 María Cielo Pasten1†
María Cielo Pasten1† José Carballo1†
José Carballo1† Jimena Gallardo1,2
Jimena Gallardo1,2 Diego Zappacosta1,2
Diego Zappacosta1,2 Juan Pablo Selva1,3
Juan Pablo Selva1,3 Juan Manuel Rodrigo1,2
Juan Manuel Rodrigo1,2 Viviana Echenique1,2
Viviana Echenique1,2 Ingrid Garbus1*
Ingrid Garbus1*- 1Centro de Recursos Naturales Renovables de la Zona Semiárida (CERZOS), Universidad Nacional del Sur-Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Bahía Blanca, Argentina
- 2Departamento de Agronomía, Universidad Nacional del Sur (UNS), Bahía Blanca, Argentina
- 3Departamento de Biología, Bioquímica y Farmacia, Universidad Nacional del Sur (UNS), Bahía Blanca, Argentina
Weeping lovegrass (Eragrostis curvula [Shrad.] Nees) is a perennial grass typically established in semi-arid regions, with good adaptability to dry conditions and sandy soils. This polymorphic complex includes both sexual and apomictic cytotypes, with different ploidy levels (2x-8x). Diploids are known to be sexual, while most polyploids are facultative apomicts, and full apomicts have also been reported. Plant breeding studies throughout the years have focused on achieving the introgression of apomixis into species of agricultural relevance, but, given the complexity of the trait, a deeper understanding of the molecular basis of regulatory mechanisms of apomixis is still required. Apomixis is thought to be associated with silencing or disruption of the sexual pathway, and studies have shown it is influenced by epigenetic mechanisms. In a previous study, we explored the role of miRNA-mRNA interactions using two contrasting E. curvula phenotypes. Here, the sexual OTA-S, the facultative Don Walter and the obligate apomictic Tanganyika cDNA and sRNA libraries were inquired, searching for miRNA discovery and miRNA expression regulation of genes related to the reproductive mode. This allowed for the characterization of seven miRNAs and the validation of their miRNA-target interactions. Interestingly, a kinesin gene was found to be repressed in the apomictic cultivar Tanganyika, targeted by a novel miRNA that was found to be overexpressed in this genotype, suggestive of an involvement in the reproductive mode expression. Our work provided additional evidence of the contribution of the epigenetic regulation of the apomictic pathway.
Introduction
Apomixis can be described as asexual reproduction through seeds, and it comprises three central stages: apomeiosis (absence of meiosis), parthenogenesis (absence of fertilization) and autonomous endosperm development or pseudogamy (Nogler, 1984; Schmidt, 2020). It has been observed that apomixis is inherited as a dominant trait, and that the three developmental stages are controlled by independent loci in several species (Grossniklaus, 2001; Vijverberg and van Dijk, 2007).
There is a wide variety of apomictic mechanisms, which can be explained by the fact that apomixis emerged independently multiple times during evolution. It is theorized that apomixis arises from spatial and temporal changes in the expression of genes involved in sexual reproduction. The regulation guided by the availability of specific expression of the genes PAR1 and PsASGR-BabyBoom-Like has been observed for parthenogenesis in Pennisetum and Taraxacum (Underwood et al., 2022, Khanday et al., 2019). However, these genes per se are insufficient to transfer apomixis to economically important crops, since no genes linked to apomeiosis were found in apomictic species.
It has been found that the reproductive state can be influenced by environmental cues (Rodrigo et al., 2017; Gao, 2018; Schmidt, 2020). In numerous species, differences in photoperiod, salt stress and heat stress result in similar responses, adding to the now accepted notion that reproduction adjustments can take place, and that they appear to have a significant role in the adaptation of facultative apomicts (Gounaris et al., 1991; Hussey et al., 1991; Carman et al., 2015; Klatt et al., 2016).
There is evidence that shows that DNA methylation, chromatin modifications and noncoding RNAs have a central role in the regulation of gene expression at transcriptional and post-transcriptional levels (Pikaard and Scheid, 2014; Wei et al., 2017; Higo et al., 2020; Brukhin and Albertini, 2021; Carballo et al., 2021). The study of epigenetics in the last years has established that these mechanisms are key to the understanding of numerous processes in plants. Among the noncoding RNAs, the most abundant and important group are the small RNAs (sRNAs), that target coding or non-coding RNAs for silencing, via guided degradation or translational repression (Borges and Martienssen, 2015; Nejat and Mantri, 2018). MicroRNAs (miRNAs) are a large class of sRNAs derived from DICER-LIKE (DCL) processing of single-stranded precursors, known to participate mostly in post-transcriptional gene silencing (PTGS) (Borges and Martienssen, 2015). miRNAs regulate gene expression by specifically targeting messenger RNAs (mRNA) in association with the RNA-induced silencing complex (RISC), consisting mainly of ARGONAUTE (AGO) proteins, mainly AGO1, AGO4 and AGO10 (Neumeier and Meister, 2021) and acting either via direct degradation or via inhibition of translation, pairing with the 3’ UTR region of the target (Melo and Melo, 2013).
miRNAs have central regulatory roles and affect processes such as reproduction, fertility, and leaf and flower morphogenesis, among others (Aukerman and Sakai, 2003; Palatnik et al., 2003; Schwab et al., 2005; Li et al., 2015; Ripoll et al., 2015). In A. thaliana, Z. bungeanum and some species of the genus Boechera, specific miRNAs have been identified (miR156/157, miR167 y miR172c) related to functions such as flower organogenesis, reproductive mode, and male and female fertility (Xing et al., 2013; Ru et al., 2006; Amiteye et al., 2011; Fei et al., 2021). Differential expression analyses of miRNAs in regards to ploidy and reproductive mode have also taken place (Amiteye et al., 2013; Ortiz et al., 2019).
Eragrostis curvula [Shrad.] Nees (weeping lovegrass), is a perennial grass belonging to the Poaceae family, native of Southern Africa, and it is commonly found in semi-arid regions around the world due to its ability to adapt to drought conditions and low fertility soils (Covas, 1991). This species constitutes a morphologically and evolutively diverse group, and it includes cytotypes with different ploidy levels (2x to 8x), which can reproduce sexually or by diplosporous apomixis (obligate or facultative) (Leigh, 1960; Voigt and Burson, 1983).
Due to its specific apomictic development, E. curvula has become one of several models for the study of apomictic mechanisms and was widely studied over the last years, obtaining information about its genome (Carballo et al., 2019), cytoembriology (Zappacosta et al., 2014), the position of the apomixis locus (Zappacosta et al., 2019), expression of candidate genes (Cervigni et al. 2008a, and Cervigni et al. 2008b; Garbus et al., 2017; Selva et al., 2017; Selva et al., 2020). More recently, the methylation status of facultative, fully sexual and fully apomictic genotypes was assessed, finding that facultative genotypes have a higher number of methylated positions (Carballo et al., 2021). Even when the methylation status could be interpreted as a consequence of the different reproduction systems rather than the cause (Mau et al., 2022), the tetraploid nature and the independent genetic origin of the three genotypes analyzed in the mentioned study is supporting the hypothesis that links the methylation status with the reproductive mode. This behavior shows the importance of epigenetic mechanisms in more dynamic reproductive conditions.
The scarce diploid plants of E. curvula (2n = 2x = 20) reproduce sexually, while polyploids mostly undergo facultative pseudogamous diplosporous apomixis (Leigh, 1960; Voigt and Burson, 1983). In this process, a megasporocyte avoids meiosis and experiences two rounds of mitotic division, generating four non-reduced nuclei at maturity: two synergid cells, an egg cell and one polar nucleus (Crane, 2001). The development of the non-reduced tetranucleate embryo sac is specific of this grass, that also undergoes parthenogenesis but requires fertilization of the polar nucleus for the development of the endosperm (Grimanelli et al., 2001; Crane, 2001).
In a previous study conducted using two contrasting E. curvula phenotypes, the sexual OTA-S and the apomictic Tanganyika, we investigated the role of miRNA-mRNA interactions in apomixis using a transcriptomic library and small RNA libraries (Garbus et al., 2019). Two genes were identified, a MADS-box transcription factor gene and a transposon, that were found to be repressed in the sexual genotype, most likely due to interactions with miRNAs, suggesting that a repressive effect on gene expression exerted by miRNAs might be involved in the expression of apomixis in E. curvula (Garbus et al., 2019).
In the current study we expanded the comprehension of the participation of miRNAs in the expression of apomixis in E. curvula, through its known function in mRNA degradation, by including the facultative apomictic material Don Walter. In this way, we analyzed the differential expression of miRNAs and their targets focusing on all the possible comparisons among the sexual cultivar OTA-S, the apomictic cultivar Tanganyika and the facultative Don Walter. There were described seven miRNAs, including their mature sequences, the pri-miRNA and pre-miRNA precursors, as well as their specific interaction with their target mRNAs. A specific regulation by genotype for the transcript for a kinesin gene that was found, being repressed in the apomictic cultivar Tanganyika, whereas the related miRNA expression was the opposite, i.e., overexpressed in the mentioned genotype. Our results support the previous evidence of the role of miRNAs in transcript regulation in apomixis.
Materials and methods
Plant materials
The experiments were carried out using three tetraploid (2n = 4x = 40) E. curvula cultivars: the sexual OTA-S (USDA: PI574506), the full apomictic Tanganyika (USDA: PI234217) and the facultative apomictic Don Walter. The plants were grown in the greenhouse in 10 L pots, under natural light conditions (38°43’0″S, 62°16’0″O), at 25˚C +/- 4°C.
cDNA and sRNA libraries
To conduct the analyses reported in this work, we made use of the cDNA libraries previously constructed by our laboratory for genotypes OTA-S, Tanganyika and Don Walter (Garbus et al., 2017; Selva et al., 2020). OTA-S and Tanganyika cDNA libraries were constructed, sequenced and assembled as reported in Garbus et al. (2017). Briefly, libraries were obtained from spikelets with basal flowers at the beginning of anthesis, containing embryo sacs at all developmental stages, in biological duplicates for each genotype, naming O2P1 and O2P2 those derived from OTA-S, whereas the ones constructed from Tanganyika were called T3P1 and T3P2.
These libraries were deposited in the Sequence Reads Archive (SRA) database at NCBI as BioProject 358210 (Biosamples SAMN06167423 and SAMN06167424), whereas the Transcriptome Shotgun Assembly (TSA) project has been deposited at DDBJ/EMBL/GenBank under the accession GFVM00000000. cDNA libraries from the cultivar Don Walter were made in triplicates from spikelets with basal flowers at the beginning of anthesis, containing embryo sacs at all developmental stages, and were named DW1, DW2 and DW3. The procedures followed for library construction are described in Selva et al. (2020). The TSA Assembly project was deposited at DDBJ/EMBL/GenBank under the accession GIQX00000000.
Concerning small RNAs, libraries from OTA-S and Tanganyika were available (Garbus et al., 2019). They were constructed starting from the small RNA fraction obtained in the separation from the large RNA fraction used for the cDNA libraries (Garbus et al., 2017; Garbus et al., 2019), and thus biological replicates for each genotype lead to the sRNA libraries called O2P1, O2P2, T3P1 and T3P2. The procedure followed for library construction, sequencing and analysis is detailed in Garbus et al. (2019).
On the other hand, sRNA libraries from Don Walter were constructed aiming at this study, as detailed in the next section.
Small RNA libraries from Don Walter construction, sequencing and processing
Starting from 30 mg of fresh tissue from each sample, ground to a fine powder using liquid nitrogen, the total RNA was extracted as two fractions, small and large RNA, which include RNA sequences smaller and larger than 200 bp, respectively, using a commercial NucleoSpin® miRNA kit (Macherey-Nagel, Düren, Germany) according to the manufacturer’s instructions (Selva et al., 2020). The small RNA fraction was sequenced in biological triplicates, DW1, DW2 and DW3, in 50 bp single end reads through Illumina MiSeq platform in the Polo d’Innovazione di Genomica, Genetica e Biologia (Polo GGB), in Siena, Italy.
