 Marta Pujol1,2
Marta Pujol1,2 Konstantinos G. Alexiou1,2
Konstantinos G. Alexiou1,2 Anne-Sophie Fontaine3
Anne-Sophie Fontaine3 Patricia Mayor4
Patricia Mayor4 Manuel Miras4Torben Jahrmann3
Manuel Miras4Torben Jahrmann3 Jordi Garcia-Mas1,2
Jordi Garcia-Mas1,2 Miguel A. Aranda4*
Miguel A. Aranda4*- 1Centre for Research in Agricultural Genomics (CRAG) CSIC-IRTA-UAB-UB, Plant and Animal Genomics Program, Barcelona, Spain
- 2Institut de Recerca i Tecnologia Agroalimentàries (IRTA), Genomics and Biotecnology Program, Barcelona, Spain
- 3Semillas Fitó S.A., Biotechnology Department, Barcelona, Spain
- 4Centro de Edafología y Biología Aplicada del Segura (CEBAS)-CSIC, Departamento de Biología del Estrés y Patología Vegetal, Murcia, Spain
Cucumber vein yellowing virus (CVYV) causes severe yield losses in cucurbit crops across Mediterranean countries. The control of this virus is based on cultural practices to prevent the presence of its vector (Bemisia tabaci) and breeding for natural resistance, which requires the identification of the loci involved and the development of molecular markers for linkage analysis. In this work, we mapped a monogenic locus for resistance to CVYV in cucumber by using a Bulked Segregant Analysis (BSA) strategy coupled with whole-genome resequencing. We phenotyped 135 F3 families from a segregating population between a susceptible pickling cucumber and a resistant Long Dutch type cucumber for CVYV resistance. Phenotypic analysis determined the monogenic and incomplete dominance inheritance of the resistance. We named the locus CsCvy-1. For mapping this locus, 15 resistant and 15 susceptible homozygous F2 individuals were selected for whole genome resequencing. By using a customized bioinformatics pipeline, we identified a unique region in chromosome 5 associated to resistance to CVYV, explaining more than 80% of the variability. The resequencing data provided us with additional SNP markers to decrease the interval of CsCvy-1 to 625 kb, containing 24 annotated genes. Markers flanking CsCvy-1 in a 5.3 cM interval were developed for marker-assisted selection (MAS) in breeding programs and will be useful for the identification of the target gene in future studies.
Introduction
Cucumber vein yellowing virus (CVYV) is an ipomovirus (family Potyviridae) that is transmitted in a semi-persistent manner by the whitefly Bemisia tabaci. CVYV was first reported in Eastern countries of the Mediterranean basin (Israel, Jordan, Turkey, and Cyprus) later expanding to Western Mediterranean countries including Spain, Portugal, France, and Tunisia (reviewed in Navas-Castillo et al., 2014). CVYV infects cucurbits, causing symptoms of variable intensity. In melon (Cucumis melo L.) and cucumber (Cucumis sativus L.), it causes a typical severe vein clearing often followed by generalized chlorosis and necrosis. In cucurbit-producing areas of heavy B. tabaci infestation, it can cause epidemics with massive yield losses and dramatic economic consequences. Although significant diversity has been reported for this virus, epidemics in Western Mediterranean countries seem to be associated to genetically uniform virus populations (Desbiez et al., 2019), perhaps as a consequence of single virus introductions followed by rapid epidemic expansions (Janssen et al., 2007). At the start of the CVYV epidemics, disease control relied heavily on early detection (Martínez-García et al., 2004) and eradication, and whitefly control. Sources of resistance were soon identified in cucumber (e.g. Picó et al., 2003) and indeed, commercial seed companies are currently selling cucumber hybrids resistant to CVYV, which represent an excellent solution for disease control. Resistant accessions, varieties and hybrids seem to share the common characteristic that resistance is partial; symptoms in inoculated plants are mild or absent, and the virus can be detected infecting systemically the so-called resistant plants, although at reduced levels as compared to susceptible controls (e.g. Galipienso et al., 2013).
Breeding for CVYV resistance of cucumber varieties requires the development of molecular markers linked to the trait of interest, as pathology tests are labor-intensive and time-consuming. For molecular breeding, several genetic and genomics resources are available for cucumber, which have substantially increased after the release of reference genomes (Huang et al., 2009; Wóycicki et al., 2011; Yang et al., 2012). Cucumber has a relatively small genome (367 Mb, 2n = 2x = 14) with a very narrow genetic base (Staub et al., 2008; Wóycicki et al., 2011). The availability of high-density consensus maps and whole genome sequencing has facilitated the identification of NB-LRR resistance genes in the cucumber genome and the map-based cloning of candidate genes (Ren et al., 2009; Yang et al., 2013). These achievements are the basis for efficient marker-assisted selection (MAS). In this sense, Bulked Segregant Analysis (BSA) was developed as a rapid method for the detection of molecular markers linked to target traits in mapping populations (Michelmore et al., 1991). The principle of BSA is the selection of a small group of individuals from a segregating population that belong to phenotypic contrasting extremes of the target trait. These individuals are then pooled in two bulks, and fingerprinted to obtain genetic polymorphisms. When the trait is monogenic, the number of individuals per bulk can be reduced to 10-20, but in the case of quantitative trait loci (QTL) this number should be increased (Sun et al., 2010; Takagi et al., 2013; Zou et al., 2016). With the improvement of technologies and the significant reduction of next generation sequencing (NGS) costs, whole-genome resequencing has been coupled to BSA. The combination of BSA with NGS (BSA-seq) has accelerated the identification of tightly linked markers for important traits, improving the resolution of maps for gene identification and QTL mapping (Zou et al., 2016). In cucumber, BSA-seq has been successfully applied for mapping traits such as early flowering (Lu et al., 2014), flesh thickness (Xu et al., 2015) and downy mildew resistance (Win et al., 2017).
The aims of this work were to study the inheritance of the resistance conferred by the resistant accession CE0749 in a segregating F2:3 population, to map the CsCvy-1 locus by using a BSA-seq approach, and to develop molecular markers that are easily transferable to cucumber breeding programs.
Materials and Methods
Plant Material and Phenotyping for CVYV Resistance
Accession CE0754 (hereafter PS), a CVYV-susceptible pickling cucumber, was crossed with accession CE0749 (hereafter PR), a CVYV-resistant Long Dutch type cucumber, to obtain the F1, F2 and F2:3 segregating populations used to perform the genetic mapping of the resistance trait. PS, PR, F1, F2, and F2:3 plants were inoculated mechanically with CVYV-AILM (Martínez-García et al., 2004) by rubbing recently-expanded cotyledons with extracts from CVYV-infected cucumber plants (cv. SMR-58) and were re-inoculated three days after. To measure virus accumulation in PS, PR and F1-inoculated plants, we followed procedures described by Marco et al. (2003); quantitative dot-blot hybridization was done using the probe described by (Martínez-García et al., 2004). Plants were sampled at 9, 16, 23, and 30 days post inoculation (dpi) taking three leaf discs measuring 8 mm in diameter per leaf sampled. Symptoms were scored using a 0–3 scale: (0) No symptoms; (1) mild chlorotic mottling in young but fully-expanded leaves in interveinal petiole-proximal leaf areas; (2) similar to (1) plus vein yellowing evident in fully-expanded leaves and incipient in young developing leaves; (3) obvious vein yellowing in all leaves, including young developing leaves, chlorotic mosaics in fully-expanded leaves and overall plant growth reduction. A minimum of nine plants were used per treatment. Plants were kept in an insect-proof glasshouse, with temperature control set at 26°C/18°C (day/night) throughout the experiments.
