mgebroff S, et al. STRIvE-01: Phase I study of EGFR806 CAR T-cell immunotherapy for recurrent/refractory solid tumors in children and young adults. J Clin Oncol. (2025) 40:2541. doi: 10.1200/JCO.2022.40.16_suppl.2541
90. Schakelaar MY, Monnikhof M, Crnko S, Pijnappel EW, Meeldijk J, ten Broeke T, et al. Cellular immunotherapy for medulloblastoma. Neuro Oncol. (2023) 25:617–27. doi: 10.1093/neuonc/noac236
91. Ciccone R, Quintarelli C, Camera A, Pezzella M, Caruso S, Manni S, et al. GD2-targeting CAR T-cell therapy for patients with GD2þ Medulloblastoma. Clin Cancer Res. (2024) 30:2545–57. doi: 10.1158/1078-0432.CCR-23-1880
92. Pal K and Sheth RA. Engineering the tumor immune microenvironment through minimally invasive interventions. Cancers (Basel). (2022) 15:196. doi: 10.3390/cancers15010196
93. Servidei T, Lucchetti D, Navarra P, Sgambato A, Riccardi R, and Ruggiero A. Cell-of-origin and genetic, epigenetic, and microenvironmental factors contribute to the intra-tumoral heterogeneity of pediatric intracranial ependymoma. Cancers (Basel). (2021) 13. doi: 10.3390/cancers13236100
94. Kouro T, Himuro H, and Sasada T. Exhaustion of CAR T cells: potential causes and solutions. J Transl Med. (2022) 20:239. doi: 10.1186/s12967-022-03442-3
95. Liu Y, An L, Huang R, Xiong J, Yang H, Wang X, et al. Strategies to enhance CAR-T persistence. biomark Res. (2022) 10:86. doi: 10.1186/s40364-022-00434-9
96. Pietrobon V, Todd LA, Goswami A, Stefanson O, Yang Z, and Marincola F. Improving CAR T-cell persistence. Int J Mol Sci. (2021) 22:10828. doi: 10.3390/ijms221910828
97. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. (2005) 2:3–14. doi: 10.1602/neurorx.2.1.3
98. Haydar D, Ibañez-Vega J, Crawford J, Children’s SJ, Hospital R, Chou C-H, et al. CAR T-cell design dependent remodeling of the brain tumor immune microenvironment identify macrophages as key players that inhibit or promote anti-tumor activity Children’s National Hospital. (USA: Cancer Research Communications, American Association for Cancer Research) (2023).
99. Perus LJM and Walsh LA. Microenvironmental heterogeneity in brain Malignancies. Front Immunol. (2019) 10:2294. doi: 10.3389/fimmu.2019.02294
100. Rojas-Quintero J, Díaz MP, Palmar J, Galan-Freyle NJ, Morillo V, Escalona D, et al. Car T cells in solid tumors: overcoming obstacles. Int J Mol Sci. (2024) 25:4170. doi: 10.3390/ijms25084170
101. Li YR, Halladay T, and Yang L. Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies. J BioMed Sci. (2024) 31:5. doi: 10.1186/s12929-024-00998-8
102. Sheng H, Li H, Zeng H, Zhang B, Lu Y, Liu X, et al. Heterogeneity and tumoral origin of medulloblastoma in the single-cell era. Oncogene. (2024) 43:839–50. doi: 10.1038/s41388-024-02967-9
103. Mansoori S, Noei A, Maali A, Seyed-Motahari SS, and Sharifzadeh Z. Recent updates on allogeneic CAR-T cells in hematological Malignancies. Cancer Cell Int. (2024) 24:304. doi: 10.1186/s12935-024-03479-y
104. Zhu X, Li Q, and Zhu X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front Cell Dev Biol. (2022) 10:1034257. doi: 10.3389/fcell.2022.1034257
105. Ahn T, Bae E-A, and Seo H. Decoding and overcoming T cell exhaustion: Epigenetic and transcriptional dynamics in CAR-T cells against solid tumors. Mol Ther. (2024) 32:1617–27. doi: 10.1016/j.ymthe.2024.04.004
106. Bulliard Y, Andersson BS, Baysal MA, Damiano J, and Tsimberidou AM. Reprogramming T cell differentiation and exhaustion in CAR-T cell therapy. J Hematol Oncol. (2023) 16:108. doi: 10.1186/s13045-023-01504-7
107. Yin X, He L, and Guo Z. T-cell exhaustion in CAR-T-cell therapy and strategies to overcome it. Immunology. (2023) 169:400–11. doi: 10.1111/imm.13642
108. Silveira CRF, Corveloni AC, Caruso SR, Macêdo NA, Brussolo NM, Haddad F, et al. Cytokines as an important player in the context of CAR-T cell therapy for cancer: Their role in tumor immunomodulation, manufacture, and clinical implications. Front Immunol. (2022) 13:947648. doi: 10.3389/fimmu.2022.947648
109. Zhang M, Kong J, Yin F, Shi J, Li J, Qiu Z, et al. Optimizing CAR-T cell Culture: Differential effects of IL-2, IL-12, and IL-21 on CAR-T cells. Cytokine. (2024) 184:156758. doi: 10.1016/j.cyto.2024.156758
110. Kong Y, Tang L, You Y, Li Q, and Zhu X. Analysis of causes for poor persistence of CAR-T cell therapy in vivo. Front Immunol. (2023) 14:1063454. doi: 10.3389/fimmu.2023.1063454
111. Liu Z, Zhou Z, Dang Q, Xu H, Lv J, Li H, et al. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics. (2022) 12:6273–90. doi: 10.7150/thno.76854