After sequencing, the adapters of the raw reads were removed and trimmed using Cutadapt (Martin, 2011) with the following parameters: Phred quality score cutoff 30, minimum read length 18. The reads of the seven samples were collapsed to remove redundancy, using the script collapse_reads.pl. These libraries were deposited in the Sequence Reads Archive (SRA) database at NCBI as BioProject PRJNA855479 (Biosamples SAMN29493390, SAMN29493391 and SAMN29493392).
miRNA prediction and target identification
The software miRDP2 was used to predict miRNAs, using the Don Walter genome assembly to discover novel and known miRNAs. The first step in miRDP2 was to align reads against the Rfam database in order to identify and discard known rRNA, tRNA, scRNA, snRNA and snoRNAs. miRDP2 reports the mature, pri-miRNA and pre-miRNAs.
The plot of the hairpins was performed using the RNAplot script from ViennaRNA package 2.0 (Lorenz et al., 2011). The predicted mature miRNA file was indexed and each sample was mapped with the software Bowtie (Langmead, 2010) with 0 mismatches to quantify mature miRNAs. A matrix with the reads mapping on the predicted mature miRNA was constructed, and filtered when a mature miRNA was absent in at least 5 samples and the sum of all the mapping reads were less than 10. The R package DESeq2 (Love et al., 2014) was used to perform the normalization and the expression analysis. A principal component analysis was conducted to reduce complexity and compare all the samples. Comparisons were made between the full apomictic cultivar Tanganyika and the sexual cultivar OTA-S, and the facultative cultivar Don Walter, hereafter referred as APOvsSEX and APOvsFAC, respectively. The facultative cultivar Don Walter was further compared to the sexual OTA-S, hereafter referred as FACvsSEX. A mature miRNA was considered to be differentially expressed when the log2FoldChange was >1 and <1 for upregulated and downregulated mature miRNAs, respectively, and when padj<0.05.
Target identification was performed by the software psRNAtarget (Dai et al., 2018) using the genome annotation and the predicted mature miRNAs as input. Finally, in order to correlate the miRNA target with the transcript expression level, a differential expression analysis was conducted using previously sequenced transcriptomic reads from the three genotypes (Garbus et al., 2017; Selva et al., 2020). For these data sets, the software hisat2 (Kim et al., 2019) was used for mapping, and DESeq2 (Love et al., 2014) was used to perform the expression analysis.
DNA and RNA extraction
DNA and RNA were extracted from the cultivars OTA-S, Tanganyika and Don Walter aiming to validate the in silico observations. To carry out this purpose, three plants from each cultivar were grown reproducing the conditions applied for the construction of the libraries. The DNA was extracted from fresh leave tissue in biological triplicates for each cultivar, following a protocol based on cetyltrimethylammonium bromide (CTAB). Briefly, plant material was initially frozen and powdered in liquid nitrogen using a plastic pestle in a 1.5 ml Eppendorf tube. A total of ~50 mg of powdered tissue was incubated at 65°C in preheated CTAB extraction buffer containing 100 mM Tris HCl pH 8, 1.4 M NaCl, 20 mM EDTA pH 8, 2% CTAB (w/v) and 0.5% (v/v) β-mercaptoethanol. Then, chloroform was added to reach the proportion 2:1, collecting the supernatant obtained after a centrifugation step. Finally, DNA was precipitated with one volume of isopropanol, washed with 70% (v/v) ethanol and resuspended in ultrapure water.
RNA extraction was conducted in biological triplicates from spikelets from the three cultivars, collected following the same criteria used to obtain the samples for library construction. Briefly, spikelets with basal flowers at the beginning of anthesis were collected based on the reproductive calendar shown in Selva et al. (2017), where all developmental stages are represented, from early archespores to mature female gametophytes, evaluated through observations under a stereoscopic microscope (Leica S APO) of several collected spikelets. RNA isolation was performed using a TRIzol based protocol. Approximately 35 mg of tissue from biological triplicates per sample was frozen and homogenized in liquid nitrogen using a plastic pestle in a 1.5 ml Eppendorf tube. Then, they were mixed with TRIzol™ Reagent (Invitrogen). Following the addition of chloroform, RNA was precipitated from the upper layer with isopropanol and washed with ethanol. The samples were resuspended in 20–25 μl of DEPC water and stored at −80°C. RNA was quantified in a DeNovix DS-11 spectrophotometer (Denovix Inc., USA), whereas its integrity was checked by agarose gel electrophoresis.
cDNA and stem-loops based cDNA synthesis
RNA was used for the synthesis of cDNA, as well as stem-loop cDNA. The cDNA synthesis was performed aiming for the validation of transcripts’ differential expression, using the ImProm II™ Reverse Transcription System (Promega) following the supplier’s instructions.
Stem-loop synthesis was aimed at miRNA quantification (Chen et al., 2005), and was also conducted using the ImProm-II™ Reverse Transcription System, but in this case instead of using random primers, a specific stem loop of ≈50 bp was designed for each of the miRNAs intended to be validated and quantified. Stem-loop primers were designed manually, as described in Garbus et al. (2019) (Table 1).

Table 1 Primers designed for stem-loop miRNAs amplification.
Reactions were conducted from 20 ng of RNA, in biological triplicates for OTA-S, Tanganyika and Don Walter. Briefly, each 20 μl reaction was incubated in a BioRad Thermocycler for 5 min at 25°C followed by 60 min at 42°C and finally 15 min at 70°C to inactivate the reverse transcriptase, and cooled at 4°C.
PCR and RT-PCR
Primers based on the sequence of the transcripts differentially regulated among libraries (Table 2), were designed using the IDT Interphase (https://www.idtdna.com/PrimerQuest/Home/Index).
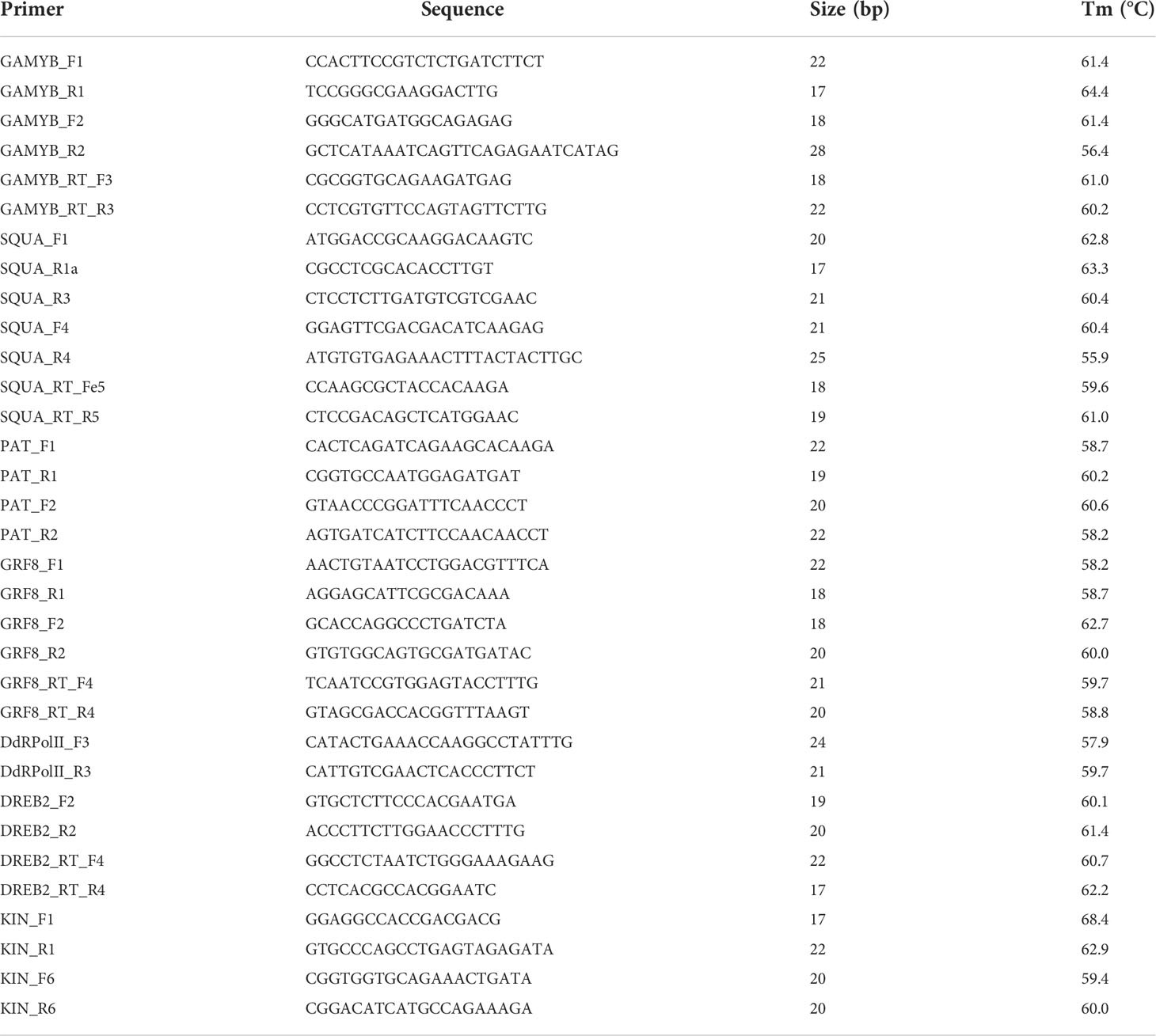
Table 2 Transcripts and/or gene based primers, designed for PCR and qPCR amplification.
Amplification of the genomic sequence of the corresponding transcripts, as well as the miRNA precursors, was conducted using DNA. The reactions were run using a MyCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA), and prepared with 0.3 μl of 40 mM dNTPs mix, 3 μl of 10× reaction buffer, 0.6 μl of each forward and reverse primer (10 pmol/μl), 0.2 μl of DNA polymerase, 1 μl of template genomic DNA (40 ng/μl), in a final reaction volume of 15 μl. The DNA polymerases used were either Taq Pegasus (PB-L) or GoTaq DNA Polymerase (Promega), both containing 5U enzyme/μl. The protocol followed for DNA amplification consisted of an initial DNA denaturation step at 95°C for 2-5 min, followed by 33 cycles at 95°C for 30 s, 30 s at the optimal annealing temperature for each primer pair, and 72°C for 30s. The final step consisted of a final extension of 3 min at 72°C. The primer annealing temperatures were optimized for each primer pair, starting from one degree below the lower melting temperature between both primers.
RT-PCR reactions were performed to verify and visualize the amplification among genotypes of the differentially regulated transcripts predicted in silico. Reactions were developed basically as described for the PCR ones, but starting from 2 μl of a 1/20 cDNA dilution.
PCR and RT-PCR amplification bands were separated by electrophoresis in a 1.5% (m/v) agarose gel and visualized using ethidium bromide in a VP High Performance UV Transilluminator.
Stem-loop miRNA amplification
The stem-loop miRNA amplification was performed from the cDNA amplified from the specific stem-loop sequences for each miRNA, using the miRNA sequences as the forward primers and an URP primer (Universal Reverse Primer), based on the stem-loop sequence, as the reverse primer (Table 1). The amplification reactions were conducted essentially as described for PCR, but starting from 2 μl of a 1/20 dilution of the retrotranscription reaction already described.
miRNAs precursor amplification
Specific primers for pri-miRNAs and pre-miRNAs were designed based on Don Walter genomic sequences, predicted by the mirDP2 software (Table 3). The amplification was carried out using a different forward primer for pri-miRNA and pre-miRNA, and the same reverse primers for both.

