 Carlos de Tomás
Carlos de Tomás Carlos M. Vicient
Carlos M. Vicient- Structure and Evolution of Plant Genomes Group, Centre for Research in Agricultural Genomics, CSIC-IRTA-UAB-UB, Edifici CRAG, Bellaterra, Barcelona, Spain
Endogenous viral elements (EVEs) are viral sequences that have been integrated into the nuclear chromosomes. Endogenous pararetrovirus (EPRV) are a class of EVEs derived from DNA viruses of the family Caulimoviridae. Previous works based on a limited number of genome assemblies demonstrated that EPRVs are abundant in plants and are present in several species. The availability of genome sequences has been immensely increased in the recent years and we took advantage of these resources to have a more extensive view of the presence of EPRVs in plant genomes. We analyzed 278 genome assemblies corresponding to 267 species (254 from Viridiplantae) using tBLASTn against a collection of conserved domains of the Reverse Transcriptases (RT) of Caulimoviridae. We concentrated our search on complete and well-conserved RT domains with an uninterrupted ORF comprising the genetic information for at least 300 amino acids. We obtained 11.527 sequences from the genomes of 202 species spanning the whole Tracheophyta clade. These elements were grouped in 57 clusters and classified in 13 genera, including a newly proposed genus we called Wendovirus. Wendoviruses are characterized by the presence of four open reading frames and two of them encode for aspartic proteinases. Comparing plant genomes, we observed important differences between the plant families and genera in the number and type of EPRVs found. In general, florendoviruses are the most abundant and widely distributed EPRVs. The presence of multiple identical RT domain sequences in some of the genomes suggests their recent amplification.
Introduction
Endogenous viral elements (EVEs) are viral sequences that have been integrated into the nuclear chromosomes, enabling their vertical transmission and potential fixation in host populations (Feschotte and Gilbert, 2012). Viral integration within eukaryotic genomes is a widely recognized phenomenon described in many species thanks to the sequencing of whole genomes. Some of these EVEs are the consequence of a mandatory genome integration stage in the life cycle of reverse-transcribing viruses, such as retroviruses (Katzourakis and Gifford, 2010), but for others, such as all plant viruses, hepadnaviruses or the SARS-CoV-2, the integration in the host genome is not part of the virus life cycle and the mechanisms of integration are few well understood (Kojima et al., 2021; Zhang et al., 2021).
The first described plant EVE was a Geminiviridae element (Bejarano et al., 1996). EVEs derived from Caulimoviridae are abundant in plants (Diop et al., 2018). EVEs derived from another non-retroviral dsRNA, ssRNA, or ssDNA viruses have also been described as, for example, from Narnaviridae (Choi et al., 2021), Partitiviridae, Betarhabdovirinae and Betaflexiviridae (Chiba et al., 2011). The genome integration mechanism of the EVEs remains largely uncharacterized and different mechanisms for the integration were proposed. The most accepted theory is that endogenization results from a non-homologous recombination between virus and host genomes, usually in the context of either a double-stranded DNA break repair or a transposon-mediated process (Richert-Pöggeler et al., 2021).
If EVEs are integrated into or near host genes, this will be generally detrimental, and they will be removed from host population by purifying selection. In the rare cases that the integration of an EVE is beneficial, it will be fixed in the host population by positive selection, in the same way that occurs with other types of genomic elements like transposons (Catlin and Josephs, 2022). However, most of the EVEs are neutral and will become degraded due to the accumulation of disruptive mutations, insertions or deletions. Due to the random nature of these mutations, it is possible to reconstruct the sequences of the infectious viruses based on the EVEs sequences, particularly for high copy number EVEs (Aiewsakun and Katzourakis, 2015). In consequence, EVEs can be considered as genomic “fossils” and be employed for investigating viral origins and diversity and become the main tool for a new emerging field called Paleovirology. Paleovirology is the study of the ancient evolution of viruses through analyzing endogenous viral elements in the host genomes (Etienne, 2017). Due to the increasing number of sequenced genomes, numerous EVEs can be uncovered, and some of them are distinct from the currently known episomal viruses (Johnson, 2019). Another important property of EVEs is that they can be used to calibrate the timing of virus evolution. If an EVE is orthologous across several species, this gives a minimum estimate for the age of the virus that integrated into the genome (Aiewsakun and Katzourakis, 2015).
Caulimoviridae is a family of double-stranded DNA (dsDNA) viruses infecting plants that contain a reverse transcription stage in their replication cycle (International Committee on Taxonomy of Viruses, ICTV, https://ictv.global/). Although integration into the genome is not an essential part of their replication cycle, there are much evidence of their presence as integrated forms among genomes of the plant kingdom (Geering et al., 2014; Diop et al., 2018) and they have been included as a new category in some repetitive DNA sequence databases like Repbase (Bao et al., 2015). Caulimoviridae can be classified into 11 genera based on their genome organization (number of open reading frames and the arrangement of protein domains within them) and the morphology of their virus particles (ICTV, https://ictv.global/). Some of these genera have been reported as EVEs in plant genomes (Endogenous pararetrovirus, EPRVs) but, in addition, many of the EPRVs belong to a genus for which so far no episomal counterparts have been described (Geering et al., 2014; Chen and Kishima, 2016; Diop et al., 2018).
The integration of an EVE into or near a gene can potentially modify gene transcription or modify mRNA processing, resulting in mutant phenotypes. Most of the described EPRVs are inserted in intergenic regions and have no apparent deleterious effect on the host. However, there are examples of EPRVs inserted inside genes with potential effects on gene expression as, for example, in the case of Vitis vinifera, which has several EPRVs inserted in introns (Geering et al., 2014).
Most of the EPRVs are transcriptionally or translationally inactive because they are partial and/or comprise rearranged sequences and/or inactivating mutations. Often EPRVs form clusters resulting from the simultaneous integration of several complete or partial copies in tandem or nested (Richert-Pöggeler et al., 2003). Infrequently, these integrated sequences are transcriptionally active and the resulting RNAs can serve as precursors of extrachromosomal viral DNA and lead to systemic and vertically transmitted infections (Hohn et al., 2008; Gayral et al., 2008). Transcriptional activation can be driven by viral promoters present within the integrated element or plant promoters in the vicinity of the EPRV sequence (Lockhart et al., 2000; Kuriyama et al., 2020). On the other hand, EPRV derived RNAs can also be inducers for RNA interference (RNAi) and gene silencing mechanisms through the generation of small interfering RNAs (siRNAs) (Bertsch et al., 2009; Ricciuti et al., 2021).
RNA-directed DNA polymerase (Reverse Transcriptase, RT) coding sequences are present in a wide variety of genetic elements and contains a relatively well conserved central domain, allowing its use for phylogenetic analyses (Hansen and Heslop-Harrison, 2004) and for searches for homologues of, for example, EPRVs in genome sequences (Diop et al., 2018).
