 Laia Capilla-PerezVictor SolierVirginie PortemerAurelie ChambonAurelie Hurel
Laia Capilla-PerezVictor SolierVirginie PortemerAurelie ChambonAurelie Hurel Alexia GuillebauxDaniel VezonLaurence CromerMathilde Grelon
Alexia GuillebauxDaniel VezonLaurence CromerMathilde Grelon Raphael Mercier*
Raphael Mercier*- CNRS, Institut Jean-Pierre Bourgin, INRA, AgroParisTech, Université Paris-Saclay, Versailles, France
Genetic screens have been crucial for deciphering many important biological processes, including meiosis. In Arabidopsis thaliana, previous forward screens have likely identified almost all the meiotic genes that when mutated lead to a pronounced decrease in fertility. However, the increasing number of genes identified in reverse genetics studies that play crucial roles in meiosis, but do not exhibit strong phenotypes when mutated, suggests that there are still many genes with meiotic function waiting to be discovered. In this study, we produced 897 A. thaliana homozygous mutant lines using Ethyl Methyl Sulfonate (EMS) mutagenesis followed by either single seed descent or haploid doubling. Whole genome sequencing of a subset of lines showed an average of 696 homozygous mutations per line, 195 of which (28%) modify a protein sequence. To test the power of this library, we carried out a forward screen looking for meiotic defects by observing chromosomes at metaphase I of male meiosis. Among the 649 lines analyzed, we identified 43 lines with meiotic defects. Of these, 21 lines had an obvious candidate causal mutation, namely a STOP or splicing site mutation in a gene previously shown to play a role in meiosis (ATM, MLH3, MLH1, MER3, HEI10, FLIP, ASY4, FLIP, PRD2, REC8, FANCL, and PSS1). Interestingly, this was the first time that six of these genes were identified in a forward screen in Arabidopsis (MLH3, MLH1, SGO1, PSS1, FANCL, and ASY4). These results illustrate the potential of this mutant population for screening for any qualitative or quantitative phenotype. Thus, this new mutant library is a powerful tool for functional genomics in A. thaliana. The HEM (Homozygote EMS Mutants) lines are available at the Versailles Arabidopsis stock center.
Introduction
Genetic screens have improved our knowledge of the genetic basis of biological processes. However, to identify all the genes involved in a biological pathway screening strategies must be sensitive, specific and feasible. In this context, the two classic approaches, which are still widely used, are forward and reverse genetics. On one hand, forward genetics can be used to decipher specific pathways without a priori knowledge of the genes involved. This is achieved by screening the phenotype of a population with random mutations throughout the genome, which alter gene function and thus give an identifiable phenotype. On the other hand, the principle of reverse genetics is to test the function of a specific candidate gene by analysing the consequences of its disruption on the process of interest.
Different types of chemical, biological and physical mutagens have been used to generate mutant collections for forward and reverse genetics (reviewed in Page and Grossniklaus, 2002; Nawaz and Shu, 2014). Ethyl Methyl Sulfonate (EMS) is the most common mutagen as it is very easy to use and can produce very high mutagenesis rates compared with other methods (reviewed in Sikora et al., 2011). EMS has an alkylating effect that mainly induces point mutations with G/C to T/A transitions, as seen for example in rice (Till et al., 2007), maize (Till et al., 2004), and Arabidopsis (McCallum et al., 2000; Martín et al., 2009). Point mutations have the potential to not only produce loss-of-function mutants but also weak or separation-of-function alleles (e.g., Séguéla-Arnaud et al., 2015). Thus analysis of these mutants can be used to functionally characterize essential genes.
Meiosis is a specialized cell division where two rounds of chromosome segregation follow one round of DNA replication leading to the production of haploid cells that are essential for sexual reproduction. The list of genes described to be involved in meiosis has steadily increased as a result of various genetic screens (see Mercier et al., 2015 and references therein, 2015). Arabidopsis first emerged as a model in the late 90s when T-DNA insertion lines with meiotic defects were first characterized (Peirson et al., 1997). Subsequently, large-scale forward screens in Arabidopsis (e.g., De Muyt et al., 2009) identified meiotic mutants by looking for mutant lines with reduced fertility as a result of meiotic defects. This strategy led to the description of a number of genes involved in meiosis, however, it was also biased toward genes whose disruption produces very pronounced meiotic defects. More recently, meiotic genes were identified in suppressor screens, for example by observing fertility restoration in zmm mutants (Crismani et al., 2012; Girard et al., 2014; Séguéla-Arnaud et al., 2015; Fernandes et al., 2018). The phenotypes of these mutants consist of an increase in crossover number, which is not easily observable at a macroscopic level, preventing identification in forward screens.
In parallel, an increasing number of important meiotic genes have been identified from reverse genetics screens such as MLH1 (Dion et al., 2007), MLH3 (Jackson et al., 2006), SGO1 (Cromer et al., 2013; Zamariola et al., 2013), PSS1 (Duroc et al., 2014), PCH2 (Lambing et al., 2015), and RPA1 (Osman et al., 2009), among others. These mutants are characterized by subtle fertility defects and have not yet been identified in any forward screens. This therefore suggests that a number of meiotic genes, whose inactivation leads to subtle meiotic defects, could have been missed in previous forward screens based on reduced fertility observable at a macroscopic level.
Here, we produced a total of 897 homozygous Arabidopsis thaliana EMS mutant lines with >170,000 mutations leading to changes in protein sequences and identified meiotic defects in 43 lines. These results demonstrate the usefulness of these HEM (Homozygote EMS Mutant) lines that can be used to detect either qualitative or quantitative phenotypes. Thus this new mutant collection is a very useful resource for functional genomics and applied research in A. thaliana.
Results
Generation of the HEM Lines: Two Collections of Homozygous Arabidopsis thaliana EMS Mutants
To produce the HEM lines we generated two collections of almost fully homozygous lines using two different strategies: (i) single seed descent (SSD) and (ii) doubled haploids (DH) (Figures 1A,B).