DNA Extraction and NGS Sequencing
Young leaves from the parental lines and the F2 population were collected, frozen in liquid nitrogen, and stored at −80°C. DNA was extracted following the CTAB method (Doyle, 1991), adding a purification step using Phenol : Chloroform:Isoamyl alcohol (25:24:1). The integrity of DNA was evaluated by agarose gel electrophoresis and quantified with the PicoGreen® dsDNA Assay Kit (Life Technologies) according to the manufacturer’s protocol. For NGS sequencing, we pooled equimolar concentrations of DNA from 15 homozygous resistant F2 plants (R-Bulk), and from 15 homozygous susceptible F2 plants (S-Bulk). Twenty µg aliquots of each bulked DNA and both parental lines were sent to the National Centre for Genomic Analysis (CNAG-CRG, Barcelona, Spain) for library construction and sequencing. Libraries of 300 bp and 500 bp average insert size for bulks and parents, respectively, were sequenced with Illumina HiSeq 2000 (Illumina, Inc. San Diego, CA, USA), generating 2 × 100 bp paired-end reads for both datasets.
Conventional Linkage Mapping
A set of 172 polymorphic cucumber SNP markers, distributed across the seven chromosomes of the cucumber genome, were selected between PR and PS from the resequencing data (Supplementary Table 1). Kompetitive Allele Specific PCR (KASP, www.lgcgroup.com) was used for genotyping a subset of 72 individuals, with the 172 SNPs converted to KASP markers, following the protocol of LGC Genomics. A genetic map was constructed using JoinMap® 5 (Kyazma, B.V.), with 172 SNP markers data and the phenotypic data used as another marker (CsCvy-1) due to the monogenic inheritance of the trait. Three F2 individuals and one SNP marker were excluded because of the high amount of missing data. Two more SNP markers were excluded for being identical (similarity value = 1.000) to other nearby markers. Linkage analysis and marker order were performed with the regression mapping algorithm, and genetic distance was calculated using the Kosambi mapping function (Crow, 1990). QTL analysis was performed using MapQTL6® (Kyazma B.V.) using both interval mapping and Kruskal-Wallis (KW) analysis.
Variant Detection and BSA-seq Analysis
Variant Detection and Functional Effect Annotation
In order to detect variants of the genome linked to the resistance against CVYV, a BSA-seq strategy was implemented in which two pools of F2 individuals were chosen depending on their phenotype. 15 homozygous resistant F2 individuals (R-bulk) and 15 homozygous susceptible F2 individuals (S-bulk) were selected for pooling and sequencing. Paired-end Illumina sequencing data from parental lines and the two bulks were trimmed (length ≥35 bp, with a mean sliding window of 4 bp phred quality score ≥20) using Trimmomatic (Bolger et al., 2014) and the output was quality checked using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Trimmed data were aligned versus the ChineseLong 9930 v3 assembly ftp://cucurbitgenomics.org/pub/cucurbit/genome/cucumber/Chinese_long/v3/) using the BWA-MEM algorithm (v0.7.16a-r1181; http://bio-bwa.sourceforge.net/bwa.shtml) with default parameters. After removal of unmapped reads and marking of PCR duplicates, variant calling was performed with samtools (v1.5; Li, 2011) using default parameters, except for the following: mapping quality ≥10 and base quality ≥20. Variant calling format (VCF) files were filtered by applying the following criteria: genotype quality ≥10, depth ≥10, biallelic sites, alternative allele frequency ≤ 0.9, no missing data.
Structural variant (SV) analysis between the two parents was conducted using DELLY (Rausch et al., 2012) and Pindel (Ye et al., 2009). Both programs were run with default parameters. Raw data underwent technical- and visual-based filtering. The technical filters applied were the following: read depth ≥10 in at least one sample, parents variable between them, variant size larger than 50 bp and smaller than 50,000 bp. Remained variants were visually inspected in IGV (Robinson et al., 2011) to avoid cases of false positives.
Annotation of the functional effect of the variants was done using snpEff (version 4.3t; (Cingolani et al., 2012).
BSA-seq Analysis
BSA-seq analysis was performed using the R package QTLseqr (Mansfeld and Grumet, 2018). More specifically, SNPs for the 2 bulks were first filtered using the function “filterSNPs”, by keeping positions with 30 ≤ total depth ≤ 150, 0.2 ≤ reference allele frequency ≤0.8 and genotype quality ≥30. For the QTL detection, the (Takagi et al., 2013) method was applied, implemented by the function “runQTLseqAnalysis”. Briefly, SNP-index was calculated for both S- and R-bulk by dividing the number of non-reference alleles with the total number of reads in a position. If the SNP-index was <0.3 in both bulks, the SNP was discarded. SNP-index values were calculated in sliding windows of 4.7 Mbp with a 10 kb step and the average SNP-index value for each window was recorded along the chromosome. In order to avoid regions that generate segregation distortion caused by other reasons other than artificial selection (Takagi et al., 2013), the SNP-index of the S-bulk was subtracted by the SNP-index of the R-bulk in order to obtain the Δ(SNP-index). If Δ(SNP-index) equaled to 1.0 then the allele originated from the R parent and if Δ(SNP-index) equaled to -1 the allele originated from the S parent. Confidence interval calculations in each SNP position (95% and 99%), considering the null hypothesis (Ho: no QTL), was done through a simulation analysis of 10,000 replications for two bulks that were randomly generated from the population and calculating, after each iteration, the SNP-index and the corresponding Δ(SNP-index) in the two simulated bulks.
Results
Phenotyping Analyses
We first analyzed symptom development and virus accumulation in PS, PR, and F1 plants in a time course experiment (Figure 1). Symptoms started to appear in inoculated PS plants as soon as 6 dpi (data not shown) and were conspicuous by 9 dpi, with plants showing obvious vein yellowing in all leaves and chlorotic mosaic in fully-expanded leaves (Figure 1A). Symptom display was delayed, and symptoms were clearly milder in PR-infected plants, although by 9 dpi all the plants showed mild interveinal chlorotic mottling in petiole-proximal leaf areas of fully expanded leaves (Figure 1A). Symptoms in F1 plants appeared at around 7 dpi, and by 9 dpi they were of intermediate severity between those of PS- and PR-infected plants; fully-expanded leaves showed chlorotic mottling but also mild vein yellowing, which was starting in young developing leaves (Figure 1A). The uniformity of symptoms in plants from each accession was remarkable, and thus each accession could be assigned to a symptom severity class without uncertainty for each observation time-point. Plants in this experiment were sampled and virus accumulation was measured, differentiating among samples from basal, medium and apical leaves (Figure 1B). Leaves from PR-infected plants accumulated significantly less virus than PS-infected leaves at all time points, except at 23 dpi in basal leaves, where no significant differences were found between both accessions. For F1-infected plants, differences in virus accumulation in leaves were less consistent, with an apparent trend suggesting intermediate levels of accumulation between those of PR and PS; statistically significant differences (P < 0.027; Kruskal-Wallis tests) were found among the three accessions at 16 dpi in basal leaves and at 23 and 30 dpi in intermediate leaves (Figure 1B). Thus, virus accumulation data was essentially consistent with data on symptom expression, and phenotyping of F1 individuals suggested incomplete dominance of the resistance trait. In any case, symptom scoring at 9 dpi appeared to be robust enough to discriminate between susceptible and resistant plants.