Table 3 Primers designed based on miRNA precursors identified in the Don Walter genome.
qPCR reactions
The experiments carried out to validate the differential regulation of transcripts and miRNAs described in silico for the three genotypes, were conducted through Quantitative Real-Time PCR (qPCR), using the thermocycler CFX Connect Real-Time PCR System (Bio-Rad). Reactions were performed in a final volume of 20 ul, and contained 50 pmol of forward and reverse primers, 3 µl of 200-fold diluted cDNA and 10 µl of Real Mix (Biodynamics, Argentina). The E. curvula housekeeping gene UBICE (Ubiquitin conjugating enzyme transcript) was used to normalize the specific gene expression levels, as in Selva et al., 2020. The thermal cycling used for these amplifications consisted of 95°C for 2 min, 40 cycles at 94°C for 10 s, 15 s at the optimal annealing temperature for each primer pair and finally at 30s at 72°C. Reactions were conducted on technical triplicates of biological duplicates for the three assayed genotypes. Dissociation curve profiles were used to verify the specificity of each reaction. Normalization of transcript levels between cDNA samples was undertaken using the 2-ΔΔCt method (Livak and Schmittgen, 2001). The statistical analysis of the qPCR fold change in the expression of genes among different treatments was done through the Student’s t-test, where a p-value of 0.05 was considered to be significant.
Results
Don Walter small RNA libraries characterization
Small RNAs libraries from inflorescences of the cultivar Don Walter were constructed for this work, whereas those from Tanganyika and OTA-S have been reported in Garbus et al., 2019. Thus, we started from a brief characterization of Don Walter sRNA libraries. There were obtained 2,541,355 raw reads for DW1, and 2,929,269 and 2,140,845 for DW2 and DW3 respectively. After trimming, a total of 2,114,363 (83.1%), 2,556,295 (87,2%) and 1,802,287 (84,1%) reads were retained for each library. As expected, reads with 21 and 24 bp were overrepresented in the three samples (Figure 1).
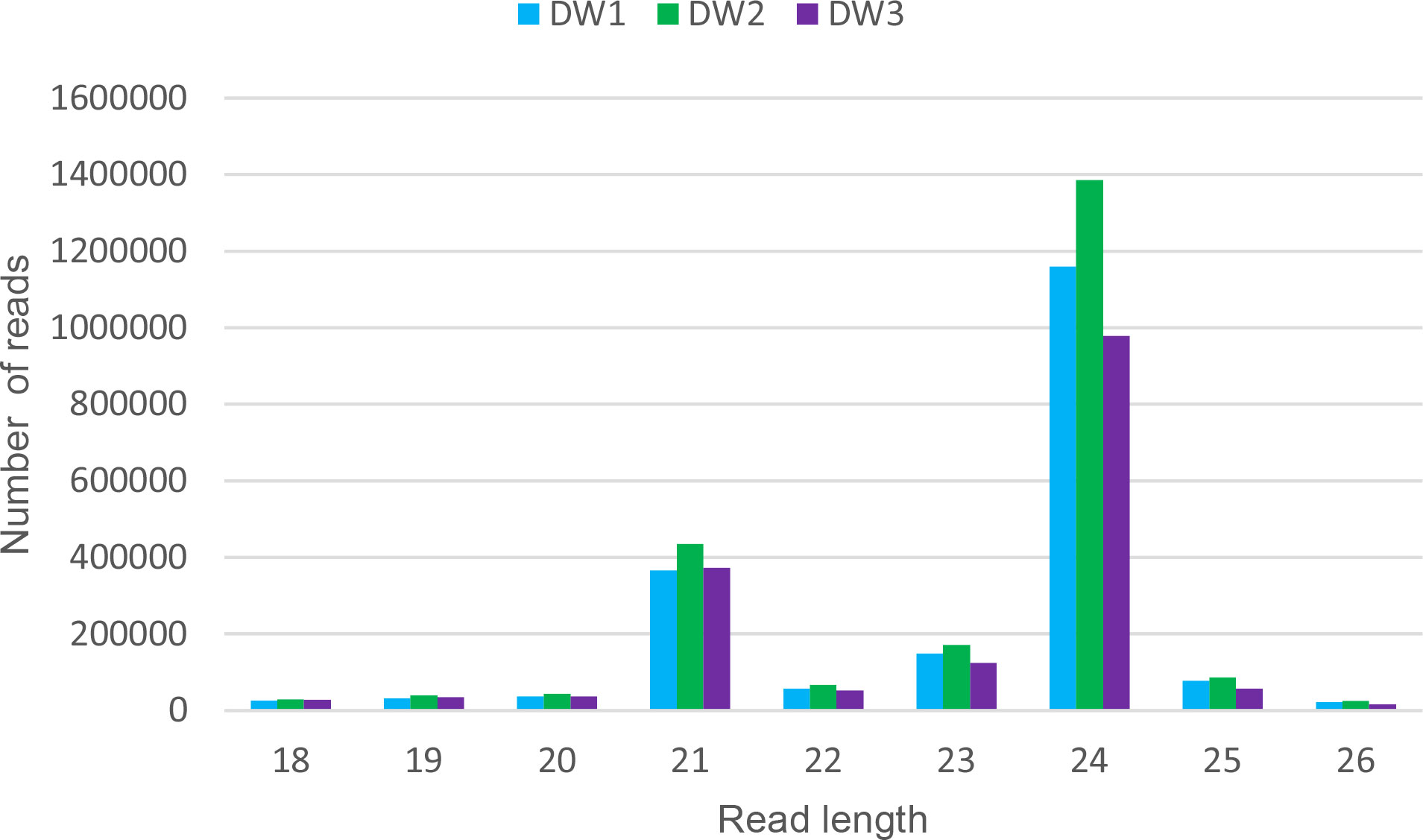
Figure 1 Read length distribution of the three Don Walter sRNA libraries. For each read length, the absolute read count per individual library is shown. The x-axis shows read lengths, and the y-axis shows the frequency of occurrence of reads of each length. The over-representation of 21 and 24 bp reads is evidenced.
miRNA prediction and expression
To identify miRNA sequences and its precursors, the reads from the seven samples, i.e., three from the facultative Don Walter, two from the sexual OTA-S and two from the full apomictic Tanganyika cultivar, were merged in a single file. After trimming and filtering, a total of 171,339,858 reads passed the quality control. Since miRDP2 pipeline requires unique sequences, the input file was collapsed into 30,124,624 unique reads attaching their frequency to the header.
The miRDP2 software predicted 953 miRNAs that were used as reference to map each sample through the Bowtie software. A count matrix was constructed to quantify the expression of the mature miRNAs in the samples. After filtering, a PCA plot and a heatmap were constructed using the normalized matrix as input, showing the similarity within samples with the same reproductive behavior (Supplementary Figures 1, 2). Finally, the same matrix was used to show the expression of all the miRNAs in each sample (Figure 2). The differential expression analysis showed that 51, 108 and 51 mature miRNAs were up-regulated in APOvsSEX, APOvsFAC and FACvsSEX comparisons, respectively. On the other hand, the down-regulated miRNAs were 52, 57 and 99 for the APOvsSEX, APOvsFAC and FACvsSEX, respectively.
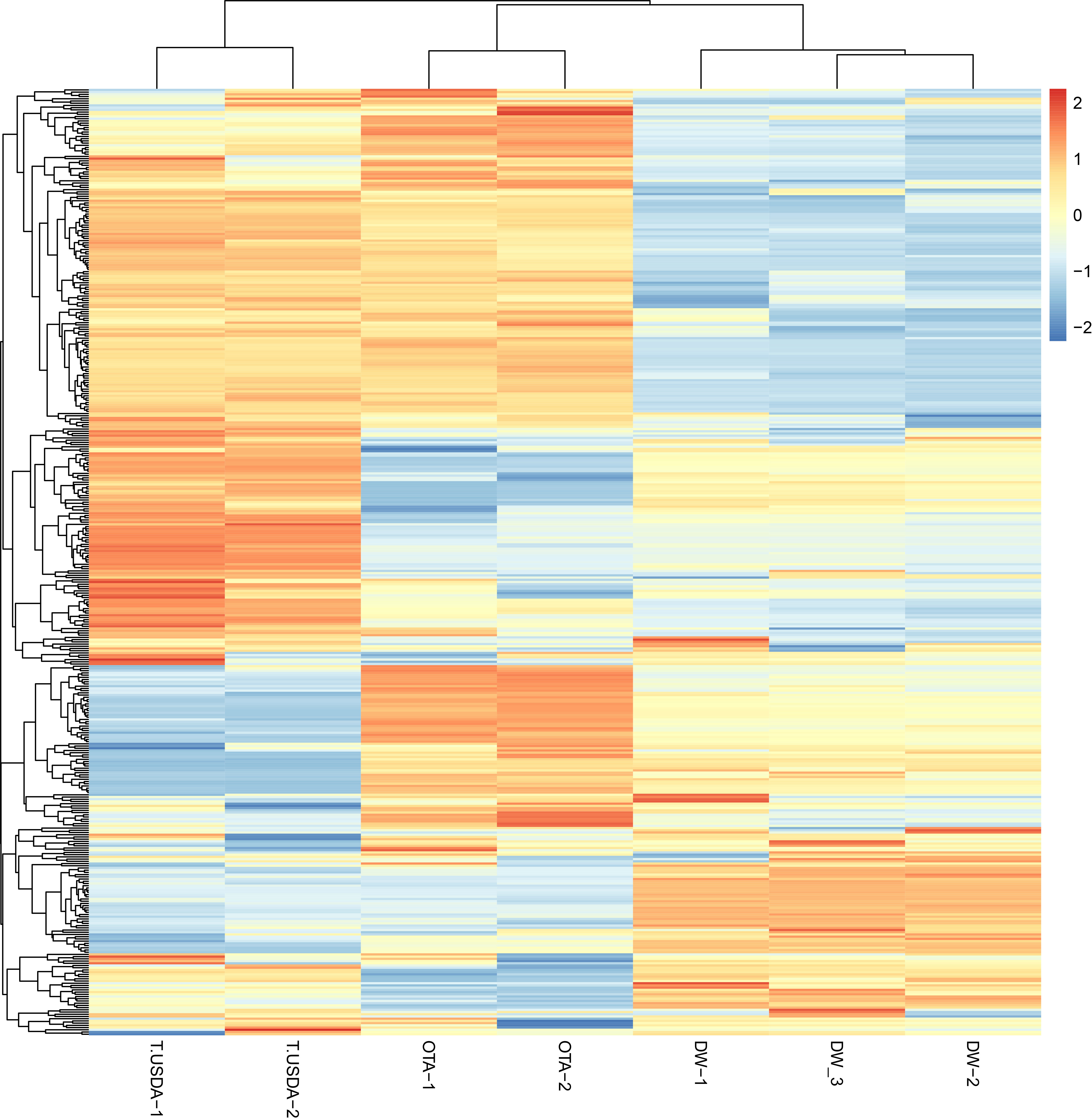
Figure 2 Heatmap showing the expression of the miRNAs in the seven samples analyzed. The count matrix was transformed in the log2 scale and normalized according to the library size. The considered samples are shown in the x-axis. In the color scale, orange and blue represent up-and down-expressed miRNAs respectively.
Target identification and transcript expression analysis
Target identification was performed through psRNATarget software (Dai et al., 2018) using the improved scoring schema V2 (2017). A total of 13,770 targets were found, which were putatively regulated by 682 unique mature miRNAs. In parallel, the expression of the transcripts was assessed. The reads were mapped against the transcripts using hisat2 (Kim et al., 2019) in order to quantify its expression. The differential expression analysis showed that 220, 2,162 and 542 transcripts were upregulated for the APOvsSEX, APOvsFAC and FACvsSEX, respectively; whereas the downregulated transcripts for the same comparisons were 195, 798 and 1,520, respectively.
Transcripts regulated by miRNAs
Based on the psRNAtarget results, we focused especially on those possible transcript-miRNA interactions in which the differential expression of transcripts and miRNAs among genotypes followed opposite direction, i.e., when transcripts showed to be up-regulated in a particular comparison, the miRNA resulted down-regulated and vice versa. Hence, 5, 14, and 16 target transcripts were found to be down-regulated while its mature miRNA was found to be up-regulated in the APOvsSEX, APOvsFAC and FACvsSEX comparisons, respectively. On the other hand, 14 and 6 targets were found up-regulated for the APOvsFAC and FACvsSEX comparison, while the mature miRNAs were found to be down-regulated. No miRNA-mRNA interactions were identified for the comparison APOvsSEX.