112. Wittling MC, Cole AC, Brammer B, Diatikar KG, Schmitt NC, and Paulos CM. Strategies for improving CAR T cell persistence in solid tumors. Cancers (Basel). (2024) 16:2858. doi: 10.3390/cancers16162858
113. López-Cantillo G, Urueña C, Camacho BA, and Ramírez-Segura C. CAR-T cell performance: how to improve their persistence? Front Immunol. (2022) 13:878209. doi: 10.3389/fimmu.2022.878209
114. Agliardi G, Dias J, Rampotas A, Garcia J, and Roddie C. Accelerating and optimising CAR T-cell manufacture to deliver better patient products. Lancet Haematol. (2025) 12:e57–67. doi: 10.1016/S2352-3026(24)00273-4
115. Gu X, Zhang Y, Zhou W, Wang F, Yan F, Gao H, et al. Infusion and delivery strategies to maximize the efficacy of CAR-T cell immunotherapy for cancers. Exp Hematol Oncol. (2024) 13:70. doi: 10.1186/s40164-024-00542-2
116. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med. (2020) 26:720–31. doi: 10.1038/s41591-020-0827-2
117. Sagnella SM, White AL, Yeo D, Saxena P, van Zandwijk N, and Rasko JEJ. Locoregional delivery of CAR-T cells in the clinic. Pharmacol Res. (2022) 182:106329. doi: 10.1016/j.phrs.2022.106329
118. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ, et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. (2024) 30:1320–9. doi: 10.1038/s41591-024-02893-z
119. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
120. Cherkassky L, Hou Z, Amador-Molina A, and Adusumilli PS. Regional CAR T cell therapy: An ignition key for systemic immunity in solid tumors. Cancer Cell. (2022) 40:569–74. doi: 10.1016/j.ccell.2022.04.006
121. Chen Y, Sun J, Liu H, Yin G, and Xie Q. Immunotherapy deriving from CAR-T cell treatment in autoimmune diseases. J Immunol Res. (2019) 2019:5727516. doi: 10.1155/2019/5727516
122. Diep YN, Kim TJ, Cho H, and Lee LP. Nanomedicine for advanced cancer immunotherapy. J Control Release. (2022) 351:1017–37. doi: 10.1016/j.jconrel.2022.10.004
123. Nawaz W, Xu S, Li Y, Huang B, Wu X, and Wu Z. Nanotechnology and Immunoengineering: How nanotechnology can boost CAR-T therapy. Acta Biomaterialia (2020) 109:21–36. doi: 10.1016/j.actbio.2020.04.015
124. Chang Y, Cai X, Syahirah R, Yao Y, Xu Y, Jin G, et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nat Commun. (2023) 14:2266. doi: 10.1038/s41467-023-37872-4
125. Marei HE. Multimodal targeting of glioma with functionalized nanoparticles. Cancer Cell Int. (2022) 22:265. doi: 10.1186/s12935-022-02687-8
126. Zhou L, Zou M, Xu Y, Lin P, Lei C, and Xia X. Nano drug delivery system for tumor immunotherapy: next-generation therapeutics. Front Oncol. (2022) 12:864301. doi: 10.3389/fonc.2022.864301
127. Kwak K, Yu B, Lewandowski RJ, and Kim DH. Recent progress in cryoablation cancer therapy and nanoparticles mediated cryoablation. Theranostics. (2022) 12:2175–204. doi: 10.7150/thno.67530
128. Saeedi M, Eslamifar M, Khezri K, and Dizaj SM. Applications of nanotechnology in drug delivery to the central nervous system. BioMed Pharmacother. (2019) 111:666–75. doi: 10.1016/j.biopha.2018.12.133
129. Pfister F, Carnell LR, Löffler L, Boosz P, Schaft N, Dörrie J, et al. Loading of CAR-T cells with magnetic nanoparticles for controlled targeting suppresses inflammatory cytokine release and switches tumor cell death mechanism. MedComm (Beijing). (2025) 6:e70039. doi: 10.1002/mco2.70039
130. Banerjee S, Sharma V, and Das Mukhopadhyay C. Exploring emerging concepts of exosomes for the diagnosis, prognosis, and therapeutics of brain cancers. Extracell Vesicle. (2024) 3:100038. doi: 10.1016/j.vesic.2024.100038
131. Lee AA, Godwin AK, and Abdelhakim H. The multifaceted roles of extracellular vesicles for therapeutic intervention with non-Hodgkin lymphoma. Extracell Vesicles Circ Nucl Acids. (2024) 5:329–43. doi: 10.20517/evcna.2024.07
132. Fu W, Lei C, Liu S, Cui Y, Wang C, Qian K, et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun. (2019) 10:4355. doi: 10.1038/s41467-019-12321-3
133. Sani F, Shojaei S, Tabatabaei SA, Khorraminejad-Shirazi M, Latifi M, Sani M, et al. CAR-T cell-derived exosomes: a new perspective for cancer therapy. Stem Cell Res Ther. (2024) 15:174. doi: 10.1186/s13287-024-03783-4
134. Jin Y, Xing J, Xu K, Liu D, and Zhuo Y. Exosomes in the tumor microenvironment: Promoting cancer progression. Front Immunol. (2022) 13:1025218. doi: 10.3389/fimmu.2022.1025218
135. Gong Y, Liu Z, Zhou P, Li J, and Miao YB. Biomimetic nanocarriers harnessing microbial metabolites usher the path for brain disease therapy. Nano TransMed. (2023) 2:100020. doi: 10.1016/J.NTM.2023.100020
136. Qin YT, Li YP, He XW, Wang X, Li WY, and Zhang YK. Biomaterials promote in vivo generation and immunotherapy of CAR-T cells. Front Immunol. (2023) 14:1165576. doi: 10.3389/fimmu.2023.1165576