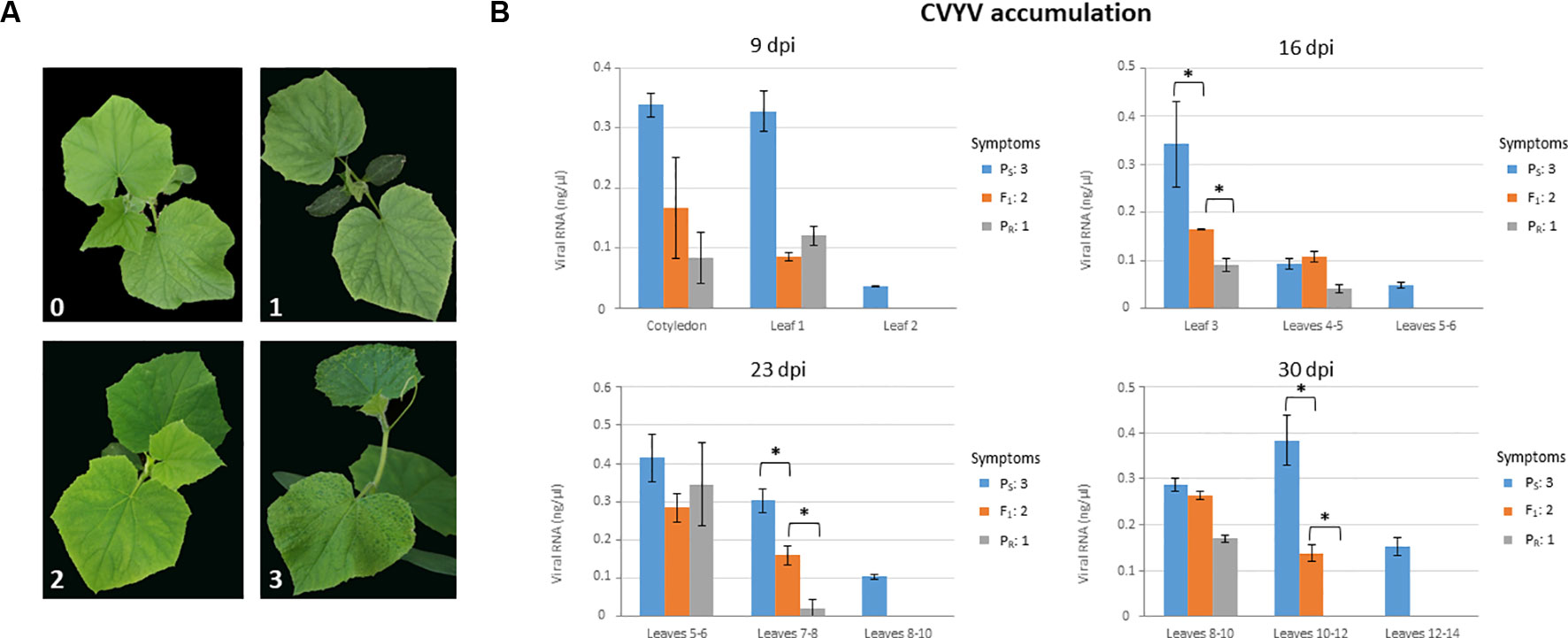
Figure 1 Phenotyping plants of the susceptible (PS) and resistant (PR) parental lines, and their F1, for CVYV susceptibility. Symptoms were scored and virus accumulation was measured at 9, 16, 23 and 30 days post-inoculation (dpi) (A) Symptoms in plants at 9 dpi. Symptoms could be assigned unequivocally to one of the following categories: (0) No symptoms; (1) mild chlorotic mottling in young but fully expanded leaves in interveinal petiole-proximal leaf areas; (2) similar to (1) plus vein yellowing evident in fully expanded leaves and incipient in young developing leaves; (3) obvious vein yellowing in all leaves, including young developing leaves, chlorotic mosaics in fully expanded leaves and overall plant growth reduction. (B) Virus accumulation was measured by quantitative dot-blot hybridization on total plant RNA extracts from basal, intermediate and apical leaves, as indicated for each graph. Three leaf discs (8 mm) were taken per leaf sampled. Virus accumulation in F1 plants was intermediate between the susceptible and resistant parental lines in basal and intermediate leaves at 16, 23 and 30 dpi, respectively; an asterisk marks statistically significant differences in Kruskal-Wallis tests (P < 0.027). A minimum of 9 plants were used per treatment. Symptom category (as in (A)) is indicated for each line on the right side of each graph for each time period after inoculation.
To determine the resistance genotype of F2 individuals, 12 individuals from each of 137 F2:3 families were inoculated and symptom scoring performed at 9 dpi (Supplementary Figure 1). Out of these, 135 families were unequivocally assigned to susceptible, resistant or segregating for resistance phenotypes and, therefore, the 135 F2 individuals could be classified as: 37 homozygous for resistance, 36 homozygous for susceptibility and 62 heterozygous, which fit well (χ2 value 0.63, P > 0.05) with a 1:2:1 segregation ratio. We propose the symbol CsCvy-1 for this monogenic incompletely-dominant resistance gene. For the BSA-seq analysis, two pools containing 15 F2 individuals homozygous for resistance and susceptibility to CVYV, respectively, were used.
Preliminary F2 Mapping
In parallel with the BSA-seq method, we performed F2 mapping with a subset of the population including the CsCvy-1 locus as a phenotypic marker. We used 172 polymorphic SNPs covering the cucumber genome: 24 SNPs in Chr01, 18 SNPs in Chr02, 34 SNPs in Chr03, 36 SNPs in Chr04, 21 SNPs in Chr05, 27 SNPs in Chr06, and 12 SNPs in Chr07 (Supplementary Table 1). We selected 72 F2 individuals (35 homozygous resistant, 34 homozygous susceptible, 2 heterozygous, and 1 individual without phenotypic data) for mapping. The 72 F2 individuals were genotyped, and most of the markers fitted the expected 1:2:1 segregation ratio. However, a group of markers on Chr05 showed a distorted segregation. This distortion was also found for the phenotypic marker CsCvy-1, due to the selection of individuals, with almost all of them being homozygous for this trait. The genetic map consisted of eight linkage groups (LGs) spanning 628 cM, with Chr03 split into two linkage groups (LG3A, LG3B) (Supplementary Figure 2, Supplementary Table 2). The average marker interval was 3.9 cM, with a maximum distance of 23.6 cM. The longest LG was LG4, with 115.1 cM, and the shortest was LG3A with 14 cM. The CsCvy-1 locus was mapped onto LG05, flanked by two markers, CVYV_121 and CVYV_122 in an interval of 5.3 cM (Figure 2A).
