 Yi-Tzu Kuo
Yi-Tzu Kuo Takayoshi Ishii
Takayoshi Ishii Jörg Fuchs
Jörg Fuchs Wei-Hsun Hsieh
Wei-Hsun Hsieh Andreas Houben
Andreas Houben Yann-Rong Lin
Yann-Rong Lin- 1Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany
- 2Department of Agronomy, National Taiwan University, Taipei, Taiwan
- 3Arid Land Research Center, Tottori University, Tottori, Japan
- 4World Vegetable Center, Tainan, Taiwan
Polyploidization is an evolutionary event leading to structural changes of the genome(s), particularly allopolyploidization, which combines different genomes of distinct species. The tetraploid species, Sorghum halepense, is assumed an allopolyploid species formed by hybridization between diploid S. bicolor and S. propinquum. The repeat profiles of S. bicolor, S. halepense, and their relatives were compared to elucidate the repeats’ role in shaping their genomes. The repeat frequencies and profiles of the three diploid accessions (S. bicolor, S. bicolor ssp. verticilliflorum, and S. bicolor var. technicum) and two tetraploid accessions (S. halepense) are similar. However, the polymorphic distribution of the subtelomeric satellites preferentially enriched in the tetraploid S. halepense indicates drastic genome rearrangements after the allopolyploidization event. Verified by CENH3 chromatin immunoprecipitation (ChIP)-sequencing and fluorescence in situ hybridization (FISH) analysis the centromeres of S. bicolor are mainly composed of the abundant satellite SorSat137 (CEN38) and diverse CRMs, Athila of Ty3_gypsy and Ty1_copia-SIRE long terminal repeat (LTR) retroelements. A similar centromere composition was found in S. halepense. The potential contribution of S. bicolor in the formation of tetraploid S. halepense is discussed.
Introduction
Large genomes are rich in various repetitive DNAs; for example, up to 85 and 90% of the maize and wheat genomes are composed of repeats (Li et al., 2004; Schnable et al., 2009). Although once thought to be “junk DNA,” repetitive DNA has been later found to be involved in regulating gene expression, maintenance of chromosomal integrity, and genome stability (Bennetzen and Wang, 2014; Mehrotra and Goyal, 2014; Garrido-Ramos, 2015). Compared to coding sequences, repetitive DNAs are considered as fast-evolving genome components. Their variable abundance, high sequence variations and distinct chromosomal distributions contribute to genome divergence among species.
Repetitive DNAs can be classified into two major types, tandem repeats (also referred to as satellite DNA) and transposable elements (TEs), according to their structural organization and sequence composition (Kubis et al., 1998). Tandem repeats, which repeat unit arrays in a head-to-tail manner, preferentially cluster at specific chromosome regions, such as (peri)centromeres, (sub)telomeres, and distinct intercalary regions. TEs tend to intersperse with other sequences and scatter throughout entire genome but can also accumulate at specific chromosomal regions, as, e.g. (peri)centromeres. TEs are further divided into DNA transposons and retrotransposons, which transpose within the genome via either cut-and-paste or copy-and-paste mechanisms, respectively. Retrotransposons are the most abundant TEs in eukaryotes. Of them, the long terminal repeat (LTR) retrotransposons, subclassified into Ty1_copia and Ty3_gypsy, were reported to be present throughout the entire plant kingdom (Kumar and Bennetzen, 1999).
Centromeric and pericentromeric regions are the hotspots for repeat accumulation. In species like rice (Cheng et al., 2002), maize (Zhong et al., 2002), and barley (Houben et al., 2007), both satellite repeats and retrotransposons are enriched in these areas and interact partly with the centromere-defining centromeric histone H3 variant CENH3. The centromeric satellite is often the most abundant tandem repeat in a genome, and its corresponding monomer unit is highly variable in sequence composition and length between species (Melters et al., 2013). The most common monomer sizes of centromeric satellites are 140 ∼ 180 and 300 ∼ 360 bp, representing mono- and dinucleosomes. However, centromeric satellite units with a length of only 20 bp as in Astragalus sinicus (Tek et al., 2011) and longer units up to 2,979 bp in Pisum fulvum (Robledillo et al., 2020) were also found.
Transposable elements not only occupy a significant portion of eukaryotic genomes but also play a role in centromere evolution (Hartley and O’Neill, 2019). The insertion of centromeric retroelements is related to the birth of new satellite families in Arabidopsis thaliana (Kapitonov and Jurka, 1999) and Aegilops speltoides (Cheng and Murata, 2003). In A. thaliana, they are also involved in the transcriptional regulation of centromeric satellite DNA (May et al., 2005). The interaction between satellite repeats and retrotransposons drives the rapid sequence changes in plant centromeres, especially after interspecific hybridization or polyploidization (Yang et al., 2018; Su et al., 2019).
The genus Sorghum belongs to the Poaceae family and is divided into five subgenera: Eusorghum, Parasorghum, Heterosorghum, Chaetosorghum, and Stiposorghum (Garber, 1950). In the subgenus Eusorghum, the cultivated species, S. bicolor (L.) Moench (2n = 2x = 20) originated from Africa and is known for its drought tolerance and broad adaptation. As one of the top five cereal crops, it is used for multiple purposes, like staple food, forage crop, and biofuel. Another cultivated species, S. bicolor var. technicum (Körn.) Stapf ex Holland, is used for making brooms. In Taiwan, the commonly discovered wild relatives of the cultivated sorghums are S. bicolor ssp. verticilliflorum (Steud.) de Wet ex Wiersema & J. Dahlb (2n = 2x = 20) and S. halepense (L.) Pers. (also called Johnsongrass, 2n = 40) (De Wet, 1978), and occasionally S. propinquum (Kunth) Hitchc as well. The tetraploid S. halepense is likely an allopolyploid species formed by hybridization between S. bicolor and S. propinquum based on the meiotic studies of S. bicolor × S. halepense hybrids (Tang and Liang, 1988), genomic sequences (Paterson et al., 2020), and it rhizomatous nature (Paterson et al., 1995). Phylogenetically S. halepense is closer to S. bicolor (Price et al., 2005), while the S. propinquum-derived rhizomes make S. halepense a noxious weed with an almost worldwide distribution.
The first S. bicolor genome was assembled by Paterson et al. (2009) and has been refined by re-sequencing and optical mapping (Deschamps et al., 2018; McCormick et al., 2018). Genome assemblies demonstrated that the heterochromatic, pericentromeric regions of S. bicolor are enriched in repetitive elements (Kim et al., 2005; Paterson et al., 2009; McCormick et al., 2018). The satellite repeat CEN38 (Miller et al., 1998b; Zwick et al., 2000) and the retrotransposon-related DNA element Sau3A9 (Miller et al., 1998a) were found to be associated with the centromeres. Nevertheless, variation in the repeat composition and distribution among different Sorghum species remain largely unknown.
In this study, we in silico identified high-copy repeats and determined their chromosomal distribution to resolve the relationship and genome diversification of diploid S. bicolor and allotetraploid S. halepense accessions. The observed polymorphic distribution of the subtelomeric satellites being more abundant in the tetraploids indicates drastic genome rearrangements after the allopolyploidization event forming S. halepense. Application of a Sorghum-specific CENH3 antibody in combination with chromatin immunoprecipitation (ChIP) sequencing and fluorescence in situ hybridization (FISH) resulted in the identification of a centromere-specific satellite and evolutionarily conserved centromere-associated TEs. The impact of dynamic repetitive DNAs in the genome of the five related Sorghum genomes is discussed.
Materials and Methods
Plant Materials
The diploid species Sorghum bicolor accession “V9,” S. bicolor ssp. verticilliflorum accession “WL” and S. bicolor var. technicum accession “YL” and two tetraploid accessions of S. halepense (accession “TT” and “US”) were grown under greenhouse condition in the Department of Agronomy, National Taiwan University, Taiwan. The collection sites or source of seeds are described in Table 1.

Table 1. Information of the Sorghum materials and the genome size.
Flow Cytometry
For nuclei isolation, approximately 0.5 cm2 of fresh leaf tissue was chopped together with equivalent amounts of leaf tissue of the internal reference standard, Glycine max (L.) Merr. convar. max var. max, Sorte Cina 5202 (Gatersleben GeneBank accession number: SOJA 392; 2.21 pg/2C), in a petri dish using the reagent kit “CyStain PI Absolute P” (Sysmex-Partec) following the manufacturer’s instructions. The resulting nuclei suspension was filtered through a 50-μm CellTrics filter (Sysmex-Partec) and measured on a BD Influx cell sorter (BD Biosciences). Six independent measurements were performed for each genotype. The absolute DNA content (pg/2C) was calculated based on the values of the G1 peak means and the corresponding genome size (Mbp/1C), according to Dolezel et al. (2003).
DNA Extraction and Genome Sequencing
The genomic DNA of Sorghum plants was extracted from the young leaves using DNeasy Plant Mini Kit (QIAGEN). Low-pass genome sequencing was performed, generating 2 × 150 bp paired-end (PE) reads using NovaSeq 6000 system (Illumina) by Novogene (China). At least 4 GB raw PE reads were generated for each Sorghum sample.
Analysis of Repetitive Genome Fractions Using RepeatExplorer and TAREAN
The quality of the PE reads was assessed by FastQC (Andrews, 2010) implanted in the RepeatExplorer pipeline1 and filtered by quality with 95% of bases equal to or above the cut-off value of 10, followed by an overlap check. Non-overlapped PE reads equivalent to 0.3× genome coverage were sampled and employed to identify, characterize, and quantify the repetitive elements in each individual genome by the graph-based clustering method using RepeatExplorer (Novák et al., 2010, 2013). Clustering was performed by default setting with 90% of similarity over 55% of the read length. The comparative clustering analysis was performed based on 0.1× genome coverage of qualified PE reads from each Sorghum sample according to the protocol in Novák et al. (2020). The automatic annotation of repeat clusters was inspected manually and revised if necessary, and the genome proportion of each repeat type was recalculated. The monomer sequence of putative satellites and LTR elements were reconstructed by TAREAN (TAndem REpeat ANalyzer) (Novák et al., 2017).
Phylogenetic Analysis
The CENH3 protein sequences of S. bicolor and other plant species were downloaded from NCBI GenBank (Supplementary Table 1). They were first aligned using ClustalW implanted in MEGA X by default setting (Thompson et al., 1994; Kumar et al., 2018), and the phylogenetic relationship was inferred by the maximum likelihood method on the IQ-Tree web server2 (Trifinopoulos et al., 2016). The tree was visualized and exported using Interactive Tree Of Life (iTOL3) (Letunic and Bork, 2007, 2019).
Probe DNA Preparation
The primers used to amplify probe DNA were designed using Primer3 based on the sequences of satellite monomer or LTR integrase domain, identified in NCBI CD-Search4 (Marchler-Bauer and Bryant, 2004; Lu et al., 2020). The sequence of primers and repeat clusters are listed in Supplementary Tables 2, 3. The probe DNAs were amplified in a mixture of 50 ng genomic DNA, 1× PCR buffer, 0.25 mM of each dNTP, 0.4 mM of each primer, 1.5 U Taq polymerase (QIAGEN), in a total of 50 μl with a program of 95°C for 5 min, 35 cycles of 95°C for 30 s, 55°C for 1 min, and 72°C for 1 min, followed by 72°C for 5 min. The purified probe DNAs were then labeled with ATTO488-dUTP or ATTO550-dUTP using the Fluorescent Nick Translation Labeling kits (Jena Bioscience).