137. Nguyen DT, Liu R, Ogando-Rivas E, Pepe A, Pedro D, Qdaisat S, et al. Bioconjugated liquid-like solid enhances characterization of solid tumor - chimeric antigen receptor T cell interactions. Acta Biomater. (2023) 172:466–79. doi: 10.1016/j.actbio.2023.09.042
138. Iqbal Z, Rehman K, Xia J, Shabbir M, Zaman M, Liang Y, et al. Biomaterial-assisted targeted and controlled delivery of CRISPR/Cas9 for precise gene editing. Biomater Sci. (2023) 11:3762–83. doi: 10.1039/d2bm01636b
139. Mellati A, Hasanzadeh E, Gholipourmalekabadi M, and Enderami SE. Injectable nanocomposite hydrogels as an emerging platform for biomedical applications: A review. Mater Sci Eng C Mater Biol Appl. (2021) 131:112489. doi: 10.1016/j.msec.2021.112489
140. Suraiya AB, Evtimov VJ, Truong VX, Boyd RL, Forsythe JS, and Boyd NR. Micro-hydrogel injectables that deliver effective CAR-T immunotherapy against 3D solid tumor spheroids. Transl Oncol. (2022) 24:101477. doi: 10.1016/j.tranon.2022.101477
141. Hu Q, Li H, Archibong E, Chen Q, Ruan H, Ahn S, et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat BioMed Eng. (2021) 5:1038–47. doi: 10.1038/s41551-021-00712-1
142. Liu C, Liao Y, Liu L, Xie L, Liu J, Zhang Y, et al. Application of injectable hydrogels in cancer immunotherapy. Front Bioeng Biotechnol. (2023) 11:1121887. doi: 10.3389/fbioe.2023.1121887
143. Liu Y, Liu F, Zeng Y, Lin L, Yu H, Zhang S, et al. Hydrogel systems for spatiotemporal controlled delivery of immunomodulators: engineering the tumor immune microenvironment for enhanced cancer immunotherapy. Front Cell Dev Biol. (2024) 12:1514595. doi: 10.3389/fcell.2024.1514595
144. Cho Y and Doh J. The extracellular matrix in solid tumor immunotherapy. Trends Immunol. (2024) 45:705–14. doi: 10.1016/j.it.2024.07.009
145. Zhao Z, Li Q, Qu C, Jiang Z, Jia G, Lan G, et al. A collagenase nanogel backpack improves CAR-T cell therapy outcomes in pancreatic cancer. Nat Nanotechnol. (2025) 1–11. doi: 10.1038/s41565-025-01924-1
146. Ogunnaike EA, Valdivia A, Yazdimamaghani M, Leon E, Nandi S, Hudson H, et al. Fibrin gel enhances the antitumor effects of chimeric antigen receptor T cells in glioblastoma. Sci Adv. (2021). doi: 10.1126/sciadv.abg5841
147. Spicer PP and Mikos AG. Fibrin glue as a drug delivery system. J Controlled Release. (2010) 148:49–55. doi: 10.1016/j.jconrel.2010.06.025
148. Wang W, Marin-Ramos NI, He H, Zeng S, Cho HY, Swenson SD, et al. NEO100 enables brain delivery of blood–brain barrier impermeable therapeutics. Neuro Oncol. (2021) 23:63–75. doi: 10.1093/neuonc/noaa206
149. Schönthal AH, Peereboom DM, Wagle N, Lai R, Mathew AJ, Hurth KM, et al. Phase i trial of intranasal NEO100, highly purified perillyl alcohol, in adult patients with recurrent glioblastoma. Neurooncol Adv. (2021) 3:1–9. doi: 10.1093/noajnl/vdab005
150. Wang W, Marin-Ramos NI, He H, Zeng S, Cho HY, Swenson SD, et al. NEO100 enables brain delivery of blood brain barrier impermeable therapeutics. Neuro Oncol. (2021) 23:63–75. doi: 10.1093/neuonc/noaa206
151. Wang W, He H, Zheng L, Zeng S, Cho HY, Kouhi A, et al. Enhancing brain entry and therapeutic activity of chimeric antigen receptor T cells with intra-arterial NEO100 in a mouse model of CNS lymphoma. J Neurosurg. (2024) 140:1549–57. doi: 10.3171/2023.10.JNS231097
152. Li J, Chen H, Xu C, Hu M, Li J, and Chang W. Systemic toxicity of CAR-T therapy and potential monitoring indicators for toxicity prevention. Front Immunol. (2024) 15:1422591. doi: 10.3389/fimmu.2024.1422591
153. Bunevicius A, McDannold NJ, and Golby AJ. Focused ultrasound strategies for brain tumor therapy. Oper Neurosurg (Hagerstown). (2020) 19:9–18. doi: 10.1093/ons/opz374
154. Shi G, Zhong M, Ye F, and Zhang X. Low-frequency HIFU induced cancer immunotherapy: tempting challenges and potential opportunities. Cancer Biol Med. (2019) 16:714–28. doi: 10.20892/j.issn.2095-3941.2019.0232
155. Sabbagh A, Beccaria K, Ling X, Marisetty A, Ott M, Caruso H, et al. Opening of the blood–brain barrier using low-intensity pulsed ultrasound enhances responses to immunotherapy in preclinical glioma models. Clin Cancer Res. (2021) 27:4325–37. doi: 10.1158/1078-0432.CCR-20-3760
156. Sheybani ND, Breza VR, Paul S, McCauley KS, Berr SS, Miller GW, et al. ImmunoPET-informed sequence for focused ultrasound-targeted mCD47 blockade controls glioma. J Control Release. (2021) 331:19–29. doi: 10.1016/j.jconrel.2021.01.023
157. Rivera J, Digklia A, Christou AS, Anibal J, Vallis KA, Wood BJ, et al. A review of ultrasound-mediated checkpoint inhibitor immunotherapy. Ultrasound Med Biol. (2024) 50:1–7. doi: 10.1016/j.ultrasmedbio.2023.08.019