Previous studies have examined the EPRVs diversity in plant genomes based on the limited number of genome sequences available in each case (Geering et al., 2014; Diop et al., 2018) Nowadays, the number of sequenced plant genomes have increased significantly, and we decided to screen them for the presence of EPRVs, obtaining a broader picture of the distribution of these endogenous elements. We identified the major EPRV lineages and analyzed their distribution in the different plant orders and genera. We also describe a new possible genus of Caulimoviridae present only as EPRVs we called Wendovirus.
Materials and methods
Discovery and analyses of recently inserted endogenous Caulimoviridae
We built a library containing an assortment of 182 RT central domain amino acid sequences (Supplementary Data 1). This collection includes one sequence from Retroviridae, 14 from Ty3/Gypsy LTR retrotransposons of the six most abundant genera in plants (Athila, CRM, Galadriel, Ogre, Reina, Retand and Tekay), 104 from the eleven genera of Caulimoviridae (Badnavirus, Caulimovirus, Vaccinivirus, Soymovirus, Cavemovirus, Solendovirus, Dioscovirus, Rosadnavirus, Tungrovirus, Petuvirus and Ruflodivirus), and 63 from six groups of exclusively endogenous Caulimoviridae (Florendovirus, Xendovirus, Yendovirus, Zendovirus, Gymnendovirus and Fernendovirus) (hereafter referred to as operational taxonomic units (OTUs) following the nomenclature proposed by Diop et al., 2018. For further analyses, we selected ten sequences representatives of the Caulimoviridae groups (Supplementary Data 2).
We selected 278 genome assemblies corresponding to 267 species (Supplementary Data 3): two from Bacteria, one from Chromista, two from Protozoa, 13 from Animal, six from Fungi and 254 from Plantae kingdom. Plantae kingdom’s genomes include three Rodophyta, seven Chlorophyta, three Bryophyta, one Marchantiophyta and 240 Tracheophyta genomes. Tracheophyta includes one Lycopodiopsida, four Pinopsida, 35 Liliopsida (11 families) and 200 Magnoliopsida (46 families) genomes. The genomes outside the Plantae kingdom were used as negative controls.
We compared the ten RT sequences with the 278 genome assemblies using tBLASTn with default parameters (except –e option set to 1e−10). Only the hits with at least 300 amino acid residues and no stop codons nor frameshifts were selected for further analysis. To avoid the inclusion in the selection of tandem duplications, we removed a hit if it was located less than 1500 bp to another (Supplementary Data 3). For each genome assembly, the selected set of RT sequences were clustered with the 182 RT selected reference domains and those having higher similarity with retrotransposons were removed from the analyses. RT sequences having higher similarity with Caulimoviridae were used for further analyses (Supplementary Data 4).
For cluster determination, the selected sequences from the genome assemblies were grouped using CD-HIT with a sequence identity cut-off of 60% (Cluster60) or of 100% (Cluster100), a bandwidth of alignment of 20 and a length of sequence to skip of 10. One sequence was then selected to be representative of each cluster60 (Supplementary Data 5). Only in the case of cluster60-8 we selected two sequences because the sequences in this cluster were clearly divided in two groups.
The cluster representative sequences were aligned with the representative sequences of episomal or endogenous Caulimoviridae (Supplementary Data 1) using MEGA-X (Kumar et al., 2018). The resulting alignment was then used to build a phylogenetic reconstruction using the maximum likelihood (ML) method and 500 bootstrap replicates using MEGA-X. The resulting tree was then used as a reference to classify the EPRV-RTs found in the genome assemblies.
The minimum ages of the integration events reported in this study were inferred by identifying the most distantly related pair of host species sharing a particular cluster of EPRVs and applying the estimated species divergence dates in TimeTree (http://www.timetree.org/) (Kumar et al., 2017).
Potential ORFs were predicted using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/) and the presence of Pfam domains in their encoded polypeptides was confirmed using MOTIF Search (https://www.genome.jp/tools/motif/).
Results
Distribution of genomic sequences encoding Reverse Transcriptase domains of recently inserted endogenous pararetroviruses (Caulimoviridae)
The objective of the work was to determine the presence of sequences encoding complete conserved RT domains corresponding to endogenous pararetrovirus (Caulimoviridae) within a collection of publicly available genome sequence assemblies from plant species and using some non-plant genome assemblies as negative controls. To identify them, we used a custom designed tBLASTn-based discovery pipeline, using as a probe a collection of 10 representative RT sequences of the different Caulimoviridae genera and OTUs (Supplementary Data 2). To give priority to the recently inserted copies, we only select sequences encoding RT domains of at least 300 amino acids that contain uninterrupted reading frames. Frequently EPRVs are inserted in tandemly arranged structures. To remove these duplications, when a RT coding region was located less than 1500 bp of another we only kept one of them. Due to their high sequence similarity, this first selection also contained RT sequences from Ty3/gypsy LTR-retrotansposons (Metaviridae). To remove them, EPRVs were confirmed by phylogenetic analyses. They were aligned with RT sequences of representative Caulimoviridae and LTR retrotransposons (Supplementary Data 1). Those sequences showing higher similarity with the Metaviridae than with Caulimoviridae were removed. Finally, we obtained 11.527 RT-EPRV sequences (Supplementary Data 4).
None of the analyzed genomes outside Plantae Kingdom contain RT- EPRV sequences and among the genomes of the Plantae kingdom, we did not find RT-EPRVs in Chlorophyta, Rodophyta, Bryophyta or Marchantiophyta. Among the Tracheophyta species, we did not find RT-EPRVs in the class Lycopodiopsida (Selaginella moellendorffii) but we found RT-EPRVs in genomes of all Tracheophyta classes (Pinopsida, Liliopsida and Magnoliopsida), confirming previous results (Gong and Han, 2018). All the four Pinopsida genomes analyzed contain RT-EPRV sequences (between 4 and 46). We included 35 genomes of species of the class Liliopsida and we found RT-EPRV sequences in 22 of them (63%) (between 1 and 63). Finally, we found RT-EPRV sequences in 180 of the 201 Magnaliopsida genomes (88%) (between 1 and 1186).
When comparing the results with the genomes of species belonging to the same genus, or varieties of the same species, the results obtained are, in general, similar. For example, the genomes of the two species of Kalanchoe contain 20 and 24, the two of Vitis contain 24 and 29 and the three of Solanum between 29 and 35. However, this is not always the case, and we can observe important differences in the number of RT-EPRVs in species of the same genus. For example, in the genera Arachis (between 56 and 473), Prunus (between 3 and 144), Rosa (between 76 and 340), Citrus (between 63 and 306) and Nicotiana (between 12 and 130). Some of these differences can be due to differences in the quality of the genome assemblies. For example, the presence of undetermined nucleotides can give rise to a reduction in the number of RT-EPRVs we detected. However, there are cases in which the best quality genome is the one with the least number of sequences. For example, we included three species of the genera Arabidopsis and the genome with the least number of sequences is the one with the best quality (Arabidopsis thaliana). All these results suggest that in some of the species there have been very recent integrations of EPRVs.
