 Su-Young Hong1†Kyeong-Sik Cheon2†Ki-Oug Yoo2Hyun-Oh Lee3
Su-Young Hong1†Kyeong-Sik Cheon2†Ki-Oug Yoo2Hyun-Oh Lee3 Kwang-Soo Cho1*Jong-Taek Suh1Su-Jeong Kim1Jeong-Hwan Nam1Hwang-Bae Sohn1Yul-Ho Kim1
Kwang-Soo Cho1*Jong-Taek Suh1Su-Jeong Kim1Jeong-Hwan Nam1Hwang-Bae Sohn1Yul-Ho Kim1- 1Highland Agriculture Research Institute (HARI), National Institute of Crop Science, Rural Development Administration, Pyeongchang, South Korea
- 2Department of Biological Sciences, Kangwon National University, Chuncheon, South Korea
- 3Phygen Genomics Institute, Seongnam, South Korea
The Chenopodium genus comprises ~150 species, including Chenopodium quinoa and Chenopodium album, two important crops with high nutritional value. To elucidate the phylogenetic relationship between the two species, the complete chloroplast (cp) genomes of these species were obtained by next generation sequencing. We performed comparative analysis of the sequences and, using InDel markers, inferred phylogeny and genetic diversity of the Chenopodium genus. The cp genome is 152,099 bp (C. quinoa) and 152,167 bp (C. album) long. In total, 119 genes (78 protein-coding, 37 tRNA, and 4 rRNA) were identified. We found 14 (C. quinoa) and 15 (C. album) tandem repeats (TRs); 14 TRs were present in both species and C. album and C. quinoa each had one species-specific TR. The trnI-GAU intron sequences contained one (C. quinoa) or two (C. album) copies of TRs (66 bp); the InDel marker was designed based on the copy number variation in TRs. Using the InDel markers, we detected this variation in the TR copy number in four species, Chenopodium hybridum, Chenopodium pumilio, Chenopodium ficifolium, and Chenopodium koraiense, but not in Chenopodium glaucum. A comparison of coding and non-coding regions between C. quinoa and C. album revealed divergent sites. Nucleotide diversity >0.025 was found in 17 regions—14 were located in the large single copy region (LSC), one in the inverted repeats, and two in the small single copy region (SSC). A phylogenetic analysis based on 59 protein-coding genes from 25 taxa resolved Chenopodioideae monophyletic and sister to Betoideae. The complete plastid genome sequences and molecular markers based on divergence hotspot regions in the two Chenopodium taxa will help to resolve the phylogenetic relationships of Chenopodium.
Introduction
Chloroplast (cp) is a plant organelle involved in photosynthesis that has originated from an ancestral endosymbiotic cyanobacteria (Cho et al., 2015). This organelle plays a role in photosynthetic carbon fixation, providing essential energy to plants (Raven and Allen, 2003). In angiosperms, the chloroplast genome consists of a circular DNA molecule with quadripartite structure comprised of a pair of inverted repeats (IRs), one large single copy region (LSC), and one small single copy region (SSC; Chaney et al., 2016; Cho et al., 2016; Fu et al., 2016). In addition to a quadripartite structure, the chloroplast genome contains about 100–130 genes with highly conserved order and sequences among most land plants (Smith, 2015). Due to its highly conserved sequence, compact size, lack of recombination, and maternal inheritance, the cp genome has been used for generating genetic markers for phylogenetic classification (Choi et al., 2016; Hu et al., 2016), divergence dating (Krak et al., 2016), and DNA barcoding system for molecular identification (Dong et al., 2012). Especially, low evolutionary rate of the cp genome in taxa that are not very young makes it an ideal system for assessing plant phylogeny (Smith, 2015). Sequencing of the complete cp DNA genome began in 1991 (Taberlet et al., 1991) and until present days, the cp genomes from 1,200 species of algae and plants have been sequenced (http://www.ncbi.nlm.nih.gov/genome/organelle/).
Chenopodium sensu lato belongs to the subfamily Chenopodioideae (Amaranthaceae, Caryophyllales), and it is the second largest and taxonomically complex genus (Rahiminejad and Gornall, 2004). The traditional family Chenopodiaceae comprised about 100 genera and 1,700 species, mainly distributed in temperate and subtropical regions. However, at present, based on molecular evidence, the family is recognized as the subfamily Chenopodioideae within Amaranthaceae and many of its genera are classified within separate subfamilies of the amaranth family (The Angiosperm Phylogeny Group, 2016). Although Chenopodium is considered monophyletic within Chenopodioideae, some researchers reported the genus polyphyletic (Fuentes-Bazan et al., 2012a,b). In addition, taxonomic identification of Chenopodium has been controversial because of the highly polymorphic leaf shape, floral structure, and seed morphology (La Duke and Crawford, 1979; Kurashige and Agrawal, 2005).
Chenopodium species are cultivated worldwide not only as pseudocereals but also as leafy vegetables. Among them, Chenopodium quinoa and Chenopodium album are most important species grown as grain and vegetable crops, respectively. C. album is an important source of vitamins and micronutrients in India (Bhargava et al., 2007), but also one of the worst weeds. Quinoa is an annual plant that originated from the Andean region and whose worldwide cultivation has been increasing rapidly (Jacobsen et al., 2003). Quinoa is recognized as a crop of great value for its high abiotic stress tolerance and high nutritious content (Repo-Carrasco et al., 2003; Choukr-Allah et al., 2016; Filho et al., 2017).
Several recent studies have attempted to elucidate the origin and polyploidization of the genome in C. album, an allohexaploid formed by hybridization between diploid and tetraploid taxa (Krak et al., 2016). The complete nuclear genome sequence of the tetraploid C. quinoa (2n = 4x = 36) was reported at 1.39 gigabases with chromosome specific scale reference genome sequences (Jarvis et al., 2017). In contrast, the chloroplast genome sequence in Chenopodium has remained incomplete until now since only a few reports provide information about chloroplast genes such as the non-coding rpl32-trnL region (Krak et al., 2016) and the rbcL (Kadereit et al., 2003) and matK/trnK genes (Fuentes-Bazan et al., 2012b).
In the present study, we report a high quality complete chloroplast genome sequences of the two important agronomic Chenopodium species, C. album and C. quinoa, obtained with the next generation sequencing technology. In addition, we conducted a comparative genomic analysis using tandem repeats, InDels, simple sequence repeats (SSRs) polymorphism, and genetic diversity to identify valuable markers for DNA barcoding and phylogenetic analysis. Additionally, we developed and applied InDel markers based on the variation in tandem repeats (TRs) copy number in trnI-GAU intron sequence as a possible DNA marker in other species of Chenopodioideae for phylogenetic analysis.