158. Arvanitis CD, Askoxylakis V, Guo Y, Datta M, Kloepper J, Ferraro GB, et al. Mechanisms of enhanced drug delivery in brain metastases with focused ultrasound-induced blood-tumor barrier disruption. Proc Natl Acad Sci U.S.A. (2018) 115:E8717–e8726. doi: 10.1073/pnas.1807105115
159. Janwadkar R, Leblang S, Ghanouni P, Brenner J, Ragheb J, Hennekens CH, et al. Focused ultrasound for pediatric diseases. Pediatrics. (2022) 149. doi: 10.1542/peds.2021-052714
160. Alkins R, Burgess A, Ganguly M, Francia G, Kerbel R, Wels WS, et al. Focused ultrasound delivers targeted immune cells to metastatic brain tumors. Cancer Res. (2013) 73:1892–9. doi: 10.1158/0008-5472.Can-12-2609
161. Mungur R, Zheng J, Wang B, Chen X, Zhan R, and Tong Y. Low-intensity focused ultrasound technique in glioblastoma multiforme treatment. Front Oncol. (2022) 12:903059. doi: 10.3389/fonc.2022.903059
162. Chen YH, Moore D, Lee CC, and Su YH. Focused ultrasound for brain metastases: an update on global clinical trials. J Neurooncol. (2023) 165:53–62. doi: 10.1007/s11060-023-04492-3
163. Seas AA, Malla AP, Sharifai N, Winkles JA, Woodworth GF, and Anastasiadis P. Microbubble-enhanced focused ultrasound for infiltrating gliomas. Biomedicines. (2024) 12:1230. doi: 10.3390/biomedicines12061230
164. Arrieta VA, Gould A, Kim KS, Habashy KJ, Dmello C, Vázquez-Cervantes GI, et al. Ultrasound-mediated delivery of doxorubicin to the brain results in immune modulation and improved responses to PD-1 blockade in gliomas. Nat Commun. (2024) 15:4698. doi: 10.1038/s41467-024-48326-w
165. Meng Y, Reilly RM, Pezo RC, Trudeau M, Sahgal A, Singnurkar A, et al. MR-guided focused ultrasound enhances delivery of trastuzumab to Her2-positive brain metastases. Sci Transl Med. (2021) 13:eabj4011. doi: 10.1126/scitranslmed.abj4011
166. Yuan J, Ye D, Chen S, and Chen H. Therapeutic ultrasound-enhanced immune checkpoint inhibitor therapy. Front Phys. (2021) 9:636985. doi: 10.3389/fphy.2021.636985
167. Rivera J, Digklia A, Christou AS, Anibal J, Vallis KA, Wood BJ, et al. A review of ultrasound-mediated checkpoint inhibitor immunotherapy. Ultrasound Med Biol. (2024) 50:1–7. doi: 10.1016/j.ultrasmedbio.2023.08.019
168. Jahangiri S and Yu F. Fundamentals and applications of focused ultrasound-assisted cancer immune checkpoint inhibition for solid tumors. Pharmaceutics. (2024) 16:411. doi: 10.3390/pharmaceutics16030411
169. Eranki A, Srinivasan P, Ries M, Kim A, Lazarski CA, Rossi CT, et al. High-intensity focused ultrasound (HIFU) triggers immune sensitization of refractory murine neuroblastoma to checkpoint inhibitor therapy. Clin Cancer Res. (2020) 26:1152–61. doi: 10.1158/1078-0432.Ccr-19-1604
170. Sanber K, Savani B, and Jain T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Br J Haematol. (2021) 195:660–8. doi: 10.1111/bjh.17544
171. Ando M, Ito M, Srirat T, Kondo T, and Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. (2020) 43:1–9. doi: 10.1080/25785826.2019.1698261
172. Choi G, Shin G, and Bae SJ. Price and prejudice? The value of chimeric antigen receptor (CAR) T-cell therapy. Int J Environ Res Public Health. (2022) 19:12366. doi: 10.3390/ijerph191912366
173. Lonez C and Breman E. Allogeneic CAR-T therapy technologies: has the promise been met? Cells. (2024) 13:146. doi: 10.3390/cells13020146
174. Depil S, Duchateau P, Grupp SA, Mufti G, and Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
175. Li W, Zhu X, Xu Y, Chen J, Zhang H, Yang Z, et al. Simultaneous editing of TCR, HLA-I/II and HLA-E resulted in enhanced universal CAR-T resistance to allo-rejection. Front Immunol. (2022) 13:1052717. doi: 10.3389/fimmu.2022.1052717
176. Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-generation manufacturing protocols enriching TSCM CAR T cells can overcome disease-specific T cell defects in cancer patients. Front Immunol. (2020) 11:1217. doi: 10.3389/fimmu.2020.01217
177. Watanabe N, Mo F, and McKenna MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. (2022) 13:876339. doi: 10.3389/fimmu.2022.876339
178. Song K. Current development status of cytokines for cancer immunotherapy. Biomol Ther (Seoul). (2024) 32:13–24. doi: 10.4062/biomolther.2023.196
179. Coppola C, Hopkins B, Huhn S, Du Z, Huang Z, and Kelly WJ. Investigation of the impact from IL-2, IL-7, and IL-15 on the growth and signaling of activated CD4(+) T cells. Int J Mol Sci. (2020) 21:7814. doi: 10.3390/ijms21217814