Classification of the RT-EPRVs present in plant genomes
To provide a classification, RT-EPRV sequences with at least 60% amino acid identity to each other were grouped, yielding a total of 57 clusters. The total number of sequences and genomes represented in each cluster varies greatly (Table 1). We performed a phylogenetic analysis using representative sequences of each cluster (Supplementary Data 5) and representatives of all Caulimoviridae genera and OTUs (Supplementary Data 1). Our phylogenetic analysis clustered together all the previous known sequences corresponding to the same genera and OTU of the Caulimoviridae, confirming the robustness of the analysis (Figure 1). This phylogenetic reconstruction allowed us to determine the diversity and nature of our collection of RT-EPRV sequences (Table 2). They were separated into 13 phyla. 30 of the clusters were associated with sequences of Caulimoviridae with episomal forms: 10 Petuvirus, 5 Dioscovirus, 5 Soymovirus, 5 Tungrovirus, 2 Badnavirus, 2 Caulimovirus and 1 Solendovirus. We did not find any representative of the genera Cavemovirus, Rosadnavirus or Vaccinivirus, and neither from the recently proposed genera Ruflodivirus. This result suggests that the virus species of these genera do not carry out endogenization, at least not recently or as frequently, or they only do it in a small range of species whose complete genomic sequence is not yet available. Of the rest, 20 clusters corresponded to OTUs from which only endogenous forms have been found: 11 Florendovirus, 3 Xendovirus, 3 Yendovirus, 3 Zendovirus and 1 Gymnendovirus. As we will describe later in detail, the remaining 6 clusters were associated with each other, forming a new OTU we called Wendovirus (Figure 1).
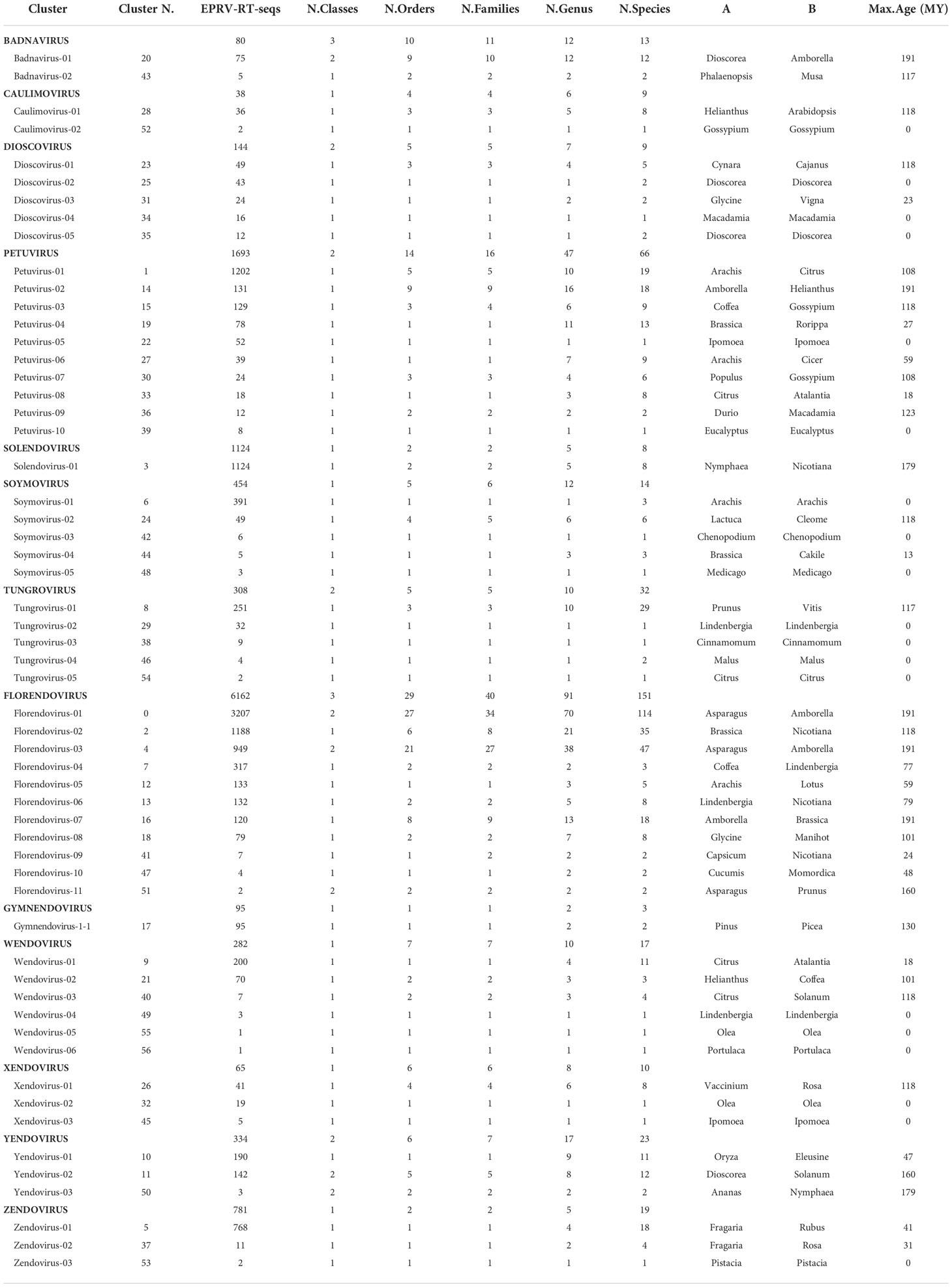
Table 1 Cluster60 statistics.
