 Catherine Damerval1*
Catherine Damerval1* Hélène Citerne1
Hélène Citerne1 Natalia Conde e Silva1
Natalia Conde e Silva1 Yves Deveaux1
Yves Deveaux1 Etienne Delannoy2Johann Joets1
Etienne Delannoy2Johann Joets1 Franck Simonnet1,3
Franck Simonnet1,3 Yannick Staedler4
Yannick Staedler4 Jürg Schönenberger4Jennifer Yansouni2Martine Le Guilloux1
Jürg Schönenberger4Jennifer Yansouni2Martine Le Guilloux1 Hervé Sauquet3,5
Hervé Sauquet3,5 Sophie Nadot3*
Sophie Nadot3*- 1GQE-Le Moulon, INRA, Univ. Paris-Sud, CNRS, AgroParisTech, Université Paris-Saclay, Gif-sur-Yvette, France
- 2Institute of Plant Sciences Paris-Saclay, CNRS, INRA, Universités Paris Diderot, Paris-Sud, Evry, Paris-Saclay, Gif-sur-Yvette, France
- 3Ecologie Systématique Evolution, AgroParisTech, CNRS, Univ. Paris-Sud, Université Paris-Saclay, Orsay, France
- 4Department of Botany and Biodiversity Research, University of Vienna, Vienna, Austria
- 5National Herbarium of New South Wales (NSW), Royal Botanic Gardens and Domain Trust, Sydney, NSW, Australia
Proteaceae are a basal eudicot family with a highly conserved floral groundplan but which displays considerable variation in other aspects of floral and inflorescence morphology. Their morphological diversity and phylogenetic position make them good candidates for understanding the evolution of floral architecture, in particular the question of the homology of the undifferentiated perianth with the differentiated perianth of core eudicots, and the mechanisms underlying the repeated evolution of zygomorphy. In this paper, we combine a morphological approach to explore floral ontogenesis and a transcriptomic approach to access the genes involved in floral organ identity and development, focusing on Grevillea juniperina, a species from subfamily Grevilleoideae. We present developmental data for Grevillea juniperina and three additional species that differ in their floral symmetry using stereomicroscopy, SEM and High Resolution X-Ray Computed Tomography. We find that the adnation of stamens to tepals takes place at early developmental stages, and that the establishment of bilateral symmetry coincides with the asymmetrical growth of the single carpel. To set a framework for understanding the genetic basis of floral development in Proteaceae, we generated and annotated de novo a reference leaf/flower transcriptome from Grevillea juniperina. We found Grevillea homologs of all lineages of MADS-box genes involved in floral organ identity. Using Arabidopsis thaliana gene expression data as a reference, we found homologs of other genes involved in floral development in the transcriptome of G. juniperina. We also found at least 21 class I and class II TCP genes, a gene family involved in the regulation of growth processes, including floral symmetry. The expression patterns of a set of floral genes obtained from the transcriptome were characterized during floral development to assess their organ specificity and asymmetry of expression.
Introduction
Proteaceae are a family of woody plants comprising approximately 1700 species in 81 genera, distributed mainly in the Southern Hemisphere, with two main centers of diversity, one in Australia and the other in South Africa. The hypogynous flowers almost invariably consist of four tepals (rarely 3 or 5), four stamens (rarely 3 or 5) opposite the tepals and with filaments that are adnate to the tepals (rarely free), and a single carpel with marginal placentation (Weston, 2007). Although this floral groundplan is highly conserved, the family displays considerable variation in other aspects of floral and inflorescence morphology. The inflorescence is basically a raceme but with various degrees of compaction. In subfamily Grevilleoideae, all but two genera are characterized by compound inflorescences consisting of racemes of flower pairs described as two-flowered short shoots sharing a common bract (Douglas and Tucker, 1996a). Early floral development has been described in detail in several species of Grevilleoideae, with emphasis on the ontogenic origin of the flower pair (Douglas and Tucker, 1996a) and carpel orientation (Douglas and Tucker, 1996b). In addition, Proteaceae have the highest number of transitions in perianth symmetry across all angiosperms (Reyes et al., 2016), with at least 10 transitions from actinomorphy (radial symmetry) to zygomorphy (bilateral symmetry) inferred throughout the family, and at least four reversals (Citerne et al., 2017). The developmental stage at which bilateral symmetry becomes visible varies across angiosperm species with a zygomorphic perianth; zygomorphy can be present from the very first stages, when the first floral organs are initiated, or can appear after all organs have been initiated and have begun to differentiate (Endress, 1999; Tucker, 1999). As in many taxa with a monomerous gynoecium, the single plicate carpel in Proteaceae becomes bilaterally symmetrical when the cleft starts to form, making the flower zygomorphic at the gynoecium level (Douglas and Tucker, 1996b; Sokoloff et al., 2017). Furthermore, in Proteaceae and more specifically in the subfamily Grevilleoideae, the high diversity of carpel orientation (dorso-ventral or oblique) adds further complexity when defining the orientation of the plane of symmetry relative to the common axis of the flower pair (Douglas and Tucker, 1996b).
The molecular bases of floral symmetry have been investigated in depth in Antirrhinum majus (Plantaginaceae). The CYCLOIDEA (CYC) gene is essential for the asymmetric development of the flower in the petal and stamen whorls (Luo et al., 1996, 1999). CYC belongs to a plant-specific transcription factor gene family, the TCP family, which is characterized by a non-canonical basic helix-loop-helix domain (the TCP domain, Cubas et al., 1999). TCP genes have been reported to be involved in various cell proliferation and growth processes that control organ development and shape (Huang and Irish, 2015; Nicolas and Cubas, 2016). In A. majus, CYC expression varies with the identity of the floral organ. Organ identity is classically determined by the combined action of four classes of genes, referred to as the A-, B-, C-, and E-classes (Coen and Meyerowitz, 1991; Causier et al., 2010). Petals are determined by the combined action of A- and B-class genes, stamens by B- and C-class genes, sepals by A-class genes only, and the carpel by C-class genes only. E-class genes act as obligate partners for all floral organ identity genes. The model is generally conserved across angiosperms with the notable exception of the A-class, whose function as defined in Arabidopsis thaliana appears to be limited to close relatives in the Brassicaceae. With the exception of one A function gene in A. thaliana, namely APETALA2, these floral organ identity genes belong to the MADS-box transcription factor gene family (Theißen et al., 1996). B- and C-class genes may play a role in maintaining expression of CYC in the second and third floral whorls in A. majus (Clark and Coen, 2002).
Asymmetric expression patterns of CYCLOIDEA homologs (hereafter CYC-like genes) have been correlated with the independent evolution of zygomorphy in many monocot and eudicot clades (e. g., Hileman, 2014; Spencer and Kim, 2018). Functional studies have confirmed the role of CYC-like genes in the asymmetric development of floral organs in distantly related core eudicot species such as Lotus japonicus, Iberis amara, a hybrid of Gerbera, and in the monocot Oryza sativa (Feng et al., 2006; Busch and Zachgo, 2007; Broholm et al., 2008; Yuan et al., 2009). In orchids, however, no TCP gene has been found to date to be involved in zygomorphy. In this large monocot family, duplication and subfunctionalization in one of the two B-class gene lineages account for the inter- and intra-whorl tepal differentiation that generates zygomorphy (reviewed in Mondragon-Palomino, 2013). B-class genes have been found to be implicated in the independent evolution of zygomorphy in other monocot taxa. For instance, in Commelina communis (Commelinaceae), it has been suggested that in addition to the asymmetric expression of a CYC-like gene, zygomorphy is mediated by the asymmetric expression of a B-class gene in the inner tepal whorl associated with a change in ventral organ identity (from petal to sepal) (Preston and Hileman, 2012). In maize, the lack of expression of the B-class genes in the dorsal domain coincides with the absence of initiation of the dorsal lodicule (Bartlett et al., 2015).
Proteaceae, with their morphological diversity and their phylogenetic position within the basal eudicots, are good candidates for understanding the evolution of floral architecture, particularly regarding the homology of the undifferentiated perianth with either the calyx or the corolla of a differentiated perianth, and the repeated evolution of zygomorphy. In addition, the origin of the tetramerous perianth remains unclear. A recent study of floral trait evolution across angiosperms suggested that the ancestral perianth of eudicots may have been dimerous and undifferentiated (Sauquet et al., 2017), in which case the tetramerous perianth of Proteaceae could be of dimerous origin inherited from the ancestor of all eudicots. Yet, Proteaceae are relatively understudied from the point of view of developmental genetics. CYC-like genes have recently been characterized in Proteaceae. In two zygomorphic species of Grevillea, expression of the same ProtCYC gene was found to be asymmetric during the late stages of floral development (Citerne et al., 2017).
This paper combines developmental and transcriptomic data in Grevillea juniperina from subfamily Grevilleoideae. We compared early floral ontogenesis in G. juniperina with three other species presenting similar floral features but differing in floral symmetry, using a range of microscopy techniques (including High Resolution X-Ray Computed Tomography). We assembled de novo a transcriptome of Grevillea juniperina from RNA-seq of leaf and flower tissues. We searched this transcriptome for homologs of genes known to be involved in floral development in Arabidopsis, as well as focusing on the entire TCP and A-, B-, C-, E-class MADS-box gene families. We characterized the expression pattern during floral development of selected genes in G. juniperina.
Materials and Methods
Plant Material
Plant material (inflorescences at various pre-anthetic stages) was collected from four species of Proteaceae belonging to tribe Embothrieae of subfamily Grevilleoideae (Weston and Barker, 2006) and endemic to Australia: Alloxylon flammeum P. H. Weston & Crisp (subtribe Embothriinae), Stenocarpus davallioides D. Foreman & B. Hyland (subtribe Stenocarpinae), Grevillea juniperina R. Br. and Grevillea petrophiloides Meisn. (subtribe Hakeinae). The first three species have zygomorphic flowers while the latter has actinomorphic flowers (Figure 1). Detailed descriptions and illustrations of these species are available in the Flora of Australia online1. A dissection of the flower of Grevillea juniperina, showing all floral organs, is shown in Citerne et al. (2017).

Figure 1. Inflorescences of the study species. All species have tetramerous flowers with stamens completely adnate to tepals. (A) Inflorescence of Grevillea juniperina with zygomorphic flowers at anthesis (arrow pointing to an open flower displaying curved tepals and style). (B) Inflorescence of Grevillea petrophiloides with actinomorphic flowers at anthesis (arrow pointing to an open flower displaying straight tepals and style). (C) Inflorescence of Alloxylon flammeum with zygomorphic flowers at anthesis (arrow pointing to an open flower displaying curved tepals and style). (D) Inflorescence of Stenocarpus davallioides with zygomorphic flowers at anthesis (arrow pointing to an open flower displaying curved tepals and style). Photographs S. Nadot (A) and H. Sauquet (B–D).