Fluorescence in situ Hybridization
Root tips were pretreated with 2 mM 8-hydroxyquinoline at room temperature (RT) for 4–5 h and fixed in freshly prepared 3:1 (v/v) ethanol: glacial acetic acid at RT, overnight. Mitotic chromosome spreads were prepared as described in Aliyeva-Schnorr et al. (2015). Slides were first treated with 45% acetic acid at RT for 10 min, followed by 0.1% pepsin/0.01 N HCl at 37°C for 10 min and post-fixed in 4% paraformaldehyde (PFA) at RT for 10 min. The hybridization mixture contained 50% (v/v) formamide, 10% (w/v) dextran sulfate, 2× SSC, and 5 ng/μl of each probe. Chromosomal DNA and probes were denatured at 75°C for 2 min, and hybridization was performed at 37°C for 15–24 h. The final stringent wash was in 2× SSC at 57°C for 20 min, followed by dehydration in 70, 90, and 100% ethanol series for 3 min each. Chromosomes were counterstained by 10 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) in Vectashield Antifade Mounting Medium (Vector Laboratories).
Indirect Immunostaining
Polyclonal antibodies against the CENH3 protein of S. bicolor were produced by using the N-ERAGGASTSATPERRNAGT-C peptide. The peptide synthesis, immunization of rabbits, and peptide affinity purification of antisera were performed by LifeTein.5 For slide preparation, root tips were fixed in 4% PFA in 1× phosphate-buffered saline (PBS) under vacuum at 4°C for 10 min, followed by 20 min at 4°C without vacuum. Roots were washed with ice-cold 1× PBS for 3 min twice, and digested with an enzyme cocktail composed of 1% (w/v) pectolyase (Sigma), 0.7% (w/v) cellulase “ONOZUKA” R-10 (Yakult), 0.7% cellulase (CalBioChem), and 1% cytohelicase (Sigma) in 1× PBS for 60 min at 37°C in a humid chamber. Roots were subsequently washed in ice-cold 1× PBS for 3 min twice. Root tips were then squashed in 1× PBS between slide and coverslip. After freezing in liquid nitrogen, coverslips were removed, and slides were kept in ice-cold 1× PBS. For immunostaining, the SbCENH3 antibody (diluted 1:2,000) was applied at 4°C overnight. Slides were washed twice in 1× PBS at 4°C. Anti-rabbit Alexa Fluor 488 (Molecular Probes) with a dilution of 1:500 was used as a secondary antibody. Finally, the slides were washed twice in 1× PBS at 4°C, dehydrated in an ethanol series (70, 90, and 99%) at RT, mounted in Vectashield antifade (Vector Laboratories) with 10 μg/ml DAPI and covered with coverslips.
Microscopy
Images were captured using an epifluorescence microscope BX61 (Olympus) equipped with a cooled CCD camera (Orca ER, Hamamatsu). Pseudocolors were applied using Adobe Photoshop CS6.
Western Blotting Analysis
Nuclear proteins from young sorghum seedlings were isolated according to Gendrel et al. (2005) and Karimi-Ashtiyani et al. (2015). For Western detection, a 1:2,000 diluted SbCENH3 antibody in 1x PBC with 5% (w/v) low-fat milk was applied at 4°C for 12 h. Proteins bound by antibodies were detected with 1:5,000 diluted anti-rabbit antibodies 800CW (925-32213, Li-COR, Lincoln, NE, United States) for 1 h at 22°C. Fluorescence signals were recorded using Odyssey (Li-COR, Lincoln, NE, United States) as recommended by the manufacturer.
CENH3 Chromatin Immunoprecipitation Sequencing
For nuclei isolation, 1 g of fresh leaf tissue was homogenized in liquid nitrogen and mixed with 10 ml of nuclei isolation buffer [1 M sucrose, 5 mM KCl, 5 mM MgCl2, 60 mM HEPES pH 8.0, 5 mM EDTA, 0.6% Triton X-100, 0.4 mM PMSF, 1 μM pepstatin A, cOmplete protease inhibitor cocktail (Roche)]. The nuclei were then fixed in 1% PFA in nuclei isolation buffer at RT and shaken at 12 rpm for 10 min. The cross-linking reaction was terminated by addition of glycine to a final concentration of 130 mM. The solution was filtrated through Miracloth (Millipore) twice and a 50-μm CellTrics filter (Sysmex) once and centrifuged at 4°C, 3,000 × g for 10 min. The pellet was resuspended in 1 ml extraction buffer [0.25 M sucrose, 10 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1% Triton X-100, 1 mM EDTA, 5 mM β-mercaptoethanol, 0.1 mM PMSF, 1 μM pepstatin A, cOmplete protease inhibitor cocktail (Roche)], transferred to a 1.5 ml tube, and followed by centrifugation at 4°C, 12,000 × g for 10 min. The supernatant was removed and nuclei were resuspended in 100 μl nuclei lysis buffer [20 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% SDS, 0.1 mM PMSF, 1 μM pepstatin A, cOmplete protease inhibitor cocktail (Roche)]. Chromatin was sonicated with a Bioruptor (Diagenode) using seven cycles of 30 s ON, 30 s OFF, for three times. The samples were then diluted 10 times with ChIP dilution buffer [16.7 mM Tris–HCl pH 8.0, 167 mM NaCl, 1.1% Triton X-100, 1 mM EDTA, cOmplete protease inhibitor cocktail (Roche)], centrifuged at 4°C, 13,000 × g for 5 min, and the supernatant was transferred to a 1.5 ml tube. The chromatin was mixed with 1:100 diluted SbCENH3 antibody and incubated at 4°C by shaking at 14 rpm for 12 h. DynabeadsTM Protein A (Invitrogen) in ChIP dilution buffer, corresponding to one-tenth volume of the chromatin solution, was added to the antibody-prebound chromatin and incubated at 4°C by shaking at 14 rpm for 1.5 h. The tube was put on a magnetic stand and all liquid was removed after the solution was cleared. Beads were then washed twice with low salt buffer (150 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0), followed by two washes with high salt buffer (500 mM NaCl, 0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0) at 4C by shaking at 14 rpm for 5 min. The bead-bound chromatin was purified by using iPure kit v2 (Diagenode) following the manual and quantified by using QubitTM dsDNA HS Assay kit (Invitrogen). The ChIP sequencing was performed using the NovaSeq 6000 system (Illumina) by Novogene (China), in the format of PE reads with 150 bp per end, and at least 6 GB raw PE reads were generated.
ChIPseq Analysis
The reads of SbCENH3-ChIPseq and input-seq were quality checked and filtered as mentioned above, using tools implanted in the Galaxy-based RepeatExplorer (see text footnote 1) portal. ChIP-Seq Mapper (Galaxy version 0.1.1) (Neumann et al., 2012) was used to evaluate the enrichment of repetitive sequences in sequencing data from CENH3-ChIP experiments, with the repeat contig sequences of S. bicolor identified by RepeatExplorer as a reference.
Results
Satellite DNA Is Less Abundant but More Diverse Than LTR Repeats Among Sorghum Genomes
To study the genome divergence among Sorghum species, the repeat composition of five related accessions was analyzed. The cultivated diploid S. bicolor “V9” (SbV9) is an early flowering accession (Hsieh et al., 2015). The other two diploid accessions, S. bicolor ssp. verticilliflorum “WL” (SbWL), which is a wild relative to S. bicolor and S. bicolor var. technicum “YL” (SbYL) which is a cultivated species, are commonly seen as feral sorghums and are morphologically distinct from S. bicolor, especially in inflorescence architectures. The two tetraploid S. halepense accessions, “TT” (ShTT) and “US” (ShUS), are wild collections in Taiwan and the United States, respectively. First, the genome sizes of the diploid (2n = 2x = 20) and tetraploid (2n = 4x = 40) accessions were 789 ∼ 800 and 1,512 ∼ 1,547 Mb/1C, respectively, determined by flow cytometry (Table 1). In between the diploid and tetraploid accessions, we did not observe severe genome size differences.
Next, to compare the genome composition between different accessions, the high-copy repeat fractions were analyzed both individually and comparatively. Irrespective of the genotype, about 55% of the genomes are composed of moderate and high-copy repeat sequences (Table 2). Among them, the retrotransposon Ty3_gypsy is the most predominant component (36.5–39.3%), followed by either Ty1_copia (4.9–6.7%) or satellite sequences (4.7–5.8%), while DNA transposons and rDNA both account for less than 1.3%. Six different clades of Ty3_gypsy retrotransposons were identified, of which Athila is the most abundant in a range of 13.7–15.3%. Out of the eight detected Ty1_copia retrotransposon classes, SIRE is the most abundant one. Although Ty1_copia is not the most abundant, it seems to be the most diverse repeat type in all five Sorghum genomes.
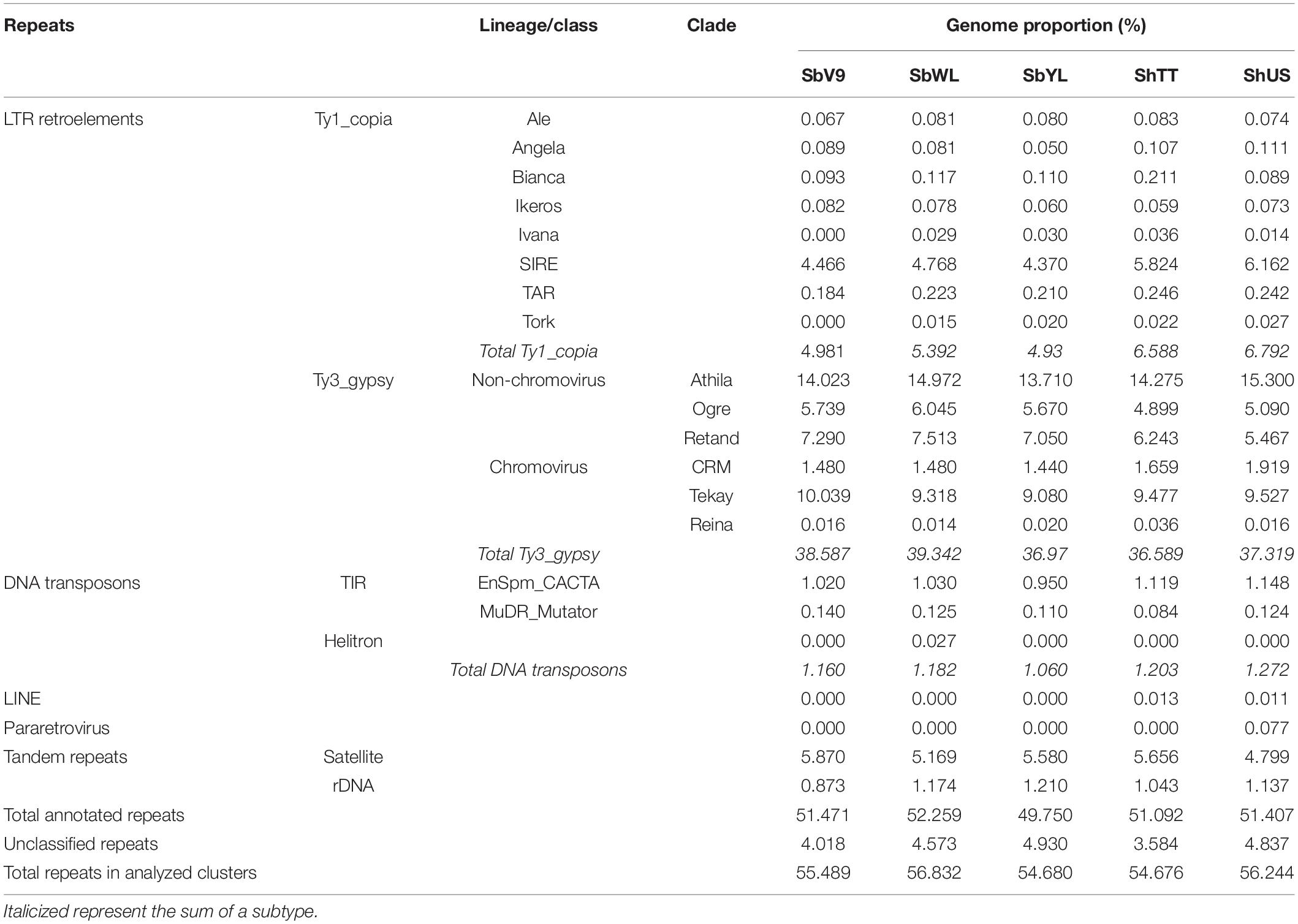
Table 2. The proportion of moderate and high-copy repetitive DNA in the five Sorghum genomes.