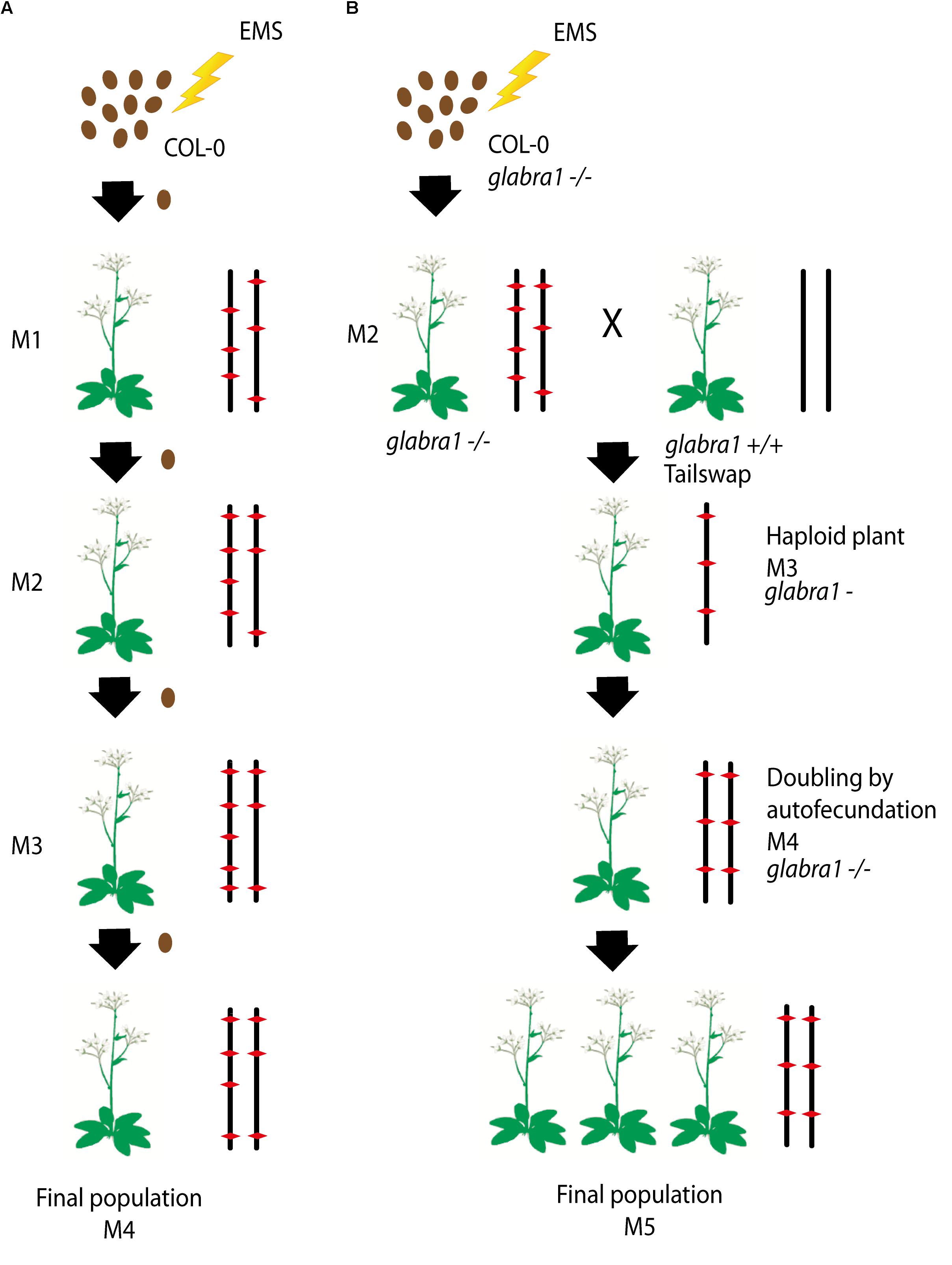
FIGURE 1. (A,B) Schematic diagram of the approaches used to generate each subset of the homozygous EMS mutant lines: (i) Single seed descent (SSD) (A) and (ii) doubled haploids (DH) (B). (A) For the SSD population, a set of Col-0 seeds were mutagenized by EMS and four different generations were grown from them by selecting a single seed in each generation. (B) The DH lines were obtained by mutagenizing a set of Col-0 GALBRA-1 seeds. M2 were then crossed to the Tailswap strain and the haploids of the next generation were selected by the absence of trichomes produced by the GLABRA- 1 mutation. Diploids were then obtained by spontaneous doubling from self-fertilization of the M3 plants. Chromosomes representing the genetic constitution (EMS mutations represented by a red diamond) are displayed for each generation.
For the SSD subset we applied EMS to wild type Col-0 seeds and produced four successive generations by self-fertilization using a single seed in each generation (SSD) (Figure 1A). Six parallel rounds of mutagenesis followed by SSD were carried out, giving a total of 698 independent mutant lines (Table 1). With this approach, we expected to obtain a level of homozygosity of 87% in the 4th generation (M4) that was screened (see below). The M5 seeds are available at the Versailles Arabidopsis stock center.
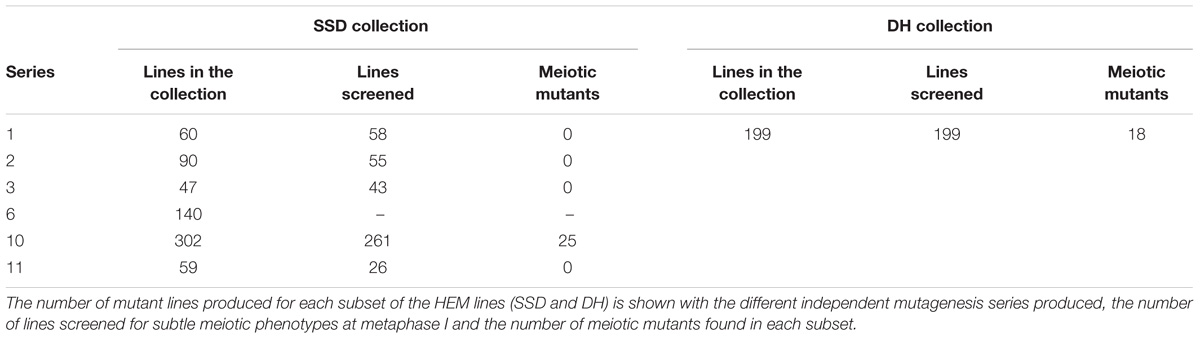
TABLE 1. The different series of independent mutagenesis carried out to generate each of the HEM subsets.