Material for Alloxylon flammeum and Stenocarpus davallioides was sampled from trees cultivated at the Royal Botanic Garden Sydney (under accessions S2003-0047 and S1986-2016, respectively; voucher specimens were made (P.H.Weston 3425 and P.H.Weston 3426) and deposited at The National Herbarium of New South Wales (NSW)) and fixed in FAA (85 mL 55% ethanol, 5 mL glacial acetic acid, 10 mL formaldehyde). Material for Grevillea petrophiloides was sampled from trees cultivated in Longueville, NSW (voucher P.H.Weston 3582: High Resolution X-ray Computed Tomography) and Oakdale, NSW (voucher P.M.Olde 18/01: light microscopy) and fixed in FAA and 70% alcohol, respectively. Five plants of Grevillea juniperina were obtained from the garden center Truffaut and planted in the Parc Botanique de Launay (Orsay, France). These plants provided fresh material for the development analysis and for RNA extraction.
Development Analysis
Light Microscopy and Scanning Electron Microscopy
Light microscopy was carried out on a Leica M125 stereomicroscope. For scanning electron microscopy, developing inflorescences and dissected tissues were dehydrated through an ethanol-acetone series, critical-point dried and sputter coated with gold. Imaging was carried out on a JEOL JSM 6390 Scanning Electron Microscope at 10 kV.
High Resolution X-Ray Computed Tomography (HRXCT) and 3D Reconstruction
For HRXCT, developing inflorescences of all four species were treated with a solution of 1% (w/v) phosphotungstic acid in FAA for 2 months (thereby ensuring saturation of the material with phosphotungstic acid), following the protocol of Staedler et al. (2013). The scans were performed on a MicroXCT-200 system (Zeiss Microscopy). X-Ray detection was performed via scintillator crystals (Zeiss Microscopy, in-house). The X-ray source was a Hammamatsu L9421-02 90 kV Microfocus X-Ray source. XMReconstructor 8.1.6599 (Zeiss Microscopy) was used to perform the 3D reconstruction from the scanning data. The AMIRA-based XM3DViewer 1.1.6 (XRadia Inc.) was used to visualize the scans. The reconstructed 3D data were then exported with XMController 8.1.6599 as a series of pictures in tiff format, typically ca. one to two thousand per sample. Scanning parameters are summarized in Supplementary Table S1.
Tissue Collection and RNA Extraction
Young leaves and flower buds of Grevillea juniperina were collected and processed as in Citerne et al. (2017). Total RNA was extracted following a 2x CTAB-based extraction buffer and lithium chloride precipitation method (Smart and Roden, 2010) and total RNA was treated with DNase (Ambion) following the manufacturer’s instructions. For RNA-seq analysis, all tissues were harvested on the same plant: young leaves and floral buds of three size ranges (1–2 mm, 2–5 mm, over 6 mm long), and processed individually.
For qPCR analyses, floral buds from different plants were combined to constitute three biological replicates for flower dissections. Dissections were carried out on freshly harvested 6–7 mm buds already exhibiting bilateral symmetry under a stereomicroscope (6–10 buds from two–three plants for each biological replicate). Three tissue types (dorsal tepals with adnate stamens, ventral tepals with adnate stamens and gynoecium) were separately frozen in liquid nitrogen.
Reference Transcriptome
Sequencing
Total RNAs of each tissue were checked for their integrity on a RNA_Nano chip, using an Agilent 2100 bioanalyzer (Agilent Technologies, Waldbronn, Germany), then pooled in equal amounts. The RNA-seq procedure was carried out at the Institute of Plant Sciences Paris-Saclay (IPS2, Saclay, France) on an IG-CNS Illumina Hiseq2000 platform. The library was constructed with the TruSeq stranded mRNA library Prep kit (Illumina®, CA, United States) with a sizing of 260 bp, and then sequenced in paired-end (PE) and a read length of 100 bases on a single lane. After quality trimming (removing the adapter, trimming of bases with a Q score <20 and removing pairs with at least one read <30 bases), 186,060,539 pairs of reads were generated. The raw data are available at SRA under the accession SRP141177.
Transcriptome Assembly
The de novo assembly of the transcriptome was performed following Roberts and Roalson (2017). Six assemblies were generated with Trinity v2.3.2 (Haas et al., 2013, with default parameters except –SS_lib_typeRF and –min_kmer_cov 3) and Velvet-Oases v1.2.09 (Schulz et al., 2012, kmers 25, 35, 45, 55, and 65) and combined with the tr2aacds.pl script (v 2014.05.15) of the Evidential Genes suite (Nakasugi et al., 2014). Only contigs classified as “main” were kept. The quality of the final assembly was assessed with BUSCO v3.0.2 (Waterhouse et al., 2018) using hmmer 3.1b2 against the embryophyta_odb9 dataset.
Homology Search and Functional Annotation
Predicted protein sequences from the Grevillea juniperina assembled transcriptome were annotated with BLASTP best hit (Camacho et al., 2009, e-value cutoff 1e-3) against a local version of the Arabidopsis thaliana protein database (TAIR 10). From BLASTP results, a gene ontology (GO) annotation was generated and extended through merging with InterProScan results using default parameters (Jones et al., 2014).
Overall homology of the G. juniperina proteome with other eudicot proteomes was assessed using OrthoVenn2 (Wang et al., 2015), selecting the proteomes of Arabidopsis thaliana and Vitis vinifera, and uploading the proteome of Nelumbo nucifera from NCBI (GCF_000365185.1_Chinese_Lotus_1.1_protein.faa.gz).
Floral Gene Mining and Phylogenetic Analyses
Phylogeny of TCP Genes
All Grevillea contigs identified as TCP genes were extracted from the G. juniperina transcriptome annotated against the proteome of Arabidopsis thaliana by BLAST homology search. Sequence alignment was done with MUSCLE3 (Edgar, 2004) based on predicted amino acid translations, then manually refined. Phylogenetic reconstruction was done using maximum likelihood as implemented in PhyML (Guindon et al., 2010), using the GTR + Γ model [substitution model selected by AIC using the SMS option in PhyML (Lefort et al., 2017)] and the NNI method of tree optimization. Branch support was calculated with the aLRT SH-like method.
Phylogeny of ABCE MADS-Box Coding Sequences
In Arabidopsis, MADS-box genes with floral organ identity function are APETALA1 (AP1) (A-class), APETALA3 (AP3) and PISTILLATA (PI) (B-class), AGAMOUS (AG) (C-class) and SEPALLATA 1/2/4 (SEP1/2/4) and SEPALLATA3 (SEP3) (E-class). We searched for homologs of these MADS-box genes in the G. juniperina transcriptome. Sixteen contigs were retrieved from the transcriptome following annotation against the Arabidopsis thaliana proteome. Sequences from transcriptomic data were translated, and seven full length protein sequences were used for subsequent analyses. MADS-box coding sequences (CDS) from G. juniperina were aligned with known floral MADS-box CDS of Arabidopsis thaliana4, Nelumbo nucifera5, Aquilegia caerulea6, and Vitis vinifera6, using MUSCLE based on predicted amino acid translations7 (protein IDs in Supplementary Table S2). Phylogenetic reconstruction was done using maximum likelihood as implemented in PhyML (Guindon et al., 2010), using the GTR + Γ + I model [substitution model selected by AIC using the SMS option in PhyML (Lefort et al., 2017)] and the NNI method of tree optimization. Branch support was calculated with the aLRT SH-like method.
Homologs of Floral Genes Co-expressed With ABCE MADS-Box Genes in Grevillea juniperina
We searched for the genes whose expression is correlated with that of the MADS-box genes implicated in the ABCE model in A. thaliana, excluding SEP4 because of its low expression specificity to reproductive tissues. The search was done with the Expression Angler program using the AtGenExpress - Plus Extended tissue Compendium data set (Toufighi et al., 2005). The Pearson correlation coefficient (r-value) cut-off was fixed at 0.5 or 0.75, and a set of unique AGI (Arabidopsis Genome Initiative) gene identifiers (AGI-ID) was created. For each AGI-ID, the frequency of occurrence was calculated for the seven A. thaliana MADS-box genes used as baits and the best r-value reported (Supplementary Table S3). Then we searched for homologs of these genes in the G. juniperina transcriptome annotated against the A. thaliana proteome. A new dataset of the best matching AGI-ID in the G. juniperina transcriptome was obtained and the corresponding numbers of G. juniperina contigs were reported (Supplementary Table S3). A list of the 25 best correlated genes was also generated for each of the seven MADS-box genes used as baits and compared to the G. juniperina transcriptome.
Quantitative RT-PCR Analyses of Floral Gene Expression
Thirteen G. juniperina contigs were selected based on their homology with genes expressed during floral development in Arabidopsis thaliana. They were renamed according to homology as follows: AGAMOUS (GjuAG2), PISTILLATA (GjuPI2), SEPALLATA (GjuSEP1 and GjuSEP3), APETALA2 (GjuAP2), APETALA1 (GjuAP1), WUSCHEL (GjuWUS), AHOOTMERISTEMLESS (GjuSTM), CUP-SHAPED COTYLEDON2 (GjuCUC2), SPATULA (GjuSPT), BEL1 (GjuBEL1), TOUSLED (GjuTSL), and CRABSCLAW (GjuCRC). In addition, we included the ProtCYC genes (GjuCYC1 and GjuCYC2) that were found to be expressed in flower buds by Citerne et al. (2017). Expression of these 15 genes was investigated in dissections of floral organs of pre-anthetic flowers (as described above).
Primer pairs were designed from the contig sequences (Supplementary Table S4). The GjuPI2 gene corresponds to a contig closely related to the full length GjuPI used in the phylogenetic reconstruction, but lacking C-terminal region. Unfortunately, we were unable to define an appropriate pair of specific primers for GjuAP1 from the available sequence. qRT-PCR reactions were performed using the Bio-Rad CFX384 touch (Bio-Rad, France) and the SYBR Premix Ex Taq (Tli RNaseH Plus) (Takara, Ozyme, France) following the supplier’s instructions. Three technical replicates were done for each of the three biological replicates of the floral dissections. A dilution series of the pooled cDNAs was used as a standard curve to validate the primer pairs and estimate the starting quantities (arbitrary units). Gene expression was normalized with the mean of the three reference genes ACT8, TUB7, and FBA1 coding for actin, tubulin and fructose bisphosphate aldolase, respectively. The normalized expression values are provided in Supplementary Table S5. Differential expression for each gene was tested with a Two-way ANOVA (tissue and replicate effects) and a Tukey test for pairwise comparisons. P-values were adjusted for multiple testing with a BH correction. Adjusted p-values lower than 0.05 were considered as significant.
Results
Floral Development and Establishment of Zygomorphy
The developing flowers in Grevillea juniperina (zygomorphic flowers at anthesis, Figure 1A, arrow) display a perianth that is actinomorphic until the tepals are formed and begin to elongate (Figures 2A–F), as in the actinomorphic species Grevillea petrophiloides (Figure 1B, distal part of the inflorescence, and Figures 3F,G). In G. juniperina, the virtual longitudinal section obtained with HRXCT shows that the carpel develops asymmetrically relative to the center of the floral bud (Figure 2D), even though the other organ whorls (tepals and stamens) are radially symmetrical. The monosymmetrical shape of the developing carpel suggests asymmetrical growth of the primordium. It can be noted that the incomplete valvate aestivation in G. juniperina (Figure 2B) makes the young flower bud slightly disymmetric rather than strictly actinomorphic. The base of the tepals and stamens is already slightly fused, indicating that the adnation of stamens to tepals occurs early during development. Early development is similar in the two other species sampled in this study, Alloxylon flammeum and Stenocarpus davallioides (both zygomorphic at anthesis; Figures 1C,D, arrows), which have flowers with an actinomorphic perianth at early stages and a gynoecium that develops asymmetrically relative to the center of the floral bud (Figures 3A–E). In Grevillea petrophiloides, the style elongates faster than the tepals before anthesis, and forces its way through the two dorsal tepals (visible in the proximal part of the inflorescence, Figure 1B), but in mature flowers, the style and the tepals are straight, contrary to the three other species.