To verify whether the high-copy repeats are shared or genotype-specific, a comparative RepeatExplorer analysis was performed using reads representing 0.1× genome coverage of each sample. The composition of the top 141 clusters with a proportion of more than 0.1% of the analyzed reads is shown in Figure 1. The read composition of satellite and LTR repeat-annotated clusters identified by TAREAN is listed in Table 3.
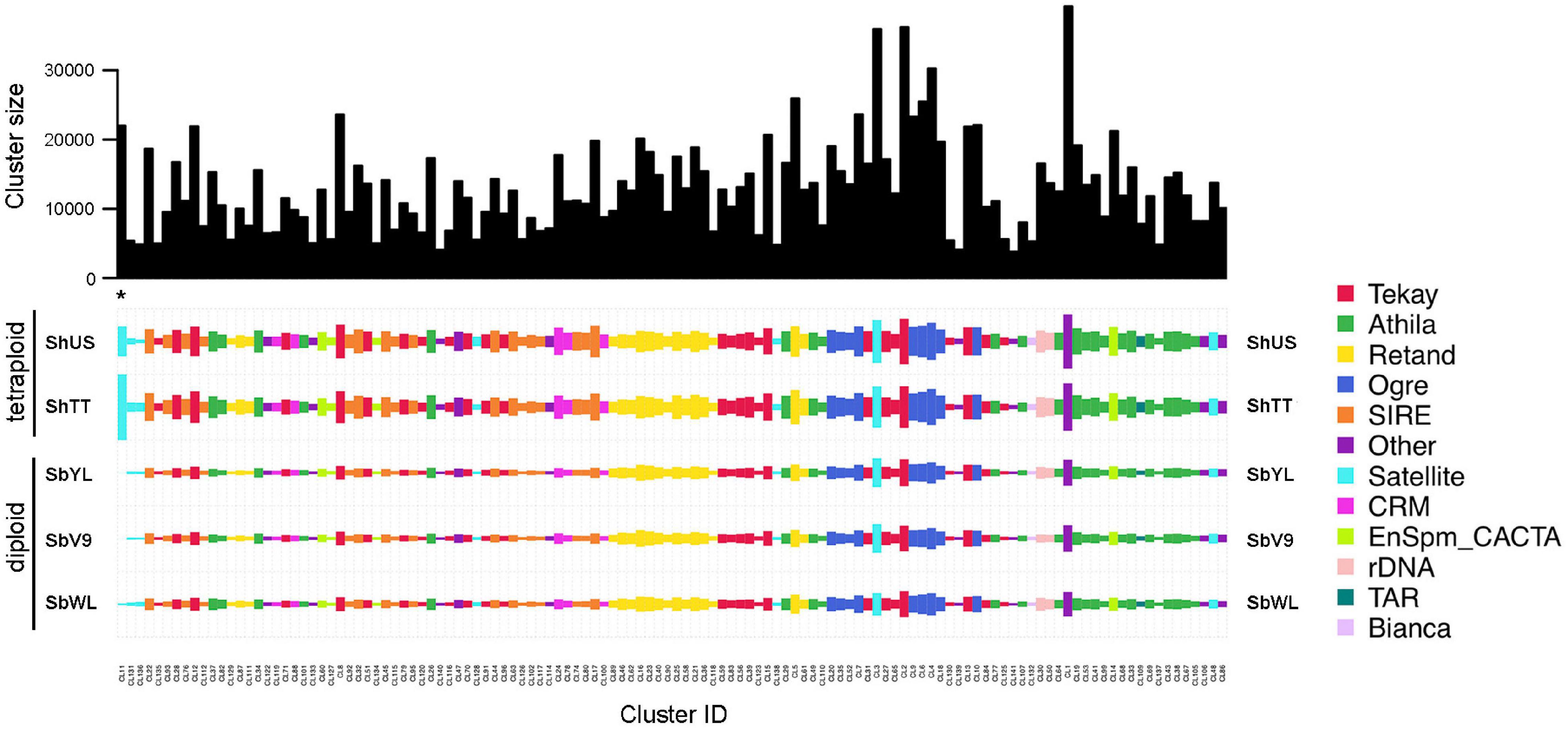
Figure 1. Comparative repeat analysis of the five Sorghum accessions. The top 141 clusters with a proportion greater than 0.1% of the analyzed reads are shown. The bar plot (top) shows the size (number of reads) of individual clusters. The color of rectangles represents the final annotation of each cluster, and the size is in proportion to the abundance of the repeat in each genome. The species and clusters are ordered using hierarchical clustering based on the correlation of quantities. The position of the satellite-type cluster CL11 (one of the two repeats with an enriched abundance in S. halepense) is marked with an asterisk.
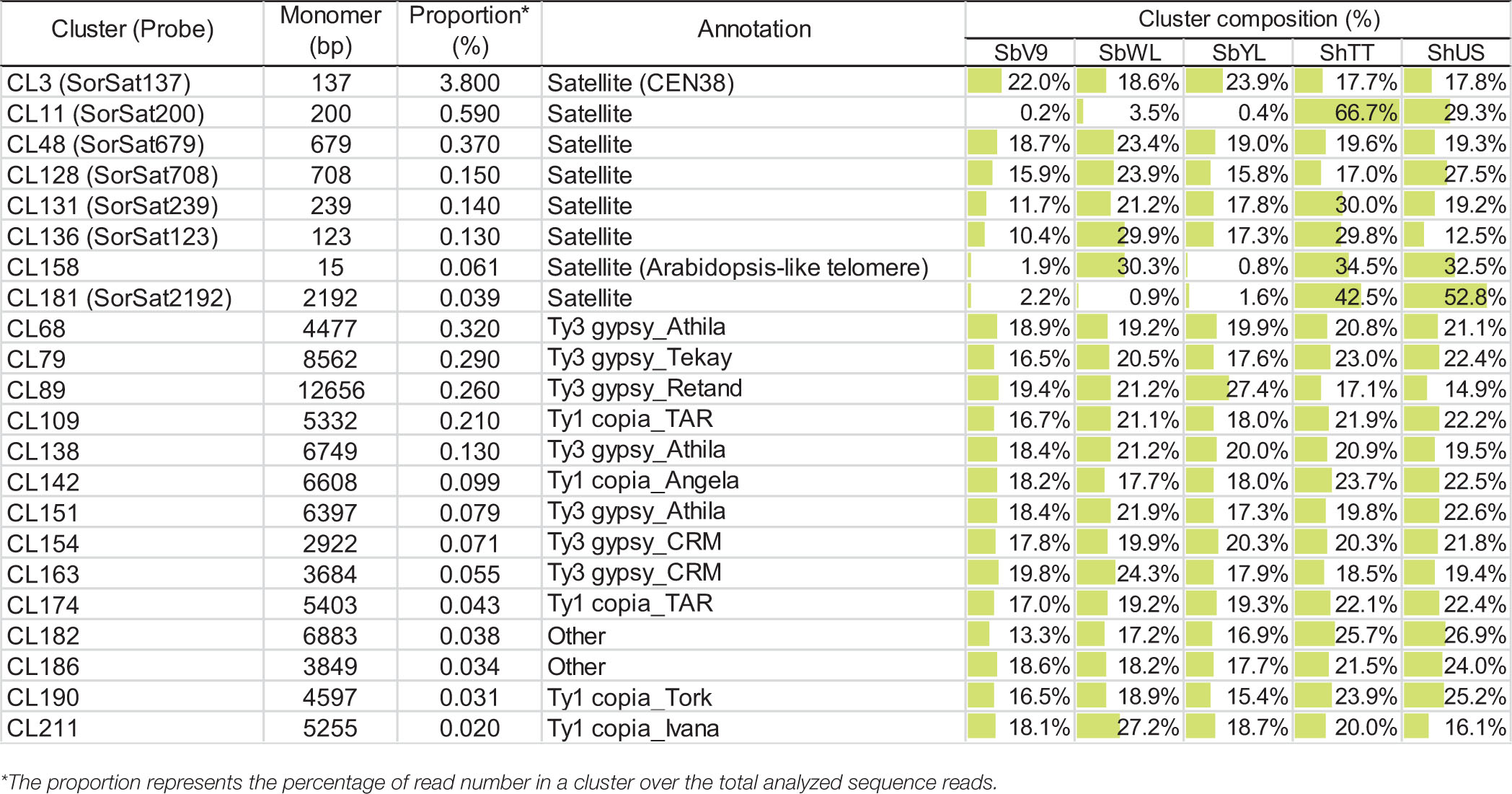
Table 3. Cluster composition of the satellite and LTR repeats in the five Sorghum genomes.
All high-copy clusters of retrotransposons were shared and almost equally enriched in all five Sorghum accessions. Nevertheless, the genome compositions of satellite repeats were variable among the accessions, specifically the satellite-type clusters CL11 and CL181, which are only abundant in the tetraploid S. halepense (Figure 1 and Table 3). The cluster CL11 with a monomer length of 200 bp was mainly represented by reads of ShTT (66.7%) and ShUS (29.3%), and only by a minor amount of SbWL (3.5%) as well as SbV9 and SbYL (less than 0.5% each) (Table 3). Similarly, over 95% of reads in the cluster CL181 were specific to the tetraploid S. halepense (42.5% from ShTT and 52.8% from ShUS). The satellite-annotated cluster CL158 with the monomer sequence (TTTAGGGTTTTAGGG), similar to the Arabidopsis-type telomere sequence, was enriched in SbWL (30.3%), ShTT (34.5%), and ShUS (32.5%) reads. In summary, most of the high-copy repeat clusters were shared and similarly abundant in the five genomes, only two satellite repeats (CL11 and CL181) showed a clear differential enrichment between the diploid and tetraploid genomes. In addition, CL158 was found to be more abundant in the tetraploid S. halepense and also in the wild diploid S. bicolor accession (SbWL).
Thus, although the total repeat frequency of all five Sorghum accessions is similar, the fast-evolving satellite repeats differ in copy number among genomes. These satellite repeats might play a key role in driving the diversification of the genomes, specifically distinguished diploid from tetraploid genomes.