180. Li W, Pan X, Chen L, Cui H, Mo S, Pan Y, et al. Cell metabolism-based optimization strategy of CAR-T cell function in cancer therapy. Front Immunol. (2023) 14:1186383. doi: 10.3389/fimmu.2023.1186383
181. Liu S, Zhao Y, Gao Y, Li F, and Zhang Y. Targeting metabolism to improve CAR-T cells therapeutic efficacy. Chin Med J (Engl). (2024) 137:909–20. doi: 10.1097/CM9.0000000000003046
182. Zhang M, Jin X, Sun R, Xiong X, Wang J, Xie D, et al. Optimization of metabolism to improve efficacy during CAR-T cell manufacturing. J Transl Med. (2021) 19:499. doi: 10.1186/s12967-021-03165-x
183. Lindner SE, Johnson SM, Brown CE, and Wang LD. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci Adv. (2020) 6:eaaz3223. doi: 10.1126/sciadv.aaz3223
184. Selli ME, Landmann JH, Terekhova M, Lattin J, Heard A, Hsu YS, et al. Costimulatory domains direct distinct fates of CAR-driven T-cell dysfunction. Blood. (2023) 141:3153–65. doi: 10.1182/blood.2023020100
185. Cappell KM and Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. (2021) 18:715–27. doi: 10.1038/s41571-021-00530-z
186. Duan Y, Chen R, Huang Y, Meng X, Chen J, Liao C, et al. Tuning the ignition of CAR: optimizing the affinity of scFv to improve CAR-T therapy. Cell Mol Life Sci. (2022) 79:14. doi: 10.1007/s00018-021-04089-x
187. Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, et al. Control of the activity of CAR-T cells within tumours via focused ultrasound. Nat BioMed Eng. (2021) 5:1336–47. doi: 10.1038/s41551-021-00779-w
188. Dimitri A, Herbst F, and Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
189. Hosseinkhani N, Derakhshani A, Kooshkaki O, Shadbad MA, Hajiasgharzadeh K, Baghbanzadeh A, et al. Immune checkpoints and car-t cells: The pioneers in future cancer therapies? Int J Mol Sci. (2020) 21:1–28. doi: 10.3390/ijms21218305
190. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
191. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol. (2021) 18:2188–98. doi: 10.1038/s41423-021-00749-x
192. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. (2021) 13. doi: 10.1126/scitranslmed.abh0272
193. Martín-Antonio B, Blanco B, González-Murillo Á, Hidalgo L, Minguillón J, and Pérez-Chacón G. Newer generations of multi-target CAR and STAb-T immunotherapeutics: NEXT CART Consortium as a cooperative effort to overcome current limitations. Front Immunol. (2024) 15:1386856. doi: 10.3389/fimmu.2024.1386856
194. Zhou D, Zhu X, and Xiao Y. Advances in CAR-T therapy for central nervous system tumors. biomark Res. (2024) 12:132. doi: 10.1186/s40364-024-00679-6
195. Schmidts A, Srivastava AA, Ramapriyan R, Bailey SR, Bouffard AA, Cahill DP, et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneous glioblastoma. Neurooncol Adv. (2023) 5:vdac185. doi: 10.1093/noajnl/vdac185
196. van der Schans JJ, van de Donk NWCJ, and Mutis T. Dual targeting to overcome current challenges in multiple myeloma CAR T-cell treatment. Front Oncol. (2020) 10:1362. doi: 10.3389/fonc.2020.01362
197. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. (2021) 13. doi: 10.1126/scitranslmed.abe7378
198. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med. (2021) 13. doi: 10.1126/scitranslmed.abd8836
199. Honikel MM and Olejniczak SH. Co-stimulatory receptor signaling in CAR-T cells. Biomolecules. (2022) 12:1303. doi: 10.3390/biom12091303
200. Abbott RC, Hughes-Parry HE, and Jenkins MR. To go or not to go? Biological logic gating engineered T cells. J Immunother Cancer. (2022) 10:e004185. doi: 10.1136/jitc-2021-004185
201. Li S, Shi L, Zhao L, Guo Q, Li J, Liu ZL, et al. Split-design approach enhances the therapeutic efficacy of ligand-based CAR-T cells against multiple B-cell Malignancies. Nat Commun. (2024) 15:9751. doi: 10.1038/s41467-024-54150-z
202. Nicolai CJ, Parker MH, Qin J, Tang W, Ulrich-Lewis JT, Gottschalk RJ, et al. In vivo CAR T-cell generation in nonhuman primates using lentiviral vectors displaying a multidomain fusion ligand . Available online at: http://ashpublications.org/blood/article-pdf/144/9/977/2240004/blood_bld-2024-024523-main.pdf. (Accessed December 1, 2024).
203. Short L, Holt RA, Cullis PR, and Evgin L. Direct in vivo CAR T cell engineering. Trends Pharmacol Sci. (2024) 45:406–18. doi: 10.1016/j.tips.2024.03.004
204. Bui TA, Mei H, Sang R, Ortega DG, and Deng W. Advancements and challenges in developing in vivo CAR T cell therapies for cancer treatment. EBioMedicine. (2024) 106:105266. doi: 10.1016/j.ebiom.2024.105266
205. Bui TA, Mei H, Sang R, Ortega DG, and Deng W. Advancements and challenges in developing in vivo CAR T cell therapies for cancer treatment (2024). Available online at: www.thelancet.com. (Accessed September 23, 2024).