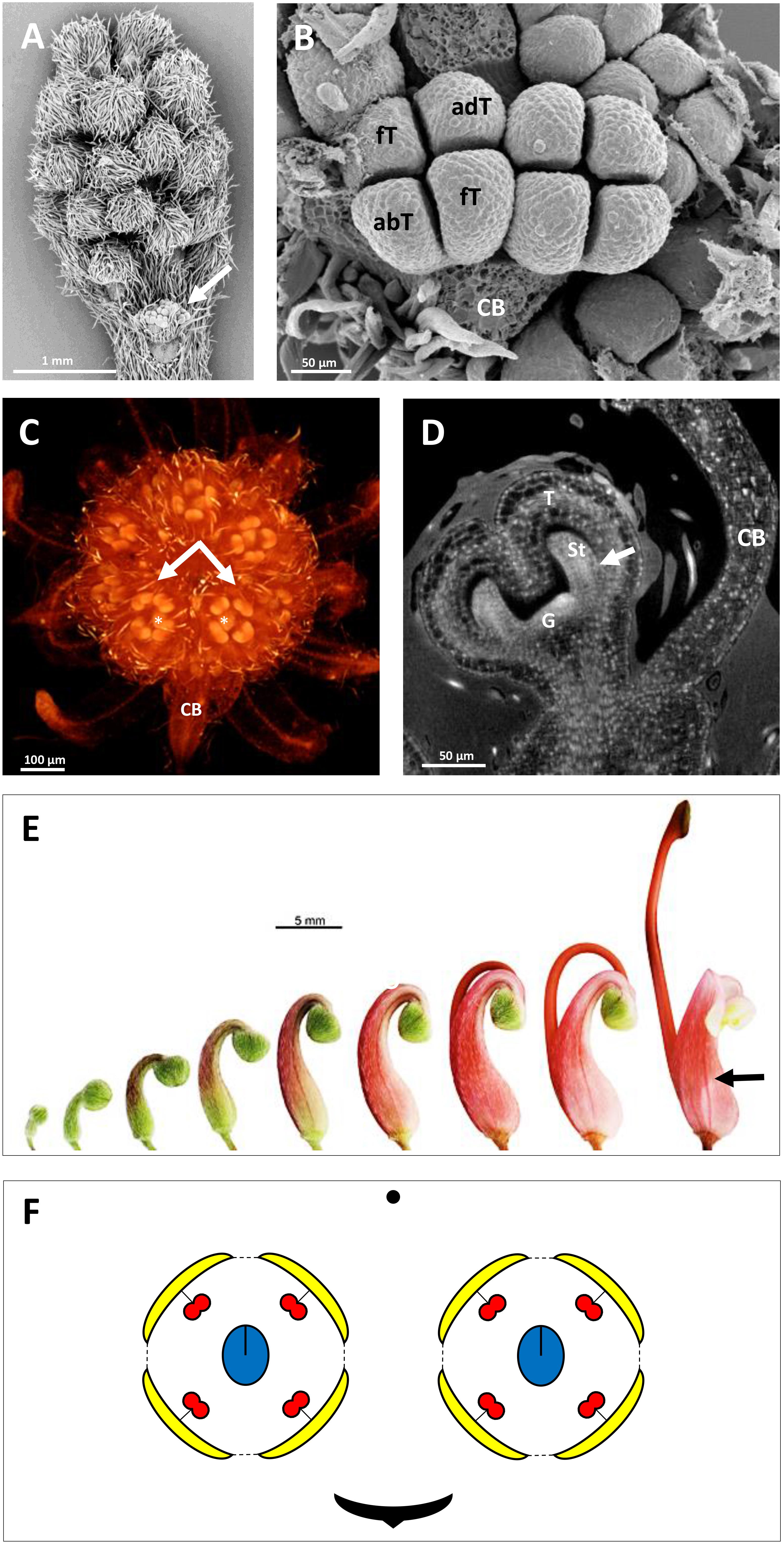
Figure 2. Inflorescence and flower pair development in Grevillea juniperina (A,B: SEM; C,D: CT-scans; E: stereomicroscope). (A) Whole inflorescence, composed of a first order axis bearing secondary order axes each surrounded by a hairy subtending bract. Each secondary axis bears pairs of flowers (conflorescences). The arrow points to one secondary order axis (subtending bract removed). (B) Flower pair, with common subtending bract (CB) removed, showing incomplete valvate aestivation of the tepals (fT, frontal tepal; adT, adaxial tepal = dorsal tepal; abT, abaxial Tepal = ventral tepal). (C) Top view of a second order axis of inflorescence, showing a flower pair from top view (arrows) with all organs initiated; the flower is made zygomorphic by the non-central position of the gynoecium (asterisks). (D) Virtual longitudinal section of a young flower bud showing the monosymmetrical gynoecium primodium (G); the arrow points to the fusion between a stamen and a tepal. (E) Floral developmental sequence, from the first stage at which zygomorphy becomes conspicuous (left) to anthetic flower. The arrow points to the strongly zygomorphic ovary visible through the perianth. (F) Floral diagram of a flower pair of Grevillea juniperina showing the orientation of each flower relatively to the common bract.
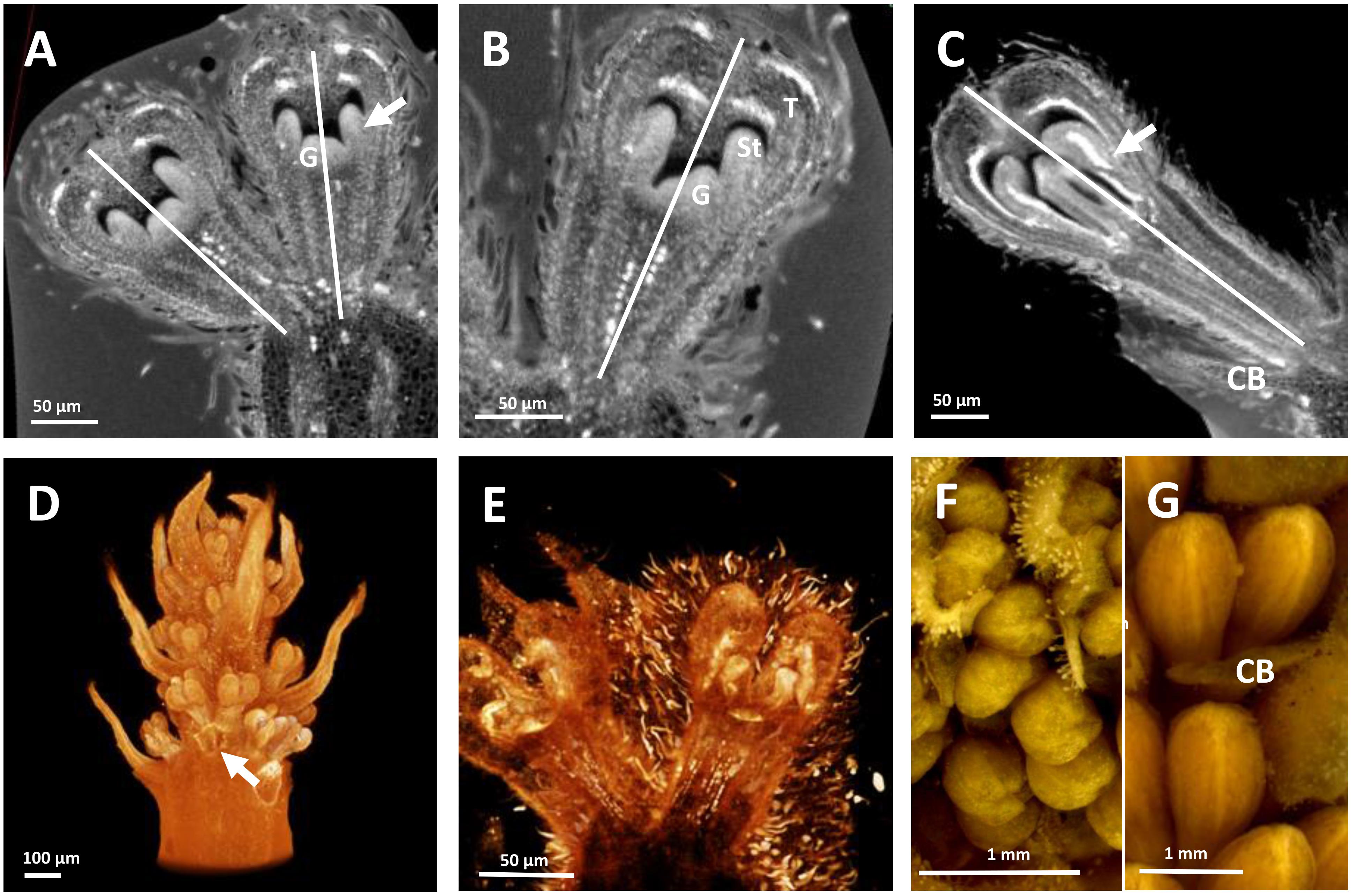
Figure 3. HRXCT images of developing flowers and flower pairs in Stenocarpus davallioides (A–C), Alloxylon flammeum (D,E) and HRXCT and stereomicroscope images of inflorescence and flower pairs in Grevillea petrophiloides (F,G). (A) Virtual longitudinal section of a flower pair (lateral view), with slight monosymmetry of the gynoecium (G) with regard to the vertical floral axis, visible on the right; the arrow points to the fusion between a stamen and a tepal. (B) Virtual longitudinal section of a flower (front view), showing the monosymmetry of the gynoecium in the vertical axis, while the tepals (T) and adnate stamens (St) are identical. (C) Virtual longitudinal section of a flower at a later stage (lateral view), showing slight asymmetry of the tepals and adnate stamens (the arrow points to the fused zone between a stamen and a tepal) with respect to the vertical axis (CB, common bract). (D) Lateral view of a whole inflorescence, with the common subtending bract of a flower pair removed (arrow). (E) Lateral view of a flower, zygomorphy is not yet apparent. (F,G) Lateral view of flower pairs at two developmental stages (CB, common bract).
Assembly and Annotation of the Reference Transcriptome
67,004 contigs were obtained from the de novo assembly of the transcriptome of G. juniperina, with lengths ranging from 180 to 9,937 bases for a mean of 877.8, a N50 of 1,491 and 59,306,330 assembled bases. The result of the BUSCO analysis was C: 84.8% [S: 74.9%, D: 9.9%], F: 8.4%, M: 6.8%, n: 1,440 demonstrating the good quality of this assembly.