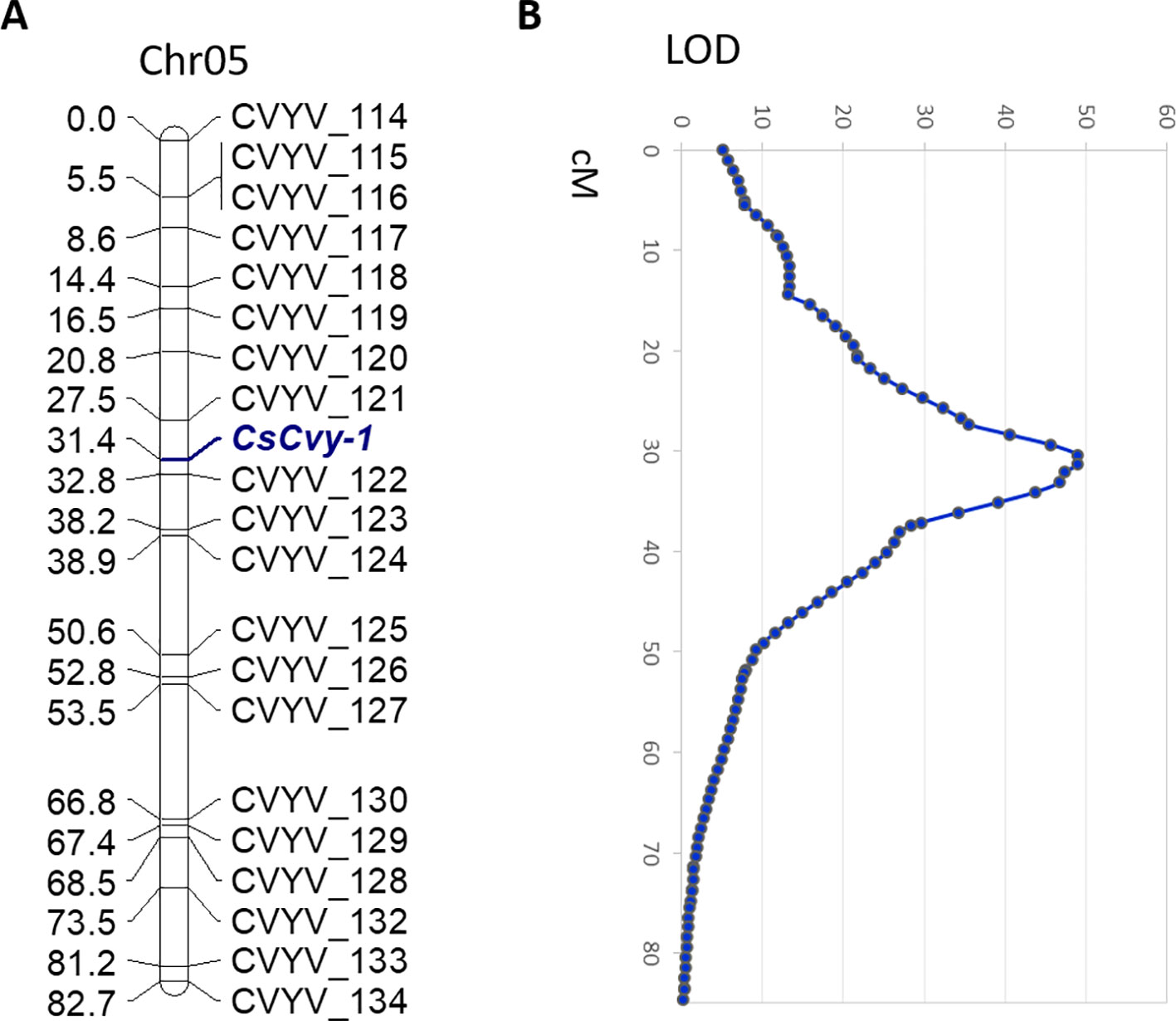
Figure 2 (A) Genetic linkage of SNP markers and the resistance locus CsCvy-1 in Chr05. Genetic distance is expressed as centiMorgans (cM). The CsCvy-1 gene is flanked by two markers in an interval of 5.3 cM. (B) Major QTL detected on Chr05 corresponding to CsCvy-1 locus.
In order to discard any other minor QTL involved in the resistance trait, a QTL analysis was performed. As expected, we obtained a single major QTL on LG05, co-localizing with CsCvy-1, with a LOD value of 49 explaining 95.8% of the variance (Figure 2B). No other significant QTLs were observed, in accordance to the monogenic inheritance of the trait.
Identification of CsCvy-1 Locus by BSA-seq
Libraries of parental lines (PS and PR), S-bulk and R-bulk were resequenced with the Illumina HiSeq2000 sequencer. In total, 274,709,874 paired-end clean reads were mapped, after trimming and adapter removal (Table 1).

Table 1 Results from the resequencing of PS, PR, S-bulk and R-bulk.
Small variant calling for the 4 datasets and subsequent variant filtering (see Material and Methods), generated 186,433; 215,735; 933,846 and 915,524 variants (SNPs and INDELs) for PS, PR, S-bulk and R-bulk, respectively, uniformly distributed throughout the genome (Supplementary Table 3). SNP variants from the 2 bulk datasets were used as input data in the QTLseqr R package for calculating SNP-index and Δ(SNP-index) for R- and S-bulks, based on the Takagi et al., 2013 method. Graphs for SNP-index of the R- and S-bulks and the Δ(SNP-index) were plotted (Figure 3). S-bulk was used as a reference dataset for the calculation of the Δ(SNP-index). BSA-seq analysis detected a single genomic region of 2,998,622 bp located in Chr05:5,088,092-8,208,448 bp, at a confidence interval higher than 0.99, associated with the CsCvy-1 locus. The highest Δ(SNP-index) value was -0.528 at position 7,678,525 bp whereas the average Δ(SNP-index) was found to be -0.523 (Figure 4A). This region contained the flanking markers selected in the preliminary mapping.
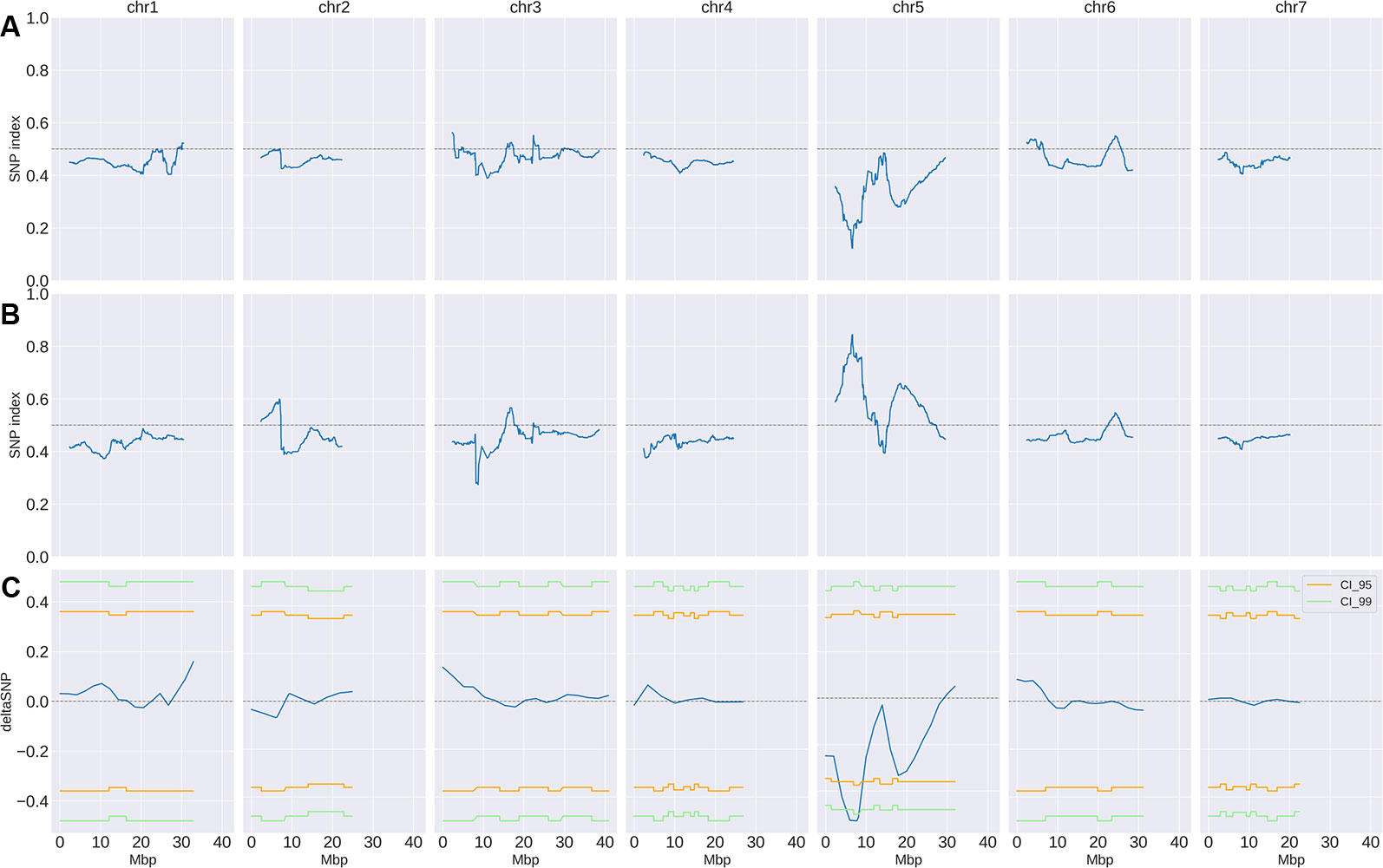
Figure 3 SNP-index and Δ(SNP-index) distribution (blue line). SNP-index for R-bulk (A) and S-bulk (B) was calculated using sliding windows of 4.7 Mbp in length with a step measuring 10 kb. The corresponding Δ(SNP-index) (C) was calculated as the difference SNP-index R-bulk – SNP-index S-bulk. Regions of statistical significance are detected as those that surpass the threshold of 0.95 (orange line) or 0.99 (green line) confidence intervals.