Genomic DNA and transcript expression analysis
The in silico analysis revealed the existence of ≈60 possible miRNA-target interactions that could be involved in the regulation of the reproductive mode in E. curvula (Supplementary Table 1). For validation purposes, after transcript annotation, seven miRNA-target interactions were selected mainly guided by their possible known function, discarding in this initial approach those transcripts without annotation and/or corresponding to hypothetical proteins (Table 4). Thus, for the comparison APOvsFAC with the target down-regulated and the miRNA up-regulated, the expression of the genes Kinesin-like protein KIN-14D, Patatin-like protein 2 and Dehydration-Responsive Element Binding Protein 2A (DREB2A) (Supplementary Figure 3A–C) was explored, whereas for the target up-regulated and miRNA down-regulated combination, the expression of Growth-regulating factor 8 (GRF8) and Squamosa-promoter binding protein-like protein 13 genes was inquired (Supplementary Figure 3D, E). The expression of the genes for DNA-Directed RNA polymerase II subunit RPB1 (DRdRNApolII) and GAMYB transcription factor were investigated because they were differential for the comparison FACvsSEX, with the target being down-regulated and miRNA up-regulated (Supplementary Figure 3F, G).
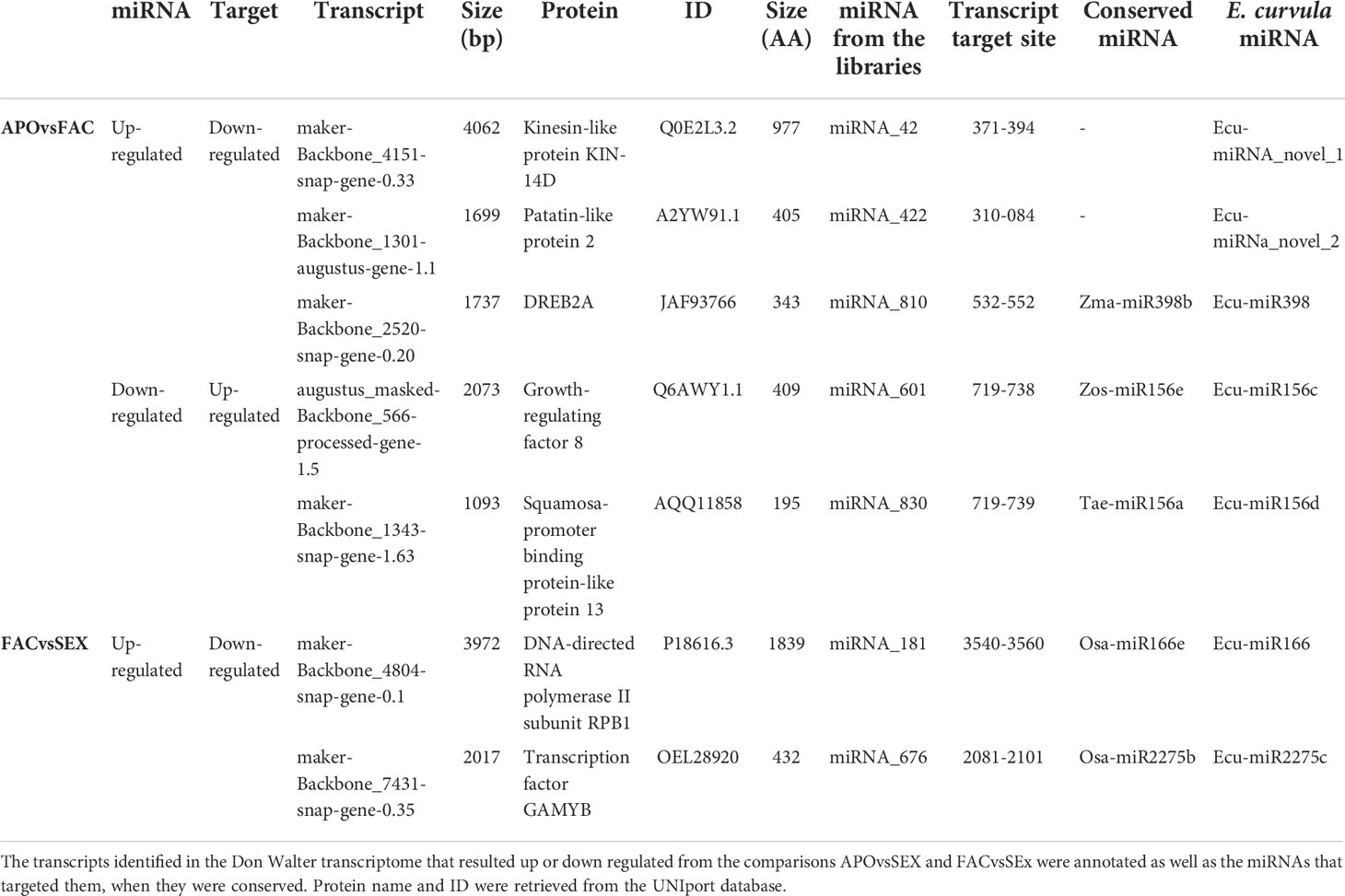
Table 4 miRNA-mRNA interactions selected for the analysis.
Initially, PCR reactions were conducted aiming to amplify the mentioned genes from genomic DNA for the three cultivars in biological triplicates, leading to the predicted amplification bands, using one or more specific primer combinations (Table 5, Figure 3). Interestingly, the kinesin transcript was differentially expressed among genotypes, being absent in the apomictic cultivar Tanganyika, in accordance with the prediction of being down-regulated in the APOvsSEX comparison (Figure 3A). However, the primer combination used for the other transcripts resulted in no amplification bands (Table 5, Figure 3B–G). In order to confirm the absence of expression, qPCR reactions were conducted. UBICE was amplified from all the cDNA samples with no significant difference in expression among them. However, the Cts obtained for the assayed cDNA samples were close to that of the negative controls, indicative of the absence of expression. Kinesin was not subjected to qPCR, since the observed amplification bands were conclusive.
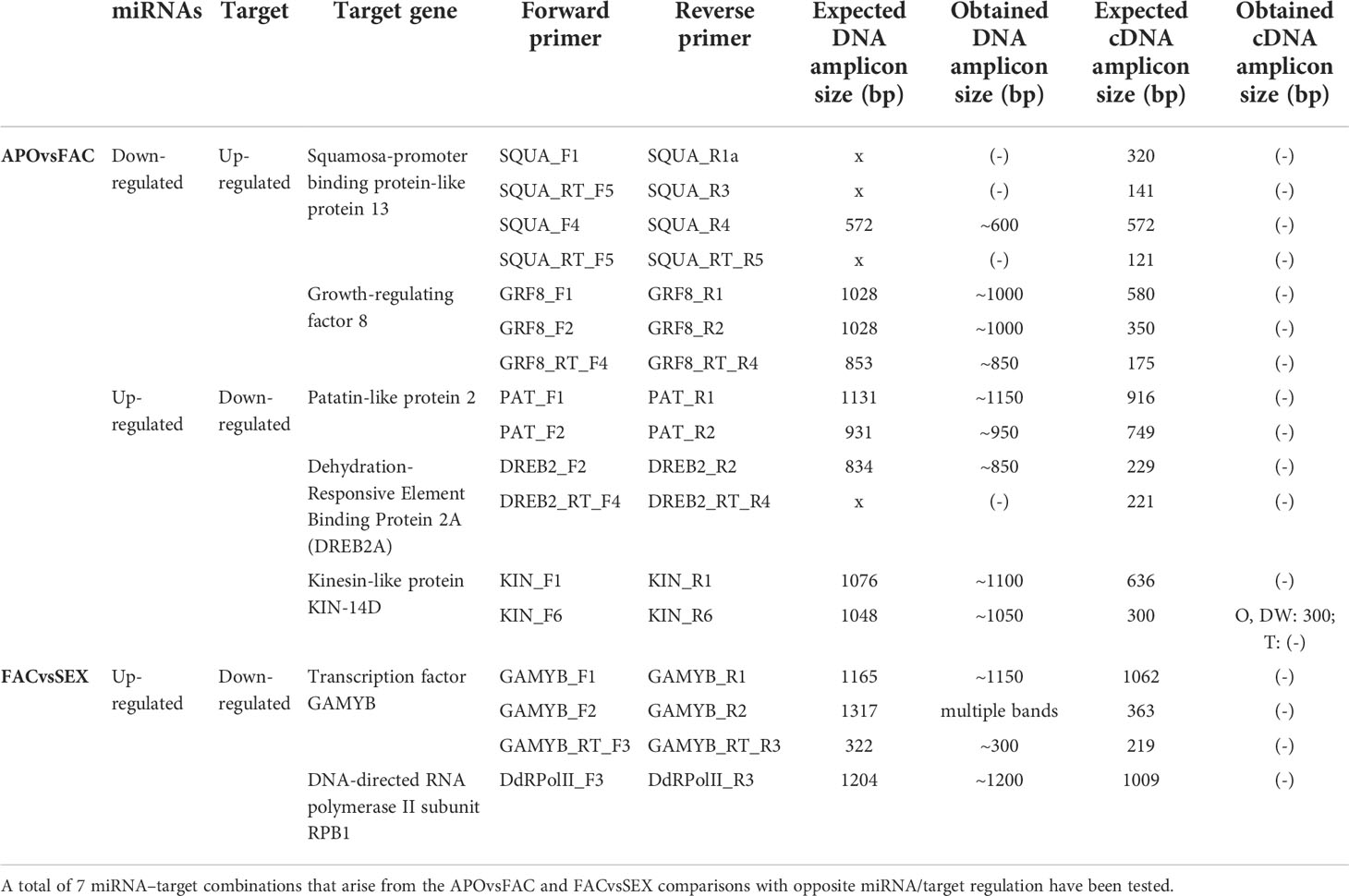
Table 5 PCR amplicons expected and predicted sizes obtained from DNA and cDNA with several combinations of transcript-based primers.