Comparative FISH of Sorghum Satellite Repeats Revealed Drastic Genome Rearrangements at Chromosomal Ends Subsequent to the S. halepense Formation
Fluorescence in situ hybridization mapping of the seven high-copy satellite repeats, except for the Arabidopsis telomere-like CL158, which intermingles with the canonical plant telomeric repeats (TTTAGGG)n (Supplementary Figure 1), was performed to elucidate their chromosomal distribution in all five Sorghum accessions. The corresponding FISH probes of individual satellite clusters were named based on their consensus monomer sizes (Table 3). SorSat137, representing the Sorghum centromeric repeat CEN38 (Zwick et al., 2000), revealed centromere-specific signals on all chromosomes of the three diploid accessions (Figures 2a1–3), as well as of the two S. halepense accessions (Figures 2a4,5). The satellite SorSat708 selectively accumulated in the pericentromeric regions of three chromosome pairs of the diploid accessions. While one chromosome pair showed very strong signals, the punctual signals on the other two pairs were severely weaker (Figures 2b1–3). In the two tetraploid S. halepense accessions, either eight signals (four strong and four weak) in ShUS (Figure 2b5) or five to six signals (one strong and four to five weak) in ShTT (Figure 2b4) were detected. In the latter case, obviously, a heteromorphic distribution of this satellite repeat occurred in at least one homologous chromosome pair. SorSat679 revealed signals in the pericentromeric regions of all SbV9 chromosomes, except for the smallest chromosome pair where this sequence was enriched in the telomeric region of one end (Figure 2c1). Nevertheless, SorSat679 displayed a relatively disperse distribution in the other accessions, with a preference in the pericentromeric regions (Figures 2c2–5).
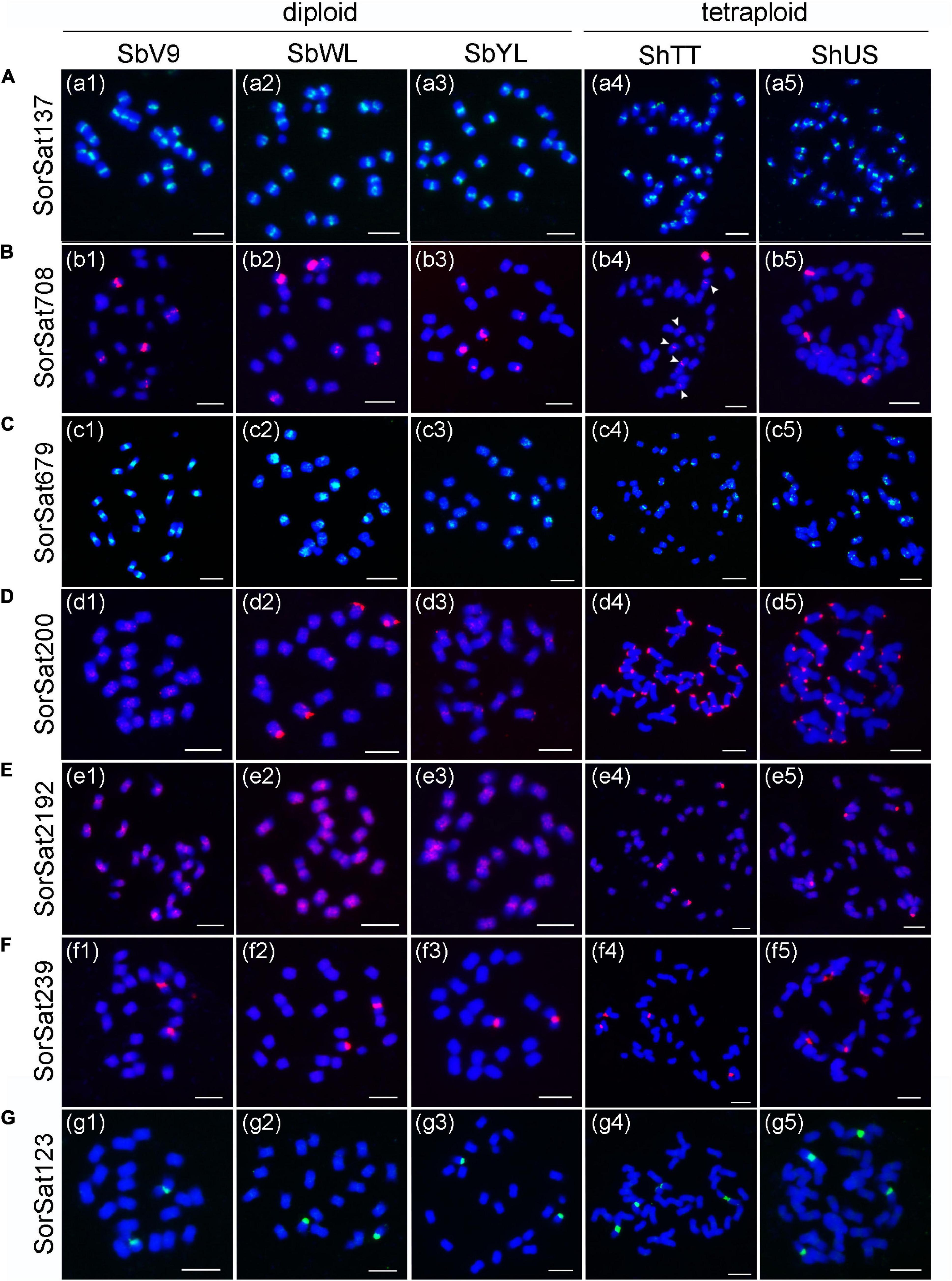
Figure 2. FISH mapping of the seven satellite repeats in the five Sorghum accessions. The satellite repeats include (A) SorSat137, (B) SorSat708, (C) SorSat679, (D) SorSat200, (E) SorSat2192, (F) SorSat239 and (G) SorSat123. Mitotic metaphase chromosomes were counterstained with DAPI. The signals of the satellite probes were pseudocolored in either red or green. The weak signals in (b4) are indicated by arrowheads. The name of Sorghum accession is labeled in abbreviation (Table 1). Bar = 5 μm.
SorSat200 was found to be accumulated at either or both ends of almost all S. halepense chromosomes, except three chromosome pairs on which no signals were detectable (Figures 2d4,5). In the diploid accessions, strong SorSat200 signals were only detected at the end of two chromosome pairs of SbWL (Figure 2d2), while the remaining chromosomes, including those of SbV9 and SbYL revealed only weak signals in the pericentromeric regions (Figures 2d1,3).
SorSat2192, the second satellite enriched in S. halepense, showed strong signals at the end of two chromosome pairs, in addition to weaker signals in pericentromeric regions in both tetraploid accessions (Figures 2e4,5). However, SorSat2192 resulted in only dispersed signals in the pericentromeric regions in diploid accessions (Figures 2e1–3). Thus, the chromosomal distributions of SorSat2192 and SorSat200 differ between the diploid and the tetraploid accessions. The satellites enriched in S. halepense tend to accumulate at the chromosome ends of S. halepense.
Both SorSat239 and SorSat123 showed signals on one chromosome pair in all diploid accessions (Figures 2f1–3, g1–3), and on two chromosome pairs in both tetraploids (Figures 2f4,5, g4,5). The colocalization of SorSat123 (Figure 3A) and SorSat239 signals (Figure 3B) with signals derived from a 45S rDNA-specific probe indicates the close proximity of both satellites to the nucleolus organizer region (NOR) in S. bicolor and S. halepense (signals in S. bicolor was shown as an example, Figures 3A,B). The 5S rDNA was detected on one and two 45S rDNA-negative chromosome pairs in diploid S. bicolor and tetraploid S. halepense, respectively (Figures 3C,D).
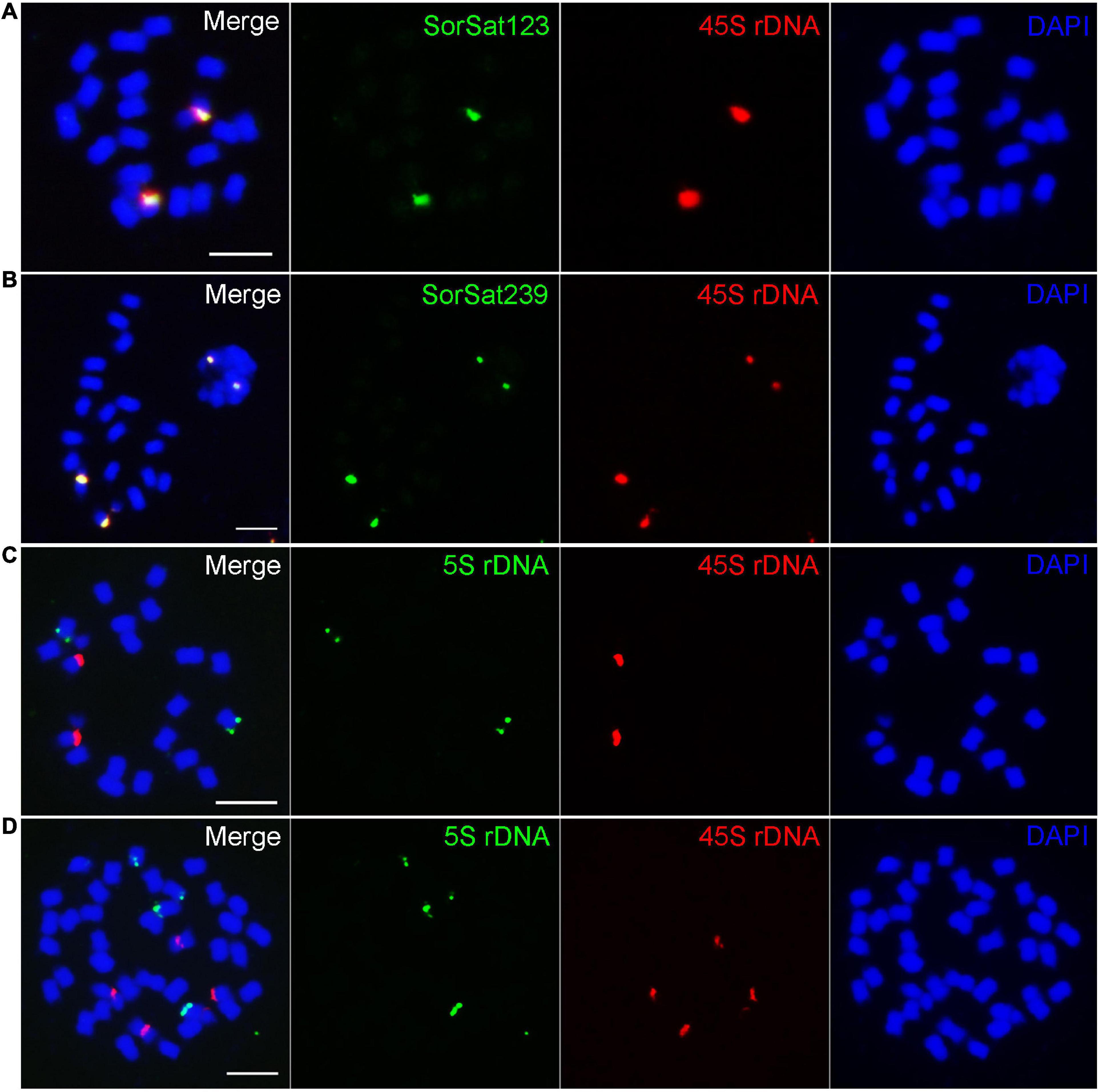
Figure 3. Fluorescence in situ hybridization mapping of the satellite repeats and rDNAs. The satellite (A) SorSat123 and (B) SorSat239 colocalize with 45S rDNA in metaphase chromosomes of S. bicolor. (C) One pair of each 5S and 45S rDNA signal is detected in diploid S. bicolor, while (D) two pairs of signals for both rDNA are present in tetraploid S. halepense. Mitotic metaphase chromosomes were counterstained with DAPI. Bar = 5 μm.