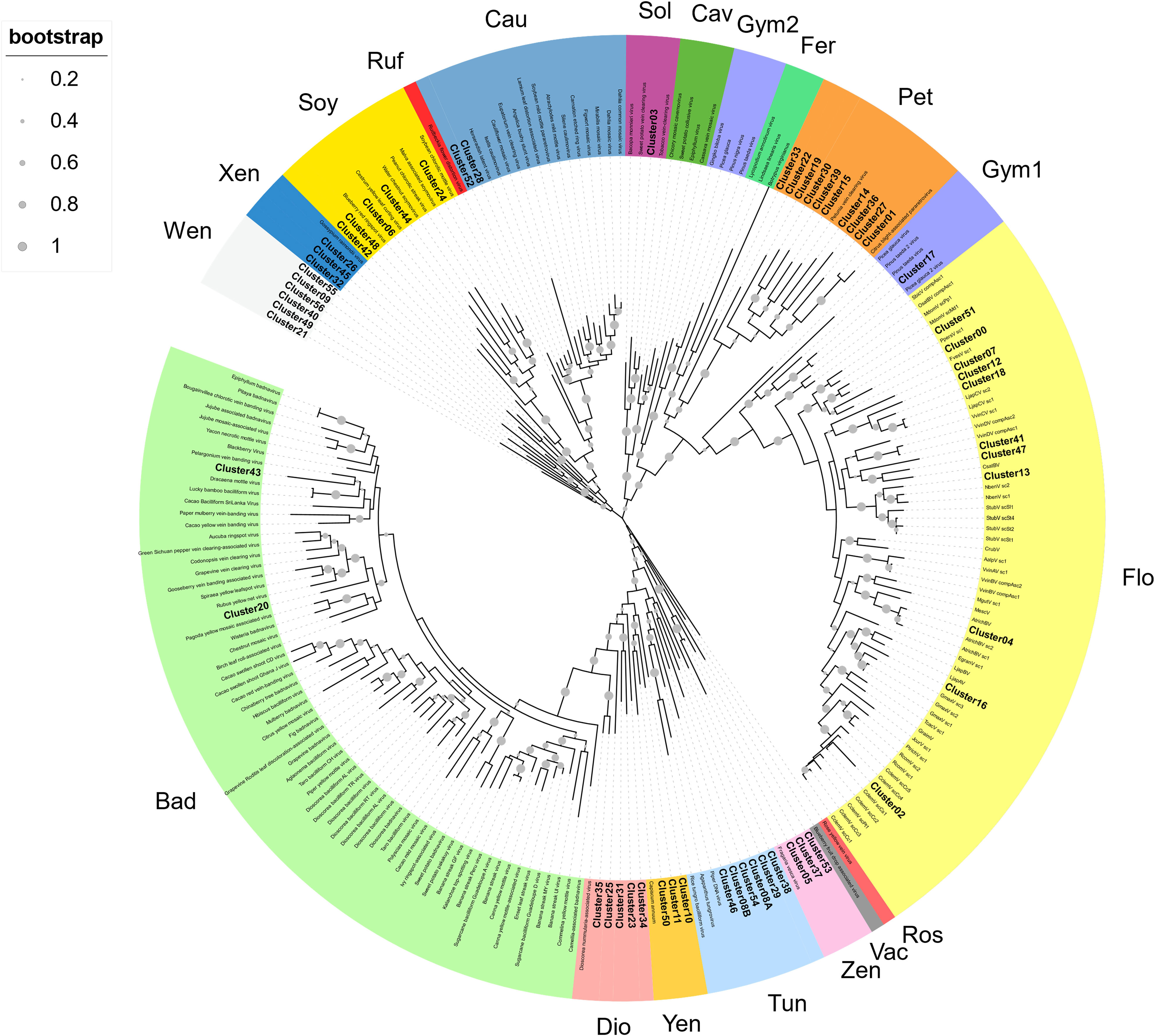
Figure 1 Phylogenetic relationships within the episomal and endogenous Caulimoviridae. Phylogram obtained from a maximum likelihood analysis with protein sequence data from RT conserved domains using 500 bootstrap replications. The size of the point indicated the bootstrap support of the tree branch. Known episomal and endogenous pararetrovirus are shown in grey and small letters. New endogenous Clusters60 are shown in bold letters. The color of the branch indicates the genus of Caulimoviridae; Bad, Badnavirus; Dio, Dioscovirus; Yen, yendovirus; Tun, tungrovirus; Zen, zendovirus; Vac, vaccinivirus; Ros, rosadnavirus; Flo, florendovirus; Gym1 and Gym2, gymnendovirus1 and 2; Pet, petuvirus; Fer, fernendovirus; Cav, cavemovirus; Sol, solendovirus; Cau, caulimovirus; Ruf, ruflodivirus; Soy, soymovirus; Xen, xendovirus; and Wen, wendovirus.
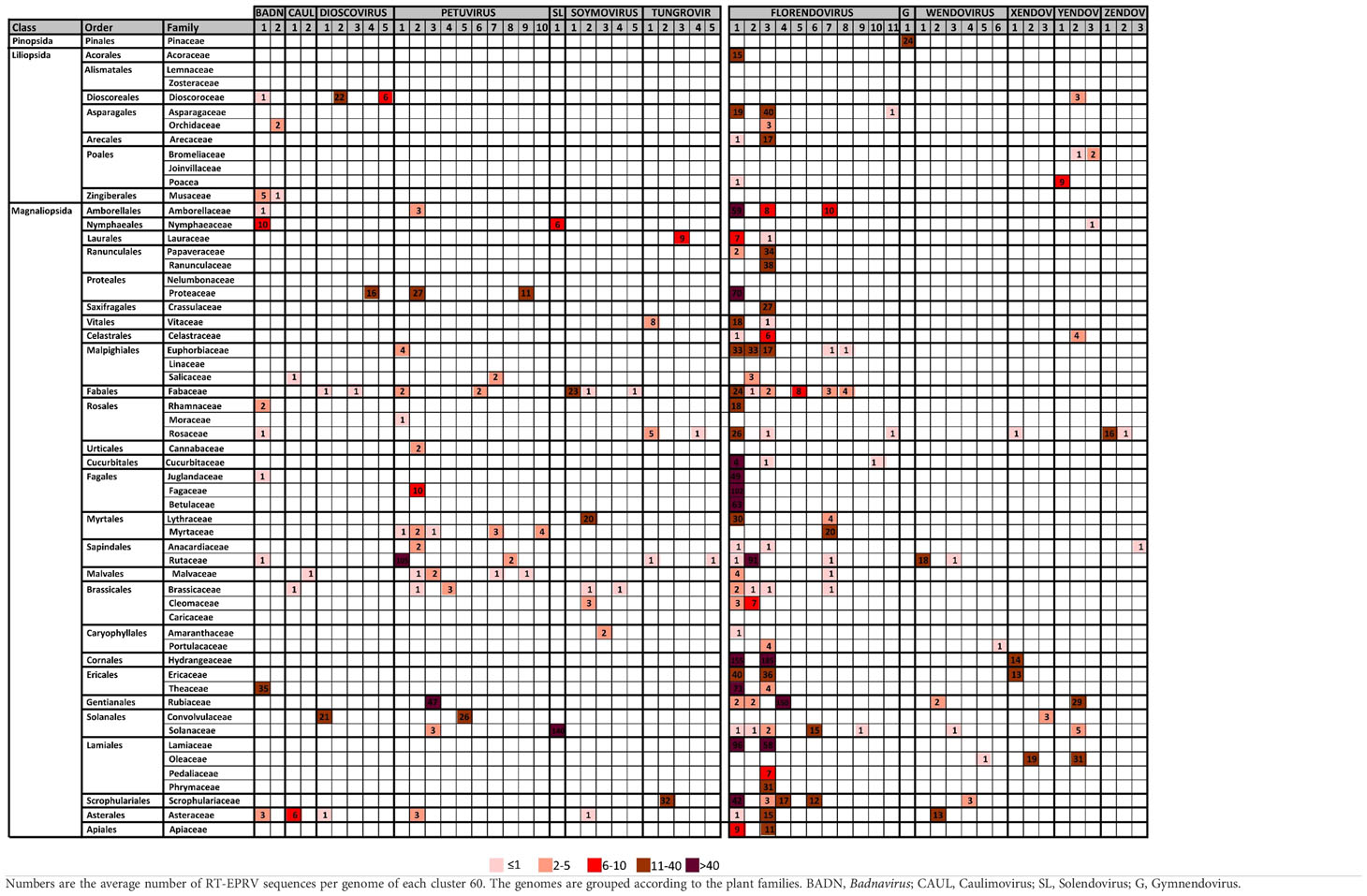
Table 2 Distribution of Cluster60 in plant families.