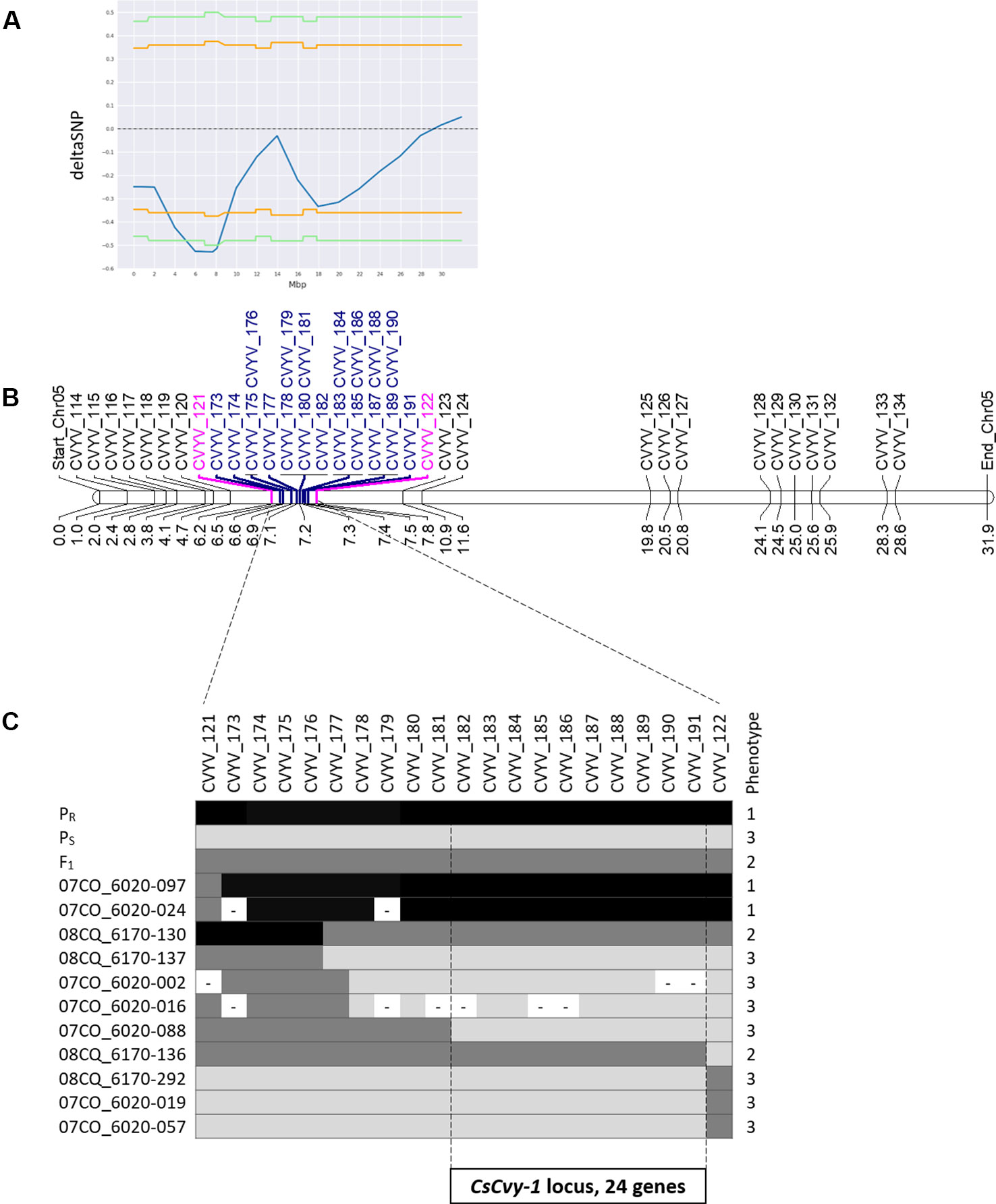
Figure 4 Scheme of fine mapping of the CsCvy-1 locus. (A) DeltaSNP graph on Chr05 with the statistical confidence interval of 0.95 (orange line) or 0.99 (green line) (B) Physical map of Chr05 with all the markers used for mapping the CsCvy-1 locus: preliminary mapping (black), flanking markers (magenta), fine mapping markers (blue). Distance is expressed in Mb on the bottom of the bar. (C) Genotyping result of recombinant individuals with the markers covering CsCvy-1 locus. Alleles are represented in three colors: black for homozygous PR allele, dark grey for heterozygous PR/PS, and light grey for homozygous PS allele.
Fine Mapping of CsCvy-1 Locus
In order to narrow down the interval of the CsCvy-1 locus, fine mapping was performed using the linked SNPs obtained by BSA-seq on Chr05 to design 19 SNP markers (Figure 4B, Supplementary Table 1). A subset of the F2 population was genotyped to identify plants with recombination events, by using the genotyping data of the preliminary mapping. Eleven recombinant informative plants delimited the candidate area of CsCvy-1 to a 626.5 kb interval, between the markers CVYV_181 and CVYV_122 (Figure 4C). This interval contains 24 annotated genes (Table 2). To further explore the genomic region containing the CsCvy-1 locus, we extended our previous small variant analysis with the detection of SVs between the parental lines. In total, we detected 2 SVs of more than 50 bp in length: one 55 bp deletion and a duplication measuring 41,644 bp (Supplementary Table 4).

Table 2 List of genes within the interval of CsCVY-1 locus in the Chinese Long 9930 v3 cucumber genome annotation, variants and structural variation analysis between PS and PR.