In summary, the chromosomal localization of the centromere-specific SorSat137, the NOR-associated SorSat123 and SorSat239 repeats is conserved in diploid S. bicolor and tetraploid S. halepense. The other four satellites SorSat200, SorSat679, SorSat708, and SorSat2192 showed distinct distribution patterns among accessions with respect to their loci number, copy number, and chromosomal localization. Especially, these satellites tend to locate in the pericentromeric regions of the diploid accessions, while the S. halepense-enriched satellites are particularly accumulated at the chromosome ends. The diversity in satellite distribution among the accessions indicates that satellite repeats might be involved in the process of genome diversification within the genus Sorghum.
The Centromeric DNA of S. bicolor Is Composed Predominantly of a Single Satellite Repeat and Diverse LTR Sequences
Although the centromere-localized satellite CEN38 was reported already before (Zwick et al., 2000), DNA sequences interacting with the CENH3-containing nucleosomes have not been determined so far in Sorghum. The CENH3 gene of S. bicolor was identified from NCBI GenBank (Supplementary Table 1), and clusters phylogenetically with the CENH3s of the closely related monocots, sugarcane (Saccharum officinarum) and maize (Zea mays) (Figure 4a). To perform Sorghum CENH3-ChIP (chromatin immunoprecipitation), part of the N-terminal sequence was used for raising a peptide antibody (anti-SbCENH3) against the CENH3 protein. The specificity of the SbCENH3 antibody was confirmed by Western blot analysis, detecting a protein of the predicted size of 17.75 kDa (Figure 4b). Immunostaining with anti-SbCENH3 antibody resulted in distinct signals in interphase nuclei as they are typical for centromeres in species without Rabl orientation and in centromere-specific signals on metaphase chromosomes of S. bicolor (Figures 4c,d).
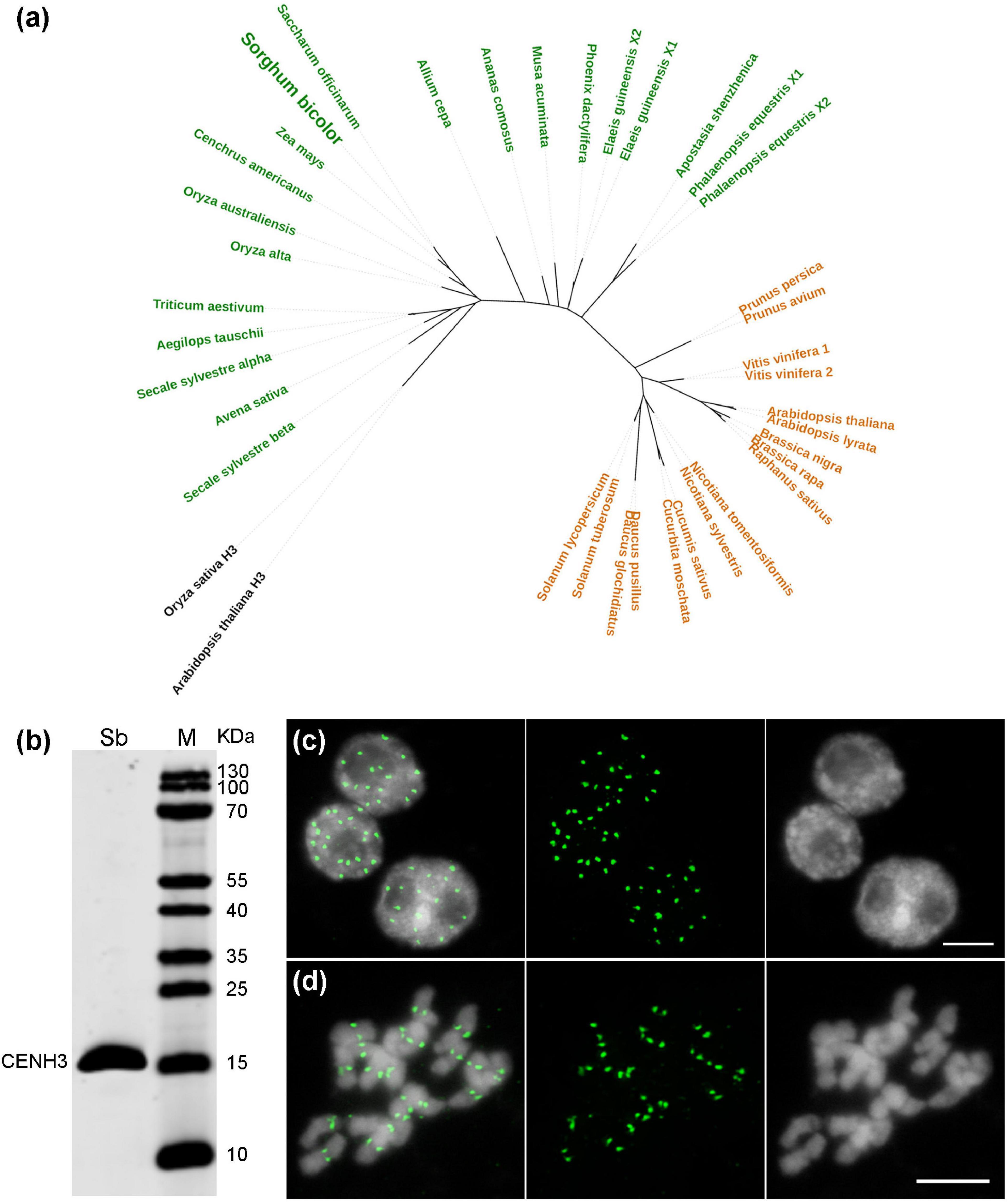
Figure 4. Phylogeny and antibody specificity of CENH3 of S. bicolor. (a) Phylogenetic relationship of CENH3 between S. bicolor and other plant species. The monocot and eudicot species are labeled in green and orange, respectively, and the canonical histone H3 used as outgroup are shown in black. The accession number of the CENH3 protein sequences are listed in Supplementary Table 1. (b) The specificity of an anti-SbCENH3 antibody probed on nuclear proteins of S. bicolor by Western blot analysis. Sb, S. bicolor. Immunodetection of SbCENH3 antibody shows distinct punctual signals (green) in interphase nuclei (c) and centromere-localized signals (green) in mitotic metaphase chromosomes (d). Nuclei and chromosomes were counterstained with DAPI. Bar = 5 μm.
After anti-SbCENH3 ChIP sequencing, 13.3 and 17.5 million of ChIP and input reads, respectively, were mapped to all the repeat clusters of S. bicolor using ChIP-Seq Mapper. The enrichment of individual repeat clusters was calculated and normalized according to the number of analyzed reads. In total, 14 clusters showed a higher ratio of ChIP/input than the threshold of four (Figure 5 and Table 4). In line with our FISH data, CL2 (SorSat137) is the only satellite repeat interacting with the centromeric nucleosomes (Figure 5). Eight LTR repeat clusters, including three Ty3_gypsy-CRM (CL72, CL143, and CL164), three Ty3_gypsy-Athila (CL96, CL128, and CL129), and two Ty1_copia-SIRE (CL59 and CL84), as well as five unclassified repeats (CL44, CL120, CL174, CL175, and CL191) were detected as centromeric candidate sequences (Table 4). Nevertheless, the genome abundances of these LTR repeats were in the range of 0.02–0.45%, at least 10 times less than the centromeric satellite repeat SorSat137 (4.7%) (Table 4).
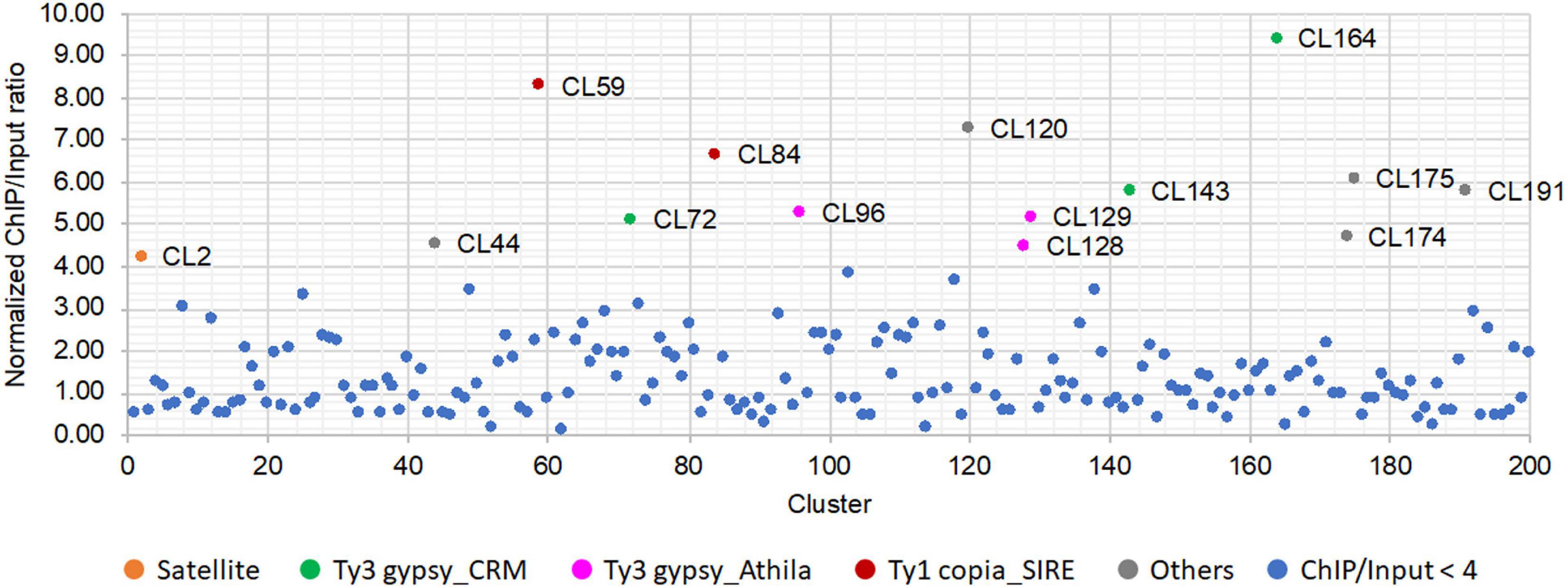
Figure 5. Relative enrichment of the repeat DNA clusters identified after CENH3-ChIP sequencing of S. bicolor. The enrichment ratio was calculated based on the read number of SbCENH3-ChIP DNA relative to the input DNA. The repeat clusters with ratio lower than four was labeled in blue and those with ratio higher than four was labeled in different colors according to their annotation.