We observed important differences between genera for both the number of RT- EPRV sequences and the diversity of species in which they were found (Table 1). Florendovirus are clearly the most abundant followed by Petuvirus, Solendovirus and Zendovirus. However, whereas Florendovirus is present in genomes of 40 families of species, Petuvirus is present in 14 and Solendovirus and Zendovirus in only two. Interestingly, although we only detected 80 RT-EPRV sequences corresponding Badnavirus, they present a wide distribution (3 Classes, 10 Orders and 11 Families). On the opposite, Gymnendovirus are only present in Pinopsida.
If we look at the different classes of plants, we observed important differences. Pinopsida only contains Gymnendovirus. Magnolids contains Badnavirus, Petuvirus, Solendovirus, Tungrovirus, Florendovirus and Yendovirus. Liliopsida contains Badnavirus, Dioscovirus, Florendovirus and Yendovirus. Finally, Magnaliopsida contains all the genera except Gymnendovirus.
If we look at the distribution of the clusters in the different plant species, we observed a wide diversity (Table 2). Some of them are exclusively present in one class. For example, Gymnendovirus-1 is only present in Pinopsida, Tungrovirus-3 is only present in Magnolids, Badnavirus-2, Dioscovirus-2 and -5 and Yendovirus-1 are only present in Liliopsida, and many clusters are only present in Magnaliopsida. On the opposite, Badnavirus-1, Florendovirus-1 and Florendovirus-3 are present in Magnolids, Liliopsida and Magnoliopsida. Looking at more detail, 31 of the 57 clusters are present in genomes of only one family of plants, whereas two are present in genomes of more than 20 plant families (both florendovirus). These differences of distribution are reflected in the Maximum Age Value (Table 1), which depends on the maximum phylogenetic distance between the species present in the cluster.
Very recent EPRV amplification in plant genomes
The above results suggest that, at least in some species, there has been a recent amplification in the number of EPRV sequences inserted in their genomes. To try to delve further into this aspect, we decided to select those cases in which 100% identical RT-EPRV sequences were present in 10 or more copies in the same genome. Using this highly restrictive criterion, we detected 31 clusters grouping a total of 1534 sequences (Table 3). These clusters (clusters100) involve 19 genomes. Only one corresponds to a Liliopsida (Hordeum vulgare) and the remaining 18 are genomic sequences of Magnaliophyta. Nine EPRV OTUs are represented in the Clusters100 including Caulimovirus, Dioscovirus, Florendovirus, Petuvirus, Solendovirus, Tungrovirus, Yendovirus, Zendovirus and the newly proposed Wendovirus.
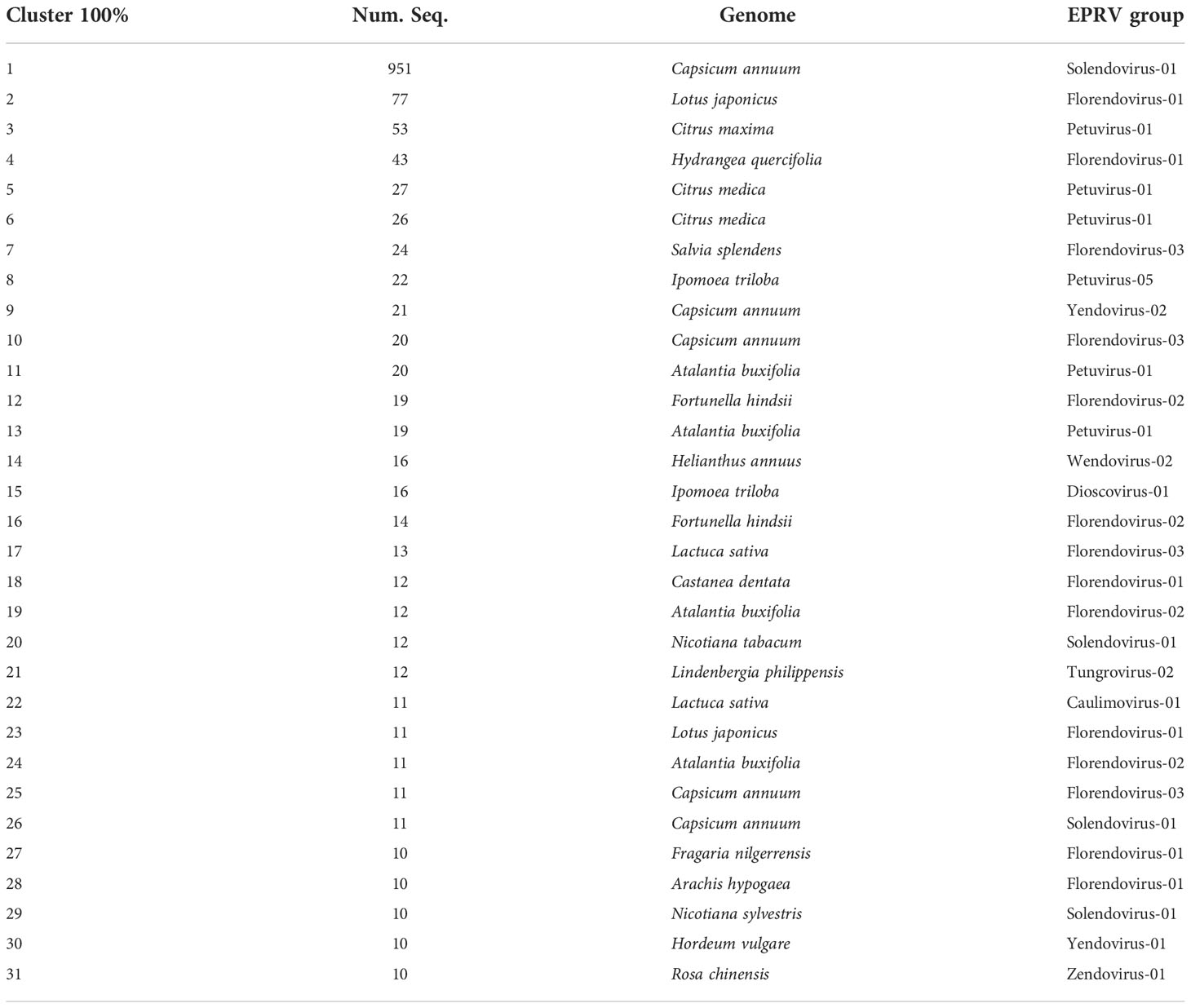
Table 3 Cluster 100 with 10 or more copies.
Cluster100-10 is particularly noteworthy as it includes 951 sequences present in the genome of pepper (Capsicum annuum). Another four groups also correspond to the same genome, with a total of 1014 sequences (962 are Solendovirus, 31 are Florendovirus and 21 Yendovirus). In total, we found 1183 RT-EPRV sequences in this genome and more than 81% are present in the Cluster100 selection. This is a very clear indication of a relatively recent proliferation of EPRVs in the pepper genome.