For the second population, the strategy was to generate homozygous mutagenized lines by haploidization followed by genome doubling (Doubled haploid, DH) (Figure 1B). We applied EMS to Col-0 seeds with a homozygous mutation in the GALBRA-1 gene (GL1), which is characterized by the absence of trichomes (Marks and Feldmann, 1989). The first generation plants (M1) were selfed and the next generation (M2) was crossed to a haploid inducing strain (the CENH3 Tailswap line) to obtain haploid descendants (Ravi and Chan, 2010). M3 haploid plants were visually identified due to the absence of trichomes conferred by the gl1 mutation (Portemer et al., 2015) and reproduced by self-fertilization, which spontaneously produced diploid seeds (M4). Using this approach, we expected to obtain virtually completely homozygous mutant lines. Finally, M4 seeds were propagated to obtain a total of 199 M5 independent lines (Figure 1B).
Analysis of Mutation Frequencies in the HEM Lines
To estimate the number of mutations in the HEM populations, we sequenced 47 mutant lines using Illumina: 25 SSD lines (series 10) and 22 DH lines. Among these, 41 lines showed a meiotic defect (see section below) and 6 lines without meiotic defects were randomly chosen among the DH lines.
The HEM populations showed a total mean number of 897 mutations per line, ranging from 500 to 1,500, with a normal distribution (Figure 2A). Of these 897 mutations, 99% were G > A or C > T transitions. When this value was compared for each of the subsets obtained, the SSD lines had more mutations (1,003 mutations per line) than the DHs (with 776 mutations per line; T-test, p < 0.05∗; Figure 2B and Table 2).
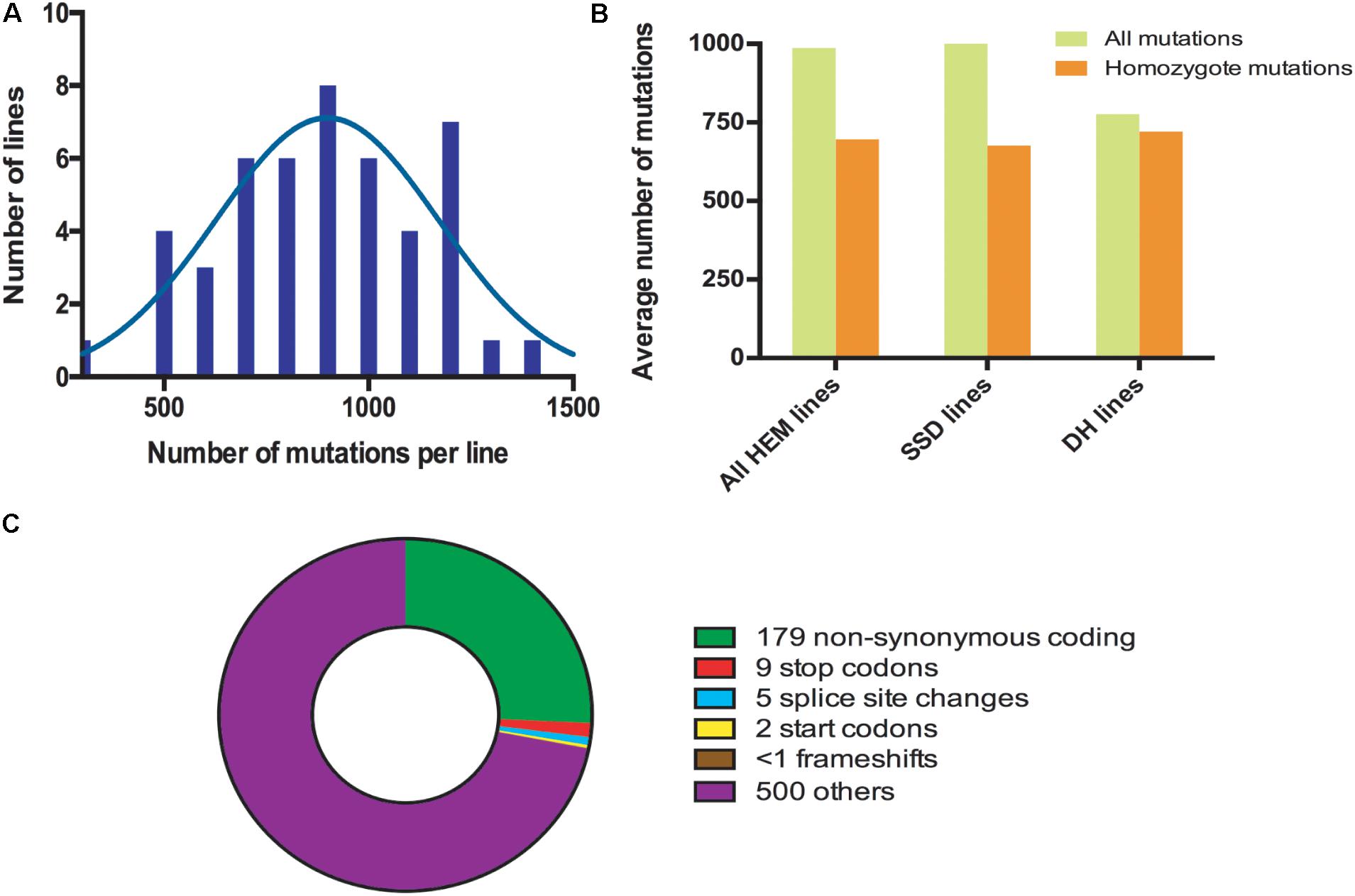
FIGURE 2. (A) Distribution frequencies of the total number of mutations per HEM line. (B) The average total number of and homozygous mutations per line, comparing the HEM lines and each of the subsets (SSDs and DHs). (C) Average number of each type of mutation predicted to change the sequence and/or knockout a protein in one HEM line. “Others” category includes: synonymous coding, intergenic, intragenic, intron, and synonymous stop mutations.