206. Uslu U, Castelli S, and June CH. CAR T cell combination therapies to treat cancer. Cancer Cell. (2024) 42:1319–25. doi: 10.1016/j.ccell.2024.07.002
207. Al-Haideri M, Tondok SB, Safa SH, Maleki AH, Rostami S, Jalil AT, et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. (2022) 22:365. doi: 10.1186/s12935-022-02778-6
208. Satapathy BP, Sheoran P, Yadav R, Chettri D, Sonowal D, Dash CP, et al. The synergistic immunotherapeutic impact of engineered CAR-T cells with PD-1 blockade in lymphomas and solid tumors: a systematic review. Front Immunol. (2024) 15:1389971. doi: 10.3389/fimmu.2024.1389971
209. He X and Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. (2020) 30:660–9. doi: 10.1038/s41422-020-0343-4
210. Twomey JD and Zhang B. Cancer immunotherapy update: FDA-approved checkpoint inhibitors and companion diagnostics. AAPS J. (2021) 23:39. doi: 10.1208/s12248-021-00574-0
211. Bagley SJ, Binder ZA, Lamrani L, Marinari E, Desai AS, Nasrallah MP, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. (2024) 5:517–31. doi: 10.1038/s43018-023-00709-6
212. Chen Q, Guo X, and Ma W. Opportunities and challenges of CD47-targeted therapy in cancer immunotherapy. Oncol Res. (2024) 32:49–60. doi: 10.32604/or.2023.042383
213. Wang SJ, Dougan SK, and Dougan M. Immune mechanisms of toxicity from checkpoint inhibitors. Trends Cancer. (2023) 9:543–53. doi: 10.1016/j.trecan.2023.04.002
214. Najafi S and Mortezaee K. Modifying CAR-T cells with anti-checkpoints in cancer immunotherapy: A focus on anti PD-1/PD-L1 antibodies. Life Sci. (2024) 338:122387. doi: 10.1016/j.lfs.2023.122387
215. Abdoli Shadbad M, Hemmat N, Khaze Shahgoli V, Derakhshani A, Baradaran F, Brunetti O, et al. A systematic review on PD-1 blockade and PD-1 gene-editing of CAR-T cells for glioma therapy: from deciphering to personalized medicine. Front Immunol. (2022) 12:788211. doi: 10.3389/fimmu.2021.788211
216. Anderson AC, Joller N, and Kuchroo VK. Lag-3, tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
217. Cai L, Li Y, Tan J, Xu L, and Li Y. Targeting LAG-3, TIM-3, and TIGIT for cancer immunotherapy. J Hematol Oncol. (2023) 16:101. doi: 10.1186/s13045-023-01499-1
218. Walsh RJ, Sundar R, and Lim JSJ. Immune checkpoint inhibitor combinations—current and emerging strategies. Br J Cancer. (2023) 128:1415–7. doi: 10.1038/s41416-023-02181-6
219. Bell M and Gottschalk S. Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol. (2021) 12:684642. doi: 10.3389/fimmu.2021.684642
220. Lee EHJ, Murad JP, Christian L, Gibson J, Yamaguchi Y, Cullen C, et al. Antigen-dependent IL-12 signaling in CAR T cells promotes regional to systemic disease targeting. Nat Commun. (2023) 14:4737. doi: 10.1038/s41467-023-40115-1
221. Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, et al. Cytokines in clinical cancer immunotherapy. Br J Cancer. (2019) 120:6–15. doi: 10.1038/s41416-018-0328-y
222. Raggi G, Roldan N, Micallef V, Rapet A, De Maddalena L, Imler T, et al. Interleukin-2 – induced vascular leak syndrome: clinically relevant in vitro recapitulation with a patient-derived lung-on-chip. Eur Respir J. (2020) 56:4326. doi: 10.1183/13993003.congress-2020.4326
223. Brog RA, Ferry SL, Schiebout CT, Messier CM, Cook WJ, Abdullah L, et al. Superkine IL-2 and IL-33 armored CAR T cells reshape the tumor microenvironment and reduce growth of multiple solid tumors. Cancer Immunol Res. (2022) 10:962–77. doi: 10.1158/2326-6066.CIR-21-0536
224. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. (2019) 7:759–72. doi: 10.1158/2326-6066.CIR-18-0466
225. Zannikou M, Duffy JT, Levine RN, Seblani M, Liu Q, Presser A, et al. IL15 modification enables CAR T cells to act as a dual targeting agent against tumor cells and myeloid-derived suppressor cells in GBM. J Immunother Cancer. (2023) 11:e006239. doi: 10.1136/jitc-2022-006239
226. Mirlekar B and Pylayeva-Gupta Y. IL-12 family cytokines in cancer and immunotherapy. Cancers (Basel). (2021) 13:1–23. doi: 10.3390/cancers13020167
227. Agliardi G, Liuzzi AR, Hotblack A, De Feo D, Núñez N, Stowe CL, et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat Commun. (2021) 12:444. doi: 10.1038/s41467-020-20599-x
228. Yang Z, Pietrobon V, Bobbin M, Stefanson O, Yang J, Goswami A, et al. Nanoscale, antigen encounter-dependent, IL-12 delivery by CAR T cells plus PD-L1 blockade for cancer treatment. J Transl Med. (2023) 21:158. doi: 10.1186/s12967-023-04014-9
229. Steven Shen by H, Sampson JH, Sanchez-Perez LA, Abraham SN, and Nair SK. IL-12 CAR T cell immunotherapy for heterogeneous brain tumors. Dissertation, Duke dept. of pathology (2023).