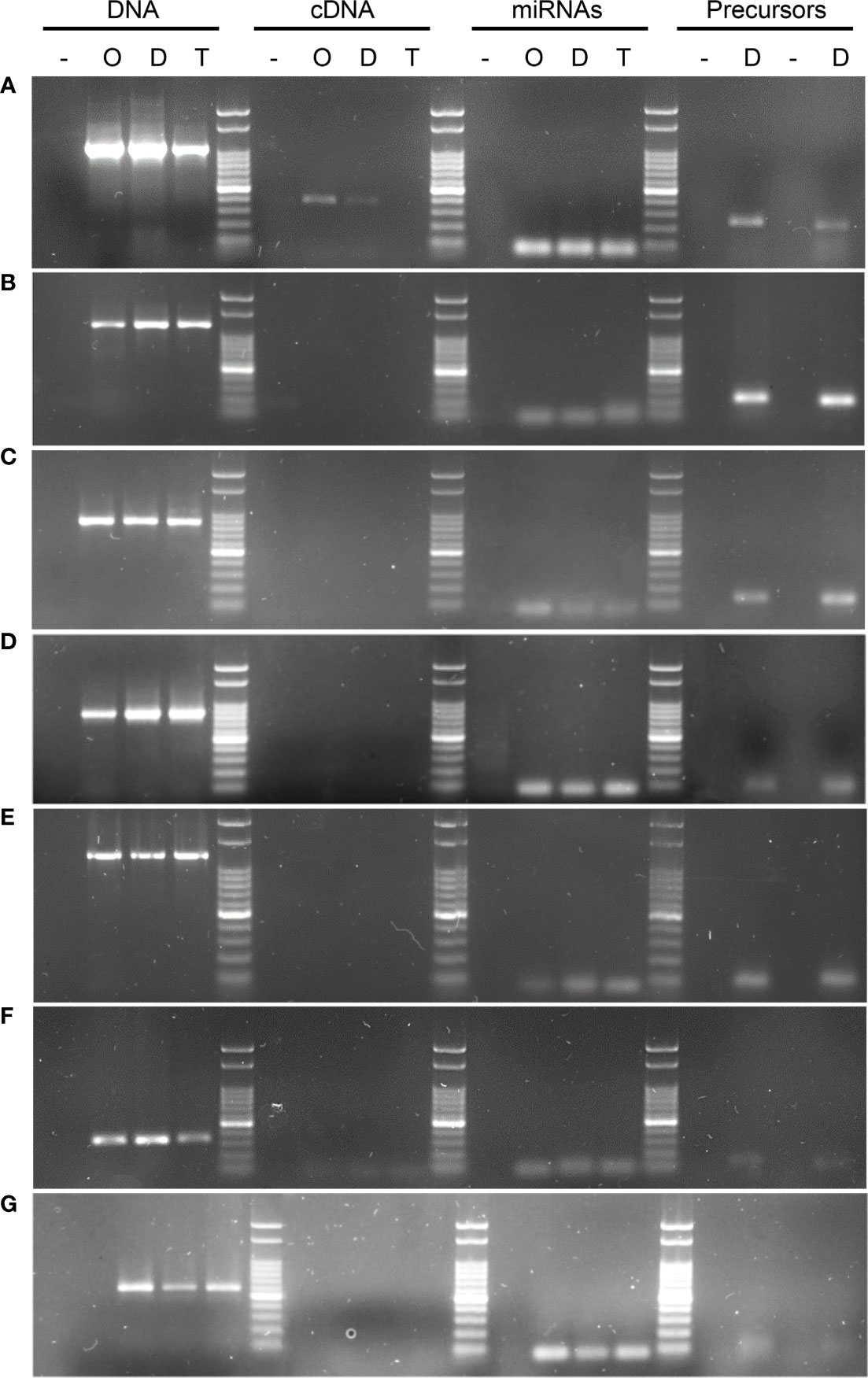
Figure 3 PCR and RT-PCR amplifications using gene, transcript, miRNA, pri-miRNA and pre-miRNA based primers. The image shows DNA from the target genes could be amplified in all of the tested samples, whereas when cDNA was the template, this led to amplification bands for 1 of the 7 transcripts tested using several primers combinations (Table 5). (A) KINESIN, which showed amplification in cDNA, and was absent only in Tanganyika, (B) PATATIN, (C) GRF8, (D) DREB2, (E) DdRNAPolII1B, (F) GAMYB, (G) SQUAMOSA. UBICE gene was used for normalization of cDNA expression. The amplification using miRNA, pri-miRNA and pre-miRNA based primers allowed the validation of the novel and reported miRNAs tested in this study.
Validation of novel and conserved miRNAs
The expression of the miRNA predicted to be interacting with the mentioned transcripts was analyzed through RT-PCR and qPCR, revealing that all the miRNAs were expressed, whereas differential expression was only detected from the miRNA that targets the kinesin transcript (Figure 4).
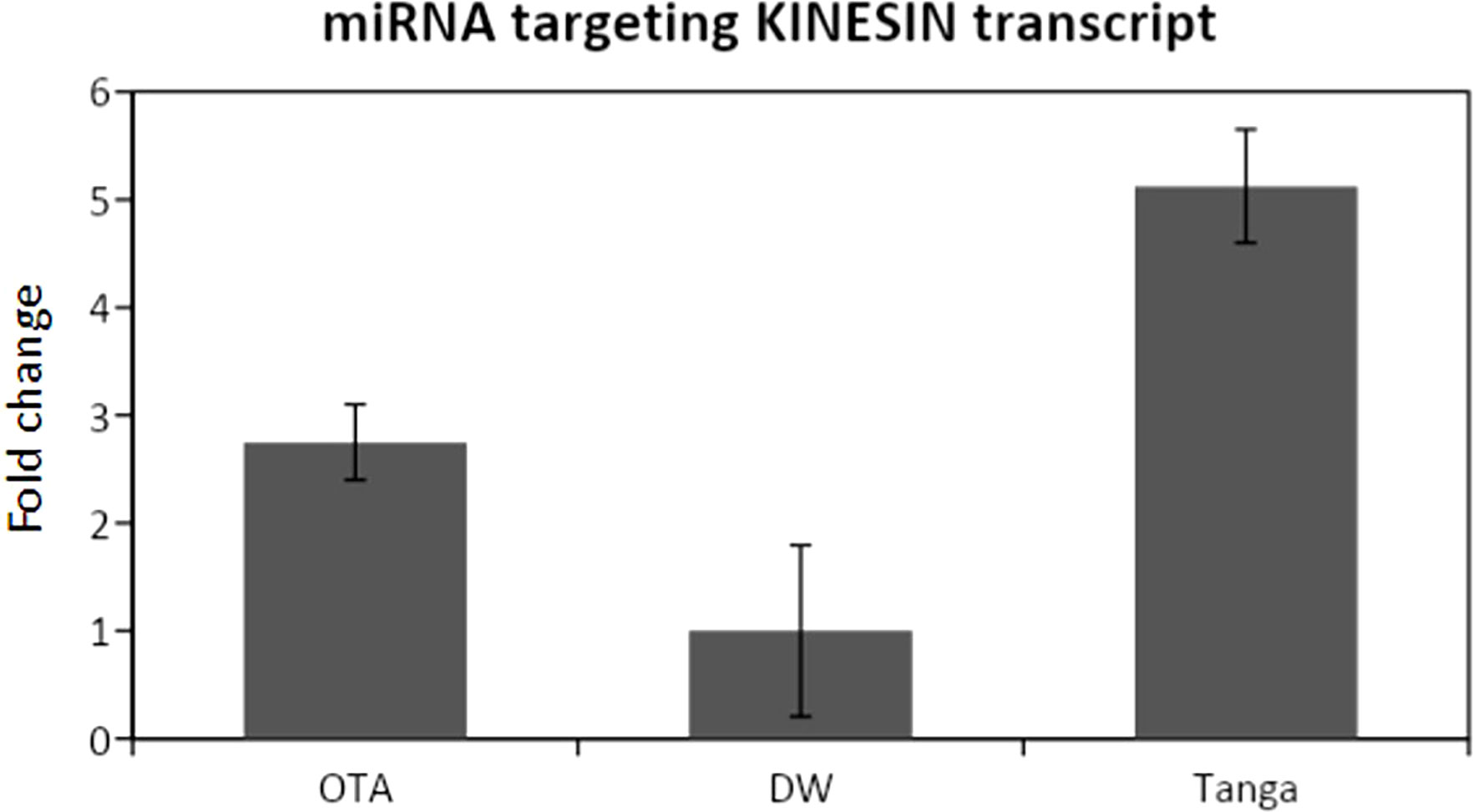
Figure 4 qPCR analysis for the miRNA that targets the Kinesin transcript. DW: Don Walter. Tanga: Tanganyika. The miRNA is overexpressed in the apomictic cultivar Tanganyika (p<0.05). Data was normalized with respect to the genotype that showed the lower expression.
miRNAs were further validated through the amplification from DNA of their pri-miRNA and pre-miRNA precursors, predicted from the Don Walter genome (Figure 3A–G), whereas their hairpin structures are shown in Figure 5. The sequences of the mature and star miRNAs as well as their precursor are reported in Supplementary Table 2.
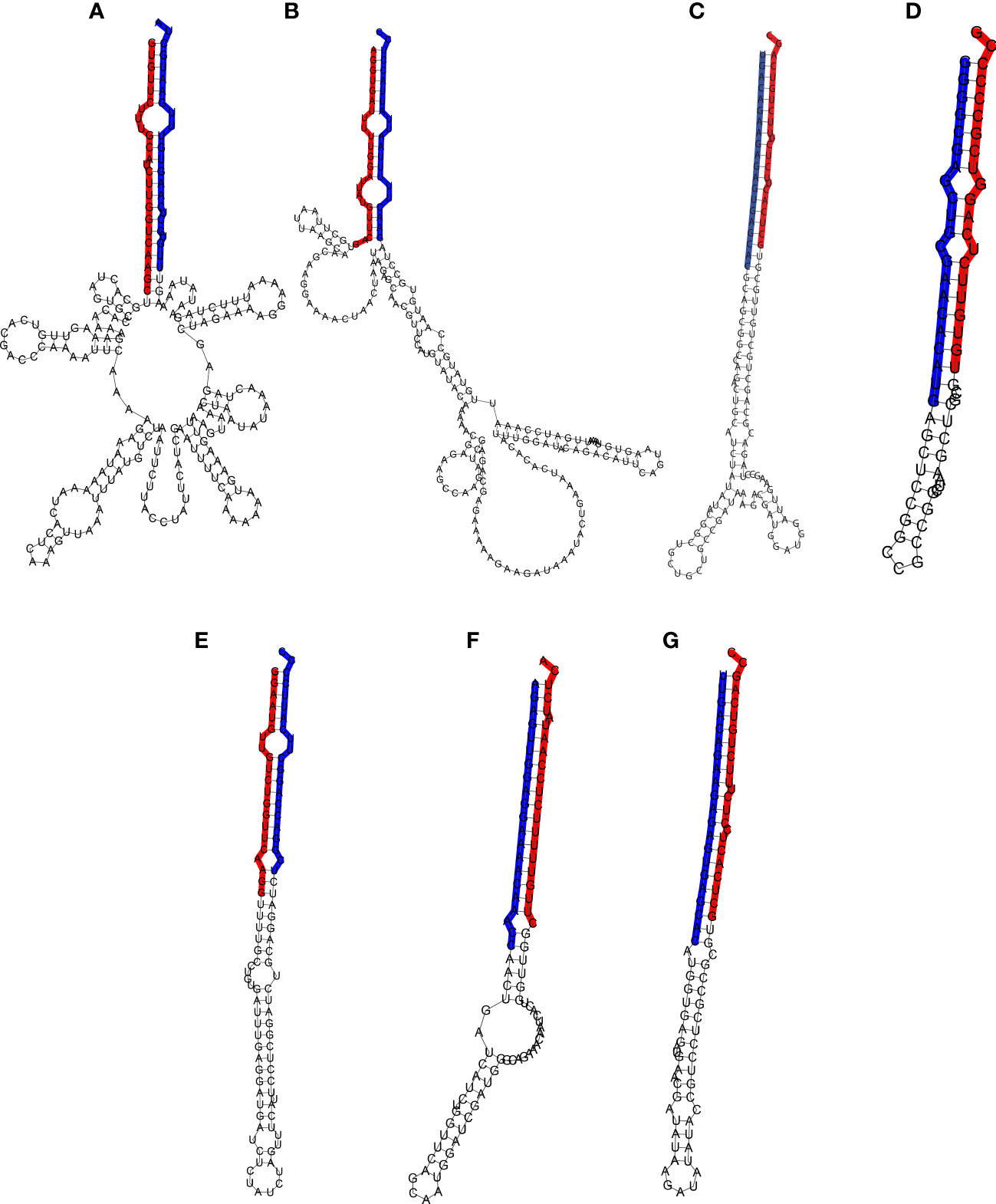
Figure 5 Hairpin structure of the miRNA precursors. The mature and star sequences are highlighted in blue and red, respectively. (A) Pre-Ecu-mir novel_1, (B) Pre-Ecu-mir novel_2, (C) Pre-Ecu-mir156c, (D) Pre-Ecu-mir398, (E) Pre-Ecu-mir166, (F) Pre-Ecu-mir 2275c, (G) Pre-Ecu-mir156d (Table 4).
The expression of all the tested miRNAs, as well as their precursors and the validation of their hairpin structures, together with the absence of amplification for most of the searched transcripts strongly suggests that a miRNA-target regulation exists for all the combinations tested.