Materials and Methods
Plant Material
Genetic resources of Chenopodium quinoa (8 accessions) were obtained from the National Agrobiodiversity Center of the Rural Development Administration (http://genebank.rda.go.kr), Korea, and cultivated and harvested in the Highland Agriculture Research Institute (800 m above sea level), Pyeongchang, Korea (Table S1). Leaves of C. album and five other Chenopodium species were collected from the specimens deposited at the Kangwon National University Herbarium (KWNU; Table S1).
Chloroplast Genome Sequence Assembly
Total genomic DNA was extracted from ~100 mg of fresh or dry leaves removed from a single plant using a NucleoSpin Plant II kit (Macherey-Nagel, GmbH, Düren, Germany) following manufacturer's instructions. Paired-end libraries of C. quinoa and C. album were constructed with an Illumina Paired-End DNA library Kit (San Diego, CA, USA) according to manufacturer's protocol and sequenced using the Illumina genome analyzer (Hiseq200) platform at Macrogen (http://www.macrogen.com/ko/). The chloroplast (cp) genome assembly was conducted by the de novo assembly protocol (Cho et al., 2015) via the Phyzen bioinformatics pipeline (http://phyzen.com). Briefly, a 500-bp paired-end library (approximate insert size 350–450 bp) generated 9,086,336 reads from C. quinoa and 6,991,000 reads form C. album. Low quality sequences (Phred score < 20) were trimmed using CLC Genomics Workbench (version 6.04; CLC Inc., Arhus Denmark). After trimming, the libraries for C. quinoa and C. album included 8,121,007 and 6,433,359 reads, respectively. Then, the de novo assembly was implemented using the CLC Genome Assembler (http://www.clcbio.com/products/clc-assembly-cell). A total of 1,190,359 and 383,862 reads were aligned and selected using nucmer tool in MUMmer (Delcher et al., 2003) and Spinacia oleracea sequence (NC_002202) as a reference. The draft cp genome contigs were merged into a single contig by joining overlapping terminal sequences of each contig. The extracted cp genomes of C. quinoa and C. album were 152,099 and 152,167 bp, with a mean coverage of 1,840 X and 645 X, respectively. The complete cp genome sequence was annotated using DOGMA (Wyman et al., 2004) and manual editing through comparison with the reported cp genomes of the reference species S. oleracea (NC_002202). Circular maps of the cp genome were generated using OGDraw v1.2 (Lohse et al., 2013).
Comparative Analysis and Divergence Hotspot Identification
mVISTA was used to compare similarities between two Chenopodium species (Mayor et al., 2000). Nucleotide and amino acids diversity was analyzed by BLASTN and BLASTP, and TRs were analyzed using Tandem Repeat Finder (Benson, 1999) with advanced parameters. The alignment parameters, match, mismatch, indels, were set to 2, 7, 7, respectively; the minimum alignment score to report repeats was 50; the minimum length was 6 bp; and the motif identity percent was 100%. The simple sequence repeats were detected using IMEx (www.mcr.org.in/IMEX; Mudunuri and Nagarajaram, 2007) with minimal repeat numbers of 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The substitution rates Ks and Ka were calculated with PAL2NAL (Suyama et al., 2006). Chloroplast genome sequences of two Chenopodium species (C. quinoa and C. album) were aligned using MAFFT (Katoh et al., 2002), and nucleotide diversity (Pi) and the total number of mutations (Eta) were determined using DnaSP (Librado and Rozas, 2009).
Phylogenetic Analysis
For phylogenetic analyses, two datasets were created. One dataset comprised sequences of 59 protein-coding genes from 25 Caryophyllales plants; the ingroup included 1 Aizoaceae, 1 Cactaceae, 11 Caryophyllaceae, and 11 Amaranthaceae, and Fagopyrum tataricum (Polygonaceae) was used as the outgroup (Table S2). The second dataset comprised the trnI-GAU intron sequences of seven Chenopodium species and one outgroup (S. oleracea). The sequences in both data matrices were compiled and aligned with MAFFT (Katoh et al., 2002). The maximum likelihood analyses of both data matrices were performed using RAxML v7.4.2 with 1,000 bootstrap replicates and the GTR+I+G model (Stamatakis, 2006). This substitution model was chosen under Akaike information criterion (AIC) and Akaike information criterion with correction (AICc) in jModeltest v. 2.1.10 (Darriba et al., 2012).
PCR Amplification Using InDel Markers
The total genomic DNA was used for PCR amplification with InDel specific primers (Table S6). The PCR reactions (20 μL) included 10 ng of genomic DNA and the AccuPower PCR PreMix (Bioneer, Daejeon, Korea) consisting of 0.2 U/μL TOP DNA polymerase, 1.5 mM Mg2+, and 250 μM of dNTP mixture with 5 pMol of each primer. The PCR amplification was performed in a thermocycler (ProFlex PCR System, Applied Biosystems, Foster City, CA, USA) using the following cycling parameters: initial denaturation at 94°C for 4 min, followed by 25 cycles of 94°C for 30 s, 65°C for 30 s, and 72°C for 1 min, and a final extension at 72°C for 7 min. The PCR products were analyzed by electrophoresis on 1.8% agarose gels and sequenced by direct sequencing at Bioneer Co. (Daejeon, Korea).
Results
Complete Chloroplast Genome Sequences
The complete cp genome of C. quinoa and C. album consisted of a single circular molecule with quadripartite structure (Figure 1). The size of the C. quinoa and C. album cp genomes was 152,099 bp and 152,167 bp, respectively. They consisted of a pair of IRs (IRa and IRb) 25,205 and 25,193 bp long, respectively, separated by the LSC (83,582 and 83,676 bp), and one SSC (18,107 and 18,105 bp) region (Table 1). The genomes contained 78 coding genes, accounting for 79,115 and 78,930 bp of the C. quinoa and C. album cp genome, respectively; of those, 62, 5, and 11 genes were located in the LSC, IR, and SSC region, respectively (Table S3). The total length of coding sequences (CDS) was 79,115 bp (the average CDS length was 849 bp) in C. quinoa and 78,930 bp (the average CDS length of 847 bp) in C. album. The total number of RNA bases was 11,906 (in C. quinoa) and 11,835 (in C. album), and the overall GC-content was similar in both species, about 37.2%. A sequence inversion was detected in the rbcL-trnV region (about 3.1 kb) compared to the S. oleracea cp genome (Figure S1). The complete cp genomes of C. quinoa and C. album are deposited in the GenBank under the accession numbers KY419706 and KY419707, respectively (Table S2).