Next, we perform a phylogenetic analysis of representatives of each Cluster-100 and from the described OTUs from Caulimoviridae (Figure 2). The sequences of some of the clusters100 are very similar and, probably, they correspond to the same virus. This is the case of clusters100-1 and -26 (Solendovirus of Capsicum annuum), clusters100-11 and -13 (Petuvirus of Atalantia buxifolia) and Clusters100-5 and -6 (Petuvirus of Citrus medica). The sequences of clusters100-12 and -16 (Florendovirus of Fortunella hindsii) and of clusters100-19 and -24 (Florendovirus of Atalantia buxifolia) are also near identical. The sequences of the Clusters100-20 and 29, that correspond to two different but closely related species (Nicotiana tabacum and Nicotiana sylvestris), are also almost identical, which suggests that they could come from the same virus capable of infecting both species. Figure 2 also shows that some of the endogenous sequences grouped in Clusters100 are very similar to the sequences of episomal virus. For example, the RT sequence of the citrus blight associated virus is highly similar to the sequences of cluster100-3, -5 and -6, all of them belonging to genomes of the genus Citrus, and the sequence of the tobacco vein clearing virus is similar to clusters100-20 and -29, belonging to genomes of the genus Nicotiana.

Figure 2 Phylogenetic relationships of representative sequences of the Cluster100. Representative sequences of the RT-EPRV Cluster100 (in red) were aligned with RT sequences of pararetroviral elements (in black), and a phylogenetic tree was constructed using the NJ method and 1000 bootstrap replications.
Wendovirus, a new group of Caulimoviridae
Six of the Cluster60 and one of the Cluster100 correspond to a new group of endogenous Caulimoviridae with distinctive characteristics that, following the nomenclature proposed by Diop et al. (2018) (Zendovirus, Xendovirus and Yendovirus), we have called them Wendovirus (Supplementary Data 4 and Table 3).
We were able to reconstruct the structure of the Wendovirus for seven genomes corresponding to Cluster60 (Figure 3; Supplementary Data 6). The structure was very similar in all of them, with four partially overlapping ORFs. Comparisons with protein motif databases allowed us to find different conserved domains (Supplementary Data 6). The ORF1 encodes for a zinc finger motif, which is typical of the Caulimoviridae coat proteins. The ORF2 encodes for a movement protein and an aspartic proteinase. The ORF3 encodes a second aspartic proteinase, the RT and the RNAseH. Finally, the ORF4 encodes a protein without significant homologies to other reference proteins and without known protein domains but that is well-conserved in all the wendovirus elements. The most noticeable aspect of these structures is the presence of two aspartic proteinase domains instead of one, as usual. They are located close to each other, but in two different ORFs (2 and 3). In the case of the HelAnn-006 element (Wendovirus2 cluster), although the domains and their order are conserved, the ORF2 is shorter and the ORF3 is divided in two. When compared to databases, the highest similarities of these two aspartic proteinase domains are with members of Caulimoviridae.

Figure 3 Schematic representation of wendovirus endogenous pararetrovirus. A scaled linear view of the genome organization of Wendovirus. The name of the sequences is the same as in Supplementary Data 4. Grey arrows mark open reading frames and colored regions within ORFs are conserved protein domains: blue, zinc finger typically present in the coat proteins; green, Movement Protein; yellow, Aspartic Proteinase; red, Reverse Transcriptase; pink, RNaseH.
Discussion
Endogenous viral elements (EVEs) are viral sequences integrated in host genomes that are inherited as host DNA sequences (Holmes, 2011). Some of the EVEs, are derived from viruses in which integration into the genome is part of their replication cycle, for example, mammalian retroviruses. However, many viruses in which integration into the genomic DNA is not a part of their normal replication cycle can also be found as EVEs, as is the case of the endogenous Caulimoviridae (Endogenous Pararetrovirus, EPRVs). The presence of EPRVs has been described in the genomes of different plant species (Hohn et al., 2008). In this work we have focused on determining the presence of EPRV sequences relatively recently integrated, based on the selection of elements with complete and conserved RT domains.
Based on the RT domain sequence similarity we detected 11.527 sequences distributed in 57 clusters corresponding to 13 OTUs. Twelve of these groups had already been described (Diop et al., 2018) and one is shown here for first time, we called Wendovirus. Contrary to what has been observed in other plant viruses as Geminivirus or Nanovirus (Nino Barreat and Katzourakis, 2021), EVEs from Caulimoviridae are exclusively present in plants. Recently integrated RT-EPRVs are present in genomes of Lycopodiopsida, Pinopsida, Liliopsida and Magnoliopsida, but not necessary in all the genomes of these groups. For example, they are not present in the genomes of Arabidopsis thaliana, Zea mays, Triticum aestivum, Phaseolus vulgaris, Theobroma cacao or Spinacia oleracea. They are also absent in the Selaginella moellendorffii (Marchantiophyta) and in Rhodophyta, Chlorophyta or Bryophyta.
We have found that, in some cases, the integration events can be considered very recent. Once in the genome, the EPRV sequences begin to accumulate point random mutations, so, if the sequences are identical that means that they probably integrated recently in the genome. We have found multiple sequences encoding identical RT domains in different species being the most extreme case Capsicum annuum in whose genome we found up to 951 sequences encoding identical RT domains. Recent genome integrations of Caulimoviridae sequences have been described in some species, such as banana (Gayral et al., 2010). It is interesting to note that, in some cases, these identical RT sequences correspond to groups that have only been detected as endogenous forms (Florendovirus, Yendovirus, Zendovirus, Wendovirus) suggesting that probably at least some of them may have their corresponding episomal virus species that have not been yet identified.
The distribution of the different clusters of EPRVs between species shows a great diversity. Some clusters are present exclusively in certain plants as, for example, Gymnendovirus in Pinopsida, Zendovirus1 in the tribus Potentilleae and Roseae, Soymovirus1 in the genus Arachis or Wendovirus1, only present in Rutaceae. In other cases, such as Florendovirus1 and 3, the distribution is very wide, including Lilipsida and Magnoliopsida. In general, the distribution of the different groups of EPRVs is consistent with the phylogeny, but not always. For example, Petuvirus2 are present in Amborella trichopoda and in eight Magnoliopsida orders, Florendovirus7 are present in Amborella trichopoda and in seven Magnoliopsida orders and Solendovirus1 are present in Nymphaea colorata and in Solanaceae. A possible explanation for these species distributions is the horizontal transmission of the virus between species. There are data suggesting multiple viral jumps between different animal species in Hepadnavirus (Dill et al., 2016), and previous data also suggests such horizontal transfers can occur for EPRVs in plants (Diop et al., 2018; Gong and Han, 2018).