InterProScan identified 30,063 contigs with at least one protein domain. Among these, 14,488 contigs could be annotated with one category (or more) of the Gene Ontology (GO). We focused on transcription factors, selecting all contigs annotated as GO:0003700 and analyzed their PFAM domains. We found 240 contigs with such transcription domain annotation (1.6% of the GO annotated contigs), most of which corresponded to AP2 (29%), WRKY DNA-binding (23%) and bZip (12%) domains (Supplementary Figure S1).
37,842 contigs had homology with 13,946 Arabidopsis proteins, while 37,817 had a GO annotation. The two best-represented categories in Biological Process were “other cellular processes” (26%) and “other metabolic processes” (22%); in Cellular Components, the four best-represented categories were “other cytoplasmic components” (18%), “other intracellular processes” (14%), “nucleus” (15%) and “other membranes” (13%); among Molecular Functions, the prominent categories were “protein binding” (20%), “other binding” (14%),“other enzyme activity” (13%), and “transferase activity” (11%) (Supplementary Figure S2). We found 937 G. juniperina contigs with a functional annotation in one or more categories identified as GO: 0009908 (flower development) and its children categories. Beyond general flower development, the most represented categories pertained to stamen (20%) and ovule and carpel (16%) development (Supplementary Figure S3).
Homology With Other Eudicot Proteomes
Proteins deduced from the contigs of G. juniperina were compared to those in the proteomes of three fully sequenced species, Arabidopsis thaliana, Nelumbo nucifera, and Vitis vinifera, using to the algorithm available from the OrthoVenn site (Figure 4). As expected because of imperfect transcriptome de novo assembly, the highest proportion of singletons was obtained from G. juniperina (58% of the number of proteins against 10–33% in the three other species). 20,787 clusters, i.e., orthologous groups were inferred, 14,362 of these included at least two species. 3,363 clusters included only sequences from G. juniperina while 1,132, 1,005, and 925 clusters were specific to A. thaliana, V. vinifera, and N. nucifera, respectively. This higher number of specific clusters could be due to various factors (distant homologs that were not identified, enrichment in specific floral symmetry genes, assembly problems, etc). A GO enrichment test on these cluster members did not provide any clues as to a possible involvement in floral symmetry. 10,041 orthologous groups included sequences from all four species. Among the three pairwise comparisons involving the inferred Grevillea proteome, Grevillea and Nelumbo, both from the order Proteales, shared the highest number of total and pair-specific orthologous groups, which is consistent with their phylogenetic position. Orthologous groups including sequences of Grevillea, Vitis, and Nelumbo were more numerous than those including sequences of Arabidopsis, Vitis, and Nelumbo.
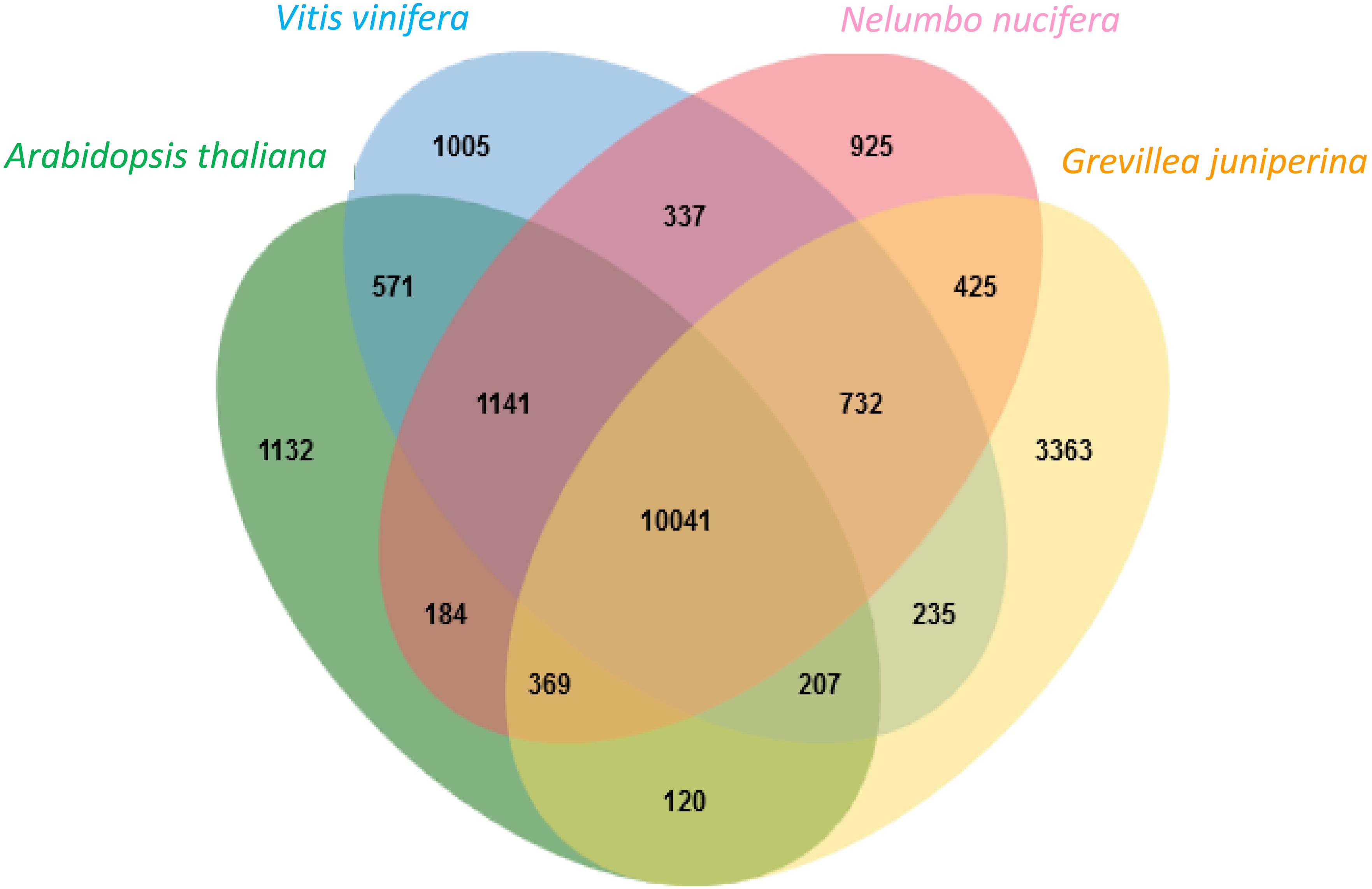
Figure 4. Distribution of clusters (orthologous groups) between the Arabidopsis thaliana, Vitis vinifera, Nelumbo nucifera proteomes and the Grevillea juniperina leaf/flower proteome.
Mining the Grevillea juniperina Transcriptome for Homologs of Genes Involved in Floral Development
We focused on two families of transcription factors involved in growth and development processes, particularly in flowers. TCP genes have been reported to be involved in various cell proliferation and growth processes that control organ development and shape, including floral symmetry (Huang and Irish, 2015; Nicolas and Cubas, 2016). MADS-box genes of A-, B-, C-, and E- classes are involved in floral organ identity and also interact with patterning and growth genes (Sablowski, 2015). In addition, we searched for homologs of genes co-expressed with the ABCE MADS-box genes as candidates for a conserved floral gene regulatory network module between A. thaliana and G. juniperina.
Phylogeny of TCP Homologous Genes
Twenty-four contigs with homology to TCP genes were identified in our G. juniperina transcriptome. Assembled fragment lengths ranged from 245 to 4,726 nucleotides; 10 fragments were predicted to contain complete ORFs (with identifiable start and stop codons) with putative ORF lengths ranging from 211 to 573 amino acids. All contigs but three had the characteristic basic helix-loop-helix TCP domain. Of the three contigs where the TCP domain could not be found, two had 76.4% sequence identity with R domains that were identified by blastx searches as homologous to TCP2. One contig lacked any recognizable domain because it corresponded to the region downstream of the TCP domain, but was found to be homologous by blastx searches to TCP8.
The phylogenetic analysis of 192 aligned nucleotide positions, mainly comprising the TCP domain, from the 21 G. juniperina (excluding the three contigs without TCP domain) and 24 A. thaliana TCP genes showed that both class I and class II TCP were recovered with high support (Figure 5). Class I genes were over-represented in G. juniperina, with at least 14 copies compared to A. thaliana (13 copies), unlike class II genes with at least seven copies compared to A. thaliana (11 copies). One CYC-like copy (homologous to the CYC-like genes TCP18/TCP12/TCP1 in A. thaliana) was found in the transcriptome of G. juniperina, corresponding to the previously characterized ProtCYC1 (Citerne et al., 2017).
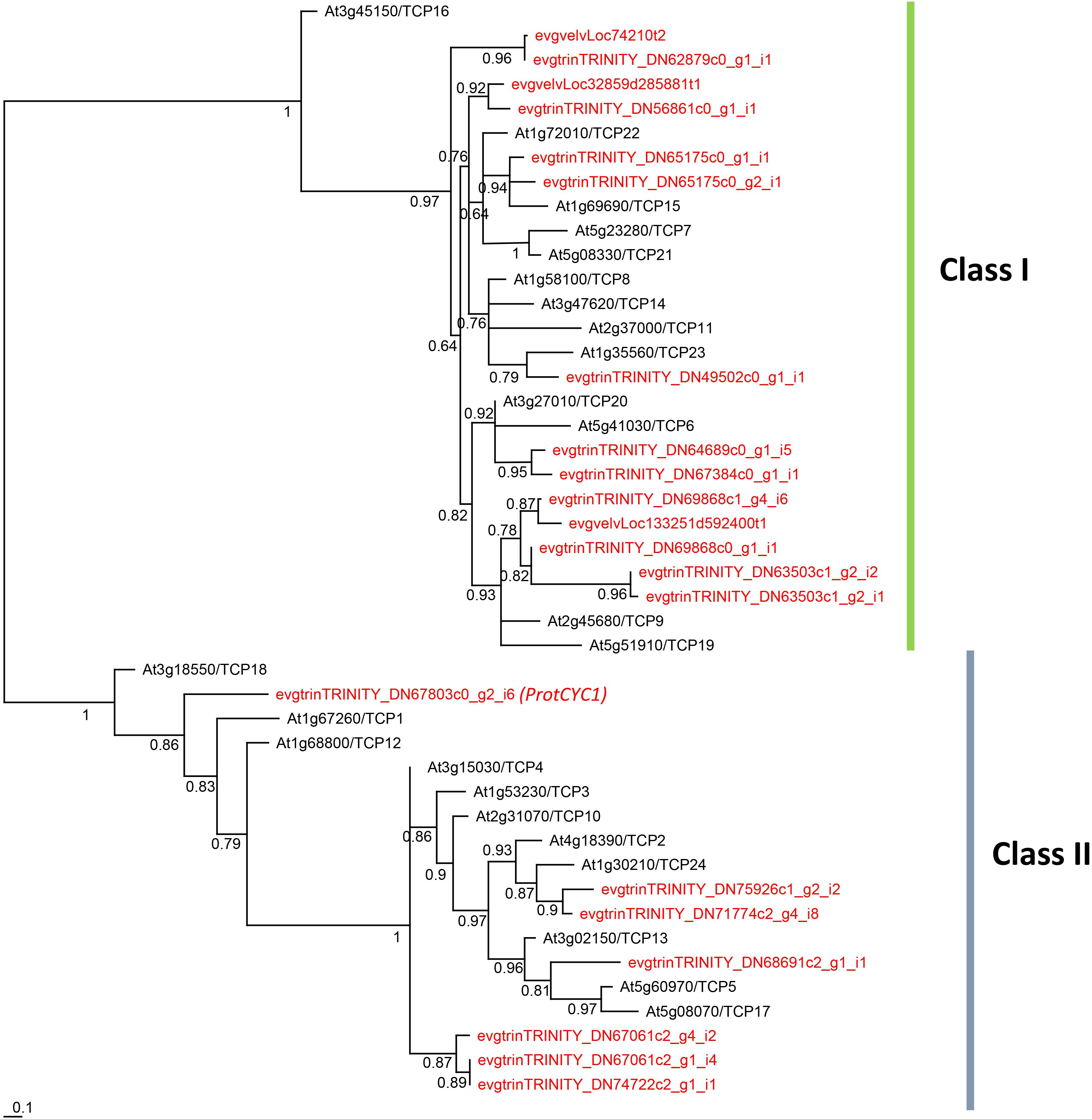
Figure 5. Unrooted phylogram of TCP genes from the Arabidopsis thaliana genome and the Grevillea juniperina transcriptome (in red). 21 of the 24 G. juniperina transcripts were included in this analysis. Branches with less than 0.5 aLRT values were collapsed. Representatives of both class I and class II TCP genes were found in G. juniperina.