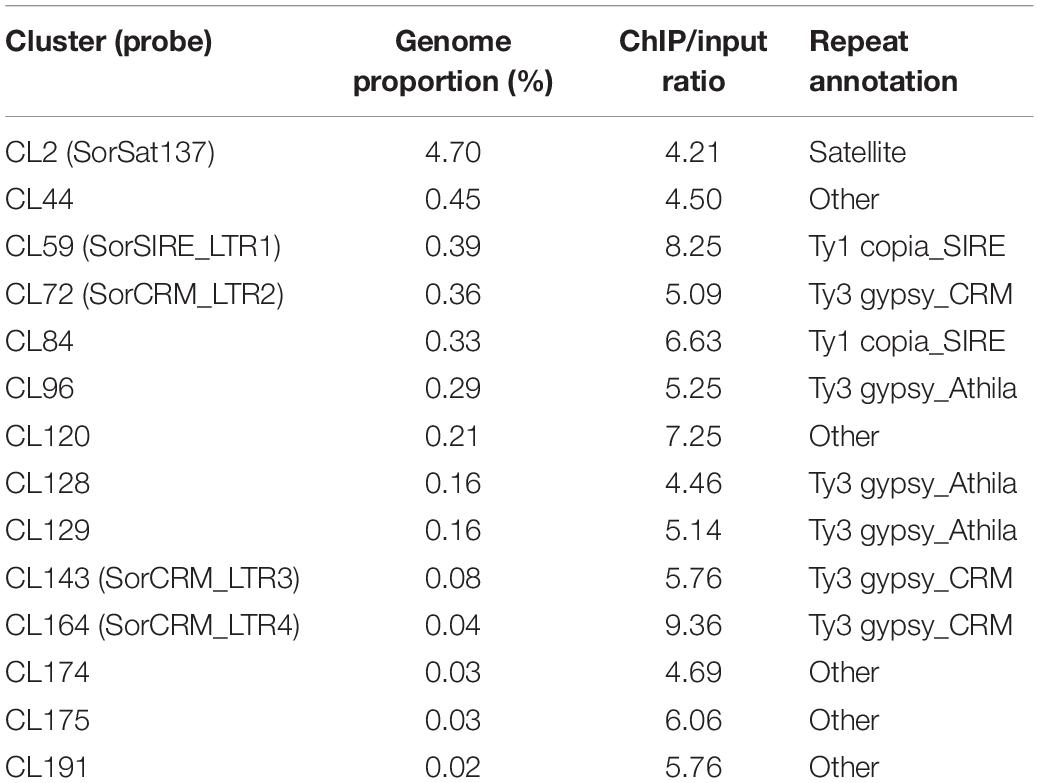
Table 4. Centromere-associated repetitive sequences in S. bicolor.
To verify the centromeric enrichment of the repeats, four repeat clusters were selected for FISH mapping. Of them, one cluster, SorSIRE_LTR1 (CL59), belongs to the most abundant Ty1_copia lineage in this genome. The other three clusters, SorCRM_LTR2 (CL72), SorCRM_LTR3 (CL143), and SorCRM_LTR4 (CL164), were annotated as CRM (centromeric retrotransposon of maize) family, which is one of the centromere-associated transposon families commonly found in plants (Jiang et al., 2003; Neumann et al., 2011).
All these four LTR repeats hybridized to the (peri)centromeric regions of S. bicolor (Figure 6), although not necessarily exclusively; for example, SorCRM_LTR3 showed a slightly more dispersed distribution in the centromere proximity (Figure 6C). The signals of the two LTRs with the highest ChIP/Input ratio (Table 4), SorSIRE_LTR1 (ChIP/Input ratio: 8.25) and SorCRM_LTR4 (ChIP/Input ratio: 9.36), overlapped strongly with the centromeric SorSat137 repeat (Figures 6A,D). The centromeric colocalization of the four tested LTRs with the predominant SorSat137 satellite indicated their association with S. bicolor centromeres, consistent with the CENH3-ChIPseq results. All these centromeric repeats seem to be conserved in the tetraploid S. halepense, as it is exemplarily shown for SorSat137 (Figures 2a4,5) as well as SorSIRE_LTR1 and SorCRM_LTR4 (Figures 6E,F).
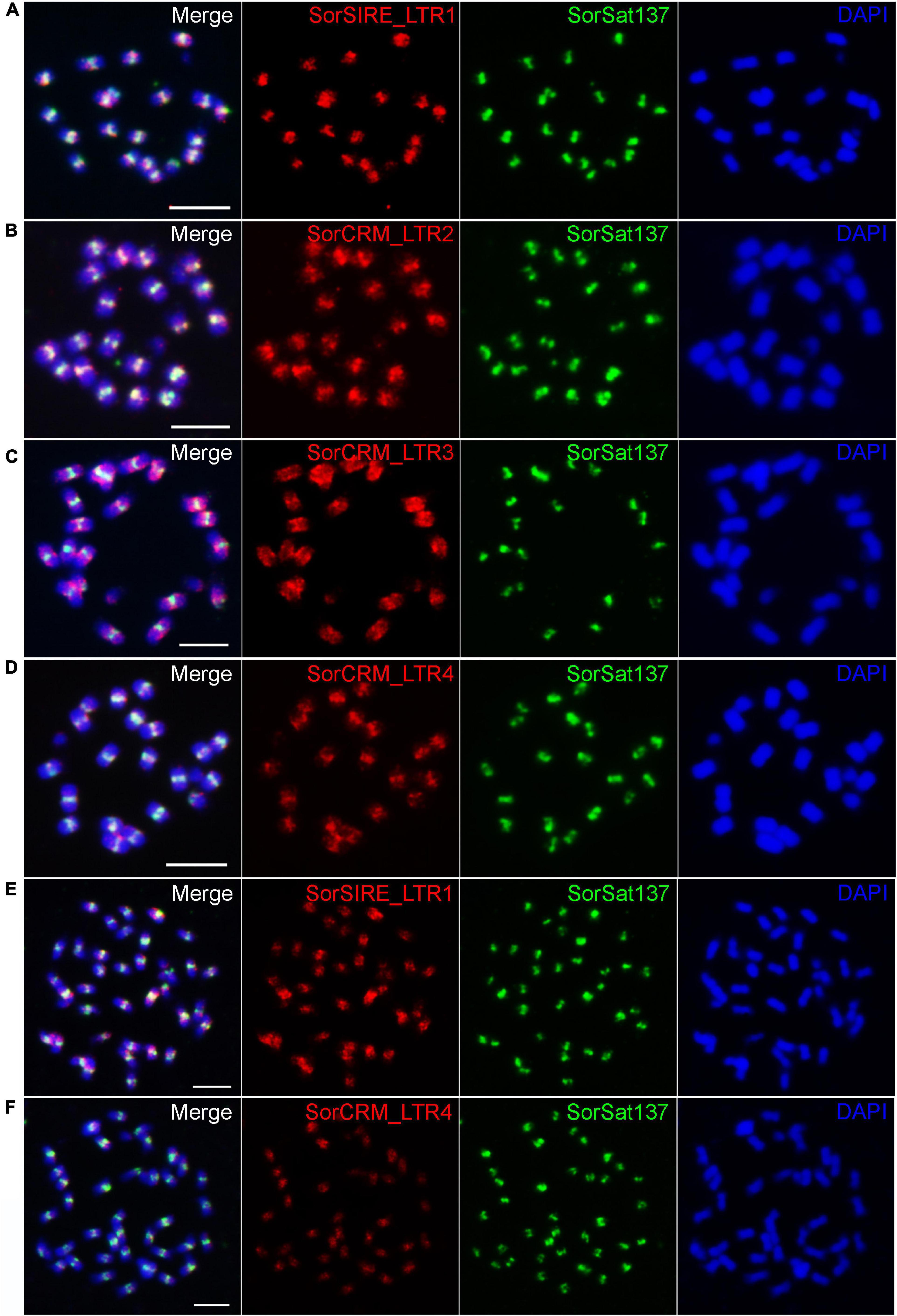
Figure 6. Fluorescence in situ hybridization mapping of centromeric repeats in S. bicolor and S. halepense. In S. bicolor, colocalization of (A) SorSIRE_LTR1, (B) SorCRM_LTR2, (C) SorCRM_LTR3, and (D) SorCRM_LTR4 with the centromeric satellite SorSat137, confirming the centromere enrichment of LTR repeats. The conserved centromeric distribution of (E) SorSIRE_LTR1 and (F) SorCRM_LTR4 in S. halepense is shown. Chromosomes were counterstained with DAPI. Bar = 5 μm.
Discussion
Divergence of Satellite Repeats Subsequent to Sorghum Polyploidization
Polyploidization has been long considered as an evolutionary event that leads to significant structural changes via genome rearrangement, especially after allopolyploidization when two distantly related genomes were combined (Otto, 2007). Many allopolyploids are superior to both parental species due to their beneficial characteristics in fitness and tolerance. The notably weedy S. halepense has been assumed to be an allotetraploid species formed by hybridization between S. bicolor and S. propinquum (Tang and Liang, 1988; Paterson et al., 1995, 2020). On the other hand, the rapid changes of fast-evolving satellite and retrotransposon DNAs alter genome composition and chromosome arrangement, leading to genome evolution. Incurred diversifying genomes pave the way for speciation. In this study, the genome-wide satellite and transposon categories and abundance were analyzed in five Sorghum accessions which were three diploid accessions, including two cultivated accessions (S. bicolor, S. bicolor var. technicum) and one weedy accession (S. bicolor ssp. verticilliflorum), and two tetraploid accessions of S. halepense.
The genome size of the tetraploid S. halepense accessions is around double of the Sorghum diploid accessions, with no genome downsizing detected in the polyploid genomes, which is similar to the example of Icelandic birch (Anamthawat-Jónsson et al., 2010). This phenomenon is opposed to the trend of genome downsizing among ancient angiosperm polyploids (Leitch and Bennett, 2004), likely indicating the recent origin of polyploidization in Sorghum.
The genomes of all Sorghum accessions comprise of about 55% of moderate and high-copy repeats, irrespective of the species or ploidy, demonstrating that no large-scale amplification or reduction of repeat sequences leading to a severe difference in total repeat content subsequent to the specification or polyploidization. Approximately 438 Mb of the S. bicolor genome is repetitive DNAs according to the RepeatExplorer-based analysis, which is very close to the estimated 460 Mb repeat fraction identified in the assembled genome of S. bicolor (Paterson et al., 2009). The comparison of genome-wide repeat profiles suggests that satellite DNAs played an important role in shaping the genome diversification in Sorghum, although their genome abundance (<6%) is much lower than that of the retroelements (>42%).
The chromosomal localization of the identified satellite DNAs revealed that drastic genome rearrangements might occur mainly in the S. halepense subtelomeric regions. Subtelomeres are known to be among the most dynamic and fast-evolving chromosomal regions (Contento et al., 2005; Garrido-Ramos, 2017) and often species-, genome-, or even chromosome-specific. For instance, diversification of subtelomeric satellite repeats between close Leymus species (Anamthawat-Jónsson et al., 2009) and between Avena subgenomes (Jiang et al., 2021) were reported. Subtelomeres are likely involved in facilitating homologous chromosome pairing during meiosis (Sadaie et al., 2003; Calderón et al., 2014) and play a role in maintaining chromosome ends and chromosomal stability (Mehrotra and Goyal, 2014; Padeken et al., 2015). In interspecific hybrids, pairing and recombination between non-homologous chromosome ends may lead to frequent sequence exchange and the birth of new subtelomeric repeats (Macas et al., 2006). The two satellite repeats enriched in S. halepense, SorSat200 and SorSat2192, are massively accumulated in the subtelomeres of S. halepense, while they preferentially reside in the pericentromeres in the diploid accessions. Hence, the accumulation of the two subtelomeric satellites, SorSat200 and SorSat2192, in the allotetraploid S. halepense might be a result of non-homologous recombination or large-scale genome rearrangement between pericentromeric and subtelomeric regions. Nevertheless, both satellite repeats are basically absent from the ends of S. bicolor chromosomes or at least only present at copy numbers not detectable by our FISH stringency. Additionally, SorSat200 shows similarity to the reported S. halepense-specific repeats XSR1 (64.7%), XSR3 (81.6%), and XSR6 (73.4%), which did not show any cross-reaction with the S. bicolor genome by Southern blot hybridization (Hoangtang et al., 1991). Hence, the accumulation of the satellite repeat(s) at the chromosomal ends must have happened after the speciation of S. halepense.
SorSat200, additionally, hybridized strongly at the four chromosome ends of diploid S. bicolor ssp. verticilliflorum (SbWL) but weakly in the pericentromeric regions of all three diploid accessions. Thus, SorSat200, although highly enriched in S. halepense, is not species-specific but exists in the diploid Sorghum species as well. SorSat200 might be a potential satellite DNA for evolutionary study of Sorghum genus due to its extraordinary diversity with respect to the chromosomal distribution.