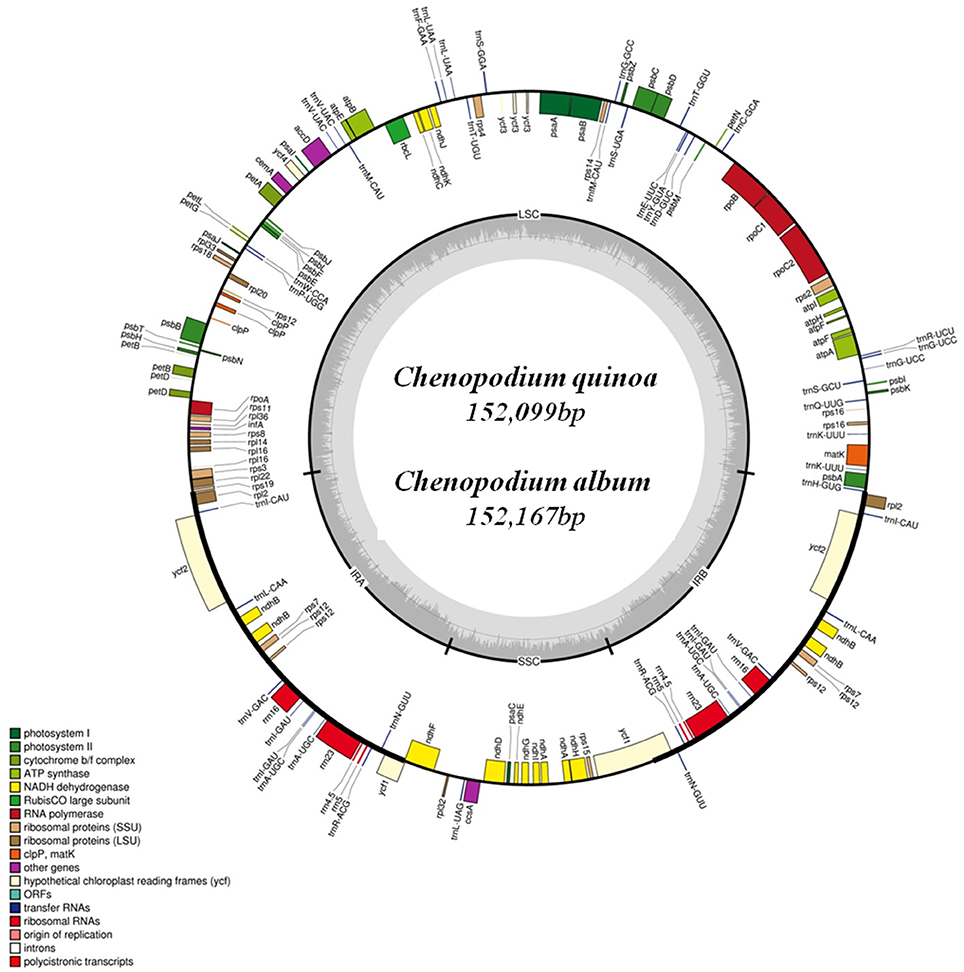
Figure 1. The chloroplast genome map of Chenopodium quinoa and C. album. Genes shown inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise.
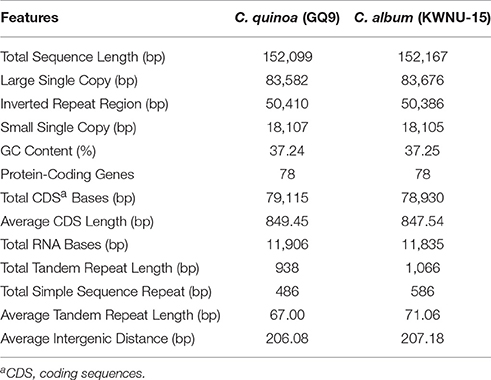
Table 1. Comparison of the complete chloroplast genome between Chenopodium quinoa and C. album.
Gene Contents and Hotspot Region in cp Genomes
The complete cp genomes of C. quinoa and C. album were compared and analyzed. The gene content, order, and orientation in the cp genomes of the two species were similar (Figure 1). The coding regions in both species were highly conserved, except for matK gene with 98.2% homology at the amino acid level (Figure S2; Table S3). The overall identity of nucleotides and amino acid sequences of coding genes was 99.8 and 99.7%, respectively, with the IR region having the lowest identity (Table S3). In general, the IR region is known to be more conservative than the LSC and SSC regions. However, this is a trend when comparing the entire IR region to the entire LSC or SSC regions. In addition, nucleotide diversity of some genes or IGS in the IR region can be higher than that of the LSC or SSC regions (Yang et al., 2016; Park et al., 2017; Song et al., 2017). Due to highly conserved coding regions, the Ka/Ks ratio was very low, approaching zero. However, the Ka/Ks values for some genes, including matK, rps16, rpoC2, ycf1, and ycf 2, were higher (Table S3). The IR/LSC and IR/SSC junction regions were compared to identify the IR expansion or contraction. The rps19, ndhF, ycf1, rpl2, and trnH genes were located in the junctions of the LSC/IRa, IRa/SSC, SSC/IRb, and IRb/LSC regions, respectively; the border position in C. quinoa was the same as that in C. album, which implied no IR expansion or contraction (Figure 2). The coding regions, introns, and intergenic spacer were compared between the two Chenopodium species. The sequence divergence between C. quinoa and C. album ranged from 0 to 0.07865. The IR region was much more conserved compared to the LSC and SSC regions. Seventeen regions, psbK-psbI, psbI-trnS, ycf3-trnS, trnS-rps4, rps4-trnT, trnT-trnL, trnM-trnV, cemA-petA, psbJ-psbL, trnW-trnP, psaJ-rpl33, petD-rpoA, rpl16-rps3, rpl22-rps19, rrn23-rrn4.5, ccsA-ndhD, and rpl32-trnL, showed high levels of sequence variation (exceeding 0.025). Of those, 14 regions were located in the LSC, one in the IR, and two in the SSC (Figure 3; Table S4).
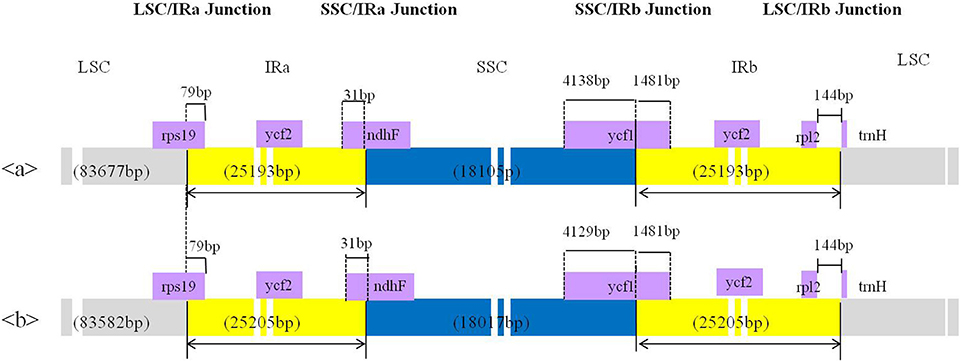
Figure 2. Comparison of the borders of the large single copy (LSC), small single copy (SSC), and inverted repeat (IR) regions of the chloroplast genome between two Chenopodium species. a, Chenopodium album; b, C. quinoa.