230. Carrara SC, Harwardt J, Grzeschik J, Hock B, and Kolmar H. TriTECM: A tetrafunctional T-cell engaging antibody with built-in risk mitigation of cytokine release syndrome. Front Immunol. (2022) 13:1051875. doi: 10.3389/fimmu.2022.1051875
231. Memari E, Khan D, Alkins R, and Helfield B. Focused ultrasound-assisted delivery of immunomodulating agents in brain cancer. J Controlled Release. (2024) 367:283–99. doi: 10.1016/j.jconrel.2024.01.034
232. Sun S, Tang Q, Sun L, Zhang J, Zhang L, Xu M, et al. Ultrasound-mediated immune regulation in tumor immunotherapy. Mater Today Adv. (2022) 14:100248. doi: 10.1016/j.mtadv.2022.100248
233. Arsiwala TA, Sprowls SA, Blethen KE, Adkins CE, Saralkar PA, Fladeland RA, et al. Ultrasound-mediated disruption of the blood tumor barrier for improved therapeutic delivery. Neoplasia. (2021) 23:676–91. doi: 10.1016/j.neo.2021.04.005
234. Joiner JB, Pylayeva-Gupta Y, and Dayton PA. Focused ultrasound for immunomodulation of the tumor microenvironment. J Immunol. (2020) 205:2327–41. doi: 10.4049/jimmunol.1901430
235. Lee S, Han H, Koo H, Na JH, Yoon HY, Lee KE, et al. Extracellular matrix remodeling in vivo for enhancing tumor-targeting efficiency of nanoparticle drug carriers using the pulsed high intensity focused ultrasound. J Controlled Release. (2017) 263:68–78. doi: 10.1016/j.jconrel.2017.02.035
236. Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, et al. Acoustogenetic control of CAR T cells via focused ultrasound. Nat Biomed Engin. (2020) 5:1336–47. doi: 10.1101/2020.02.18.955005
237. Song Y, Liu Q, Zuo T, Wei G, and Jiao S. Combined antitumor effects of anti-EGFR variant III CAR-T cell therapy and PD-1 checkpoint blockade on glioblastoma in mouse model. Cell Immunol. (2020) 352:104112. doi: 10.1016/j.cellimm.2020.104112
238. Hawkins ER, D’Souza RR, and Klampatsa A. Armored CAR T-cells: the next chapter in T-cell cancer immunotherapy. Biologics. (2021) 15:95–105. doi: 10.2147/btt.S291768
239. Liu K, Cui JJ, Zhan Y, Ouyang QY, Lu QS, Yang DH, et al. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Mol Cancer. (2022) 21:98. doi: 10.1186/s12943-022-01561-5
240. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. (2020) 12. doi: 10.1126/SCITRANSLMED.AAZ1863
241. Aalipour A, Le Boeuf F, Tang M, Murty S, Simonetta F, Lozano AX, et al. Viral delivery of CAR targets to solid tumors enables effective cell therapy. Mol Ther Oncol. (2020) 17:232–40. doi: 10.1016/j.omto.2020.03.018
242. Ponterio E, Haas TL, and De Maria R. Oncolytic virus and CAR-T cell therapy in solid tumors. Front Immunol. (2024) 15:1455163. doi: 10.3389/fimmu.2024.1455163
243. Qin VM, Haynes NM, D’Souza C, Neeson PJ, and Zhu JJ. CAR-T plus radiotherapy: A promising combination for immunosuppressive tumors. Front Immunol. (2022) 12:813832. doi: 10.3389/fimmu.2021.813832
244. Szlasa W, Sztuder A, Kaczmar-Dybko A, Maciejczyk A, and Dybko J. Efficient combination of radiotherapy and CAR-T – A systematic review. Biomed Pharmacother. (2024) 174:116532. doi: 10.1016/j.biopha.2024.116532
245. Abou-El-Enein M, Elsallab M, Feldman SA, Fesnak AD, Heslop HE, Marks P, et al. Scalable manufacturing of CAR T cells for cancer immunotherapy. Blood Cancer Discov. (2021) 2:408–22. doi: 10.1158/2643-3230.BCD-21-0084
246. Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat BioMed Eng. (2022) 6:118–28. doi: 10.1038/s41551-021-00842-6
247. Selim AG, Minson A, Blombery P, Dickinson M, Harrison SJ, and Anderson MA. CAR-T cell therapy: practical guide to routine laboratory monitoring. Pathology. (2021) 53:408–15. doi: 10.1016/j.pathol.2021.02.002
248. Pfeifer R, Henze J, Wittich K, Gosselink A, Kinkhabwala A, Gremse F, et al. A multimodal imaging workflow for monitoring CAR T cell therapy against solid tumor from whole-body to single-cell level. Theranostics. (2022) 12:4834–50. doi: 10.7150/THNO.68966
249. Bulte JWM. Direct versus indirect labeling for chimeric antigen receptor T-cell tracking using PET. Radiology. (2024) 310:e240241. doi: 10.1148/radiol.240241
250. Krebs S, Ponomarev V, Slovin S, and Schöder H. Imaging of CAR T-cells in cancer patients: Paving the way to treatment monitoring and outcome prediction. J Nucl Med. (2019) 60:879–81. doi: 10.2967/jnumed.119.227561
251. Paterson K, Paterson S, Mulholland T, Coffelt SB, and Zagnoni M. Assessment of CAR-T cell-mediated cytotoxicity in 3D microfluidic cancer co-culture models for combination therapy. IEEE Open J Eng Med Biol. (2022) 3:86–95. doi: 10.1109/OJEMB.2022.3178302
252. Kiru L, Zlitni A, Tousley AM, Nicol As Dalton G, Wu W, Lafortune F, et al. In vivo imaging of nanoparticle-labeled CAR T cells. PNAS: Immunology and Inflammation. (2022) doi: 10.1073/pnas.2102363119/-/DCSupplemental