Discussion
Eragrostis curvula (Schrad.) Nees, commonly known as weeping lovegrass, is a perennial grass from the Poaceae family, widely cultivated in the semiarid region of Argentina as a forage grass (Covas, 1991). E. curvula has a basic chromosome number of x=10, and it includes genotypes with different ploidy levels. While the diploid cytotypes are scarce and sexual in nature, tetraploid plants and plants with higher ploidy levels reproduce by pseudogamous diplosporous apomixis, defined as asexual reproduction through seeds (Leigh, 1960; Voigt and Burson, 1983; Noirot et al., 1997). Apomixis has been described in over 400 angiosperms, and it leads to the creation of clones of the mother plant (Nogler, 1984). However, this attribute is absent in most species of agricultural importance. The study of the genetic regulation of this mechanism would be important for the development of tools that could enable its transference to key species, such as wheat, rice or maize, in order to fixate important agronomic traits through generations (Hanna and Bashaw, 1987). The purpose of this work was focused on analyzing the epigenetic mechanisms that could be playing a role in diplosporous apomictic reproduction in this species.
Studies suggest that, instead of an independently originated trait, apomixis arises as the result of modifications in the temporal or spatial regulation of the sexual pathway in seed development, which produces variations or omissions in critical developmental stages (Koltunow and Grossniklaus, 2003; Schmidt, 2020). With this theory in mind, the expression of a gene or genes in specific cells from the nucellus could determine if sexual or apomictic processes, or both, will take place in that ovule, proposing that apomixis-related genes must be absent or unexpressed for the sexual processes to take place (Koltunow, 1993; Koltunow and Grossniklaus, 2003).
Genomic and transcriptomic studies were performed in the past to discover the molecular mechanisms involved in apomixis. Even though several advances were made in the last years, candidate genes related to meiosis could not be found through these approaches so far. Moreover, the involvement of epigenetic factors is being studied, as they can occur rapidly and are particularly frequent as a result of polyploidization and hybridization, and they could also help explain the parallel and independent occurrence of apomixis several times during evolution (Koltunow and Grossniklaus, 2003; Comai, 2005). In the year 2010, García-Aguilar et al., 2010 observed that a deregulation of DNA methylation produced apomeiotic-like phenotypes in reproductive cells of maize. Also, in apomictic genotypes of Boechera subjected to stress, Taraxacum-type diplospory arised in the female gametophyte. In studies with the same genotypes, the suppression of DNA methylation before the formation of the MMC triggered Antennaria-type mitotic diplospory (Gao, 2018). In E. curvula, it has been observed that environmental signals have an influence over the number of sexual embryo sacs (Rodrigo et al., 2017), and that polyploidization involves an increase in genome methylation, and it could lead to gene silencing, in order to preserve the functional characteristics of the diploid (Ochogavía et al., 2009; Zappacosta et al., 2014; Carballo et al., 2021).
As mentioned previously, miRNAs are known to play central regulatory roles during plant development, and they are part of diverse biological processes such as flower and leaf morphogenesis, response to stress, reproductive cell identity, fertility, etc. (Aukerman and Sakai, 2003; Palatnik et al., 2003; Sunkar and Zhu, 2004; Schwab et al., 2005; Ru et al., 2006; Olmedo-Monfil et al., 2010; Amiteye et al., 2011; Li et al., 2015; Ripoll et al., 2015; Fei et al., 2021).
Our work established that the gene kinesin is differentially expressed among genotypes, being absent in the apomictic genotype Tanganyika. A BLAST analysis of the transcript revealed that it is homolog of Kinesin14D. Moreover, the expression of the miRNA postulated to be regulating kinesin, was found to be upregulated in the genotype Tanganyika. This is a novel miRNA identified in this work, referred in the manuscript Ecu-miRNA_novel_1, and its existence was confirmed by the amplification of the precursors pri-miRNA and pre-miRNA and the predicted hairpin structure. Kinesins act as microtubule-based molecular motors involved in several biological processes such as cell division, intracellular transport, and cell shape determination (Zhu and Dixit, 2012; Ali and Yang, 2020). Kinesin genes are distributed in 14 families, with Kinesin7 and Kinesin14 being the most represented in land plants (Richardson et al., 2006). It is proposed that land plants may have evolved novel Kinesin14 motors to account for microtubule-based functions that are plant-specific or are typically performed by Dynein in other eukaryotes, as is spindle formation and the expansion of the phragmoplast (Zhu and Dixit, 2012; Gicking et al., 2018).
The Kinesin14 family includes many members that are up-regulated during mitosis and function during cell division and localization of plastids, thus influencing plant development (Gicking et al., 2018). ATK1 and ATK5 are two minus-end-directed Kinesin14 proteins that localize in the spindle apparatus. A. thaliana atk1 mutants exhibit abnormalities in meiosis, such as extended spindle poles and reduced male fertility (Chen et al., 2002), as well as mild spindle bipolarity defects during mitosis (Marcus et al., 2003). The lack of spindle bipolarity in atk1-1 leads to abnormal chromosome segregation during meiosis, whereas in mitosis this defect is corrected by anaphase, perhaps due to functional compensation by other minus-end-directed kinesins such as ATK5 (Nebenführ and Dixit, 2018). Mitotic spindles in the atk5-1 mutant are abnormally broad (Ambrose et al., 2005). Furthermore, maize mutants in a Kinesin14 member, that is likely a functional homolog of ATK1, showed extended spindle poles and altered spindle length and width during meiosis (Higgins et al., 2016).
A study conducted in Citrullus lanatus, showed that 38 Kinesin genes were overexpressed in the reproductive organs, being 20 preferentially expressed in the female flower, and 9 specifically expressed in the male flowers, thus suggesting that they play critical roles in the growth and development of reproductive tissues (Tian et al., 2021). Interestingly ClKIN14D, homologous to Kinesin14D, was found to be overexpressed in male flowers and repressed in female flowers (Tian et al., 2021).
The analysis of protein motifs of the Kinesin translated from Don Walter transcriptome, revealed that it contains a calponin-homology domain (CH), thus classified as KCH, a unique subset of Kinesin14 that can bind to both microtubules and actin filaments in land plants (Umezu et al., 2011). KCHs are most abundant in developing tissues and are thought to participate in cell division and elongation (Frey et al., 2010). Identified KCHs showed to be mostly expressed in mature pollen, being proposed that they are the main actors for the movement of sperm cells in the pollen tube (Schattner et al., 2021). The expression of OsKCH1 was quantified in rice, revealing that the highest abundance of transcripts was found in primary leaf, primary root, and the developing flower (Frey et al., 2010).
In this bibliographic context it is clear that kinesins are involved in plant reproduction and, thus, is plausible that the miRNA-target interaction that impedes the translation of the kinesin transcript is contributing to a defective sexual expression and leading to an apomictic phenotype in Tanganyika. Moreover, Kinesins were reported to be targeted by miR1863 (Smita et al., 2021) in grapevine and by sly-miR171a in Solanum lycopersicum (López-Galiano et al., 2019). It is not unexpected that kinesins, with a main role in mitosis and meiosis, are determinant in plant growth and fertility.
The expression of the orthologous gene from Arabidopsis, At1g73860, was demonstrated to be tissue-specific for the microgametogenesis, specifically expressed in mature pollen (Honys and Twell, 2004; Klepikova et al., 2016), thus arguing in favor of a specific role in the reproductive pathway. In addition, the rice orthologous Os02g0229600, was found highly expressed in mature pollen as well as in pre-flowering panicles (Sakai et al., 2011).
Whether the epigenetic mechanisms are the cause or the result of changes in the reproductive behavior is still under discussion (Hand and Koltunow, 2014; Mau et al., 2022), but under the light of our results, the fact that the kinesin gene is involved in apomixis in E. curvula cannot be denied. Thus, this gene is disclosed as a strong candidate for further studies allowing the design of strategies to inquire into apomixis mechanisms in this or other species.
Concerning the other analyzed genes, the absence of mRNA expression combined with the observed miRNA expression demonstrated miRNA-target interactions for all of them. Out of the analyzed transcripts, DNA-directed RNA polymerase II subunit RPB1 (DdRNAPolII1B), Squamosa-promoter binding protein-like 13 (SBP13), Growth-regulating factor 8 (GRF8) and Dehydration-Responsive Element Binding Protein 2A (DREB2A) are targeted by the conserved miRNAs with identity to Osa-miR166e, Tae-miR156a, Zma-miR396d and Zma-miR398b respectively, whereas the other transcripts are regulated by miRNAs firstly described in this work, and validated through the amplification of the pri-miRNAs and pre-miRNAs (Table 4).
Interestingly, several of our analyzed transcripts were previously demonstrated to be regulated by miRNAs. miR156 was reported to target SBP genes (Poethig, 2010; Wang, 2014; Garbus et al., 2019), and was shown to be highly conserved, playing an important role in plant development (Poethig, 2010). It is highly expressed during the juvenile phase and declines during vegetative phase change (Poethig, 2010), leading to an increased expression of SBPs genes that act by promoting the development of adult vegetative traits and floral induction (Wang, 2014). Overexpression of miRNA156 in A. thaliana produces nearly full sterility, and the rescue of SPL function brings back normal reproductive behavior (Zheng et al., 2019). It is also known that miRNA156’s expression is fine-tuned since its temporal and spatial expression change during the reproductive development (Wu and Poethig, 2006; Zheng et al., 2019). We can hypothesize that the expression of miRNA156 could have different expression patterns related to the reproductive mode. Additional analyses testing its expression in different developmental stages of apomictic and sexual plants should be conducted to confirm this assumption.
The transcription factor GAMYB (Gibberellin-and-Abscisic acid-regulated MYB), involved in flowering time and flower organ development (Sunkar and Zhu, 2004), was also reported to be involved in another development via regulating the expression of phenylalanine ammonia lyase (PAL) (Woodger et al., 2003). It was proposed that miR319a-regulated GAMYB might also be involved in the browning inhibition of fresh-cut apples by H2S treatment (Chen et al., 2020). Other study states that, in Oryza sativa, miR1026 and miR159 target DdRNAPolII1B and GAMYB, respectively, being both up-regulated under stress conditions. The gene Patatin-like protein 2 (PLP2), related to lipid metabolism, was proposed to be targeted by miR6478 (Chen et al., 2020). DNA-directed RNA polymerase II has been reported to be targeted by miR172 in the genus Mimulus (Barozai et al., 2011).
This study provided small RNA libraries available for the scientific community for further analysis of miRNA regulation, at the time that it postulates and validates the presence in the genome of the facultative apomictic cultivar Don Walter of five conserved and two novel miRNAs, given by their evidenced pri-miRNA, pre-miRNA and hairpin structures, being functional as mRNA expression repressors. Moreover, a Kinesin gene expression could be related to miRNA differential regulation in the obligate apomictic genotype Tanganyika.
This work contributed new supporting evidence about the participation of miRNAs in the regulation of genes that are determinant for reproduction in E. curvula.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
Author contributions
Conception: IG, VE. Experimental design: IG, JC. Materials: JS, JR, DZ, JC. Experimental procedures: MP, JG, JC. Bioinformatic data processing: JC, IG. Analysis and interpretation: IG, JC, MP. Writing: IG, JC, MP. Critical review: VE. All authors contributed to the article and approved the submitted version.
Funding
PIP 2021 – 2023 (Code: 11220200100306CO) Identificación de interacciones miRNA-mRNA basada en el análisis del degradoma orientada a la caracterización de su rol en la regulación epigenética de la apomixis diplospórica en pasto llorón (Eragrostis curvula) to IG: laboratory supplies financement. PICT Raíces 2017 - 0879. Genómica estructural para acceder a la región condicionante de la apomixis en Eragrostis curvula. to VE and Mario Cáccamo (NIAB, UK): laboratory supplies and sequencing services financement H2020-MSCA-RISE-2019, Mechanisms of Apomictic Developments (MAD) to VE in Argentina: sequencing services and open access publication fees. PGI 24/A261 Estudio de las bases genéticas y epigenéticas asociadas a la apomixis en Eragrostis curvula (Shrad.) Nees to V.E: laboratory supplies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1012682/full#supplementary-material
Supplementary Figure 1 | Heatmap of the clustering of samples based on distance matrix. Heatmap showing the distance matrix obtained on logarithm transformed counts, showing similarities and dissimilarities between the seven samples DW-1, DW-2, DW-3, T.USDA-1, T.USDA-2, OTA-1, OTA-2. In the color scale, red and blue represent more and less distance between samples, respectively.