We have detected differences in the number of EPRVs in the different genomes. Sometimes the differences are also observed comparing the genomes of species of the same genus or varieties of the same species. The number of EPRVs observed results from the combination of the virus integration and the mechanisms of amplification or reduction of the integrated sequences. First, Caulimoviridae integration requires the presence of viruses that are infectious for the species and that the defense mechanisms of the plant are not able to eliminate, or not completely. Second, the main integration mechanism is thought to involve illegitimate recombination, which requires the existence of DNA double-strand breaks and subsequent repair mechanisms (Richert-Pöggeler et al., 2021). Furthermore, to be transmitted, integration must occur in reproductive cells. Third, once integrated, EPRVs, copies are inactivated by sequence degeneration or fragmentation, or by the insertion of transposable elements, and subjected to epigenetic silencing (reviewed by Richert-Pöggeler et al., 2021). All these processes lead to the degeneration of the coding sequences. Finally, it has also been proposed that once integrated, the sequences can be amplified, and different mechanisms have been suggested such as transposition like retroelements, rolling circle amplification, unequal meiotic crossing-over of tandem arrays, or ectopic recombination between EPRV clusters on non-homologous chromosomes (reviewed by Richert-Pöggeler et al., 2021). Variations in any of these processes together with the time elapsed since the last event of integration could explain the observed differences in the number of EPRVs in the analyzed genomes. Nor can we rule out that the different quality of the genome assemblies may also affect.
We have identified a new putative genus of the Caulimoviridae, tentatively named ‘Wendovirus’. Wendovirus genomes are about 7,7 Kb long and are present in the genomes of different Magnaliopsida species, especially in Rutaceae and in sunflower. Our phylogenetic analysis shows that wendovirus are related to Xendovirus and Soymovirus. They contain four ORFs that encode the typical protein domains in Caulimoviridae: Zinc-Finger, Movement Protein, Aspartic Proteinase, Reverse Transcriptase and RNAseH. A remarkable feature of wendovirus is the presence of two protease coding domains located in two different ORFs (Figure 3). Although both encode aspartyl proteases, the domains are different (PF13975 in ORF2 and PF00077 in ORF3), so the hypothesis that their origin was a genomic duplication can be discarded. When compared to protein bases, all these described domains, including the two aspartic proteinase domains, show the greatest similarities against other members of Caulimoviridae. Therefore, it seems to be ruled out that the second proteinase domain could come from some other families of viruses. Recombination between EPRV fragments has been observed (Chabannes and Iskra-Caruana, 2013) and many viruses have modularly acquired domains and ORFs (Smyshlyaev et al., 2013; Koonin et al., 2015). Encapsidation of genomes (or genome fragments) of different species of Caulimoviriridae in the same capsid can lead to recombination and formation of chimeric genomes. Virus-like particles (VLPs) containing host RNAs were found to be produced during agroinfiltration of cucumber necrosis virus, some of them corresponding to retrotransposon or retrotransposon-like RNA sequences (Ghoshal et al., 2015). On the other hand, template switching between two RNA molecules during reverse transcription has been shown for retroviruses, LTR retrotransposons and is proposed for Caulimoviridae (Froissart et al., 2005; Tromas et al., 2014; Sanchez et al., 2017; Richert-Pöggeler et al., 2021). Such an acquisition of ORFs likely contributed to the evolution of the Wendovirus, although the possible functions of this second proteinase domain remain unknown.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
All authors contributed equally to the design and processing data. All authors have read and approved the final manuscript.
Funding
This work was funded by grants from the Spanish Ministerio de Economía, Industria y Competitividad (AGL2016-78992-R/FEDER), Ministerio de Ciencia e Innovación (PID2019-106374RB-I00/AEI/10.13039/501100011033), by “ERDF A way of making Europe” and by the CERCA Programme of the Generalitat de Catalunya. We also acknowledge financial support from the Spanish Ministerio de Economía y Competitividad through the “Severo Ochoa Programme for Centres of Excellence in R&D” 2016-2019 (SEV-2015-0533) and CEX2019-000902-S funded by MCIN/AEI/10.13039/501100011033.
Acknowledgments
We thank all groups members whose comments contributed to this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1011565/full#supplementary-material
Supplementary Data Sheet 1 | RT domain sequences of Caulimoviridae, retrovirus and LTR-retrotransposons used in this study.
Supplementary Data Sheet 2 | Caulimoviridae RT sequences used in the tBLASTn analyses.
Supplementary Data Sheet 3 | Genome assemblies used in the tBLASTn analyses.
Supplementary Data Sheet 4 | RT-EPRV sequences found.
Supplementary Data Sheet 5 | Representative RT-EPRV sequences for the clusters60 used in the phylogenetic analysis.
Supplementary Data Sheet 6 | Wendovirus sequences and polypeptides encoded.
Abbreviations
EPRV, Endogenous pararetrovirus; RT, Reverse Transcriptase; EVEs, Endogenous viral elements; OUT, operational taxonomic unit.
References
Aiewsakun, P., Katzourakis, A. (2015). Endogenous viruses: Connecting recent and ancient viral evolution. Virology 479-480, 26–37. doi: 10.1016/j.virol.2015.02.011
Bao, W., Kojima, K. K., Kohany, O. (2015). Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11. doi: 10.1186/s13100-015-0041-9
Bejarano, E. R., Khashoggi, A., Witty, M., Lichtenstein, C. (1996). Integration of multiple repeats of geminiviral DNA into the nuclear genome of tobacco during evolution. Proc. Natl. Acad. Sci. U.S.A. 93, 759–764. doi: 10.1073/pnas.93.2.759
Bertsch, C., Beuve, M., Dolja, V. V., Wirth, M., Pelsy, F., Herrbach, E., et al. (2009). Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol. Direct 4, 21. doi: 10.1186/1745-6150-4-21
Catlin, N. S., Josephs, E. B. (2022). The important contribution of transposable elements to phenotypic variation and evolution. Curr. Opin. Plant Biol. 65, 102140. doi: 10.1016/j.pbi.2021.102140
Chabannes, M., Iskra-Caruana, M. L. (2013). Endogenous pararetroviruses–a reservoir of virus infection in plants. Curr. Opin. Virol. 3, 615–620. doi: 10.1016/j.coviro.2013.08.012
Chen, S., Kishima, Y. (2016). Endogenous pararetroviruses in rice genomes as a fossil record useful for the emerging field of palaeovirology. Mol. Plant Path. 17, 1317–1320. doi: 10.1111/mpp.12490
Chiba, S., Kondo, H., Tani, A., Saisho, D., Sakamoto, W., Kanematsu, S., et al. (2011). Widespread endogenization of genome sequences of non-retroviral RNA viruses into plant genomes. PloS Pathog. 7, e1002146. doi: 10.1371/journal.ppat.1002146