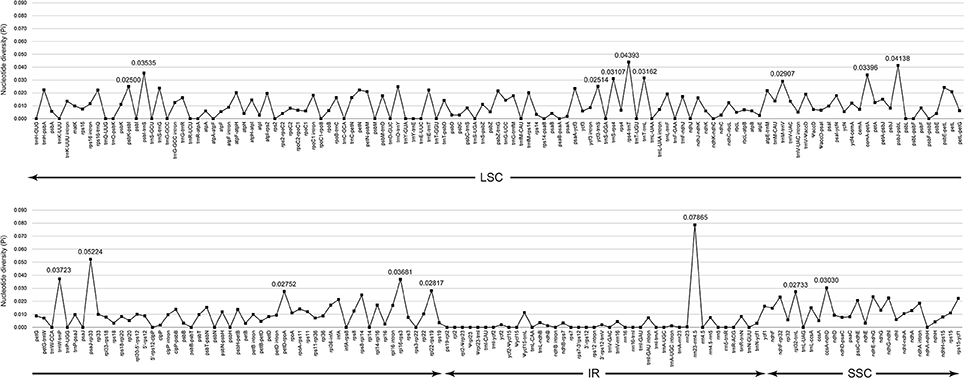
Figure 3. Comparison of the nucleotide diversity (Pi) values between Chenopodium quinoa and C. album.
Tandem Repeats, InDels, and SSR Characteristics
The number, length, and repeat unit of TRs were similar and highly conserved in both species, except for the copy number variation. A total of 14 and 15 TRs, 938 bp and 1,066 bp in length, were identified in the cp genomes of C. quinoa and C. album, respectively (Table 1). The average TR length was 71 bp in C. album, 4 bp longer than that of TRs in C. quinoa. Among TRs, nine TRs were located in the IR, four within the LSC, and three in the SSC region (Table 2) in C. album. One specific TR (24 bp) detected in intergenic sequences between rps12 and petB of the LSC region in C. album was absent in C. quinoa; the two species shared 14 TRs in their cp genomes; one TR (64 bp) was only found in C. quinoa between rrn4.5 and rrn5 intergenic sequences (Table S5). We identified one more copy number in three TRs (TR2, TR8, and TR10) in the C. album cp genome compared to that of C. quinoa (Table 2).

Table 2. Variations in tandem repeat number of chloroplast genome sequences between Chenopodium quinoa and C. album.
Most of the InDels were found in the IR region; two InDels (both longer than 60 bp) in the two species were located in the coding sequences of ycf2 and trnI-GAU and were 90 and 66 bp long, respectively (Table S6). We detected quite an interesting variation in the copy number of the trnI-GAU intron sequence between exon 1 and exon 2. Namely, C. quinoa and C. album had the same copies of TR11, both 95 bp long, whereas C. album had two copies of TR10 within the trnI-GAU intron compared to only one copy in C. quinoa, which accounted for the 66 bp long InDel designated InDel_QA_02 (Figure 4). We designed InDel specific primers to confirm the InDel in the trnI-GAU intron sequence by PCR amplification in both species (Table S6). The size variation of the resulting amplicons showed an exact 66 bp difference between the two species (Figure 4) and dot-plot analysis of the aligned sequences of InDel_QA_02 confirmed a 66 bp InDel in trnI-GAU intron sequences (Figure S3).
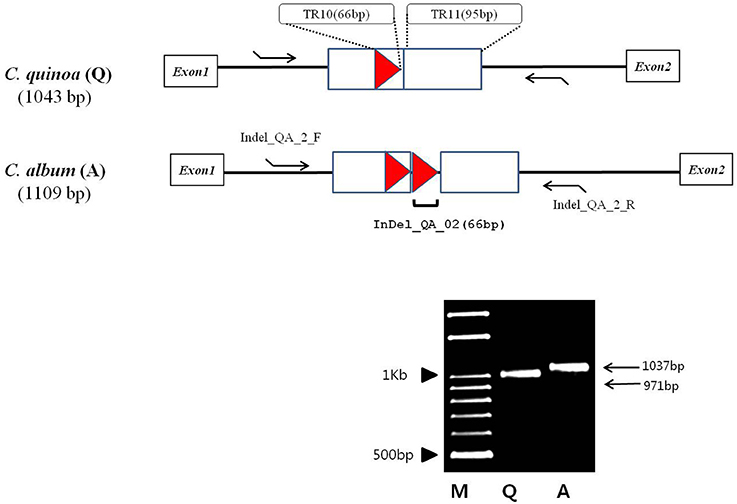
Figure 4. Schematic diagram of the alignment of the Chenopodium quinoa (Q) and C. album (A) trnI-GAU gene sequences. Tandem repeats, 95 and 66 bp long, are designated with a rectangle and a triangle, respectively. Tandem repeat motives and copy numbers are shown in Table S5. InDel_QA_02 primers (Table S6) that amplify the 66 bp tandem repeat region are shown as arrows. M, 100 bp DNA ladder; Q, C. quinoa; A, C. album.
We identified 44 and 53 SSRs in the cp genome of C. quinoa and C. album, respectively (Table S7). The most abundant SSRs motifs were mononucleotides, accounting for about 62 and 66% of the SSRs motifs in C. quinoa and C. album, respectively, and the majority repeat sequence was A/T. A total of 28 SSRs were shared by both species and they were mostly detected in the LSC region, inter-genic sequences, and mononucleotides (Figure 5).
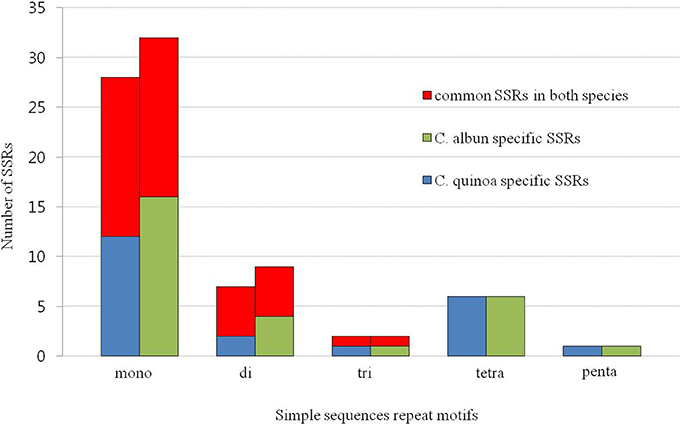
Figure 5. Frequency of simple sequence repeats (SSRs) in the chloroplast genome of two Chenopodium species.