Supplementary Figure 2 | Principal component analysis plot. PCA plot showing the similarity within samples with the same reproductive behavior. The variance of principal components PC1 and PC2 are represented in the x and y axis, respectively.
Supplementary Figure 3 | Alignment between miRNAs and their targeted transcripts. miRNA - target interaction between. (A) Ecu-miRNA_novel_1 and kinesin-like protein KIN-14D, (B) Ecu-miRNA_novel_2 and patatin-like protein 2, (C) Ecu-miR156c and Growth-regulating factor 8, (D) Ecu-miR398 and DREB2A, (E) Ecu-miR166 and DNA-directed RNA polymerase II subunit RPB1, (F) Ecu-miR2275c and Transcription factor GAMYB, and (G) Ecu-miR156d and Squamosa-promoter binding protein-like protein 13 (Table 4).
References
Ali, I., Yang, W.-C. (2020). The functions of kinesin and kinesin-related proteins in eukaryotes. Cell Adhesion Migration 14 (1), 139–152. doi: 10.1080/19336918.2020.1810939
Ambrose, J. C., Li, W., Marcus, A., Ma, H., Cyr, R. (2005). A Minus-End–directed kinesin with plus-end tracking protein activity is involved in spindle morphogenesis. Mol. Biol. Cell 16 (4), 1584–1592. doi: 10.1091/mbc.e04-10-0935
Amiteye, S., Corral, J. M., Vogel, H., Kuhlmann, M., Mette, M. F., Sharbel, T. F. (2013). Novel MicroRNAs and microsatellite-like small RNAs in sexual and apomictic boechera species. MicroRNA 2 (1), 46–63. doi: 10.2174/2211536611302010006
Amiteye, S., Corral, J. M., Vogel, H., Sharbel, T. F. (2011). Analysis of conserved microRNAs in floral tissues of sexual and apomictic boechera species. BMC Genomics 12 (1), 1–16. doi: 10.1186/1471-2164-12-500
Aukerman, M. J., Sakai, H. (2003). Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell 15 (11), 2730–2741. doi: 10.1105/tpc.016238
Barozai, M. Y. K., Din, M., Baloch, I. A. (2011). Identification of microRNAs in ecological model plant Mimulus. J. Biophys. Chem. 02 (03), 322–331. doi: 10.4236/jbpc.2011.23037
Borges, F., Martienssen, R. A. (2015). The expanding world of small RNAs in plants. Nat. Rev. Mol. Cell Biol. 16 (12), 727–pp.741. doi: 10.1038/nrm4085
Brukhin, V., Albertini, E. (2021). Epigenetic modifications in plant development and reproduction. Epigenomes 5 (4), 25. doi: 10.3390/epigenomes5040025
Carballo, J., Santos, B. A. C. M., Zappacosta, D., Garbus, I., Selva, J. P., Gallo, C. A., et al. (2019). A high-quality genome of eragrostis curvula grass provides insights into poaceae evolution and supports new strategies to enhance forage quality. Sci. Rep. 9 (1), 1–15. doi: 10.1038/s41598-019-46610-0
Carballo, J., Zappacosta, D., Marconi, G., Gallardo, J., Di Marsico, M., Gallo, C. A., et al. (2021). Differential methylation patterns in apomictic vs. sexual genotypes of the diplosporous grass eragrostis curvula. Plants 10 (5), 946. doi: 10.3390/plants10050946
Carman, J. G., de Arias, M. M., Nelson, S. M., Zhao, X., Gao, L., Srivastava, M., et al. (2015). “Hot on the trail of the sex-apomixis switch in boechera (Brassicaceae),” in In Proceedings of the Plant and Animal Genome Conference (San Diego, USA: PAG XXIV). Available at: https://pag.confex.com/pag/xxiii/webprogram/Paper14382.html
Cervigni, G. D. L., Paniego, N., Díaz, M., Selva, J. P., Zappacosta, D., Zanazzi, D., et al. (2008b). Expressed sequence tag analysis and development of gene associated markers in a near-isogenic plant system of eragrostis curvula. Plant Mol. Biol. 67 (1-2), 1–10. doi: 10.1007/s11103-007-9282-4
Cervigni, G. D. L., Paniego, N., Pessino, S., Selva, J. P., Díaz, M., Spangenberg, G., et al. (2008a). Gene expression in diplosporous and sexual eragrostis curvula genotypes with differing ploidy levels. Plant Mol. Biol. 67 (1-2), 11–23. doi: 10.1007/s11103-008-9305-9
Chen, C., Liu, C., Jiang, A., Zhao, Q., Zhang, Y., Hu, W. (2020). miRNA and degradome sequencing identify miRNAs and their target genes involved in the browning inhibition of fresh-cut apples by hydrogen sulfide. J. Agric. Food Chem. 68 (31), 8462–8470.
Chen, C., Marcus, A., Li, W., Hu, Y., Calzada, J.-P. V., Grossniklaus, U., et al. (2002). The arabidopsis ATK1 gene is required for spindle morphogenesis in male meiosis. Development 129 (10), 2401–2409. doi: 10.1242/dev.129.10.2401
Chen, C., Ridzon, D. A., Broomer, A. J., Zhou, Z., Lee, D. H., Nguyen, J. T., et al. (2005). Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res. 33 (20), e179–e179. doi: 10.1093/nar/gni178
Comai, L. (2005). The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6 (11), 836–846. doi: 10.1038/nrg1711
Covas, G. (1991). “Introducción del pasto llorón en la Argentina in El pasto llorón, su biología y manejo,” in Cerzos y dpto. de agronomía (Bahía Blanca: Universidad Nacional del Sur), 1–6.
Crane, C. F. (2001). ““Classification of apomictic mechanisms”,” in The flowering of apomixis: from mechanisms to genetic engineering, vol. 24 . Eds. Savidan, Y., Carman, J. G., Dresselhaus, T. (Mexico: CIMMYT, IRD, EU DG VI (FAIR), 43.
Dai, X., Zhuang, Z., Zhao, P. X. (2018). psRNATarget: a plant small RNA target analysis serve Release. Nucleic Acids Res. 46 (W1), 49–54. doi: 10.1093/nar/gky316
Fei, X., Lei, Y., Qi, Y., Wang, S., Hu, H., Wei, A. (2021). Small RNA sequencing provides candidate miRNA-target pairs for revealing the mechanism of apomixis in zanthoxylum bungeanum. BMC Plant Biol. 21 (1), 1–13. doi: 10.1186/s12870-021-02935-5
Frey, N., Klotz, J., Nick, P. (2010). A kinesin with calponin-homology domain is involved in premitotic nuclear migration. J. Exp. Bot. 61 (12), 3423–3437. doi: 10.1093/jxb/erq164
Gao, L. (2018). Pharmacologically induced meiosis apomeiosis interconversions in boechera, arabidopsis and vigna (Utah: Utah State University).
Garbus, I., Romero, J. R., Selva, J. P., Pasten, M. C., Chinestra, C., Carballo, J., et al. (2017). De novo transcriptome sequencing and assembly from apomictic and sexual eragrostis curvula genotypes. PloS One 12 (11), e0185595. doi: 10.1371/journal.pone.0185595
Garbus, I., Selva, J. P., Pasten, M. C., Bellido, A. M., Carballo, J., Albertini, E., et al. (2019). Characterization and discovery of miRNA and miRNA targets from apomictic and sexual genotypes of eragrostis curvula. BMC Genomics 20 (1), 1–13. doi: 10.1186/s12864-019-6169-0
García-Aguilar, M., Michaud, C., Leblanc, O., Grimanelli, D. (2010). Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 22 (10), 3249–3267. doi: 10.1105/tpc.109.072181
Gicking, A. M., Swentowsky, K. W., Dawe, R. K., Qiu, W. (2018). Functional diversification of the kinesin-14 family in land plants. FEBS Lett. 592 (12), 1918–1928. doi: 10.1002/1873-3468.13094
Gounaris, E. K., Sherwood, R. T., Gounaris, I., Hamilton, R. H., Gustine, D. L. (1991). Inorganic salts modify embryo sac development in sexual and aposporous cenchrus ciliaris. Sexual Plant Reprod. 4 (3), 188–192. doi: 10.1007/bf00190003
Grimanelli, D., Leblanc, O., Perotti, E., Grossniklaus, U. (2001). Developmental genetics of gametophytic apomixis. Trends Genet. 17 (10), 597–604. doi: 10.1016/s0168-9525(01)02454-4
Grossniklaus, U. (2001). “From sexuality to apomixis: molecular and genetic approaches,” in The flowering of apomixis: From mechanisms to genetic engineering, 168–211. Edited by Savidan, Y., Carman, J. G., Dresselhaus, T., Mexico D.F: CIMMYT, IRD, European Comission DG VI (FAIR).
Hand, M. L., Koltunow, A. M. G. (2014). The genetic control of apomixis: Asexual seed formation. Genetics 197 (2), 441–450. doi: 10.1534/genetics.114.163105
Hanna, W. W., Bashaw, E. C. (1987). Apomixis: Its identification and use in plant breeding 1. Crop Sci. 27 (6), 1136–1139. doi: 10.2135/cropsci1987.0011183x002700060010x
Higo, A., Saihara, N., Miura, F., Higashi, Y., Yamada, M., Tamaki, S., et al. (2020). DNA Methylation is reconfigured at the onset of reproduction in rice shoot apical meristem. Nat. Commun. 11 (1), 1–12. doi: 10.1038/s41467-020-17963-2
Higgins, D. M., Nannas, N. J., Dawe, R. K. (2016). The maize divergent spindle-1 (dv1) gene encodes a kinesin-14A motor protein required for meiotic spindle pole organization. Front. Plant Sci. 7, 1277.
Honys, D., Twell, D. (2004). Transcriptome analysis of haploid male gametophyte development in arabidopsis. Genome Biol. 5 (11), R85. doi: 10.1186/gb-2004-5-11-r85
Hussey, M. A., Bashaw, E. C., Hignight, K. W., Dahmer, M. L. (1991). Influence of photoperiod on the frequency of sexual embryo saes in facultative apomictic buffelgrass. Euphytica 54 (2), 141–145. doi: 10.1007/bf00039600
Khanday, I., Skinner, D., Yang, B., Mercier, R., Sundaresan, V. (2019). A male-expressed rice embryogenic trigger redirected for asexual propagation through seeds. Nature 565 (7737), 91–95. doi: 10.1038/s41586-018-0785-8
Kim, D., Paggi, J. M., Park, C., Bennett, C., Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi: 10.1038/s41587-019-0201-4
Klatt, S., Hadacek, F., Hodač, L., Brinkmann, G., Eilerts, M., Hojsgaard, D., et al. (2016). Photoperiod extension enhances sexual megaspore formation and triggers metabolic reprogramming in facultative apomictic ranunculus auricomus. Front. Plant Sci. 7. doi: 10.3389/fpls.2016.00278;
Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., Logacheva, M. D., Penin, A. A. (2016). A high resolution map of the arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88 (6), 1058–1070. doi: 10.1111/tpj.13312
Koltunow, A. M., Grossniklaus, U. (2003). Apomixis: A developmental perspective. Annu. Rev. Plant Biol. 54 (1), 547–574. doi: 10.1146/annurev.arplant.54.110901.160842
Koltunow, A. M. (1993). Apomixis: embryo sacs and embryos formed without meiosis or fertilization in ovules. Plant Cell 5 (10), 1425.
Langmead, B. (2010). Aligning short sequencing reads with bowtie. Curr. Protoc. Bioinf. 32, 11.7.1–11.7.14. doi: 10.1002/0471250953.bi1107s32
Leigh, J. H. (1960). Temperature, moisture and daylength effects in lovegrass (Eragrostis curvula (Schrad) nees). S. Afr. J. Sci. 56 (11), 268–269.