With the purpose of detecting alterations in potential candidate genes that could be responsible for the resistance phenotype, we performed an annotation of the effect of small variations in the 24 genes found in the CsCyv-1 region. In total, we annotated 45 SNPs/INDELs, of which 17 were highlighted for being within or close to coding sequences of 10 genes (Supplementary Table 5). The vast majority of variations (14 out of 17) were non-synonymous, and were annotated as modifiers or with low impact. Among the remaining four variants, three SNPs caused synonymous changes, and one insertion of 2 nucleotides caused a frameshift resulting in a premature stop codon (Supplementary Table 5). The three missense variants (moderate impact) were located in the coding sequence of CsaV3_5G011200, CsaV3_5G011220 and CsaV3_5G011240 genes, whereas the frameshift variant (high impact) was located in CsaV3_5G11180 gene. The frameshift caused the disruption of the CsaV3_5G11180 gene, coding for a serine/arginine repetitive matrix protein (SARMP) 2-like, at position 607 (Supplementary Figure 3). A search for conserved motifs detected a Constitutive Photomorphogenic 1 (COP1)-interacting protein signature. With regards to structural variations, we also detected a single duplication event in PR measuring 41,644 bp in length located in Chr05:7,195,565-7,237,209 that contained genes CsaV3_5G011170 to CsaV3_5G011220 (Table 2 and Supplementary Figure 4). Genes CsaV3_5G011170 and CsaV3_5G011190 encode for unknown proteins, CsaV3_5G11180 encodes the above-mentioned SARMP and genes CsaV3_5G011200 and CsaV3_5G011210 encode two RNA-dependent RNA polymerases (RDRs) 1a and 1b, respectively. In contrast, the deletion of 55 bp was located in an intergenic region.
Discussion
In cucumber, the disease caused by CVYV is a limiting factor for production in areas with high pressure from viruliferous whiteflies. In this work, we phenotyped an F2:3 segregating population identifying a single monogenic locus, CsCvy-1, that controls resistance to CVYV. The resistance conferred by this locus is partial, as PR plants showed viral accumulation in systemically infected leaves. However, the progression of the disease was very much reduced as compared to susceptible controls, the growth of the plants was not affected at all, and viral accumulation was significantly reduced as compared to PS and F1 plants. In cucumber, the accession C.sat-10 (Picó et al., 2003) was described as having partial resistance against CVYV, and a segregating F2 population was obtained to study the genetic control of this resistance (Picó et al., 2008); the segregation fitted a monogenic control with dominance. These features are very similar to what we have described here; however, in the case of C.sat-10, no mapping studies were performed to determine the localization of the locus in the cucumber genome. Thus, although our resistance data fit well with those from Picó et al. (2003; 2008), we cannot rule out that both resistances to CVYV characterized in cucumber are independent. By comparing with closely related species such as melon, Pitrat et al. (2012) evaluated a collection of 1,188 accessions for resistance against CVYV, and studied the inheritance in F1, F2 and BC progenies. Three loci were detected in their work: Cvy-1, controlling resistance in PI 164323 and necrosis in HSD 93-20-A; cvy-2, showing recessive tolerance in HSD 2458, and Cvy-3, showing dominance for severe mosaic symptoms in Ouzbèque 2.
Breeding for resistance requires the availability of highly linked markers that can be utilized for performing rapid and specific introgressions of the desired trait. For marker development, conventional gene mapping is based on the phenotyping and genotyping of a large number of individuals in a population, and it is time-consuming, costly and limiting in terms of the size of the population (Takagi et al., 2013). One way to improve conventional mapping is to use BSA-seq, in which the number of individuals to be analyzed can be narrowed down to two representative bulks, and at the same time as the mapping, the resequencing data offers the possibility of obtaining a high number of markers linked to the trait (Yang et al., 2013). BSA-seq has been successfully applied for the mapping of important agronomical traits in many crops such as rice (Abe et al., 2012; Takagi et al., 2013; Yang et al., 2013; Sun et al., 2018), lettuce (Huo et al., 2016), potato (Kaminski et al., 2016), soybean (Song et al., 2017), broccoli (Shu et al., 2018; Branham and Farnham, 2019) or sorghum (Han et al., 2015). Moreover, in cucurbits BSA-seq has enabled the identification of candidate genes for dwarfism (Dong et al., 2018), yellow skin (Dou et al., 2018) and light rind color (Oren et al., 2019) in watermelon; mapping flavor traits in melon (Zhang et al., 2016); or the identification of candidate genes for flesh thickness (Xu et al., 2015), aphid resistance (Liang et al., 2016), early flowering QTL (Lu et al., 2014), two major QTLs for downy mildew resistance (Win et al., 2017), and three major QTLs conferring subgynoecy (Win et al., 2019) in cucumber. In the present study, through the use of a BSA-seq strategy, the CsCvy-1 locus has been successfully mapped to a region of 2.9 Mb in chromosome 5, whereas no other regions of the genome exhibited significant association with the resistance. These results confirmed previous analysis performed with the conventional mapping approach and were later used for fine mapping. The BSA-seq analysis provided enough SNPs to fine map the trait within a narrow interval of 625 kb, containing only 24 annotated genes.
In order to identify candidate genes for CsCvy-1, we performed an analysis of small variants and structural variation around this locus. Most of the variations had a small, if any, predicted impact, except for the insertion of 2 nucleotides in the gene encoding a SARMP2-like, which causes a frameshift mutation in the coding sequence with the subsequent truncation of the protein. A functional analysis of the SARMP-like protein sequence detected a COP1-interacting protein signature. Interestingly, COP1 has been associated with plant pathogen resistance (Lim et al., 2018) and the Arabidopsis thaliana COP1-interacting protein is a positive regulator of ABA response (Ren et al., 2016), which is an essential regulator of plant immunity (Berens et al., 2017). Nevertheless, perhaps the most appealing modification within the CsCvy-1 region is the duplication of the 41 Kb fragment containing the genes encoding RDRs 1a and 1b (CsaV3_5G011200 and CsaV3_5G011210). RDRs are critical players in RNA silencing pathways; they are the key enzymes in the process of amplification of double-stranded RNAs that activate gene silencing after nuclease processing. A role for RDRs in antiviral immunity has long been acknowledged, in particular for members of the RDR1 and RDR6 clades (Willmann et al., 2012). For instance, the absence of a functional RDR1 in Nicotiana benthamiana can explain enhanced susceptibility to many viruses in this species (Yang et al., 2004). In relation to virus resistance in crop species, it has been recently demonstrated that the tomato genes Ty-1 and Ty-3 for resistance to tomato yellow leaf curl virus are alleles of an RDR gene (Verlaan et al., 2013). In cucumber, a recent report shows that the RDR1a and 1b genes have enhanced expression in natural or engineered lines showing broad virus resistance; importantly, one of the viruses tested was CVYV (Leibman et al., 2018). Taking these data together, a mechanistic explanation for our observations may consist of enhanced antiviral activity in the PR line as a consequence of enhanced RDR1a and/or 1b expression; this, in turn, would be the consequence of the described gene duplication. This is an attractive hypothesis that awaits further testing, although at least a good alternative candidate gene (i.e., the gene encoding a SARMP2-like protein) exists. From the point of view of resistance stability when confronted to different CVYV strains (Desbiez et al., 2019), the potential implication of RDR1a/b represents an optimistic perspective, given its broad spectrum of action (Leibman et al., 2018).
In conclusion, in this work we identified the monogenic locus CsCvy-1, inherited under incomplete dominance, in a short interval of 5.3 cM containing 24 genes. This is the first report where a CVYV resistance locus has been mapped in cucumber, and valuable molecular markers for MAS breeding programs have been developed. Moreover, our findings will be the basis for further map-based cloning and functional validation of the resistance gene.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher. Variation data were deposited to European Variation Archive (EVA: https://www.ebi.ac.uk/eva) with the project accession number PRJEB34274.