Phylogeny of ABCE MADS-Box Genes
Sixteen contigs annotated as one of the Arabidopsis thaliana ABCE MADS-box AGI gene identifiers were extracted from the G. juniperina transcriptome after annotation. Seven fragments were predicted to contain full length ORFs (with identifiable start and stop codons) with putative lengths ranging from 209 to 247 amino acids. All full-length contigs had the characteristic MIKC domains. Among the nine incomplete contigs, four lacked the N-terminal coding sequence and five lacked the C-terminal region. Among these incomplete sequences, one, three, and five contigs aligned partially with GjuAP1, GjuSEP, and GjuPI, respectively, suggesting other alleles or paralogs.
A phylogenetic tree including full length G. juniperina coding sequences and A-, B-, C-, and E-class MADS-box CDS from Vitis vinifera, Nelumbo nucifera, Arabidopsis thaliana, and Aquilegia caerulea was reconstructed using PhyML (Figure 6). We retrieved the A-, B-, C-, and E-class gene lineages with good support, and at least one G. juniperina sequence falls in each of these clades. Two AG-like G. juniperina sequences were found in the C lineage, one of which is closely related to the N. nucifera sequences. Three G. juniperina sequences were found in the E lineage. One of these fell in the SEP3 clade; the other two grouped together and might correspond to closely related paralogs. Surprisingly in the B-class, no G. juniperina sequence was found in the AP3 lineage, while one sequence was found in the PI one, closely related to the two other basal eudicot sequences.
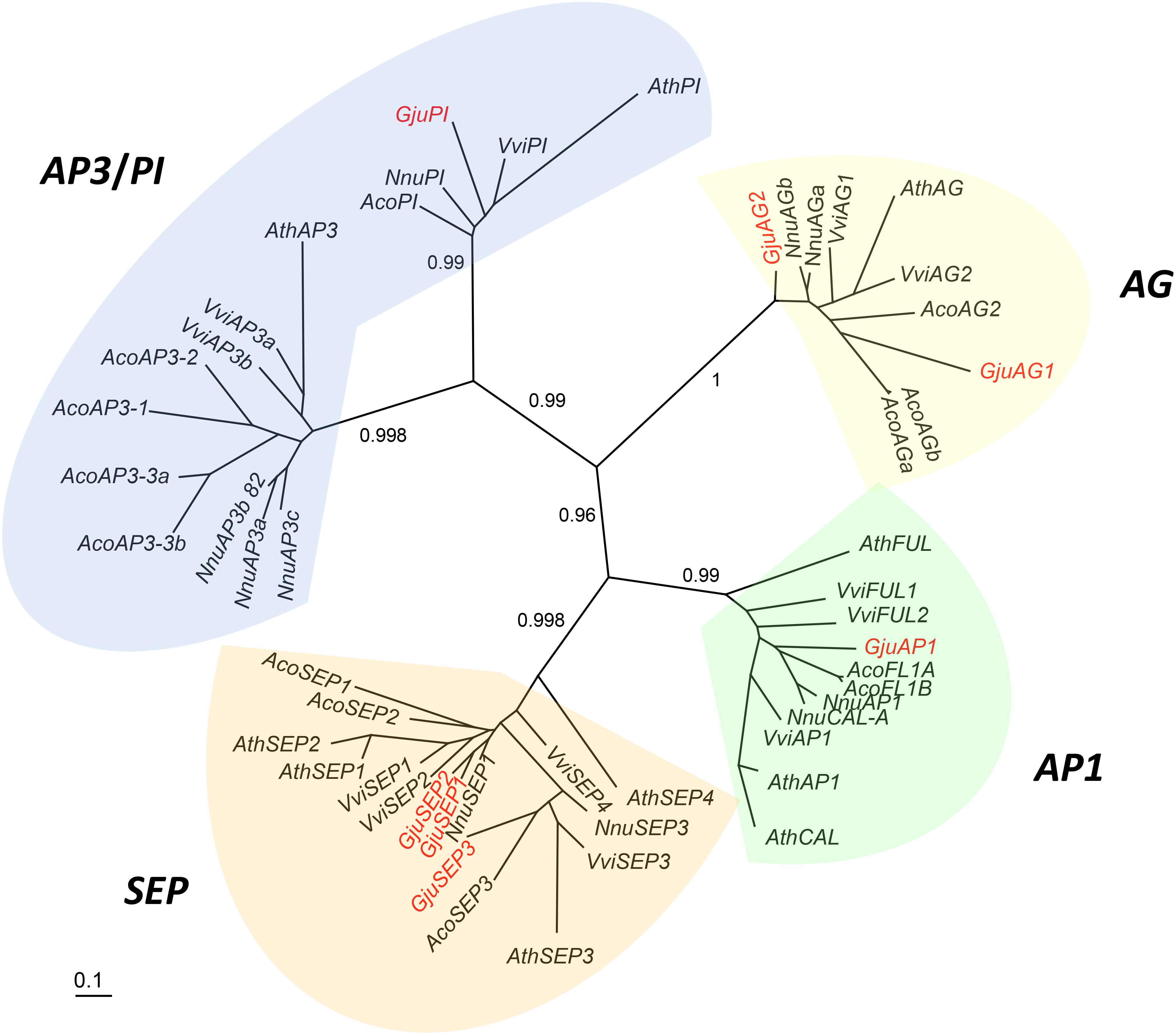
Figure 6. Unrooted phylogram of A-, B-, C-, E-class MADS-box genes. Seven G. juniperina full-length transcripts were included in this analysis. Branches with less than 0.5 aLRT values were collapsed. “Gju” is the abbreviation for Grevillea juniperina, “Vvi” for Vitis vinifera, “Nnu” for Nelumbo nucifera, “Ath” for Arabidopsis thaliana and “Aco” for Aquilegia caerulea genes. Protein IDs are specified in Supplementary Table S2.
Homologs of Genes Co-expressed With ABCE MADS-Box Genes
To investigate the extent to which the gene network involved in floral organ development is conserved between Arabidopsis and Grevillea, we searched for genes with expression patterns that are correlated with that of ABCE MADS-box genes in Arabidopsis thaliana using the Expression Angler Website, and then searched for their homologs in the annotated G. juniperina transcriptome. In Arabidopsis, 822 and 34 genes were found with a Pearson correlation coefficient cut-off (r-value) of, respectively, 0.5 and 0.75 (Supplementary Table S3). Analysis of the annotated G. juniperina transcriptome indicates global conservation of the gene network between the two species (Figure 7A) although with some variations (Table 1). Homologs of ABCE genes and of their highest correlated genes were found in the G. juniperina transcriptome, suggesting they are conserved between the two species (Figure 7A and Table 1). Indeed, 10 out of the 14 Arabidopsis genes with an r-value of 0.8 have at least one homologous contig in the G. juniperina transcriptome.
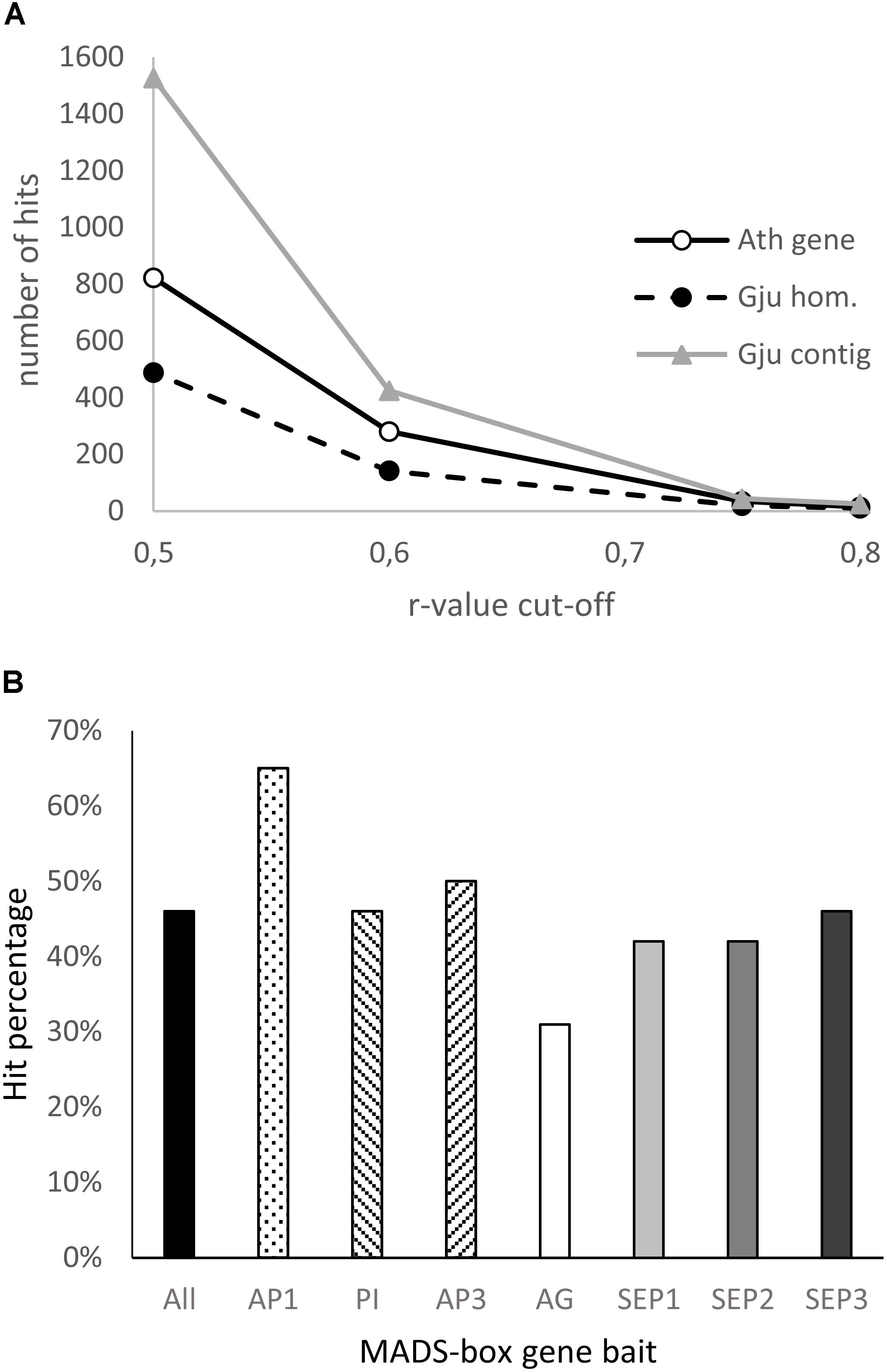
Figure 7. Analysis of the Grevillea juniperina homologs of genes co-expressed with the ABCE model MADS-box genes in Arabidopsis thaliana. (A) Number of A. thaliana genes (At gene) of the GRN and their homologous sequences in G. juniperina (Gju hom) plotted relative to the Pearson correlation coefficient (r-value) cut-off. The number of corresponding contigs for each homolog (Gju contig) are also plotted. (B) Percentage of the 25 A. thaliana MADS-box best-correlated genes having at least one homolog in G. juniperina. The list of the 25 best correlated genes was done using each of the seven A. thaliana MADS-box genes as a bait, or a compilation of the best correlated to all the ABCE MADS-box genes (all).