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Li, Z.-F., Zhang, Y.-C., Chen, Y.-Q. (2015). miRNAs and lncRNAs in reproductive development. Plant Sci. 238, 46–52. doi: 10.1016/j.plantsci.2015.05.017
López-Galiano, M. J., Sentandreu, V., Martínez-Ramírez, A. C., Rausell, C., Real, M. D., Camañes, G., et al. (2019). Identification of stress associated microRNAs in Solanum lycopersicum by high-throughput sequencing. Genes 10 (6), 475. doi: 10.3390/genes10060475
Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F., et al. (2011). ViennaRNA package 2.0. Algorithms Mol. Biol. 6 (1), 1–14. doi: 10.1186/1748-7188-6-26
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 1–14. doi: 10.1186/s13059-014-0550-8
Marcus, A. I., Li, W., Ma, H., Cyr, R. J. (2003). A kinesin mutant with an atypical bipolar spindle undergoes normal mitosis. Mol. Biol. Cell 14 (4), 1717–1726. doi: 10.1091/mbc.e02-09-0586
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17 (1), p.10. doi: 10.14806/ej.17.1.200
Mau, M., Mandáková, T. M., Ma, X., Ebersbach, J., Zou, L., Lysak, M. A., et al. (2022). Evolution of an apomixis-specific allele class in supernumerary chromatin of apomictic boechera. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.890038
Melo, C. A., Melo, S. A. (2013). Biogenesis and physiology of MicroRNAs, Chapter 2, pp 5–24, in Non-coding RNAs and Cancer, Editor: Fabbri, M., Editorial: Springer, New York, NY doi: 10.1007/978-1-4614-8444-8_2
Nebenführ, A., Dixit, R. (2018). Kinesins and myosins: Molecular motors that coordinate cellular functions in plants. Annu. Rev. Plant Biol. 69 (1), 329–361. doi: 10.1146/annurev-arplant-042817-040024
Nejat, N., Mantri, N. (2018). Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit. Rev. Biotechnol. 38 (1), 93–105. doi: 10.1080/07388551.2017.1312270
Neumeier, J., Meister, G. (2021). siRNA specificity: RNAi mechanisms and strategies to reduce off-target effects. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.526455
Nogler, G. A. (1984). Gametophytic apomixis. In: Johri, B.M. (eds) Embryology of Angiosperms, (Berlin, Heidelberg: Springer). 475–518. doi: 10.1007/978-3-642-69302-1_10
Noirot, M., Couvet, D., Hamon, S. (1997). Main role of self-pollination rate on reproductive allocations in pseudogamous apomicts. Theor. Appl. Genet. 95 (3), 479–483. doi: 10.1007/s001220050586
Ochogavía, A. C., Cervigni, G., Selva, J. P., Echenique, V. C., Pessino, S. C. (2009). Variation in cytosine methylation patterns during ploidy level conversions in eragrostis curvula. Plant Mol. Biol. 70 (1-2), 17–29. doi: 10.1007/s11103-009-9454-5
Olmedo-Monfil, V., Durán-Figueroa, N., Arteaga-Vázquez, M., Demesa-Arévalo, E., Autran, D., Grimanelli, D., et al. (2010). Control of female gamete formation by a small RNA pathway in arabidopsis. Nature 464 (7288), 628–632. doi: 10.1038/nature08828
Ortiz, J. P. A., Leblanc, O., Rohr, C., Grisolia, M., Siena, L. A., Podio, M., et al. (2019). Small RNA-seq reveals novel regulatory components for apomixis in Paspalum notatum. BMC Genomics 20 (1), 1–17. doi: 10.1186/s12864-019-5881-0
Palatnik, J. F., Allen, E., Wu, X., Schommer, C., Schwab, R., Carrington, J. C., et al. (2003). Control of leaf morphogenesis by microRNAs. Nature 425 (6955), 257–263. doi: 10.1038/nature01958
Pikaard, C. S., Scheid, M. O. (2014). Epigenetic regulation in plants. Cold Spring Harbor Perspect. Biol. 6 (12), a019315–a019315. doi: 10.1101/cshperspect.a019315
Poethig, R. S. (2010). The past, present, and future of vegetative phase change. Plant Physiol. 154 (2), 541–544. doi: 10.1104/pp.110.161620
Richardson, D. N., Simmons, M. P., Reddy, A. S. (2006). Comprehensive comparative analysis of kinesins in photosynthetic eukaryotes. BMC Genomics 7 (1), 1–37. doi: 10.1186/1471-2164-7-18
Ripoll, J. J., Bailey, L. J., Mai, Q.-A., Wu, S. L., Hon, C. T., Chapman, E. J., et al. (2015). microRNA regulation of fruit growth. Nat. Plants 1 (4), 1–9. doi: 10.1038/nplants.2015.36
Rodrigo, J. M., Zappacosta, D. C., Selva, J. P., Garbus, I., Albertini, E., Echenique, V. (2017). Apomixis frequency under stress conditions in weeping lovegrass (Eragrostis curvula). PloS One 12 (4), e0175852. doi: 10.1371/journal.pone.0175852
Ru, P., Xu, L., Ma, H., Huang, H. (2006). Plant fertility defects induced by the enhanced expression of microRNA167. Cell Res. 16 (5), 457–465.
Sakai, H., Mizuno, H., Kawahara, Y., Wakimoto, H., Ikawa, H., Kawahigashi, H., et al. (2011). Retrogenes in rice (Oryza sativa l. ssp. japonica) exhibit correlated expression with their source genes. Genome Biol. Evol. 3, 1357–1368. doi: 10.1093/gbe/evr111
Schattner, S., Schattner, J., Munder, F., Höppe, E., Walter, W. J. (2021). A tug-of-War model explains the saltatory sperm cell movement in Arabidopsis thaliana pollen tubes by kinesins with calponin homology domain. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.601282
Schmidt, A. (2020). Controlling apomixis: Shared features and distinct characteristics of gene regulation. Genes 11 (3), 329. doi: 10.3390/genes11030329
Schwab, R., Palatnik, J. F., Riester, M., Schommer, C., Schmid, M., Weigel, D. (2005). Specific effects of MicroRNAs on the plant transcriptome. Dev. Cell 8 (4), 517–527. doi: 10.1016/j.devcel.2005.01.018
Selva, J. P., Siena, L., Rodrigo, J. M., Garbus, I., Zappacosta, D., Romero, J. R., et al. (2017). Temporal and spatial expression of genes involved in DNA methylation during reproductive development of sexual and apomictic. Eragrostis curvula. Sci. Rep. 7 (1), 1–11. doi: 10.1038/s41598-017-14898-5
Selva, J. P., Zappacosta, D., Carballo, J., Rodrigo, J. M., Bellido, A., Gallo, C. A., et al. (2020). Genes modulating the increase in sexuality in the facultative diplosporous grass Eragrostis curvula under water stress conditions. Genes 11 (9), 969. doi: 10.3390/genes11090969
Smita, S., Robben, M., Deuja, A., Accerbi, M., Green, P. J., Subramanian, S., et al. (2021). Integrative analysis of gene expression and miRNAs reveal biological pathways associated with bud paradormancy and endodormancy in grapevine. Plants 10 (4), p.669. doi: 10.3390/plants10040669
Sunkar, R., Zhu, J.-K. (2004). Novel and stress-regulated MicroRNAs and other small RNAs from Arabidopsis[W]. Plant Cell 16 (8), 2001–2019. doi: 10.1105/tpc.104.022830
Tian, S., Jiang, J., Xu, G., Wang, T., Liu, Q., Chen, X., et al. (2021). Genome wide analysis of kinesin gene family in Citrullus lanatus reveals an essential role in early fruit development. BMC Plant Biol. 21 (1), 1–16. doi: 10.1186/s12870-021-02988-6
Umezu, N., Umeki, N., Mitsui, T., Kondo, K., Maruta, S. (2011). Characterization of a novel rice kinesin O12 with a calponin homology domain. J. Biochem. 149 (1), 91–101. doi: 10.1093/jb/mvq122
Underwood, C. J., Vijverberg, K., Rigola, D., Okamoto, S., Oplaat, C., den Camp, R. H. M. O., et al. (2022). A PARTHENOGENESIS allele from apomictic dandelion can induce egg cell division without fertilization in lettuce. Nat. Genet. 54 (1), 84–93. doi: 10.1038/s41588-021-00984-y
Vijverberg, K., van Dijk, P. J. (2007). Genetic linkage mapping of apomixis loci, In: Hörandl, E., Grossniklaus, U., van Dijk, P. J., Sharbel, T. F., eds. Apomixis: evolution, mechanisms and perspectives. Vienna, Austria: International Association for Plant Taxonomy, 137–158.
Voigt, P. W., Burson, B. L. (1983). “Breeding of apomictic eragrostis curvula,” in Proceedings of the XIV international grassland congress (CRC Press), 160–163.
Wang, J.-W. (2014). Regulation of flowering time by the miR156-mediated age pathway. J. Exp. Bot. 65 (17), 4723–4730. doi: 10.1093/jxb/eru246
Wei, J.-W., Huang, K., Yang, C., Kang, C.-S. (2017). Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 37 (1), 3–9. doi: 10.3892/or.2016.5236
Woodger, F. J., Millar, A., Murray, F., Jacobsen, J. V., Gubler, F. (2003). The role of GAMYB transcription factors in GA-regulated gene expression. J. Plant Growth Regul. 22 (2), 176–184. doi: 10.1007/s00344-003-0025-8
Wu, G., Poethig, R. S. (2006). Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 133 (18), 3539–3547. doi: 10.1242/dev.02521
Xing, S., Salinas, M., Garcia‐Molina, A., Höhmann, S., Berndtgen, R., Huijser, P. (2013). SPL 8 and mi R 156‐targeted SPL genes redundantly regulate A rabidopsis gynoecium differential patterning. Plant J. 75 (4), 566–577.
Zappacosta, D., Gallardo, J., Carballo, J., Meier, M., Rodrigo, J. M., Gallo, C. A., et al. (2019). A high-density linkage map of the forage grass Eragrostis curvula and localization of the diplospory locus. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00918
Zappacosta, D. C., Ochogavía, A. C., Rodrigo, J. M., Romero, J. R., Meier, M. S., Garbus, I., et al. (2014). Increased apomixis expression concurrent with genetic and epigenetic variation in a newly synthesized Eragrostis curvula polyploid. Sci. Rep. 4 (1), 1–7. doi: 10.1038/srep04423
Zheng, C., Ye, M., Sang, M., Wu, R. (2019). A regulatory network for miR156-SPL module in Arabidopsis thaliana. Int. J. Mol. Sci. 20 (24), 6166. doi: 10.3390/ijms20246166
Keywords: Eragrostis curvula, apomixis, sRNA libraries, miRNA, miRNA-mRNA interaction
Citation: Pasten MC, Carballo J, Gallardo J, Zappacosta D, Selva JP, Rodrigo JM, Echenique V and Garbus I (2022) A combined transcriptome - miRNAome approach revealed that a kinesin gene is differentially targeted by a novel miRNA in an apomictic genotype of Eragrostis curvula. Front. Plant Sci. 13:1012682. doi: 10.3389/fpls.2022.1012682
Received: 05 August 2022; Accepted: 14 September 2022;
Published: 30 September 2022.
Edited by:
Fulvio Pupilli, National Research Council (CNR), ItalyReviewed by:
Kitty Vijverberg, Naturalis Biodiversity Center, NetherlandsRoss Andrew Bicknell, The New Zealand Institute for Plant and Food Research Ltd, New Zealand
Vladimir Brukhin, Theodosius Dobzhansky Center for Genome Bioinformatics, Russia
Copyright © 2022 Pasten, Carballo, Gallardo, Zappacosta, Selva, Rodrigo, Echenique and Garbus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingrid Garbus, aWdhcmJ1c0BjcmliYS5lZHUuYXI=
†These authors have contributed equally to this work