Author Contributions
MA, JG-M, TJ, and MP conceived and designed the research. PM, MM and MA provided the plant material and performed the tests with CVYV. A-SF conducted the conventional mapping. KA performed the bioinformatics analysis of the BSA-seq. MP conducted marker development, mapping analysis, and wrote the manuscript with important contributions from MA and KA. All authors read and approved the final manuscript.
Conflict of Interest
Authors A-SF and TJ were employed by company Semillas Fitó S.A.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Vanessa Alfaro and Daniel Alonso for their technical assistance. This work was supported by Semillas Fitó S.A., and by the CERCA Programme/Generalitat de Catalunya. The CRAG and CEBAS acknowledge financial support from the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa Programme for Centres of Excellence in R&D” 2016–2019 (SEV-2015-0533)” and grant AGL2015-72804-EXP, respectively.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01583/full#supplementary-material
References
Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., et al. (2012). Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. doi: 10.1038/nbt.2095
Berens, M. L., Berry, H. M., Mine, A., Argueso, C. T., Tsuda, K. (2017). Evolution of hormone signaling networks in plant defense. Annu. Rev. Phytopathol. 55, 401–425. doi: 10.1146/annurev-phyto-080516-035544
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Branham, S. E., Farnham, M. W. (2019). Identification of heat tolerance loci in broccoli through bulked segregant analysis using whole genome resequencing. Euphytica 215, 1–9. doi: 10.1007/s10681-018-2334-9
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 6, 80–92. doi: 10.4161/fly.19695
Desbiez, C., Caciagli, P., Wipf-Scheibel, C., Millot, P., Ruiz, L., Marian, et al. (2019). Evidence for long-term prevalence of cucumber vein yellowing virus in Sudan and genetic variation of the virus in Sudan and the Mediterranean Basin. Plant Pathol. 68, 1268–1275. doi: 10.1111/ppa.13055
Dong, W., Wu, D., Li, G., Wu, D., Wang, Z. (2018). Next-generation sequencing from bulked segregant analysis identifies a dwarfism gene in watermelon. Sci. Rep. 8, 1–7. doi: 10.1038/s41598-018-21293-1
Dou, J., Lu, X., Ali, A., Zhao, S., Zhang, L., He, N., et al. (2018). Genetic mapping reveals a marker for yellow skin in watermelon (Citrullus lanatus l.). PloS One 13, 1–15. doi: 10.1371/journal.pone.0200617
Doyle, J. (1991). “DNA Protocols for Plants,” in Molecular Techniques in Taxonomy. NATO ASI Series (Series H: Cell Biology), vol. 57. Eds. Hewitt, G. M., Johnston, A. W. B., Young, J. P. W. (Berlin, Heidelberg: Springer). doi: 10.1007/978-3-642-83962-7_18
Galipienso, L., Janssen, D., Rubio, L., Aramburu, J., Velasco, L. (2013). Cucumber vein yellowing virus isolate-specific expression of symptoms and viral RNA accumulation in susceptible and resistant cucumber cultivars. Crop Prot. 43, 141–145. doi: 10.1016/j.cropro.2012.08.004
Han, Y., Lv, P., Hou, S., Li, S., Ji, G., Ma, X., et al. (2015). Combining next generation sequencing with bulked segregant analysis to fine map a stem moisture locus in sorghum (Sorghum bicolor L. Moench). PloS One 10, 1–14. doi: 10.1371/journal.pone.0127065
Huang, S., Li, R., Zhang, Z., Li, L., Gu, X., Fan, W., et al. (2009). The genome of the cucumber, Cucumis sativus L. Nat. Genet. 41, 1275–1281. doi: 10.1038/ng.475
Huo, H., Henry, I. M., Coppoolse, E. R., Verhoef-Post, M., Schut, J. W., de Rooij, H., et al. (2016). Rapid identification of lettuce seed germination mutants by bulked segregant analysis and whole genome sequencing. Plant J. 88, 345–360. doi: 10.1111/tpj.13267
Janssen, D., Velasco, L., Martín, G., Segundo, E., Cuadrado, I. M. (2007). Low genetic diversity among Cucumber vein yellowing virus isolates from Spain. Virus Genes 34, 367–371. doi: 10.1007/s11262-006-0026-3
Kaminski, K. P., Kørup, K., Andersen, M. N., Sønderkær, M., Andersen, M. S., Kirk, H. G., et al. (2016). Next generation sequencing bulk segregant analysis of potato support that differential flux into the cholesterol and stigmasterol metabolite pools is important for steroidal glycoalkaloid content. Potato Res. 59, 81–97. doi: 10.1007/s11540-015-9314-4
Leibman, D., Kravchik, M., Wolf, D., Haviv, S., Weissberg, M., Ophir, R., et al. (2018). Differential expression of cucumber RNA-dependent RNA polymerase 1 genes during antiviral defence and resistance. Mol. Plant Pathol. 19, 300–312. doi: 10.1111/mpp.12518
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Liang, D., Chen, M., Qi, X., Xu, Q., Zhou, F., Chen, X. (2016). QTL Mapping by SLAF-seq and expression analysis of candidate genes for aphid resistance in cucumber. Front. Plant Sci. 7, 1–8. doi: 10.3389/fpls.2016.01000
Lim, G.-H., Hoey, T., Zhu, S., Clavel, M., Yu, K., Navarre, D., et al. (2018). COP1, a negative regulator of photomorphogenesis, positively regulates plant disease resistance via double-stranded RNA binding proteins. PloS Pathog. 14, e1006894. doi: 10.1371/journal.ppat.1006894
Lu, H., Lin, T., Klein, J., Wang, S., Qi, J., Zhou, Q., et al. (2014). QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor. Appl. Genet. 127, 1491–1499. doi: 10.1007/s00122-014-2313-z
Mansfeld, B. N., Grumet, R. (2018). QTLseqr: an R package for bulk segregant analysis with next-generation sequencing. Plant Genome 11, 0. doi: 10.3835/plantgenome2018.01.0006
Marco, C. F., Aguilar, J. M., Abad, J., Gómez-Guillamón, M. L., Aranda, M. A. (2003). Melon Resistance to Cucurbit yellow stunting disorder virus Is Characterized by Reduced Virus Accumulation. Phytopathology 93, 844–852. doi: 10.1094/phyto.2003.93.7.844
Martínez-García, B., Marco, C. F., Goytia, E., López-Abella, D., Serra, M. T., Aranda, M. A., et al. (2004). Development and use of detection methods specific for Cucumber vein yellowing virus (CVYV). Eur. J. Plant Pathol. 110, 811–821. doi: 10.1007/s10658-004-2491-7
Michelmore, R. W., Paran, I., Kesseli, R. V. (1991). Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. U. S. A. 88, 9828–9832. doi: 10.1073/pnas.88.21.9828
Navas-Castillo, J., López-Moya, J. J., Aranda, M. A. (2014). Whitefly-transmitted RNA viruses that affect intensive vegetable production. Ann. Appl. Biol. 165, 155–171. doi: 10.1111/aab.12147
Oren, E., Tzuri, G., Vexler, L., Dafna, A., Meir, A., Faigenboim, A., et al. (2019). Multi-allelic APRR2 Gene is associated with fruit pigment accumulation in melon and watermelon. J. Exp. Bot. 70, 3781–3798. doi: 10.1093/jxb/erz182
Picó, B., Villar, C., Nuez, F. (2003). Screening Cucumis sativus landraces for resistance to cucumber vein yellowing virus. Plant Breed. 122, 426–430. doi: 10.1046/j.1439-0523.2003.00882.x
Picó, B., Sifres, A., Martinez-Perez, E., Leiva-Brondo, M., Nuez, F. (2008). Genetics of the resistance to CVYV in cucumber. In Modem variety breeding for present and future needs. Proceedings of the 18th EUCARPIA general congress, (Prohens, J, Badenes, ML, eds), Valencia (Spain), pp 452–456.