TABLE 2. Number of mutations detected in the HEM lines and in the SSD and DH subsets.
The percentage of fixed mutations, was different for each of the subsets due to the different approaches used (SSD and DH; Figure 2B and Table 2): in the SSD lines 70% of the mutations were fixed (676 mutations per line on average), which was lower than the 87% expected for those lines, perhaps due to counter-selection. In the case of the DH lines, we obtained an average of 94% homozygous mutations (720 mutations per line on average). The six randomly chosen lines had a similar number of mutations than the lines having a meiotic phenotype (690 vs. 697 mutations per line, respectively).
Considering that the DH lines were produced by doubling the genome of haploid plants, we expected complete fixation of the mutations and this is what we observed in 16 of the 22 lines analyzed (>95% of detected mutation). However, in six lines the percentage of homozygous mutations was 80% (on average) suggesting that the haploidization was not successful or that cross-pollination occurred during their production. Regardless, these lines still show a very high level of homozygosity, equivalent to that in the SSD lines.
In all the HEM lines (SSD and DH), 28% of the fixed mutations (an average of 195 mutations per line) modified a protein sequence (Figure 2C and Table 2). This category includes: non-synonymous coding mutations that change a single AA (179 mutations per line, on average); small indels leading to frame shifts (<1 per line, on average); new stop codons (nine per line, on average); new start codons (two per line, on average) and splice site changes (five per line, on average). Considering only the mutations resulting in frameshifts, new stop codons or splice site changes, 14 mutations per line should severely disrupt gene functions (Figure 2C and Table 2).
In summary, when a single line of the HEM library is screened, the effect of 195 homozygous mutations causing an amino acid change can be examined, at least 14 of which are predicted to knock out the function of the protein.
A Screen for Subtle Meiotic Defects
The HEM populations were then tested in a forward genetic screen targeting subtle meiotic phenotypes with the aim of identifying new genes involved in the meiotic process. For this we used two different approaches: (i) Alexander staining to detect dead pollen grains (Alexander, 1969) and then observations of meiotic chromosomal behavior (using chromosome spreads) in the selected lines, or (ii) direct observation of meiotic chromosomal spreads without a pre-screen.
All of the 199 M5 mutant lines from the DH subset were first pre-screened by Alexander staining. The meiotic chromosomal behavior was then examined in lines with more than 10% dead pollen grains. In the case of the SSD lines, we screened 539 M4 mutants (77% of the total 698 lines produced) by directly observing meiotic chromosomal behavior at metaphase I. Of these, the lines with a minimum of 20 cells at metaphase I captured were considered as screened, which resulted in a total of 450 mutant lines. For each of the mutants with a meiotic defect at metaphase I, we verified that the same phenotype was observed in the next generation. To optimize the screening procedure, we focused on metaphase I: (i) cells at that stage are relatively easy to find and (ii) most meiotic defects can be detected at metaphase I (e.g., modifications in crossover number or distribution, chromosome alignment defects and DSB repair defects). A drawback is that some defects cannot be observed at that stage (e.g., premature loss of cohesion, meiosis II spindle defects, cell cycle defects) and would be missed in this screen. However, we occasionally detected meiosis II defects that were included in the study.
We identified a total of 43 lines with various meiotic defects (18 DH lines and 25 SSD lines), representing 9% (18/199) of the screened DH lines and 6% (25/450) of the SSD lines. However, we observed an important difference among the six different series of mutagenesis used to produce the SSD subset (Table 1): 10% of the lines (25/261; Table 1) had meiotic defects in series 10, whereas no meiotic defects were observed in the other series produced (1–3, 6 and 11; 0/189). This variability could reflect differences in the efficiency of the mutagenesis due to slight variations in experimental conditions (e.g., room temperature, age of the seeds…) that may influence the final outcome of EMS mutagenesis. Therefore, the high number of meiotic mutants observed in the SSD series 10 and the HD lines suggests that these series are especially suitable for carrying out other forward genetic screens.
Overall, after screening 80% of the HEM lines the rate of meiotic mutant was high, with 9.4% of the lines showing different types of meiotic phenotypes in the DH and SSD series 10 (43 mutants with a robust meiotic phenotype among 199 DH lines + 261 series 10 SSD lines). The phenotypes described in the 43 HEM lines identified cover a variety of meiotic defects at metaphase I, compared to wild type (Figure 3A): (i) Different levels of fragmentation (suggesting a failure to complete the recombination process and leading in some cases to reduced fertility; observed in 10 lines; Figure 3B), (ii) bivalent shape defects (observed in five lines; Figure 3C), (iii) the presence of univalent chromosomes at different frequencies suggesting a lack of crossovers (ranging from 0.1 to 6 pair of univalent chromosomes per cell; observed in 26 lines; Figures 3D,E), and (iv) bivalent alignment abnormalities (observed in 2 lines; Figure 3F).