trnI-GAU Intron Sequence Variation in Chenopodioideae
The copy number variation of TRs in trnI-GAU intron sequences among Chenopodioideae was also investigated (Figure 6). The total length of the trnI-GAU intron in eight species, seven Chenopodium species and one outgroup, ranged from 805 bp (S. oleracea) to 1,109 bp (C. album and Chenopodium koraiense); the length of aligned sequences was 996 bp (Table S8; Figure S4). C. album and C. koraiense possessed two copies of TR10 (66 bp), four species (C. quinoa, Chenopodium hybridum, Chenopodium pumilio, Chenopodium ficifolium) had one copy, and Chenopodium glaucum had no TR10 in the trnI-GAU sequences. All Chenopodium species, except for C. glaucum, contained two copies of TR11 (95 bp) in the trnI-GAU sequences (Table 3). The maximum likelihood analysis resolved Chenopodium monophyletic. C. glaucum was the earliest diverging lineage and sister to other species. C. album and C. koraiense formed a clade that was sister to the C. pumilio and C. ficifolium clade. C. quinoa clustered together with C. hybridum in a strongly supported clade (boostrap support = 100; Figure 7).
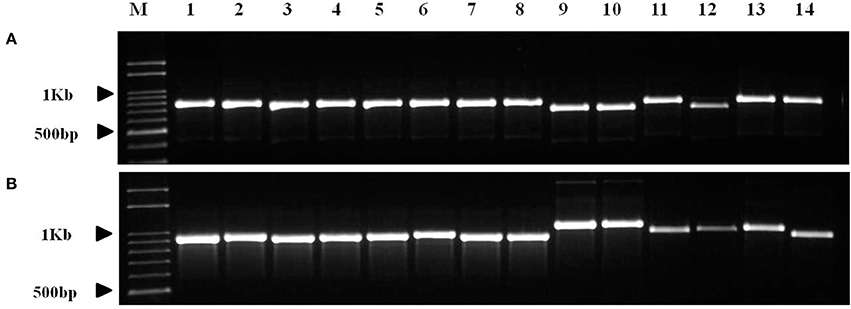
Figure 6. PCR amplification of Chenopodium quinoa germplasm and seven Chenopodium species using InDel markers. (A) InDel_QA_01; (B) InDel_QA_02. Details of the germplasm list are shown in Table S1. 1–8, Chenopodium quinoa; 9, C. album; 10, C. koraiense; 11, C. glaucum; 12, C. ficifolium; 13, C. hybridum; 14, C. pumilio.
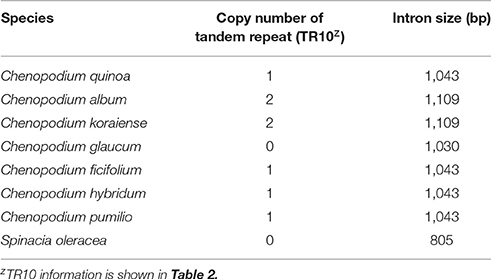
Table 3. Copy number variation of tandem repeats and intron size of trnI-GAU gene in chloroplast genome sequences of the seven Chenopodium taxa with out-group (Spinacia olreacea).
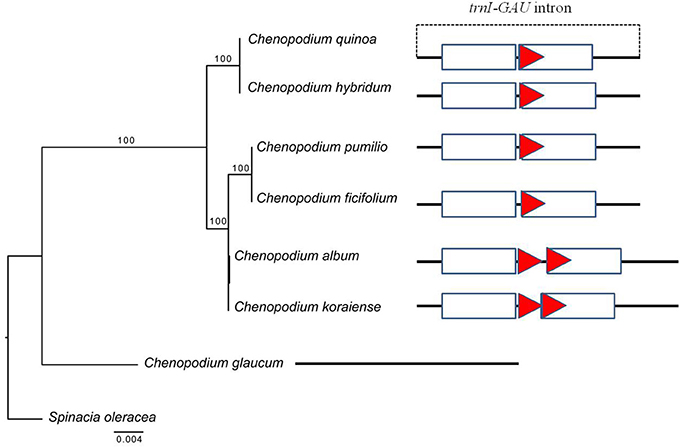
Figure 7. Phylogenetic tree reconstruction and copy number variation of tandem repeats in eight taxa using maximum likelihood analysis based on trnI-GAU sequences. Bootstrap values >50% are given at the nodes. The triangle indicates tandem repeat (66 bp) and sequence information for each taxon is shown in Figure S3. The rectangle represents tandem repeats (95 bp) in the trnI-GAU gene.
Phylogenetic Relationship of 59 Protein-Coding Genes in the cp Genome
The maximum likelihood analysis was conducted based on 59 protein-coding genes from 25 taxa (Figure 8). The length of aligned protein-coding gene sequences was 48,361 bp. In the phylogenetic tree, the Core Caryophyllales were monophyletic and formed four clades. Aizoaceae (Mesembryanthemum crystallinum) occupied the most basal position, followed by Cactaceae (Carnegiea gigantea). In the Caryophyllaceae clade, Alsinoideae (Colobanthus quitensis) were a sister to Caryophylleae. Amaranthaceae formed three subclades: Amaranthoideae (Amaranthus hypochondriacus) were the most basal and sister to the remaining five subfamilies; Salicornioideae, Suaedoideae, and Salsoloideae formed a clade; and Betoideae (Beta vulgaris) was sister to Chenopodioideae. Within Chenopodioideae, the sister relationship between S. oleracea and Chenopodium (C. quinoa and C. album) was highly supported (bootstrap support = 100).
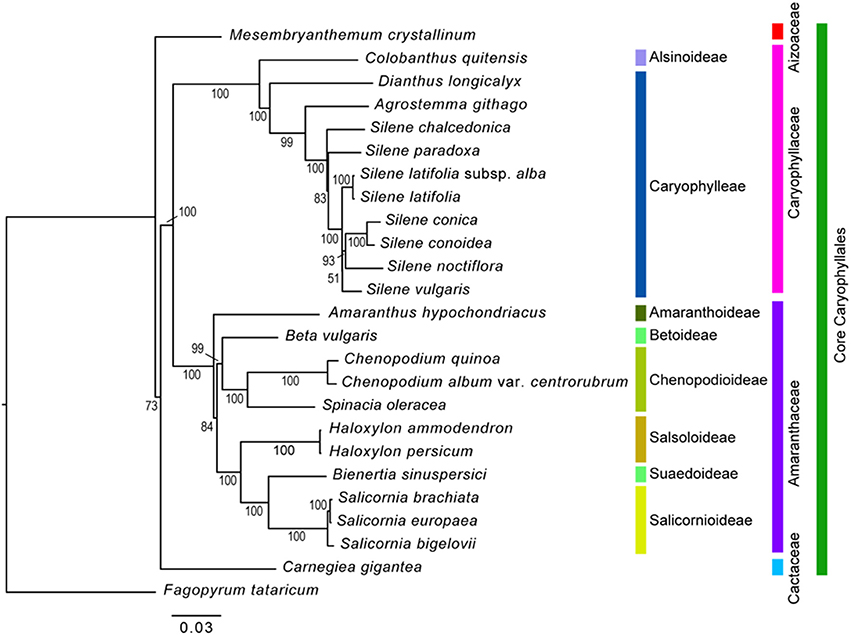
Figure 8. Phylogenetic tree reconstruction of 25 taxa using maximum likelihood based on 59 protein-coding genes. Bootstrap values >50% are given at the nodes.