Pitrat, M., Wipf-Scheibel, C., Besombes, D., Desbiez, C., Lecoq, H. (2012). Resistance of melon to Cucumber Vein Yellowing Virus (CVYV). Cucurbitaceae 2012, Proceedings of the Xth EUCARPIA Meeting on Genetics and Breeding of Cucurbitaceae (eds. Sari, Solmaz, Aras) Antalya (Turkey) pp 157–164.
Rausch, T., Zichner, T., Schlattl, A., Stütz, A. M., Benes, V., Korbel, J. O. (2012). DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, 333–339. doi: 10.1093/bioinformatics/bts378
Ren, Y., Zhang, Z., Liu, J., Staub, J. E., Han, Y., Cheng, Z., et al. (2009). An integrated genetic and cytogenetic map of the cucumber genome. PLoS ONE 4 (6), e5795. doi: 10.1371/journal.pone.0005795
Ren, C., Zhu, X., Zhang, P., Gong, Q. (2016). Arabidopsis COP1-interacting protein 1 is a positive regulator of ABA response. Biochem. Biophys. Res. Commun. 477, 847–853. doi: 10.1016/j.bbrc.2016.06.147
Robinson, J., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E., Getz, G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Shu, J., Liu, Y., Zhang, L., Li, Z., Fang, Z., Yang, L., et al. (2018). QTL-seq for rapid identification of candidate genes for flowering time in broccoli × cabbage. Theor. Appl. Genet. 131, 917–928. doi: 10.1007/s00122-017-3047-5
Song, J., Li, Z., Liu, Z., Guo, Y., Qiu, L.-J. (2017). Next-generation sequencing from bulked-segregant analysis accelerates the simultaneous identification of two qualitative genes in soybean. Front. Plant Sci. 8, 1–11. doi: 10.3389/fpls.2017.00919
Staub, J. E., Robbins, M. D., Wehner, T. C. (2008). “Cucumber,” in Vegetables I. Handbook of Plant Breeding, vol. 1 . Eds. Prohens, J., Nuez, F. (New York, NY: Springer). doi: 10.1007/978-0-387-30443-4_8
Sun, Y., Wang, J., Crouch, J. H., Xu, Y. (2010). Efficiency of selective genotyping for genetic analysis of complex traits and potential applications in crop improvement. Mol. Breed. 26, 493–511. doi: 10.1007/s11032-010-9390-8
Sun, J., Yang, L., Wang, J., Liu, H., Zheng, H., Xie, D., et al. (2018). Identification of a cold-tolerant locus in rice (Oryza sativa L.) using bulked segregant analysis with a next-generation sequencing strategy. Rice 11, 1–12. doi: 10.1186/s12284-018-0218-1
Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., et al. (2013). QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 74, 174–183. doi: 10.1111/tpj.12105
Verlaan, M. G., Hutton, S. F., Ibrahem, R. M., Kormelink, R., Visser, R. G. F., Scott, J. W., et al. (2013). The Tomato Yellow Leaf Curl Virus Resistance Genes Ty-1 and Ty-3 Are Allelic and Code for DFDGD-Class RNA-Dependent RNA Polymerases. PLoS Genet. 9 (3), e1003399. doi: 10.1371/journal.pgen.1003399
Wóycicki, R., Witkowicz, J., Gawroński, P., Dabrowska, J., Lomsadze, A., Pawełkowicz, M., et al. (2011). The genome sequence of the North-European Cucumber (Cucumis sativus l.) unravels evolutionary adaptation mechanisms in plants. PLoS ONE 6 (7), e22728. doi: 10.1371/journal.pone.0022728
Willmann, M. R., Endres, M. W., Cook, R. T., Gregory, B. D. (2012). The functions of RNA-dependent RNA polymerases in Arabidopsis. Arab. B. 9, e0146. doi: 10.1199/tab.0146
Win, K. T., Vegas, J., Zhang, C., Song, K., Lee, S. (2017). QTL mapping for downy mildew resistance in cucumber via bulked segregant analysis using next-generation sequencing and conventional methods. Theor. Appl. Genet. 130, 199–211. doi: 10.1007/s00122-016-2806-z
Win, K. T., Zhang, C., Silva, R. R., Lee, J. H., Kim, Y. C., Lee, S. (2019). Identification of quantitative trait loci governing subgynoecy in cucumber. Theor. Appl. Genet. 132, 1505–1521. doi: 10.1007/s00122-019-03295-3
Xu, X., Lu, L., Zhu, B., Xu, Q., Qi, X., Chen, X. (2015). QTL mapping of cucumber fruit flesh thickness by SLAF-seq. Sci. Rep. 5, 1–9. doi: 10.1038/srep15829
Yang, S.-J., Carter, S. A., Cole, A. B., Cheng, N.-H., Nelson, R. S. (2004). A natural variant of a host RNA-dependent RNA polymerase is associated with increased susceptibility to viruses by Nicotiana benthamiana. Proc. Natl. Acad. Sci. 101, 6297–6302. doi: 10.1073/pnas.0304346101
Yang, L., Koo, D. H., Li, Y., Zhang, X., Luan, F., Havey, M. J., et al. (2012). Chromosome rearrangements during domestication of cucumber as revealed by high-density genetic mapping and draft genome assembly. Plant J. 71, 895–906. doi: 10.1111/j.1365-313X.2012.05017.x
Yang, Z., Huang, D., Tang, W., Zheng, Y., Liang, K., Cutler, A. J., et al. (2013). Mapping of quantitative trait loci underlying cold tolerance in rice seedlings via high-throughput sequencing of pooled extremes. PLoS ONE 8 (7), e68433. doi: 10.1371/journal.pone.0068433
Ye, K., Schulz, M. H., Long, Q., Apweiler, R., Ning, Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. doi: 10.1093/bioinformatics/btp394
Zhang, H., Yi, H., Wu, M., Zhang, Y., Zhang, X., Li, M., et al. (2016). Mapping the flavor contributing traits on “Fengwei melon” (Cucumis melo L.) chromosomes using parent resequencing and super bulked-segregant analysis. PloS One 11, 1–24. doi: 10.1371/journal.pone.0148150
Keywords: Cucumber vein yellowing virus, cucumber, resistance, mapping, BSA-seq, breeding, marker-assisted selection
Citation: Pujol M, Alexiou KG, Fontaine A-S, Mayor P, Miras M, Jahrmann T, Garcia-Mas J and Aranda MA (2019) Mapping Cucumber Vein Yellowing Virus Resistance in Cucumber (Cucumis sativus L.) by Using BSA-seq Analysis. Front. Plant Sci. 10:1583. doi: 10.3389/fpls.2019.01583
Received: 04 July 2019; Accepted: 12 November 2019;
Published: 03 December 2019.
Edited by:
Jaime Prohens, Polytechnic University of Valencia, SpainReviewed by:
Dirk Janssen, IFAPA Centro La Mojonera, SpainZhiyong Liu, Institute of Genetics and Developmental Biology, China
Xingfang Gu, Chinese Academy of Agricultural Sciences, China
Copyright © 2019 Pujol, Alexiou, Fontaine, Mayor, Miras, Jahrmann, Garcia-Mas and Aranda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Aranda, bS5hcmFuZGFAY2ViYXMuY3NpYy5lcw==