Besides the disperse signals of SorSat2192 in the pericentromeric regions of all analyzed Sorghum accessions, selective accumulation at distal regions of two chromosome pairs was detected in the tetraploid S. halepense. A similar heterogeneous chromosomal distribution was found for the satellite Khipu in Phaseolus vulgaris (Richard et al., 2013). Khipu repeats were detected in both subtelomeres and centromeres, and even different copy numbers and sequence variants were present among chromosome ends. The availability of an assembled genome of S. halepense might help to understand the heterogeneous distribution of subtelomeric satellites in Sorghum.
In addition to the subtelomeric repeats, the satellite repeats SorSat679 and SorSat708, which preferentially locate in the pericentromeres, displayed chromosome-specific amplification in an accession-specific manner. Depending on the accession, different numbers of chromosomes showed enrichment of these repeats at distinct loci. Particularly, for SorSat708 a heteromorphic pattern on one chromosome pair of S. halepense “TT” was observed. Such heteromorphic distributions of repeat DNAs between homologous chromosomes were reported in other plant species, such as Allium subvillosum (Jamilena et al., 1990), Brachycome dichromosomatica (Houben et al., 2000), Secale cereale (Viinikka and Kavander, 1986), and Triticum araraticum (Badaeva et al., 1994). SorSat708 might reflect repeat composition diversity within the species S. halepense.
In contrast to the polymorphic distributions of pericentromeric and subtelomeric satellite repeats, the chromosomal association of SorSat123 and SorSat239 with 45S rDNA are conserved among the diploid and tetraploid Sorghum accessions. These repeats are likely originated from the intergenic 45S rDNA regions as also described for satellite repeats in other species (Macas et al., 2003; Garrido-Ramos, 2015) and might arose before the diversification of the studied Sorghum species.
The analysis of genome-wide repetitive profiles in the Sorghum accessions demonstrates the importance of satellite DNAs in shaping the genome divergence in Sorghum. In diploid Sorghum genomes, especially the differential amplifications of the pericentromeric satellite repeats, such as SorSat679 and SorSat708, among chromosomes contributed to their diversification. Among Sorghum genotypes, the observed polymorphic distribution of the subtelomeric satellites enriched in the tetraploids indicates drastic genome rearrangements after the allopolyploidization event forming S. halepense.
The Sequence Composition of the Satellite-Dominated Centromere in S. bicolor Is Conserved in Eusorghum
Our CENH3 ChIP-seq analysis revealed that the centromeres of S. bicolor are mainly comprised of the satellite repeat SorSat137 (CEN38, Miller et al., 1998b; Zwick et al., 2000) with a genome proportion of 4.7% in combination with diverse retroelements. These retroelements include CRM as well as Athila of Ty3_gypsy and Ty1_copia-SIRE sequences with a total genome proportion of only 1.81%.
A comparison of the consensus sequences of the SorSat137 family members in the five Eusorghum genomes revealed a similarity of more than 92%. This repeat also exists in most centromeres of S. officinarum × spontaneum but with a lower sequence homology (Zwick et al., 2000), as well as in the Sorghum subgenera: Chaetosorghum, Heterosorghum, Parasorghum, and Stiposorghum (Anderson, 2005). Most likely, SorSat137 (CEN38, Miller et al., 1998b; Zwick et al., 2000) preexisted in the common ancestor of Sorghum and Saccharum and diverged during the evolution of Sorghum except in the subgenus Eusorghum.
The similarity of the centromeric composition of Sorghum and Saccharum is not restricted to the satellite repeat SorSat137. The centromeric Ty3_gypsy-CRM retroelements were also identified in the wild Saccharum species, S. spontaneum (Zhang et al., 2017). While Ty3_gypsy-CRM retroelements are commonly found in plant centromeres (Miller et al., 1998a; Neumann et al., 2011), Ty3_gypsy-Athila elements are less frequently detected in centromeric regions except in the centromere core of Arabidopsis (Kumekawa et al., 2000), Festuca, and Lolium species (Zwyrtkova et al., 2020).
The other S. bicolor CENH3 nucleosome interacting retroelement belongs to the SIRE class (or Sirevirus), which is the most abundant Ty1_copia retrotransposon of 4.5% in this genome. In most plant species, SIRE retrotransposons tend to show a disperse distribution along chromosomes or are preferentially enriched in pericentromeric heterochromatin (Weber et al., 2010; de Souza et al., 2018).
The distribution patterns of the identified centromeric satellite and retrotransposon repeats in the allotetraploid S. halepense are similar to those in the diploid S. bicolor. This high similarity in the centromeric composition implies the close relationship of both species and supports the assumption that S. bicolor might have been involved in the formation of S. halepense.
Data Availability Statement
The data presented in the study are deposited in the European Nucleotide Archive (ENA) repository, accession number PRJEB46549 (http://www.ebi.ac.uk/ena/data/view/PRJEB46549).
Author Contributions
Y-TK performed DNA extraction, repeat analysis, FISH, ChIPseq experiments, phylogenetic analysis, and wrote the manuscript. TI identified CENH3, performed western blot, and immunostaining. JF measured the genome size. W-HH collected the plant materials. AH and Y-RL designed the research. Y-TK, JF, Y-RL, and AH revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Ministry of Science and Technology (MOST 106-2313-B-002-034-MY3; MOST 108-2811-B-002-608; and MOST 109-2917-I-564-022) and Taiwan and a sub-award from the CSIRO for the grant “Capturing Heterosis” from the BMGF (United States).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Anne Fiebig (IPK, Gatersleben, Germany) for submitting the sequencing datasets to the public repository.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.729734/full#supplementary-material
Footnotes
- ^ https://repeatexplorer-elixir.cerit-sc.cz/galaxy/
- ^ http://iqtree.cibiv.univie.ac.at
- ^ https://itol.embl.de/
- ^ https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
- ^ www.lifetein.com
References
Aliyeva-Schnorr, L., Ma, L., and Houben, A. (2015). A fast air-dry dropping chromosome preparation method suitable for FISH in plants. J. Vis. Exp. 106:e53470. doi: 10.3791/53470
Anamthawat-Jónsson, K., Thórsson, A. T., Temsch, E. M., and Greihuber, J. (2010). Icelandic birch polyploids- The case of a perfect fit in genome size. J. Bot. 2010:347254. doi: 10.1155/2010/347254
Anamthawat-Jónsson, K., Wenke, T., Thórsson, A. T., Sveinsson, S., Zakrzewski, F., and Schmidt, T. (2009). Evolutionary diversification of satellite DNA sequences from Leymus (Poaceae: Triticeae). Genome 52, 381–390. doi: 10.1139/g09-013
Anderson, J. C. (2005). The Use of CEN38 in Assessing Evolutionary Relationships in the Genus Sorghum. Master’s thesis. College Station, TX: Texas A&M University.
Andrews, S. (2010). FastQC: A Quality Control Tool For High Throughput Sequence Data. Berlin: Springer.
Badaeva, E. D., Badaev, N. S., Gill, B. S., and Filatenko, A. A. (1994). Intraspecific karyotype divergence in Triticum araraticum (Poaceae). Plant Syst. Evol. 192, 117–145. doi: 10.1007/Bf00985912
Bennetzen, J. L., and Wang, H. (2014). The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 65, 505–530. doi: 10.1146/annurev-arplant-050213-035811
Calderón, M. D. C., Rey, M.-D., Cabrera, A., and Prieto, P. (2014). The subtelomeric region is important for chromosome recognition and pairing during meiosis. Sci. Rep. 4:6488. doi: 10.1038/srep06488
Cheng, Z., Dong, F., Langdon, T., Ouyang, S., Buell, C. R., Gu, M., et al. (2002). Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 14, 1691–1704. doi: 10.1105/tpc.003079
Cheng, Z. J., and Murata, M. (2003). A centromeric tandem repeat family originating from a part of Ty3/gypsy-retroelement in wheat and its relatives. Genetics 164, 665–672. doi: 10.1093/genetics/164.2.665
Contento, A., Heslop-Harrison, J. S., and Schwarzacher, T. (2005). Diversity of a major repetitive DNA sequence in diploid and polyploid Triticeae. Cytogenet. Genome Res. 109, 34–42. doi: 10.1159/000082379
de Souza, T. B., Chaluvadi, S. R., Johnen, L., Marques, A., González-Elizondo, M. S., Bennetzen, J. L., et al. (2018). Analysis of retrotransposon abundance, diversity and distribution in holocentric Eleocharis (Cyperaceae) genomes. Ann. Bot. 122, 279–290. doi: 10.1093/aob/mcy066
De Wet, J. (1978). Special paper: systematic and evolution of sorghum sect. Sorghum (Gramineae). Am. J. Bot. 65, 477–484. doi: 10.1002/j.1537-2197.1978.tb06096.x
Deschamps, S., Zhang, Y., Llaca, V., Ye, L., Sanyal, A., King, M., et al. (2018). A chromosome-scale assembly of the sorghum genome using nanopore sequencing and optical mapping. Nat. Commun. 9:4844. doi: 10.1038/s41467-018-07271-1
Dolezel, J., Bartos, J., Voglmayr, H., and Greilhuber, J. (2003). Nuclear DNA content and genome size of trout and human. Cytometry A 51, 127–128. doi: 10.1002/cyto.a.10013
Garber, E. D. (1950). Cytotaxonomic Studies in the Genus Sorghum. Berkeley: University of California Press.
Garrido-Ramos, M. A. (2015). Satellite DNA in plants: more than just rubbish. Cytogenet. Genome Res. 146, 153–170. doi: 10.1159/000437008
Garrido-Ramos, M. A. (2017). Satellite DNA: an evolving topic. Genes 8:230. doi: 10.3390/genes8090230
Gendrel, A. V., Lippman, Z., Martienssen, R., and Colot, V. (2005). Profiling histone modification patterns in plants using genomic tiling microarrays. Nat. Methods 2, 213–218. doi: 10.1038/nmeth0305-213
Hartley, G., and O’Neill, R. J. (2019). Centromere repeats: hidden gems of the genome. Genes 10:223. doi: 10.3390/genes10030223
Hoangtang, Dube, S. K., Liang, G. H., and Kung, S. D. (1991). Possible repetitive DNA markers for Eusorghum and Parasorghum and their potential use in examining phylogenetic hypotheses on the origin of Sorghum species. Genome 34, 241–250. doi: 10.1139/g91-038
Houben, A., Schroeder-Reiter, E., Nagaki, K., Nasuda, S., Wanner, G., Murata, M., et al. (2007). CENH3 interacts with the centromeric retrotransposon cereba and GC-rich satellites and locates to centromeric substructures in barley. Chromosoma 116, 275–283. doi: 10.1007/s00412-007-0102-z
Houben, A., Wanner, G., Hanson, L., Verlin, D., Leach, C. R., and Timmis, J. N. (2000). Cloning and characterisation of polymorphic heterochromatic segments of Brachycome dichromosomatica. Chromosoma 109, 206–213. doi: 10.1007/s004120050430
Hsieh, W. H., Hwu, K. K., Wu, Y. P., Chen, C. H., and Lin, Y. R. (2015). The assessment of genetic diversity of sorghum germpplasm by using SSR markers. J. Agric. Assoc. Taiwan 16, 85–110.