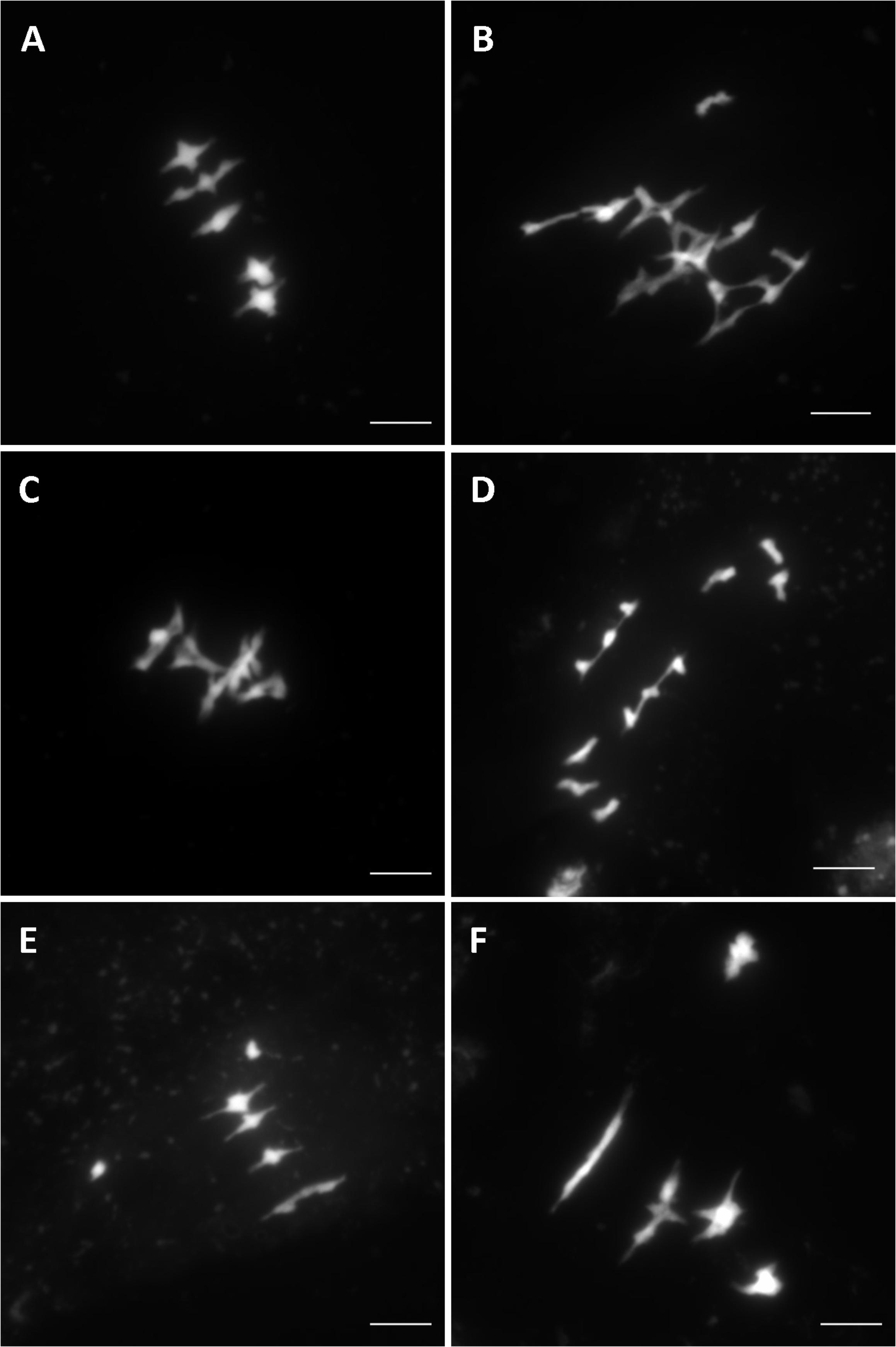
FIGURE 3. Different meiotic phenotypes observed in the HEM lines at metaphase I showing: (A) Wild type meiosis (B) Chromosome fragmentation, (C) Bivalent shape defects, (D,E) Different levels of univalent chromosomes (ranging from a mean of 6 to 0.1 per cell) and (F) Bivalent alignment defects.
Identification of Candidate Causal Mutations
Of the 41 lines sequenced with meiotic defects, 18 had an obvious candidate mutation (Table 3). We considered a candidate mutation to be causal when the mutation was predicted to strongly affect a protein (i.e., stop codon, frame shift or a splice site change) with a described role in meiosis. Additionally, the observed phenotype had to be consistent with the previously described phenotype. An additional missense mutation in ATM was shown to be causal by genetic mapping (Table 3). In all these lines, the presence of the candidate mutation was confirmed by Sanger sequencing. In addition, Sanger sequencing of the candidate gene SGO1 in two lines that showed premature loss of sister chromatid cohesion identified a stop and a splice site mutation (lines HDGem3 and HD479; Table 3).
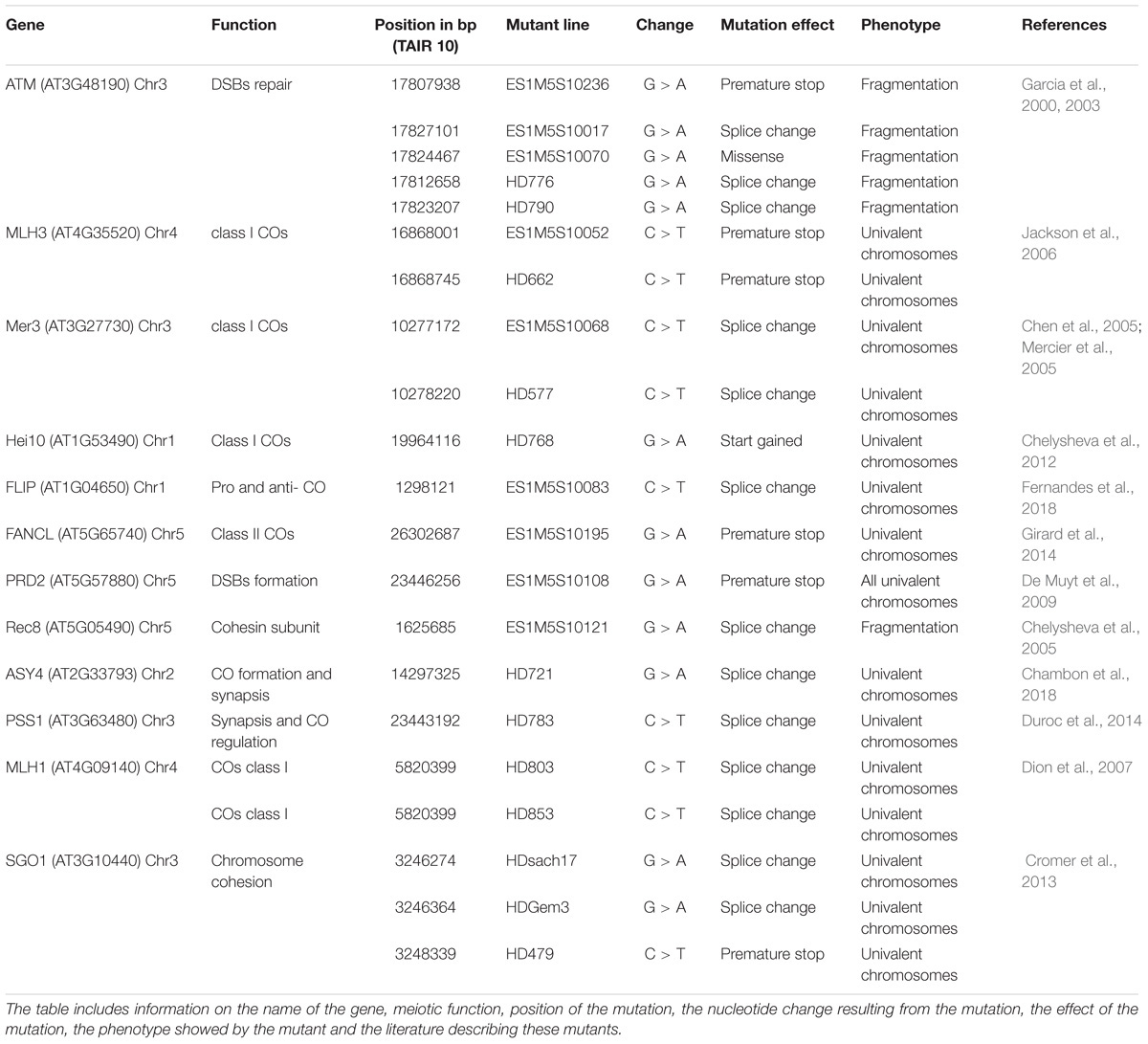
TABLE 3. List of the genes identified as candidate causal mutations in the HEM lines.