253. Song X, Zhang Y, Lv X, Xu Z, Long Y, Gai Y, et al. Noninvasive longitudinal PET/CT imaging of CAR T cells using PSMA reporter gene. Eur J Nucl Med Mol Imaging. (2024) 51:965–77. doi: 10.1007/s00259-023-06508-6
254. Miyazaki T, Aiyama H, and Ishikawa E. CAR-T cell in vivo tracking method using PET scan with the reporter gene and new investigational tracer [18F] FHBG. Transl Cancer Res. (2017) 6:S1003–5. doi: 10.21037/tcr.2017.06.50
255. Volpe A, Lang C, Lim L, Man F, Kurtys E, Ashmore-Harris C, et al. Spatiotemporal PET imaging reveals differences in CAR-T tumor retention in triple-negative breast cancer models. Mol Ther. (2020) 28:2271–85. doi: 10.1016/j.ymthe.2020.06.028
256. Rubin DB, Danish HH, Ali AB, Li K, Larose S, Monk AD, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain. (2019) 142:1334–48. doi: 10.1093/brain/awz053
257. Brudno JN and Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood. (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
258. Perna F, Parekh S, Diorio C, Smith M, Subklewe M, Mehta R, et al. CAR T-cell toxicities: From bedside to bench, how novel toxicities inform laboratory investigations. Blood Adv. (2024) 8:4348–58. doi: 10.1182/bloodadvances.2024013044
259. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, and Sadelain M. CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
260. Cosenza M, Sacchi S, and Pozzi S. Cytokine release syndrome associated with T-cell-based therapies for hematological Malignancies: Pathophysiology, clinical presentation, and treatment. Int J Mol Sci. (2021) 22:7652. doi: 10.3390/ijms22147652
261. Brudno JN and Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. (2019) 34:45–55. doi: 10.1016/j.blre.2018.11.002
262. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med. (2016) 375:2561–9. doi: 10.1056/nejmoa1610497
263. Wang Z and Han W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomark Res. (2018) 6:4. doi: 10.1186/s40364-018-0116-0
264. Tallantyre EC, Evans NA, Parry-Jones J, Morgan MPG, Jones CH, and Ingram W. Neurological updates: neurological complications of CAR-T therapy. J Neurol. (2021) 268:1544–54. doi: 10.1007/s00415-020-10237-3
265. Gust J, Ponce R, Liles WC, Garden GA, and Turtle CJ. Cytokines in CAR T cell–associated neurotoxicity. Front Immunol. (2020) 11:577027. doi: 10.3389/fimmu.2020.577027
266. Harvey K, Madsen PJ, Smith T, Griffin C, Patterson L, Vitanza NA, et al. Intracranial cannula implantation for serial locoregional chimeric antigen receptor (CAR) T cell infusions in mice. J Visualized Experiments. (2023) e64886. doi: 10.3791/64886
267. Jain MD, Smith M, and Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy . Available online at: http://ashpublications.org/blood/article-pdf/141/20/2430/2051666/blood_bld-2022-017414-c-main.pdf. (Accessed November 9, 2024).
268. Pratta M, Burke L, DiPersio JF, Maziarz RT, Feldman P, Brodeur T, et al. JAK1 inhibition during CAR T-cell treatment does not affect CAR T-cell proliferation, persistence, or function. Blood. (2022) 140:10350–1. doi: 10.1182/BLOOD-2022-169382
269. Mulvey A, Trueb L, Coukos G, and Arber C. Novel strategies to manage CAR-T cell toxicity. Nat Rev Drug Discov. (2025) 1–19. doi: 10.1038/s41573-024-01100-5
270. Quintarelli C, Vera JF, Savoldo B, Attianese GMPG, Pule M, Foster AE, et al. Co-expression of cytokine and suicide genes to enhance the activity and safety of tumor-specific cytotoxic T lymphocytes. Blood. (2007) 110:2793–802. doi: 10.1182/blood-2007-02-072843
271. Huang S, Xu J, Baran N, and Ma W. Advancing the next generation of cancer treatment with circular RNAs in CAR-T cell therapy. Biomed Pharmacother. (2024) 181:117753. doi: 10.1016/j.biopha.2024.117753
Keywords: CAR-T cells, pediatric brain tumors, trafficking, persistence, cellular exhaustion, tumor microenvironment, focused ultrasound, targeted therapy
Citation: Yaacoub S, Vannoy E, Maslova S, Haffey A, Khorsandi K, Sheybani N and Haydar D (2025) CAR-T cell therapy in brain malignancies: obstacles in the face of cellular trafficking and persistence. Front. Immunol. 16:1596499. doi: 10.3389/fimmu.2025.1596499
Received: 19 March 2025; Accepted: 29 May 2025;
Published: 19 June 2025.
Edited by:
Wenxue Ma, University of California, San Diego, United StatesReviewed by:
Sarah Tettamanti, Fondazione Matilde Tettamanti Menotti De Marchi, ItalyZhijin Fan, Sun Yat-sen University, China
Copyright © 2025 Yaacoub, Vannoy, Maslova, Haffey, Khorsandi, Sheybani and Haydar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dalia Haydar, REhBWURBUkBjaGlsZHJlbnNuYXRpb25hbC5vcmc=