Table 1. List of genes co-expressed with the ABCE MADS-box genes in Arabidopsis thaliana (r-value ≥ 0.75, in bold the 14 AGI-ID with an r-value ≥ 0.8; AGI ID, Arabidopsis Genome Initiative, gene identifier).
Analyses of the 25 best correlated genes to each of the seven A. thaliana ABCE MADS-box genes showed an overrepresentation of homologs of the AP1-correlated genes and an underrepresentation of the AG-correlated genes in the G. juniperina annotated transcriptome. The analysis confirmed that no homologs of AP3 were found in the G. juniperina transcriptome (Table 1). Nevertheless, the percentage of G. juniperina homologs of AP3-correlated genes was similar to the percentage of the homologs of PI-correlated genes, or to the percentage computed over all the best ABCE MADS-box correlated genes (Figure 7B).
Gene Expression During Floral Development
Homologs of genes that play a role in floral meristem termination, organ identity/development and boundaries between organs in Arabidopsis thaliana were identified in the G. juniperina transcriptome annotated against the Arabidopsis proteome. In particular, genes expressed in carpel and/or ovule development in A. thaliana (Arnaud and Pautot, 2014; Maugarny et al., 2016) and in other distantly related taxa (Fourquin et al., 2005; Reymond et al., 2012) were targeted, since the gynoecium can easily be separated from perianth with adnate stamens tissues, and its growth pattern is a major component of floral symmetry. In addition, the two previously characterized ProtCYC1 and ProtCYC2 genes (Citerne et al., 2017) respectively, GjuCYC1 and GjuCYC2, were included in the expression analysis.
The expression pattern of these genes was examined in dissected organs from 6 to 7 mm buds using qRT-PCR. GjuCRC and GjuSTM had a predominant expression in the gynoecium compared to the dorsal and ventral organs, while GjuAP2, GjuBEL1, GjuCYC2 and GjuPI2 were predominantly expressed in the dorsal and ventral organs. GjuSEP1 and GjuSEP3 were significantly induced in the ventral organs compared to the gynoecium. Finally, GjuCYC1 was predominantly expressed in the ventral organs (Figure 8).
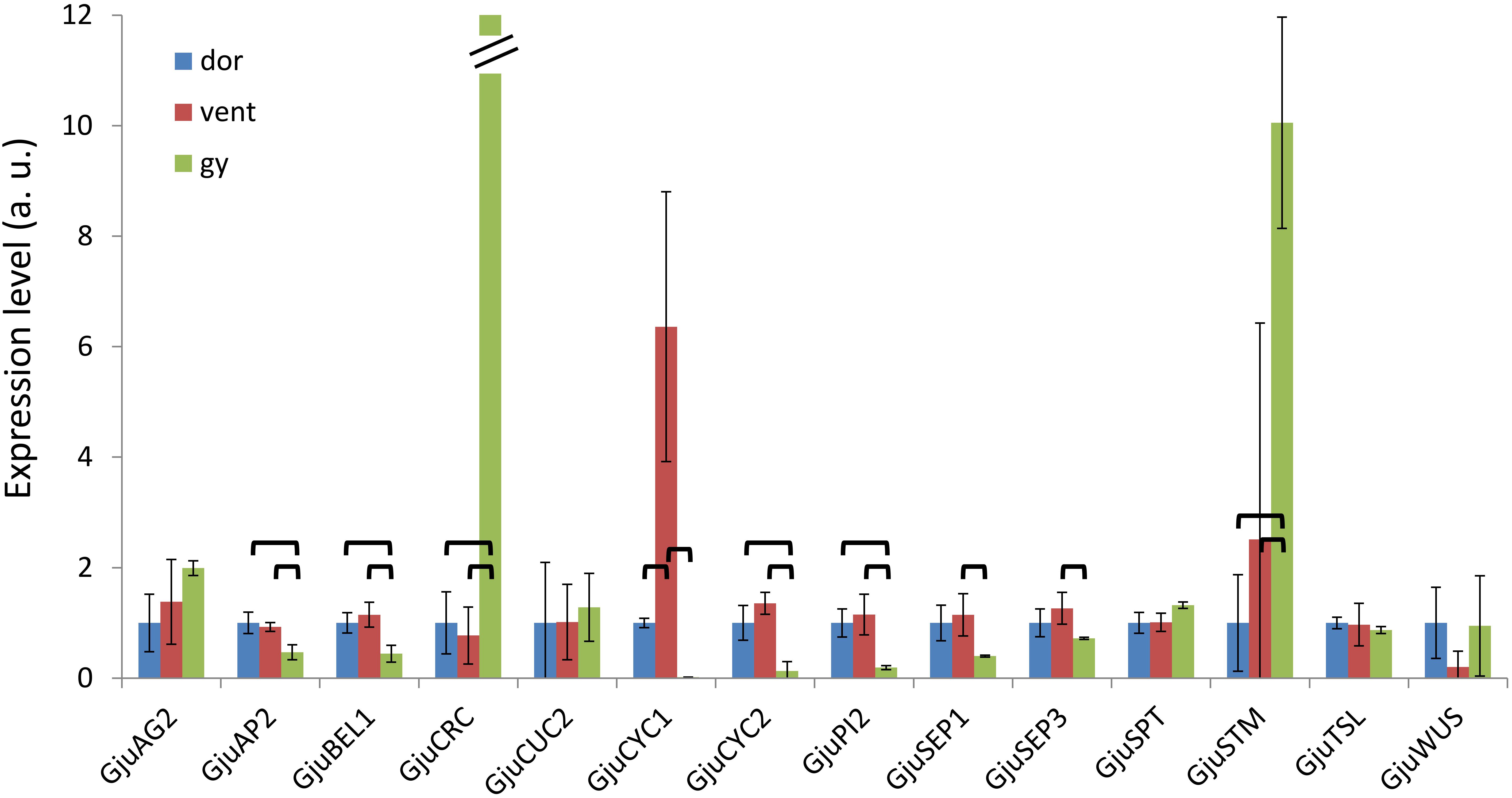
Figure 8. Expression patterns of 14 floral genes assessed by qRT-PCR in floral dissections of Grevillea juniperina. Organs were dissected from 6–7 mm buds; dor, dorsal tepals and adnate stamens; vent, ventral tepals and adnate stamens; gy, gynoecium. For a better display, the normalized expression levels of each gene in this figure are shown relative to the dor sample. In this figure, the expression level of GjuCRC in the gynoecium is 844. The error bars are calculated from three biological replicates. The statistically significant pairwise comparisons are indicated with brackets.
Discussion
By describing specific developmental features of flowers within Proteaceae, we found that the adnation of stamens to tepals takes place at early developmental stages, and that the establishment of bilateral symmetry coincides with asymmetrical growth of the single carpel. The transcriptome data obtained in parallel provides a wide array of genes expressed during floral development, opening the way for future studies to look at the expression of selected candidates within specific organs and/or at specific developmental stages, as discussed below.
Floral Development
In angiosperms, the initiation of floral organs typically follows a highly conserved pattern, with a centripetal initiation of the organs from the outermost to the innermost (generally the gynoecium) (Endress, 2006). However, there is no general rule as to when symmetry is established during development (Endress, 1999). In species with zygomorphic flowers, bilateral symmetry can be visible very early on during development; alternatively, floral buds can remain actinomorphic until late stages of development. Furthermore, species with actinomorphic flowers at anthesis may undergo transitory bilateral symmetry of the floral bud during development (reviewed in Reyes et al., 2016). At the gynoecium level, the type of symmetry is conditioned by the number of carpels and their closure pattern, which can be either ascidiate or plicate (Endress, 2015; Sokoloff et al., 2017). In species with a single plicate carpel presenting a marked cleft during development, as it is the case in Proteaceae (Douglas and Tucker, 1996b), the gynoecium becomes zygomorphic as soon as the cleft begins to form. In most subfamilies of Proteaceae, the cleft is typically oriented along the dorso-ventral axis, facing the adaxial tepal. In Grevilloideae, however, the situation is more complex, as described in detail by Douglas and Tucker (1996b) (see also Johnson and Briggs, 1975), who suggested that the variation in carpel orientation could be related to the space left after stamen initiation. Our observations suggest asymmetrical growth of the carpel primordium from the very early stages, resulting in early zygomorphy of the gynoecium. The orientation of flowers within the flower pair is also variable (Douglas and Tucker, 1996a), making the interpretation of axes of symmetry even more complex. Such variation in carpel and flower orientation has been suggested to be related to the inflorescence structure and degree of compaction that are both variable across the family (Johnson and Briggs, 1975). In Embothrieae, the tribe to which our four study species belong, zygomorphy is widespread and has been inferred as the ancestral condition (Citerne et al., 2017). The different degrees of floral bilateral symmetry at anthesis in the species examined here could be related to differences in inflorescence morphology. How the shape and compaction of the inflorescence may condition flower symmetry remains to be explored. The fact that ProtCYC genes are expressed (although faintly in the case of ProtCYC1) in the gynoecium of G. juniperina (Citerne et al., 2017 and this study), and asymmetrically in the tepals and/or adnate stamens (ProtCYC1 only), suggests that these genes could play a role in the genetic control of floral zygomorphy in Proteaceae, as already suggested by Citerne et al. (2017).