The genes described in the 21 lines are involved in a wide range of meiotic mechanisms: ATM (found in five different mutant lines) plays a role in DSBs repair (Garcia et al., 2000, 2003); MLH1, MLH3, MER3 (each identified in two different mutant lines), and HEI10 are involved in class I crossover formation (Mercier et al., 2005; Jackson et al., 2006; Dion et al., 2007; Chelysheva et al., 2012), FLIP is both an anti- and pro-crossover factor (Fernandes et al., 2018); FANCL promotes crossover formation (Girard et al., 2014); PRD2 is required for DSBs formation (De Muyt et al., 2009); REC8 and SGO1 (found in 3 different mutant lines) are both involved in sister chromatid cohesion (Chelysheva et al., 2004; Cromer et al., 2013); ASY4 is involved in crossover and synaptonemal complex formation (Chambon et al., 2018); and PSS1 plays a role in chromosome synapsis and crossover distribution (Duroc et al., 2014; Table 3). Interestingly, most of these mutations have only a mild impact on fertility (e.g., MLH1, MLH3, ASY4, FANCL, or PSS1).
In addition, among the 43 lines identified with meiotic phenotypes, in 22 there was no obvious candidate mutation, according to the criteria described above. These lines displayed different meiotic phenotypes and further work is needed to identify their causal mutation.
Discussion
We have described the HEM collections, two libraries of almost fully homozygous Arabidopsis EMS mutants. These mutants show a high mutation rate per line, 897 mutations per line on average, of which most are fixed. Thus, due to the fixed nature of the mutations, these libraries can be used to repeatedly screen for a specific phenotype and therefore, to analyze either quantitative or qualitative traits.
Of all the mutations produced, we estimate that the HEM lines contain, >170,000 mutations (195 per line, on average) with an effect in the protein sequence. Among these >12,000 (14 per line, on average) are mutations that likely knock out the protein’s function (new stop codons, splice site changes and frameshifts).
In this study, the HEM lines were used in a forward screen targeting subtle meiotic phenotypes as a new approach to identify novel meiotic mutants. Nine Percent of the mutant lines screened in the DH and SSD series 10 collections showed defects in meiosis (43 lines), which is a direct evidence of the efficiency of mutagenesis in the HEM lines.
Within these mutants, there are 12 clear candidate genes (ATM, MLH3, MLH1, MER3, HEI10, SGO1, ASY4, FLIP, FANCL, PRD2, REC8, and PSS1) involved in different meiotic mechanisms. Interestingly, six of these identified genes (MLH3, MLH1, SGO1 PSS1, FANCL, and ASY4) have been found here for the first time in a forward genetic screen. These mutants have only moderate defects in chromosome distribution at meiosis, leading to a subtle reduction in fertility, which is under the threshold of detection by visual examination of fruit length.
In addition, the finding that in 22 mutant lines there is no obvious causal mutation among the previously described meiotic genes, suggests that these lines may be mutated in novel meiotic genes that will require genetic mapping to be identified. Thus, these results are a proof of concept and support the usefulness of the HEM lines to decipher various biological processes. The two collections are available at the Versailles Arabidopsis stock center.
Materials and Methods
EMS Mutagenesis and Plant Growth
To generate the single seed descent (SSD) collection, we applied ethyl methanesulfonate (EMS) to wild type A. thaliana accession Col-0 as described in (Portemer et al., 2015). Seeds were incubated for 17 h at room temperature with gentle agitation in 5 mL of 0.3% (v/v) EMS. Neutralization was performed by adding 5 mL of sodium thiosulfate 1 M for 5 min. Three milliliter of water was added to make the seeds sink. The supernatant was removed and the seeds were washed three times for 20 min with 15 mL of water. Mutagenized seeds were grown and carried through to the fourth generation using only one seed each time. The M4 seeds were used to screen for meiotic defects.
To generate the DH collection, mutagenesis was performed as in the SSD in Col-0 plants with an existing T-DNA insertion in GLABRA1 (GL1). Mutagenized seeds were grown and then crossed as male to the tailswap line (TS) to obtain haploid plants that could be identified due to their lack of trichomes. Diploids were obtained by self-fertilization. M4 seeds were multiplied to obtain the final mutant population of the collection. All plants were cultivated in greenhouses with a 16 h/day and 8 h/night photoperiod, at 20°C and 70% humidity.