Choi, I. S., Wojciechowski, M. F., Ruhlman, T. A., Jansen, R. K. (2021). In and out: Evolution of viral sequences in the mitochondrial genomes of legumes (Fabaceae). Mol. Phylog. Evol. 17, 107236. doi: 10.1016/j.ympev.2021.107236
Dill, J. A., Camus, A. C., Leary, J. H., Di Giallonardo, F., Holmes, E. C., Ng., T. F. F. (2016). Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of hepadnaviruses. J. Virol. 90, 7920–7933. doi: 10.1128/JVI.00832-16
Diop, S. I., Geering, A. D. W., Alfama-Depauw, F., Loaec, M., Teycheney, P. Y., Maumus, F. (2018). Tracheophyte genomes keep track of the deep evolution of the caulimoviridae. Sci. Rep. 8, 572. doi: 10.1038/s41598-017-16399-x
Etienne, L. (2017). Paleovirology: looking back in time to better understand and control modern viral infections. Virologie (Montrouge) 21, 245–246. doi: 10.1684/vir.2017.0715
Feschotte, C., Gilbert, C. (2012). Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13, 283–296. doi: 10.1038/nrg3199
Froissart, R., Roze, D., Uzest, M., Galibert, L., Blanc, S., Michalakis, Y. (2005). Recombination every day: Abundant recombination in a virus during a single multi-cellular host infection. PloS Biol. 3, e89. doi: 10.1371/journal.pbio.0030089
Gayral, P., Blondin, L., Guidolin, O., Carreel, F., Hippolyte, I., Perrier, X., et al. (2010). Evolution of endogenous sequences of banana streak virus: What can we learn from banana (Musa sp.) evolution? J. Virol. 84, 7346–7359. doi: 10.1128/JVI.00401-10
Gayral, P., Noa-Carrazana, J. C., Lescot, M., Lheureux, F., Lockhart, B. E. L., Matsumoto, T., et al. (2008). A single banana streak virus integration event in the banana genome as the origin of infectious endogenous pararetrovirus. J. Virol. 82, 6697–6710. doi: 10.1128/JVI.00212-08
Geering, A. D., Maumus, F., Copetti, D., Choisne, N., Zwickl, D. J., Zytnicki, M., et al. (2014). Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat. Commun. 10, 5269. doi: 10.1038/ncomms6269
Ghoshal, K., Theilmann, J., Reade, R., Maghodia, A., Rochon, D. (2015). Encapsidation of host RNAs by cucumber necrosis virus coat protein during both agroinfiltration and infection. J. Virol. 89, 10748–10761. doi: 10.1128/jvi.01466-15
Gong, Z., Han, G. Z. (2018). Euphyllophyte paleoviruses illuminate hidden diversity and macroevolutionary mode of Caulimoviridae. J. Virol. 92, e02043-17. doi: 10.1128/JVI.02043-17
Hansen, C., Heslop-Harrison, J. S. (2004). Sequences and phylogenies of plant pararetroviruses, viruses, and transposable elements. Adv. Bot. Res. 41, 165–193. doi: 10.1016/S0065-2296(04)41004-0
Hohn, T., Richert-Pöggeler, K. R., Staginnus, C., Harper, G., Schwarzacher, T., Teo, C. H., et al. (2008). ““Evolution of integrated plant viruses.”,” in Plant virus evolution. Ed. Roossinck, ,. M. J. (Berlin: Springer), 53–81. doi: 10.1007/978-3-540-75763-4_4
Holmes, E. C. (2011). The evolution of endogenous viral elements. Cell Host Microbe 10, 368–377. doi: 10.1016/j.chom.2011.09.002
Johnson, W. E. (2019). Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 17, 355–370. doi: 10.1038/s41579-019-0189-2
Katzourakis, A., Gifford, R. J. (2010). Endogenous viral elements in animal genomes. PloS Genet. 6, e1001191. doi: 10.1371/journal.pgen.1001191
Kojima, S., Yoshikawa, K., Ito, J., Nakagawa, S., Parrish, N. F., Horie, M., et al. (2021). Virus-like insertions with sequence signatures similar to those of endogenous nonretroviral RNA viruses in the human genome. Proc. Natl. Acad. Sci. U.S.A. 118, e2010758118. doi: 10.1073/pnas.2010758118
Koonin, E. V., Dolja, V. V., Krupovic, M. (2015). Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology 479-480, 2–25. doi: 10.1016/j.virol.2015.02.039
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kumar, S., Stecher, G., Suleski, M., Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Kuriyama, K., Tabara, M., Moriyama, H., Kanazawa, A., Koiwa, H., Takahashi, H., et al. (2020). Disturbance of floral colour pattern by activation of an endogenous pararetrovirus, petunia vein clearing virus, in aged petunia plants. Plant J. 103, 497–511. doi: 10.1111/tpj.14728
Lockhart, B. E., Menke, J., Dahal, G., Olszewski, N. E. (2000). Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 81, 1579–1585. doi: 10.1099/0022-1317-81-6-1579
Nino Barreat, J. G., Katzourakis, A. (2021). Paleovirology of the DNA viruses of eukaryotes. Trends Microbiol. 30, 281–292. doi: 10.1016/j.tim.2021.07.004
Ricciuti, E., Laboureau, N., Noumbissié, G., Chabannes, M., Sukhikh, N., Pooggin, M. M., et al. (2021). Extrachromosomal viral DNA produced by transcriptionally active endogenous viral elements in non-infected banana hybrids impedes quantitative PCR diagnostics of banana streak virus infections in banana hybrids. J. Gen. Virol. 102, 11. doi: 10.1099/jgv.0.001670
Richert-Pöggeler, K. R., Noreen, F., Schwarzacher, T., Harper, G., Hohn, T. (2003). Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. EMBO J. 22, 4836–4845. doi: 10.1093/emboj/cdg443
Richert-Pöggeler, K. R., Vijverberg, K., Alisawi, O., Chofong, G. N., Heslop-Harrison, J. S., Schwarzacher, T. (2021). Participation of multifunctional RNA in replication, recombination and regulation of endogenous plant pararetroviruses (EPRVs). Front. Plant Sci. 21(12). doi: 10.3389/fpls.2021.689307
Sanchez, D. H., Gaubert, H., Drost, H. G., Zabet, N. R., Paszkowski, J. (2017). High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat. Commun. 8, 1374. doi: 10.1038/s41467-017-01374-x
Smyshlyaev, G., Voigt, F., Blinov, A., Barabas, O., Novikova, O. (2013). Acquisition of an archaea-like ribonuclease h domain by plant L1 retrotransposons supports modular evolution. Proc. Nat. Acad. Sci. U.S.A. 110, 20140–20145. doi: 10.1073/pnas.1310958110
Tromas, N., Zwart, M. P., Poulain, M., Elena, ,. S. F. (2014). Estimation of the in vivo recombination rate for a plant RNA virus. J. Gen. Virol. 95, 724–732. doi: 10.1099/vir.0.060822-0
Zhang, L., Richards, A., Barrasa, M. I., Hughes, S. H., Young, R. A., Jaenisch, R. (2021). Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. U.S.A. 118, e2105968118. doi: 10.1073/pnas.2105968118
Keywords: pararetrovirus, Reverse Transcriptase (RT), Caulimoviridae, endogenous, virus
Citation: de Tomás C and Vicient CM (2022) Genome-wide identification of Reverse Transcriptase domains of recently inserted endogenous plant pararetrovirus (Caulimoviridae). Front. Plant Sci. 13:1011565. doi: 10.3389/fpls.2022.1011565
Received: 04 August 2022; Accepted: 15 November 2022;
Published: 14 December 2022.
Edited by:
Gennady I. Karlov, All-Russia Research Institute of Agricultural Biotechnology, RussiaReviewed by:
Katja R. Richert-Pöggeler, Julius Kühn-Institut, GermanyOsamah Alisawi, University of Kufa, Iraq
Copyright © 2022 de Tomás and Vicient. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos M. Vicient, Y2FybG9zLnZpY2llbnRAY3JhZ2Vub21pY2EuZXM=