The position of stamens, opposite the tepals, questions the identity of the perianth. In whorled flowers, the stamens usually alternate with adjacent perianth parts when in equal number. In the order Ericales, obhaplostemony (petal-opposed stamens) is believed to be derived from diplostemony (Schönenberger et al., 2005), with the loss of a stamen whorl. Detailed expression studies of the homologs of floral organ identity genes and their correlated genes found in the G. juniperina transcriptome may provide clues as to the homology of the perianth. Furthermore, the transcriptomic data may also help to investigate the genetic control of organ fusion.
A Transcriptomic Tool to Investigate the Genetic Bases of Floral Development
Transcriptomic data from Proteaceae species are scarce and have mostly been obtained from leaves for specific purposes: in Leucadendron to derive phylogenetic markers (Tonnabel et al., 2014), in Protea repens to study population differentiation in relation to local adaptation (Akman et al., 2016), in Banksia hookeriana to derive SSR markers (Lim et al., 2017) and in Gevuina avellana to study heteroblastic development under different light conditions (Ostria-Gallardo et al., 2016). In Macadamia integrifolia, a reference transcriptome was built from flowers, shoots and leaves, in parallel to a draft genome (Nock et al., 2016). Our Grevillea juniperina transcriptome was built from an equal mix of RNAs from young leaves and flower buds at three developmental stages, all posterior to organ inception. The general metrics of this transcriptome such as N50, mean length of contigs, percentage of annotated contigs, all fall within the range of values obtained in other Proteaceae species (e.g., Akman et al., 2016). The sequencing effort and mix of tissues were designed to cover a large panel of expressed genes including transcription factors, with a higher proportion of floral samples in the library used for RNA-seq to obtain a good coverage of transcripts expressed during floral development. We probably missed some rare floral transcripts, such as the ProtCYC2 gene that we previously found to be expressed in floral buds of the same species of Grevillea (Citerne et al., 2017), and whose expression was confirmed here. Equally, the absence of a homolog of the B-class gene AP3 is intriguing, because we were able to find at least one homologous contig for all the other A-, B-, C-, and E-class genes. The fact that we found G. juniperina homologs of A. thaliana genes with correlated expression with AP3 would rather be in favor of a technical bias. Gene expression analysis showed expected patterns for the homologs of the identity genes tested, including GjuAP2, a homolog of the A. thaliana AP2 gene, which is one of the two A-class genes and the only ABCE model gene that is not from the MADS-box gene family. TCP genes are also well represented in the transcriptome, with 14 and seven homologs belonging to class I and class II subfamilies, respectively, which is comparable to the size of the family in many eudicots. One sequence matched the previously characterized ProtCYC1 gene, which is homologous to the Arabidopsis CYC-like genes (TCP18/TCP12/TCP1). This gene is the only one exhibiting an asymmetric expression pattern in the floral dissection that could be related to the asymmetry of the perianth and stamens, as already observed by Citerne et al. (2017) in this species and another zygomorphic Grevillea species.
ABCE MADS-box genes are master regulators of the floral organ development Gene Regulatory Network (GRN) that are subjected to strong functional constraints, in contrast with the great flexibility of organ morphogenesis (Davila-Velderrain et al., 2013; Becker and Ehlers, 2016). Genes co-expressed with these regulatory genes in Arabidopsis could serve as baits to identify conserved modules of connected genes in other species and analyze their evolution and expression patterns. A large proportion of genes with expression patterns that are highly correlated with floral organ identity gene expression in Arabidopsis thaliana have homologs in the G. juniperina transcriptome (10 out of 14). Available functional studies in A. thaliana indicate that these genes code for transcription factors and/or hormonal signaling pathway proteins, and are important regulators of flower organ development, sexual reproduction, and seed fertility (Ge et al., 2005; Szecsi et al., 2006; Hou et al., 2008; Varaud et al., 2011; Li et al., 2012; Hong et al., 2017). A less stringent correlation threshold gives a lesser proportion of homologous genes in the G. juniperina transcriptome. This may suggest that beyond a core set of genes whose expression patterns are strongly parallel to that of organ identity genes, other genes involved in flower development have diverged in the two species. It is worth noting the relative overrepresentation in G. juniperina of homologs of AP1-correlated genes. It remains to be seen whether this finding may be relevant for assessing the homology of undifferentiated perianth organs with sepals or petals. Among the genes expressed during carpel and ovule development in Arabidopsis, only GjuCRC and GjuSTM exhibit a predominant expression in the gynoecium of G. juniperina, while GjuAG2 was also found to be expressed in perianth and stamen tissues (both ventral and dorsal organs). Detailed expression analysis of these genes at the tissue level (using in situ hybridization for example) may provide insight into the processes involved in the monosymmetric growth of the gynoecium during floral ontogeny.
Floral evo–devo questions that concern Proteaceae are many, among which the homology of the undifferentiated perianth to a calyx or a corolla, the adnation of stamens, or the chronology and developmental processes of the acquisition of zygomorphy. Such issues could be addressed by combining developmental and molecular approaches, for example using RNA-seq at different floral developmental stages in a panel of well-chosen species. The G. juniperina reference transcriptome will be a valuable resource for all these future studies. It will be a reference for identifying differentially expressed genes related to the establishment of symmetry at appropriate developmental stages in different floral organs, and for investigating the gene network underlying tepal development, providing insights into the homology of the colored perianth organs. Choosing appropriate developmental stages may be facilitated by High Resolution X-Ray Computed Tomography giving access to early developmental stages without dissection, which is especially valuable for species with young compact and tough inflorescences.
Author Contributions
SN and CD designed the project. HS, FS, YS, JS, and SN performed the developmental analysis. ED and JY generated RNA-seq data and transcriptome assembly. JJ, NCS, YD, HC, CD, and FS annotated and mined transcriptomic data. ED and MLG performed the expression analysis. All authors contributed to the manuscript.
Funding
This work was funded by a PRES UNIVERSUD grant for the SPAM project, a 2012 grant from IFR 87 ‘La Plante et son Environnement’ and by the Agence Nationale de la Recherche (ANR-07-BLAN-0112 grant). HC and FS were funded by ANR and PRES grants, respectively.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge Peter Weston and Peter Olde for providing some of the plant material, Véronique Brunaud for her work on the first version of the transcriptome assembly, Florian Jabbour and Dmitry Sokoloff for helpful discussions on floral developmental, and Véronique Normand for her technical assistance in the lab.
The GQE-Le Moulon and Institute of Plant Sciences Paris Saclay benefit from the support of the LabExSaclay Plant Sciences-SPS (ANR-10-LABX-0040-SPS).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00018/full#supplementary-material
Footnotes
- ^http://www.environment.gov.au/science/abrs/online-resources/flora-of-australia-online
- ^http://www.bioinfogenome.net/OrthoVenn/
- ^https://www.ebi.ac.uk/Tools/msa/muscle/
- ^https://www.arabidopsis.org/
- ^https://www.ncbi.nlm.nih.gov/
- ^https://phytozome.jgi.doe.gov/
- ^http://translatorx.co.uk/
References
Akman, M., Carlson, J. E., Holsinger, K. E., and Latimer, A. M. (2016). Transcriptome sequencing reveals population differentiation in gene expression linked to functional traits and environmental gradients in the South African shrub Protea repens. New Phytol. 210, 295–309. doi: 10.1111/nph.13761
Arnaud, N., and Pautot, V. (2014). Ring the BELL and tie the KNOX: roles for TALES in gynoecium development. Front. Plant Sci. 5:93. doi: 10.3389/fpls.2014.00093
Bartlett, M. E., Williams, S. K., Taylor, Z., DeBlasio, S., Goldshmidt, A., Hall, D. H., et al. (2015). The maize PI/GLO ortholog Zmm16/sterile tassel silky ear1 interacts with the zygomorphy and sex determination pathways in flower development. Plant Cell 27, 3081–3098. doi: 10.1105/tpc.15.00679
Becker, A., and Ehlers, K. (2016). Arabidopsis flower development—of protein complexes, targets, and transport. Protoplasma 253, 219–230. doi: 10.1007/s00709-015-0812-7
Broholm, S. K., Tätiharju, S., Laitinen, R. A. E., Albert, V. A., Heeri, T. H., and Elomaa, P. (2008). A TCP domain transcription factor controls flower type specification along the radial axis of the Gerbera (Asteraceae) inflorescence. Proc. Nat. Acad. Sci. U.S.A. 105, 9117–9122. doi: 10.1073/pnas.0801359105
Busch, A., and Zachgo, S. (2007). Control of corolla monosymmetry in the Brassicaceae Iberis amara. Proc. Nat. Acad. Sci. U.S.A. 104, 16714–16719. doi: 10.1073/pnas.0705338104
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Causier, B., Schwarz-Sommer, Z., and Davies, B. (2010). Floral organ identity: 20 years of ABCs. Sem. Cell Dev. Biol. 21, 73–79. doi: 10.1016/j.semcdb.2009.10.005
Citerne, H. L., Reyes, E., Le Guilloux, M., Delannoy, E., Simonnet, F., Sauquet, H., et al. (2017). Characterization of CYCLOIDEA-like genes in Proteaceae, a basal eudicot family with multiple shifts in floral symmetry. Ann. Bot. 119, 367–378. doi: 10.1111/nph.13761
Clark, J. I., and Coen, E. S. (2002). The cycloidea gene can respond to a common dorsoventral prepattern in Antirrhinum. Plant J. 30, 639–648. doi: 10.1046/j.1365-313X.2002.01310.x
Coen, E., and Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37. doi: 10.1038/353031a0
Cubas, P., Lauter, N., Doebley, J., and Coen, E. (1999). The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 18, 215–222. doi: 10.1046/j.1365-313X.1999.00444.x