Plant Phenotyping
Alexander staining for pollen viability was performed as described in Alexander (1969). Meiotic chromosomal spreads were prepared and stained with DAPI as described in Ross et al. (1996). Observations were made using a Zeiss Axio Observer epifluorescence microscope and photographs were taken using an AxioCam MRm (Zeiss) camera driven by ZEN 2 Software (Carl Zeiss Microscopy, GmbH). Plots and statistical analysis were made using the GraphPad software Prism61.
Whole Genome Sequencing and Mutation Analysis
Genome sequencing was performed with Illumina Hiseq3000 HWIJ00115 with > 8X coverage. The resulting fastq files were analyzed using the Mutdetect pipeline (version 0.0.6-e3ef10e) 1http://www.graphpad.com
(Girard et al., 2015) using TAIR10 COL-0 genome as the reference genome. The FileMatch package was used to eliminate false positives by comparing each muther two mutant lines as controls. Mutations were considered after quality filtering (>80) and the presence of 0 or only one read with wild type allele was considered to indicate a homozygous mutations. Additionally, to differentiate real mutations from false positives, we compared the total number of reads with the coverage, discarding mutations that showed unmapped reads as a proxy for repetitive regions. The sequencing raw data of fully characterized lines is available in the sequence read archive at NCBI SRA accession: SRP156100) and we encourage future users of the collection to do the same.
Author Contributions
LC-P, VP, MG, and RM contributed to the conception and design of the study. LC-P, VS, AC, AH, AG, VP, DV, and LC performed the experiments. LC and RM analyzed the sequencing data. LC-P wrote the first draft of the manuscript.
Funding
The authors thank Rijk Zwaan for funding this study (Xmas project). The IJPB benefits from the support of the LabEx Saclay Plant Sciences-SPS (ANR-10-LABX-0040-SPS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Versailles Arabidopsis Resource center for technical support.
Footnotes
References
Alexander, M. P. (1969). Differential staining of aborted and nonaborted pollen. Stain Technol. 44, 117–122. doi: 10.3109/10520296909063335
Chambon, A., West, A., Vezon, D., Horlow, C., De Muyt, A., Chelysheva, L., et al. (2018). Identification of ASYNAPTIC4, a component of the meiotic chromosome axis. Plant Physiol. 178, 233–246. doi: 10.1104/pp.17.01725
Chelysheva, L., Diallo, S., Vezon, D., Gendrot, G., Vrielynck, N., Belcram, K., et al. (2004). AtREC8 and AtSCC3 are essential to the monopolar orientation of the kinetochores during meiosis. J. Cell Sci. 117, 4017–4023. doi: 10.1242/jcs.01352
Chelysheva, L., Diallo, S., Vezon, D., Gendrot, G., Vrielynck, N., Belcram, K., et al. (2005). AtREC8 and AtSCC3 are essential to the monopolar orientation of the kinetochores during meiosis. J. Cell Sci. 118, 4621–32. doi: 10.1242/jcs.02583
Chelysheva, L., Vezon, D., Chambon, A., Gendrot, G., Pereira, L., Lemhemdi, A., et al. (2012). The Arabidopsis HEI10 is a new ZMM protein related to Zip3. PLoS Genet. 8:e1002799. doi: 10.1371/journal.pgen.1002799
Chen, C., Zhang, W., Timofejeva, L., Gerardin, Y., and Ma, H. (2005). The Arabidopsis ROCK-N-ROLLERS gene encodes a homolog of the yeast ATP-dependent DNA helicase MER3 and is required for normal meiotic crossover formation. Plant J. 43, 321–334. doi: 10.1111/j.1365-313X.2005.02461.x
Crismani, W., Girard, C., Froger, N., Pradillo, M., Santos, J. L., Chelysheva, L., et al. (2012). FANCM limits meiotic crossovers. Science 336, 1588–1590. doi: 10.1126/science.1220381
Cromer, L., Jolivet, S., Horlow, C., Chelysheva, L., Heyman, J., De Jaeger, G., et al. (2013). Centromeric cohesion is protected twice at meiosis, by SHUGOSHINs at anaphase i and by PATRONUS at interkinesis. Curr. Biol. 23, 2090–2099. doi: 10.1016/j.cub.2013.08.036
De Muyt, A., Pereira, L., Vezon, D., Chelysheva, L., Gendrot, G., Chambon, A., et al. (2009). A high throughput genetic screen identifies new early meiotic recombination functions in Arabidopsis thaliana. PLoS Genet. 5:e1000654. doi: 10.1371/journal.pgen.1000654
Dion, E., Li, L., Jean, M., and Belzile, F. (2007). An Arabidopsis MLH1 mutant exhibits reproductive defects and reveals a dual role for this gene in mitotic recombination. Plant J. 51, 431–40. doi: 10.1111/j.1365-313X.2007.03145.x
Duroc, Y., Lemhemdi, A., Larchevêque, C., Hurel, A., Cuacos, M., Cromer, L., et al. (2014). The kinesin AtPSS1 promotes synapsis and is required for proper crossover distribution in meiosis. PLoS Genet. 10:e1004674. doi: 10.1371/journal.pgen.1004674
Fernandes, J. B., Duhamel, M., Seguéla-Arnaud, M., Froger, N., Girard, C., Choinard, S., et al. (2018). FIGL1 and its novel partner FLIP form a conserved complex that regulates homologous recombination. PLoS Genet. 14:e1007317. doi: 10.1371/journal.pgen.1007317
Garcia, V., Bruchet, H., Camescasse, D., Granier, F., Bouchez, D., and Tissier, A. (2003). AtATM Is essential for meiosis and the somatic response to dna damage in plants. Plant Cell 15, 119–132. doi: 10.1105/tpc.006577
Garcia, V., Salanoubat, M., Choisne, N., and Tissier, A. (2000). An ATM homologue from Arabidopsis thaliana: complete genomic organisation and expression analysis. Nucleic Acids Res. 28, 1692–1699. doi: 10.1093/nar/28.8.1692