Discussion
Comparative Analysis of the Chenopodium Chloroplast Genome
The complete cp genome sequences provide valuable information in plant phylogenies due to their highly conserved genome structure and higher evolutionary rate as compared to that of the mitochondrial genome (Chaney et al., 2016). Although, the cp genome has a nearly collinear gene order in most land plants, the changes in the genome such as sequence inversion (Cho et al., 2015), gene loss (Fu et al., 2016), and expansion at the borders of the LSC, SSC, and IR regions (Choi et al., 2016) occur in the course of evolution. We found a 3.1 kb inversion in the rbcL to trnV region of the Chenopodium cp genome when its sequences were compared to the sequences of S. oleracea; this inversion may have been facilitated by tRNA activity (Walker et al., 2014) or by high G + C content (Fullerton et al., 2001). The flanking region of the inversion contained a tRNA gene, including intron sequences with similar G + C content (37.98%), indicating that the 3.1 kb inversion may be promoted by the presence of the tRNA. The border regions between two IR regions and the SSC region have contributed to genome size variation by expansion or contraction among land plants (Cho and Park, 2016; Hu et al., 2016; Ni et al., 2016). Although, the genome size differs between C. album and C. quinoa, the results of the present study revealed that the junction areas were highly conserved.
Repeat sequences such as TRs and SSRs play an important role in the rearrangement and stabilization of cp genome sequences (Vieira et al., 2014) and the copy number variation in different species, even in the same species (Kim et al., 2015), which characteristics render them suitable molecular markers for authentication (Cho et al., 2015, 2016) and phylogenetic analysis (Yang et al., 2013; Williams et al., 2016). The occurrence of the repeats is more prevalent in the intergenic sequence than it is in the CDS, which was also confirmed in this study (Table 2; Table S7). TRs and SSRs are possibly related to cp genome size variation and divergence because of the recombination (Ogihara et al., 1988; Marshall et al., 2001). In this study, the SSRs and TRs were prevalent in the LSC region and contributed to 68 bp longer genome of C. album compared to that of C. quinoa.
Divergence Region of the Chenopodium Chloroplast Genome
In previous molecular phylogenetic studies, Chenopodium formed a polyphyletic group and phylogenetic relationships of some of the taxa were unclear (Kadereit et al., 2003, 2010; Fuentes-Bazan et al., 2012b). These studies were based on the ITS sequences of the nuclear ribosomal DNA and trnL-trnF, matK-trnK, atpB, atpB-rbcL, and rbcL sequences of the cp genome. In the present study, the nucleotide diversity of the cp regions was relatively low (trnL-trnF, 0.01918; matK, 0.00982; trnK-UUU intron, 0.01359; atpB, 0.00601; atpB-rbcL, 0.00689; rbcL, 0.00493). Based on our study, high sequence divergence was detected in the following regions: psbK-psbI, psbI-trnS, ycf3-trnS, trnS-rps4, rps4-trnT, trnT-trnL, trnM-trnV, cemA-petA, psbJ-psbL, trnW-trnP, psaJ-rpl33, petD-rpoA, rpl16-rps3, rpl22-rps19, rrn23-rrn4.5, ccsA-ndhD, and rpl32-trnL (Figure 3; Table S4). Therefore, these regions are considered useful markers for elucidating the phylogenetic relationship within Chenopodium. However, when selecting suitable molecular markers, the length of amplified regions must also be considered. The length of nine regions, psbI-trnS, trnM-trnV, psbJ-psbL, trnW-trnP, petD-rpoA, rpl16-rps3, rpl22-rps19, rrn23-rrn4.5, and ccsA-ndhD, is considered relatively short and insufficient to reproduce the nucleotide variation in various taxa. In contrast, the remaining eight regions (psbK-psbI, ycf3-trnS, trnS-rps4, rps4-trnT, trnT-trnL, cemA-petA, psaJ-rpl33, and rpl32-trnL) are judged suitable for phylogenetic analysis of Chenopodium and helpful to evaluate unresolved phylogenetic relationships.
Intron Sequence Variation in Chenopodium Species
Introns in cp genomes are generally conserved, but structural variations such as sequence loss or variations (SNP), have been reported in several species. Structural intron variation is known to occur in ATP synthetase (atpF), RNA polymerase (rpoC2), and ribosomal proteins (rpl2, rps12, and rps16; Daniell et al., 2016; He et al., 2017). Introns have important roles in gene expression regulation by alternative splicing or stabilization of transcripts and they are gained or lost over evolutionary time (Daniell et al., 2008). Intron variations are also often implemented in phylogenetic and evolutionary analyses. In the present study, we identified 10 proteins and 6 tRNAs with introns in cp genes (Table S3). Although intron sequence variation such as transversion, transition, and small InDels (3–10 bp) have been reported in proteins (Cho et al., 2016; Devi and Chrungoo, 2017), the present study is the first report of the variations in TR copy number in tRNA introns. The changes in highly conserved cp genes have been used to resolve phylogenetic relationships in angiosperm families. To test whether our findings can be applied in phylogenetic analysis, we investigate the copy number variation of the trnI-GAU intron in other Chenopodium species in Korea. All the seven Chenopodium species, except C. glaucum, contained the same TR motifs and copy number variations. These results implied that trnI-GAU intron sequences provide valuable information about Chenopodium phylogenetic relationships. Additional studies should examine whether the copy number variation is present in other Chenopodium species and explore other properties such as transcript stability of the cp genome among different Chenopodium species.
Comparison of Phylogenetic Relationships with Previous Studies
The results of the phylogenetic analysis using 59 protein-coding genes of 24 Core Caryophyllales species and one outgroup resulted in a well-resolved topology in which the monophyly of the tested families and subfamilies was supported. However, our results showed a slight difference from the APG IV system (The Angiosperm Phylogeny Group, 2016). Specifically, Aizoaceae were placed in the most basal clade and Cactaceae formed a sister clade to Caryophyllaceae and Amaranthaceae. In contrast, Caryophyllaceae and Amaranthaceae are in a clade sister to other two families in the APG IV system. In addition, the phylogenetic relationships among Amaranthaceae species in the present study did not corroborate the results of the previous study based on rbcL sequences (Kadereit et al., 2010): (1) Amaranthoideae formed a basal clade within the Amaranthaceae; (2) Betoideae were sister to Chenopodioideae, but they formed an unresolved paraphyletic clade in the previous study; and (3) Chenopodioideae were more closely related to Betoideae, instead to Salsoloideae, Suaedoideae, and Salicornioideae reported in the previous study. We believe that these differences are due to increased resolution resulting from the addition of more gene regions. However, the present study analyzed a limited number of species. Therefore, further studies should include various species to further elucidate the phylogenetic relationships of Caryophyllales and Amaranthaceae.