Jamilena, M., Rejon, C. R., and Rejon, M. R. (1990). Variation in the heterochromatin and nucleolar organizing regions of Allium subvillosum L. (Liliaceae). Genome 33, 779–784. doi: 10.1139/g90-116
Jiang, J., Birchler, J. A., Parrott, W. A., and Kelly Dawe, R. (2003). A molecular view of plant centromeres. Trends Plant Sci. 8, 570–575. doi: 10.1016/j.tplants.2003.10.011
Jiang, W., Jiang, C., Yuan, W., Zhang, M., Fang, Z., Li, Y., et al. (2021). A universal karyotypic system for hexaploid and diploid Avena species brings oat cytogenetics into the genomics era. BMC Plant Biol. 21:213. doi: 10.1186/s12870-021-02999-3
Kapitonov, V. V., and Jurka, J. (1999). Molecular paleontology of transposable elements from Arabidopsis thaliana. Genetica 107, 27–37. doi: 10.1023/A:1004030922447
Karimi-Ashtiyani, R., Ishii, T., Niessen, M., Stein, N., Heckmann, S., Gurushidze, M., et al. (2015). Point mutation impairs centromeric CENH3 loading and induces haploid plants. Proc. Natl. Acad. Sci. U.S.A. 112, 11211–11216. doi: 10.1073/pnas.1504333112
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, J. S., Islam-Faridi, M. N., Klein, P. E., Stelly, D. M., Price, H. J., Klein, R. R., et al. (2005). Comprehensive molecular cytogenetic analysis of sorghum genome architecture: distribution of euchromatin, heterochromatin, genes and recombination in comparison to rice. Genetics 171, 1963–1976. doi: 10.1534/genetics.105.048215
Kubis, S., Schmidt, T., and Heslop-Harrison, J. S. (1998). Repetitive DNA elements as a major component of plant genomes. Ann. Bot. 82, 45–55.
Kumar, A., and Bennetzen, J. L. (1999). Plant retrotransposons. Ann. Rev. Genet. 33, 479–532. doi: 10.1146/annurev.genet.33.1.479
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumekawa, N., Hosouchi, T., Tsuruoka, H., and Kotani, H. (2000). The size and sequence organization of the centromeric region of Arabidopsis thaliana chromosome 5. DNA Res. 7, 315–321. doi: 10.1093/dnares/7.6.315
Leitch, I. J., and Bennett, M. D. (2004). Genome downsizing in polyploid plants. Biol. J. Linn. Soc. 82, 651–663. doi: 10.1111/j.1095-8312.2004.00349.x
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, W., Zhang, P., Fellers, J. P., Friebe, B., and Gill, B. S. (2004). Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 40, 500–511. doi: 10.1111/j.1365-313x.2004.02228.x
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Macas, J., Navratilova, A., and Koblizkova, A. (2006). Sequence homogenization and chromosomal localization of VicTR-B satellites differ between closely related Vicia species. Chromosoma 115, 437–447. doi: 10.1007/s00412-006-0070-8
Macas, J., Navratilova, A., and Meszaros, T. (2003). Sequence subfamilies of satellite repeats related to rDNA intergenic spacer are differentially amplified on Vicia sativa chromosomes. Chromosoma 112, 152–158. doi: 10.1007/s00412-003-0255-3
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331. doi: 10.1093/nar/gkh454
May, B. P., Lippman, Z. B., Fang, Y. D., Spector, D. L., and Martienssen, R. A. (2005). Differential regulation of strand-specific transcripts from Arabidopsis centromeric satellite repeats. PLoS Genet. 1:e0010079. doi: 10.1371/journal.pgen.0010079
McCormick, R. F., Truong, S. K., Sreedasyam, A., Jenkins, J., Shu, S., Sims, D., et al. (2018). The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 93, 338–354. doi: 10.1111/tpj.13781
Mehrotra, S., and Goyal, V. (2014). Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genomics Proteomics Bioinformatics 12, 164–171. doi: 10.1016/j.gpb.2014.07.003
Melters, D. P., Bradnam, K. R., Young, H. A., Telis, N., May, M. R., Ruby, J. G., et al. (2013). Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 14:R10. doi: 10.1186/gb-2013-14-1-r10
Miller, J. T., Dong, F., Jackson, S. A., Song, J., and Jiang, J. (1998a). Retrotransposon-related DNA sequences in the centromeres of grass chromosomes. Genetics 150:1615. doi: 10.1093/genetics/150.4.1615
Miller, J. T., Jackson, S., Nasuda, S., Gill, B., Wing, R. A., and Jiang, J. (1998b). Cloning and characterization of a centromere-specific repetitive DNA element from Sorghum bicolor. Theor. Appl. Genet. 96, 832–839. doi: 10.1007/s001220050809
Neumann, P., Navratilova, A., Koblizkova, A., Kejnovsky, E., Hribova, E., Hobza, R., et al. (2011). Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mob. DNA 2:4. doi: 10.1186/1759-8753-2-4
Neumann, P., Navratilova, A., Schroeder-Reiter, E., Koblizkova, A., Steinbauerova, V., Chocholova, E., et al. (2012). Stretching the rules: monocentric chromosomes with multiple centromere domains. PLoS Genet. 8:e1002777. doi: 10.1371/journal.pgen.1002777
Novák, P., Avila Robledillo, L., Koblizkova, A., Vrbova, I., Neumann, P., and Macas, J. (2017). TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45:e111. doi: 10.1093/nar/gkx257
Novák, P., Neumann, P., and Macas, J. (2010). Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11:378. doi: 10.1186/1471-2105-11-378
Novák, P., Neumann, P., and Macas, J. (2020). Global analysis of repetitive DNA from unassembled sequence reads using repeatexplorer2. Nat. Protoc. 15, 3745–3776. doi: 10.1038/s41596-020-0400-y
Novák, P., Neumann, P., Pech, J., Steinhaisl, J., and Macas, J. (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793. doi: 10.1093/bioinformatics/btt054
Otto, S. P. (2007). The evolutionary consequences of polyploidy. Cell 131, 452–462. doi: 10.1016/j.cell.2007.10.022
Padeken, J., Zeller, P., and Gasser, S. M. (2015). Repeat DNA in genome organization and stability. Curr. Opi. Genet. Dev. 31, 12–19. doi: 10.1016/j.gde.2015.03.009
Paterson, A. H., Bowers, J. E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., et al. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556. doi: 10.1038/nature07723
Paterson, A. H., Kong, W. Q., Johnston, R. M., Nabukalu, P., Wu, G. H., Poehlman, W. L., et al. (2020). The evolution of an invasive plant, Sorghum halepense L. (’Johnsongrass’). Front. Genet. 11:317. doi: 10.3389/fgene.2020.00317
Paterson, A. H., Schertz, K. F., Lin, Y. R., Liu, S. C., and Chang, Y. L. (1995). The weediness of wild plants - Molecular analysis of genes influencing dispersal and persistence of johnsongrass, Sorghum halepense (L) Pers. Proc. Natl. Acad. Sci U.S.A. 92, 6127–6131. doi: 10.1073/pnas.92.13.6127
Price, H. J., Dillon, S. L., Hodnett, G., Rooney, W. L., Ross, L., and Johnston, J. S. (2005). Genome evolution in the genus Sorghum (Poaceae). Ann. Bot. 95, 219–227. doi: 10.1093/aob/mci015
Richard, M. M. S., Chen, N. W. G., Thareau, V., Pflieger, S., Blanchet, S., Pedrosa-Harand, A., et al. (2013). The subtelomeric khipu satellite repeat from Phaseolus vulgaris: lessons learned from the genome analysis of the Andean genotype G19833. Front. Plant Sci. 4:109. doi: 10.3389/fpls.2013.00109
Robledillo, L. A., Neumann, P., Koblizkova, A., Novak, P., Vrbova, I., and Macas, J. (2020). Extraordinary sequence diversity and promiscuity of centromeric satellites in the Legume Tribe Fabeae. Mol. Biol. Evol. 37:90. doi: 10.1093/molbev/msaa090
Sadaie, M., Naito, T., and Ishikawa, F. (2003). Stable inheritance of telomere chromatin structure and function in the absence of telomeric repeats. Genes Dev. 17, 2271–2282. doi: 10.1101/gad.1112103
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115.
Su, H., Liu, Y., Liu, C., Shi, Q., Huang, Y., and Han, F. (2019). Centromere satellite repeats have undergone rapid changes in polyploid wheat subgenomes. Plant Cell 31, 2035–2051. doi: 10.1105/tpc.19.00133
Tang, H., and Liang, G. (1988). The genomic relationship between cultivated sorghum [Sorghum bicolor (L.) Moench] and Johnsongrass [S. halepense (L.) Pers.]: a re-evaluation. Theor. Appl. Genet. 76, 277–284. doi: 10.1007/bf00257856
Tek, A. L., Kashihara, K., Murata, M., and Nagaki, K. (2011). Functional centromeres in Astragalus sinicus include a compact centromere-specific histone H3 and a 20-bp tandem repeat. Chromosome Res. 19, 969–978. doi: 10.1007/s10577-011-9247-y
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Viinikka, Y., and Kavander, T. (1986). C-band polymorphism in the inbred lines showing neocentric activity in rye. Hereditas 104, 203–207. doi: 10.1111/j.1601-5223.1986.tb00533.x
Weber, B., Wenke, T., Frommel, U., Schmidt, T., and Heitkam, T. (2010). The Ty1-copia families SALIRE and Cotzilla populating the Beta vulgaris genome show remarkable differences in abundance, chromosomal distribution, and age. Chromosome Res. 18, 247–263. doi: 10.1007/s10577-009-9104-4
Yang, X., Zhao, H., Zhang, T., Zeng, Z., Zhang, P., Zhu, B., et al. (2018). Amplification and adaptation of centromeric repeats in polyploid switchgrass species. New Phytol. 218, 1645–1657. doi: 10.1111/nph.15098
Zhang, W., Zuo, S., Li, Z., Meng, Z., Han, J., Song, J., et al. (2017). Isolation and characterization of centromeric repetitive DNA sequences in Saccharum spontaneum. Sci. Rep. 7:41659. doi: 10.1038/srep41659
Zhong, C. X., Marshall, J. B., Topp, C., Mroczek, R., Kato, A., Nagaki, K., et al. (2002). Centromeric retroelements and satellites interact with maize kinetochore protein CENH3. Plant Cell 14, 2825–2836. doi: 10.1105/tpc.006106
Zwick, M. S., Islam-Faridi, M. N., Zhang, H. B., Hodnett, G. L., Gomez, M. I., Kim, J. S., et al. (2000). Distribution and sequence analysis of the centromere-associated repetitive element CEN38 of Sorghum bicolor (Poaceae). Am. J. Bot. 87, 1757–1764. doi: 10.2307/2656825
Keywords: satellite DNA, centromere, genome evolution, CENH3, long terminal repeat (LTR), dryland crop, Sorghum
Citation: Kuo Y-T, Ishii T, Fuchs J, Hsieh W-H, Houben A and Lin Y-R (2021) The Evolutionary Dynamics of Repetitive DNA and Its Impact on the Genome Diversification in the Genus Sorghum. Front. Plant Sci. 12:729734. doi: 10.3389/fpls.2021.729734
Received: 23 June 2021; Accepted: 23 July 2021;
Published: 12 August 2021.
Edited by:
Tony Heitkam, Technische Universität Dresden, GermanyReviewed by:
Kesara Anamthawat-Jonsson, University of Iceland, IcelandGernot Presting, University of Hawaii, United States
Copyright © 2021 Kuo, Ishii, Fuchs, Hsieh, Houben and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Houben, aG91YmVuQGlway1nYXRlcnNsZWJlbi5kZQ==; Yann-Rong Lin, eWFubi1yb25nLmxpbkB3b3JsZHZlZy5vcmc=
†These authors share last authorship