Girard, C., Chelysheva, L., Choinard, S., Froger, N., Macaisne, N., Lemhemdi, A., et al. (2015). AAA-ATPase FIDGETIN-LIKE 1 and helicase FANCM antagonize meiotic crossovers by distinct mechanisms. PLoS Genet. 11:e1005369. doi: 10.1371/journal.pgen.1005369
Girard, C., Crismani, W., Froger, N., Mazel, J., Lemhemdi, A., Horlow, C., et al. (2014). FANCM-associated proteins MHF1 and MHF2, but not the other fanconi anemia factors, limit meiotic crossovers. Nucleic Acids Res. 42,9087–9095. doi: 10.1093/nar/gku614
Jackson, N. P., Sanchez-Moran, E., Buckling, E., Armstrong, S. J., Jones, G. H., and Franklin, F. C. H. (2006). Reduced meiotic crossovers and delayed prophase I progression in AtMLH3-deficient Arabidopsis. EMBO J. 25, 1315–23. doi: 10.1038/sj.emboj.7600992
Lambing, C., Osman, K., Nuntasoontorn, K., West, A., Higgins, J. D., Copenhaver, G. P., et al. (2015). Arabidopsis PCH2 Mediates Meiotic Chromosome Remodeling and Maturation of Crossovers. PLoS Genet. 11:e1005372. doi: 10.1371/journal.pgen.1005372
Marks, M. D., and Feldmann, K. A. (1989). Trichome development in Arabidopsis thaliana. I. T-DNA tagging of the GLABROUS1 gene. Plant Cell 1, 1043–1050. doi: 10.1105/tpc.1.11.1043
Martín, B., Ramiro, M., Martínez-Zapater, J. M., and Alonso-Blanco, C. (2009). A high-density collection of EMS-induced mutations for TILLING in Landsberg erecta genetic background of Arabidopsis. BMC Plant Biol. 9:147. doi: 10.1186/1471-2229-9-147
McCallum, C. M., Comai, L., Greene, E. A., and Henikoff, S. (2000). Targeted screening for induced mutations. Nat. Biotechnol. 18, 455–457. doi: 10.1038/74542
Mercier, R., Jolivet, S., Vezon, D., Huppe, E., Chelysheva, L., Giovanni, M., et al. (2005). Two meiotic crossover classes cohabit in Arabidopsis: one is dependent on MER3,whereas the other one is not. Curr. Biol. 15, 692–701. doi: 10.1016/j.cub.2005.02.056
Mercier, R., Mezard, C., Jenczewski, E., Macaisne, N., and Grelon, M. (2015). The molecular biology of meiosis in plants. Annu. Rev. Plant Biol. 66, 297–327. doi: 10.1146/annurev-arplant-050213-035923
Nawaz, Z., and Shu, Q. (2014). Molecular nature of chemically and physically induced mutants in plants: a review. Plant Genet. Res. 12, S74–S78. doi: 10.1017/S1479262114000318
Osman, K., Sanchez-Moran, E., Mann, S. C., Jones, G. H., and Franklin, F. C. H. (2009). Replication protein A (AtRPA1a) is required for class I crossover formation but is dispensable for meiotic DNA break repair. EMBO J. 28, 394–404. doi: 10.1038/emboj.2008.295
Page, D. R., and Grossniklaus, U. (2002). The art and design of genetic screens: Arabidopsis thaliana. Nat. Rev. Genet. 3, 124–136. doi: 10.1038/nrg730
Peirson, B. N., Bowling, S. E., and Makaroff, C. A. (1997). A defect in synapsis causes male sterility in a T-DNA-tagged Arabidopsis thaliana mutant. Plant J. 11, 659–669. doi: 10.1046/j.1365-313X.1997.11040659.x
Portemer, V., Renne, C., Guillebaux, A., and Mercier, R. (2015). Large genetic screens for gynogenesis and androgenesis haploid inducers in Arabidopsis thaliana failed to identify mutants. Front. Plant Sci. 6:147. doi: 10.3389/fpls.2015.00147
Ravi, M., and Chan, S. W. L. (2010). Haploid plants produced by centromere-mediated genome elimination. Nature 464, 615–618. doi: 10.1038/nature08842
Ross, K. J., Fransz, P., and Jones, G. H. (1996). A light microscopic atlas of meiosis in Arabidopsis thaliana. Chromosom Res. 4, 507–516. doi: 10.1007/BF02261778
Séguéla-Arnaud, M., Crismani, W., Larchevêque, C., Mazel, J., Froger, N., Choinard, S., et al. (2015). Multiple mechanisms limit meiotic crossovers: TOP3α and two BLM homologs antagonize crossovers in parallel to FANCM. Proc. Natl. Acad. Sci. U.S.A. 112, 4713–4718. doi: 10.1073/pnas.1423107112
Sikora, P., Chawade, A., Larsson, M., Olsson, J., and Olsson, O. (2011). Mutagenesis as a tool in plant genetics, functional genomics, and breeding. Int. J. Plant Genomics 2011:314829. doi: 10.1155/2011/314829
Till, B. J., Cooper, J., Tai, T. H., Colowit, P., Greene, E. A., Henikoff, S., et al. (2007). Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 7:19. doi: 10.1186/1471-2229-7-19
Till, B. J., Reynolds, S. H., Weil, C., Springer, N., Burtner, C., Young, K., et al. (2004). Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biol. 4:12. doi: 10.1186/1471-2229-4-12
Keywords: forward screens, mutagenesis, meiosis, EMS, mutant collection, Arabidopsis
Citation: Capilla-Perez L, Solier V, Portemer V, Chambon A, Hurel A, Guillebaux A, Vezon D, Cromer L, Grelon M and Mercier R (2018) The HEM Lines: A New Library of Homozygous Arabidopsis thaliana EMS Mutants and its Potential to Detect Meiotic Phenotypes. Front. Plant Sci. 9:1339. doi: 10.3389/fpls.2018.01339
Received: 28 June 2018; Accepted: 24 August 2018;
Published: 19 September 2018.
Edited by:
Changbin Chen, University of Minnesota Twin Cities, United StatesReviewed by:
Junhua Li, Henan Normal University, ChinaBradlee Nelms, Stanford University, United States
Copyright © 2018 Capilla-Perez, Solier, Portemer, Chambon, Hurel, Guillebaux, Vezon, Cromer, Grelon and Mercier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raphael Mercier, cmFwaGFlbC5tZXJjaWVyQGlucmEuZnI=