Author Contributions
SH and JS conceived the design of the study, analyzed the data and drafted the manuscript. KC and HL performed the bioinformatics work. KY collected and identified samples. SK, JN, HS, YK grew and collected samples of Chenopodium quinoa germplasm in HARI. KC was responsible for data analysis and writing of the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01135402),” Rural Development Administration, Republic of Korea.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.01696/full#supplementary-material
Figure S1. BLASTZ analysis of Chenopodium quinoa chloroplast genome against Spinacia oleracea (NC_002202) chloroplast sequences. The inversion region is delimited with the red rectangular line. Blue and yellow bars indicate contigs matching the reference sequence in forward and reverse orientation, respectively.
Figure S2. Comparison of the chloroplast genomes between Chenopodium quinoa and C. album using mVISTA LAGAN program. Blue block: conserved gene; sky blue: tRNA and rRNA; red block: intergenic region. White regions indicate sequence divergence between two chloroplast sequences.
Figure S3. Dot-plot analysis and sequence comparison of InDel_QA_02 region between Chenopodium quinoa and C. album. The Indel_QA_02 region is shown in Figure 4. Tandem repeats are underlined. C. album has two tandem repeat units, whereas C. quinoa has one unit.
Figure S4. ClustalW alignment of trnI-GAU gene intron sequences of the chloroplast genome from seven Chenopodium species.
Abbreviations
CDS, coding sequences; cp, chloroplast; IRs, inverted repeats; LSC, large single copy region; SSRs, simple sequence repeats.
References
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bhargava, A., Shukla, S., and Ohri, D. (2007). Evaluation of foliage yield and leaf quality traits in Chenopodium spp. in multiyear trials. Euphytica 153, 199–213. doi: 10.1007/s10681-006-9255-8
Chaney, L., Mangelson, R., Ramaraj, T., Jellen, E. N., and Maughan, P. J. (2016). The complete chloroplast genome sequences for four Amaranthus species (Amaranthaceae). Appl. Plant Sci. 4:1600063. doi: 10.3732/apps.1600063
Cho, K.-S., and Park, T.-H. (2016). Complete chloroplast genome sequence of Solanum nigrum and development of markers for the discrimination of S. nigrum. Horticult. Environ. Biotechnol. 57, 69–78. doi: 10.1007/s13580-016-0003-2
Cho, K.-S., Cheon, K.-S., Hong, S.-Y., Cho, J.-H., Im, J.-S., Mekapogu, M., et al. (2016). Complete chloroplast genome sequences of Solanum commersonii and its application to chloroplast genotype in somatic hybrids with Solanum tuberosum. Plant Cell Rep. 35, 2113–2123. doi: 10.1007/s00299-016-2022-y
Cho, K. S., Yun, B. K., Yoon, Y. H., Hong, S. Y., Mekapogu, M., Kim, K. H., et al. (2015). Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum) and comparative analysis with common buckwheat (F. esculentum). PLoS ONE 10:e0125332. doi: 10.1371/journal.pone.0125332
Choi, K. S., Chung, M. G., and Park, S. (2016). The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Front. Plant Sci. 7:355. doi: 10.3389/fpls.2016.00355
Choukr-Allah, R., Rao, N. K., Hirich, A., Shahid, M., Alshankiti, A., Toderich, K., et al. (2016). Quinoa for marginal environments: toward future food and nutritional security in MENA and central asia regions. Front. Plant Sci. 7:346. doi: 10.3389/fpls.2016.00346
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. doi: 10.1186/s13059-016-1004-2
Daniell, H., Wurdack, K. J., Kanagaraj, A., Lee, S. B., Saski, C., and Jansen, R. K. (2008). The complete nucleotide sequence of the cassava (Manihot esculenta) chloroplast genome and the evolution of atpF in Malpighiales: RNA editing and multiple losses of a group II intron. Theor. Appl. Genet. 116, 723–737. doi: 10.1007/s00122-007-0706-y
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Delcher, A. L., Salzberg, S. L., and Phillippy, A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10; Unit: 10.3. doi: 10.1002/0471250953.bi1003s00
Devi, R. J., and Chrungoo, N. K. (2017). Evolutionary divergence in Chenopodium and validation of SNPs in chloroplast rbcL and matk genes by allele-specific PCR for development of Chenopodium quinoa-specific markers. Crop J. 5, 32–42. doi: 10.1016/j.cj.2016.06.019
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7:e35071. doi: 10.1371/journal.pone.0035071
Filho, A. M. M., Pirozi, M. R., Borges, J. T. D. S., Pinheiro Sant'Ana, H. M., Chaves, J. B. P., and Coimbra, J. S. D. R. (2017). Quinoa: nutritional, functional, and antinutritional aspects. Crit. Rev. Food Sci. Nutr. 57, 1618–1630. doi: 10.1080/10408398.2014.1001811
Fu, P. C., Zhang, Y. Z., Geng, H. M., and Chen, S. L. (2016). The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 4:e2540. doi: 10.7717/peerj.2540
Fuentes-Bazan, S., Mansion, G., and Borsch, T. (2012a). Towards a species level tree of the globally diverse genus Chenopodium (Chenopodiaceae). Mol. Phylogenet. Evol. 62, 359–374. doi: 10.1016/j.ympev.2011.10.006
Fuentes-Bazan, S., Uotila, P., and Borsch, T. (2012b). A novel phylogeny-based generic classification for Chenopodium sensu lato, and a tribal rearrangement of Chenopodioideae (Chenopodiaceae). Willdenowia 42, 5–24. doi: 10.3372/wi.42.42101
Fullerton, S. M., Bernardo Carvalho, A., and Clark, A. G. (2001). Local rates of recombination are positively correlated with GC content in the human genome. Mol. Biol. Evol. 18, 1139–1142. doi: 10.1093/oxfordjournals.molbev.a003886
He, L., Qian, J., Li, X., Sun, Z., Xu, X., and Chen, S. (2017). Complete chloroplast genome of medicinal plant lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 22:E249. doi: 10.3390/molecules22020249
Hu, Y., Woeste, K. E., and Zhao, P. (2016). Completion of the chloroplast genomes of five chinese juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Jacobsen, S. E., Mujica, A., and Jensen, C. R. (2003). The resistance of quinoa (Chenopodium quinoaWilld.) to adverse abiotic factors. Food Rev. Int. 19, 99–109. doi: 10.1081/FRI-120018872