Davila-Velderrain, J., Servin-Marquez, A., and Alvarez-Buylla, E. R. (2013). Molecular evolution constraints in the floral organ specification gene regulatory network module across 18 angiosperm genomes. Mol. Biol. Evol. 31, 560–573. doi: 10.1093/molbev/mst223
Douglas, A. W., and Tucker, S. C. (1996a). Inflorescence ontogeny and floral organogenesis in Grevilloideae (Proteaceae), with emphasis on the nature of the flower pairs. Int. J. Plant Sci. 157, 341–372. doi: 10.1086/297354
Douglas, A. W., and Tucker, S. C. (1996b). The developmental basis of diverse carpel orientations in Grevilleoideae. Int. J. Plant Sci. 157, 373–397. doi: 10.1086/297355
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Endress, P. K. (1999). Symmetry in flowers: diversity and evolution. Int. J. Plant Sci. 160, S3–S23. doi: 10.1086/314211
Endress, P. K. (2006). Angiosperm floral evolution: morphological developmental framework. Adv. Bot. Res. 44, 1–61. doi: 10.1016/S0065-2296(06)44001-5
Endress, P. K. (2015). Patterns of angiospermy development before carpel: sealing across living angiosperms: diversity, and morphological and systematic aspects. Bot. J. Lin. Soc. 178, 556–591. doi: 10.1111/boj.12294
Feng, X., Zhao, Z., Tian, Z., Xu, S., Luo, Y., Cai, Z., et al. (2006). Control of petal shape and floral zygomorphy in Lotus japonicus. Proc. Natl. Acad. Sci. U.S.A. 103, 49670–44975. doi: 10.1073/pnas.0600681103
Fourquin, C., Vinauger-Douard, M., Folgliani, B., Dumas, C., and Scutt, C. P. (2005). Evidence that CRABS CLAW and TOUSLED have conserved their roles in carpel development since the ancestor of the extant angiosperms. Proc. Natl. Acad. Sci. U.S.A. 102, 4649–4654. doi: 10.1073/pnas.0409577102
Ge, X., Dietrich, C., Matsuno, M., Li, G., Berg, H., and Xia, Y. (2005). An Arabidopsis aspartic protease functions as an anti-cell-death component in reproduction and embryogenesis. EMBO Rep. 6, 282–288. doi: 10.1038/sj.embor.7400357
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hileman, L. C. (2014). Trends in flower symmetry evolution revealed through phylogenetic and developmental genetic advances. Philos. Trans. R. Soc. B 369:20130348. doi: 10.1098/rstb.2013.0348
Hong, L., Brown, J., Segerson, N. A., Rose, J. K., and Roeder, A. H. (2017). CUTIN SYNTHASE 2 maintains progressively developing cuticular ridges in Arabidopsis sepals. Mol. Plant 10, 560–574. doi: 10.1016/j.molp.2017.01.002
Hou, X., Hu, W. W., Shen, L., Lee, L. Y., Tao, Z., Han, J. H., et al. (2008). Global identification of DELLA target genes during Arabidopsis flower development. Plant Physiol. 147, 1126–1142. doi: 10.1104/pp.108.121301
Huang, T., and Irish, V. (2015). Temporal control of plant organ growth by TCP transcription factors. Curr. Biol. 25, 1765–1770. doi: 10.1016/j.cub.2015.05.024
Johnson, L. A., and Briggs, B. G. (1975). On the Proteaceae – the evolution and classification of a southern family. Bot. J. Lin. Soc. 70, 83–182. doi: 10.1111/j.1095-8339.1975.tb01644.x
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: smart Model Selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Li, X. C., Zhu, J., Yang, J., Zhang, G. R., Xing, W. F., Zhang, S., et al. (2012). Glycerol-3-phosphate acyltransferase 6 (GPAT6) is important for tapetum development in Arabidopsis and plays multiple roles in plant fertility. Mol. Plant 5, 131–142. doi: 10.1093/mp/ssr057
Lim, L. S., D’Agui, H. M., Enright, N. J., and He, T. (2017). Characterization of leaf transcriptome in Banksia hookeriana. Genomics Proteomics Bioinformatics 15, 49–56. doi: 10.1016/j.gpb.2016.11.001
Luo, D., Carpenter, R., Copsey, L., Vincent, C., Clark, J., and Coen, E. (1999). Control of organ asymmetry in flowers of Antirrhinum. Cell 99, 367–376. doi: 10.1016/S0092-8674(00)81523-8
Luo, D., Carpenter, R., Vincent, C., Copsey, L., and Coen, E. (1996). Origin of floral asymmetry in Antirrhinum. Nature 383, 794–799. doi: 10.1038/383794a0
Maugarny, A., Gonçalves, B., Arnaud, N., and Laufs, P. (2016). “CUC transcription factors: to the meristem and beyond,” in Plant Transcription Factors. Evolutionary, Structural, and Functional Aspects, ed. D. H. Gonzalez (Cambridge, MA: Academic Press), 229–247. doi: 10.1016/B978-0-12-800854-6.00015-4
Mondragon-Palomino, M. (2013). Perspectives on MADS-box expression during orchid flower evolution and development. Front. Plant Sci. 4:377. doi: 10.3389/fpls.2013.00377
Nakasugi, K., Crowhurst, R., Bally, J., and Waterhouse, P. (2014). Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS One 9:e91776. doi: 10.1371/journal.pone.0091776
Nicolas, M., and Cubas, P. (2016). TCP factors: new kids on the signaling block. Curr. Opin. Plant Biol. 33, 33–41. doi: 10.1016/j.pbi.2016.05.006
Nock, C. J., Baten, A., Barkla, B. J., Furtado, A., Henry, R. J., and King, G. J. (2016). Genome and transcriptome sequencing characterizes the gene space of Macadamia integrifolia (Proteaceae). BMC Genomics 17:937. doi: 10.1186/s12864-016-3272-3
Ostria-Gallardo, E., Ranjan, A., Zumstein, K., Chitwood, D. H., Kumar, R., Townsley, B. T., et al. (2016). Transcriptomic analysis suggests a key role for SQUAMOSA PROMOTER BINDING PROTEIN LIKE, NAC and YUCCA genes in the heteroblastic development of the temperate rainforest tree Gevuina avellana (Proteaceae). New Phytol. 210, 694–708. doi: 10.1111/nph.13776
Preston, J. C., and Hileman, L. C. (2012). Parallel evolution of TCP and B-class genes in Commelinaceae flower bilateral symmetry. EvoDevo 3:6. doi: 10.1186/2041-9139-3-6
Reyes, E., Sauquet, H., and Nadot, S. (2016). Perianth symmetry changed at least 199 times in angiosperm evolution. Taxon 65, 945–964. doi: 10.12705/655.1
Reymond, M. C., Brunoud, G., Chauvet, A., Martinez-Garcia, J. F., Martin-Magniette, M.-L., Monéger, F., et al. (2012). A light-regulated genetic module was recruited to carpel development in Arabidopsis following a structural change to SPATULA. Plant Cell 24, 2812–2825. doi: 10.1105/tpc.112.097915
Roberts, W. R., and Roalson, E. H. (2017). Comparative transcriptome analyses of flower development in four species of Achimenes (Gesneriaceae). BMC Genomics 18:240. doi: 10.1186/s12864-017-3623-8
Sablowski, R. (2015). Control of patterning, growth, and differentiation by floral organ identity genes. J. Exp. Bot. 66, 1065–1073. doi: 10.1093/jxb/eru514
Sauquet, H., von Balthazar, M., Magallon, S., Doyle, J. A., Endress, P. K., Bailes, E. J., et al. (2017). The ancestral flower of angiosperms and its early diversification. Nat. Commun. 8:16047. doi: 10.1038/ncomms16047
Schönenberger, J., Anderberg, A. A., and Sytsma, K. J. (2005). Molecular phylogenetics and patterns of floral evolution in the Ericales. Int. J. Plant Sci. 166, 265–288. doi: 10.1086/427198
Schulz, M. H., Zerbino, D. R., Vingron, M., and Birney, E. (2012). Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092. doi: 10.1093/bioinformatics/bts094
Smart, M., and Roden, L. C. (2010). A small-scale RNA isolation protocol useful for high-throughput extractions from recalcitrant plants. S. Afr. J. Bot. 76, 375–379. doi: 10.1016/j.sajb.2010.01.002
Sokoloff, D. D., Nuraliev, M. S., Oskolski, A. A., and Remizowa, M. V. (2017). Gynoecium evolution in angiosperms: monomery, pseudomonomery, and mixomery. Moscow Univ. Biol. Sci. Bulletin 72, 97–108. doi: 10.3732/ajb.1600269
Spencer, V., and Kim, M. (2018). Re”CYC”ling molecular regulators in the evolution and development of flower symmetry. Semin. Cell Dev. Biol. 79, 16–26. doi: 10.1016/j.semcdb.2017.08.052
Staedler, Y. M., Masson, D., and Schönenberger, J. (2013). Plant tissues in 3D via X-ray tomography: simple contrasting methods allow high resolution imaging. PLoS One 8:e75295. doi: 10.1371/journal.pone.0075295
Szecsi, J., Joly, C., Bordji, K., Varaud, E., Cock, J. M., Dumas, C., et al. (2006). BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J. 25, 3912–3920. doi: 10.1038/sj.emboj.7601270
Theißen, G., Kim, J. T., and Saedler, H. (1996). Classification and phylogeny of the MADS-box multigene family suggest defined roles of MADS-box gene subfamilies in the morphological evolution of eukaryotes. J. Mol. Evol. 43, 484–516. doi: 10.1007/BF02337521
Tonnabel, J., Olivieri, I., Mignot, A., Rebelo, A., Justy, F., Santoni, S., et al. (2014). Developing nuclear DNA phylogenetic markers in the angiosperm genus Leucadendron (Proteaceae): a next-generation sequencing transcriptomic approach. Mol. Phyl. Evol. 70, 37–46. doi: 10.1016/j.ympev.2013.07.027
Toufighi, K., Brady, S. M., Austin, R., Ly, E., and Provart, N. J. (2005). The Botany Array Resource: e-Northern, expression angling, and promoter analyses. Plant J. 43, 153–163. doi: 10.1111/j.1365-313X.2005.02437.x
Tucker, S. C. (1999). Evolutionary lability of symmetry in early floral development. Int. J. Plant Sci. 160(6 Suppl.), S25–S39. doi: 10.1086/314212
Varaud, E., Brioudes, F., Szecsi, J., Leroux, J., Brown, S., Perrot-Rechenmann, C., et al. (2011). AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell 23, 973–983. doi: 10.1105/tpc.110.081653
Wang, Y., Coleman-Derr, D., Chen, G., and Gu, Y. Q. (2015). OrthoVenn : a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 43, W78–W84. doi: 10.1093/narlgkv487
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol 35, 543–548. doi: 10.1093/molbev/msx319
Weston, P. H. (2007). “Proteaceae,” in The Families and Genera of Vascular Plants, Vol. 9, ed. K. Kubitski (Berlin: Springer), 364–404. doi: 10.1007/978-3-540-32219-1_42
Weston, P. H., and Barker, N. P. (2006). A new suprageneric classification of the Proteaceae, with an annotated checklist of genera. Telopea 11, 314–344. doi: 10.7751/telopea20065733
Keywords: Proteaceae, flower, development, floral symmetry, High Resolution X-Ray Computed Tomography, transcriptome, MADS-box genes, TCP genes
Citation: Damerval C, Citerne H, Conde e Silva N, Deveaux Y, Delannoy E, Joets J, Simonnet F, Staedler Y, Schönenberger J, Yansouni J, Le Guilloux M, Sauquet H and Nadot S (2019) Unraveling the Developmental and Genetic Mechanisms Underpinning Floral Architecture in Proteaceae. Front. Plant Sci. 10:18. doi: 10.3389/fpls.2019.00018
Received: 12 June 2018; Accepted: 08 January 2019;
Published: 25 January 2019.
Edited by:
Zhongchi Liu, University of Maryland, College Park, United StatesReviewed by:
Oriane Hidalgo, Royal Botanic Gardens, Kew, United KingdomHongzhi Kong, Institute of Botany (CAS), China
Verónica S. Di Stilio, University of Washington, United States
Copyright © 2019 Damerval, Citerne, Conde e Silva, Deveaux, Delannoy, Joets, Simonnet, Staedler, Schönenberger, Yansouni, Le Guilloux, Sauquet and Nadot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine Damerval, Y2F0aGVyaW5lLmRhbWVydmFsQHUtcHN1ZC5mcg== Sophie Nadot, c29waGllLm5hZG90QHUtcHN1ZC5mcg==