Jarvis, D. E., Ho, Y. S., Lightfoot, D. J., Schmockel, S. M., Li, B., Borm, T. J., et al. (2017). The genome of Chenopodium quinoa. Nature 542, 307–312. doi: 10.1038/nature21370
Kadereit, G., Borsch, T., Weising, K., and Freitag, H. (2003). Phylogeny of Amaranthaceae and Chenopodiaceae and the evolution of C4 photosynthesis. Int. J. Plant Sci. 164, 959–986. doi: 10.1086/378649
Kadereit, G., Mavrodiev, E. V., Zacharias, E. H., and Sukhorukov, A. P. (2010). Molecular phylogeny of Atripliceae (Chenopodioideae, Chenopodiaceae): implications for systematics, biogeography, flower and fruit evolution, and the origin of C4 photosynthesis. Am. J. Bot. 97, 1664–1687. doi: 10.3732/ajb.1000169
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kim, K., Lee, S.-C., Lee, J., Lee, H. O., Joh, H. J., Kim, N.-H., et al. (2015). Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng Species. PLoS ONE 10:e0117159. doi: 10.1371/journal.pone.0117159
Krak, K., Vít, P., Belyayev, A., Douda, J., Hreusová, L., and Mandák, B. (2016). Allopolyploid origin of Chenopodium album s. str. (Chenopodiaceae): a molecular and cytogenetic insight. PLoS ONE 11:e0161063. doi: 10.1371/journal.pone.0161063
Kurashige, N. S., and Agrawal, A. A. (2005). Phenotypic plasticity to light competition and herbivory in Chenopodium album (Chenopodiaceae). Am. J. Bot. 92, 21–26. doi: 10.3732/ajb.92.1.21
La Duke, J., and Crawford, D. J. (1979). Character compatibility and phyletic relationships in several closely related species of chenopodium of the Western United States. Taxon 28, 307–314. doi: 10.2307/1219738
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Mayor, C., Brudno, M., Schwartz, J. R., Poliakov, A., Rubin, E. M., Frazer, K. A., et al. (2000). VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16, 1046–1047. doi: 10.1093/bioinformatics/16.11.1046
Marshall, H. D., Newton, C., and Ritland, K. (2001). Sequence-repeat polymorphisms exhibit the signature of recombination in lodgepole pine chloroplast DNA. Mol. Biol. Evol. 18, 2136–2138. doi: 10.1093/oxfordjournals.molbev.a003757
Mudunuri, S. B., and Nagarajaram, H. A. (2007). IMEx: Imperfect Microsatellite Extractor. Bioinformatics 23, 1181–1187. doi: 10.1093/bioinformatics/btm097
Ni, L., Zhao, Z., Xu, H., Chen, S., and Dorje, G. (2016). The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the sino-himalayan subregion. Gene 577, 281–288. doi: 10.1016/j.gene.2015.12.005
Ogihara, Y., Terachi, T., and Sasakuma, T. (1988). Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. U.S.A. 85, 8573–8577.
Park, I., Kim, W. J., Yeo, S. M., Choi, G., Kang, U. M., Piao, R., et al. (2017). The complete chloroplast genome sequences of Fritillaria ussuriensis maxim. and Fritillaria cirrhosa D. don, and comparative analysis with other Fritillaria species. Molecules 22:982. doi: 10.3390/molecules22060982
Rahiminejad, M. R., and Gornall, R. J. (2004). Flavonoid evidence for allopolyploidy in the Chenopodium album aggregate (Amaranthaceae). Plant Sys. Evol. 246, 77–87. doi: 10.1007/s00606-003-0108-9
Raven, J. A., and Allen, J. F. (2003). Genomics and chloroplast evolution: what did cyanobacteria do for plants? Genome Biol. 4, 209. doi: 10.1186/gb-2003-4-3-209
Repo-Carrasco, R., Espinoza, C., and Jacobsen, S. E. (2003). Nutritional value and use of the andean crops Quinoa (Chenopodium quinoa) and Ka-iwa (Chenopodium pallidicaule). Food Rev. Int. 19, 179–189. doi: 10.1081/FRI-120018884
Smith, D. R. (2015). Mutation rates in plastid genomes: they are lower than you might think. Genome Biol. Evol. 7, 1227–1234. doi: 10.1093/gbe/evv069
Song, Y., Wang, S., Ding, Y., Xu, J., Li, M. F., Zhu, S., et al. (2017). Chloroplast genome resource of Paris for species discrimination. Sci. Rep. 7, 3427. doi: 10.1038/s41598-017-02083-7
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612. doi: 10.1093/nar/gkl315
Taberlet, P., Gielly, L., Pautou, G., and Bouvet, J. (1991). Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 17, 1105–1109. doi: 10.1007/BF00037152
The Angiosperm Phylogeny Group (2016). An update of The Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Vieira, L. D. N., Faoro, H., Rogalski, M., Fraga, H. P. D. F., Cardoso, R. L. A., de Souza, E. M., et al. (2014). The complete chloroplast genome sequence of podocarpus lambertii: genome structure, evolutionary aspects, gene content and SSR detection. PLoS ONE 9:e90618. doi: 10.1371/journal.pone.0090618
Walker, J. F., Zanis, M. J., and Emery, N. C. (2014). Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 101, 722–729. doi: 10.3732/ajb.1400049
Williams, A. V., Miller, J. T., Small, I., Nevill, P. G., and Boykin, L. M. (2016). Integration of complete chloroplast genome sequences with small amplicon datasets improves phylogenetic resolution in Acacia. Mol. Phylogenet. Evol. 96, 1–8. doi: 10.1016/j.ympev.2015.11.021
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Keywords: Chenopodioideae, chloroplast genome, phylogenetic tree, InDel, tandem repeats
Citation: Hong S-Y, Cheon K-S, Yoo K-O, Lee H-O, Cho K-S, Suh J-T, Kim S-J, Nam J-H, Sohn H-B and Kim Y-H (2017) Complete Chloroplast Genome Sequences and Comparative Analysis of Chenopodium quinoa and C. album. Front. Plant Sci. 8:1696. doi: 10.3389/fpls.2017.01696
Received: 29 May 2017; Accepted: 15 September 2017;
Published: 06 October 2017.
Edited by:
Jun Yu, Beijing Institute of Genomics, ChinaReviewed by:
Yingjuan Su, Sun Yat-sen University, ChinaPerla Hamon, Institute of Research for Development, France
Copyright © 2017 Hong, Cheon, Yoo, Lee, Cho, Suh, Kim, Nam, Sohn and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kwang-Soo Cho, a3NjaG9sb3ZlQGtvcmVhLmty
†These authors have contributed equally to this work.